

Abstract
Vaccines have made gratifying progress in preventing the 2019 coronavirus disease (COVID-19) pandemic. However, the emergence of variants, especially the latest delta variant, has brought considerable challenges to human health. Hence, the development of robust therapeutic approaches, such as anti-COVID-19 drug design, could aid in managing the pandemic more efficiently. Some drug design strategies have been successfully applied during the COVID-19 pandemic to create and validate related lead drugs. The computational drug design methods used for COVID-19 can be roughly divided into (i) structure-based approaches and (ii) artificial intelligence (AI)-based approaches. Structure-based approaches investigate different molecular fragments and functional groups through lead drugs and apply relevant tools to produce antiviral drugs. AI-based approaches usually use end-to-end learning to explore a larger biochemical space to design antiviral drugs. This review provides an overview of the two design strategies of anti-COVID-19 drugs, the advantages and disadvantages of these strategies and discussions of future developments.
Keywords: COVID-19, SARS-CoV-2, computational drug design, structure-based, artificial intelligence
1 Introduction
The emerging coronavirus disease 2019 (COVID-19) is a disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which has structural similarities with SARS-CoV-1 and poses a massive crisis to global public health. SARS-CoV-2 can be spread through air droplets, etc. [1]. Due to the high infection rate of COVID-19, a dramatic increase occurred in terms of case and death numbers. As of 10 June 2021, there were 174 061 995 confirmed cases and 3 758 560 deaths, according to the World Health Organization.
Vaccines are considered one of the most effective methods to help human society return to normal. Several vaccines have been authorized for emergency use. Although vaccines have produced very positive effects in many countries, they still face several significant challenges. Studies have reported that a small number of patients presented with venous thrombosis and thrombocytopenia after receiving the first dose of the SARS-CoV-2 vaccine against COVID-19 [2–5]. Moreover, the vaccine protection effect is not 100%. For example, the efficacy of the two whole-virus inactivated vaccines designed by the China National Biotech Group Company Limited on symptomatic COVID-19 cases was 72.8% and 78.1% in the third phase, separately [6, 7]. The last and most important concern is whether the vaccines are still effective against the emerging SARS-CoV-2 variants. At present, delta has quickly become the dominant SARS-CoV-2 variant. Research suggests that vaccines offer slightly reduced protection against delta, and vaccinated people with breakthrough infections can spread the delta variant [8, 9]. Whether vaccines can effectively slash the spread of delta remains unknown.
Drug development is another important way to defend against viruses. Drug repositioning has been a critical direction in the field of drug research. In recent months, researchers have searched for drugs to treat COVID-19 by finding new therapeutic targets and discovering often unknown relationships among apparently distant diseases. However, most drugs, such as remdesivir, dexamethasone and hydroxychloroquine, fail to display efficacy in treating COVID-19 [10].
The design of exclusive therapeutic drugs is a trend from the perspective of structural biology and molecular physics [11, 12]. Drug design may have a more therapeutic effect than drug repositioning and drug combinations in the disease of complex, rare or chronic, etc. [13, 14]. Currently, many specific drugs have been designed by virtual screening techniques based on specific pharmacological insights or an end-to-end framework to control the generation of molecules [15–20].
With the in-depth study of diseases and exploration of the structure of target proteins [21–25], many new technologies have been applied to drug design in extensive practice of medicinal chemistry by improving potency, altering physical properties and eliminating or modifying toxicophores [26–30]. All drug designs are the interaction between a drug and its target (usually proteins). Therefore, improving methods of predicting the magnitude of protein–ligand interactions can improve the efficiency of drug development [31–34]. Drug discovery steps require structural optimization of lead compounds to establish the highest possible level of selectivity, potency, and appropriate physicochemical and pharmacokinetic characteristics [26, 35, 36]. Critically, surveying binding hotspots in protein surfaces can help guide the exploration of potential ligand-binding regions [37–41].
The world is plagued by the emergence of the SARS-CoV-2 virus. Unfortunately, many methods face data scarcity when designing drugs for new targets [42]. With the development of big data and computer technology, the application of machine learning and deep learning as drug design algorithms has grown in recent decades [43–46]. Deep learning has become more active in the preparation and process of drug design, such as predicting molecular properties and activity [47, 48], identifying drug–target interactions [49–51] and planning chemical syntheses [44, 52–54]. Therefore, the potential of deep learning and molecular modeling methods helps develop drug design pipelines, especially where there are limited or unavailable target-specific ligand datasets [55, 56]. The designed drug must have an excellent inhibitory effect on the disease. Nevertheless, the prediction of the pharmacokinetics and toxicity characteristics of the scheduled drug can avoid the failure of clinical trials [57, 58]. Now, a set of tools incorporating in silico and deep learning are used to advance sequence-based or structure-based drug design problems in the computer-aided drug design area [19, 59–64].
Therefore, the rapid application of drug design on an increasingly broader scale with the advancement of biometrics and bioinformatics [65–67]. Efficient tools are now available for systematically designing compounds with biological activity as preliminary drug candidates [68–70]. Drug design strategies have been applied to various epidemic diseases [38, 71–74]. For example, to avoid the early infection of HIV, a generative adversarial autoencoder[75], which combines a neural network with virtual screening of a chemical database, was developed to design potential HIV-1 entry inhibitors. However, as COVID-19 is raging on a global scale, researchers have focused their attention on drug design against SARS-COV-2.
SARS-CoV-2 is a single-stranded RNA virus that includes two types of proteins: small envelope (E) glycoprotein, structural [spike (S) glycoprotein, membrane (M) glycoprotein, and nucleocapsid (N) protein] and nonstructural (NSP) protein (i.e. nsp1-16), which has a genome size of approximately 30 000 bp [76]. All SARS-CoV-2 proteins play an essential role in pathogenesis and virus replication. For example, S is a promising drug target because it attaches to human cells and participates in entering the cells [77]. NSP is contained in ORF1a and ORF1ab, and they produce two polyproteins, Pp1a and Pp1ab [78, 79]. The latter protein is produced by ribosomal transfer, enabling continuous translation of ORF1a and ORF1ab [80]. Specifically, ORF1ab is the largest protein of SARS-CoV-2, and the ORF1ab gene of human  -coronavirus (HBC) species has a signature of a strong positive selection site in the genome analysis of SARS-CoV-2. The positively selected sites of ORF1ab could justify some clinical features of SARS-CoV-2 compared with other HBCs [81]. Moreover, mutational spectra should be considered when designing drugs [82]. The Pp1a protein contains two viral proteases, 3C-like main protease (Mpro, corresponding to nsp5) and papain-like protease (PLpro, a domain of nsp3)[83]. The main protease (M
-coronavirus (HBC) species has a signature of a strong positive selection site in the genome analysis of SARS-CoV-2. The positively selected sites of ORF1ab could justify some clinical features of SARS-CoV-2 compared with other HBCs [81]. Moreover, mutational spectra should be considered when designing drugs [82]. The Pp1a protein contains two viral proteases, 3C-like main protease (Mpro, corresponding to nsp5) and papain-like protease (PLpro, a domain of nsp3)[83]. The main protease (M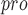 ) of SARS-CoV-2 is a crucial enzyme of coronaviruses and has a pivotal role in mediating viral replication and transcription [20, 84–86]. PL
) of SARS-CoV-2 is a crucial enzyme of coronaviruses and has a pivotal role in mediating viral replication and transcription [20, 84–86]. PL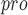 is also critical to SARS-CoV-2 replication and represents a promising goal for drug design and development [87–89]. Among nonstructural proteins, the large, multidomain Nsp3 is encoded by SARS-CoV-2. One of its units is the ADP-ribose phosphatase domain (ADRP; also known as the macro domain, MacroD), which interferes with the host immune response [90].
is also critical to SARS-CoV-2 replication and represents a promising goal for drug design and development [87–89]. Among nonstructural proteins, the large, multidomain Nsp3 is encoded by SARS-CoV-2. One of its units is the ADP-ribose phosphatase domain (ADRP; also known as the macro domain, MacroD), which interferes with the host immune response [90].
It is well known that molecular inhibitors such as drugs can achieve inhibitory effects by targeting cancer or virus expression pathways. For example, seasonal and pandemic influenza have a substantial impact on global public health [91]. A study found that endonuclease activity exists in the independently folded N-terminal domain of PA (PAN) [92, 93], where PA is a subunit of RNA-dependent RNA polymerase (RdRp) that can catalyze viral transcription. Subsequently, the molecular mechanism of the inhibitor was structurally confirmed, and the interaction sites in the crystal structure of the virus strain and the inhibitor were obtained [94]. This information also provides a basis for drug discovery and design. The molecular mechanisms involved in immune regulation during the new coronavirus infection have played a major role in resisting the new coronavirus. Mitogen-activated protein kinase (MAPK) affects cell defense and apoptosis [95]. Azithromycin has been shown to control activation of the MAPK cascade, one of the molecular mechanisms involved in virus infection, thereby reducing virus replication [96].
In general, SARS-CoV-2-related proteins have been suggested as targets for drug design [97]. In particular, the proteins considered in the entire design of anti-coronavirus drugs are also related to cancer treatment and other diseases [98–100]. Indeed, potentially suitable drugs against this virus essentially affect signal transduction and the synthesis of macromolecules, which strongly interfere with the host immune response, particularly the proteins associated with COVID-19 [13].
2 Drug design strategies for COVID-19
Computational drug design approaches applied to COVID-19 can be broadly categorized as (i) structure-based approaches and (ii) AI-based approaches [101–105]. Some methods consist of both structure-based and AI-based approaches [106–109]. The drug design process is shown in Figure 1.
Figure 1.

Structure-based approaches and AI-based approaches for designing drugs. The decisive part of structure-based and AI-based approaches is the identification of the interaction between molecules and target proteins. Structure-based approaches of designing drugs rely on the three-dimensional structure of the target protein and its active site to identify the interaction between protein and molecule, while AI-based approaches rely on the knowledge of protein and molecule to understand the interaction between them through machine learning or deep learning algorithms.
2.1 Structure-based approaches
Insertion of halogen atoms on hit or lead compounds has been used to exploit their steric effects because the formation of halogen groups in ligand–target complexes favorably contributes to the stability of the protein–ligand complex [28, 110, 111]. The use of carbamates in medicinal chemistry has increased, and many derivatives are specifically designed to form drug–target interactions through their carbamate moiety [29, 112, 113]. Fragment-based drug discovery is an effective strategy for generating small-molecule protein inhibitors and drug candidates, which has led to three FDA-approved drugs and clinical trials for nearly 50 molecules [114]. To enable the screening of virtual libraries in search of active compounds, combinatorial chemistry and structure-based design need to be combined, which can exploit ligands and targets’ fundamental structural and physicochemical properties [115–117]. We believe that these approaches have been helpful for designing antiviral compounds.
2.1.1 Structure-based drug design for target CoV main proteases
The genus Coronavirus contains approximately 25 coronaviruses (CoVs), which are essential pathogens causing highly prevalent diseases [38]. By comparing four crystal structures and homologous models representing all three genetic clusters of the genus Coronavirus, it was found that the CoV main proteases (M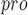 or 3CL
or 3CL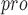 ) are critical enzymes in viral gene expression and replication and share a highly conserved substrate recognition pocket [118, 119]. In addition, the active sites of M
) are critical enzymes in viral gene expression and replication and share a highly conserved substrate recognition pocket [118, 119]. In addition, the active sites of M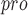 , S1
, S1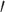 , S1, S2 and S4 are highly conserved among all coronaviruses [38]. It is possible to design and synthesize inhibitors that target SARS-CoV-2 M
, S1, S2 and S4 are highly conserved among all coronaviruses [38]. It is possible to design and synthesize inhibitors that target SARS-CoV-2 M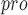 by analyzing the substrate-binding pocket of SARS-CoV M
by analyzing the substrate-binding pocket of SARS-CoV M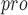 (PDB ID 2H2Z). When designing a new inhibitor, Dai et al. chose an aldehyde as a new warhead to form a covalent bond with cysteine. In addition, they introduced cyclohexyl or 3-fluorophenyl in P2 to enhance the activity and introduced indole groups in P3 to form new hydrogen bonds with S4, which improves the drug-like properties [103]. UCI-1 is a cyclic peptide inhibitor that was designed based on the crystal structure of an inactive SARS-CoV M
(PDB ID 2H2Z). When designing a new inhibitor, Dai et al. chose an aldehyde as a new warhead to form a covalent bond with cysteine. In addition, they introduced cyclohexyl or 3-fluorophenyl in P2 to enhance the activity and introduced indole groups in P3 to form new hydrogen bonds with S4, which improves the drug-like properties [103]. UCI-1 is a cyclic peptide inhibitor that was designed based on the crystal structure of an inactive SARS-CoV M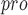 (C145A) variant. The purpose of designing UCI-1 is to mimic the conformation of a C-terminal autolytic cleavage site of the SARS-CoV M
(C145A) variant. The purpose of designing UCI-1 is to mimic the conformation of a C-terminal autolytic cleavage site of the SARS-CoV M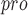 , a naturally occurring M
, a naturally occurring M substrate. In UCI-1, the carboxy-terminus of the P2’ residue is linked to the amino-terminus of the P2 residue with a [4-(2-aminoethyl)phenyl]-acetic acid (AEPA) group, creating a cyclophane. The (2-aminoethyl)phenyl group of AEPA is designed to act as a surrogate for a phenylalanine side chain at position P3’ and fill the S3’ pocket. Furthermore, research shows that UCI-1 tends to be nontoxic toward human embryonic kidney cells at concentrations that inhibit M
substrate. In UCI-1, the carboxy-terminus of the P2’ residue is linked to the amino-terminus of the P2 residue with a [4-(2-aminoethyl)phenyl]-acetic acid (AEPA) group, creating a cyclophane. The (2-aminoethyl)phenyl group of AEPA is designed to act as a surrogate for a phenylalanine side chain at position P3’ and fill the S3’ pocket. Furthermore, research shows that UCI-1 tends to be nontoxic toward human embryonic kidney cells at concentrations that inhibit M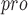 , but compared with other M
, but compared with other M inhibitors, it shows lower activity [86].
inhibitors, it shows lower activity [86].
Owing to the structural elucidation of the target, the application of structure-based drug design (SBDD) software is flourishing [120–122]; the basic steps of SBDD are described in detail in Figure 2. Using different complementary virtual screening and docking approaches can identify nonapproved active compounds as new potential inhibitors of 3CLpro from the ZINC15 library. Then, these compounds could be further optimized by using SBDD [123]. On the other hand, one study started with the X-ray crystal structure of SARS-CoV M [86] and then used UCSF Chimera software [124] to modify the substrate to create a cyclic peptide inhibitor within the M
[86] and then used UCSF Chimera software [124] to modify the substrate to create a cyclic peptide inhibitor within the M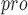 active site. Finally, AutoDock Vina [125] was used to evaluate this model by docking the inhibitor to SARS-CoV-2 M
active site. Finally, AutoDock Vina [125] was used to evaluate this model by docking the inhibitor to SARS-CoV-2 M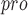 .
.
Figure 2.

The basic steps of SBDD. First, the preparation of the target macromolecule structure is fundamental to SBDD approaches, which can be obtained by structure identification techniques such as X-ray and NMR analysis, searching the PDB database or using other calculation methods. Next, the binding site of the target macromolecule is identified to determine the protein–ligand interaction, and ligands or drug-like compounds with the limitation of Lipinski’s rule of five’ are selected from a chemical database to construct a ligand library. When all preparations are ready, molecule docking programs are applied, and scoring functions are mostly used in the post-docking analysis. Finally, the top-ranked molecules are chemically synthesized, and biological evaluation is necessary.
2.1.2 SBDD for target PL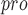
PL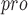 aids coronaviruses in their evasion of host innate immune responses because it has the additional function of stripping ubiquitin and interferon-stimulated gene 15 (ISG15) from host-cell proteins [126]. Inhibition of SARS-CoV-2 PL
aids coronaviruses in their evasion of host innate immune responses because it has the additional function of stripping ubiquitin and interferon-stimulated gene 15 (ISG15) from host-cell proteins [126]. Inhibition of SARS-CoV-2 PL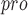 with GRL-0617 has three main tasks: impair the virus-induced cytopathogenic effect, maintain the antiviral interferon pathway and reduce viral replication in infected cells [127]. A recent investigation attempted to design potential SARS-CoV PL
with GRL-0617 has three main tasks: impair the virus-induced cytopathogenic effect, maintain the antiviral interferon pathway and reduce viral replication in infected cells [127]. A recent investigation attempted to design potential SARS-CoV PL inhibitors containing naphthalene and 3,4-dihydro-2H-pyran moieties connected via-NHCO-linker [89].
inhibitors containing naphthalene and 3,4-dihydro-2H-pyran moieties connected via-NHCO-linker [89].
Another study first used HyCoSuL, a novel chemical approach to perform comprehensive activity profiling of SARS-CoV-2 PL , and revealed the molecular rules governing PL
, and revealed the molecular rules governing PL substrate specificity [128]. Then, compared with other proteases, potent inhibitors (VIR250 and VIR251) were designed and biochemically characterized were shown to be more effective against SARS-CoV-2 PL
substrate specificity [128]. Then, compared with other proteases, potent inhibitors (VIR250 and VIR251) were designed and biochemically characterized were shown to be more effective against SARS-CoV-2 PL and related SARS-CoV-1 PL
and related SARS-CoV-1 PL , presenting high selectivity. In addition, it was surprisingly discovered that the P4 amino acids of VIR250 and VIR251 occupy both sides of the wide S4 pocket of SARS-CoV-2 PL
, presenting high selectivity. In addition, it was surprisingly discovered that the P4 amino acids of VIR250 and VIR251 occupy both sides of the wide S4 pocket of SARS-CoV-2 PL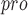 , which will make contributions to future drug discovery [87]. It is worth noting that there is not enough information about SARS-CoV-2-PL
, which will make contributions to future drug discovery [87]. It is worth noting that there is not enough information about SARS-CoV-2-PL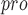 , and HyCoSul is based on a hypothesis that SARS-CoV-2-PL
, and HyCoSul is based on a hypothesis that SARS-CoV-2-PL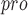 is highly similar to SARS-CoV-PL
is highly similar to SARS-CoV-PL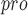 . The crystal structure of the SARS-CoV-2 ADP-ribose phosphatase domain (ADRP) in multiple states was determined by many studies: in the apo form and in complexes with 2-(N-morpholino) ethane sulfonic acid (MES), ADP-ribose (ADPr) and AMP. Researchers have proposed a robust system to identify potential small-molecule inhibitors with apo crystals diffracting to atomic resolution based on structure-based experiments [90].
. The crystal structure of the SARS-CoV-2 ADP-ribose phosphatase domain (ADRP) in multiple states was determined by many studies: in the apo form and in complexes with 2-(N-morpholino) ethane sulfonic acid (MES), ADP-ribose (ADPr) and AMP. Researchers have proposed a robust system to identify potential small-molecule inhibitors with apo crystals diffracting to atomic resolution based on structure-based experiments [90].
2.1.3 SBDD for target spike glycoprotein
A coronavirus was identified as the causative agent of SARS as early as 2003 [129]. Current research shows that the latest SARS-CoV-2 in 2019 has a very similar structure and function to SARS [130–132]. The COVID-19 genomes from isolates of China, India, Italy, Nepal and the USA have a sequence similarity of approximately 60% with the human SARS-CoV German isolate, but it has sequence similarity of 79–80% with bat SARS-CoV [133]. Spike glycoprotein (S) is crucial in the attachment of SARS-CoV-2 to host receptors and cell entry, leading to COVID-19 infection [134, 135]. An in silico pharmacophore modeling and virtual screening approach has been used to explore drugs against receptor-binding domain (RBD) of SARS-CoV-2. First, the 3D structure of RBD is modelled, the conservative area is used as a template and LigandScout is used to design the pharmacophore. Then, the Cambridge, DrugBank, ZINC and TIMBLE databases are used to screen lead compounds. Finally, AutoDock Vina Is used to dock the shortlisted lead compounds molecularly and visualize the interacting residues [77]. All the resulting lead compounds are preliminary findings, and clinical and investigational trials are still required. New research points out that the cell membrane receptor angiotensin-converting enzyme 2 (ACE2) plays a key role in the entry of SARS-CoV-2 into cells [136]. SARS-CoV-2 S interacts with ACE2 through the RBD [137]. To this end, a structure-based ACE2 variant dataset was combined with the SARS-CoV-2 RBD, resulting in a total of 242 structural models. These models can be used as a starting point for drug design and be used further to understand the recognition of SARS-CoV-2 S protein by ACE2 [138]. Research has identified a pair of key salt bridges formed by the side chains of K537 and E619. Drugs designed to prevent the formation of these salt bridges can effectively treat COVID-19, but for the simplicity of the protein complex, it is concluded to be applicable to the trimeric S protein [139].
For the different target proteins of SARS-COV-2, SBDD studies against the COVID-19 pandemic are summarized in Table 1. In addition, small peptides can be transported across the cell membrane by amino acid and peptide transporters [140]. A variety of small peptide compounds have been developed to inhibit ACE [141, 142]. Another promising method is to select and combine the substructures of highly active compounds to form new peptide analogs [143–146].
Table 1.
Structure-based approaches for drug design
| Reference | Target protein | Description | Advantages | Possible designing drugs or pharmacophore |
|---|---|---|---|---|
| 77 | Spike glycoprotein | The conserved region was used as a template to design a pharmacophore using LigandScout. | Interacting residues were visualized. | 6-(1H-imidazol-1-yl)-N-[1-(3,4,5-trifluorophenyl)ethyl]pyrimidin-4-amine |
| 138 | Spike glycoprotein | 242 structural models of variants of human ACE2 bound to the receptor binding domain (RBD) of the S protein was built and refined their interfaces with HADDOCK. | The effects of these variations on the 3D structure of the protein and its complex with RBD have been systematic studied. | https://kastritislab.github.io/human-ace2-variants/ |
| 139 | Spike glycoprotein | Mutations of K537Q and E619D reduced their side chain lengths and eliminated this pair of salt bridges. | The screened molecule is capable of blocking the formation of the key pair of salt bridges. | The side chains of K537 and E619. |
| 103 | M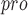
|
The aldehyde groups of 11a and 11b are covalently bound to cysteine 145 of M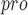 . . |
Both compounds showed good pharmacokinetic properties in vivo, among which 11a has lower toxicity. | 11a and 11b. |
| 86 | M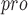
|
The cyclic peptide inhibitor was designed to mimic the conformation of a substrate at a C-terminal autolytic cleavage site of M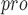 . . |
The inhibitor is active against M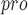 in vitro and is nontoxic toward human cells in culture. in vitro and is nontoxic toward human cells in culture. |
A first-in-class cyclic peptide inhibitor. |
| 123 | M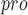
|
3CL proteases have a central role in polyprotein processing during replication. | The period up to approval as a therapeutic against SARS-CoV-2 could hopefully be shortened. | Hydrogen-bond acceptors or donors of pocket amino acids. |
| 89 | PL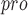
|
Potential SARS-CoV PLpro inhibitors containing naphthalene and 3,4-dihydro-2H-pyran moieties connected via -NHCO- linker have been designed. | The designed ligands against the receptor SARS CoV-2 Papain-like protease (PL ) have strong binding affinity and inhibition potential. ) have strong binding affinity and inhibition potential. |
naphthalene based SARS-CoV PL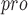 inhibitors. inhibitors. |
| 87 | PL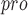
|
Viral papain-like cysteine protease (PL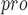 , NSP3) represents a promising target for the development of antiviral drugs. , NSP3) represents a promising target for the development of antiviral drugs. |
SARS-CoV-2-PL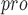 harbours deISGylating activities similar to SARS-CoV-1-PL harbours deISGylating activities similar to SARS-CoV-1-PL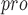 , but its ability to hydrolyse K48-linked Ub chains is diminished , but its ability to hydrolyse K48-linked Ub chains is diminished |
VIR250 and VIR251. |
| 90 | PL
|
Macrodomains may help to reduce the viral load and facilitate recovery. | A robust system has been developed to identify potential small-molecule inhibitors for structure-based experiments. | The anomeric C atom. |
2.2 AI-based approaches
Designing novel drugs for new diseases is a complicated process necessary to find molecules that bind to specific biomolecular targets and have good physical and chemical properties over a broad chemical space [147]. First, knowledge of the virus and target is necessary. A number of methods have used machine learning or deep learning algorithms to identify the structure of the protein, but most of them could not obtain an accurate structure until AlphaFold [148] appeared. The input of AlphaFold is the given protein sequence, which can be more easily obtained than the structure through experiments. Then, it learns protein-specific potential by training a deep neural network and makes an accurate prediction of the structure by minimizing the potential by gradient descent. Another high-quality prediction of protein structure is C-I-TASSER [149]; it is an improvement based on I-TASSER [150], with three modules added on the original basis, which are a new multivariate sequence comparison protocol, an improved meta method NeBcon and an optimized contact potential.
Then, with the ever-increasing number of potential lead compounds derived through virtual screening studies, it is helpful to screen extensive virtual libraries of compounds with improved biological properties such as explicitness and selectivity toward the respective target, lower toxicity or reduced cost to discover drugs with essential biological features. It is laborious for drug designers to extract synthetic information from substantial compound libraries to devise drugs with important biological features; machine learning has become an effective tool to solve this problem [151–155]. For example, random forest analyses coupled with a unique approach to bioactivity and chemical data curation have led to a series of target-specific and cross-validated predictive feature fingerprints [156]. Moreover, in the study of reference [157], the desire to map small molecules to target interactions across multiple stages of SARS-CoV-2 infection drives the initial target selection.
Deep learning can be applied to extract complex and deeper features from simple representations [158]. For example, given a protein pocket and using deep generative modeling for compound design, potential binding compounds can be generated [159]. We know that effective noncovalent inhibitors of the major proteases of SARS-CoV and SARS-CoV-2 should have the same structural and chemical characteristics [84, 160, 161]. The ligand generative adversarial network (LIGANN) is a novel virtual screening technique that is applied for multimodal structure-based ligand design [61]. The ligand is generated in LIGANN to match the shape and chemical properties of the binding pocket and then is decoded into its SMILES sequence, thereby directly realizing AI-based drug design [73].
CogMol (controlled generation of molecules) is an end-to-end framework that was proposed to design new drug-like small molecules targeting novel viral proteins with high affinity and off-target selectivity [162]. CogMol combines the variational autoencoder in the molecular SMILES format and the multi-attribute controlled sampling scheme and applies this approach to three SARS-CoV-2 target proteins: the main protease, the RBD of the spike protein and the nonstructural protein. Compared with earlier deep-learning molecular methods, the new approach explores new molecules by adding a meaningful molecular fragment one by one rather than a single atom at a time [163, 164]. However, due to the deviation of the training data or the inaccuracy of the predictor used to control the generation, there may be a certain degree of unreliability and inability to meet the needs of generating molecules with specific properties. ADQN-FBDD [165] is another AI-based drug design approach against SARS-CoV-2. First, an initial molecular database targeting SARS-CoV-2 3CL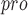 is built by collecting SARS-CoV 3CL
is built by collecting SARS-CoV 3CL inhibitors (284 molecules). Then, this set of molecules is split into fragments, and the molecular weight of each fragment does not exceed 200 Daltons. Finally, the features of the fragments are input into the deep Q-learning network to generate potential lead compounds. Most of the computation-aided drug design methods need to be experimentally verified, and ADQN-FBDD is no exception. In conclusion, the major steps of AI-based drug design are described in Figure 3.
inhibitors (284 molecules). Then, this set of molecules is split into fragments, and the molecular weight of each fragment does not exceed 200 Daltons. Finally, the features of the fragments are input into the deep Q-learning network to generate potential lead compounds. Most of the computation-aided drug design methods need to be experimentally verified, and ADQN-FBDD is no exception. In conclusion, the major steps of AI-based drug design are described in Figure 3.
Figure 3.

The basic steps of AI-based drug design. The first step is target discovery, which means identifying the target protein that interacts with coronavirus. Next, a virtual screening technique is applied to search ligands from chemical databases, and knowledge of target protein–ligand interactions can be obtained from databases or calculated by other calculation methods. Then, AI methods, including machine learning and deep learning, such as generators and predictors, can be used to discover novel drugs. However, before machine learning or deep learning algorithms are used to generate lead molecules, the molecule fed into the learning system should be transformed into vectors, such as molecular descriptors, fingerprints, SMILES strings or grids.
3 Discussion and conclusion
Although vaccines against SARS-CoV-2 have been developed to help prevent COVID-19, there are still some potential problems. Some vaccines developed internationally continue to reveal potential risks and cannot effectively prevent the virus after vaccination. Drug relocation is also a popular way to treat COVID-19, but previous drugs often do not have a significant inhibitory effect on SARS-CoV-2. Therefore, the development of drugs against SARS-CoV-2 is more reasonable and attractive and can provide an effective first line of defense for emerging coronavirus-related drugs in the future. In recent years, technological developments have enabled the determination of more protein structures and expanded structural biology, thereby accelerating the progress of drug design.
It is estimated that the traditional pharmaceutical industry spends US$2.6 billion to design a new drug, with the failure rate reaching 90% between clinical trials and approval [169]. The emergence of structure-based and AI-based drug design has greatly reduced the cost and time and is more creative. Moreover, the key to structure-based and AI-based drug design is target–protein determination and target–drug interactions. However, SBDD relies more on the three-dimensional structure to optimize the drug compounds, which has a larger chemical space, whereas AI-based structures use machine learning and deep learning algorithms in combination with virtual screening to find the lead molecule. Finally, predicts the binding models between COVID-19 targets and designed molecules by [170].
We also found that structure-based approaches are more abundant than purely AI-based methods because SBDD methods have been developed for decades. As a result, the entire design–experiment–verification process is flawless, and the corresponding tools are also more comprehensive. We summarize the tools of drug design in Table 2. On the other hand, the contribution of AI-based methods to drug design may not achieve the expected results. The possible reason is that these methods are limited to our knowledge. Currently, we do not have enough knowledge to understand a virus that was only discovered less than 2 years ago. Therefore, basic research is essential to the analysis of research applied to biomedicine because basic research is an indispensable foundation for knowledge growth, but it is often undervalued. Nevertheless, powerful and effective calculation methods and pipelines for designing compounds can provide beneficial drug candidates for the treatment of SARS-CoV-2 infection, and these candidates or their variants are likely to produce effective anti-COVID-19 lead drugs. Therefore, we believe that these advances will help make more meaningful contributions to the fight against COVID-19 in the future.
Table 2.
The tools of drug design
| Reference | Tools name & Website | Description |
|---|---|---|
| 101 | GOLEM N/A | GOLEM from the field of inductive logic programming was applied to the drug design problem of modelling structure–activity relationships. |
| 121 | Drug Guru N/A | Drug Guru (drug generation using rules) is a new web-based computer software program for medicinal chemists that applies a set of transformations, that is, rules, to an input structure. |
| 59 | ReLeaSE https://github.com/isayev/ReLeaSE | ReLeaSE is a novel computational strategy for de novo design of molecules with desired properties. |
| 60 | GuacaMol https://www.benevolent.com/guacamol | GuacalMol is a de novo design tool that seeks to generate molecules with required property profiles by virtual design-make-test cycles. |
| 166 | DeepScaffold https://github.com/deep-scaffold/deep_scaffold | DeepScaffold is proposed based on a wide spectrum of scaffold definitions, including Bemis-Murcko scaffolds, cyclic skeletons, and scaffolds with specifications on side-chain properties. |
| 167 | Cov_FB3D http://202.114.32.71:10280/wandox/Cov_FB3D/home.html | Cov_FB3D involves the in silico assembly of potential novel covalent inhibitors by identifying the active fragments in the covalently binding site of the target protein. |
| 73 | LIGANN N/A | LIGANN is developed by using a combination of virtual screening, docking and molecular dynamics techniques for noncovalent inhibition of the main protease 3CL of SARS-CoV-2. of SARS-CoV-2. |
| 114 | FBDD https://github.com/PatWalters/fragment_expansion | FBDD has reviewed the different types of linkers published and obtained the lead compound by designing these linkers. |
| 124 | UCSF ChimeraX https://www.rbvi.ucsf.edu/chimerax | UCSF ChimeraX is the next-generation interactive visualization program from the Resource for Biocomputing, Visualization, and Informatics (RBVI), following UCSF ChimeraX. |
| 171 | PerSpect ML N/A | PerSpect ML considers 11 persistent spectral variables and feeds them into machine learning models to predict the protein–ligand affinity for drug design. |
| 55 | OpenChem https://github.com/Mariewelt/OpenChem | OpenChem offers easy and fast model development, modular software design, and several data preprocessing modules. |
| 63 | Target2DeNovoDrug https://github.com/bengeof/Target2DeNovoDrug | Target2DeNovoDrug offers researchers a fresh set of tools to approach SBDD problems by integrating data science and artificial intelligence (AI). |
| 64 | MolAICal https://molaical.github.io | MolAICal software is introduced to supply a way for generating 3D drugs in the 3D pocket of protein targets by combining the merits of a deep learning model and classical algorithm. |
| 168 | Target2DeNovoDrugPropMax https://github.com/bengeof/QPoweredTarget2DeNovoDrugPropMax | Target2DeNovoDrugPropMax includes a variety of AI methods along with incorporated in silico techniques to provide a holistic tool for automated drug discovery. |
N/A: N/A means that the tool does not provide a link in their article, due to copyright or other reasons.
Key Points
Drug design is rapidly being applied on an increasingly broader scale with the advancement of biometrics and bioinformatics. In particular, the design of anti-COVID-19 drugs is meaningful work for COIVD-19.
Structure-based approaches use the basic structure, physicochemical properties of ligands and targets to screen virtual libraries to find active anti-COVID-19 drugs. In summary, develop corresponding design strategies of anti-COVID-19 drugs according to the characteristics of different target proteins.
AI-based approaches can extract complex and deeper features from simple drug molecular representations and generate potential binding compounds. It is essential to extract features from compounds or proteins using interpretable methods.
5 Author contributions statement
J.W., Y.Z., Y.L. and W.N. analyze the data and wrote the manuscript. L.D. reviewed and revised the manuscript.
Jinxian Wang received the Bachelor’s degree from Hunan Agricultural University in 2019, and at present is studying for a master degree at Central South University supervised by Prof. Lei Deng. His study focuses on machine learning and bioinformatics.
Ying Zhang is a pharmacist at Department of Pharmacy, Heilongjiang Province land reclamation headquarters general hospital, Harbin, China. His study focuses on drug development.
Wenjuan Nie is a graduate student at School of Computer, Central South University, Changsha, China. Her research interests is using machine learning algorithms to predict protein–NA binding residues.
Yi Luo received BE degree in Computer Science from School of Computer Science and Technology, South West University, ChongQing,China. Currently,he is a MS candidate in Data Science from School of Science.the University of Auckland,Auckland, New Zealand. His research interests include data mining, bioinformatics and data analysis.
Lei Deng is a professor at School of Computer Science and Engineering, Central South University, Changsha, China. His research interests include data mining, bioinformatics and systems biology.
Contributor Information
Jinxian Wang, School of Computer Science and Engineering, Central South University,410075, Changsha, China.
Ying Zhang, Department of Pharmacy, Heilongjiang Province Land Reclamation Headquarters General Hospital, 150001, Harbin, China.
Wenjuan Nie, School of Computer Science and Engineering, Central South University,410075, Changsha, China.
Yi Luo, School of Science, The University of Auckland,Auckland 1010, Auckland, New Zealand.
Lei Deng, School of Computer Science and Engineering, Central South University,410075, Changsha, China.
Funding
National Natural Science Foundation of China (grant No. 61972422).
References
- 1. Klompas M, Baker MA, Rhee C. Airborne transmission of sars-cov-2: theoretical considerations and available evidence. JAMA 2020;324(5):441–42. [DOI] [PubMed] [Google Scholar]
- 2. Schultz NH, Sørvoll IH, Michelsen AE, et al. Thrombosis and thrombocytopenia after chadox1 ncov-19 vaccination. N Engl J Med 2021;384(22):2124–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Scully M, Singh D, Lown R, et al. Pathologic antibodies to platelet factor 4 after chadox1 ncov-19 vaccination. N Engl J Med 2021;384(23):2202–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Greinacher A, Thiele T, Warkentin TE, et al. Thrombotic thrombocytopenia after chadox1 ncov-19 vaccination. N Engl J Med 2021;384(22):2092–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cines DB, Bussel JB. SARS-CoV-2 Vaccine-Induced Immune Thrombotic Thrombocytopenia. N Engl J Med 2021;384(23):2254–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kaabi, Zhang Y, Xia S, et al. Effect of 2 inactivated sars-cov-2 vaccines on symptomatic covid-19 infection in adults: A randomized clinical trial. JAMA 2021;326(1):35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kyriakidis NC, López-Cortés A, González EV, et al. Sars-cov-2 vaccines strategies: a comprehensive review of phase 3 candidates. npj Vaccines 2021;6(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Layan M, Gilboa M, Gonen T, et al. Impact of bnt162b2 vaccination and isolation on sars-cov-2 transmission in Israeli households: an observational study. medRxiv 2021. 10.1101/2021.07.12.21260377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Prunas O, Warren JL, Crawford FW, et al. Vaccination with bnt162b2 reduces transmission of sars-cov-2 to household contacts in Israel. medRxiv 2021. 10.1101/2021.07.13.21260393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dotolo S, Marabotti A, Facchiano A, et al. A review on drug repurposing applicable to covid-19. Brief Bioinform 2021;22(2):726–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen X. Customized cancer therapy based on the dynamic analysis of the tumor-immune-drug system interaction. 2021.
- 12. Lynch ML, Snell EH, Bowman SEJ. Structural biology in the time of covid-19: perspectives on methods and milestones. IUCrJ 2021;8(3):335–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Card GL, Blasdel L, England BP, et al. A family of phosphodiesterase inhibitors discovered by cocrystallography and scaffold-based drug design. Nat Biotechnol 2005;23(2):201–7. [DOI] [PubMed] [Google Scholar]
- 14. Brown N, Ertl P, Lewis R. Artificial intelligence in chemistry and drug design, 2020;709–15. [DOI] [PubMed]
- 15. Meanwell NA. Synopsis of some recent tactical application of bioisosteres in drug design. J Med Chem 2011;54(8):2529–91. [DOI] [PubMed] [Google Scholar]
- 16. Scior T, Bender A, Tresadern G, et al. Recognizing pitfalls in virtual screening: a critical review. J Chem Inf Model 2012;52(4):867–81. [DOI] [PubMed] [Google Scholar]
- 17. Cheng T, Li Q, Zhou Z, et al. Structure-based virtual screening for drug discovery: a problem-centric review. AAPS J 2012;14(1):133–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pyzer-Knapp EO, Suh C, Gómez-Bombarelli R, et al. What is high-throughput virtual screening? A perspective from organic materials discovery. Annu Rev Mat Res 2015;45:195–216. [Google Scholar]
- 19. Gómez-Bombarelli R, Wei JN, Duvenaud D, et al. Automatic chemical design using a data-driven continuous representation of molecules. ACS Central Sci 2018;4(2):268–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang S, Krumberger M, Morris MA, et al. Structure-based drug design of an inhibitor of the sars-cov-2 (covid-19) main protease using free software: a tutorial for students and scientists. Eur J Med Chem 2021;218:113390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr 2006;62(4):439–50. [DOI] [PubMed] [Google Scholar]
- 22. Meisburger SP, Thomas WC, Watkins MB, et al. X-ray scattering studies of protein structural dynamics. Chem Rev 2017;117(12):7615–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Meisburger SP, Case DA, Ando N. Diffuse x-ray scattering from correlated motions in a protein crystal. Nat Commun 2020;11(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shanmugaraj B, Siriwattananon K, Wangkanont K, et al. Perspectives on monoclonal antibody therapy as potential therapeutic intervention for coronavirus disease-19 (covid-19). Asian Pac J Allergy Immunol 2020;38(1):10–8. [DOI] [PubMed] [Google Scholar]
- 25. Huo L, Li JJ, Chen L, et al. Single-cell multi-omics sequencing: application trends, covid-19, data analysis issues and prospects. Brief Bioinform 2021;22(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thompson KH, Orvig C. Metal complexes in medicinal chemistry: new vistas and challenges in drug design. Dalton Trans 2006;(6):761–4. 10.1039/B513476E. [DOI] [PubMed] [Google Scholar]
- 27. Andricopulo AD, Salum LB, Abraham DJ. Structure-based drug design strategies in medicinal chemistry. Curr Top Med Chem 2009;9(9):771–90. [DOI] [PubMed] [Google Scholar]
- 28. Hernandes MZ, Cavalcanti SMT, Moreira DRM, et al. Halogen atoms in the modern medicinal chemistry: hints for the drug design. Curr Drug Targets 2010;11(3):303–14. [DOI] [PubMed] [Google Scholar]
- 29. Ghosh AK, Brindisi M. Organic carbamates in drug design and medicinal chemistry. J Med Chem 2015;58(7):2895–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Meyers J, Fabian B, Brown N. De novo molecular design and generative models. Drug Discov Today 2021. 10.1016/j.drudis.2021.05.019. [DOI] [PubMed] [Google Scholar]
- 31. Mavromoustakos T, Durdagi S, Koukoulitsa C, et al. Strategies in the rational drug design. Curr Med Chem 2011;18(17):2517–30. [DOI] [PubMed] [Google Scholar]
- 32. Cournia Z, Allen B, Sherman W. Relative binding free energy calculations in drug discovery: recent advances and practical considerations. J Chem Inf Model 2017;57(12):2911–37. [DOI] [PubMed] [Google Scholar]
- 33. Williams-Noonan BJ, Yuriev E, Chalmers DK. Free energy methods in drug design: Prospects of "alchemical perturbation" in medicinal chemistry: miniperspective. J Med Chem 2018;61(3):638–49. [DOI] [PubMed] [Google Scholar]
- 34. Wan S, Bhati AP, Zasada SJ, et al. Rapid, accurate, precise and reproducible ligand–protein binding free energy prediction. Interface Focus 2020;10(6):20200007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ooms F. Molecular modeling and computer aided drug design. examples of their applications in medicinal chemistry. Curr Med Chem 2000;7(2):141–58. [DOI] [PubMed] [Google Scholar]
- 36. Noble MEM, Endicott JA, Johnson LN. Protein kinase inhibitors: insights into drug design from structure. Science 2004;303(5665):1800–5. [DOI] [PubMed] [Google Scholar]
- 37. Gane PJ, Dean PM. Recent advances in structure-based rational drug design. Curr Opin Struct Biol 2000;10(4):401–4. [DOI] [PubMed] [Google Scholar]
- 38. Yang H, Xie W, Xue X, et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol 2005;3(10):e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Clasman JR, Báez-Santos YM, Mettelman RC, et al. X-ray structure and enzymatic activity profile of a core papain-like protease of MERS coronavirus with utility for structure-based drug design. Sci Rep 2017;7(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mirza MU. Drug design strategies against newly emerging viral diseases, 2020.
- 41. Curran PR, Radoux CJ, Smilova MD, et al. Hotspots api: a python package for the detection of small molecule binding hotspots and application to structure-based drug design. J Chem Inf Model 2020;60(4):1911–6. [DOI] [PubMed] [Google Scholar]
- 42. Qian Y, Xing Y, Dong L. Deep learning for a low-data drug design system. In: 2020 IEEE International Conference on E-health Networking, Application & Services (HEALTHCOM). Shenzhen Guangdong, China: IEEE, 2021, 1–4. [Google Scholar]
- 43. Gertrudes JC, Maltarollo VG, Silva RA, et al. Machine learning techniques and drug design. Curr Med Chem 2012;19(25):4289–97. [DOI] [PubMed] [Google Scholar]
- 44. Segler MHS, Preuss M, Waller MP. Planning chemical syntheses with deep neural networks and symbolic ai. Nature 2018;555(7698):604–10. [DOI] [PubMed] [Google Scholar]
- 45. Schneider P, Walters WP, Plowright AT, et al. Rethinking drug design in the artificial intelligence era. Nat Rev Drug Discov 2020;19(5):353–64. [DOI] [PubMed] [Google Scholar]
- 46. Serafim MSM, dos Santos Júnior VS, Gertrudes JC, et al. Machine learning techniques applied to the drug design and discovery of new antivirals: a brief look over the past decade. Expert Opin Drug Discovery 2021;16:1–15. [DOI] [PubMed] [Google Scholar]
- 47. Aliper A, Plis S, Artemov A, et al. Deep learning applications for predicting pharmacological properties of drugs and drug repurposing using transcriptomic data. Mol Pharm 2016;13(7):2524–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Coley CW, Barzilay R, Green WH, et al. Convolutional embedding of attributed molecular graphs for physical property prediction. J Chem Inf Model 2017;57(8):1757–72. [DOI] [PubMed] [Google Scholar]
- 49. Ragoza M, Hochuli J, Idrobo E, et al. Protein–ligand scoring with convolutional neural networks. J Chem Inf Model 2017;57(4):942–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jiménez J, Doerr S, Martínez-Rosell G, et al. Deepsite: protein-binding site predictor using 3d-convolutional neural networks. Bioinformatics 2017;33(19):3036–42. [DOI] [PubMed] [Google Scholar]
- 51. Gomes J, Ramsundar B, Feinberg EN, et al. Atomic convolutional networks for predicting protein-ligand binding affinity. arXiv preprint arXiv:1703.10603. 2017.
- 52. Liu B, Ramsundar B, Kawthekar P, et al. Retrosynthetic reaction prediction using neural sequence-to-sequence models. ACS central science 2017;3(10):1103–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Segler MHS, Waller MP. Neural-symbolic machine learning for retrosynthesis and reaction prediction. Chemistry 2017;23(25):5966–71. [DOI] [PubMed] [Google Scholar]
- 54. Jin W, Coley CW, Barzilay R, et al. Predicting organic reaction outcomes with Weisfeiler-Lehman network. arXiv preprint arXiv:1709.04555. 2017.
- 55. Korshunova M, Ginsburg B, Tropsha A, et al. Openchem: a deep learning toolkit for computational chemistry and drug design. J Chem Inf Model 2021;61(1):7–13. [DOI] [PubMed] [Google Scholar]
- 56. Krishnan SR, Bung N, Bulusu G, et al. Accelerating de novo drug design against novel proteins using deep learning. J Chem Inf Model 2021;61(2):621–30. [DOI] [PubMed] [Google Scholar]
- 57. Yang H, Sun L, Li W, et al. In silico prediction of chemical toxicity for drug design using machine learning methods and structural alerts. Front Chem 2018;6:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Youjun X, Lin K, Wang S, et al. Deep learning for molecular generation. Future Med Chem 2019;11(6):567–97. [DOI] [PubMed] [Google Scholar]
- 59. Popova M, Isayev O, Tropsha A. Deep reinforcement learning for de novo drug design. Sci Adv 2018;4(7):eaap7885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Brown N, Fiscato M, Segler MHS, et al. Guacamol: benchmarking models for de novo molecular design. J Chem Inf Model 2019;59(3):1096–108. [DOI] [PubMed] [Google Scholar]
- 61. Skalic M, Sabbadin D, Sattarov B, et al. From target to drug: generative modeling for the multimodal structure-based ligand design. Mol Pharm 2019;16(10):4282–91. [DOI] [PubMed] [Google Scholar]
- 62. Kim M, Park K, Kim W, et al. Target-specific drug design method combining deep learning and water pharmacophore. J Chem Inf Model 2020;61(1):36–45. [DOI] [PubMed] [Google Scholar]
- 63. Madaj R, Geoffrey B, Sanker A, et al. Target2denovodrug: a novel programmatic tool for in silico-deep learning based de novo drug design for any target of interest. J Biomol Struct Dyn 2021;1–6. 10.1080/07391102.2021.1898474. [DOI] [PubMed] [Google Scholar]
- 64. Bai Q, Tan S, Xu T, et al. Molaical: a soft tool for 3d drug design of protein targets by artificial intelligence and classical algorithm. Brief Bioinform 2021;22(3):bbaa161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ciccone L, Tonali N, Nencetti S, et al. Natural compounds as inhibitors of transthyretin amyloidosis and neuroprotective agents: Analysis of structural data for future drug design. J Enzyme Inhib Med Chem 2020;35(1):1145–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Li R, Wu K, Yu L, et al. Integrative pharmacological mechanism of vitamin c combined with glycyrrhizic acid against covid-19: findings of bioinformatics analyses. Brief Bioinform 2021;22(2):1161–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cannataro M, Harrison A. Bioinformatics helping to mitigate the impact of covid-19–editorial, 2021;22(2):613–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kuriwaki I, Kameda M, Hisamichi H, et al. Structure-based drug design of 1, 3, 5-triazine and pyrimidine derivatives as novel fgfr3 inhibitors with high selectivity over vegfr2. Bioorg Med Chem 2020;28(10):115453. [DOI] [PubMed] [Google Scholar]
- 69. Xu L, Yu J, Zhang Z, et al. Network bioinformatics analysis provides insight into drug repurposing for covid-19. Med Drug Discov 2021;10:100090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nocentini A, Angeli A, Carta F, et al. Reconsidering anion inhibitors in the general context of drug design studies of modulators of activity of the classical enzyme carbonic anhydrase. J Enzyme Inhib Med Chem 2021;36(1):561–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mouchlis VD, Melagraki G, Zacharia LC, et al. Computer-aided drug design of β-secretase, γ-secretase and anti-tau inhibitors for the discovery of novel Alzheimer’s therapeutics. Int J Mol Sci 2020;21(3):703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gilles A, Frechin L, Natchiar K, et al. Targeting the human 80s ribosome in cancer: from structure to function and drug design for innovative adjuvant therapeutic strategies. Cell 2020;9(3):629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Macchiagodena M, Pagliai M, Procacci P. Identification of potential binders of the main protease 3clpro of the covid-19 via structure-based ligand design and molecular modeling. Chem Phys Lett 2020;750:137489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Veeravarapu H, Malkhed V, Mustyala KK, et al. Structure-based drug design, synthesis and screening of mmaa1 inhibitors as novel anti-tb agents. Mol Divers 2021;25(1):351–66. [DOI] [PubMed] [Google Scholar]
- 75. Nikolaev GI, Shuldov NA, Anischenko AI, et al. Development of a neural network-based approach for prediction of potential hiv-1 entry inhibitors using deep learning and molecular modeling methods. In: International Symposium on Bioinformatics Research and Applications. Moscow, Russia: Springer, 2020, 304–11. [Google Scholar]
- 76. Ibrahim IM, Abdelmalek DH, Elfiky AA. Grp78: a cell’s response to stress. Life Sci 2019;226:156–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Shehroz M, Zaheer T, Hussain T. Computer-aided drug design against spike glycoprotein of sars-cov-2 to aid covid-19 treatment. Heliyon 2020;6(10):e05278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Cui J, Li F, Shi Z-L. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol 2019;17(3):181–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kelly JA, Olson AN, Neupane K, et al. Structural and functional conservation of the programmed- 1 ribosomal frameshift signal of SARS coronavirus 2 (SARS-cov-2). J Biol Chem 2020;295(31):10741–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bredenbeek PJ, Pachuk CJ, Noten AFH, et al. The primary structure and expression of the second open reading frame of the polymerase gene of the coronavirus mhv-a59; a highly conserved polymerase is expressed by an efficient ribosomal frameshifting mechanism. Nucleic Acids Res 1990;18(7):1825–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Emam M, Oweda M, Antunes A, et al. Positive selection as a key player for sars-cov-2 pathogenicity: insights into orf1ab, s and e genes. Virus Res 2021;302:198472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Banerjee S, Seal S, Dey R, et al. Mutational spectra of sars-cov-2 orf1ab polyprotein and signature mutations in the United States of America. J Med Virol 2021;93(3):1428–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Snijder EJ, Decroly E, Ziebuhr J. The nonstructural proteins directing coronavirus RNA synthesis and processing. Adv Virus Res 2016;96:59–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Yang H, Yang M, Ding Y, et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc Natl Acad Sci 2003;100(23):13190–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Jin Z, Xiaoyu D, Yechun X, et al. Structure of m pro from sars-cov-2 and discovery of its inhibitors. Nature 2020;582(7811):289–93. [DOI] [PubMed] [Google Scholar]
- 86. Kreutzer AG, Krumberger M, Chelsea Marie T, et al. Structure-based design of a cyclic peptide inhibitor of the sars-cov-2 main protease. bioRxiv 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Rut W, Lv Z, Zmudzinski M, et al. Activity profiling and crystal structures of inhibitor-bound sars-cov-2 papain-like protease: a framework for anti–covid-19 drug design. Sci Adv 2020;6(42):eabd4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Amin SA, Ghosh K, Gayen S, et al. Chemical-informatics approach to covid-19 drug discovery: Monte Carlo based qsar, virtual screening and molecular docking study of some in-house molecules as papain-like protease (plpro) inhibitors. J Biomol Struct Dyn 2020;39(13):4764–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Bhati S. Structure-based drug designing of naphthalene based SARS-cov plpro inhibitors for the treatment of covid-19. Heliyon 2020;6(11):e05558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Michalska K, Kim Y, Jedrzejczak R, et al. Crystal structures of sars-cov-2 ADP-ribose phosphatase: from the apo form to ligand complexes. IUCrJ 2020;7(5):814–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Reid AH, Taubenberger JK, Fanning TG. The 1918 Spanish influenza: integrating history and biology. Microbes Infect 2001;3(1):81–7. [DOI] [PubMed] [Google Scholar]
- 92. Tomassini J, Selnick H, Davies ME, et al. Inhibition of cap (m7gpppxm)-dependent endonuclease of influenza virus by 4-substituted 2, 4-dioxobutanoic acid compounds. Antimicrob Agents Chemother 1994;38(12):2827–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Dias A, Bouvier D, Crépin T, et al. The cap-snatching endonuclease of influenza virus polymerase resides in the pa subunit. Nature 2009;458(7240):914–8. [DOI] [PubMed] [Google Scholar]
- 94. Kowalinski E, Zubieta C, Wolkerstorfer A, et al. Structural analysis of specific metal chelating inhibitor binding to the endonuclease domain of influenza ph1n1 (2009) polymerase. PLoS Pathog 2012;8(8):e1002831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Mohanta TK, Sharma N, Arina P, et al. Molecular insights into the mapk cascade during viral infection: potential crosstalk between hcq and hcq analogues. Biomed Res Int, 2020;2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Mohanta TK, Arina P, Sharma N, et al. Role of azithromycin in antiviral treatment: enhancement of interferon-dependent antiviral pathways and mitigation of inflammation may rely on inhibition of the mapk cascade? Am J Transl Res 2020;12(12):7702. [PMC free article] [PubMed] [Google Scholar]
- 97. Yuki K, Fujiogi M, Koutsogiannaki S. Covid-19 pathophysiology: a review. Clin Immunol 2020;215:108427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hernández-Alcoceba R, Saniger L, Campos J, et al. Choline kinase inhibitors as a novel approach for antiproliferative drug design. Oncogene 1997;15(19):2289–301. [DOI] [PubMed] [Google Scholar]
- 99. Rahuel J, Rasetti V, Maibaum J, et al. Structure-based drug design: the discovery of novel nonpeptide orally active inhibitors of human renin. Chem Biol 2000;7(7):493–504. [DOI] [PubMed] [Google Scholar]
- 100. McKenna R, Supuran CT. Carbonic anhydrase inhibitors drug design. Carbonic anhydrase: mechanism, regulation, links to disease, and industrial applications 2014;75:291–323. [DOI] [PubMed] [Google Scholar]
- 101. King RD, Muggleton S, Lewis RA, et al. Drug design by machine learning: the use of inductive logic programming to model the structure-activity relationships of trimethoprim analogues binding to dihydrofolate reductase. Proc Natl Acad Sci 1992;89(23):11322–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Douangamath A, Fearon D, Gehrtz P, et al. Crystallographic and electrophilic fragment screening of the sars-cov-2 main protease. Nat Commun 2020;11(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Dai W, Zhang B, Jiang X-M, et al. Structure-based design of antiviral drug candidates targeting the sars-cov-2 main protease. Science 2020;368(6497):1331–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Ferraro M, Decherchi S, De Simone A, et al. Multi-target dopamine d3 receptor modulators: Actionable knowledge for drug design from molecular dynamics and machine learning. Eur J Med Chem 2020;188:111975. [DOI] [PubMed] [Google Scholar]
- 105. Mouchlis VD, Afantitis A, Serra A, et al. (eds). Advances in de novo drug design: From conventional to machine learning methods. Int J Mol Sci 2021;22(4):1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Elton DC, Boukouvalas Z, Fuge MD, et al. Deep learning for molecular design-a review of the state of the art. Mol Syst Design Eng 2019;4(4):828–49. [Google Scholar]
- 107. Schneider G. Mind and machine in drug design. Nat Mach Intell 2019;1(3):128–30. [Google Scholar]
- 108. Terzopoulos P. Deep Learning Protein and Molecule Representations for Drug Design, 2020.
- 109. Madaj R, Geoffrey B, Sanker A, et al. Target2denovodrugpropmax: a novel programmatic tool incorporating deep learning and in silico methods for automated de novo drug design for any target of interest. bioRxiv. 2021;2020–12.
- 110. Yunxiang L, Shi T, Wang Y, et al. Halogen bonding a novel interaction for rational drug design? J Med Chem 2009;52(9):2854–62. [DOI] [PubMed] [Google Scholar]
- 111. Yunxiang L, Liu Y, Zhijian X, et al. Halogen bonding for rational drug design and new drug discovery. Expert Opin Drug Discovery 2012;7(5):375–83. [DOI] [PubMed] [Google Scholar]
- 112. Chaturvedi D, Brahmachari G. Role of organic carbamates in anticancer drug design. In: Chemistry and Pharmacology of Naturally Occurring Bioactive Compounds, 2013, 117–40.
- 113. Li D, Deng Y, Achab A, et al. Carbamate and n-pyrimidine mitigate amide hydrolysis: Structure-based drug design of tetrahydroquinoline ido1 inhibitors. ACS Med Chem Lett 2021;12(3):389–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Bancet A, Raingeval C, Lomberget T, et al. Fragment linking strategies for structure-based drug design. J Med Chem 2020;63(20):11420–35. [DOI] [PubMed] [Google Scholar]
- 115. Shakeel A, Altaf AA, Qureshi AM, et al. Thiourea derivatives in drug design and medicinal chemistry: a short review. J Drug Des Med Chem 2016;2(1):10. [Google Scholar]
- 116. Vázquez J, López M, Gibert E, et al. Merging ligand-based and structure-based methods in drug discovery: an overview of combined virtual screening approaches. Molecules 2020;25(20):4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Xiong G-L, Zhao Y, Lu L, et al. Computational bioactivity fingerprint similarities to navigate the discovery of novel scaffolds. J Med Chem 2021;64(11):7544–54. [DOI] [PubMed] [Google Scholar]
- 118. Bacha U, Barrila J, Velazquez-Campoy A, et al. Identification of novel inhibitors of the SARS coronavirus main protease 3clpro. Biochemistry 2004;43(17):4906–12. [DOI] [PubMed] [Google Scholar]
- 119. Jain RP, Pettersson HI, Zhang J, et al. Synthesis and evaluation of keto-glutamine analogues as potent inhibitors of severe acute respiratory syndrome 3clpro. J Med Chem 2004;47(25):6113–6. [DOI] [PubMed] [Google Scholar]
- 120. Guida WC. Software for structure-based drug design. Curr Opin Struct Biol 1994;4(5):777–81. [DOI] [PubMed] [Google Scholar]
- 121. Stewart KD, Shiroda M, James CA. Drug guru: a computer software program for drug design using medicinal chemistry rules. Bioorg Med Chem 2006;14(20):7011–22. [DOI] [PubMed] [Google Scholar]
- 122. Bisht N, Sah AN, Bisht S, et al. Emerging need of today: significant utilization of various databases and softwares in drug design and development. Mini Rev Med Chem 2020;21(8):1025–32. [DOI] [PubMed] [Google Scholar]
- 123. Meyer-Almes F-J. Repurposing approved drugs as potential inhibitors of 3cl-protease of sars-cov-2: virtual screening and structure based drug design. Comput Biol Chem 2020;88:107351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Pettersen EF, Goddard TD, Huang CC, et al. Ucsf chimerax: structure visualization for researchers, educators, and developers. Protein Sci 2021;30(1):70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Trott O, Olson AJ. Autodock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010;31(2):455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Harcourt BH, Jukneliene D, Kanjanahaluethai A, et al. Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain-like protease activity. J Virol 2004;78(24):13600–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Shin D, Mukherjee R, Grewe D, et al. Papain-like protease regulates sars-cov-2 viral spread and innate immunity. Nature 2020;587(7835):657–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Poreba M, Salvesen GS, Drag M. Synthesis of a hycosul peptide substrate library to dissect protease substrate specificity. Nat Protoc 2017;12(10):2189. [DOI] [PubMed] [Google Scholar]
- 129. Lee N, Hui D, Wu A, et al. A major outbreak of severe acute respiratory syndrome in Hong Kong. N Engl J Med 2003;348(20):1986–94. [DOI] [PubMed] [Google Scholar]
- 130. Giannis D, Ziogas IA, Gianni P. Coagulation disorders in coronavirus infected patients: Covid-19, sars-cov-1, mers-cov and lessons from the past. J Clin Virol 2020;127:104362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Van Doremalen N, Bushmaker T, Morris DH, et al. Aerosol and surface stability of sars-cov-2 as compared with sars-cov-1. N Engl J Med 2020;382(16):1564–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Cevik M, Tate M, Lloyd O, et al. SARS-CoV-2, SARS-CoV, and MERS-CoV viral load dynamics, duration of viral shedding, and infectiousness: a systematic review and meta-analysis. The lancet microbe, 2020. [DOI] [PMC free article] [PubMed]
- 133. Mohanta TK, Mohanta YK. Corona virus (covid19) genome: genomic and biochemical analysis revealed its possible synthetic origin. J Appl Biotechnol Bioeng 2020;7(5):200–13. [Google Scholar]
- 134. Belouzard S, Millet JK, Licitra BN, et al. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 2012;4(6):1011–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Walls AC, Park Y-J, Tortorici MA, et al. Structure, function, and antigenicity of the sars-cov-2 spike glycoprotein. Cell 2020;181(2):281–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Hoffmann M, Kleine-Weber H, Schroeder S, et al. Sars-cov-2 cell entry depends on ace2 and tmprss2 and is blocked by a clinically proven protease inhibitor. Cell 2020;181(2):271–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Tai W, He L, Zhang X, et al. Characterization of the receptor-binding domain (rbd) of 2019 novel coronavirus: implication for development of rbd protein as a viral attachment inhibitor and vaccine. Cell Mol Immunol 2020;17(6):613–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Sorokina M, Teixeira JMC, Barrera-Vilarmau S, et al. Structural models of human ace2 variants with sars-cov-2 spike protein for structure-based drug design. Sci Data 2020;7(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Mao R, Bie L, Maofeng X, et al. Antiviral drug design based on the opening mechanism of spike glycoprotein in sars-cov-2. Phys Chem Chem Phys 2021;23(22):12549–58. [DOI] [PubMed] [Google Scholar]
- 140. Steffansen B, Nielsen CU, Frokjaer S. Delivery aspects of small peptides and substrates for peptide transporters. Eur J Pharm Biopharm 2005;60(2):241–5. [DOI] [PubMed] [Google Scholar]
- 141. Gruenfeld N, Stanton JL, Yuan AM, et al. Angiotensin converting enzyme inhibitors: 1-glutarylindoline-2-carboxylic acid derivatives. J Med Chem 1983;26(9):1277–82. [DOI] [PubMed] [Google Scholar]
- 142. Bai JPF, Amidon GL. Structural specificity of mucosal-cell transport and metabolism of peptide drugs: implication for oral peptide drug delivery. Pharm Res 1992;9(8):969–78. [DOI] [PubMed] [Google Scholar]
- 143. Silva DG, Freitas MP, da Cunha EFF, et al. Rational design of small modified peptides as ace inhibitors. Med Chem Comm 2012;3(10):1290–3. [Google Scholar]
- 144. Pinheiro JR, Bitencourt M, da Cunha EFF, et al. Novel anti-hiv cyclotriazadisulfonamide derivatives as modeled by ligand-and receptor-based approaches. Bioorg Med Chem 2008;16(4):1683–90. [DOI] [PubMed] [Google Scholar]
- 145. Antunes JE, Freitas MP, da Cunha EFF, et al. In silico prediction of novel phosphodiesterase type-5 inhibitors derived from sildenafil, vardenafil and tadalafil. Bioorg Med Chem 2008;16(16):7599–606. [DOI] [PubMed] [Google Scholar]
- 146. Goodarzi M, Da Cunha EFF, Freitas MP, et al. Qsar and docking studies of novel antileishmanial diaryl sulfides and sulfonamides. Eur J Med Chem 2010;45(11):4879–89. [DOI] [PubMed] [Google Scholar]
- 147. Matthew AN, Leidner F, Lockbaum GJ, et al. Drug design strategies to avoid resistance in direct-acting antivirals and beyond. Chem Rev 2021;121(6):3238–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Senior AW, Evans R, Jumper J, et al. Improved protein structure prediction using potentials from deep learning. Nature 2020;577(7792):706–10. [DOI] [PubMed] [Google Scholar]
- 149. Zheng W, Li Y, Zhang C, et al. Deep-learning contact-map guided protein structure prediction in casp13. Proteins 2019;87(12):1149–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Zhang Y. I-tasser server for protein 3d structure prediction. BMC Bioinformatics 2008;9(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Burbidge R, Trotter M, Buxton B, et al. Drug design by machine learning: support vector machines for pharmaceutical data analysis. Comput Chem 2001;26(1):5–14. [DOI] [PubMed] [Google Scholar]
- 152. Lo Y-C, Rensi SE, Torng W, et al. Machine learning in chemoinformatics and drug discovery. Drug Discov Today 2018;23(8):1538–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Limeng P, Naderi M, Liu T, et al. e toxpred: a machine learning-based approach to estimate the toxicity of drug candidates. BMC Pharmacol Toxicol 2019;20(1):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Akhtar S, Khan MKA, Osama K. Machine learning approaches to rational drug design. In: Computer-Aided Drug Design. Singapore: Springer, 2020, 279–306. [Google Scholar]
- 155. Lalmuanawma S, Hussain J, Chhakchhuak L. Applications of machine learning and artificial intelligence for covid-19 (sars-cov-2) pandemic: a review. Chaos Solitons Fractals 2020;110059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Cooper K, Baddeley C, French B, et al. Novel development of predictive feature fingerprints to identify chemistry-based features for the effective drug design of sars-cov-2 target antagonists and inhibitors using machine learning. ACS Omega 2021;6(7):4857–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Gordon DE, Jang GM, Bouhaddou M, et al. A sars-cov-2 protein interaction map reveals targets for drug repurposing. Nature 2020;583(7816):459–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Arús-Pous J, Patronov A, Bjerrum EJ, et al. Smiles-based deep generative scaffold decorator for de-novo drug design. J Chem 2020;12:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Skalic M, et al. Deep learning for drug design: modeling molecular shapes. PhD thesis. Catalonia, The Kingdom of Spain: Universitat Pompeu Fabra, 2019. [Google Scholar]
- 160. Martínez-Rosell G, Giorgino T, De Fabritiis G. Playmolecule proteinprepare: a web application for protein preparation for molecular dynamics simulations. J Chem Inf Model 2017;57(7):1511–6. [DOI] [PubMed] [Google Scholar]
- 161. Zhavoronkov A, Aladinskiy V, Zhebrak A, et al. Potential covid-2019 3c-like protease inhibitors designed using generative deep learning approaches. Insilico Med Hong Kong Ltd A 2020;307:E1. [Google Scholar]
- 162. Chenthamarakshan V, Das P, Hoffman SC, et al. Cogmol: target-specific and selective drug design for covid-19 using deep generative models. arXiv preprint arXiv:2004.01215. 2020.
- 163. Jin W, Barzilay R, Jaakkola T. Junction tree variational autoencoder for molecular graph generation. In: International Conference on Machine Learning (PMLR), 2018, 2323–32.
- 164. You J, Liu B, Ying R, et al. Graph convolutional policy network for goal-directed molecular graph generation. arXiv preprint arXiv:1806.02473. 2018.
- 165. Tang B, He F, Liu D, et al. Ai-aided design of novel targeted covalent inhibitors against sars-cov-2. bioRxiv 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Li Y, Jianxing H, Wang Y, et al. Deepscaffold: a comprehensive tool for scaffold-based de novo drug discovery using deep learning. J Chem Inf Model 2019;60(1):77–91. [DOI] [PubMed] [Google Scholar]
- 167. Wei L, Wen W, Rao L, et al. Cov_fb3d: a de novo covalent drug design protocol integrating the ba-samp strategy and machine-learning-based synthetic tractability evaluation. J Chem Inf Model 2020;60(9):4388–402. [DOI] [PubMed] [Google Scholar]
- 168. Geoffrey B, Madaj R, Valluri PP, et al. QPoweredTarget2DeNovoDrugPropMax: a novel programmatic tool incorporating deep learning and in silico methods for automated de novo drug design for any target of interest. 2021.
- 169. Ferreira LLG, Andricopulo AD. From chemoinformatics to deep learning: an open road to drug discovery. Future Med Chem 2019;11(5):371–4. [DOI] [PubMed] [Google Scholar]
- 170. Kong R, Yang G, Xue R, et al. Covid-19 docking server: a meta server for docking small molecules, peptides and antibodies against potential targets of covid-19. Bioinformatics 2020;36(20):5109–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Meng Z, Xia K. Persistent spectral based machine learning (PerSpect ML) for drug design. arXiv preprint arXiv:2002.00582 2020 Feb 3. [Google Scholar]