

Abstract
Background
Vaccines that are shelf stable and easy to administer are crucial to improve vaccine access and reduce severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) transmission around the world.
Methods
In this study, we demonstrate that an oral, adenovirus-based vaccine candidate protects against SARS-CoV-2 in a Syrian hamster challenge model.
Results
Hamsters administered 2 doses of VXA-CoV2-1 showed a reduction in weight loss and lung pathology and had completely eliminated infectious virus 5 days postchallenge. Oral immunization induced antispike immunoglobulin G, and neutralizing antibodies were induced upon oral immunization with the sera, demonstrating neutralizing activity.
Conclusions
Overall, these data demonstrate the ability of oral vaccine candidate VXA-CoV2-1 to provide protection against SARS-CoV-2 disease.
Keywords: COVID, hamster, mucosal, oral vaccine, SARS-CoV-2
Vaccinating Syrian hamsters with an oral vaccine against SARS-CoV-2 protects against disease-associated symptoms of weight loss and lung pathology. Spike-specific IgG was detected in the sera and nasal wash postvaccination, and viral loads were decreased upon challenge with SARS-CoV-2.
The global pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has led to almost 4 million deaths and hundreds of millions of cases worldwide. Vaccination is the best way to prevent widespread death and illness, and fortunately several vaccines have been approved for Emergency Use Authorization (EUA). Currently, the SARS-CoV-2 vaccines authorized are all given by the intramuscular (IM) route, with some requiring −80°C or −20°C storage. This has been a major barrier for vaccine rollout during the pandemic, because not all areas have the infrastructure to support the rollout of an injectable vaccine.
Although vaccination rates are increasing and cases are decreasing in wealthy countries, many low- and middle-income countries are experiencing widespread SARS-CoV-2 transmission and an excess of deaths due to coronavirus disease 2019 without proper vaccine access. Unmitigated SARS-CoV-2 transmission puts pressure on the constantly evolving virus, resulting in the emergence of viral variants that may evade vaccine-induced immunity, requiring boosters and variant-specific vaccines. Vaccinating the globe once is difficult; vaccinating every 6 to 12 months by injection will likely be impossible. A global pandemic needs an easy-to-deliver, efficacious, shelf-stable, oral vaccine solution that can protect against SARS-CoV-2.
There are no approved oral vaccines against SARS-CoV-2, and despite that the EUA- approved IM vaccines are very efficacious against the original parental SARS-CoV-2 strain, some vaccines may not be as potent, may not block transmission, or may fail against variants that circumvent antibodies to the receptor binding domain [1]. For respiratory viruses, the mucosal surfaces of entry, such as the upper respiratory tract (URT), are the first line of defense. Local immunity at these sites of infection involves immunoglobulin (Ig) A and IgG antibodies that can block infection directly at the site of viral replication and have been shown to be cross-reactive [2, 3]. In addition, secretory IgA antibodies that actively transfer to the mucosal surface are polymeric in nature and may result in more potent neutralization against SARS-CoV-2 [4] compared with IgG antibodies. Furthermore, the mucosal immune system shares trafficking immune cells that “home” between mucosal sites [5]. Antibody-secreting plasmablasts and plasma cells stimulated in the gut can traffic to other mucosal sites including the nose, trachea, lung, mammary gland, and respiratory tract [6]; thus, targeting the gut for immunization can lead to trafficking of antibody-secreting cells to the URT for secretion of potently neutralizing antibodies. Generating a robust mucosal IgA “and” systemic IgG response is an ideal strategy to provide multipoint protection against a virus as highly transmissible as SARS-CoV-2.
Our oral vaccine platform is a replicationdefective adenovirus type5-vectored vaccine that expresses antigen along with a novel Tolllike receptor 3 agonist as an adjuvant. More than 500 human subjects have been administered these oral vaccines, which have been well tolerated and able to generate robust humoral and cellular immune responses to the expressed antigens [7–9]. We have previously published studies on the selection of our clinical candidate and the immunogenicity achieved in mice [10], but there are limits to the mouse models for SARS-CoV-2 infection because SARS-CoV-2 only binds to human angiotensin-converting enzyme 2 (ACE2) but not mouse ACE2 [11].
However, hamsters are a good model of SARS-CoV-2 infection, because they have ACE2 sequence homologous to human ACE2, are permissible for infection, and show robust viral replication. They also display moderate to severe clinical signs such as weight loss and develop strong pulmonary pathology similar to that of humans [12]. At least one of the vaccines currently under EUA also used a Syrian hamster model to demonstrate vaccine efficacy [13].
In this study, we demonstrate the ability of our SARS-CoV-2 clinical candidate vaccine VXA-COV2-1 to protect hamsters from SARS-CoV-2 challenge. We show that oral and intranasal vaccination elicits both binding and neutralizing IgG and protects against lung pathology, pulmonary viral load, and clinical disease. These data add to our preclinical evidence that oral vaccination can protect against SARS-CoV-2.
METHODS
Vaccine Constructs
VXA-CoV2-1 is an adenovirus type 5 (rAd5) vector containing full-length SARS-CoV-2 S gene under control of the cytomegalovirus promoter and full-length SARS-CoV-2N gene under control of the human beta-actin promoter. The published amino acid sequences (GenBank accession no. MN908947.3) of the SARS-CoV-2 spike (S) and the SARS-CoV-2 nucleocapsid (N) were used to create recombinant plasmids containing transgenes cloned into the E1 region of rAd5 [14], using the same vector backbone used in prior clinical trials for oral rAd tablets [7]. All vaccines were grown in the Expi293F suspension cell line (Thermo Fisher Scientific) and purified by cesium chloride density centrifugation.
Animals and Study Design
Male Syrian hamsters (Mesocricetus auratus) approximately 12–14 weeks old with a weight of 106–136 grams were sourced from Charles River Laboratory. Animal work was performed at Lovelace Biomedical, with approval from the Institutional Animal Care and Use Committee. Hamsters were singly housed in filter-topped cage systems and were supplied with a certified diet, filtered municipal water, and dietary and environmental enrichment.
Forty-eight hamsters were randomly divided into 6 groups of 8 per group and received either 1 or 2 doses of Vaxart’s vaccine candidate, VXA-CoV2-1 (Figure 1). All vaccinations were given at a dose of 1e9 IU. Oral vaccine was delivered by gavage in 300 μL phosphate-buffered saline (PBS) subsequent to delivery of 300 μL 7.5% bicarbonate buffer. Intranasal vaccination was delivered in PBS by pipette, 50 μL/nostril. The control group received PBS via oral gavage. One group received recombinant SARS-CoV-1 S protein, made in insect cells, by the IM immunization group (catalog no. NR-722, BEI). This approach should induce a nonneutralizing serum antibody response to the spike protein due to the mismatched antigen and nonhuman cell glycosylation [15], which allows us to potentially observe any vaccine-dependent enhancement of disease. All animals were challenged by intranasal inoculation of SARS-CoV-2 at approximately 1E + 05 TCID50 (50%tissue culture infective dose)/animal 8 weeks after the initial vaccination and were killed 5 days later for terminal assays.
Figure 1.

Schematic of Syrian hamster challenge study. Hamsters were immunized on weeks 0 and 4 (or just week 0 for 1 dose groups) and challenged intranasally (i.n.) with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) on week 8. VXA-COV2-1 was given at 1e9 IU per hamster (either orally or by i.n.). As a control, 5 µg of SARS-CoV protein was given to each animal in the protein group. Untreated animals were given no vaccine, but they were challenged at the same time as the vaccine groups. Serum was taken at day 0, 4 weeks, 8 weeks, and day 5 postchallenge. Lung tissue was taken at day 5 postchallenge. N = 8 per group. Created with BioRender.com.
Enzyme-Linked Immunosorbent Assay
Microtiter plates (Nunc MaxiSorp) were coated with 1 μg/mL SARS-CoV-2 S1 protein (GenScript) in carbonate buffer ([pH 9.4] Thermo Scientific) and incubated overnight at 4°C before blocking with 100 µL PBS-0.05% Tween (PBST) + 1% bovine serum albumin (BSA) for 1 hour. Serum samples were serially diluted in PBST. After 2 hours at room temperature (RT), the plates were washed 3 times with PBST, then 100 μL per well of 1:3000 goat antihamster IgG-horseradish peroxidase (HRP) (Thermo Fisher) in PBST + 1% BSA was added. After 1 hour at RT, 50 µL/well of 3,3’,5,5’-Tetramethylbenzidine (TMB) substrate (Rockland) was added after 3× wash step with PBST. After 10 minutes of development, 50 µL/well of 2 M sulfuric acid was used to stop the reaction. Optical densities (ODs) were measured at 450nm with a Spectra Max M2 microplate reader.
Severe Acute Respiratory Syndrome Coronavirus 2 Surrogate Neutralization Assay
Neutralizing antibodies were measured using a SARS-CoV-2 surrogate virus neutralization test kit (GenScript). Hamster sera diluted from 1:20 to 1:500 was incubated at a 1:1 ratio with HRP-conjugated SARS-CoV-2 RBD protein for 30 minutes at 37°C. After incubation, 100 µL was added to human ACE2 precoated plates and incubated for 15 minutes at 37°C. Plates were washed 4 times with 260 µL per well of supplied wash solution, followed by the addition of 100 μL per well of TMB. Plates were developed for 15 minutes at RT in the dark before development was stopped with 50 µL per well of supplied stop solution. The OD was measured at 450nm with a Spectra Max M2 microplate reader.
Severe Acute Respiratory Syndrome Coronavirus 2 Challenge
All animals were challenged by intranasal inoculation of 1E + 05 TCID50/animal SARS-CoV-2, isolate USA-WA1/2020 (BEI Resources) at 100 µL/nostril 8 weeks postinitial vaccination. Virus was propagated in Vero E6 cells (catalog no. N596, BEI) at the University of Texas Medical Branch, a biosafety level 3 facility. (The following reagent was obtained through BEI Resources, National Institute of Allergy and Infectious Diseases, National Institutes of Health: SARS-CoV Spike (S) Protein deltaTM, Recombinant from Baculovirus, NR-722.)
Assessment of Infectious Severe Acute Respiratory Syndrome Coronavirus 2 Load in Lung Homogenate
Lung tissue samples from euthanized hamsters infected with SARS-CoV-2 were collected, weighed, and homogenized with beads using a Tissue Lyser (QIAGEN). Each sample was serially diluted 10-fold in Dulbecco’s modified Eagle’s medium containing 2% fetal bovine serum and 1% Pen-Strep solution. VeroE6 cells that were plated in monolayers at >90% confluency, in 96-well plates, were rinsed with PBS before being inoculated with 100 µL of each sample dilution in 5 replicates. Negative control wells contained medium only. The plates were incubated at 37°C and 5% CO2 for 72 hours. Cytopathic effect was scored after fixing the cell monolayers with 10% formalin and staining with 0.5% crystal violet. The Reed and Muench method [16] was used to determine the tissue culture infectious dose 50%/mL of lung homogenate (TCID50). Infectious virus titers were expressed as TCID50/gram of tissue.
Detection of Severe Acute Respiratory Syndrome Coronavirus 2 in Lung Homogenates by Reverse-Transcription Quantitative Polymerase Chain Reaction
Lung samples were weighed and homogenized with beads using a Tissue Lyser in 1mL TRI reagent, then ribonucleic acid (RNA) was purified using the Direct-zol-96 RNA kit (Zymo Research). Copies of SARS-CoV-2N were measured by reverse-transcription quantitative polymerase chain reaction TaqMan Fast Virus 1-Step assay (Applied Biosystems). The SARS-CoV-2-specific primers and probes from the 2019-nCoV RUO Assay kit (Integrated DNA Technologies) were used: L primer, TTACAAACATTGGCCGCAAA; R primer, GCGCGACATTCCGAAGAA; probe, 6FAM-ACAATTTGCCCCCAGCGCTTCAG-BHQ-1. Reactions were carried out on a StrataGene MX3005P or BioRad CFX384 Touch instrument according to the manufacturer’s specifications. A semilogarithmic standard curve of synthesized SARS-CoV-2N gene RNA (LBRI) was obtained by plotting the cycle threshold values against the logarithm of cDNA concentration and used to calculate SARS-CoV-2N gene in copies/gram tissue.
Histopathology
The lungs were collected, examined, and weighed, with representative samples preserved for histopathology. Right lung lobes were instilled via major airway(s) with 10% neutral-buffered formalin for fixation, then trimmed, processed, paraffin embedded, sectioned at 4 µm, and stained with hematoxylin and eosin for microscopic examination.
Histopathologic examination was conducted with findings for a given tissue graded subjectively and semiquantitatively by a single pathologist on a scale of 1–5 (1 = minimal, 2 = mild, 3 = moderate, 4 = marked, 5 = severe). The Provantis v10.2.3 (Instem LSS Ltd) computer software/database was used for data acquisition, reporting, and analysis. Lung sections were evaluated for mixed cell inflammation and associated epithelial hypertrophy/hyperplasia focused on the peribronchovascular and centriacinar areas of the lungs, alveolar hemorrhage, and hypertrophy/hyperplasia in the bronchi.
Immunoglobulin G Nasal Wash MSD
V-plex SARS-CoV-2 panel 2 plates (MSD) were blocked with Blocker A solution (MSD) for 1 hour before washing and the addition of nasal wash samples at neat and 1:10 dilutions. After 2 hours at RT, plates were washed and 1 µg/mL sulfo-tagged goat antihamster IgG detection antibody (MSD, Invitrogen) was added. After 1 hour and a wash step, Gold Read Buffer B (MSD) was added and plates were analyzed using MSD QuickPlex.
Statistics
Statistical analyses were performed using GraphPad Prism v9 software. Each specific test is indicated in the figure legends. P ≤ .05 were considered significant.
RESULTS
Oral and Intranasal VXA-CoV-2-1 Reduced Weight Loss and Lung Inflammation
To assess the ability of our vaccine candidate to protect against SARS-CoV-2 challenge, hamsters were vaccinated at 4 weeks and 8 weeks before intranasal challenge with SARS-CoV-2 (Figure 1). All hamsters that were twice vaccinated with VXA-COV2-1, either orally or intranasally, were protected from SARS-CoV-2 disease. Hamsters were weighed and observed daily for weight loss. Unvaccinated control hamsters lost up to 10% total weight loss by day 5, and the twice-vaccinated hamsters showed no significant weight loss over the 5 days (Figure 2A).
Figure 2.

Vaccinated hamsters are protected from severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) associated weight loss and lung pathology. (A) Animals were monitored for weight for 5 days after challenge. Means (±standard error of the means [SEMs]) are shown for each group. (B) Lung weights on day 5 were taken and normalized by the actual animal weight to calculate a percentage of body weight. Means (±SEMs) are shown for each group, one-way analysis of variance statistical analysis was performed among the groups, with all groups compared with the no vaccine group. (C) Histology scores of lungs. (D) Histopathology of the lungs. Lung lobes were collected, trimmed, and processed routinely, paraffin embedded, sectioned at 4 µm, and stained with hematoxylin and eosin for microscopic examination.
Pulmonary infection via SARS-CoV-2 induces pulmonary inflammation, which is characterized by cellular infiltrates and edema. This in turn causes an increase in lung weights, which can be quantified as an indirect measure of pulmonary inflammation at necropsy. The lung weight as a percentage of total body weight was analyzed to quantify pulmonary inflammation in each animal (Figure 2B). Lungs from unvaccinated animals and those that received a single IM injection with protein or a single oral vaccination were approximately 1.3% of the total body weight (Figure 2B). In contrast, animals that received the vaccine twice by the oral or intranasal routes had significantly lower lung weights (Figure 2B). This indicates that administration to the oral or respiratory mucosa has a significant protective effect on lung inflammation. In addition, in the 1-dose oral group, 37.5% (3 of 8) of animals maintained their body weight and had no increase in lung weight, suggesting partial protection (Figure 2A and B).
Histopathological analysis of lung samples (Figure 2C and D) demonstrated a reduction in the severity of the inflammatory response in the lungs in the groups given 2 doses of VXA-CoV2-1, either given orally or via the intranasal route. Two oral doses of VXA-CoV2-1 also prevented epithelial hypertrophy/hyperplasia in the peribronchovascular and centriacinar areas of the lungs (Figure 2C). Although 2 IM doses of SARS-CoV-1 protein moderately protected animals from weight loss and increased lung inflammation after challenge, histology scores showed increased epithelial hypertrophy/hyperplasia in these animals (Figure 2C) and alveolar hemorrhage similar to animals that received a single dose of VXA-CoV2-1. No evidence of vaccine enhanced disease was seen in any group, including the 2-dose protein group. In summary, these results demonstrate that the mucosal vaccine administered intranasally or orally is protective against SARS-CoV-2 homologous challenge in hamsters.
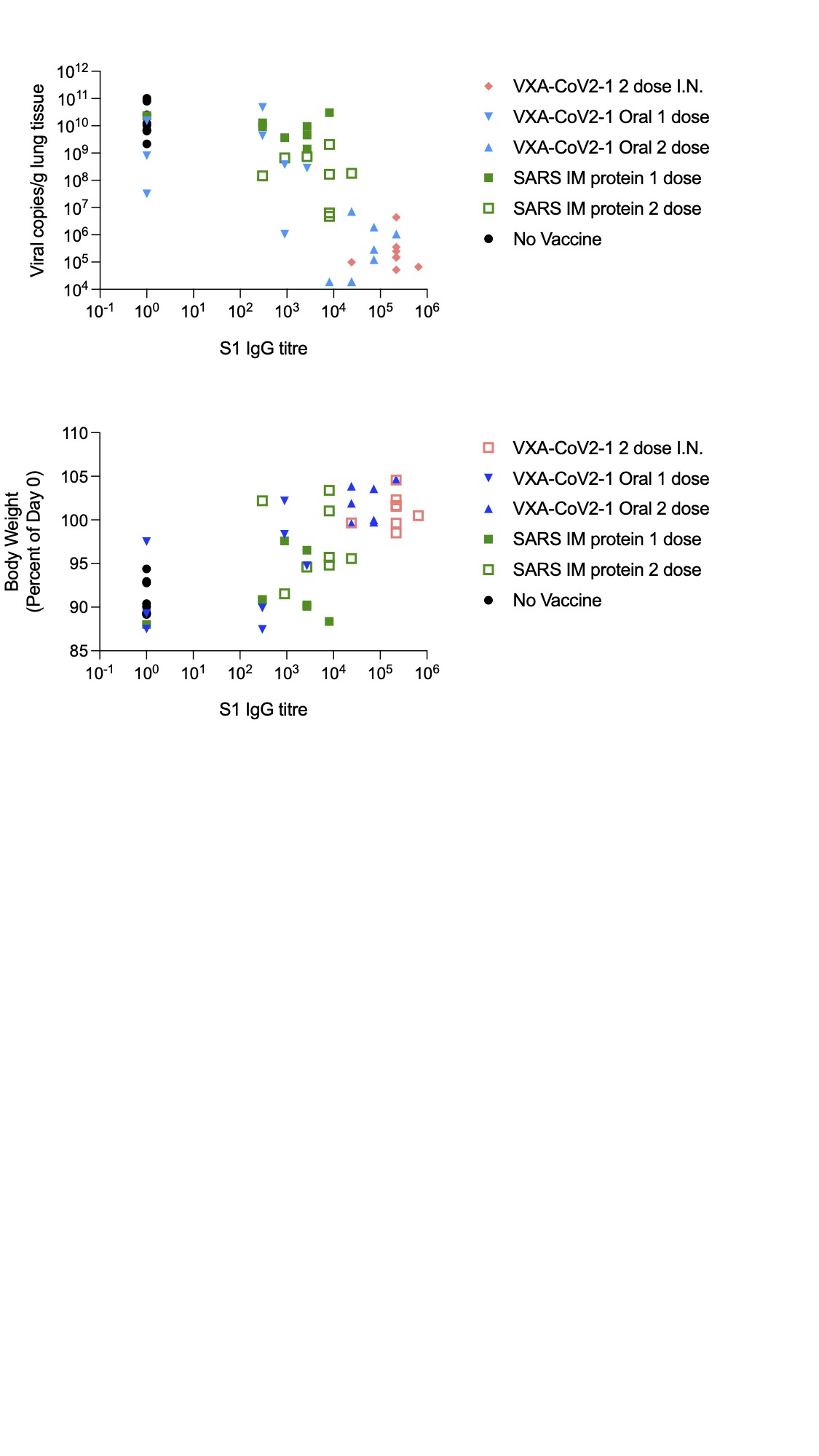
One of the goals of a vaccine is not only to reduce disease but also to reduce viral load. Copies of SARS-CoV-2 in the lungs were measured by quantitative polymerase chain reaction (qPCR) to determine whether oral vaccination decreased viral load at day 5 postchallenge. A reduction of >4 logs in viral load was observed in animals immunized twice by the oral or intranasal routes (Figure 3A). Due to the nature of qPCR, viral copies that are noninfectious can be detected. To test whether the detectable copies of the virus are infectious, a TCID50 assay was run, which measures the amount of live, infectious virus in a sample. Although more variable, similar trends across the groups were detected in infectious viral loads as with the molecular viral titers (Figure 3B). It is striking that animals that were immunized twice by the oral or intranasal routes were devoid of detectable infectious virus. Four of the 8 hamsters that received only 1 oral immunization also did not have any detectable infectious virus at day 5 postinfection.
Figure 3.

Two-dose oral or intranasal vaccination generates high titer antibodies that are neutralizing. (A) S-specific immunoglobulin G in the serum at day 0, 28, and 54 postvaccination and 5 days postchallenge was measured by enzyme-linked immunosorbent assay (ELISA) and graphed over time. All vaccinated groups generated antibodies. (B) The ability for the antibodies in serum taken at week 8 to neutralize virus was measured in a surrogate viral neutralizing assay (SVNT). Antibodies in the oral and intranasal groups were able to block the binding of ACE-2 to S1 in the competitive ELISA SVNT. One-way analysis of variance statistical analysis was performed among the groups with all groups compared with the no vaccine group.
Oral and Intranasal VXA-CoV-2-1 Induced Robust Immunoglobulin G and Neutralizing Antibody Responses
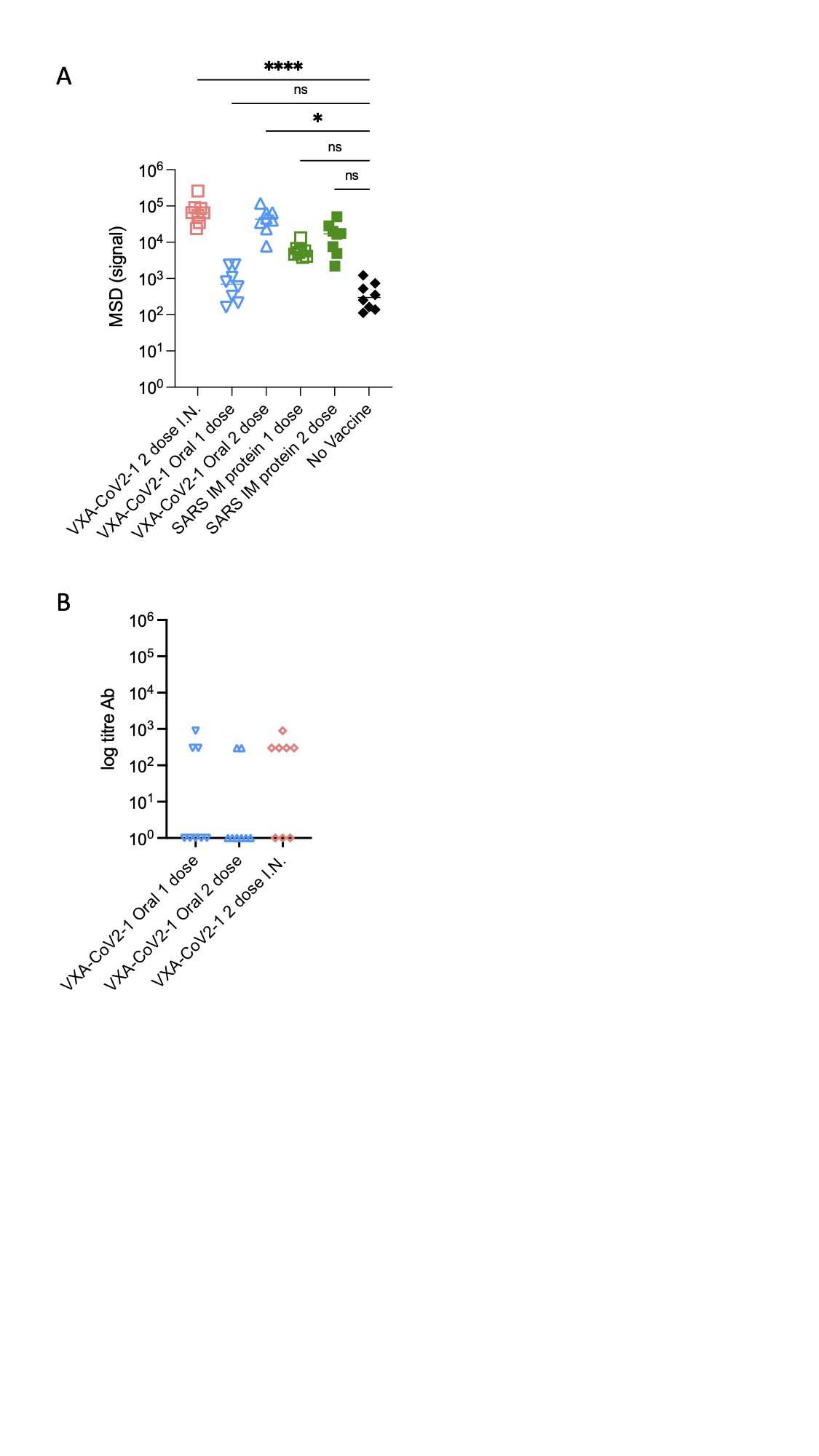
To determine whether VXA-CoV2-1 induced an adaptive immune response that could correlate with disease protection, serum and nasal washes were taken from the hamsters at 4 and 8 weeks postvaccination and 5 days postchallenge. Immunoglobulin G specific for the S1 region of the SARS-CoV-2 spike protein was measured via enzyme-linked immunosorbent assay. Hamsters given VXA-COV2-1 intranasally or orally demonstrated anti-S1 IgG titers in serum (Figure 4A), with the titers increasing after boost vaccination. The SARS-CoV-2 S-specific IgG measured in nasal washes was significantly increased compared with the unvaccinated control and the intramuscular protein group (Supplementary Figure 1A). In addition, 2 immunizations with VXA-CoV2-1 by the oral or intranasal route induced neutralizing antibodies in the serum, as demonstrated by a surrogate viral neutralizing assay where neutralizing antibodies blocked the binding of the SARS-CoV-2 receptor binding domain to ACE2 (Figure 4B). The animals injected intramuscularly with SARS-CoV-1 protein showed modest antibody titers; however, only 4 of the 8 animals immunized twice with protein had neutralizing antibodies. In the single oral VAX-COV2-1 group, 3 animals had modest binding antibody responses at 8 weeks postimmunization that increased to similar levels as the 2-dose group after viral challenge. These were the same animals that also showed partial protection with a reduction in lung inflammation and weight loss. The hamsters that received 1 dose but did not show protection had no persistence of antibodies at 8 weeks and substantially reduced viral titers after challenge, demonstrating that protection and antibody levels were linked (Supplementary Figure 2). It is worth noting that although our VXA-CoV-2-1 contains transgenes for both S and N, the promoter for the nucleocapsid gene is the human β-actin, which may not act as well in rodent cells. Consistent with this, the N-specific antibody responses were weak in hamsters (Supplementary Figure 1B).
Figure 4.

Oral and intranasal vaccination decreases virus in the lungs. (A) Lung severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) titers as measured by quantitative reverse-transcription polymerase chain reaction (PCR) on day 5 postchallenge. Lung tissues were homogenized, and RNA was extracted and run on a quantitative PCR to detect SARS-CoV-2. Viral titers are substantially reduced in the 2-dose oral and intranasal groups. (B) The 50%tissue culture infective dose (TCID-50) per gram/lung tissue. The lung tissue extract was added to Vero cells in serial dilutions, and wells with cytopathic effect were counted as a measure of infectious dose. One-way analysis of variance statistical analysis was performed among the groups with all groups compared with the no vaccine group.
DISCUSSION
This study shows hamsters that received 2 doses of VXA-CoV2-1 by either intranasal or oral route were protected from homologous strain challenge. Animals receiving 2 doses of VXA-CoV2-1 showed minimal weight loss and full recovery by day 5 postinfection. These animals had minimal to no lung weight gain, minimal pulmonary pathology, and highly significant reduced viral titers in the lungs 5 days postinfection. In particular, oral vaccination with VXA-CoV2-1 reduced the viral titers in the lungs by 4–5 orders of magnitude, with no infectious virus detected. VXA-COV2-1 administered mucosally induced systemic antibody responses, as evidenced by serum IgG and neutralizing antibodies in the serum of vaccinated animals. Two doses of VXA-COV2-1 were required for eliciting neutralizing antibody responses; however, some protection, in half of the animals, was induced after a single immunization. Thirty-seven percent of animals in the single oral immunization arm also showed moderate levels of protection as assessed by weight loss and lung histopathology; the partially protected animals elicited higher antibody titers than the nonprotected hamsters.
A key limitation to this study is that oral tablets cannot be given to hamsters, and gavage as a delivery method in animals is a difficult technique to deliver accurate volumes [17]; therefore, intranasal delivery was used as a mucosal positive control in this experiment. The oral vaccine arm immune responses and lung pathology after challenge were similar to intranasal delivery of the same vaccine construct, which suggests that oral immunization could be just as potent as respiratory-based immunization. Intranasal delivery, although lacking needles, would still require using a device used by qualified healthcare providers, sterile fill finish, and cold chain; thus, this does not really solve many of the logistical issues associated with vaccinating during a pandemic. In contrast, a tablet vaccine handed out at room temperature (or potentially shipped by mail) solves the key logistical issues involved with a pandemic response.
There have been multiple other studies immunizing small animals with vaccines against SARS-CoV-2, including using adenoviral vectors intranasally [13, 18–20]. The SARS-CoV-2 challenge in Syrian hamsters has been used as proof-of-concept for other vaccination studies because of the similar clinical manifestations in the lungs of hamsters compared with human lungs exposed to SARS-CoV-2 [12]. These studies have shown protection against challenge [13], which concurs with our data, and some have shown decreased shedding with intranasal delivery [18–20]. Although viral shedding over time was not measured in this study, other studies have correlated lack of weight loss and decreased lung pathology with a reduction in viral titers in the airway [12], and we would expect these to decrease in both the lungs and the nasal cavity.
Despite the plethora of SARS-CoV-2 literature, studies on mucosal immunity have been lacking. The SARS-CoV-2-specific IgA and IgG have been found in mucosal fluids from convalescent patients [21] and dominates the early immune response [22], and SARS-CoV-2- specific secretory IgA has been found in human milk [23], concurring with what we know about respiratory infections; however, all the vaccines approved for EUA are injected and unlikely to induce a robust mucosal response [24]. Vaxart’s oral platform is designed to elicit mucosal IgG and IgA as well as α4β7 homing B and T cells [7, 8, 25]. We demonstrated levels of IgG in the nasal washes postvaccination, but a key limitation to this study is the lack of reagents that allow immune measurements of hamster IgA [26]; therefore, the contribution of mucosal IgA elicited after VXA-CoV2-1 administration to protection in hamsters is unknown. VXA-CoV2-1 is currently in phase 1 trials (https://clinicaltrials.gov/show/NCT04563702), and an increase in SARS-CoV-2-specific nasal and saliva IgA was seen in our clinical trial subjects (data not shown), suggesting that we should expect an increase in IgA in the vaccinated hamsters as the same construct elicited IgA in human subjects. Efforts are ongoing to create reagents that will allow us to address this question directly. As a vaccine strategy, local IgA could play important roles that are different than that induced by an injected vaccine. We expect that mucosal IgA may reduce the spread of the virus in the respiratory tract, creating a lower ability to shed virus. Indeed, intranasal vaccination has been shown to decrease transmission in cohoused animals [20], compared with IM inoculation of the same vaccine. We recently showed that oral vaccination with this adenovirus-based vaccine candidate induced mucosal IgA and reduced transmission of SARS-CoV-2 in hamsters [27]. Creating respiratory-specific IgA has consistently been observed with the platform in different animal models including humans [28, 29].
One concern is that incomplete protection afforded by injected vaccines may allow for replication in the nasal passages, and these could lead to transmission of virus even from subjects that are asymptomatic; this has been seen with the delta variant, which is more transmissible. There are already reports of increased breakthrough infections in highly vaccinated nations such as the Seychelles, and in the United States and United Kingdom cases are once again rising. Mucosal IgA is more likely to hinder transmission; a vaccine that could decrease asymptomatic transmission could alleviate this issue and support societal and economic openings.
CONCLUSIONS
In conclusion, we describe an oral vaccine candidate that elicits anti-S antibodies and provides protection from SARS-CoV-2 pathology in a Syrian hamster challenge model. Studies to further investigate transmission, viral shedding, and mucosal IgA are underway. Clinical trials with the same vaccine candidate are currently in phase I trials; an oral vaccine that can be easily administered could provide protection to many countries that are struggling with vaccine administration.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
{kind=link}
{kind=link}
Notes
Acknowledgments. We thank Becca Flitter, Anne Moore, and Stephanie Langel for helpful comments and editing suggestions for the manuscript.
Author contributions. A. D. W. supervised the study; S. M. B. performed the histopathology; A. E. K. performed the virological analysis; N. P. and E. G. D. created the vaccine material; S. N. Tu designed the study; S. J., C. I. M., and S. N. Te performed immunological analysis. S. J. wrote the manuscript.
Potential conflicts of interest. S. J., C. I. M., N. P., E. G. D., S. N. Te., and S. N. Tu. were employees of Vaxart during the study and received stock options and/or compensation as part of their employment. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1. Wang P, Nair MS, Liu L, et al. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature 2021; 593:130–5. [DOI] [PubMed] [Google Scholar]
- 2. Seibert CW, Rahmat S, Krause JC, et al. Recombinant IgA is sufficient to prevent influenza virus transmission in guinea pigs. J Virol 2013; 87:7793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lowen AC, Steel J, Mubareka S, Carnero E, García-Sastre A, Palese P. Blocking interhost transmission of influenza virus by vaccination in the guinea pig model. J Virol 2009; 83:2803–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang Z, Lorenzi JCC, Muecksch F, et al. Enhanced SARS-CoV-2 neutralization by dimeric IgA. Sci Transl Med 2021; 13:eabf1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Farstad IN, Halstensen TS, Lazarovits AI, Norstein J, Fausa O, Brandtzaeg P. Human intestinal B-cell blasts and plasma cells express the mucosal homing receptor integrin alpha 4 beta 7. Scand J Immunol 1995; 42:662–72. [DOI] [PubMed] [Google Scholar]
- 6. Toapanta FR, Simon JK, Barry EM, et al. Gut-homing conventional plasmablasts and CD27(-) plasmablasts elicited after a short time of exposure to an oral live-attenuated shigella vaccine candidate in humans. Front Immunol 2014; 5:374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim L, Liebowitz D, Lin K, et al. Safety and immunogenicity of an oral tablet norovirus vaccine, a phase I randomized, placebo-controlled trial. JCI Insight 2018; 3:e121077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liebowitz D, Lindbloom JD, Brandl JR, Garg SJ, Tucker SN. High titre neutralising antibodies to influenza after oral tablet immunisation: a phase 1, randomised, placebo-controlled trial. Lancet Infect Dis 2015; 15:1041–8. [DOI] [PubMed] [Google Scholar]
- 9. Peters W, Brandl JR, Lindbloom JD, et al. Oral administration of an adenovirus vector encoding both an avian influenza A hemagglutinin and a TLR3 ligand induces antigen specific granzyme B and IFN-γ T cell responses in humans. Vaccine 2013; 31:1752–8. [DOI] [PubMed] [Google Scholar]
- 10. Moore A, Dora E, Peinovich N, et al. Pre-clinical studies of a recombinant adenoviral mucosal vaccine to prevent SARS-CoV-2 infection [preprint]. bioRxiv 2021. [Google Scholar]
- 11. Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol 2020; 5:562–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Imai M, Iwatsuki-Horimoto K, Hatta M, et al. Syrian hamsters as a small animal model for SARS-CoV-2 infection and countermeasure development. Proc Natl Acad Sci U S A 2020; 117:16587–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tostanoski LH, Wegmann F, Martinot AJ, et al. Ad26 vaccine protects against SARS-CoV-2 severe clinical disease in hamsters. Nat Med 2020; 26:1694–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A 1998; 95:2509–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harrison RL, Jarvis DL. Protein N-glycosylation in the baculovirus-insect cell expression system and engineering of insect cells to produce “mammalianized” recombinant glycoproteins. Adv Virus Res 2006; 68:159–91. [DOI] [PubMed] [Google Scholar]
- 16. Reed LJ, Muench H. A simple method of estimating fifty percent endpoints. Am J Epidemiol 1938; 27:493–7. [Google Scholar]
- 17. Turner PV, Brabb T, Pekow C, Vasbinder MA. Administration of substances to laboratory animals: routes of administration and factors to consider. J Am Assoc Lab Anim Sci 2011; 50:600–13. [PMC free article] [PubMed] [Google Scholar]
- 18. Bricker TL, Darling TL, Hassan AO, et al. A single intranasal or intramuscular immunization with chimpanzee adenovirus vectored SARS-CoV-2 vaccine protects against pneumonia in hamsters. Cell Rep 2021; 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marsh GA, McAuley AJ, Au GG, et al. ChAdOx1 nCoV-19 (AZD1222) vaccine candidate significantly reduces SARS-CoV-2 shedding in ferrets. NPJ Vaccines 2021; 6:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. van Doremalen N, Purushotham J, Schulz J, et al. Intranasal ChAdOx1 nCoV-19/AZD1222 vaccination reduces shedding of SARS-CoV-2 D614G in rhesus macaques. Sci Transl Med 2021; 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Isho B, Abe KT, Zuo M, et al. Persistence of serum and saliva antibody responses to SARS-CoV-2 spike antigens in COVID-19 patients. Sci Immunol 2020; 5:eabe5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sterlin D, Mathian A, Miyara M, et al. IgA dominates the early neutralizing antibody response to SARS-CoV-2. Sci Transl Med 2021; 13:eabd2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fox A, Marino J, Amanat F, et al. Robust and specific secretory IgA against SARS-CoV-2 detected in human milk. iScience 2020; 23:101735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Boyaka PN. Inducing mucosal IgA: a challenge for vaccine adjuvants and delivery systems. J Immunol 2017; 199:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liebowitz D, Gottlieb K, Kolhatkar NS, et al. Efficacy, immunogenicity, and safety of an oral influenza vaccine: a placebo-controlled and active-controlled phase 2 human challenge study. Lancet Infect Dis 2020; 20:435–44. [DOI] [PubMed] [Google Scholar]
- 26. Warner BM, Safronetz D, Kobinger GP. Syrian hamsters as a small animal model for emerging infectious diseases: advances in immunologic methods. Adv Exp Med Biol 2017; 972:87–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Langel SN, Johnson S, Martinez CI, et al. Oral and intranasal Ad5 SARS-CoV-2 vaccines decrease disease and viral transmission in a golden hamster model. bioRxiv 2021: 10.03.462919. [Google Scholar]
- 28. Kim L, Martinez CJ, Hodgson KA, et al. Systemic and mucosal immune responses following oral adenoviral delivery of influenza vaccine to the human intestine by radio controlled capsule. Sci Rep 2016; 6:37295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Joyce C, Scallan CD, Mateo R, Belshe RB, Tucker SN, Moore AC. Orally administered adenoviral-based vaccine induces respiratory mucosal memory and protection against RSV infection in cotton rats. Vaccine 2018; 36:4265–77. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.