

Abstract
We conducted next generation DNA sequencing on 335 biliary tract cancers and characterized the genomic landscape by anatomic site within the biliary tree. In addition to frequent FGFR2 fusions among patients with intrahepatic cholangiocarcinoma (IHCC), we identified FGFR2 extracellular domain in-frame deletions (EIDs) in 5 of 178 (2.8%) patients with IHCC, including two patients with FGFR2 p.H167_N173del. Expression of this FGFR2 EID in NIH3T3 cells resulted in constitutive FGFR2 activation, oncogenic transformation, and sensitivity to FGFR inhibitors. Three patients with FGFR2 EIDs were treated with Debio 1347, an oral FGFR-1/2/3 inhibitor, and all showed partial responses. One patient developed an acquired L618F FGFR2 kinase domain mutation at disease progression and experienced a further partial response for 17 months to an irreversible FGFR2 inhibitor, futibatinib. Together, these findings reveal FGFR2 EIDs as an alternative mechanism of FGFR2 activation in IHCC that predict sensitivity to FGFR inhibitors in the clinic.
Keywords: cholangiocarcinoma, biliary tract, FGFR2
INTRODUCTION
Biliary tract cancers are a heterogeneous group of malignancies originating from the intrahepatic bile ducts (intrahepatic cholangiocarcinoma, IHCC), extrahepatic bile ducts (extrahepatic cholangiocarcinoma, EHCC), gallbladder (GBC), and ampulla of Vater (AMP). Recent genomic studies have revealed multiple targetable alterations in biliary cancers (1,2), and promising data have emerged from clinical trials in genetically defined patient subsets, including those with FGFR2 fusions (3–5), IDH1 mutations (6), BRAF mutations (7), mismatch repair deficiency, HER2 (ERBB2) amplification/mutation and ALK, ROS1, or NTRK translocations (1,2).
FGFR2 activating fusions (~15%) and point mutations (3–5%) are particularly common in IHCC, where they are present at higher frequencies than in any other major cancer type (1,2,4,8–13). Recently, an ATP-competitive FGFR kinase inhibitor, pemigatinib, became the first FDA-approved targeted therapy for cholangiocarcinoma, specifically among patients with an FGFR2 fusion (3). In addition to pemigatinib, other FGFR kinase inhibitors, including Debio 1347, erdafitinib, infigratinib (BGJ398), derazantinib (ARQ 087), and a covalent inhibitor, futibatinib (TAS-120), have demonstrated clinical activity in early trials, with median progression-free survival of approximately 6 months in patients with FGFR2 fusions (3,5,14–17). IHCCs with FGFR2 point mutations or amplifications appear to be less sensitive to FGFR inhibitors, highlighting the importance of additional genotype-phenotype analysis of FGFR2 variants (18). Studies dissecting the molecular mechanisms of FGFR inhibitor resistance in patients with FGFR2 fusions have revealed the emergence of secondary mutations in the FGFR2 kinase domain (5,19). Notably, the irreversible FGFR inhibitor futibatinib has been effective in treating several patients with acquired resistance to other FGFR inhibitors (5).
The importance of FGFR2 in the pathogenesis of many IHCCs led us to hypothesize that other FGFR2 activating alterations might predict sensitivity to FGFR inhibitors (11). Here, we examined the genomic features of 335 biliary tract tumors, including 178 IHCCs. We identified FGFR2 extracellular domain in-frame deletions (EIDs) as an unexpected mechanism of FGFR2 activation in IHCC, and we validated the transforming activity of these alterations in preclinical models. Importantly, all three patients with FGFR2 EIDs treated with FGFR inhibitors demonstrated tumor response, suggesting a new genomically-defined cohort of patients who may benefit from FGFR-targeted therapies.
RESULTS
Genomic Landscape of Biliary Tract Cancers
Between October 2013 and April 2019, next generation DNA sequencing (NGS) in a CLIA-certified laboratory was performed for 335 patients with biliary tract cancer, including 178 patients with IHCC (Table 1). Nearly 60% of patients presented with distant metastases at the time of their cancer diagnosis, and DNA sequencing was performed from the primary tumor (69%) or a metastatic lesion (31%). Mean tumor cellularity was 44%, and mean tumor coverage for NGS was 315X.
Table 1:
Characteristics of patients with biliary tract cancer
| Characteristic | AMP (N=30) |
EHCC (N=49) |
GBC (N=73) |
IHCC (N=178) |
Mixed (N=5) | Full Population (N=335) |
|---|---|---|---|---|---|---|
| Gender | ||||||
| Female | 11 (37%) | 17 (35%) | 53 (73%) | 90 (51%) | 3 (60%) | 174 (52%) |
| Male | 19 (63%) | 32 (65%) | 20 (27%) | 88 (49%) | 2 (40%) | 161 (48%) |
| Race | ||||||
| White | 27 (90%) | 43 (88%) | 67 (92%) | 156 (88%) | 4 (80%) | 297 (89%) |
| Asian | 0 (0%) | 4 (8%) | 2 (3%) | 5 (3%) | 1 (20%) | 12(4%) |
| Black | 1 (3%) | 0 (0%) | 0 (0%) | 6 (3%) | 0 (0%) | 7 (2%) |
| Other/Unknown | 2 (7%) | 2 (4%) | 4 (5%) | 11 (6%) | 0 (0%) | 19 (6%) |
| Age at diagnosis | ||||||
| Median age (range) | 63 [41–81] | 63 [21–89] | 63 [38–86] | 64 [18–88] | 63 [52–69] | 64 [18–89] |
| BMI at diagnosis | ||||||
| Median BMI (range) | 26 [17–36] | 27 [15–42] | 27 [18–48] | 27 [16–56] | 29 [26–35] | 27 [15–56] |
| Biopsy site | ||||||
| Primary/Local Recurrence | 24 (80%) | 37(76%) | 39 (53%) | 127 (71%) | 4 (80%) | 231 (69%) |
| Distant Metastasis | 6 (20%) | 12 (24%) | 34 (47%) | 51 (29%) | 1 (20%) | 104 (31%) |
| Resection of primary tumor | ||||||
| Yes | 18 (60%) | 27 (55%) | 26 (36%) | 40 (22%) | 2 (40%) | 113 (34%) |
| Recurrence | ||||||
| Yes | 13 (72%) | 19 (70%) | 20 (77%) | 32 (80%) | 2 (100%) | 86 (76%) |
| No | 5 (28%) | 8 (30%) | 6 (23%) | 8 (20%) | 0 (0%) | 27 (24%) |
| Metastatic disease | ||||||
| Metastatic disease at diagnosis | 10 (33%) | 18 (37%) | 41 (56%) | 117 (66%) | 3 (60%) | 189 (56%) |
| Progressed to metastatic disease | 14 (47%) | 22 (45%) | 23 (32%) | 45 (25%) | 2 (40%) | 106 (32%) |
| Total incidence of metastatic disease | 24 (80%) | 40 (82%) | 64 (88%) | 162 (91%) | 5 (100%) | 295 (88%) |
Abbreviations: AMP, ampullary carcinoma; EHCC, extrahepatic cholangiocarcinoma; GBC, gallbladder adenocarcinoma; IHCC, intrahepatic cholangiocarcinoma; Mixed, mixed hepatocellular cholangiocarcinoma; BMI, body mass index.
As seen in prior sequencing efforts (2,8–10,12,13), a large diversity of genetic alterations were identified in the biliary cancer cohort, with differences by anatomic origin within the biliary tree (Figure 1A). As expected, several alterations that predict sensitivity to targeted therapies were enriched within IHCC patients, including FGFR2 rearrangements (12%) and mutations in IDH1 (19%), IDH2 (5%), and BRAF (3%) (Supplemental Table 1). FGFR2 fusions typically involved exons 1–17 of FGFR2, and the most common fusion partners were BICC1 (3/21, 14%) and AHCYL1 (2/21, 9.5%) (Supplemental Table 2). BRAF V600E mutations, FGFR2 alterations, KRAS mutations, and NRAS mutations were all mutually exclusive in the full biliary cancer cohort. IDH1 mutations co-occurred in 1 of 21 FGFR2 rearranged tumors, 4 of 74 KRAS mutated tumors, and 2 of 10 NRAS mutated tumors. We also note that of the 74 patients with KRAS mutations identified in our cohort, 4 patients had KRAS G12C mutations, which are of high clinical interest given ongoing trials of KRAS inhibitors (20).
Figure 1: Genomic alterations in 335 biliary tract cancers identified by targeted next generation sequencing.
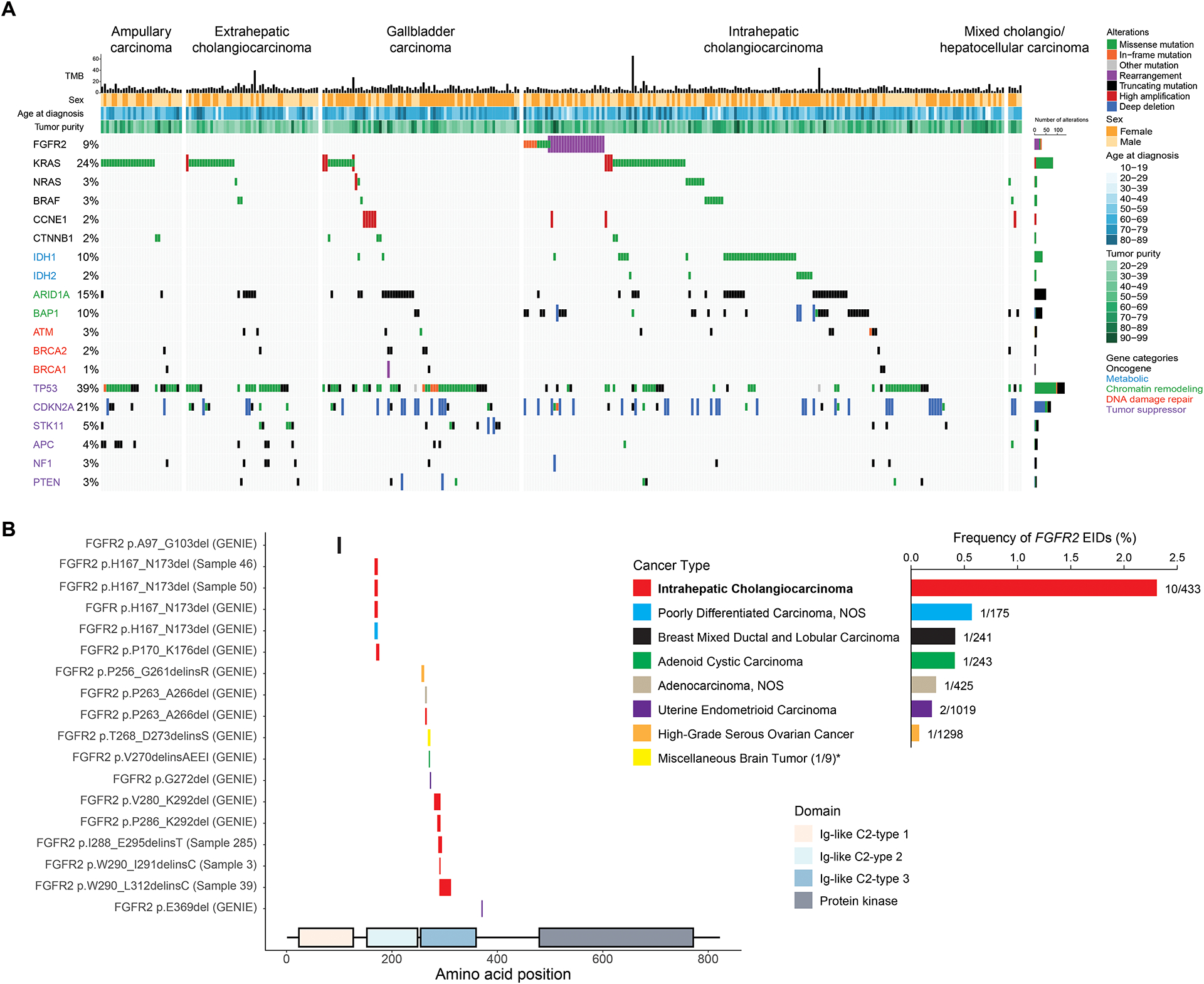
A. Co-mutation plot highlighting frequently altered genes in biliary tract cancers. Columns represent individual patients with biliary tract cancer, and rows indicate somatic genomic alterations. Each sample’s mutation per megabase (TMB) is represented by a histogram. The types of genomic alterations are color coded. Genes are listed according to the following functional classes: oncogenes, metabolic chromatin remodeling, DNA damage repair, and tumor suppressors (from top to bottom, color coded). B. Schematic depiction of the intragenic location of extracellular domain FGFR2 EIDs and their alteration frequencies by tumor type in the GENIE database. FGFR2 EIDs are enriched in samples from IHCC patients. * indicates that the “Miscellaneous brain tumor” cancer type was not included in the frequency graph due to low sample number.
Deleterious mutations of genes involved in the DNA homologous recombination (HR) pathway were identified in all biliary subtypes, most frequently involving ATM (3.9% IHCC, 4.1% EHCC, and 2.7% GBC), but with mutations in BRCA1, BRCA2, and PALB2 also observed (Supplemental Table 3). Across the full cohort, 12.2% of patient specimens had at least one alteration in HR pathway genes.
Preclinical data have suggested that MTAP deficient tumors can be therapeutically targeted by inhibitors of PRMT5 and MAT2A (21). We observed two-copy MTAP deletions in all biliary cancer subtypes (10.4% IHCC, 7.7% EHCC, 16.7% GBC, 12.5% AMP), and these deletions were associated with co-deletion of the neighboring CDKN2A and CDKN2B genes. However, only 26/46 (56.5%) of CDNK2A biallelic deletions were accompanied by two-copy MTAP deletions, suggesting the need to specifically assay for MTAP loss.
The median estimated tumor mutational burden (TMB) of the entire biliary cohort was 6.8 mutations/Mb (range 0.0–65.4) (Supplemental Figure 1). Using previously validated criteria (22), 2/178 (1.1%) IHCCs and 1/49 (2%) EHCCs were suspicious for mismatch repair deficiency based on genomic analysis. Analysis of mismatch repair proteins by immunohistochemistry confirmed that two of the three patients were mismatch repair deficient. Further genomic analysis of the third case, which had intact MLH1, MSH2, MSH6, and PMS2 by immunohistochemistry, revealed that this tumor had an estimated TMB of 44 mut/Mb and two loss of function events in MLH3 (frameshift mutation and low-level deletion). Seven additional biliary tumors had an estimated TMB greater than 15 mut/Mb (Supplemental Figure 1). None of these tumors harbored a POLE mutation, indicating other causes for their higher mutational load.
Association of Genomic Events with Patient Outcomes
We performed Cox proportional hazards regression, controlling for age, gender, presence of metastatic disease at diagnosis, and surgical resection of the primary tumor, to determine whether the main alterations identified in biliary cancer—ARID1A, BRAF, CDKN2A, FGFR2, IDH1, KRAS, and TP53— were associated with patient outcomes. Inactivating mutations or two copy deletions of CDKN2A were associated with reduced overall survival (hazard ratio [HR], 1.38; 95% confidence interval [CI], 1.01–1.88; p-value 0.04) among all patients with biliary cancer (Supplemental Figure 2A). When examining only patients with IHCC, V600E BRAF mutations were also strongly associated with reduced overall survival (HR, 4.41; 95% CI, 1.75 – 11.10; p-value 0.002) (Supplemental Figure 2B). None of the other gene alterations (in ARID1A, FGFR2, IDH1, KRAS, and TP53) were associated with overall survival time in the full biliary cancer population.
We next examined whether alterations in ARID1A, BRAF, CDKN2A, FGFR2, IDH1, KRAS, and TP53 were associated with real-world progression-free survival (rwPFS) on first-line chemotherapy in patients who presented with metastatic disease. Using data abstracted from clinical records, rwPFS measures how long patients are treated with a chemotherapy regimen and is derived by using the date of completion of chemotherapy or death as its events. Controlling for age and gender, TP53 alterations were associated with decreased rwPFS on first-line chemotherapy (HR, 2.21; 95% CI, 1.36–3.59; p-value 0.001) and decreased OS (HR, 2.36; 95% CI, 1.40–4.00; p-value 0.001) among patients with metastatic IHCC (Supplemental Figure 3A and 3B).
Identification of Extracellular Domain FGFR2 In-Frame Deletions
In addition to the 12% of IHCC patients whose tumor harbored an FGFR2 fusion, we found four oncogenic FGFR2 extracellular activating mutations: p.F276C (n=1), p.C382R (n=2), and p.Y375C (n=1). However, upon further examination of this cohort for other types of FGFR2 alterations, we also identified 5 (2.8%) patients with extracellular domain FGFR2 in-frame deletions (Table 2). Two IHCCs harbored an identical exon 5 deletion, FGFR2 p.H167_N173del, while the other three FGFR2 indels were located in exon 7 (Figure 1B). Activating extracellular domain alterations have been identified in other oncogenic receptor tyrosine kinases, such as EGFR, HER2, PDGFRA and RET (23–26). In addition, FGFR2 extracellular domain activating mutations, and rarely small in-frame FGFR2 deletions, are known to cause autosomal dominant congenital craniosynostosis syndromes (27–31). Thus, we investigated whether these extracellular domain FGFR2 in-frame deletions (EIDs) might be oncogenic drivers in cholangiocarcinoma and amenable to inhibition by FGFR inhibitors.
Table 2:
FGFR2 Extracellular Activating In-Frame Deletions Identified in Intrahepatic Cholangiocarcinoma Patients by Oncopanel and in any Malignancy by GENIE
| Tumor Type | Amino Acid Change | Database | Prediction | Confidencea | Domain | Role of WT Amino Acid | Figure | Mapping #b |
|---|---|---|---|---|---|---|---|---|
| Intrahepatic Cholangiocarcinoma | p.H167_N173del | Oncopanel Sample 46 | Pathogenic | High | Ig-like C2-type D2 | Extended recognition of FGF Dimerization Beta-strand structure |
2A, 2B | 1 |
| Intrahepatic Cholangiocarcinoma | p.H167_N173del | Oncopanel Sample 50 | Pathogenic | High | Ig-like C2-type D2 | Extended recognition of FGF Dimerization Beta-strand structure |
2A, 2B | 1 |
| Intrahepatic Cholangiocarcinoma | p.W290_L312delinsC | Oncopanel Sample 39 | Pathogenic | Moderate | Ig-like C2-type D3 | Beta-sheet and turn, folding of D3 domain. | Supp. 8 | 2 |
| Intrahepatic Cholangiocarcinoma | p.I288_E295delinsT | Oncopanel Sample 285 | Pathogenic | High | Ig-like C2-type D3 | Recognition of FGF Beta-strand structure, folding of D3 domain |
Supp. 9 | 3 |
| Intrahepatic Cholangiocarcinoma | p.W290_I291WI>C | Oncopanel Sample 3 | Pathogenic | Reported (43) | Ig-like C2-type D3 | Beta-strand structure, folding of D3 domain | Supp. 10 | 4 |
| Breast Mixed Ductal and Lobular Carcinoma | p.A97_G103del | GENIE MSK-P-0011392-T01-IM5 | Pathogenic | Low (not in X-ray) |
Ig-like C2-type D1 | Beta-strand structure, folding of D1 domain (not in X-ray, inferred from FGFR1 NMR) | Supp. 11 | 5 |
| Intrahepatic Cholangiocarcinoma | p.H167_N173del | GENIE MSK-P-0032563-T01-IM6 | Pathogenic | High | Ig-like C2-type D2 | Extended recognition of FGF Dimerization Beta-strand structure |
2A, 2B | 1 |
| Poorly Differentiated Carcinoma, NOS | p.H167_N173del | GENIE MSK-P-0020056-T01-IM6 | Pathogenic | High | Ig-like C2-type D2 | Extended recognition of FGF Dimerization Beta-strand structure |
2A, 2B | 1 |
| Intrahepatic Cholangiocarcinoma | p.P170_K176del | GENIE VICC-482349-unk-1 | Pathogenic | High | Ig-like C2-type D2 | Recognition of FGF Dimerization Beta-strand structure |
Supp. 12 | 6 |
| High-Grade Serous Ovarian Cancer | p.P256_G261delinsR | GENIE UHN-OCT998164-ARC1 | Pathogenic | Moderate | Ig-like C2-type D3 | Extended non-structural region linking D3 to D2 | Supp. 13 | 7 |
| Intrahepatic Cholangiocarcinoma | p.P263_A266del | GENIE MSK-P-0033098-T01-M6 | Pathogenic | Moderate | Ig-like C2-type D3 | Beta-strand structure, folding of D3 domain | Supp. 14 | 8 |
| Adenocarcinoma, unspecified | p.P263_A266del | GENIE NKI-9NDA-J5ML | Pathogenic | Moderate | Ig-like C2-type D3 | Beta-strand structure, folding of D3 domain | Supp. 14 | 8 |
| Miscellaneous Brain Tumor | p.T268_D273delinsS | GENIE MSK-P-0001890-T01-IM3 | Pathogenic | Moderate | Ig-like C2-type D3 | Beta-strand and turn structure, folding of D3 domain | Supp. 15 | 9 |
| Adenoid Cystic Carcinoma | p.V270delinsAEEI | GENIE VICC-349321-unk-1 | Non-pathogenic | Moderate | Ig-like C2-type D3 | Single position in turn | Supp. 16 | 10 |
| Uterine Endometrioid Carcinoma | p.G272del | GENIE VICC-580559-unk-1 | Non-pathogenic | Moderate | Ig-like C2-type D3 | Single position in turn | Supp. 17 | 11 |
| Intrahepatic Cholangiocarcinoma | p.V280_K292del | GENIE MSK-P-0018110-T01-IM6 | Pathogenic | High | Ig-like C2-type D3 | Extended recognition of FGF Beta-strand and turn structure, folding of D3 domain |
Supp. 18 | 12 |
| Intrahepatic Cholangiocarcinoma | p.P286_K292del | GENIE MSK-P-0036277-T01-IM6 | Pathogenic | High | Ig-like C2-type D3 | Recognition of FGF Beta-strand structure, folding of D3 domain |
Supp. 19 | 13 |
| Uterine Endometrioid Carcinoma | p.E369del | GENIE MSK-P-0014582-T01-IM6 | Non-pathogenic | Low (not in X-ray) | - | Single position in a non-structured region (not in X-ray), not conserved among FGFRs | - | 14 |
Abbreviations: GENIE, Genomics evidence neoplasia information exchange; NOS, Not otherwise specified.
Confidence on the prediction of pathogenicity based on mapping to protein sequence, overlap with reference mutations (see Supplemental Figure 7) (36–51) and structural role in the three-dimensional structure of FGFR2 (see corresponding Figures, last column).
As depicted on the map in Supplemental Figure 7 of the FGFR2 the protein sequence.
We first sought to further define the prevalence of FGFR2 EIDs by examining the Genomics Evidence Neoplasia Information Exchange (GENIE) database, a resource generated by an international consortium of 19 institutions and consisting of large numbers of genomic profiles generated by multiple NGS platforms (32). We identified 13 additional tumors that carried FGFR2 EIDs in the GENIE database (Table 2, Figure 1B). Notably, we found two additional FGFR2 p.H167_N173del EIDs in an IHCC and a poorly differentiated carcinoma of unknown primary. Other FGFR2 EIDs occurred in patients with breast, ovarian, brain, adenoid cystic and endometrioid carcinomas, although at much lower frequency than in IHCC (Figure 1B). Next, we considered the other genomic events in tumors with FGFR2 rearrangements or FGFR2 EIDs and noted that tumors with these two different types of FGFR2 alterations exhibited similar patterns of mutational co-occurrence (Supplemental Figure 4). We then examined the ClinVar database (33) and identified four FGFR2 EIDs overlapping with those detected in IHCC that were reported to be pathogenic for congenital craniosynostosis syndromes (Supplemental Figure 5). Studies from congenital craniosynostosis syndromes have demonstrated that interference with one of the three disulfide cysteine bonds in the extracellular domain of FGFR2, or the insertion of an additional cysteine which can lead to the creation of rogue disulfide bridges, can be activating (34). Consistent with this, two of the FGFR2 EIDs we identified, p.W290_I291WI>C and p.W201_L223delinsC, result in the addition of an extracellular cysteine.
Finally, we examined the isoform expression of FGFR2 in one patient for whom extra tumor tissue was available. FGFR2 has two major isoforms with distinct FGF ligand affinities that are encoded via alternative splicing of exon 6 into exons 7b or 7c (generating FGFR2 IIIb (NM_001144913.1) or FGFR2 IIIc (NM_000141.5) isoforms) (35). Using RNA extracted from tumor tissue from IHCC Patient 46 with the FGFR2 p.H167_N173del alteration, we found that this FGFR2 EID was expressed in the context of the FGFR2 IIIb isoform as has been reported for all FGFR2 fusions described to date in IHCC (Supplemental Figure 6).
In Silico Evaluation of FGFR2 EID Oncogenicity
Using the crystal structure of wild-type FGFR2 (pdb:1EV2), we modeled the structural impact of FGFR2 EIDs in silico. FGFR2 p.H167_N173del is an extended deletion in the Ig-like C2-type D2 extracellular domain, with most of its sequence (His167 to Pro170) structured as a beta-strand and Ala171, Ala172 and Asn173 within a turn (Figure 2A, 2B and Supplemental Figure 7). Consequently, deletion of this region is predicted to markedly alter the three-dimensional structure of an important region that forms a beta-sheet within the D2 domain and is involved in both FGF ligand binding and FGFR2 dimerization (Figure 2A, 2B, beige ribbon with green highlighted amino acids). Several of the deleted amino acids interact with FGF residues (blue ribbon in Figure 2B), including (1) a hydrogen-bond network including the FGF Tyr103 side chain, the D2 Asn173 side chain, the D2 Ala168 backbone oxygen and two water molecules, (2) direct hydrogen-bonding of the FGF Tyr24 side chain to the D2 Ala168 backbone nitrogen, and (3) extensive van der Waals contacts between FGF and the side chains of Ala168, Pro170 and Asn173. The deleted region also likely plays a role in FGFR2 dimerization, as the Asn173 side chain forms a hydrogen-bond with the Ser220 side chain of the other FGFR2 monomer (red ribbon in Figure 2B), and Ala172 and Asn173 of both monomers are in van der Waals contact. Taken together, these structural findings suggest that deletion of p.H167_N173 from the extracellular domain of FGFR2 may cause either ligand-independent FGFR2 activation or increased ligand affinity and dimerization, both of which would result in oncogenic activity and cellular transformation.
Figure 2: FGFR2 EIDs are oncogenic and confer sensitivity to FGFR inhibitors.
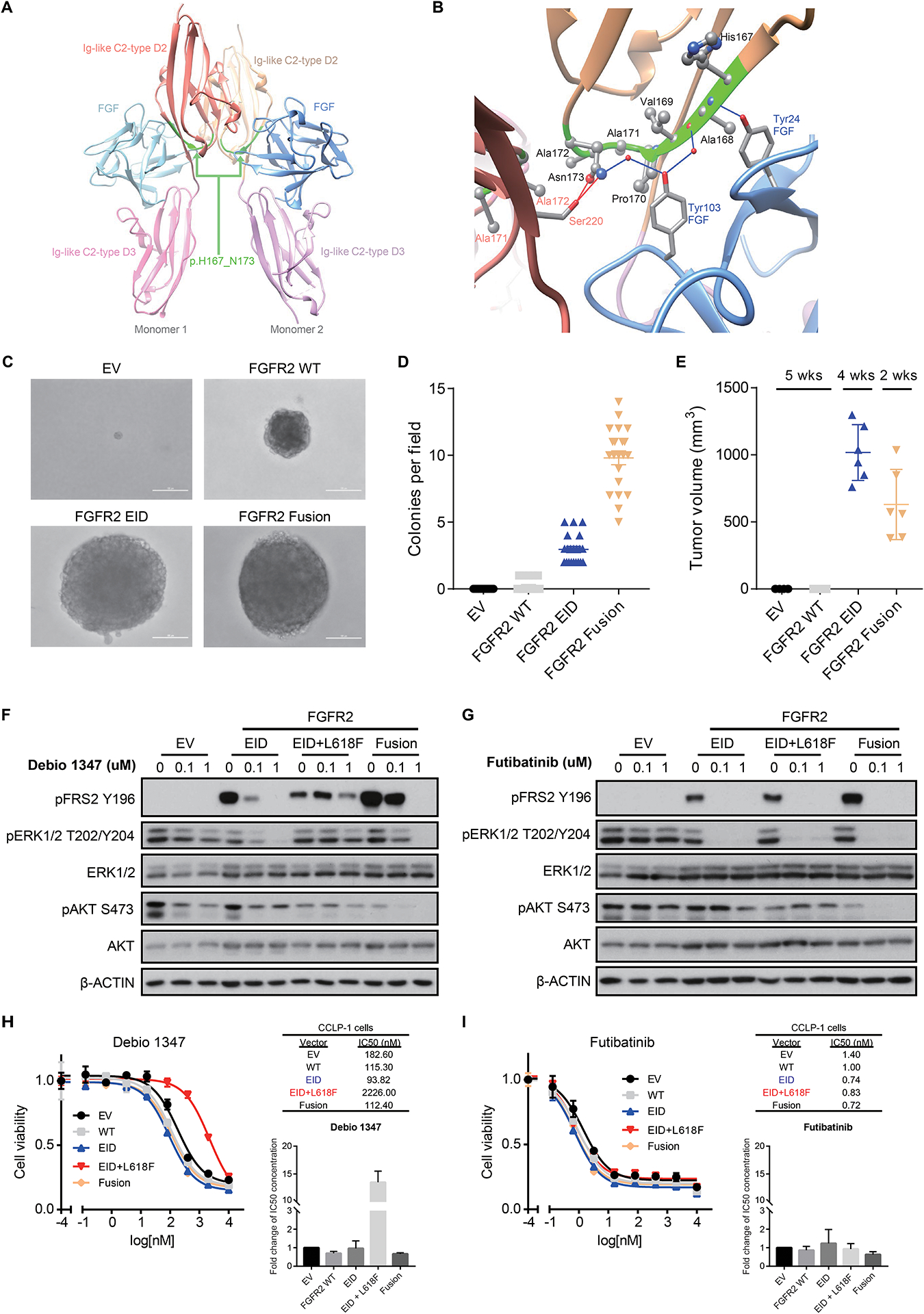
A, B. Modeling alteration p.H167_N173del within the three-dimensional structure of FGFR2. Panel A shows a global view of the p.H167_N173 deletion (ribbon highlighted in green) located in both Ig-like C2-type D2 extracellular domains of FGFR2 dimeric structure; refer to Supplemental Figure 7 (deletion #1) for mapping onto the protein sequence. As shown more closely in panel B, p.H167_N173 (ribbon highlighted in green) is an extended deletion, mostly included in a beta-strand of the C2-type D2 domain (ribbon in beige), and makes several intermolecular interactions with both the FGF ligand (ribbon and residue names in blue) and the C2-type D2 domain of the other monomer (ribbon and residue names in red). Intermolecular hydrogen-bonds (direct or water-mediated) between p.H167_N173 residues and FGF or the other monomer are shown as blue and red lines, respectively. Van der Waals contacts are not displayed explicitly. C, D. Expression of FGFR2 p.H167_N173del in NIH3T3 cells is sufficient for transformation in soft agar colony formation assays. Cells were transduced with retroviral vectors containing empty vector control (EV), wild-type FGFR2 (FGFR2 WT), FGFR2 p.H167_N173del (FGFR2 EID), or FGFR2-OPTN fusion (FGFR2 Fusion). Representative images (C) and quantification of number of colonies per field of view (D) are shown. Line and bars indicate mean with standard error, with 21 fields of view assessed across three biologic replicates. E. Volume of subcutaneous tumors forming in NSG mice following implantation of NIH3T3 cells expressing the indicated constructs (n=6 mice per condition). Both the FGFR2 p.H167_N173del EID and the FGFR2-PHGDH fusion (FGFR2 Fusion) induce tumor formation. Expression of the FGFR2-PHGDH fusion led to slightly faster tumor growth necessitating euthanasia at an earlier time point. Line and bars indicate mean with standard deviation. F, G. Expression of FGFR2 p.H167_N173del (EID), the EID with an L618F mutant kinase domain (EID+L618F), and FGFR2-PHGDH fusions (Fusion) lead to constitutive FGFR signaling (induction of FRS2) in NIH3T3 cells. FGFR2 EIDs and fusions are sensitive to treatment with Debio1347 (reduction in pFRS2 and pERK), whereas the L618F mutation causes resistance (F); all three of these FGFR2 alterations are inhibited by futibatinib (TAS-120) (G). H, I. Cell viability assays in CCLP-1 cells harboring empty vector control (EV), wild-type FGFR2 (WT), FGFR2 p.H167_N173del (EID), the co-occurring FGFR2 kinase mutation L618F (EID+L618F), or FGFR2-PHGDH fusion (Fusion) and treated with Debio 1347 (H) or futibatinib (I). Drug response measurements were performed in three independent experiments with each consisting of two technical replicates. Inset graphs demonstrate the average fold change in IC50 between conditions. Representative dose response curves and IC50 values are shown from a single experiment, with error bars on the curves representing standard deviation from 2 technical replicates.
Using a similar in silico approach, we evaluated the pathogenicity of the other FGFR2 EIDs (Table 2). The level of confidence for each prediction was based on mapping to protein sequence, overlap with reference mutations (Supplemental Figure 7) (36–51) and assessing each alteration’s role within FGFR2’s three-dimensional X-ray structure (pdb:1EV2, Supplemental Figures 8–19). As a second example, FGFR2 p.I288_E295delinsT represents a deletion in the Ig-like C2-type D3 extracellular domains (Supplemental Figure 9). Most of its sequence (Ile288 to His293) is structured as a beta-strand while Val294 and Glu295 are within a less structured region, likely representing a turn. Similar to the FGFR2 p.H167_N173del, deletion of this region would likely profoundly modify the three-dimensional structure of an important beta-sheet within the D3 domain (pink ribbon in Supplemental Figure 9), and might also alter recognition of the FGF ligand (cyan ribbon in Supplemental Figure 9) since the deleted D3 Ile288 hydrophobic side chain is in van der Waals contact with the Phe17 side chain of FGF.
Overall, eleven of the 14 identified sequence-unique FGFR2 EIDs were predicted to be pathogenic, five with high confidence, based upon their location within the FGFR2 protein. Longer lengths of the altered sequences correlated with their predicted impact on protein structure and consequently were an influential determinant of inferred pathogenicity. As in the case of FGFR2 p.H167_N173del, the remaining FGFR2 EIDs identified as high confidence for pathogenicity were located in one or more critical structural regions responsible for protein folding, FGF binding, or FGFR2 dimerization. Two FGFR2 EIDs were not resolved within the FGFR2 crystal structure and consequently were not clearly evaluable for pathogenicity.
FGFR2 EIDs are Transforming In Vitro and In Vivo
We next tested the transforming capacity of the recurrent FGFR2 p.H167_N173del variant in functional studies. To this end, we transduced 3T3 mouse embryonic fibroblast cells with viral vectors carrying FGFR2 p.H167_N173del and used wild-type FGFR2, the FGFR2-OPTN fusion, and an empty vector as controls. Expression of FGFR2 p.H167_N173del promoted cellular transformation, anchorage independent colony formation and tumor formation upon subcutaneous implantation in immunodeficient mice, to levels comparable to the FGFR2 fusion (Figure 2C–E). Immunoblot analysis demonstrated that FGFR2 p.H167_N173del exhibited constitutive FGFR2 kinase activity, as reflected by phosphorylation of its substrate, the docking/scaffolding adaptor protein FRS2 (Figure 2F, lanes 1 and 4, “EID” represents FGFR2 p.H167_N173del).
In Vitro Response of FGFR2 EIDs to FGFR Inhibition
Notably, the FGFR inhibitors Debio 1347 and futibatinib both diminished phosphorylation of FRS2 in 3T3 cells transfected with FGFR2 p.H167_N173del (Figure 2F and 2G), and this reduction was similar to that observed in cells expressing the FGFR2-PHGDH fusion protein. We also observed coordinate reduction in downstream MAP kinase pathway activity, as assessed by ERK1/2 phosphorylation. Furthermore, the above FGFR inhibitors potently inhibited the growth of FGFR-dependent human CCLP-1 IHCC cells (5) expressing either FGFR2 p.H167_N173del or the FGFR2-PHGDH fusion (Figure 2H and 2I). Thus, FGFR2 p.H167_N173del is a novel constitutively active oncogenic form of FGFR2 that is effectively inhibited by FGFR2 kinase inhibitors.
Clinical cases of IHCCs harboring FGFR2 EIDs treated with FGFR inhibitors
Given the known importance of FGFR2 to cholangiocarcinoma pathogenesis, we treated three patients with advanced FGFR2 EID-positive IHCC with the investigational agent Debio 1347, an orally available FGFR-1,2,3 ATP competitive inhibitor, and observed a partial response in all three patients.
Patient 46 is a woman who presented at 48 years of age with right upper quadrant pain emanating from a large liver mass (oncological history summarized in Figure 3A). Partial hepatectomy removed a 9.6 cm T1N0 well differentiated IHCC, and she was subsequently treated with 6 months of adjuvant gemcitabine. After 5 years of surveillance, a recurrent mass was identified, and repeat partial hepatectomy removed an isolated 4.2 cm hepatic recurrence. The patient was treated with 6 months of adjuvant gemcitabine/cisplatin. The cancer again recurred in the liver, but after an ablation procedure, local therapies were no longer possible due to the development of multiple liver lesions.
Figure 3: Treatment course for Patient 46 with intrahepatic cholangiocarcinoma and FGFR2 p.H167_N173del EID.
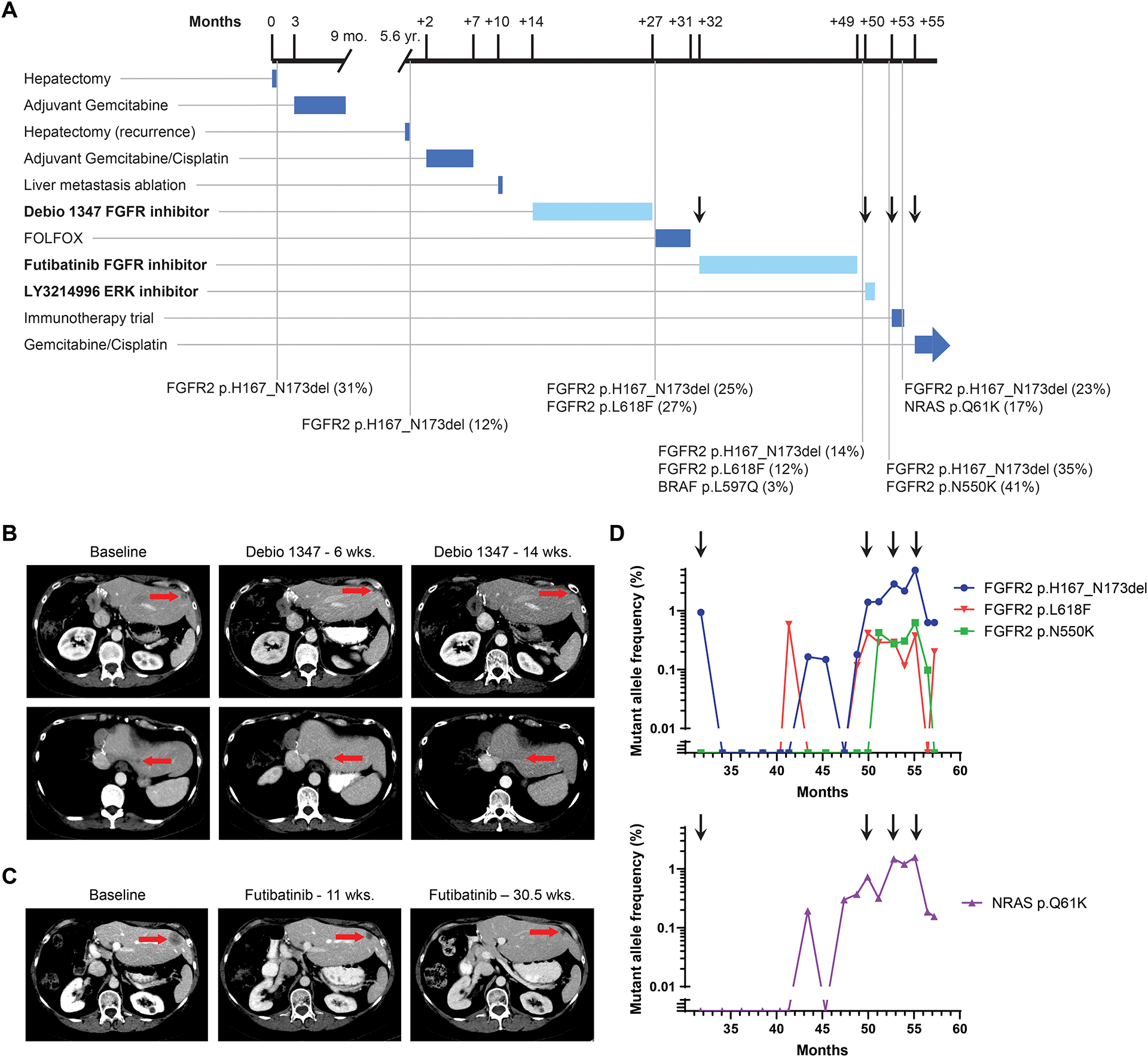
A. Timeline depicts patient’s treatment course and duration of therapy. Notable somatic mutations detected on liver biopsies (black vertical lines) are listed below the timeline. The mutation allele fraction (MAF) is listed as a percentage next to the genomic alteration. Arrows correspond to time points when a new treatment was started, in reference to droplet digital PCR analysis in panel D. Treatments with FGFR and MAPK targeted agents are highlighted in bold and duration delineated with a lighter blue color. B. Computed tomography (CT) scans demonstrating the patient’s liver lesions at baseline and after 6 and 14 weeks of Debio 1347 treatment. C. CT scans evaluating the patient’s liver lesions at baseline and after 11 and 30.5 weeks of futibatinib (TAS-120) treatment. D. Selected alterations identified retrospectively in circulating cell free DNA by droplet digital PCR of serially banked plasma samples. Arrows correspond to time points when a new treatment was started in the clinical timeline in panel A.
NGS testing in a CLIA-certified laboratory was performed on DNA isolated from her second hepatectomy specimen, and FGFR2 p.H167_N173del (mutation allele fraction [MAF], 12%) was identified without another clear driver event (Figure 3A). She was enrolled and treated on a phase 1 trial of Debio 1347 (NCT01948297)(14) resulting in a 51% tumor reduction (RECISTv1.1) and a progression-free survival time of 13 months (Figure 3B).
Tumor biopsy at the time of progression on Debio 1347 revealed persistence of FGFR2 p.H167_N173del (MAF, 25%) and development of an FGFR2 p.L618F kinase domain mutation (MAF, 27%; Figure 3A). This mutation was previously undescribed, although other FGFR2 p.L618 variants (e.g., p.L618V and p.L618M) cause acquired resistance to infigratinib, another FGFR-1,2,3 ATP competitive inhibitor, in patients with FGFR2 fusion-positive IHCC (19,52). We expressed the p.L618F mutation, in the context of FGFR2 p.H167_N173del, in NIH3T3 cells. In vitro studies demonstrated that this mutation conferred resistance to Debio 1347, as reflected by sustained FGFR2 signaling with persistent FRS2 and ERK1/2 phosphorylation (Figure 2F). In contrast, the irreversible FGFR1–3 inhibitor futibatinib —which has been shown to overcome multiple mutations that cause resistance to ATP-competitive FGFR inhibitors in vitro and in patients (5)—retained activity against this mutation in NIH3T3 cells (Figure 2G). We also tested this mutation in the FGFR-dependent IHCC cell line, CCLP-1. FGFR2 p.L618F caused a >13-fold increase in IC50 for Debio 1347 as compared to wild-type, whereas futibatinib efficacy was unaffected (Figure 2H and 2I).
As her tumor sample underwent repeat NGS, the patient was treated with FOLFOX (5-fluorouracil, leucovorin, oxaliplatin), a standard chemotherapy regimen for patients with advanced cholangiocarcinoma (53). The best tumor response was stable disease, and this regimen was stopped after disease progression was observed on imaging after 5 months of therapy (Figure 3A). At the time of tumor progression, a single-patient investigational new drug (IND) application was pursued to enable treatment with futibatinib. Treatment with futibatinib resulted in a partial response, with a 61% tumor reduction and a response duration of 17 months (Figure 3C). The same liver lesion that was biopsied after development of acquired resistance to Debio 1347 was biopsied again after the development of resistance on futibatinib. In addition to the FGFR2 p.H167_N173del (MAF, 14%) and p.L618F (MAF, 12%) alterations, NGS revealed a BRAF p.L597Q mutation at a low allele frequency (MAF, 3%) (Figure 3A).
In light of the BRAF p.L597Q mutation, we hypothesized that treating the patient with an ERK inhibitor might provide further disease control. However, her disease progressed after 2 months of treatment with the LY3214996 ERK inhibitor. Tumor biopsy of a different liver lesion, performed during treatment with the LY3214996 ERK inhibitor, revealed FGFR2 p.H167_N173del (MAF, 35%) and a new FGFR2 p.N550K (MAF, 41%) kinase domain mutation, which has previously been observed in the context of acquired resistance to FGFR inhibitors and has reduced sensitivity to Debio 1347 and futibatinib in vitro (Figure 3A) (5,19). Circulating cell free DNA (cfDNA) analysis (Guardant360) performed one week after cessation of ERK inhibitor treatment identified the FGFR2 p.H167_N173del (MAF, 1.3%) and FGFR2 p.N550K (MAF, 0.2%) kinase domain mutations and also revealed an NRAS p.Q61K (MAF, 0.9%) mutation, suggesting development of several mechanisms of resistance to FGFR2 and MAPK-directed therapy. A subsequent tumor biopsy done before starting treatment on an investigational immunotherapy study, and which targeted the same lesion that was biopsied during ERK inhibitor therapy, revealed FGFR2 p.H167_N173del (MAF, 23%) and also demonstrated NRAS p.Q61K (MAF, 17%), which was previously identified in cfDNA testing.
Retrospectively, we applied droplet digital polymerase chain reaction (ddPCR) to circulating cfDNA extracted from 17 serially collected blood samples to identify the temporal variation in tumor-associated alterations (Figure 3D). Data are from samples collected at the start of futibatinib and every 4–8 weeks thereafter; samples collected before this time did not have sufficient tumor DNA for mutation detection, presumably because of insufficient shedding of tumor DNA. As expected, the FGFR2 p.H167_N173del allele fraction decreased during treatment response to futibatinib, increased at the time of resistance, and decreased again with cancer response to gemcitabine/cisplatin. Allele fractions for both FGFR2 p.L618F and FGFR2 p.N550K remained low during futibatinib therapy, rose afterward, and then declined during gemcitabine/cisplatin treatment. In contrast, the NRAS p.Q61K allele fraction increased as tumor resistance developed to futibatinib, decreased modestly during ERK inhibitor therapy, and decreased again during the treatment response to gemcitabine/cisplatin. These data are consistent with FGFR2 p.H167_N173del conferring strong oncogene-addiction in IHCC and with the RAS/MEK/ERK pathway being a major output of FGFR signaling. The identification of polyclonal secondary kinase domain mutations upon resistance to FGFR inhibition in FGFR2 p.H167_N173del-driven IHCC suggests similar resistance mechanisms to those in FGFR2-fusion-positive tumors (19). By contrast, acquired mutations in MAPK pathway proteins have not previously been reported in the context of FGFR2 fusion tumors, although a more comprehensive analysis is required in both settings.
Patient 285 is a 65-year-old man with a history of hepatitis C who was found to have a solitary asymptomatic liver mass during radiological surveillance for an HPV+ squamous cell carcinoma of the head and neck. A partial hepatectomy removed a 5.2 cm T1NX poorly differentiated adenocarcinoma consistent with intrahepatic cholangiocarcinoma. The patient declined adjuvant therapy and was placed on surveillance. One year later, he was found to have a peritoneal nodule that was biopsied and confirmed to be recurrent cholangiocarcinoma. The patient declined cytotoxic chemotherapy, but his tumor was found to harbor an FGFR2 p.I288_E295delinsT alteration (MAF, 15%) by NGS of his peritoneal nodule biopsy. He did not qualify for clinical trials and was treated with Debio 1347 on a single patient IND. Treatment with Debio 1347 resulted in a partial response, with a 50% reduction in tumor burden (Figure 4A). The response is ongoing after 24 months.
Figure 4: Patients with FGFR2 EIDs have prolonged clinical responses to FGFR inhibitors.
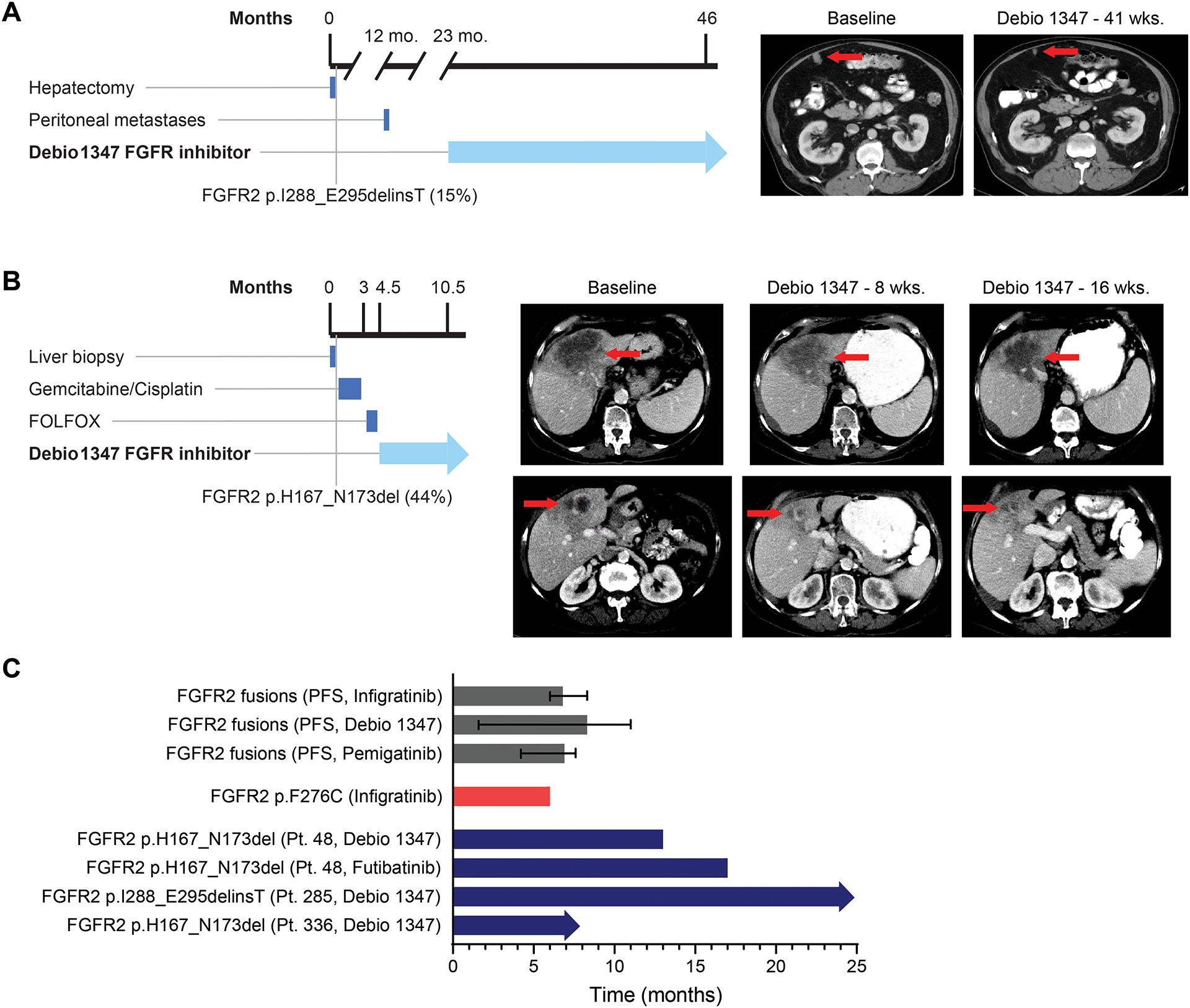
A. Timeline and computed tomography scans demonstrating Patient 285’s peritoneal lesion at baseline and after 41 weeks of Debio 1347 treatment. B. Timeline and computed tomography scans demonstrating Patient 336’s liver mass at baseline and after 8 and 16 weeks of treatment with Debio 1347. C. Bar graph depicting progression-free survival (PFS) of patients with cholangiocarcinoma harboring an FGFR2 fusion treated with infigratinib (17), Debio 1347, and pemigatinib (3). Error bars indicate 95% confidence intervals. Graph also shows time on treatment for a cholangiocarcinoma patient with an FGFR2 F276C extracellular domain mutation who was treated with infigratinib (41), and time on treatment for three IHCC FGFR2 EID patients described in this manuscript. Arrows indicate that treatment is ongoing.
Patient 336 was identified with FGFR2 p.H167_N173del (MAF, 44%), although was not included in our patient cohort described above due to diagnosis after the closure date for the cohort. This patient is a 75-year-old woman with a history of stage IA marginal zone lymphoma of the neck (treated with radiation) who was found to have a 10 cm liver mass during imaging for lymphoma surveillance. Liver biopsy showed a CK7+/CK20− adenocarcinoma consistent with IHCC. A PET/CT scan showed an FDG-avid liver mass and multiple pulmonary nodules. She was initially treated with gemcitabine/cisplatin. However, after three cycles of treatment over 9 weeks, she developed new liver lesions, and her lung lesions increased in size. She was then treated with two cycles of FOLFOX but had difficulty tolerating this treatment. Due to the FGFR2 p.H167_N173del EID, she began treatment on a single patient IND application with Debio 1347. The patient experienced a partial response with a 36% decrease in tumor burden within liver lesions, lung lesions, and lymphadenopathy observed in restaging scans at 16 weeks and she continues to respond after 7 months of treatment (Figure 4B).
We next examined the relative responses of patients with FGFR2 rearrangements, point mutations, and EIDs to the FGFR-targeted therapies pemigatinib, infigratinib, Debio 1347, and futibatinib. While a limited number of patients have been treated, the duration of response in patients with FGFR2 EIDs compares favorably to the response times for patients with fusions or point mutations (Figure 4C) (3,17,41). Likewise, partial responses observed in each of these patients compares favorably with the overall response rate of approximately 25–35% reported for the different FGFR inhibitors in patients with IHCC harboring FGFR2 fusions (3,17). These findings suggest that the degree of oncogenic addiction to FGFR2 may vary across these mutational contexts and support further clinical evaluation of FGFR inhibitors in patients with FGFR2 EIDs.
DISCUSSION
Insights into the genomics of biliary tract cancer have led to new therapy options for patients with these aggressive malignancies (1–3,6,7,54). Our data, along with previous reports, underscore the high frequency of targetable alterations in IHCC and, accordingly, the importance of clinical genomic sequencing to guide therapy choices (2,8–10,12,13). These studies also highlight the substantial molecular heterogeneity of IHCC, as evidenced by the range of driver mutations observed and the observation that no single gene was altered in more than 40% of our patients (8). Thus, even with the initial success of targeting several molecular subtypes of IHCC, identifying additional druggable alterations remains a critical need and important goal. In addition to the well-known targets in IHCC such as FGFR2 rearrangements, IDH1 mutations, and BRAF V600E mutations, we identified alterations in homologous recombination repair pathway genes, KRAS G12C mutations, and 2-copy MTAP deletions that are targets of emerging and investigational therapies (20,21,55).
Our genomic studies of biliary tract tumors highlight the particularly prominent role of the FGFR2 pathway in driving IHCC. We identified FGFR2 fusions, EIDs and point mutations in 30 of 178 IHCCs (16.9%), whereas none of the 73 GBCs or 49 EHCCs showed FGFR2 alterations. The role of FGFR2 as an oncogenic driver is further emphasized by the fact that none of the 30 IHCCs with FGFR2 alterations had co-occurring KRAS, NRAS, or BRAF V600E mutations. Notably, we identified FGFR2 EIDs as a new class of recurrent FGFR2 alterations in IHCC that are transforming in vitro and in vivo and show sensitivity to FGFR inhibitors. Importantly, these FGFR2 EIDs were present in 3% of our patients with IHCC and all three patients with FGFR2 EIDs treated with an FGFR inhibitor demonstrated deep and durable disease responses. Furthermore, while FGFR2 EIDs were enriched in IHCC, they are also present in multiple other malignancies, including breast, lung, and endometrioid cancers, suggesting additional potential treatment opportunities beyond IHCC. Future studies are needed to refine the incidence estimates of FGFR2 EIDs in both IHCC and other malignancies. While we found that 3% of our IHCC patients had FGFR2 EIDs, this frequency may be influenced by referral patterns to a large tertiary cancer center. In addition, the commonly low tumor purity of cholangiocarcinoma specimens may reduce sensitivity for detecting FGFR2 EIDs, and the OncoPanel assay used in the current study has not been validated for clinical reporting of specimens with <20% tumor purity.
The FGFR inhibitor pemigatinib was recently FDA approved for the treatment of IHCC harboring FGFR2 rearrangements (3). FGFR inhibitors have an objective response rate of 20% – 35% and a median PFS of 6 – 8 months in IHCC patients with FGFR2 rearrangements (3,4,56). This activity of FGFR inhibitors in IHCC patients with FGFR2 fusions stands in contrast to the efficacy of FGFR inhibitors in our small cohort of patients with FGFR2 EIDs, who all experienced a partial response on Debio 1347. One of the patients progressed after 13 months of Debio 1347, whereas the other two patients remain on treatment after 7 and 24 months, respectively, suggesting that FGFR2 EIDs may confer enhanced sensitivity to FGFR inhibitors.
Our data indicate that both FGFR2 rearrangements and FGFR2 EIDs are transforming and constitutively activate the receptor. However, the mechanisms of kinase activation are likely distinct. In the case of fusions, the FGFR2 kinase domain is preserved and the C-terminus regulatory domains of the FGFR2 protein are replaced by different fusion partners that commonly contain an intracellular dimerization domain which promotes FGFR2 dimerization and activation (57). By contrast, the wild-type intracellular domains remain intact in FGFR2 EIDs. However, FGFR2 extracellular domain alterations frequently involve loss or gain of cysteine residues, which disrupt inhibitory intramolecular disulfide bonds or create aberrant intermolecular disulfide bonds, respectively, to promote receptor dimerization. Furthermore, our molecular modeling indicates that the FGFR2 p.H167_N173del EID alters a region involved in both FGFR2 dimerization and FGF ligand binding. Whether FGFR2 EIDs render the receptor completely ligand-independent or also affect ligand sensitivity and selectivity in addition to influencing receptor dimerization will require further study. Moreover, it will be important to determine whether FGFR2 fusions and EIDs exhibit downstream differences in signaling outputs, particularly since FGFR2 EIDs appear to be sensitive to FGFR inhibition.
As NGS testing in CLIA-certified laboratories becomes more widespread in the era of molecular medicine, standardizing variant detection and subsequent functional evaluation of these variants in a clinically meaningful time frame remains challenging. Sequencing platforms across different centers may vary in the transcripts that they detect, and clinical assessment of variant functionality and pathogenicity remains inexact. While experimental validation remains the gold standard, in silico modeling may enable more rapid estimation of the potential oncogenicity of molecular alterations, and could thereby assist clinicians in identifying new treatment options for patients. As a proof of principle of this approach, 5 of our 14 sequence-unique FGFR2 EIDs, including FGFR2 p.H167_N173del and p.I288_E295delinsT, were predicted to have a high likelihood of pathogenicity by in silico modeling, and IHCCs harboring several of these FGFR2 EIDs proved to be highly sensitive to FGFR inhibition in the clinic. Similar approaches may be applicable for the analysis of mutational variants including EIDs identified in other receptor tyrosine kinases.
Acquired resistance continues to limit the effectiveness of FGFR monotherapy in cancers with FGFR2 rearrangements and FGFR2 EIDs. Polyclonal FGFR2 kinase domain mutations have been demonstrated as mechanisms of acquired resistance in FGFR2 translocated cholangiocarcinoma (5,19). After a 13-month response to the Debio 1347 FGFR inhibitor, one of our patients developed two secondary FGFR2 kinase domain mutations. Despite these mutations, the patient’s cancer responded to subsequent treatment with an irreversible FGFR inhibitor, futibatinib. The potential of futibatinib treatment to overcome acquired resistance to Debio 1347 and other reversible FGFR inhibitors has been reported in several patients with FGFR2 translocated cholangiocarcinoma (5). Although switching FGFR inhibitors can potentially overcome FGFR2 kinase domain resistance mutations, the development of MAPK pathway mutations presents a further challenge. After a 17-month response to futibatinib, our patient developed BRAF p.L597Q and NRAS p.Q61K mutations. Treatment with the LY3214996 ERK inhibitor did not lead to disease response, which highlights the challenge of therapeutically targeting subclonal resistance populations and the likely need for combinatorial strategies to overcome this resistance. Since the MAPK pathway and SHP2 protein play important roles in the regulation and signal transduction emanating from FGFR2, additional investigation into combinations of FGFR2 inhibitors with MEK/ERK or SHP2 inhibitors is greatly needed.
In sum, we characterized the genomic landscape of biliary tract cancer in a large cohort of patients with both localized and advanced disease. Targetable alterations were identified in 48% of IHCC patient tumors, supporting clinical sequencing for all patients who present with these aggressive malignancies. In a subset of patients with IHCC, we identified extracellular domain FGFR2 in-frame deletions that were activating, transforming, and sensitive to FGFR inhibitors in the laboratory and in patients. These alterations were also identified in other tumor types, suggesting a potentially important new treatment opportunity for patients with IHCC and other malignancies.
METHODS
Patient data
Patients treated at the Dana-Farber Cancer Institute (DFCI) and Brigham and Women’s Hospital were offered enrollment into Profile, a genomic profiling initiative (54). After patients signed an Institutional Review Board (IRB)-approved written informed consent form, their tumor specimens were obtained, evaluated by a pathologist to ensure at least 20% malignant cells, subjected to DNA extraction, and analyzed by NGS testing, called the OncoPanel assay, in a CLIA-certified laboratory (CLIA certificate: 22D2040971) (54). This assay uses hybridization-based capture for 277 (OncoPanel version 1), 302 (version 2), or 447 (version 3) cancer-associated genes, corresponding to a total target coverage of 753334 bp to 1315078 bp. Computational analysis of the sequences was performed with MuTect v1.1.46 to identify single nucleotide variants, GATK Indelocator to identify insertions and deletions, and the RobustCNV and BreaKmer algorithms to identify copy number and structural variants (58,59). We included all structural variants involving oncogenes and known oncogenic fusion partners in OncoKB (60) and in the literature, as well as those involving tumor suppressor genes and intergenic regions likely to be detrimental to protein function.
The OncoPanel pipeline performs germline filtering by using the dbSNP, the Exome Variant Server, the NHLBI GO Exome Sequencing Project, and an in-house panel of normal population DNA samples. We further removed likely germline variants that were present at >0.1% in gnomAD version r2.0 (61), or were annotated as benign or likely benign in the ClinVar database (doi 10.1093/nar/gkx1153)(33). Variants were annotated for oncogenicity with the OncoKb knowledgebase through OncoKB API v2.4, and variants categorized as oncogenic, likely oncogenic, or predicted oncogenic, along with those found in manual literature review, were retained (60). MSI-High samples were determined on the basis of elevated TMB and homopolymer indel rates, calculated as previously described (62). cfDNA analysis was performed either through a CLIA approved commercially available assay (Guardant 360) or via ddPCR with previously described methods (19).
Clinical data from patients treated at DFCI were collected from their medical records with a standardized data capture approach (PRISSMM) (63), and imaging was reviewed with an attending radiologist for RECISTv1.1 measurement. Actionable alterations were defined as alterations with either an FDA approved or targeted therapy available. The GENIE database was generated by an international consortium of 19 institutions (32). The alteration frequencies for FGFR2 EIDs across different tumor types in GENIE were estimated using only samples that were assessed by an assay that covered exons 2–9 of FGFR2 (i.e. assays that effectively covered the exons encoding the extracellular domain of FGFR2); approximately half of the assays in GENIE cover these exons. Samples were organized by tumor type based on GENIE’s “Cancer_type_detailed” field.
For terminology describing FGFR2 rearrangements, we followed the convention used by Silverman and colleagues (18), whereby FGFR2 rearrangements are referred to as a “fusion” if the breakpoint is within the intron 17 or exon 18 hotspot and the gene partner is previously described or the novel gene is predicted to be an in-frame partner.
Treatment of Patients with FGFR2 EIDs
Patients were treated with Debio 1347 either as part of a phase 1 clinical trial (NCT01948297) or through a single-patient IND application. Futibatinib was obtained through a single patient IND application, whereas treatment with the LY3214996 ERK inhibitor was performed in a phase 1 trial (NCT02857270). The clinical trials were conducted according to the principles of the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice guidelines. All patients provided signed IRB-approved written informed consent forms before enrollment.
Statistical Analysis
Comparisons between categorial variables were made with Fisher’s exact tests. Kaplan-Meier methodology was used to obtain survival estimates. Overall survival was defined as the time between the date of diagnosis and date of death. Those patients who did not have a date of death were censored at their last known date alive. Real-world progression-free survival (rwPFS) was calculated as previously described (64). rwPFS was measured from the beginning of the specified treatment until either death from any cause or when the clinician noted disease progression on an imaging study. For rwPFS, patients still receiving treatment were censored at the date of last patient contact. For multivariate analyses, the Wald adjusted p-value for the coefficient of interest was reported (65). Two-sided p-values < 0.05 were considered statistically significant. Survival analysis was performed with the survival and survminer packages in R (66).
Genomic alterations were tested for statistical co-occurrence with a binomial test. The expected probability of co-occurrence was the product of the prevalence of each of the two events in the pair (p1 * p2). The expected probability of mutual exclusivity was defined as ((1−p1) * p2) + (p1 * (1−p2)). One-tailed binomial p-values for each hypothesis are reported. P-values less than 0.05 were considered statistically significant.
Constructs and viral infections
Wild-type FGFR2, FGFR2–PHGDH fusion and FGFR2-OPTN fusions were all amplified from reverse-transcribed cDNAs from ICC patient samples and inserted into the pMSCV vector using the NEBuilder HiFi DNA Assembly (New England Biolabs). FGFR2 in-frame deletions and mutations were introduced into the constructs using the Q5 Site-Directed Mutagenesis Kit (New England Biolabs). Targeted Sanger sequencing was performed to confirm the mutations generated. Retrovirus was generated by transfecting the pMSCV constructs and packaging plasmids into 293T cells. Viral infections of NIH3T3 and CCLP-1 cells were performed in the presence of polybrene. Infected cells were selected in blasticidin (15–20 μg/mL) for one week. For both cell lines, the period of time in culture between thawing, infection, selection, recovery, and experimental setup and completion was less than 10 passages.
In vitro functional studies
CCLP-1 cells were kind gifts of Dr. P.J. Bosma (Academic Medical Center, Amsterdam, the Netherlands). These cells were authenticated by short tandem repeat (STR) DNA profiling through the American Type Culture Collection (ATCC). NIH3T3 cells were obtained from the ATCC. Cell lines were routinely checked to be mycoplasma free. For soft agar assays, NIH3T3 cells engineered with indicated retroviral constructs were seeded at a density of 5000 cells and grown with complete DMEM media in 0.4% top layer agarose on top of 1% bottom layer agarose. After 3 weeks, photomicrographs of colonies were taken using a Nikon Eclipse Ti microscope at 20X magnification for representative images and 2X magnification for quantitative analysis. Three biologic replicates were performed for each condition, and the number of colonies per field across 21 fields of view (7 fields of view per biologic replicate) were quantified using ImageJ software. For cell viability assays, CCLP-1 cells engineered with indicated retroviral constructs were plated at a density of 3000 cells per well in 96-well plates with duplicates for each dose point. 24 hours later, therapeutic agents were added to the plate and cells were cultured for another 3 days. Cell viability was then assessed by MTT assay. Each dose point was normalized to DMSO controls to estimate relative viability. IC50 values were determined by GraphPad Prism 7 using a 3-parameter dose–response model. Experiments were repeated 3 times and the relative fold change in IC50 was quantified across these three biologic replicates.
Immunoblot analysis
NIH3T3 cells engineered with indicated constructs were serum-starved for 24 hours, followed by 6 hours of drug treatment. Cell protein lysates were prepared in Thermo Scientific™ RIPA Lysis and Extraction Buffer (PI89900) containing Pierce™ Protease Inhibitor (A32965) and Calbiochem phosphatase inhibitor cocktail set I and II. Protein concentration was determined by Pierce BCA Protein Assay. 20μg protein was then subject to immunoblot analysis with specific antibodies. The following antibodies were used: from Cell Signaling Technology (all at 1:1000 dilution), phospho-FRS2 Y196 (3864S), phospho-ERK1/2 T202/Y204 (4370S), ERK1/2 (4695S), phospho-AKT S473 (4060S), AKT (9272S); from Sigma (1:5000 dilution), β-actin (A5316).
Animal studies
Mice were housed in a specific pathogen-free environment. All experiments were conducted under protocol 2005N000148 approved by the Subcommittee on Research Animal Care at MGH. 6- to 8-week-old male NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, 00557, The Jackson Laboratory) were used in the experiments. NIH3T3 cells engineered with indicated constructs were subcutaneously injected into NSG mice (1×10^6 cells per mouse, 6 mice per condition). Tumors were monitored and mice were sacrificed after 2 to 5 weeks.
FGFR2 isoform assessment
RNA was extracted from cryopreserved tissue obtained via core needle biopsy of a metastatic IHCC lesion from patient 46 under IRB-approved protocols using the Qiagen AllPrep DNA/RNA/miRNA Universal kit. RNA was reverse transcribed into first-strand cDNA according to the manufacturer’s instructions using RevertAidFirst Strand cDNA Synthesis Kit (Fermentas). Quantitative RT-PCR (qRT-PCR) was performed on a CFX384 Real-Time PCR Detection System (Bio-Rad) with SYBR-Green Master PCR mix (Roche). GAPDH (human) served as the internal control. Specific primers used for detecting human FGFR2 isoforms and GAPDH were as follows: FGFR2, 5’-GGACCCAAAATGGGAGTTTC-3’ and 5’-ACCACTTGCCCAAAGCAA-3’; FGFR2 IIIb, 5’-TGCTGGCTCTGTTCAATGTG-3’ and 5’-GGCGATTAAGAAGACCCCTA-3’; FGFR2 IIIc, 5’-ACACCACGGACAAAGAGATT-3’ and 5’-GGCGATTAAGAAGACCCCTA-3’; GAPDH, 5’-AGGTGAAGGTCGGAGTCAAC-3’ and 5’-AGTTGAGGTCAATGAAGGGG-3’.
Molecular Modeling
To estimate the pathogenicity of each FGFR2 EID, the sequences of alterations found in patients were mapped onto the protein sequence of FGFR2 (UniProt P21802-3, Supplemental Figure 7). Further analyses were performed to localize alterations within the three-dimensional dimeric structure of the extracellular domains D2-D3 of FGFR2 complexed with FGF, modeled by overlapping both monomers of the X-ray structure (pdb:1EV2) on the biological dimer of FGFR1-FGF (pdb:1CVS). Overlap with reference mutations (36–51), secondary structure elements, and regions involved in recognition of the FGF ligand or in dimerization increased the confidence level of the prediction.
Supplementary Material
SIGNIFICANCE.
FGFR2 extracellular domain in-frame deletions (EIDs) are transforming genomic alterations that occur predominantly in patients with intrahepatic cholangiocarcinoma. These FGFR2 EIDs are sensitive to FGFR inhibition in vitro, and patients with these alterations benefited from treatment with FGFR inhibitors in the clinic.
Funding:
This study was supported by the Evan Schumacher Fund for Rare Cancer Research, TargetCancer Foundation, Team Evan Schumacher, Haya Linde Memorial Fund, and Marilyn and Lionel Lumaghini Family Fund for Cholangiocarcinoma Research. The work was also supported by a grant from the National Institute of Health (P50CA127003), the Hope Funds for Cancer Research, and a V Foundation Translational Research Award. J.M.C. is also supported by Stand Up To Cancer and the Lustgarten Foundation.
Disclosures
James Cleary received research funding to his institution from Abbvie, Merus, Roche, and Bristol Myers Squib. He received research funding from Merck, Astrazeneca, Esperas Pharma, and Tesaro, received consulting fees from Bristol Myers Squibb, and received travel funding from Bristol Myers Squib.
Nabeel Bardeesy has received research support from Taiho Pharmaceuticals.
Lipika Goyal is scientific advisory board member/consultant for Agios, Alentis, AstraZeneca, Debiopharm, H3 Biomedicine, Incyte, QED Therapeutics, Sirtex, and Taiho.
Ryan Sullivan received research funding from Merck. He reports Consulting/Scientific Advisory Board fees from Asana Biosciences, AstraZeneca, Bristol-Myers Squibb, Eisai, Iovance, Merck, Novartis, Oncosec, Pfizer, and Replimune.
Geoffrey Shapiro has received research funding from Eli Lilly, Merck KGaA/EMD-Serono, Merck, and Sierra Oncology. He has served on advisory boards for Pfizer, Eli Lilly, G1 Therapeutics, Roche, Merck KGaA/EMD-Serono, Sierra Oncology, Bicycle Therapeutics, Fusion Pharmaceuticals, Cybrexa Therapeutics, Astex, Almac, Ipsen, Bayer, Angiex, Daiichi Sankyo, Seattle Genetics, Boehringer Ingelheim, ImmunoMet , Asana, Artios, Atrin, Concarlo Holdings, Syros, Zentalis and CytomX Therapeutics. In addition, he holds a patent entitled, “Dosage regimen for sapacitabine and seliciclib,” also issued to Cyclacel Pharmaceuticals, and a pending patent, entitled, “Compositions and Methods for Predicting Response and Resistance to CDK4/6 Inhibition,” together with Liam Cornell.
Franck Brichory, Anne Vaslin Chessex, Anna Pokorska-Bocci, and Claudio Zanna are employees of Debiopharm.
Kimmie Ng reports institutional research funding from Pharmavite, Revolution Medicines, and Evergrande Group. She is on the advisory board for Array Biopharma and has received consulting fees from X-Biotix Therapeutics.
Pasi Jänne has received consulting fees from AstraZeneca, Boehringer-Ingelheim, Pfizer, Roche/Genentech, Takeda Oncology, ACEA Biosciences, Eli Lilly and Company, Araxes Pharma, Ignyta, Mirati Therapeutics, Novartis, LOXO Oncology, Daiichi Sankyo, Sanofi Oncology, Voronoi, SFJ Pharmaceuticals, Takeda Oncology, Silicon Therapeutics, Transcenta, and Biocartis; receives post-marketing royalties from DFCI owned intellectual property on EGFR mutations licensed to Lab Corp; has sponsored research agreements with AstraZeneca, Daichi-Sankyo, PUMA, Boehringer Ingelheim, Eli Lilly and Company, Revolution Medicines and Astellas Pharmaceuticals; and has stock ownership in LOXO Oncology and Gatekeeper Pharmaceuticals.
Vincent Zoete received consulting fees from Cellestia Biotech. He received service fees to his institution from DebioPharm.
William Hahn is a consultant for Thermo Fisher, Solasta Ventures, MPM Capital, Tyra Biosciences, iTeos, Frontier Medicines, and Paraxel. He is a founder and member of the scientific advisory board for KSQ Therapeutics.
Brian Wolpin received research funding from Celgene and Eli Lilly, and consulting fees from BioLineRx, Celgene, and Grail.
References
- 1.Rizvi S, Khan SA, Hallemeier CL, Kelley RK, Gores GJ. Cholangiocarcinoma - evolving concepts and therapeutic strategies. Nat Rev Clin Oncol 2018;15(2):95–111 doi 10.1038/nrclinonc.2017.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Valle JW, Lamarca A, Goyal L, Barriuso J, Zhu AX. New Horizons for Precision Medicine in Biliary Tract Cancers. Cancer Discov 2017;7(9):943–62 doi 10.1158/2159-8290.Cd-17-0245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abou-Alfa GK, Sahai V, Hollebecque A, Vaccaro G, Melisi D, Al-Rajabi R, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol 2020;21(5):671–84 doi 10.1016/s1470-2045(20)30109-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Javle M, Lowery M, Shroff RT, Weiss KH, Springfeld C, Borad MJ, et al. Phase II Study of BGJ398 in Patients With FGFR-Altered Advanced Cholangiocarcinoma. J Clin Oncol 2018;36(3):276–82 doi 10.1200/JCO.2017.75.5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goyal L, Shi L, Liu LY, Fece de la Cruz F, Lennerz JK, Raghavan S, et al. TAS-120 Overcomes Resistance to ATP-Competitive FGFR Inhibitors in Patients with FGFR2 Fusion-Positive Intrahepatic Cholangiocarcinoma. Cancer Discov 2019;9(8):1064–79 doi 10.1158/2159-8290.CD-19-0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abou-Alfa GK, Macarulla T, Javle MM, Kelley RK, Lubner SJ, Adeva J, et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): a multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol 2020;21(6):796–807 doi 10.1016/s1470-2045(20)30157-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Subbiah V, Lassen U, Élez E, Italiano A, Curigliano G, Javle M, et al. Dabrafenib plus trametinib in patients with BRAF(V600E)-mutated biliary tract cancer (ROAR): a phase 2, open-label, single-arm, multicentre basket trial. Lancet Oncol 2020;21(9):1234–43 doi 10.1016/s1470-2045(20)30321-1. [DOI] [PubMed] [Google Scholar]
- 8.Jiao Y, Pawlik TM, Anders RA, Selaru FM, Streppel MM, Lucas DJ, et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat Genet 2013;45(12):1470–3 doi 10.1038/ng.2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farshidfar F, Zheng S, Gingras M-C, Newton Y, Shih J, Robertson AG, et al. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell Reports 2017;18(11):2780–94 doi 10.1016/j.celrep.2017.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakamura H, Arai Y, Totoki Y, Shirota T, Elzawahry A, Kato M, et al. Genomic spectra of biliary tract cancer. Nat Genet 2015;47(9):1003–10 doi 10.1038/ng.3375. [DOI] [PubMed] [Google Scholar]
- 11.Arai Y, Totoki Y, Hosoda F, Shirota T, Hama N, Nakamura H, et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014;59(4):1427–34 doi 10.1002/hep.26890. [DOI] [PubMed] [Google Scholar]
- 12.Lowery MA, Ptashkin R, Jordan E, Berger MF, Zehir A, Capanu M, et al. Comprehensive Molecular Profiling of Intrahepatic and Extrahepatic Cholangiocarcinomas: Potential Targets for Intervention. Clin Cancer Res 2018;24(17):4154–61 doi 10.1158/1078-0432.Ccr-18-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jusakul A, Cutcutache I, Yong CH, Lim JQ, Huang MN, Padmanabhan N, et al. Whole-Genome and Epigenomic Landscapes of Etiologically Distinct Subtypes of Cholangiocarcinoma. Cancer Discov 2017;7(10):1116–35 doi 10.1158/2159-8290.Cd-17-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Voss MH, Hierro C, Heist RS, Cleary JM, Meric-Bernstam F, Tabernero J, et al. A Phase I, Open-Label, Multicenter, Dose-escalation Study of the Oral Selective FGFR Inhibitor Debio 1347 in Patients with Advanced Solid Tumors Harboring FGFR Gene Alterations. Clin Cancer Res 2019;25(9):2699–707 doi 10.1158/1078-0432.Ccr-18-1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bahleda R, Italiano A, Hierro C, Mita A, Cervantes A, Chan N, et al. Multicenter Phase I Study of Erdafitinib (JNJ-42756493), Oral Pan-Fibroblast Growth Factor Receptor Inhibitor, in Patients with Advanced or Refractory Solid Tumors. Clin Cancer Res 2019;25(16):4888–97 doi 10.1158/1078-0432.Ccr-18-3334. [DOI] [PubMed] [Google Scholar]
- 16.Mazzaferro V, El-Rayes BF, Droz Dit Busset M, Cotsoglou C, Harris WP, Damjanov N, et al. Derazantinib (ARQ 087) in advanced or inoperable FGFR2 gene fusion-positive intrahepatic cholangiocarcinoma. Br J Cancer 2019;120(2):165–71 doi 10.1038/s41416-018-0334-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Javle M, Kelley RK, Roychowdhury S, Weiss KH, Abou-Alfa GK, Macarulla T, et al. LBA28 - Updated results from a phase II study of infigratinib (BGJ398), a selective pan-FGFR kinase inhibitor, in patients with previously treated advanced cholangiocarcinoma containing FGFR2 fusions. Annals of Oncology 2018;29:viii720 doi 10.1093/annonc/mdy424.030. [DOI] [Google Scholar]
- 18.Silverman IM, Hollebecque A, Friboulet L, Owens S, Newton RC, Zhen H, et al. Clinicogenomic Analysis of FGFR2-Rearranged Cholangiocarcinoma Identifies Correlates of Response and Mechanisms of Resistance to Pemigatinib. Cancer Discov 2021;11(2):326–39 doi 10.1158/2159-8290.CD-20-0766. [DOI] [PubMed] [Google Scholar]
- 19.Goyal L, Saha SK, Liu LY, Siravegna G, Leshchiner I, Ahronian LG, et al. Polyclonal Secondary FGFR2 Mutations Drive Acquired Resistance to FGFR Inhibition in Patients with FGFR2 Fusion-Positive Cholangiocarcinoma. Cancer Discov 2017;7(3):252–63 doi 10.1158/2159-8290.CD-16-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discovery 2020;10(1):54–71 doi 10.1158/2159-8290.Cd-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mavrakis KJ, McDonald ER 3rd, Schlabach MR, Billy E, Hoffman GR, deWeck, et al. Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science 2016;351(6278):1208–13 doi 10.1126/science.aad5944. [DOI] [PubMed] [Google Scholar]
- 22.Christakis AG, Papke DJ, Nowak JA, Yurgelun MB, Agoston AT, Lindeman NI, et al. Targeted Cancer Next-Generation Sequencing as a Primary Screening Tool for Microsatellite Instability and Lynch Syndrome in Upper Gastrointestinal Tract Cancers. Cancer Epidemiology Biomarkers & Prevention 2019;28(7):1246–51 doi 10.1158/1055-9965.Epi-18-1250. [DOI] [PubMed] [Google Scholar]
- 23.Structure Plaza-Menacho I. and function of RET in multiple endocrine neoplasia type 2. Endocr Relat Cancer 2018;25(2):T79–T90 doi 10.1530/ERC-17-0354. [DOI] [PubMed] [Google Scholar]
- 24.Ip CKM, Ng PKS, Jeong KJ, Shao SH, Ju Z, Leonard PG, et al. Neomorphic PDGFRA extracellular domain driver mutations are resistant to PDGFRA targeted therapies. Nature communications 2018;9(1):4583- doi 10.1038/s41467-018-06949-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Casolari DA, Nguyen T, Butcher CM, Iarossi DG, Hahn CN, Bray SC, et al. A novel, somatic, transforming mutation in the extracellular domain of Epidermal Growth Factor Receptor identified in myeloproliferative neoplasm. Sci Rep 2017;7(1):2467 doi 10.1038/s41598-017-02655-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joshi SK, Keck JM, Eide CA, Bottomly D, Traer E, Tyner JW, et al. ERBB2/HER2 mutations are transforming and therapeutically targetable in leukemia. Leukemia 2020;34(10):2798–804 doi 10.1038/s41375-020-0844-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Azoury SC, Reddy S, Shukla V, Deng CX. Fibroblast Growth Factor Receptor 2 (FGFR2) Mutation Related Syndromic Craniosynostosis. International journal of biological sciences 2017;13(12):1479–88 doi 10.7150/ijbs.22373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cornejo-Roldan LR, Roessler E, Muenke M. Analysis of the mutational spectrum of the FGFR2 gene in Pfeiffer syndrome. Human genetics 1999;104(5):425–31 doi 10.1007/s004390050979. [DOI] [PubMed] [Google Scholar]
- 29.Slavotinek A, Crawford H, Golabi M, Tao C, Perry H, Oberoi S, et al. Novel FGFR2 deletion in a patient with Beare-Stevenson-like syndrome. American journal of medical genetics Part A 2009;149a(8):1814–7 doi 10.1002/ajmg.a.32947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Priolo M, Lerone M, Baffico M, Baldi M, Ravazzolo R, Cama A, et al. Pfeiffer syndrome type 2 associated with a single amino acid deletion in the FGFR2 gene. Clinical genetics 2000;58(1):81–3 doi 10.1034/j.1399-0004.2000.580116.x. [DOI] [PubMed] [Google Scholar]
- 31.Paznekas WA, Cunningham ML, Howard TD, Korf BR, Lipson MH, Grix AW, et al. Genetic Heterogeneity of Saethre-Chotzen Syndrome, Due to TWIST and FGFR Mutations. The American Journal of Human Genetics 1998;62(6):1370–80 doi 10.1086/301855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov 2017;7(8):818–31 doi 10.1158/2159-8290.Cd-17-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic acids research 2014;42(Database issue):D980–D5 doi 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robertson SC, Meyer AN, Hart KC, Galvin BD, Webster MK, Donoghue DJ. Activating mutations in the extracellular domain of the fibroblast growth factor receptor 2 function by disruption of the disulfide bond in the third immunoglobulin-like domain. Proceedings of the National Academy of Sciences of the United States of America 1998;95(8):4567–72 doi 10.1073/pnas.95.8.4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gallo LH, Nelson KN, Meyer AN, Donoghue DJ. Functions of Fibroblast Growth Factor Receptors in cancer defined by novel translocations and mutations. Cytokine Growth Factor Rev 2015;26(4):425–49 doi 10.1016/j.cytogfr.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 36.Reintjes N, Li Y, Becker A, Rohmann E, Schmutzler R, Wollnik B. Activating somatic FGFR2 mutations in breast cancer. PLoS One 2013;8(3):e60264 doi 10.1371/journal.pone.0060264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kan SH, Elanko N, Johnson D, Cornejo-Roldan L, Cook J, Reich EW, et al. Genomic screening of fibroblast growth-factor receptor 2 reveals a wide spectrum of mutations in patients with syndromic craniosynostosis. Am J Hum Genet 2002;70(2):472–86 doi 10.1086/338758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barnett CP, Nataren NJ, Klingler-Hoffmann M, Schwarz Q, Chong CE, Lee YK, et al. Ectrodactyly and Lethal Pulmonary Acinar Dysplasia Associated with Homozygous FGFR2 Mutations Identified by Exome Sequencing. Hum Mutat 2016;37(9):955–63 doi 10.1002/humu.23032. [DOI] [PubMed] [Google Scholar]
- 39.Ng PK, Li J, Jeong KJ, Shao S, Chen H, Tsang YH, et al. Systematic Functional Annotation of Somatic Mutations in Cancer. Cancer Cell 2018;33(3):450–62.e10 doi 10.1016/j.ccell.2018.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gartside MG, Chen H, Ibrahimi OA, Byron SA, Curtis AV, Wellens CL, et al. Loss-of-function fibroblast growth factor receptor-2 mutations in melanoma. Mol Cancer Res 2009;7(1):41–54 doi 10.1158/1541-7786.Mcr-08-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Egan JB, Marks DL, Hogenson TL, Vrabel AM, Sigafoos AN, Tolosa EJ, et al. Molecular Modeling and Functional Analysis of Exome Sequencing-Derived Variants of Unknown Significance Identify a Novel, Constitutively Active FGFR2 Mutant in Cholangiocarcinoma. JCO Precis Oncol 2017;2017 doi 10.1200/PO.17.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liao RG, Jung J, Tchaicha J, Wilkerson MD, Sivachenko A, Beauchamp EM, et al. Inhibitor-sensitive FGFR2 and FGFR3 mutations in lung squamous cell carcinoma. Cancer Res 2013;73(16):5195–205 doi 10.1158/0008-5472.Can-12-3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanizaki J, Ercan D, Capelletti M, Dodge M, Xu C, Bahcall M, et al. Identification of Oncogenic and Drug-Sensitizing Mutations in the Extracellular Domain of FGFR2. Cancer Res 2015;75(15):3139–46 doi 10.1158/0008-5472.CAN-14-3771. [DOI] [PubMed] [Google Scholar]
- 44.Dutt A, Salvesen HB, Chen TH, Ramos AH, Onofrio RC, Hatton C, et al. Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc Natl Acad Sci U S A 2008;105(25):8713–7 doi 10.1073/pnas.0803379105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robertson SC, Meyer AN, Hart KC, Galvin BD, Webster MK, Donoghue DJ. Activating mutations in the extracellular domain of the fibroblast growth factor receptor 2 function by disruption of the disulfide bond in the third immunoglobulin-like domain. Proceedings of the National Academy of Sciences of the United States of America 1998;95(8):4567–72 doi 10.1073/pnas.95.8.4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sarabipour S, Hristova K. Pathogenic Cysteine Removal Mutations in FGFR Extracellular Domains Stabilize Receptor Dimers and Perturb the TM Dimer Structure. J Mol Biol 2016;428(20):3903–10 doi 10.1016/j.jmb.2016.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mansukhani A, Bellosta P, Sahni M, Basilico C. Signaling by fibroblast growth factors (FGF) and fibroblast growth factor receptor 2 (FGFR2)-activating mutations blocks mineralization and induces apoptosis in osteoblasts. J Cell Biol 2000;149(6):1297–308 doi 10.1083/jcb.149.6.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eswarakumar VP, Horowitz MC, Locklin R, Morriss-Kay GM, Lonai P. A gain-of-function mutation of Fgfr2c demonstrates the roles of this receptor variant in osteogenesis. Proc Natl Acad Sci U S A 2004;101(34):12555–60 doi 10.1073/pnas.0405031101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suh YJ, Bae HS, Choi JY, Lee JH, Kim MJ, Kim S, et al. A novel FGFR2 mutation in tyrosine kinase II domain, L617F, in Crouzon syndrome. J Cell Biochem 2014;115(1):102–10 doi 10.1002/jcb.24637. [DOI] [PubMed] [Google Scholar]
- 50.Pollock PM, Gartside MG, Dejeza LC, Powell MA, Mallon MA, Davies H, et al. Frequent activating FGFR2 mutations in endometrial carcinomas parallel germline mutations associated with craniosynostosis and skeletal dysplasia syndromes. Oncogene 2007;26(50):7158–62 doi 10.1038/sj.onc.1210529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Y, Zhou X, Oberoi K, Phelps R, Couwenhoven R, Sun M, et al. p38 Inhibition ameliorates skin and skull abnormalities in Fgfr2 Beare-Stevenson mice. J Clin Invest 2012;122(6):2153–64 doi 10.1172/jci62644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krook MA, Lenyo A, Wilberding M, Barker H, Dantuono M, Bailey KM, et al. Efficacy of FGFR Inhibitors and Combination Therapies for Acquired Resistance in FGFR2-Fusion Cholangiocarcinoma. Mol Cancer Ther 2020;19(3):847–57 doi 10.1158/1535-7163.MCT-19-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lamarca A, Palmer DH, Wasan HS, Ross PJ, Ma YT, Arora A, et al. ABC-06 | A randomised phase III, multi-centre, open-label study of active symptom control (ASC) alone or ASC with oxaliplatin / 5-FU chemotherapy (ASC+mFOLFOX) for patients (pts) with locally advanced / metastatic biliary tract cancers (ABC) previously-treated with cisplatin/gemcitabine (CisGem) chemotherapy. Journal of Clinical Oncology 2019;37(15_suppl):4003- doi 10.1200/JCO.2019.37.15_suppl.4003. [DOI] [Google Scholar]
- 54.Sholl LM, Do K, Shivdasani P, Cerami E, Dubuc AM, Kuo FC, et al. Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight 2016;1(19):e87062 doi 10.1172/jci.insight.87062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schram AM, O’Reilly EM, Somwar R, Benayed R, Shameem S, Chauhan T, et al. Abstract PR02: Clinical proof of concept for MCLA-128, a bispecific HER2/3 antibody therapy, in NRG1 fusion-positive cancers. Molecular Cancer Therapeutics 2019;18(12 Supplement):PR02–PR doi 10.1158/1535-7163.Targ-19-pr02. [DOI] [Google Scholar]
- 56.Meric-Bernstam F, Arkenau H, Tran B, Bahleda R, Kelley R, Hierro C, et al. O-001 - Efficacy of TAS-120, an irreversible fibroblast growth factor receptor (FGFR) inhibitor, in cholangiocarcinoma patients with FGFR pathway alterations who were previously treated with chemotherapy and other FGFR inhibitors. Annals of Oncology 2018;29:v100 doi 10.1093/annonc/mdy149. [DOI] [Google Scholar]
- 57.Li F, Peiris MN, Donoghue DJ. Functions of FGFR2 corrupted by translocations in intrahepatic cholangiocarcinoma. Cytokine & Growth Factor Reviews 2020;52:56–67 doi 10.1016/j.cytogfr.2019.12.005. [DOI] [PubMed] [Google Scholar]
- 58.do Valle ÍF, Giampieri E, Simonetti G, Padella A, Manfrini M, Ferrari A, et al. Optimized pipeline of MuTect and GATK tools to improve the detection of somatic single nucleotide polymorphisms in whole-exome sequencing data. BMC Bioinformatics 2016;17(Suppl 12):341- doi 10.1186/s12859-016-1190-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abo RP, Ducar M, Garcia EP, Thorner AR, Rojas-Rudilla V, Lin L, et al. BreaKmer: detection of structural variation in targeted massively parallel sequencing data using kmers. Nucleic acids research 2015;43(3):e19–e doi 10.1093/nar/gku1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis Oncol 2017;2017 doi 10.1200/PO.17.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581(7809):434–43 doi 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Papke DJ, Nowak JA, Yurgelun MB, Frieden A, Srivastava A, Lindeman NI, et al. Validation of a targeted next-generation sequencing approach to detect mismatch repair deficiency in colorectal adenocarcinoma. Modern Pathology 2018;31(12):1882–90 doi 10.1038/s41379-018-0091-x. [DOI] [PubMed] [Google Scholar]
- 63.Kehl KL, Elmarakeby H, Nishino M, Van Allen EM, Lepisto EM, Hassett MJ, et al. Assessment of Deep Natural Language Processing in Ascertaining Oncologic Outcomes From Radiology Reports. JAMA Oncol 2019;5(10):1421–9 doi 10.1001/jamaoncol.2019.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Griffith SD, Miksad RA, Calkins G, You P, Lipitz NG, Bourla AB, et al. Characterizing the Feasibility and Performance of Real-World Tumor Progression End Points and Their Association With Overall Survival in a Large Advanced Non-Small-Cell Lung Cancer Data Set. JCO Clin Cancer Inform 2019;3:1–13 doi 10.1200/cci.19.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yu Z, Demetriou M, Gillen DL. Genome-Wide Analysis of Gene-Gene and Gene-Environment Interactions Using Closed-Form Wald Tests. Genet Epidemiol 2015;39(6):446–55 doi 10.1002/gepi.21907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Borgan Ø Modeling Survival Data: Extending the Cox Model. Therneau Terry M. and Grambsch Patricia M., Springer-Verlag, New York, 2000. No. of pages: xiii + 350. Price: $69.95. ISBN 0-387-98784-3. Statistics in Medicine 2001;20(13):2053–4 doi 10.1002/sim.956. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.