

Abstract
Background/Aim:
Tyrosine kinase inhibitors (TKIs) are used for the treatment of both wild type and mutant non-small cell lung cancer (NSCLC); however, acquired resistance is a major clinical challenge. Herein, we aimed to investigate the effects of telmisartan (Tel), CFM 4.16 and sorafenib combination in rociletinib resistant NSCLC tumors.
Materials and Methods:
3D spheroid cultures and western blotting were used for evaluating cytotoxic effects and protein expression. An in vivo rociletinib resistant H1975 xenograft model of NSCLC was developed by subcutaneous injection of rociletinib resistant H1975 cells into nude mice.
Results:
Tel, CFM 4.16 and sorafenib combination displayed superior anti-cancer effects in 3D spheroid cultures and a rociletinib resistant H1975 xenograft model of NSCLC by decreasing the protein expression of oncogenic and cancer stem cell markers (Nanog, Sox2 and Oct4).
Conclusion:
Tel facilitates effective penetration of CFM 4.16 and sorafenib in rociletinib resistant H1975 models of NSCLC.
Keywords: Non-small cell lung cancer, telmisartan, CFM 4.16, sorafenib, rociletinib, resistance
Lung cancer is the leading cause of cancer-related deaths worldwide for both men and women (1). 50% of the lung cancer patients die within one year of diagnosis and 5-year survival rate is less than 18% (2). Non-small cell lung cancer (NSCLC) accounts for 85% of lung cancers and around 70% of the lung cancer patients are in advanced metastatic stage at the time of diagnosis (3). Multiple studies have demonstrated superior efficacy with first generation (gefitinib and erlotinib), second generation (afatinib) and third generation (osimertinib and rociletinib) tyrosine kinase inhibitors (TKIs) in advanced metastatic lung cancer tumors harbouring epidermal growth factor receptor (EGFR) mutations when compared to standard chemotherapy of cisplatin, paclitaxel and gemcitabine (4-8).
Rociletinib, an irreversible third-generation tyrosine kinase inhibitor (TKI) is demonstrated to be effective against EGFR T790M mutations in NSCLC through covalent binding with cysteine (C) 797 in the ATP binding pocket of EGFR kinase domain and strong hydrophobic interactions with the gatekeeper methionine (M) residue in T790M (9). However, preclinical trials have shown minimal efficacy of rociletinib against EGFR wild type NSCLC (10). It has been well demonstrated to show better therapeutic effects than first generation TKIs (i.e., erlotinib or gefitinib) for NSCLC harbouring T790M and L858R mutations and brain metastasis (11, 12). However, the majority of NSCLC patients develop resistance to rociletinib due to HER2 amplification, MET amplification, BRAF mutation, epithelial-mesenchymal transition (EMT), over-expression of basal phosphorylated AKT levels, ERK activation, EGFR mutations involving substitution of cysteine at position 797 with serine (C797S), lysine at position 844 with valine (K844V), and leucine at position 718 with glutamine (L718Q) (13-16).
Sorafenib, approved by the FDA for kidney and liver cancers, is a multi-kinase inhibitor of serine/threonine kinases CRAF, BRAF (wild type), as well as the mutant BRAFV600E type (17-19). A growing body of evidence demonstrates that sorafenib inhibits the activation of VEGR, PDGFR, FGFR, c-KIT, MET, MAPK, and angiogenesis (17, 18, 20). Sorafenib induces apoptosis and inhibits MEK and ERK phosphorylation in a variety of cancers harbouring BRAF and/or KRAS or NRAS mutations (18, 21). Sorafenib, in combination with various molecules targeting EGFR and MET, have shown good anti-cancer activity in wild type and drug resistant lung cancers (19, 22).
Cell cycle and apoptosis regulator protein 1/CCAR1 (CARP-1) is a peri-nuclear phosphoprotein that regulates the cell cycle and tumor progression by co-activating the anaphase-promoting complex (APC/C), an E3 ubiquitin ligase (23, 24). It also regulates chemotherapy-induced apoptosis through p53 co-activation (25). CARP-1 functional mimetics (CFMs) induce apoptosis and inhibit cell growth in a variety of cancer cells by decreasing the binding of CARP-1 with APC/C subunit APC2 (26). CFM 4.16, a CARP-1 functional mimetic in combination with sorafenib has shown good anti-cancer activity in TNBC, NSCLC, and renal cancers. This combination also showed excellent anti-cancer efficacy in rociletinib resistant H1975 NSCLC xenograft model (22).
Clinical efficacy of various anti-cancer drugs is limited because of poor diffusion and penetration of drugs into solid tumors owing to high interstitial pressure or tumor stromal barriers (i.e., collagen rich network embedment in hyaluronan of tumor stroma) (27-29). The role of telmisartan in decreasing tumor interstitial fibrosis and promoting intratumoral distribution of nanoparticles and liposomes is well documented (30-32). Telmisartan also shows anti-cancer effects in NSCLC by inhibition of PI3K signalling and activation of peroxisome proliferator activated receptor-γ (PPARγ) pathways (33, 34).
In this study, we hereby hypothesize that sorafenib (i.e., which targets mutant BRAF, decreases MET amplification) in combination with CFM 4.16 (i.e., which improves the sensitivity of sorafenib) and telmisartan (i.e., which disrupts tumor stroma and helps in effective permeation of CFM 4.16) might provide good anti-cancer effects in rociletinib resistant models of NSCLC.
Materials and Methods
CFM 4.16 was received as a kind gift from Dr. Arun Rishi (Wayne State University, Detroit, MI, USA). RPMI-1640 medium-high glucose, Fetal bovine serum, Antibiotic (Penicillin, Streptomycin, and Neomycin) solution, Fluorescein isothiocyanate (FITC), 3-(4, 5- dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT), trypsin-EDTA, Tel, sorafenib and crystal violet dye were procured from Sigma-Aldrich (St. Louis, MO, USA). Dimethyl sulfoxide (DMSO) and phosphate buffered saline (PBS) were purchased from VWR international (Radnor, PA, USA). Cultrex and 96-well suspension culture plates were purchased from Bio-techne (Minneapolis, MN, USA). All the primary antibodies used in our study were purchased from Cell Signaling Technology (Danvers, MA, USA). Secondary antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX, USA).
Cell culture.
Non-small cell lung cancer cell lines: HCC827, HCC827-R (T790M EGFR ERL Resistant) and H1975 (T790M EGFR ERL Resistant) cells were received from Dr. Arun Rishi (Wayne State University, Detroit, MI, USA). Rociletinib resistant H1975 (ROC-Res-H1975) cells were developed in our laboratory by continuous exposure of H1975 cells to rociletinib, starting from a dose of 150 nM and further escalating to 4 μM over a period of 8-10 months. All cells were grown in RPMI media supplemented with 10% FBS, 100 units per ml penicillin, and 100 mg/ml streptomycin under standard conditions of 5% CO2 and 37°C in a controlled humidified (95% relative humidity) incubator.
3D spheroid cell culture.
Cells were cultured in RPMI media supplemented with heat inactivated FBS. Upon reaching 70% confluency, cells were routinely passaged using 0.05% trypsin-EDTA and collected according to standard cell culture procedures. Cells were then counted and reseeded on low attachment plates in heat inactivated media supplemented with 2% cultrex, under standard conditions of 5% CO2 and 37°C in a controlled humidified (95% relative humidity) incubator. After the formation of spheroids within 3 days, cells were subjected to different treatments. Media were changed every two days.
Cell viability assays.
Cytotoxicity of CFM 4.16, sorafenib and telmisartan (Tel) was investigated either alone or in combinations in wild type and resistant lung cancer cell lines. Cells were seeded in 96-well plates (approximately 8,000 cells per well). Cells were treated with different concentrations of CFM 4.16, sorafenib, and Tel for 48 h to determine the individual cytotoxic effects and IC50 concentration of the drugs. For the CFM 4.16 and sorafenib combination, cells were pre-treated with CFM 4.16 for 12 h and further subjected to treatment with different concentrations of sorafenib for 36 h. For the Tel, CFM 4.16 and sorafenib combination, cells were pre-treated with Tel and CFM 4.16 for 12 h and later treated with different concentrations of sorafenib for 36 h. Cell viability was assessed by using MTT assay as previously described (35).
Cell uptake studies.
HCC827, HCC827-ERL-R, H1975, and ROC-Res-H1975 cells suspended in RPMI medium containing 2% cultrex reagent were seeded in Scivax low attachment 96-well plates and incubated at 37°C and 5% CO2 for 3 days before treatment. After the formation of spheroids, cells were pre-treated with Tel followed by incubation with FITC solution for 4 h. Cells were washed with PBS and then subjected to trypsinization using 0.25% trypsin EDTA solution for 5-10 min. Centrifugation was performed and the cell pellet was washed twice with PBS before resuspending at a density of 2×105 cells/ 500 μl in PBS for analysis using the BD FACS-Calibur flow cytometer (BD Bioscience, Franklin Lakes, NJ, USA).
Western blot analysis.
Whole cell protein lysates of HCC827, HCC827-ERL-R, H1975 and ROC-Res-H1975 cells treated with CFM 4.16 (10 μM), Tel (25 μM) and sorafenib (5 μM) either alone or in combinations were prepared by using RIPA buffer (Cell Signaling, Danvers, MA, USA) according to previously described methods (36). Protein estimation was carried out by using the bicinchoninic acid (BCA) assay. Briefly, 40 μg protein was loaded into each well of a 10% SDS-PAGE gel (Mini-PROTEAN® TGX™ Precast Gels). Protein was transferred to a PVDF membrane (Bio-Rad Laboratories, Hercules, CA, USA) after electrophoresis. After transfer, membranes were blocked for 4-6 h using 3% bovine serum albumin in PBS containing 0.1% Tween-20 (PBST). Blots were then incubated with primary antibodies (1:1,000) overnight followed by washing thrice with PBST for 5 min each, and finally incubated with appropriate HRP-conjugated secondary antibodies [rabbit anti-mouse IgG (catalog number: 7076S; Cell Signaling Technology)]; goat anti-rabbit IgG (catalog number: 7074S; Cell Signaling Technology) for 1 h at room temperature. After washing with PBST again thrice for 5 min each, the blots were incubated with SuperSignal West Pico Chemiluminescent substrate and their images were captured using Chemidoc Instrument (Bio-Rad). The immunoblots were quantified by densitometry scanning using NIH ImageJ software.
CFM 4.16 SNEDD formulation.
CFM 4.16 NLF was prepared and characterized according to the methods previously described (37, 38). Briefly, suitable quantity of CFM-4.16 was initially mixed with Compritol 888ATO, Miglyol 812N, and Geleol, followed by melting of this mixture at 70°C to form a uniform and clear oil phase. The aqueous phase containing surfactant Tween 80 and D-α-tocopheryl polyethylene glycol succinate (Vitamin E TPGS) dispersed in double distilled water was added drop wise to the oil phase at 70°C. Afterwards, NanoDebee was used for homogenising the coarse emulsion for 15 min under high pressure for about 5 cycles.
Rociletinib resistant H1975 xenograft model of non-small cell lung cancer.
Female athymic nude mice (Foxn1nu; 3-5 weeks old, 20-25 grams’ body weight, n=4) were procured from Envigo, housed in controlled climate conditions with a 12 h light/12 h dark cycle with free access to water and food intake, and acclimatized for 7-10 days. All the animal experiments carried out were reviewed and approved by the Institutional Animal Use and Care Committee of Florida Agricultural and Mechanical University and carried out according to the NIH guidelines (Guide for the care and use of laboratory animals) and applicable national laws. All animal experiments complied with the ARRIVE guidelines (39). Rociletinib resistant H1975 xenograft model of NSCLC was developed by subcutaneous injection of 3 million ROC-Res-H1975 cells, suspended in matrigel in 1:1 ratio into the right flank of nude mice. The digital Vernier caliper instrument was used for measuring the length and width of the tumors. Tumor volume (TV) was then calculated by using the formula TV=½ ab2, where ‘a’ and ‘b’ represent the length and width of the tumors, respectively. After the animals showed a tumor volume of around 400 mm3, they were randomly divided into six groups. Animals were treated with Tel (10 mg/kg body weight), CFM4.16 NLF (40 mg/kg body weight) and sorafenib (30 mg/kg body weight) alone for 2 weeks. For CFM 4.16 and sorafenib combination, animals were pre-treated with CFM 4.16 followed by treatment with sorafenib for 2 weeks. For Tel, CFM 4.16 and sorafenib combination, animals were pre-treated with Tel (10 mg/kg body weight) thrice a week followed by treatment with CFM4.16 NLF (40 mg/kg body weight) and sorafenib (30 mg/kg body weight) for 2 weeks. Tumor volume was measured twice a week during the duration of drug treatments.
Statistical analysis.
All the data are presented as the mean±standard error of mean (SEM). Significant difference among the treatment groups was determined by using either Student’s t-test or one-way ANOVA through using GraphPad prism version 5.0 (San Diego, CA, USA). A p-Value less than 0.05 was considered to be statistically significant.
Results
Combinatorial anticancer effects of sorafenib, CFM 4.16 and telmisartan in NSCLC and drug resistant NSCLC cells
Cytotoxicity assays were performed to determine initially the individual anti-cancer potential of sorafenib, CFM 4.16 and Tel in 2D and 3D cultures of HCC827, erlotinib resistant HCC827 (HCC827-ERL-R), H1975 and rociletinib resistant H1975 (ROC-Res-H1975) cells. The same number of cells (8×103 cells/well) was used for comparing cytotoxic data in 2D and 3D cultures. CFM 4.16 and Tel were treated with concentrations ranging from 6.25 to 100 μM. Sorafenib concentrations ranged from 0.78 to 100 μM. We observed a dose-dependent decrease in the viability of all tested cell lines with the treatments after 48 h. IC50 values of sorafenib in 3D cultures of HCC827, HCC827-ERL-R, H1975 and ROC-Res-H1975 cells were found to be 5.77-fold, 3.73-fold, 3.30-fold, and 3.18-fold higher when compared to 2D cultures of these cells, respectively (Table I). IC50 values of CFM 4.16 in 3D cultures of HCC827, HCC827-ERL-R, H1975 and ROC-Res-H1975 cells were found to be 2.27-fold, 2.49-fold, 1.98-fold, and 2.05-fold higher when compared to 2D cultures of these cells, respectively (Table I). IC50 values of Tel in 3D cultures of HCC827, HCC827-ERL-R, H1975 and ROC-Res-H1975 cells were found to be 1.67-fold, 1.67-fold, 1.34-fold, and 1.28-fold higher when compared to 2D cultures of these cells, respectively (Table I). IC50 values in 3D cultures with all the treatments were higher in comparison to 2D monolayers, suggesting the influence of the 3D tumor microenvironment in determining anti-cancer efficacy. Based on our cytotoxicity assay observations in 2D and 3D cultures, we selected 10 μM of CFM 4.16 and 25 μM of Tel for combination studies with sorafenib. CFM 4.16 pre-treatment for 12 h increased the sensitivity of sorafenib in both 2D and 3D cultures of HCC827, HCC827-ERL-R, H1975 and ROC-Res-H1975 cells (Table I). Tel and CFM 4.16 combination pre-treatment for 12 h did not lead to an improved sensitivity to sorafenib in 2D cultures of HCC827, HCC827-ERL-R, H1975 and ROC-Res-H1975 cells when compared to the IC50 values of CFM 4.16 and sorafenib combination (Table I). However, Tel and CFM 4.16 pre-treatment significantly improved the sensitivity of sorafenib in 3D cultures of all the tested cell lines (Table I), suggesting that tumor stromal disruption by Tel was responsible for the higher anti-cancer effects of the combination treatment in 3D cultures.
Table I.
Cytotoxicity of sorafenib, CFM 4.16 and Tel either alone or in combination in non-small cell lung cancer (NSCLC) and drug resistant NSCLC cells.
| Treatment | HCC827 | HCC827-ERL-R | H1975 | ROC-Res-H1975 | ||||
|---|---|---|---|---|---|---|---|---|
| 2D | 3D | 2D | 3D | 2D | 3D | 2D | 3D | |
| Sorafenib IC50 (μM | 6.3±0.3 | 36.3±1.8 | 15.7±0.8 | 58.7±2.9 | 17.1±0.4 | 55.1±2.8 | 18.3±0.9 | 58.3±2.9 |
| CFM 4.16 IC50 (μM) | 21±1.0 | 47.7±2.4 | 21±1.1 | 52.6±2.6 | 25±1.3 | 49.7±2.5 | 27±1.4 | 55.6±2.8 |
| Tel IC50 (μM) | 53±2.6 | 88.9±4.4 | 51±2.6 | 85.7±4.3 | 62±3.1 | 83.5±4.2 | 64±2.7 | 81.6±4.1 |
| CFM 4.16 (10 μM) + Sorafenib IC50 (μM) | 3.2±0.2 | 16.6±0.8 | 3.8±0.2 | 21.6±1.1 | 6.7±0.3 | 19.9±1.0 | 16.7±0.2 | 21.3±1.1 |
| CFM 4.16 (10 μM) + Tel (25 μM) + Sorafenib IC50 (μM) | 3.1±0.1 | 8.9±0.4 | 4.04±0.1 | 6.7±0.3 | 6.6±0.2 | 7.4±0.4 | 17.4±0.1 | 11.2±0.6 |
All the data (IC50 values) shown are representative of three independent experiments and expressed as mean±SEM. Tel: Telmisartan; CFM 4.16: CARP-1 functional mimetic 4.16; μM: Micromolar; IC50: Concentration of a drug that is required for 50% inhibition in vitro; HCC827-ERL-R: erlotinib resistant HCC827 cells; ROC-Res-H1975: Rociletinib resistant H1975 cells.
CFM 4.16 pre-treatment improved the sensitivity of sorafenib in H1975 and ROC-Res-H1975 cells.
Resistance to tyrosine kinase inhibitors (TKIs) is majorly associated with the activation of oncogenic drivers such as MET and EGFR in non-small cell lung cancer cells. We observed a significant decrease in the expression of p-MET and p-EGFR with sorafenib treatment in H1975 cells. CFM 4.16 pre-treatment resulted in a higher increase in the sensitivity to sorafenib by decreasing the phosphorylation of MET and EGFR in H1975 cells. Sorafenib did not decrease the phosphorylation of MET and EGFR in ROC-Res-H1975 cells. However, CFM 4.16 pre-treatment (10 μM) for 12 h significantly improved the sensitivity of sorafenib in ROC-Res-H1975 cells by decreasing the phosphorylation of MET and EGFR when compared to the sorafenib alone group (Figure 1). Tel (25 μM) pre-treatment in combination with CFM 4.16 did not improve the sensitivity of sorafenib in 2D cultures of H1975 and ROC-Res-H1975 cells when compared to CFM 4.16 and sorafenib combination treatment (Figure 1). This might be due to the absence of the tumor microenvironment in 2D cultures.
Figure 1.
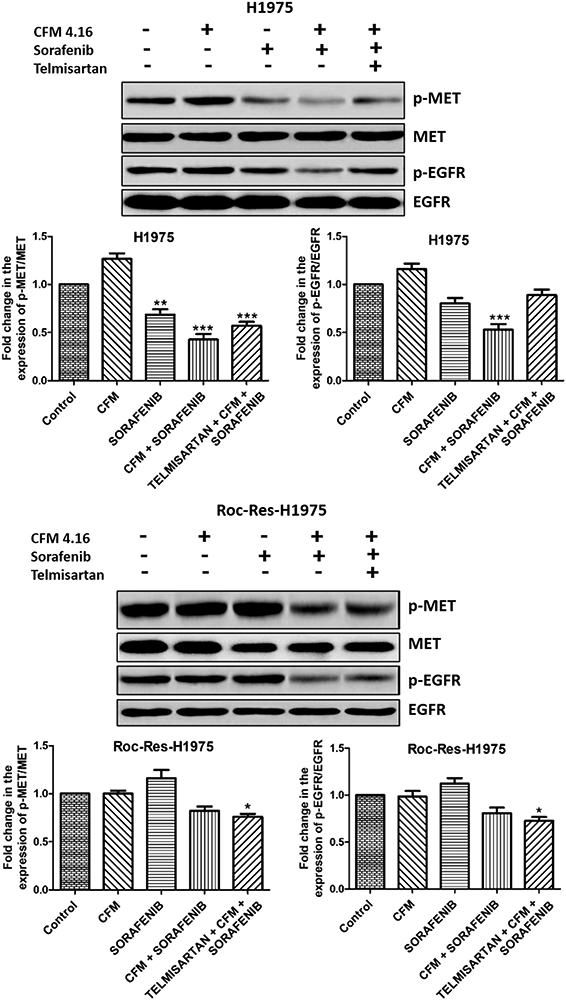
Western blots and densitometric analysis of p-MET, MET, p-EGFR and EGFR in H1975 and rociletinib resistant H1975 cells. Data are representative of three different experiments and were presented as mean, and error bars refer to SEM. *p<0.05, **p<0.01, ***p<0.001 was considered significant when compared to control. MET: MET proto-oncogene, receptor tyrosine kinase; EGFR: epidermal growth factor receptor; Roc-Res-H1975: rociletinib resistant H1975.
CFM 4.16, sorafenib and Tel combination decreased the protein expression of lung cancer stem cell markers.
As cancer stem cells are majorly involved in the development of drug resistance, we investigated the effects of CFM 4.16, sorafenib and Tel combination on lung cancer stem cell markers such as Sox2, Nanog and Oct4. It was observed that CFM 4.16 alone did not decrease the expression of cancer stem cell markers in both H1975 and ROC-Res-H1975 cells. However, sorafenib alone decreased the expression of Nanog, Oct4 and Sox2 in both H1975 and ROC-Res-H1975 cells. CFM 4.16 pre-treatment followed by sorafenib treatment significantly decreased the expression of lung cancer stem cell markers (Figure 2). The combination with Tel treatment further improved the sensitivity of sorafenib in 2D cultures of H1975 and ROC-Res-H1975 cells (Figure 2). This might be due to the Tel effect on lung cancer stem cell markers in H1975 and ROC-Res-H1975 cells. Recent reports have shown that Tel alone can decrease the expression of lung cancer stem cell markers such as Oct4 and Nanog (40).
Figure 2.
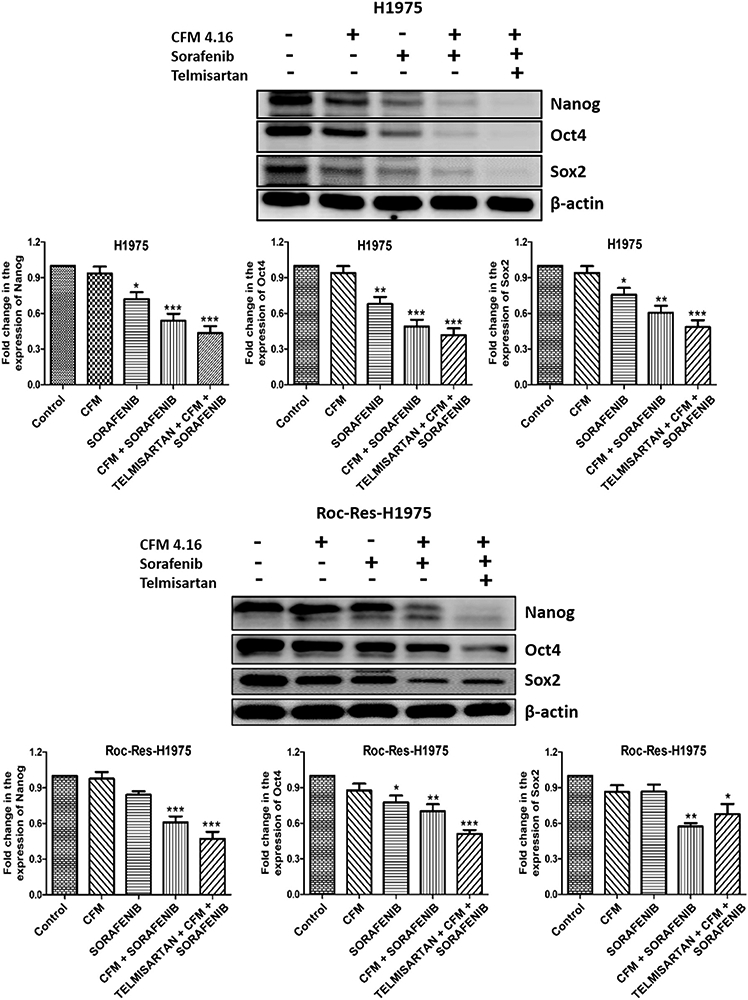
Western blots and densitometric analysis of Nanog, Oct4 and Sox2 in H1975 and Rociletinib resistant H1975 cells. Data are representative of three different experiments and were presented as mean, and error bars refer to SEM. *p<0.05, **p<0.01, ***p<0.001 was considered significant when compared to control.
Telmisartan pre-treatment increased the uptake of FITC in 3D spheroids of lung cancer cells.
Telmisartan pre-treatment improved the cellular uptake of FITC in 3D spheroids of HCC827, HCC827-ERL-R, H1975 and ROC-Res-H1975 cells (Figure 3) as depicted by the increased rightward shift in the flow cytometry analysis, which revealed that Tel treated FITC stained 3D spheroids displayed increased FITC ratios (Stained: Unstained) with values of 21, 24, 27 and 22 in HCC827, HCC827-ERL-R, H1975 and ROC-Res-H1975 cells, respectively. Free FITC stained 3D spheroids showed decreased FITC ratios of 8, 11, 7 and 13 in HCC827, HCC827-ERL-R, H1975 and ROC-Res-H1975 cells, respectively (Figure 3). This suggests that Tel disruption of tumour stromal barriers present in 3D spheroids facilitated increased uptake of FITC in all these cells when compared to free FITC stained cells.
Figure 3.

Flow cytometry analysis of uptake of FITC in control (untreated) and Tel pre-treated 3D spheroids. Data shown are representative of three independent experiments and presented as mean, and error bars refer to SEM. FITC: Fluorescein isothiocyanate.
Tel, CFM 4.16 NLF and sorafenib combination displays superior anti-cancer effect in rociletinib resistant H1975 NSCLC xenograft model.
As CFM 4.16, sorafenib and Tel combination showed good anti-cancer effects in H1975 and drug resistant H1975 cells in vitro, we further investigated their combinatorial effects in rociletinib resistant H1975 NSCLC xenograft model to ensure efficacy. We developed rociletinib resistant H1975 cells in our laboratory by continuous exposure of H1975 cells to rociletinib, starting from a dose of 150 nM, and further escalating to 4 μM over a period of 8-10 months. H1975-rociletinib resistant NSCLC xenograft model was developed by subcutaneous injection of rociletinib resistant H1975 cells on to the right side of nude mice. Tumor volume was checked twice weekly, once the palpable tumor was observed. The tumours were allowed to grow to the desired tumor volume (i.e., approximately around 400 mm3), and thereafter we divided the animals into five groups (n=6), so as to minimize the weight and tumour size difference among the groups. Control mice received 0.9% saline solution. Tel pre-treatment at a dose of 10 mg/kg body weight for three times a week in combination with CFM4.16 NLF (40 mg/kg body weight) and sorafenib (30 mg/kg body weight) significantly decreased tumor burden after 14-days in comparison to control and CFM 4.16 treated tumors (Figure 4). Tumor volume was checked twice a week during 2 weeks of treatment by using a digital Vernier caliper.
Figure 4.
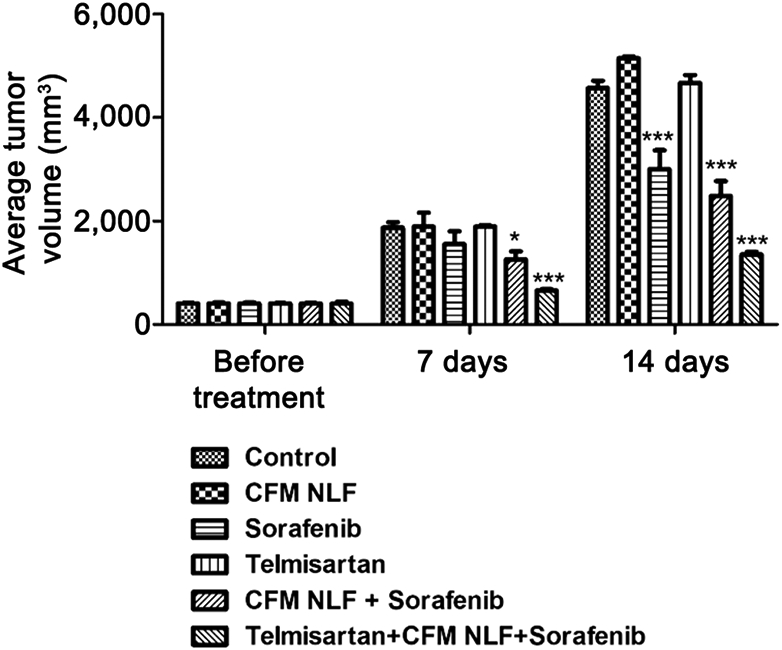
Tel oral administration followed by CFM 4.16 NLF and sorafenib combination significantly inhibited the tumor growth in the rociletinib resistant H1975 xenograft model. Data represent mean±S.E.M. of 4 mice per group. ***p<0.001 and *p<0.05 significant vs. control.
CFM 4.16 NLF, Sorafenib and Tel combination decreased the protein expression of p-MET and p-EGFR in rociletinib resistant H1975 NSCLC xenograft model.
Since we have observed that CFM 4.16, sorafenib and Tel combination significantly decreased the expression of oncogenic proteins (p-MET/MET and p-EGFR/EGFR) in vitro, we also investigated their expression in the ROC-Res-H1975 xenograft model. It was observed that sorafenib alone decreased the expression of p-MET/MET but not p-EGFR/EGFR in ROC-Res-H1975 tumor lysate. Similarly, CFM 4.16 NLF and sorafenib combination decreased the expression of p-MET/MET but not p-EGFR/EGFR in ROC-Res-H1975 tumor lysates. However, the combination of Tel, CFM 4.16 NLF and sorafenib induced a significant decrease in the expression of phosphorylated-MET (p<0.001) and- EGFR (p<0.001) proteins, which are oncogenic and responsible for drug resistance in cancer cells (Figure 5).
Figure 5.
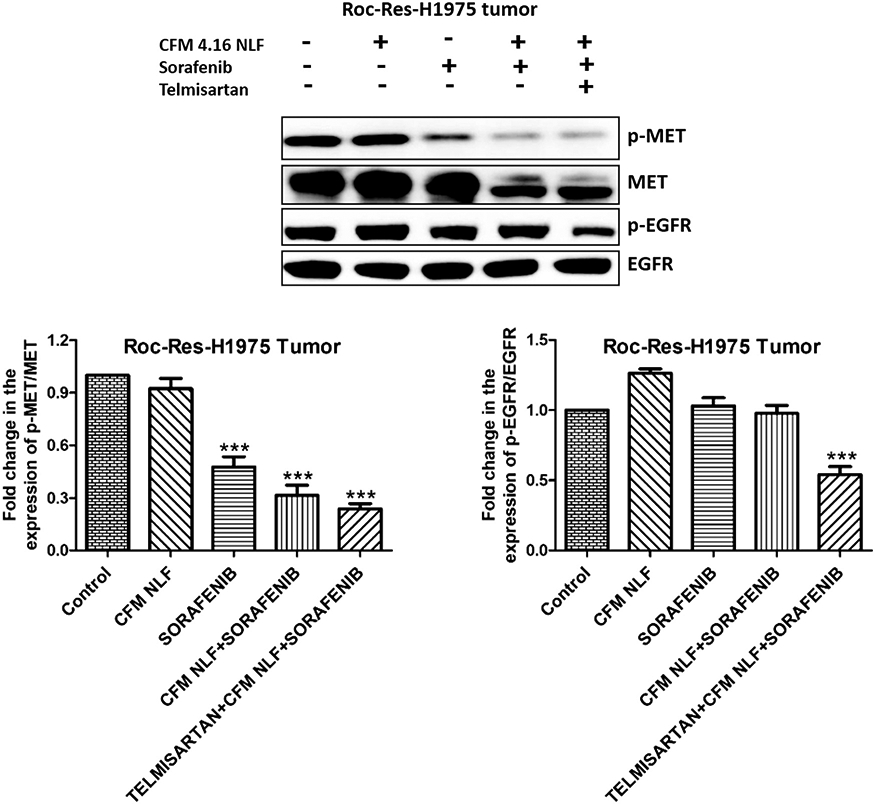
Western blots and densitometric analysis of p-EGFR and p-MET in rociletinib resistant H1975 xenograft model of lung cancer. Data are representative of three different experiments and presented as mean, and error bars refer to SEM. ***p<0.001 was considered significant when compared to control. MET: MET proto-oncogene, receptor tyrosine kinase; EGFR: epidermal growth factor receptor.
CFM 4.16 NLF, sorafenib and Tel combination decreased the protein expression of fibrotic, migration and cancer stem cell markers in rociletinib resistant H1975 xenograft model of NSCLC.
We investigated the effects of CFM 4.16 NLF, sorafenib and Tel combination on the fibrosis (TGF-β), migration (MMP9 and E-Cadherin) and cancer stem cell markers (Nanog and Sox2) in ROC-Res-H1975 tumor lysate. It was observed that Tel alone, significantly decreased the expression of TGF-β (p<0.001) and increased the expression of E-cadherin, respectively in ROC-Res-H1975 tumor lysate. However, Tel alone did not induce any alterations in the exepression of cancer stem cell markers in the ROC-Res-H1975 xenograft model of NSCLC. CFM 4.16 NLF and sorafenib combination decreased TGF- β (p<0.05), MMP9 (p<0.001) and Sox2 levels (p<0.01) in ROC-Res-H1975 xenograft model of NSCLC. This combination also increased the expression of E-cadherin (p<0.001), thereby decreasing migration (Figure 6). We also observed that CFM 4.16, sorafenib and Tel combination significantly decreased the expression of TGF-β (p<0.001), MMP-9 (p<0.001), and lung cancer stem cell markers (p<0.001), which contribute to the sorafenib poor uptake and resistance in lung cancer cells (Figure 6). The combination therapy also significantly increased the expression of E-cadherin (p<0.001) in the ROC-Res-H1975 xenograft model of NSCLC (Figure 6).
Figure 6.
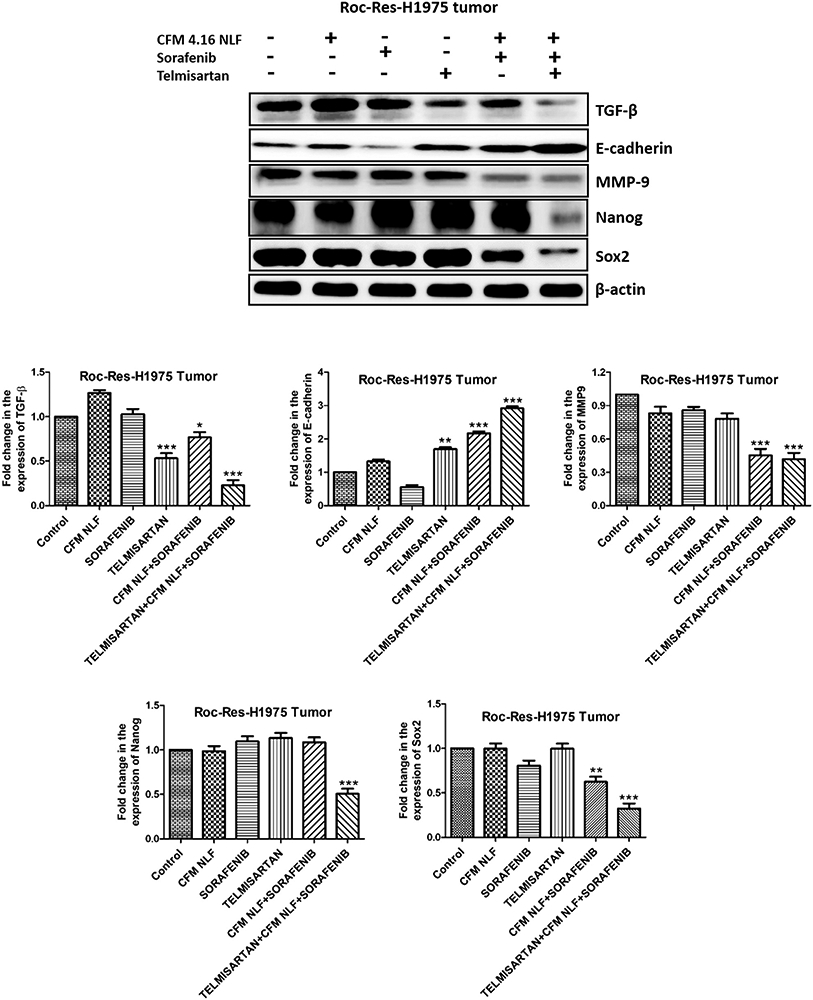
Western blots and densitometric analysis of TGF-β, E-cadherin, MMP9, Nanog and Sox2 in rociletinib resistant H1975 xenograft model of lung cancer. Data are representative of three different experiments and presented as mean, and error bars refer to SEM. *p<0.05, **p<0.01, ***p<0.001 was considered significant when compared to control. TGF-: Transforming growth factor β; MMP9: matrix metalloproteinase 9.
Discussion
Lung cancers with epidermal growth factor receptor (EGFR) gene mutations account for approximately 40% of lung adenocarcinomas in East Asians and around 20% in Caucasians and African Americans (41). TKIs are considered as the first line chemotherapy for NSCLC. Resistance to TKIs such as gefitinib, erlotinib or sorafenib involves hyperactivation/mutations in oncogenic proteins such as EGFR, MET, vascular endothelial growth factor receptor (VEGFR), fibroblast growth factor receptor (FGFR), BRAF, Alk, and Src in various cancers (42-44).
Different EGFR-dependent and EGFR-independent mechanisms have been demonstrated to be responsible for the development of third generation TKI (i.e., osimertinib and rociletinib) resistance in EGFR mutant NSCLC (45-47). Inhibition of AKT (i.e., by using MK-2206 and GDC-0068) and VEGFR/MET/AXL axis (i.e., by XL-880) have been reported to improve the sensitivity of rociletinib in rociletinib resistant models of NSCLC (14). Due to the paucity of therapeutic options readily available for secondary resistance after rociletinib treatment, there is an urgent unmet clinical challenge for the identification of therapeutics to combat rociletinib resistance in NSCLC. To the best of our knowledge, this study investigated for the first time the anti-cancer potential of telmisartan (Tel), CFM 4.16 and sorafenib combination in both in vivo and in vitro models of rociletinib resistant H1975 cells.
Herein, we investigated the anti-cancer efficacy of sorafenib, Tel, CFM 4.16, CFM 4.16 + sorafenib combination, and Tel + CFM 4.16 + sorafenib combination in 2D and 3D spheroid cultures of HCC827, erlotinib resistant HCC827, H1975 and rociletinib resistant H1975 cells. We observed higher IC50 values in 3D spheroid cultures when compared to 2D cultures of these cells, indicating the influence of the tumor microenvironment in determining the anti-cancer effects of chemotherapeutics. 3D spheroids serve as excellent physiologic tumor models in mimicking in vivo the tumour microenvironment for evaluating the anticancer effects of various chemotherapeutics (48, 49).
Multiple evidence has demonstrated the low permeability of free FITC across the cell membranes (50, 51). Our cell viability data has shown that TEL + CFM 4.16+ sorafenib combination decreased the proliferation of 3D spheroids. This is further supported by a cell uptake study, which showed that Tel pre-treatment significantly increased the uptake of FITC in 3D spheroids when compared to free FITC stained 3D spheroids as analyzed by flow cytometry. Decreased diffusion of chemotherapeutics through the spheroids contribute to resistance in 3D spheroid cultures, and tumor penetration remains a vital barrier inhibiting treatment response (52).
Rociletinib resistant H1975 xenograft model was developed by subcutaneous injection of rociletinib resistant H1975 cells into the right flank of nude mice. It was observed that Tel + CFM 4.16 NLF + sorafenib combination significantly decreased tumor volume when compared to control.
To elucidate the molecular changes involved behind the enhanced anticancer effects of Tel + CFM 4.16 + sorafenib combination in our study, we investigated the protein expression of p-EGFR/EGFR and p-MET/MET in 2D cultures of wild type and rociletinib resistant H1975 cells. It was observed that telmisartan (Tel), CFM 4.16 and sorafenib combination decreased p-MET/MET expression in both wild type and rociletinib resistant H1975 cells. However, this combination significantly decreased p-EGFR/EGFR in rociletinib resistant H1975 cells when compared to wild type H1975 cells. This demonstrates the efficacy of combination therapy in rociletinib resistant H1975 cells. Moreover, this combination therapy significantly decreased the protein expression of p-EGFR/EGFR and p-MET/MET in rociletinib resistant H1975 xenograft model, indicating its therapeutic efficacy. The superior effects of this combination in rociletinib resistant H1975 xenograft model may be due to the disruption of tumor stromal barriers by telmisartan (i.e., an anti-fibrotic agent), which facilitates CFM 4.16 and sorafenib to penetrate more into the deeper layers of tumors, to produce more anti-cancer effects when administered in vivo.
Cancer stem cells (CSCs) are involved in the initiation, maintenance, progression, metastasis and drug resistance of cancer cells (53). Various factors such as EMT, alterations in the metabolism of tumor cells, epigenetic modifications, and the tumor microenvironment are involved in the resistance of cancer cells through activation of various signaling pathways associated with cancer stemness (54). TGF- β is involved in the transformation of cells from a non-CSC to an CSC phenotype through activation of Zinc finger E-box-binding homeobox 1 (ZEB1), a key mediator of EMT (55). EMT is well demonstrated to be related with the stemness and maintenance of CSCs, which display stem cell-like properties by expressing Sox2, Oct4 and Nanog genes (56). Deregulated EGFR is involved in CSCs stemness and drug resistance of cancer cells (57). RAF/MEK/ERK/STAT3/AKT signaling is also associated with stemness of CSCs, drug resistance and recurrence of cancers (58). CSCs stemness (i.e., increased expression of Sox2 and CD24) is one of the factors responsible for resistance of cancer cells to BRAF inhibitors (59). A study by Cheriyan et al., has demonstrated that CARP-1 functional mimetic (CFM 4.16) in combination with sorafenib significantly decreased tumor growth of rociletinib-resistant H1975 NSCLC cells by inducing CARP-1 expression and facilitating apoptosis (22). Herein, we investigated the expression of CSC markers such as Nanog, Oct4 and Sox2 and observed that Tel, CFM 4.16 and sorafenib combination significantly decreased the levels of these proteins in 2D cultures of wild type and rociletinib resistant H1975 cells as well as in rociletinib resistant H1975 tumor cell lysates. This suggests the efficacy of this combination in decreasing drug resistance and improving anti-cancer effects.
Matrix metalloproteinases (MMPs) are involved in the pathophysiology of several cancers through zinc-dependent degradation and remodeling of extracellular matrix (ECM) proteins (60, 61). MMP-9 plays a crucial role in the invasion, metastasis, angiogenesis and progression of various cancers (62). It is negatively correlated with immune regulation of cancers through transforming growth factor-β (TGF-β) activation and shedding of intercellular adhesion molecule-1 (ICAM-1). MMP-9 is over-expressed and considered as a biomarker for the prognosis of breast cancer (63). TGF-β mediates not only the proliferation, differentiation, apoptosis, migration, adhesion, immune surveillance, and survival of cancer cells, but also favors the production of extracellular matrix proteins (64, 65). Tel, CFM 4.16 NLF and sorafenib combination significantly decreased the protein expression of MMP-9 in rociletinib resistant H1975 tumor model.
Decreased expression of E-cadherin, a well-known tumor suppressor and epithelial cell-cell adhesion molecule is associated with loss of differentiation and increased EMT in various cancers (66, 67). E-cadherin serves as a prognostic marker in breast, endometrial, prostate, gastric and lung cancers (68-72). A study by Wang et al., has demonstrated that TGF-β induces morphological and phenotypic changes associated with increased EMT and decreased E-cadherin levels (73). However, it was recently reported that loss of E-cadherin causes up-regulation of TGF-β signaling related genes in breast cancer cells (74). E-cadherin suppresses tumorigenicity and metastasis through inhibition of β-catenin and EGFR proteins, which are oncogenic in nature (75, 76). Tel, CFM 4.16 NLF and sorafenib combination significantly increased the protein expression of E-cadherin in rociletinib resistant H1975 tumor model. This indicates that the combination therapy not only decreases migration and invasion, but also increases the tumor-suppressive effects in the rociletinib resistant H1975 xenograft model.
Conclusion
In summary, the combination of Tel, CFM 4.16 and sorafenib displays superior anti-cancer effects in 2D and 3D spheroid cultures of rociletinib resistant H1975 cells, along with an excellent therapeutic potential in decreasing tumor growth in a rociletinib resistant H1975 xenograft model of NSCLC by decreasing the protein expression of p-EGFR/EGFR, p-MET/MET, CSC markers (Nanog, Sox2 and Oct4), TGF-β (i.e., involved in fibrosis) and MMP9 (involved in migration) and increasing the expression of E-cadherin (i.e., a tumor suppressor and involved in EMT). This study provides preliminary insights of the therapeutic efficacy of Tel, CFM 4.16 and sorafenib combination in decreasing rociletinib resistance of NSCLC.
Acknowledgements
The Authors are thankful to the National Institute on Minority Health and Health Disparities of National Institutes of Health, Grant/Award Number: U54 MD007582 and NSF-CREST center for Complex Material Design and also for Multidimensional Additive processing (CoMand) award #1735968 for providing the funding for this research work.
Footnotes
Conflicts of Interest
The Authors declare that they have no conflicts of interest associated with this study.
References
- 1.Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, Shafi S, Johnson DH, Mitter R, Rosenthal R, Salm M, Horswell S, Escudero M, Matthews N, Rowan A, Chambers T, Moore DA, Turajlic S, Xu H, Lee SM, Forster MD, Ahmad T, Hiley CT, Abbosh C, Falzon M, Borg E, Marafioti T, Lawrence D, Hayward M, Kolvekar S, Panagiotopoulos N, Janes SM, Thakrar R, Ahmed A, Blackhall F, Summers Y, Shah R, Joseph L, Quinn AM, Crosbie PA, Naidu B, Middleton G, Langman G, Trotter S, Nicolson M, Remmen H, Kerr K, Chetty M, Gomersall L, Fennell DA, Nakas A, Rathinam S, Anand G, Khan S, Russell P, Ezhil V, Ismail B, Irvin-Sellers M, Prakash V, Lester JF, Kornaszewska M, Attanoos R, Adams H, Davies H, Dentro S, Taniere P, O’Sullivan B, Lowe HL, Hartley JA, Iles N, Bell H, Ngai Y, Shaw JA, Herrero J, Szallasi Z, Schwarz RF, Stewart A, Quezada SA, Le Quesne J, Van Loo P, Dive C, Hackshaw A, Swanton C and TRACERx Consortium: Tracking the evolution of non-small-cell lung cancer. N Engl J Med 376(22): 2109–2121, 2017. PMID: 28445112. DOI: 10.1056/NEJMoa1616288 [DOI] [PubMed] [Google Scholar]
- 2.Zappa C and Mousa SA: Non-small cell lung cancer: current treatment and future advances. Transl Lung Cancer Res 5(3): 288–300, 2016. PMID: 27413711. DOI: 10.21037/tlcr.2016.06.07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Molina JR, Yang P, Cassivi SD, Schild SE and Adjei AA: Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc 83(5): 584–594, 2008. PMID: 18452692. DOI: 10.4065/83.5.584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Halliday PR, Blakely CM and Bivona TG: Emerging targeted therapies for the treatment of non-small cell lung cancer. Curr Oncol Rep 21(3): 21, 2019. PMID: 30806814. DOI: 10.1007/s11912-019-0770-x [DOI] [PubMed] [Google Scholar]
- 5.Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I, Fujita Y, Okinaga S, Hirano H, Yoshimori K, Harada T, Ogura T, Ando M, Miyazawa H, Tanaka T, Saijo Y, Hagiwara K, Morita S, Nukiwa T and North-East Japan Study Group: Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med 362(25): 2380–2388, 2010. PMID: 20573926. DOI: 10.1056/NEJMoa0909530 [DOI] [PubMed] [Google Scholar]
- 6.Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, Zhang S, Wang J, Zhou S, Ren S, Lu S, Zhang L, Hu C, Hu C, Luo Y, Chen L, Ye M, Huang J, Zhi X, Zhang Y, Xiu Q, Ma J, Zhang L and You C: Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 12(8): 735–742, 2011. PMID: 21783417. DOI: 10.1016/S1470-2045(11)70184-X [DOI] [PubMed] [Google Scholar]
- 7.Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, Li W, Hou M, Shi JH, Lee KY, Xu CR, Massey D, Kim M, Shi Y and Geater SL: Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol 15(2): 213–222, 2014. PMID: 24439929. DOI: 10.1016/S1470-2045(13)70604-1 [DOI] [PubMed] [Google Scholar]
- 8.Mok TS, Wu Y-L, Ahn M-J, Garassino MC, Kim HR, Ramalingam SS, Shepherd FA, He Y, Akamatsu H, Theelen WS, Lee CK, Sebastian M, Templeton A, Mann H, Marotti M, Ghiorghiu S, Papadimitrakopoulou VA and AURA3 Investigators: Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med 376(7): 629–640, 2017. PMID: 27959700. DOI: 10.1056/NEJMoa1612674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tran PN and Klempner SJ: Profile of rociletinib and its potential in the treatment of non-small-cell lung cancer. Lung Cancer (Auckl) 7: 91–97, 2016. PMID: 28210165. DOI: 10.2147/LCTT.S94337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sequist LV, Rolfe L and Allen AR: Rociletinib in EGFR-mutated non-small-cell lung cancer. N Engl J Med 373(6): 578–579, 2015. PMID: 26244318. DOI: 10.1056/NEJMc1506831 [DOI] [PubMed] [Google Scholar]
- 11.Wang S, Cang S and Liu D: Third-generation inhibitors targeting EGFR T790M mutation in advanced non-small cell lung cancer. J Hematol Oncol 9: 34, 2016. PMID: 27071706. DOI: 10.1186/s13045-016-0268-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Proto C, Imbimbo M, Gallucci R, Brissa A, Signorelli D, Vitali M, Macerelli M, Corrao G, Ganzinelli M, Greco FG, Garassino MC and Lo Russo G: Epidermal growth factor receptor tyrosine kinase inhibitors for the treatment of central nervous system metastases from non-small cell lung cancer: the present and the future. Transl Lung Cancer Res 5(6): 563–578, 2016. PMID: 28149752. DOI: 10.21037/tlcr.2016.10.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ercan D, Choi HG, Yun CH, Capelletti M, Xie T, Eck MJ, Gray NS and Jänne PA: EGFR mutations and resistance to irreversible pyrimidine-based EGFR inhibitors. Clin Cancer Res 21(17): 3913–3923, 2015. PMID: 25948633. DOI: 10.1158/1078-0432.CCR-14-2789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walter AO, Sjin RT, Haringsma HJ, Ohashi K, Sun J, Lee K, Dubrovskiy A, Labenski M, Zhu Z, Wang Z, Sheets M, St Martin T, Karp R, van Kalken D, Chaturvedi P, Niu D, Nacht M, Petter RC, Westlin W, Lin K, Jaw-Tsai S, Raponi M, Van Dyke T, Etter J, Weaver Z, Pao W, Singh J, Simmons AD, Harding TC and Allen A: Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov 3(12): 1404–1415, 2013. PMID: 24065731. DOI: 10.1158/2159-8290.CD-13-0314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ercan D, Xu C, Yanagita M, Monast CS, Pratilas CA, Montero J, Butaney M, Shimamura T, Sholl L, Ivanova EV, Tadi M, Rogers A, Repellin C, Capelletti M, Maertens O, Goetz EM, Letai A, Garraway LA, Lazzara MJ, Rosen N, Gray NS, Wong KK and Jänne PA: Reactivation of ERK signaling causes resistance to EGFR kinase inhibitors. Cancer Discov 2(10): 934–947, 2012. PMID: 22961667. DOI: 10.1158/2159-8290.CD-12-0103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mizuuchi H, Suda K, Murakami I, Sakai K, Sato K, Kobayashi Y, Shimoji M, Chiba M, Sesumi Y, Tomizawa K, Takemoto T, Sekido Y, Nishio K and Mitsudomi T: Oncogene swap as a novel mechanism of acquired resistance to epidermal growth factor receptor-tyrosine kinase inhibitor in lung cancer. Cancer Sci 107(4): 461–468, 2016. PMID: 26845230. DOI: 10.1111/cas.12905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, Cao Y, Shujath J, Gawlak S, Eveleigh D, Rowley B, Liu L, Adnane L, Lynch M, Auclair D, Taylor I, Gedrich R, Voznesensky A, Riedl B, Post LE, Bollag G and Trail PA: BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 64(19): 7099–7109, 2004. PMID: 15466206. DOI: 10.1158/0008-5472.CAN-04-1443 [DOI] [PubMed] [Google Scholar]
- 18.Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, Schwartz B, Simantov R and Kelley S: Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov 5(10): 835–844, 2006. PMID: 17016424. DOI: 10.1038/nrd2130 [DOI] [PubMed] [Google Scholar]
- 19.Al-Marrawi MY, Saroya BS, Brennan MC, Yang Z, Dykes TM and El-Deiry WS: Off-label use of cetuximab plus sorafenib and panitumumab plus regorafenib to personalize therapy for a patient with V600E BRAF-mutant metastatic colon cancer. Cancer Biol Ther 14(8): 703–710, 2013. PMID: 23792568. DOI: 10.4161/cbt.25191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM and Lynch M: Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther 7(10): 3129–3140, 2008. PMID: 18852116. DOI: 10.1158/1535-7163.MCT-08-0013 [DOI] [PubMed] [Google Scholar]
- 21.Takezawa K, Okamoto I, Yonesaka K, Hatashita E, Yamada Y, Fukuoka M and Nakagawa K: Sorafenib inhibits non-small cell lung cancer cell growth by targeting B-RAF in KRAS wild-type cells and C-RAF in KRAS mutant cells. Cancer Res 69(16): 6515–6521, 2009. PMID: 19638574. DOI: 10.1158/0008-5472.CAN-09-1076 [DOI] [PubMed] [Google Scholar]
- 22.Cheriyan VT, Alsaab H, Sekhar S, Venkatesh J, Mondal A, Vhora I, Sau S, Muthu M, Polin LA, Levi E, Bepler G, Iyer AK, Singh M and Rishi AK: A CARP-1 functional mimetic compound is synergistic with BRAF-targeting in non-small cell lung cancers. Oncotarget 9(51): 29680–29697, 2018. PMID: 30038713. DOI: 10.18632/oncotarget.25671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Puliyappadamba VT, Wu W, Bevis D, Zhang L, Polin L, Kilkuskie R, Finley RL Jr, Larsen SD, Levi E, Miller FR, Wali A and Rishi AK: Antagonists of anaphase-promoting complex (APC)-2-cell cycle and apoptosis regulatory protein (CARP)-1 interaction are novel regulators of cell growth and apoptosis. J Biol Chem 286(44): 38000–38017, 2011. PMID: 21903591. DOI: 10.1074/jbc.M111.222398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lehman NL, Tibshirani R, Hsu JY, Natkunam Y, Harris BT, West RB, Masek MA, Montgomery K, van de Rijn M and Jackson PK: Oncogenic regulators and substrates of the anaphase promoting complex/cyclosome are frequently overexpressed in malignant tumors. Am J Pathol 170(5): 1793–1805, 2007. PMID: 17456782. DOI: 10.2353/ajpath.2007.060767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim JH, Yang CK, Heo K, Roeder RG, An W and Stallcup MR: CCAR1, a key regulator of mediator complex recruitment to nuclear receptor transcription complexes. Mol Cell 31(4): 510–519, 2008. PMID: 18722177. DOI: 10.1016/j.molcel.2008.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muthu M, Cheriyan VT and Rishi AK: CARP-1/CCAR1: a biphasic regulator of cancer cell growth and apoptosis. Oncotarget 6(9): 6499–6510, 2015. PMID: 25894788. DOI: 10.18632/oncotarget.3376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Curnis F, Sacchi A and Corti A: Improving chemotherapeutic drug penetration in tumors by vascular targeting and barrier alteration. J Clin Invest 110(4): 475–482, 2002. PMID: 12189241. DOI: 10.1172/JCI15223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marcucci F and Corti A: How to improve exposure of tumor cells to drugs: promoter drugs increase tumor uptake and penetration of effector drugs. Adv Drug Deliv Rev 64(1): 53–68, 2012. PMID: 21983328. DOI: 10.1016/j.addr.2011.09.007 [DOI] [PubMed] [Google Scholar]
- 29.Chauhan VP, Martin JD, Liu H, Lacorre DA, Jain SR, Kozin SV, Stylianopoulos T, Mousa AS, Han X, Adstamongkonkul P, Popović Z, Huang P, Bawendi MG, Boucher Y and Jain RK: Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun 4: 2516, 2013. PMID: 24084631. DOI: 10.1038/ncomms3516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Godugu C, Patel AR, Doddapaneni R, Marepally S, Jackson T and Singh M: Inhalation delivery of Telmisartan enhances intratumoral distribution of nanoparticles in lung cancer models. J Control Release 172(1): 86–95, 2013. PMID: 23838154. DOI: 10.1016/j.jconrel.2013.06.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patel K, Doddapaneni R, Chowdhury N, Boakye CH, Behl G and Singh M: Tumor stromal disrupting agent enhances the anticancer efficacy of docetaxel loaded PEGylated liposomes in lung cancer. Nanomedicine (Lond) 11(11): 1377–1392, 2016. PMID: 27171485. DOI: 10.2217/nnm.16.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Q, Liu G, Liu S, Su H, Wang Y, Li J and Luo C: Remodeling the tumor microenvironment with emerging nanotherapeutics. Trends Pharmacol Sci 39(1): 59–74, 2018. PMID: 29153879. DOI: 10.1016/j.tips.2017.10.009 [DOI] [PubMed] [Google Scholar]
- 33.Li J, Chen L, Yu P, Liu B, Zhu J and Yang Y: Telmisartan exerts anti-tumor effects by activating peroxisome proliferator-activated receptor-γ in human lung adenocarcinoma A549 cells. Molecules 19(3): 2862–2876, 2014. PMID: 24603556. DOI: 10.3390/molecules19032862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang S and Wang Y: Telmisartan inhibits NSCLC A549 cell proliferation and migration by regulating the PI3K/AKT signaling pathway. Oncol Lett 15(4): 5859–5864, 2018. PMID: 29552215. DOI: 10.3892/ol.2018.8002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Surapaneni SK, Bashir S and Tikoo K: Gold nanoparticles-induced cytotoxicity in triple negative breast cancer involves different epigenetic alterations depending upon the surface charge. Sci Rep 8(1): 12295, 2018. PMID: 30115982. DOI: 10.1038/s41598-018-30541-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arthur P, Patel N, Surapaneni SK, Mondal A, Gebeyehu A, Bagde A, Kutlehria S, Nottingham E and Singh M: Targeting lung cancer stem cells using combination of Tel and Docetaxel liposomes in 3D cultures and tumor xenografts. Toxicol Appl Pharmacol 401: 115112, 2020. PMID: 32540278. DOI: 10.1016/j.taap.2020.115112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheriyan VT, Muthu M, Patel K, Sekhar S, Rajeswaran W, Larsen SD, Polin L, Levi E, Singh M and Rishi AK: CARP-1 functional mimetics are novel inhibitors of drug-resistant triple negative breast cancers. Oncotarget 7(45): 73370–73388, 2016. PMID: 27687593. DOI: 10.18632/oncotarget.12333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muthu M, Somagoni J, Cheriyan VT, Munie S, Levi E, Ashour AE, Yassin AE, Alafeefy AM, Sochacki P, Polin LA, Reddy KB, Larsen SD, Singh M and Rishi AK: Identification and testing of novel CARP-1 functional mimetic compounds as inhibitors of non-small cell lung and triple negative breast cancers. J Biomed Nanotechnol 11(9): 1608–1627, 2015. PMID: 26485930. DOI: 10.1166/jbn.2015.2099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG and NC3Rs Reporting Guidelines Working Group: Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160(7): 1577–1579, 2010. PMID: 20649561. DOI: 10.1111/j.1476-5381.2010.00872.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Green R, Howell M, Khalil R, Nair R, Yan J, Foran E, Katiri S, Banerjee J, Singh M, Bharadwaj S, Mohapatra SS and Mohapatra S: Actinomycin D and telmisartan combination targets lung cancer stem cells through the Wnt/Beta catenin pathway. Sci Rep 9(1): 18177, 2019. PMID: 31796785. DOI: 10.1038/s41598-019-54266-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DeSantis CE, Siegel RL, Sauer AG, Miller KD, Fedewa SA, Alcaraz KI and Jemal A: Cancer statistics for African Americans, 2016: Progress and opportunities in reducing racial disparities. CA Cancer J Clin 66(4): 290–308, 2016. PMID: 26910411. DOI: 10.3322/caac.21340 [DOI] [PubMed] [Google Scholar]
- 42.Rosenzweig SA: Acquired resistance to drugs targeting tyrosine kinases. Adv Cancer Res 138: 71–98, 2018. PMID: 29551130. DOI: 10.1016/bs.acr.2018.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang S, Song Y, Yan F and Liu D: Mechanisms of resistance to third-generation EGFR tyrosine kinase inhibitors. Front Med 10(4): 383–388, 2016. PMID: 27770386. DOI: 10.1007/s11684-016-0488-1 [DOI] [PubMed] [Google Scholar]
- 44.Botting GM, Rastogi I, Chhabra G, Nlend M and Puri N: Mechanism of resistance and novel targets mediating resistance to EGFR and c-Met tyrosine kinase inhibitors in non-small cell lung cancer. PLoS One 10(8): e0136155, 2015. PMID: 26301867. DOI: 10.1371/journal.pone.0136155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E and Tiseo M: Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br J Cancer 121(9): 725–737, 2019. PMID: 31564718. DOI: 10.1038/s41416-019-0573-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van Der Steen N, Caparello C, Rolfo C, Pauwels P, Peters GJ and Giovannetti E: New developments in the management of non-small-cell lung cancer, focus on rociletinib: what went wrong? Onco Targets Ther 9: 6065–6074, 2016. PMID: 27785053. DOI: 10.2147/OTT.S97644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Minari R, Bordi P and Tiseo M: Third-generation epidermal growth factor receptor-tyrosine kinase inhibitors in T790M-positive non-small cell lung cancer: review on emerged mechanisms of resistance. Transl Lung Cancer Res 5(6): 695–708, 2016. PMID: 28149764. DOI: 10.21037/tlcr.2016.12.02 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tung YC, Hsiao AY, Allen SG, Torisawa YS, Ho M and Takayama S: High-throughput 3D spheroid culture and drug testing using a 384 hanging drop array. Analyst 136(3): 473–478, 2011. PMID: 20967331. DOI: 10.1039/c0an00609b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fang Y and Eglen RM: Three-dimensional cell cultures in drug discovery and development. SLAS Discov 22(5): 456–472, 2017. PMID: 28520521. DOI: 10.1177/1087057117696795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hu CS, Chiang CH, Hong PD and Yeh MK: Influence of charge on FITC-BSA-loaded chondroitin sulfate-chitosan nanoparticles upon cell uptake in human Caco-2 cell monolayers. Int J Nanomedicine 7: 4861–4872, 2012. PMID: 23028215. DOI: 10.2147/IJN.S34770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kommineni N, Saka R, Bulbake U and Khan W: Cabazitaxel and thymoquinone co-loaded lipospheres as a synergistic combination for breast cancer. Chem Phys Lipids 224: 104707, 2019. PMID: 30521787. DOI: 10.1016/j.chemphyslip.2018.11.009 [DOI] [PubMed] [Google Scholar]
- 52.Gebeyehu A, Surapaneni SK, Huang J, Mondal A, Wang VZ, Haruna NF, Bagde A, Arthur P, Kutlehria S, Patel N, Rishi AK and Singh M: Polysaccharide hydrogel based 3D printed tumor models for chemotherapeutic drug screening. Sci Rep 11(1): 372, 2021. PMID: 33431915. DOI: 10.1038/s41598-020-79325-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abad E, Graifer D and Lyakhovich A: DNA damage response and resistance of cancer stem cells. Cancer Lett 474: 106–117, 2020. PMID: 31968219. DOI: 10.1016/j.canlet.2020.01.008 [DOI] [PubMed] [Google Scholar]
- 54.Khan AQ, Rashid K, AlAmodi AA, Raza SS and Uddin S: Recent developments in unraveling signaling mechanisms underlying drug resistance due to cancer stem-like cells. Curr Opin Pharmacol 54: 130–141, 2020. PMID: 33166909. DOI: 10.1016/j.coph.2020.09.012 [DOI] [PubMed] [Google Scholar]
- 55.Kim BN, Ahn DH, Kang N, Yeo CD, Kim YK, Lee KY, Kim TJ, Lee SH, Park MS, Yim HW, Park JY, Park CK and Kim SJ: TGF-β induced EMT and stemness characteristics are associated with epigenetic regulation in lung cancer. Sci Rep 10(1): 10597, 2020. PMID: 32606331. DOI: 10.1038/s41598-020-67325-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shibue T and Weinberg RA: EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol 14(10): 611–629, 2017. PMID: 28397828. DOI: 10.1038/nrclinonc.2017.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lv Y, Cang W, Li Q, Liao X, Zhan M, Deng H, Li S, Jin W, Pang Z, Qiu X, Zhao K, Chen G, Qiu L and Huang L: Erlotinib overcomes paclitaxel-resistant cancer stem cells by blocking the EGFR-CREB/GRβ-IL-6 axis in MUC1-positive cervical cancer. Oncogenesis 8(12): 70, 2019. PMID: 31772161. DOI: 10.1038/s41389-019-0179-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nagathihalli NS, Castellanos JA, Lamichhane P, Messaggio F, Shi C, Dai X, Rai P, Chen X, VanSaun MN and Merchant NB: Inverse correlation of STAT3 and MEK signaling mediates resistance to RAS pathway inhibition in pancreatic cancer. Cancer Res 78(21): 6235–6246, 2018. PMID: 30154150. DOI: 10.1158/0008-5472.CAN-18-0634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hüser L, Sachindra S, Granados K, Federico A, Larribère L, Novak D, Umansky V, Altevogt P and Utikal J: SOX2-mediated upregulation of CD24 promotes adaptive resistance toward targeted therapy in melanoma. Int J Cancer 143(12): 3131–3142, 2018. PMID: 29905375. DOI: 10.1002/ijc.31609 [DOI] [PubMed] [Google Scholar]
- 60.Gialeli C, Theocharis AD and Karamanos NK: Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J 278(1): 16–27, 2011. PMID: 21087457. DOI: 10.1111/j.1742-4658.2010.07919.x [DOI] [PubMed] [Google Scholar]
- 61.Kessenbrock K, Plaks V and Werb Z: Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141(1): 52–67, 2010. PMID: 20371345. DOI: 10.1016/j.cell.2010.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nazir SU, Kumar R, Singh A, Khan A, Tanwar P, Tripathi R, Mehrotra R and Hussain S: Breast cancer invasion and progression by MMP-9 through Ets-1 transcription factor. Gene 711: 143952, 2019. PMID: 31265880. DOI: 10.1016/j.gene.2019.143952 [DOI] [PubMed] [Google Scholar]
- 63.Li H, Qiu Z, Li F and Wang C: The relationship between MMP-2 and MMP-9 expression levels with breast cancer incidence and prognosis. Oncol Lett 14(5): 5865–5870, 2017. PMID: 29113219. DOI: 10.3892/ol.2017.6924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jakowlew SB: Transforming growth factor-beta in cancer and metastasis. Cancer Metastasis Rev 25(3): 435–457, 2006. PMID: 16951986. DOI: 10.1007/s10555-006-9006-2 [DOI] [PubMed] [Google Scholar]
- 65.Dennler S, Goumans MJ and ten Dijke P: Transforming growth factor beta signal transduction. J Leukoc Biol 71(5): 731–740, 2002. PMID: 11994497. [PubMed] [Google Scholar]
- 66.Baranwal S and Alahari SK: Molecular mechanisms controlling E-cadherin expression in breast cancer. Biochem Biophys Res Commun 384(1): 6–11, 2009. PMID: 19379710. DOI: 10.1016/j.bbrc.2009.04.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Umbas R, Isaacs WB, Bringuier PP, Schaafsma HE, Karthaus HF, Oosterhof GO, Debruyne FM and Schalken JA: Decreased E-cadherin expression is associated with poor prognosis in patients with prostate cancer. Cancer Res 54(14): 3929–3933, 1994. PMID: 7518346. [PubMed] [Google Scholar]
- 68.Heimann R and Hellman S: Clinical progression of breast cancer malignant behavior: what to expect and when to expect it. J Clin Oncol 18(3): 591–599, 2000. PMID: 10653874. DOI: 10.1200/JCO.2000.18.3.591 [DOI] [PubMed] [Google Scholar]
- 69.Mell LK, Meyer JJ, Tretiakova M, Khramtsov A, Gong C, Yamada SD, Montag AG and Mundt AJ: Prognostic significance of E-cadherin protein expression in pathological stage I-III endometrial cancer. Clin Cancer Res 10(16): 5546–5553, 2004. PMID: 15328195. DOI: 10.1158/1078-0432.CCR-0943-03 [DOI] [PubMed] [Google Scholar]
- 70.van Oort IM, Tomita K, van Bokhoven A, Bussemakers MJ, Kiemeney LA, Karthaus HF, Witjes JA and Schalken JA: The prognostic value of E-cadherin and the cadherin-associated molecules alpha-, beta-, gamma-catenin and p120ctn in prostate cancer specific survival: a long-term follow-up study. Prostate 67(13): 1432–1438, 2007. PMID: 17639504. DOI: 10.1002/pros.20626 [DOI] [PubMed] [Google Scholar]
- 71.Chan AO, Lam SK, Chu KM, Lam CM, Kwok E, Leung SY, Yuen ST, Law SY, Hui WM, Lai KC, Wong CY, Hu HC, Lai CL and Wong J: Soluble E-cadherin is a valid prognostic marker in gastric carcinoma. Gut 48(6): 808–811, 2001. PMID: 11358900. DOI: 10.1136/gut.48.6.808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang YL, Chen MW and Xian L: Prognostic and clinicopathological significance of downregulated E-cadherin expression in patients with non-small cell lung cancer (NSCLC): a meta-analysis. PLoS One 9(6): e99763, 2014. PMID: 24978478. DOI: 10.1371/journal.pone.0099763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang B, Herman-Edelstein M, Koh P, Burns W, Jandeleit-Dahm K, Watson A, Saleem M, Goodall GJ, Twigg SM, Cooper ME and Kantharidis P: E-cadherin expression is regulated by miR-192/215 by a mechanism that is independent of the profibrotic effects of transforming growth factor-beta. Diabetes 59(7): 1794–1802, 2010. PMID: 20393144. DOI: 10.2337/db09-1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Padmanaban V, Krol I, Suhail Y, Szczerba BM, Aceto N, Bader JS and Ewald AJ: E-cadherin is required for metastasis in multiple models of breast cancer. Nature 573(7774): 439–444, 2019. PMID: 31485072. DOI: 10.1038/s41586-019-1526-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jeanes A, Gottardi CJ and Yap AS: Cadherins and cancer: how does cadherin dysfunction promote tumor progression? Oncogene 27(55): 6920–6929, 2008. PMID: 19029934. DOI: 10.1038/onc.2008.343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.van Roy F: Beyond E-cadherin: roles of other cadherin superfamily members in cancer. Nat Rev Cancer 14(2): 121–134, 2014. PMID: 24442140. DOI: 10.1038/nrc3647 [DOI] [PubMed] [Google Scholar]