


Abstract
SCoV2-MD (www.scov2-md.org) is a new online resource that systematically organizes atomistic simulations of the SARS-CoV-2 proteome. The database includes simulations produced by leading groups using molecular dynamics (MD) methods to investigate the structure-dynamics-function relationships of viral proteins. SCoV2-MD cross-references the molecular data with the pandemic evolution by tracking all available variants sequenced during the pandemic and deposited in the GISAID resource. SCoV2-MD enables the interactive analysis of the deposited trajectories through a web interface, which enables users to search by viral protein, isolate, phylogenetic attributes, or specific point mutation. Each mutation can then be analyzed interactively combining static (e.g. a variety of amino acid substitution penalties) and dynamic (time-dependent data derived from the dynamics of the local geometry) scores. Dynamic scores can be computed on the basis of nine non-covalent interaction types, including steric properties, solvent accessibility, hydrogen bonding, and other types of chemical interactions. Where available, experimental data such as antibody escape and change in binding affinities from deep mutational scanning experiments are also made available. All metrics can be combined to build predefined or custom scores to interrogate the impact of evolving variants on protein structure and function.
Lay Summary
SCoV2-MD is a new online resource that systematically organizes atomistic simulations of the SARS-CoV-2 proteome. The database includes simulations produced by leading groups using molecular dynamics (MD) methods to investigate the structure-dynamics-function relationships of viral proteins. SCoV2-MD cross-references the molecular data with the pandemic evolution by tracking all available variants sequenced during the pandemic and deposited in the GISAID resource. SCoV2-MD enables the interactive analysis of the deposited trajectories through a web interface, which enables users to search by viral protein, isolate, phylogenetic attributes, or specific point mutation. Each mutation can then be analyzed interactively combining static (e.g. a variety of amino acid substitution penalties) and dynamic (time-dependent data derived from the dynamics of the local geometry) scores.
Graphical Abstract
Graphical Abstract.
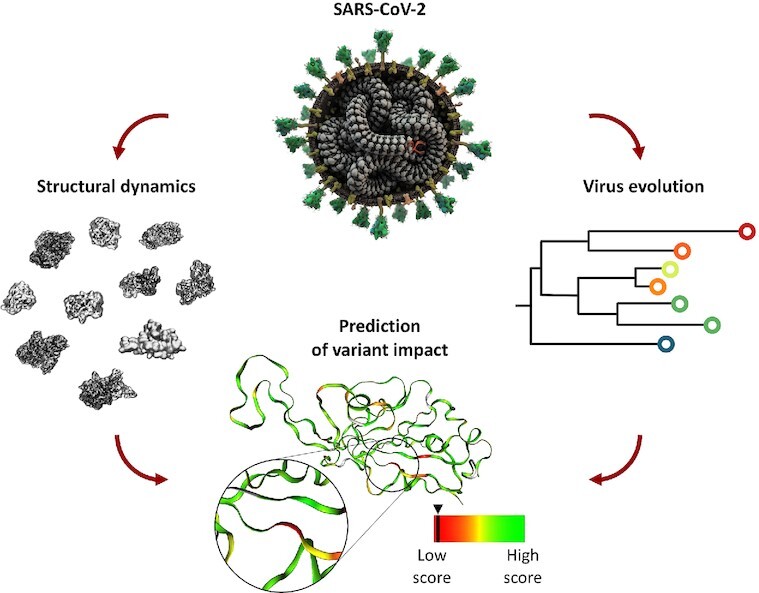
SCoV2-MD integrates structural dynamics of SARS-CoV-2 proteins with mutation events from virus evolution. Static and dynamic properties of each mutation can be combined to obtain predictions of its impact on viral proteins.
INTRODUCTION
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the causative agent of the coronavirus disease 2019 (COVID-19), which already accounts for >4.2 million deaths globally, as of 10 August 2021 (WHO, Coronavirus (COVID-19) Dashboard, covid19.who.int). The diffusion of the COVID-19 pandemic has produced emerging variants (1), which have been tracked through massive sequencing efforts at an unprecedented rate soon surpassing that of any other pathogen and phylogenetic analysis (2–4). Thus, as of July 2021, almost 2 million full genomes are available via the Global Initiative on Sharing All Influenza Data (GISAID), one of the main pandemic genome databases (3,5).
SARS-CoV-2 is a single-stranded RNA beta-coronavirus enveloped by an outer membrane and expressing 16 non-structural, 9 accessory and 4 structural proteins (Figure 1). While the spike, membrane, and envelope structural proteins are embedded in the membrane and involved in cell recognition and entry, one structural protein, the nucleocapsid, interacts inside the membrane with viral RNA to form a ribonucleoprotein complex that works as a scaffold for genome replication and virion assembly. The four structural proteins make up approximately one third of the viral genome (6). The remaining two-thirds of the viral genome encodes for the non-structural proteins (nsp) 1 to 16 (Figure 1). Some nsps are critical enzymes for virus replication such as proteases (nsp3, nsp5), RNA-dependent RNA polymerases (consisting of nsp7, two copies of nsp8, and nsp12), the RNA helicase (nsp13), and the proofreading exonuclease (nsp14) (7).
Figure 1.

Structure-based overview of the SARS-CoV-2 proteome and protein-based entry point to the database. (A) Three-dimensional model of the virion, displaying the structural proteins’ assembly; (B) Three-dimensional models of the available nsp proteins. The diagrams provide a unified overview of the available viral structures and provide one of the entry points for browsing the proteome. Selecting the proteins leads to a list of the related simulation data. (C) Coverage of SARS-CoV-2 proteome plotting experimentally available structures (pink) and simulated proteins in SCoV2-MD (green). Nsp14 (grey) was simulated based on a theoretical model using a structure of SARS-CoV-1 nsp14 (PDB ID 5C8S) as template.
Unveiling the structural basis of SARS-CoV-2 infection has been a key priority since the emergence of the COVID-19 disease. In the wake of the increased availability of structural information of SARS-CoV-2 proteins, numerous groups have tackled the study of SARS-CoV-2 proteins using molecular dynamics (MD) simulations, often after Herculean modeling and computational efforts, with the goal of supporting pandemic response efforts (8–10). Obtained MD data are highly relevant to understand the functional dynamics of the viral proteome, which cannot often be deduced from the static structure that has been experimentally solved. In addition, it can help rationalize the structural/functional impact of sequence variability in the viral proteome. This is particularly useful when the relationship between mutation location and activity is not obvious (e.g. the mutation is distant from the protein's active center).
However, while computational scientists are urgently aware of the need to share the resulting data (11), these are usually hosted at disparate sites, hardly discoverable, and not amenable to systematic analysis. In practice, this limits the ability of computational structural biologists to reuse these trajectories in large-scale efforts, e.g. for dynamic docking (12,13), discovering transient pockets (14), or associating them with phenotypes (15).
Here, we present SCoV2-MD (www.scov2-md.org), a cross-disciplinary database developed to investigate diverse questions on the interplay between the structural biology of the viral 3D proteome, its dynamics, and viral variants’ phenotypes. The platform focuses on the dynamics of protein non-covalent contacts, supporting the interpretation of allostery, the exploration of individual subunits, interfaces, and protein-ligand contacts, and the mapping of external information. An important asset of SCoV2-MD is that it provides tools to interrogate the impact of variant substitutions using a combination of static and time-resolved structural descriptors.
So far, numerous resources have been dedicated to track, monitor, and classify the phylogenetic diversity of SARS-CoV-2, such as GISAID (16), ViruSurf (17), or PANGO lineage (18). Other efforts aimed at predicting potential functional effects of SARS-CoV-2 variants based on evolutionary considerations (19), static structures of proteins and complexes, or experimental data (e.g. MutFunc (20), COV3D (21) and COVID-3D (22)). Our database builds upon previous approaches, extending them through the integration of time-resolved MD data with available information on variants to improve the prediction of their potential functional impact.
MATERIALS AND METHODS
Data source overview
The SCoV2-MD platform includes, at the time of manuscript preparation, simulations of >250 different systems, covering all SARS-CoV-2 proteins with known structure. A large part of the simulations is collated from public databases, mainly BioExcel-CV19 (https://bioexcel-cv19.bsc.es/), COVID-19 Molecular Structure and Therapeutics Hub (https://covid.molssi.org/), the CHARMM-GUI simulation archive (23), and the Exscalate4Cov project (https://www.exscalate4cov.eu/). Additionally, we generated in-house simulations to achieve complete coverage of all SARS-CoV-2 proteins with known structures (Supplementary Note S1 and Supplementary Table S1). Individual researchers can contribute their simulation data upon request. The whole dataset is accessible for free and without registration at www.scov2-md.org.
Database schema and infrastructure
The data model of the database (Supplementary Figure S1) is based on five main entities, namely: protein objects, identified by their sequence and their relationship with UniprotKB entries; final protein objects, representing the viral proteins after the transcribed poly-proteins are cleaved by its proteinases; model objects, describing the three-dimensional structures identified by the Protein Data Bank (PDB, rcsb.org) (24) identifier; dynamics objects representing the MD simulations; and dynamics components with details of the molecules in the simulated systems. The database integrates experimental data from GISAID (16) and Mutfunc: SARS-CoV-2 (20).
Similar to the GPCRmd platform (25), the Workbench page of SCoV2-MD builds on a WebGL-based structure viewer, NGL version 2.0.0, (26,27) with the MDsrv 0.3.5 (28) backend, which allows efficient streaming and sharing of trajectories online. Intuitive selection capabilities enable the creation of various 3D representations using the NGL selection language (27). The Workbench integrates annotation data from UniprotKB (29) and variant data from GISAID (16). UniprotKB data is used to extract domains and annotations of the protein represented and map them to the structure, while GISAID (16) data is referenced to display and cross-reference known protein variants on the structure. The SCoV2-MD database and web interface are based upon the Django Web Framework (v.1.9), PostgreSQL (v.9), Python (v.3.4) and JavaScript libraries jQuery 1.9, jQuery UI 1.11.2.
Variant impact scoring
The database's web portal provides the Variant Impact toolkit for interactively referencing the MD simulations with SARS-CoV-2 sequences obtained from GISAID (16), which can be found in the Workbench page. Each of the variants is annotated with the corresponding static (mutation-dependent) and time-dependent (computed on the basis of the simulation dynamics) descriptors of their impact on multiple aspects of the protein's structure and dynamics and the viral function (Figure 4 and Supplementary Note S2). The descriptors are further combined in an impact score, defined as a weighted sum of descriptors:
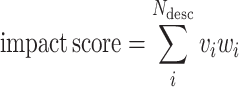 |
where Ndesc is the number of descriptors, vi the value of descriptor i, and wi the corresponding weight. Users may assign either predefined (see next section) or custom weight combinations to the descriptors to reflect various aspects of the structural impact of the variant. The obtained score is presented together with a q value, showing its normalized rank in the distribution of impact scores of all the variant-associated substitutions occurring in residues modeled in the simulation considered (e.g. in a simulation of the receptor-binding domain (RBD) of the spike protein, an histogram of impact scores is built on the basis of the amino-acid substitutions associated to known variants located in the RBD). In other words, q = 0, 0.5 and 1, respectively mean that the selected amino acid variant achieves the minimum, median and maximum effect score with respect to the other variants observed in the sequence of the simulated protein.
Figure 4.

Prediction of variant impact. (A) Sequence viewer with color-coded impact score across the protein sequence and information on reported variants. The sequence viewer is interactive in two ways: (i) the color-coded scale updates on the fly with the defined impact score, and (ii) selected residues are automatically shown in (B) the 3D viewer of the structural dynamics. (C) Variant impact score. The user can combine over 30 different descriptors into a user-defined score. We also provide default scores, such as RMSD, contacts, or scores fitted to experimental data. (D) The impact score for a specific variant is highlighted within the score distribution for the entire protein. A significant shift of the variant score to the right or left from the distribution peak reflects a variant with a high propensity to disturb protein function based on the selected descriptor combination.
Model-based predictions of variant impact for the spike protein's receptor-binding domain (RBD)
We developed simple predictive models able to qualitatively estimate the impact of each variant, in terms of (a) the change in binding affinity between SARS-CoV-2′s spike RBD and the angiotensin-converting enzyme 2 (ACE2) receptor (30); (b) the change in expression of the RBD on yeast cells (30) and (c) the potential for antibody evasion (31). The three models are based on regularized regression models (LASSO) (Supplementary Note S3, Supplementary Table S2) trained on 23 per-variant covariates, used as predictors, computed based on the 12 MD trajectories containing the RBD available at the time of writing. Each of the models was trained to fit the corresponding quantity, measured experimentally per-variant in deep scanning mutagenesis experiments. The three pre-computed models can be enabled via the web interface, in the Variant Impact section of the Workbench page, with buttons that load the corresponding sets of coefficients in the ‘weight’ sliders.
RESULTS & DISCUSSION
Tracking of structural virus evolution
A key aspect in understanding the diversity and impact of SARS-CoV-2 is monitoring the emergence of variants. Since the outbreak in 2019, many viral mutations have occurred. Some of them reach high regional frequencies with the ability to rapidly spread worldwide and potentially evade immunization and antibody treatment (e.g. B.1.1.7 (Alpha), B.1.351 (Beta), B.1.617.2 (Delta) and P.1 (Gamma) variants). Our database provides a visualization tool for viral phylogenetic data from the worldwide collaborative effort hosted at GISAID (16). This tool allows viral infections to be tracked by region, gender, age, etc. as well as following the emergence of mutations as a function of time (Figure 2). Each small circle is interactive and represents one sequenced viral genome. When clicking on a viral genome that includes novel variants/mutations, the user can directly investigate the variant location on the 3D structure of implicated viral proteins as well as stream time-resolved dynamics on the fly (see next section).
Figure 2.

Phylogenetic tree of SARS-CoV-2 viral evolution. Sequenced samples (isolates) are mapped onto the tree. Each interactive circle represents one sequenced viral genome and is linked to the simulations of the proteins mutated with respect to the reference sequence. The rectangular inset (left) tags a subset of isolates with some of the available descriptors (regions, sex, age and mutations).
Structural dynamics of the viral 3D proteome
Visualize and stream viral proteins’ dynamics
The database of time-resolved dynamics of SARS-CoV-2 currently consists of over 250 simulations covering the entire viral 3D proteome (i.e. experimentally solved 3D structures) with at least one simulation entry for each protein (Figure 3A). Data entries have been either simulated by us or collected from public resources. The user can intuitively select a simulation of interest from an interactive graphical representation of the SARS-CoV-2 virus (Figure 3B) or from a dedicated search tool (Figure 3C). Once a simulation has been selected, one can easily view (Figure 3D) and modify its graphical representation using either the quick or customized selection (Figure 3E). In addition, we implemented the option to highlight domains relevant for protein function (e.g. binding motif of the spike protein to the human ACE2 host cell receptor) which have been retrieved from the Uniprot database (Figure 3E). The most important merit of the SCoV2-MD resource is that viral proteins are not static, instead one can stream the time-resolved dynamics at atomistic resolution on-the-fly using the simulation viewer (Figure 3D).
Figure 3.

Structural dynamics of the SARS-CoV-2 proteome. (A) SCoV2-MD is a database of time-resolved dynamics for SARS-CoV-2 proteins. (B) The user can intuitively select a simulation of interest from an interactive graphical representation of the SARS-CoV-2 virus, or (C) use our dedicated search tool. (D) The viewer module enables interactive visualization and streaming of the MD simulations. (E) For that, we provide a selection panel including quick and custom selection. (F) Our platform includes interactive analysis tools such as an interaction network displaying intermolecular and intramolecular contacts. (G) The user can also check common structural stability metrics such as RMSD and RMSF.
General analysis of the wild-type (WT) simulation
SCoV2-MD provides analysis tools that allow for an overall understanding of the structural dynamics for a specific protein (Supplementary Note S4). This includes intramolecular interaction networks including hydrogen bonds, π-stacking and T-stacking, among others, which can be shown for the current frame or as an average over the entire trajectory. In the average mode, line thickness indicates the contact strength, which enables quick identification of relevant contacts. For instance, one can easily identify the non-covalent contacts (electrostatics, dispersion effects, etc.) corresponding to π-cation interactions in the binding interface between the RBD from the spike protein and the ACE2 host cell receptor (Figure 3F). Moreover, a RMSD (root mean squared deviation) plot shows the evolution of the viral protein along the trajectory with respect to the initial structure (Figure 3G). Furthermore, a RMSF (root mean square fluctuation) plot provides a quick overview of stable and highly flexible regions in the protein (Figure 3G).
Variant descriptors and phenotype analysis tool
An important goal of SCoV2-MD is the ability to predict the impact of variant substitution on the viral proteome based on static and time-resolved descriptors via an ‘impact score’ (Figure 4). The database integrates over 30 static and time-dependent descriptors (Supplementary Note S2). Static descriptors reflect substitution (e.g. BLOSUM, charge differences, and so on), conservation (frequency, SIFT score (32), etc.), structural impacts (post-translational modification, surface accessibility, etc.) and, if available, experimental observations (e.g. antibody escape, binding affinity, expression changes). Conservation, structural and experimental descriptors are collected from the Mutfunc database (20). Time-dependent descriptors are extracted from atomistic simulations and include RMSF and a large variety of non-covalent contact types (such as Van der Waals, hydrogen bonding, etc.) among others. They are computed on the basis of the MDtraj (33) and GetContacts libraries (34). The list of descriptors provides the user with a rich repertoire to score and interrogate variant substitutions on-the-fly.
Search for variant substitutions with impact on protein function
A basic search for critical regions impacted by variant substitutions can be as follows. Regions of high structural stability are expected to be crucial for the overall function of the viral protein. Unfavorable variant substitutions (e.g. Cys to Arg) in these regions can significantly disturb protein stability. To detect a combination of such events in the protein, the user needs to ‘turn on’ RMSF in addition to any of the provided substitution scores (e.g. BLOSUM) in the impact score panel (Figure 4C). The user-defined impact score is plotted across the entire protein in the sequence highlighting regions that are predicted to be affected by reported variant substitutions (orange to red, Figure 4A). In our example (https://submission.gpcrmd.org/covid19/29/), we observe hotspots of high stability with unfavorable substitutions in C488 in the RBD of the spike protein. Interestingly, structural visualization in the MD viewer reveals that C488 forms a disulfide bridge with C480. Without a doubt, unfavorable variant substitutions such as C488R will disrupt the disulfide bridge introducing flexibility into this region. This in turn can alter the propensity of the SARS-CoV-2 virus to attach to the ACE2 host cell receptor.
Finally, the user can validate the customized impact score for a C488R substitution in the context of all reported substitutions in the viral protein of interest (the score distribution across the entire protein is shown on Figure 4D). Overall, high or low impact scores that are significantly shifted from the distribution peak can be expected to significantly alter protein function. Once such variants have been detected, experimental validation is required to determine if the function of the viral protein is enhanced or diminished by the structural alterations.
Case study: assessing the impact of SARS-CoV-2 variability on drug binding
An important viral threat is that newly emerging variants can develop resistance against antiviral agents or antibodies. Sites of high mutational frequency ‘in’ or ‘adjacent to’ the binding sites of antibodies/antiviral agents in the viral proteome can impact the therapeutic efficacy. In a case study, we interrogated one of these highly variable positions (T24) located in the SARS-CoV-2 main protease, Mpro (3CLpro), with around 1000 detected cases to date (35). Position 24 is adjacent to the binding site of the protease inhibitor ML188 (Figure 5) with an antiviral SARS-CoV-2 inhibition activity at micromolar range (2.5 μM) (36,37). T24A (T3287A in orf1a) is also a characteristic mutation of lineage B.1.524 (Malaysian strain, variant of concern, which peaked around November 2020) (B.1.524 Lineage Report, outbreak.info). We have simulated the WT (https://submission.gpcrmd.org/covid19/255/) and the T24A mutant (https://submission.gpcrmd.org/covid19/257/) in complex with the protease inhibitor ML188 for 1 μs in three replicates (Supplementary Note S1 and Supplementary Table S1). Interestingly, we observe that the ML188 inhibitor unbinds in both the WT as well as the T24A mutant (Supplementary Note S5, Supplementary Table S3). One important finding is that the ML188 inhibitor visits along its unbinding pathway an intermediate state that is in contact with position 24 (Figure 5). Therefore, we can expect that structural alteration of position 24 from a threonine to an alanine will alter (un)binding kinetics. In fact, we observe that T24A tends to unbind at shorter time scales compared to the WT (Supplementary Table S3). Of note, another polar-to-hydrophobic mutation at the same site, T24I, is present in ∼43% of samples in the C.1.2 strain, which peaked around July 2021, hinting at a selective advantage (C.1.2 Lineage Report, outbreak.info). This example highlights the relevance of variability within the viral proteome for drug action but can also serve as a guide for the rational design of antiviral drugs/antibodies that are more resistant to virus evolution by avoiding these regions.
Figure 5.

Unbinding pathway of the ML188 inhibitor from the SARS-CoV-2 main protease, Mpro (3CLpro). The inhibitor is in its crystallized binding pose at time T = 0 ns (yellow). The inhibitor leaves its original binding pose towards an intermediate state which is in contact with the mutated position T24A at T = 200 ns (red). From here, it moves to a second intermediate state with contacts to E47 at T = 201 ns (green) before it completely unbinds.
CONCLUSION
Enormous research efforts have resulted in high-resolution structural information on most of the SARS-CoV-2 3D-proteome, widely accessible via the PDB (rcsb.org) (24). The experimental techniques employed, namely X-ray crystallography and cryogenic electron microscopy, provide structural data which has been an excellent starting point for further efforts launched using MD simulations to gather time-resolved information about the functional dynamics of viral proteins. The SCoV2-MD database has the objective to organize, cross-reference, and share MD dynamics data for the entire viral proteome. In particular, interactive streaming and analysis tools provide a rapid and intuitive way to explore viral protein flexibility, and enable checking of hypotheses on the fly (19). Importantly, such dynamics data is not only of high value for understanding protein function but also allows for an improved insight on the structural determinants of variant impact as demonstrated in this work. We expect to periodically update the SCoV2-MD with new simulation data for multimeric complexes and missing regions in the viral proteome when they are experimentally solved and released.
DATA AVAILABILITY
SCoV2-MD is an open-source collaborative initiative. The database is freely accessible without registration at www.scov2-md.org. Its source code is available in the GitHub repository https://github.com/GPCRmd/SCoV2-md.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to the groups who shared and made reusable their simulations, including Vito Genna (IRB Barcelona), Adam Hospital (IRB Barcelona), the Chang group (University of California, Riverside), D.E. Shaw Research, Dmitry Morozov (University of Jyväskylä, Finland), Dr Im's research team (Lehigh University, Bethlehem), the Amaro Lab (University of California San Diego), the Onufriev group (California State University, Los Angeles and Virginia Tech). We are grateful to the Coronavirus Structural Task Force (www.insidecorona.net) for making available curated protein structures, which we used in our simulations. We would like to thank the GISAID Initiative and are grateful to all of the data contributors, i.e. the Authors, the Originating laboratories responsible for obtaining the specimens, and the Submitting laboratories for generating the genetic sequence and metadata and sharing via the GISAID Initiative, on which this research is based. We thank scistyle.com for the SARS-CoV-2 virion rendering. We are grateful to the anonymous reviewers of NAR for their extensive and thoughtful suggestions.
Contributor Information
Mariona Torrens-Fontanals, Research Programme on Biomedical Informatics, Hospital del Mar Medical Research Institute—Department of Experimental and Health Sciences, Pompeu Fabra University, Barcelona 08003, Spain.
Alejandro Peralta-García, Research Programme on Biomedical Informatics, Hospital del Mar Medical Research Institute—Department of Experimental and Health Sciences, Pompeu Fabra University, Barcelona 08003, Spain.
Carmine Talarico, EXSCALATE, Dompé Farmaceutici S.p.A., Via Tommaso De Amicis, 95, Napoli, 80131, Italy.
Ramon Guixà-González, Laboratory of Biomolecular Research, Paul Scherrer Institute, CH-5232 Villigen PSI, Switzerland; Condensed Matter Theory Group, Paul Scherrer Institute, CH-5232 Villigen PSI, Switzerland.
Toni Giorgino, Biophysics Institute (CNR-IBF), National Research Council of Italy, Milan 20133, Italy; Department of Biosciences, University of Milan, Milan 20133, Italy.
Jana Selent, Research Programme on Biomedical Informatics, Hospital del Mar Medical Research Institute—Department of Experimental and Health Sciences, Pompeu Fabra University, Barcelona 08003, Spain.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Spanish Ministry of Science, Innovation and Universities [FPU16/01209 to M.T.-F.]; Health department of the Government of Catalonia [DGRIS to A.P.G. and J.S.]; EU H2020-SC1-PHE-CORONAVIRUS-2020 call [101003551 ‘EXaSCale smArt pLatform Against paThogEns for Corona Virus-Exscalate4CoV’ to C.T.]; CINECA for the availability of high performance computing resources and support under the ISCRA initiative [GSNSDr3 and GSNSDr2 to T.G.]; Swiss National Science Foundation [192780 to R.G.-G.]. Funding for open access charge: Health department of the Government of Catalonia [DGRIS].
Conflict of interest statement. None declared.
REFERENCES
- 1.Harvey W.T., Carabelli A.M., Jackson B., Gupta R.K., Thomson E.C., Harrison E.M., Ludden C., Reeve R., Rambaut A., Peacock S.J.et al.. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021; 19:409–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hadfield J., Megill C., Bell S.M., Huddleston J., Potter B., Callender C., Sagulenko P., Bedford T., Neher R.A.. Nextstrain: real-time tracking of pathogen evolution. Bioinforma. Oxf. Engl. 2018; 34:4121–4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.du Plessis L., McCrone J.T., Zarebski A.E., Hill V., Ruis C., Gutierrez B., Raghwani J., Ashworth J., Colquhoun R., Connor T.R.et al.. Establishment and lineage dynamics of the SARS-CoV-2 epidemic in the UK. Science. 2021; 371:708–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hodcroft E.B., Zuber M., Nadeau S., Vaughan T.G., Crawford K.H.D., Althaus C.L., Reichmuth M.L., Bowen J.E., Walls A.C., Corti D.et al.. Spread of a SARS-CoV-2 variant through Europe in the summer of 2020. Nature. 2021; 595:707–712. [DOI] [PubMed] [Google Scholar]
- 5.Shu Y., McCauley J.. GISAID: Global initiative on sharing all influenza data – from vision to reality. Eurosurveillance. 2017; 22:30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Payne S. Family coronaviridae. Viruses. 2017; Elsevier; 149–158. [Google Scholar]
- 7.Denison M.R., Graham R.L., Donaldson E.F., Eckerle L.D., Baric R.S.. Coronaviruses: an RNA proofreading machine regulates replication fidelity and diversity. RNA Biol. 2011; 8:270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Casalino L., Dommer A.C., Gaieb Z., Barros E.P., Sztain T., Ahn S.-H., Trifan A., Brace A., Bogetti A.T., Clyde A.et al.. AI-driven multiscale simulations illuminate mechanisms of SARS-CoV-2 spike dynamics. Int. J. High Perform. Comput. Appl. 2021; 35:432–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu A., Pak A.J., He P., Monje-Galvan V., Casalino L., Gaieb Z., Dommer A.C., Amaro R.E., Voth G.A.. A multiscale coarse-grained model of the SARS-CoV-2 virion. Biophys. J. 2021; 120:1097–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zimmerman M.I., Porter J.R., Ward M.D., Singh S., Vithani N., Meller A., Mallimadugula U.L., Kuhn C.E., Borowsky J.H., Wiewiora R.P.et al.. SARS-CoV-2 simulations go exascale to predict dramatic spike opening and cryptic pockets across the proteome. Nat. Chem. 2021; 13:651–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amaro R.E., Mulholland A.J.. A community letter regarding sharing biomolecular simulation data for COVID-19. J. Chem. Inf. Model. 2020; 60:2653–2656. [DOI] [PubMed] [Google Scholar]
- 12.Gioia D., Bertazzo M., Recanatini M., Masetti M., Cavalli A.. Dynamic docking: a paradigm shift in computational drug discovery. Mol. Basel Switz. 2017; 22:E2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Basciu A., Malloci G., Pietrucci F., Bonvin A.M.J.J., Vargiu A.V.. Holo-like and druggable protein conformations from enhanced sampling of binding pocket Volume and Shape. J. Chem. Inf. Model. 2019; 59:1515–1528. [DOI] [PubMed] [Google Scholar]
- 14.Yuan J.-H., Han S.B., Richter S., Wade R.C., Kokh D.B.. Druggability assessment in TRAPP using machine learning approaches. J. Chem. Inf. Model. 2020; 60:1685–1699. [DOI] [PubMed] [Google Scholar]
- 15.Cagiada M., Johansson K.E., Valanciute A., Nielsen S.V., Hartmann-Petersen R., Yang J.J., Fowler D.M., Stein A., Lindorff-Larsen K.. Understanding the origins of loss of protein function by analyzing the effects of thousands of variants on activity and abundance. Mol. Biol. Evol. 2021; 38:3235–3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elbe S., Buckland-Merrett G.. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017; 1:33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Canakoglu A., Pinoli P., Bernasconi A., Alfonsi T., Melidis D.P., Ceri S.. ViruSurf: an integrated database to investigate viral sequences. Nucleic Acids Res. 2021; 49:D817–D824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rambaut A., Holmes E.C., O’Toole Á., Hill V., McCrone J.T., Ruis C., du Plessis L., Pybus O.G.. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020; 5:1403–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lubin J.H., Zardecki C., Dolan E.M., Lu C., Shen Z., Dutta S., Westbrook J.D., Hudson B.P., Goodsell D.S., Williams J.K.et al.. Evolution of the SARS-CoV-2 proteome in three dimensions (3D) during the first six months of the COVID-19 pandemic. 2020; bioRxiv doi:01 December 2020, preprint: not peer reviewed 10.1101/2020.12.01.406637. [DOI] [PMC free article] [PubMed]
- 20.Dunham A., Jang G.M., Muralidharan M., Swaney D., Beltrao P.. A missense variant effect prediction and annotation resource for SARS-CoV-2. 2021; bioRxiv doi:24 February 2021, preprint: not peer reviewed. 10.1101/2021.02.24.432721. [DOI]
- 21.Gowthaman R., Guest J.D., Yin R., Adolf-Bryfogle J., Schief W.R., Pierce B.G.. CoV3D: a database of high resolution coronavirus protein structures. Nucleic Acids Res. 2021; 49:D282–D287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Portelli S., Olshansky M., Rodrigues C.H.M., D’Souza E.N., Myung Y., Silk M., Alavi A., Pires D.E.V., Ascher D.B.. Exploring the structural distribution of genetic variation in SARS-CoV-2 with the COVID-3D online resource. Nat. Genet. 2020; 52:999–1001. [DOI] [PubMed] [Google Scholar]
- 23.Jo S., Kim T., Iyer V.G., Im W.. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 2008; 29:1859–1865. [DOI] [PubMed] [Google Scholar]
- 24.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E.. The Protein Data Bank. Nucleic Acids Res. 2000; 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodríguez-Espigares I., Torrens-Fontanals M., Tiemann J.K.S., Aranda-García D., Ramírez-Anguita J.M., Stepniewski T.M., Worp N., Varela-Rial A., Morales-Pastor A., Medel-Lacruz B.et al.. GPCRmd uncovers the dynamics of the 3D-GPCRome. Nat. Methods. 2020; 17:777–787. [DOI] [PubMed] [Google Scholar]
- 26.Rose A.S., Bradley A.R., Valasatava Y., Duarte J.M., Prlic A., Rose P.W.. Web-based molecular graphics for large complexes. Proceedings of the 21st International Conference on Web3D Technology - Web3D ’16. 2016; New York, New York, USA: ACM Press; 185–186. [Google Scholar]
- 27.Rose A.S., Hildebrand P.W.. NGL Viewer: a web application for molecular visualization. Nucleic Acids Res. 2015; 43:W576–W579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tiemann J.K.S., Guixà-González R., Hildebrand P.W., Rose A.S.. MDsrv: Viewing and sharing molecular dynamics simulations on the web. Nat. Methods. 2017; 14:1123–1124. [DOI] [PubMed] [Google Scholar]
- 29.The UniProt Consortium UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021; 49:D480–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Starr T.N., Greaney A.J., Hilton S.K., Ellis D., Crawford K.H.D., Dingens A.S., Navarro M.J., Bowen J.E., Tortorici M.A., Walls A.C.et al.. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell. 2020; 182:1295–1310.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greaney A.J., Starr T.N., Gilchuk P., Zost S.J., Binshtein E., Loes A.N., Hilton S.K., Huddleston J., Eguia R., Crawford K.H.D.et al.. Complete mapping of mutations to the SARS-CoV-2 spike receptor-binding domain that escape antibody recognition. Cell Host Microbe. 2021; 29:44–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vaser R., Adusumalli S., Leng S.N., Sikic M., Ng P.C.. SIFT missense predictions for genomes. Nat. Protoc. 2016; 11:1–9. [DOI] [PubMed] [Google Scholar]
- 33.McGibbon R.T.T., Beauchamp K.A.A., Harrigan M.P.P., Klein C., Swails J.M.M., Hernández C.X.X., Schwantes C.R.R., Wang L.-P., Lane T.J.J., Pande V.S.S.. MDTraj: a modern open library for the analysis of molecular dynamics trajectories. Biophys. J. 2015; 109:1528–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Venkatakrishnan A.J., Fonseca R., Ma A.K., Hollingsworth S.A., Chemparathy A., Hilger D., Kooistra A.J., Ahmari R., Babu M.M., Kobilka B.K.et al.. Uncovering patterns of atomic interactions in static and dynamic structures of proteins. 2019; bioRxiv doi:13 November 2019, preprint: not peer reviewed 10.1101/840694. [DOI]
- 35.Singer J., Gifford R., Cotten M., Robertson D.. CoV-GLUE: a web application for tracking SARS-CoV-2 genomic variation. 2020; 18 June 2020, preprint: not peer reviewed 10.20944/preprints202006.0225.v1. [DOI]
- 36.Jacobs J., Grum-Tokars V., Zhou Y., Turlington M., Saldanha S.A., Chase P., Eggler A., Dawson E.S., Baez-Santos Y.M., Tomar S.et al.. Discovery, synthesis, and structure-based optimization of a series of N-(tert-butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. J. Med. Chem. 2013; 56:534–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lockbaum G.J., Reyes A.C., Lee J.M., Tilvawala R., Nalivaika E.A., Ali A., Kurt Yilmaz N., Thompson P.R., Schiffer C.A.. Crystal structure of SARS-CoV-2 main protease in complex with the non-covalent inhibitor ML188. Viruses. 2021; 13:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
SCoV2-MD is an open-source collaborative initiative. The database is freely accessible without registration at www.scov2-md.org. Its source code is available in the GitHub repository https://github.com/GPCRmd/SCoV2-md.