

Abstract
Organisms in nature have evolved into proficient synthetic chemists, utilizing specialized enzymatic machinery to biosynthesize an inspiring diversity of secondary metabolites. Often serving to boost competitive advantage for their producers, these secondary metabolites have widespread human impacts as antibiotics, anti-inflammatories, and antifungal drugs. The natural products discovery field has begun a shift away from traditional activity-guided approaches and is beginning to take advantage of increasingly available metabolomics and genomics datasets to explore undiscovered chemical space. Major strides have been made and now enable -Omics-informed prioritization of chemical structures for discovery, including the prospect of confidently linking metabolites to their biosynthetic pathways. Over the last decade, more integrated strategies now provide researchers with pipelines for simultaneous identification of expressed secondary metabolites and their biosynthetic machinery. However, continuous collaboration by the natural products community will be required to optimize strategies for effective evaluation of natural product biosynthetic gene clusters to accelerate discovery efforts. Here, we provide an evaluative guide to scientific literature as it relates to studying natural product biosynthesis using genomics, metabolomics, and their integrated datasets. Particular emphasis is placed on the unique insights that can be gained from large-scale integrated strategies, and we provide source organism-specific considerations to evaluate the gaps in our current knowledge.
Graphical Abstract

Here we provide a comprehensive guide for studying natural product biosynthesis using genomics, metabolomics, and their integrated datasets. We emphasize integrated strategies and provide a critical outlook on remaining challenges in the field.
1. Introduction
Natural medicines have been used since the beginning of human history, providing treatments for a wide variety of diseases.1, 2 Indeed, plants, fungi, and bacteria have a centuries-old connection with humankind, producing both beneficial and harmful metabolites with profound impacts on human health. These secondary metabolites, also known as specialized metabolites or natural products, are formed by diverse enzymatic machinery evolutionarily primed over millennia to help the producing organism secure its environmental niche,3 and have become an invaluable source of inspiration for innovative pharmaceutical drugs active against a variety of diseases.1, 4, 5 Although hundreds of life-saving drugs have been discovered from and inspired by nature, the fact remains that the majority of secondary metabolites contributing to the biological activity of these organisms has not yet been discovered.6, 7
Historically, researchers have relied on bioactivity- or chemical signature-guided fractionation approaches to isolate and identify individual constituents from complex natural product mixtures.8–14 These methodologies have found widespread use, particularly bioactivity-guided fractionation, leading to the discovery of life-saving drugs including taxol, camptothecin,10, 11 vinblastine,13 and artemisinin.12 Despite their popularity, the use of such fractionation approaches for natural products discovery is becoming less fruitful, with scientists often re-discovering known constituents in a given mixture.15, 16 In recent years, scientists have begun to shift away from bioactivity-guided fractionation as the gold standard approach for natural product discovery, turning instead to genomics, metabolomics, and other Big Data approaches to guide isolation efforts towards uncharted chemical space.17, 18
Now, natural products chemists face a new challenge, no longer held back by a lack of data. Instead, they find themselves in an age where thousands of genome sequences and metabolite profiles of phylogenetically diverse organisms are readily available.19–22 The ever-decreasing costs of DNA sequencing have revealed the untapped biosynthetic potential of even well-studied organisms, which encode an abundance of biosynthetic genes that have yet to be linked to metabolite products.23 Moreover, analytical instrumentation continues to become more sophisticated and efficient, enabling facile detection and comparison of thousands of secondary metabolites.24–26 To keep up with the pace at which new samples are being analyzed both chemically and genetically, technological tools are being developed at an accelerated pace, enabling scientists to use this information to successfully navigate biosynthetic dark matter. Using automated predictive algorithms for identifying genes involved in secondary metabolism,27–39 scientists have begun to unravel the patterned nature of biosynthetic pathways, enabling targeted discovery of novel chemistry using genome mining.17, 40 At the same time, metabolomics analyses are gaining popularity for visualizing metabolite relatedness and predicting chemical substructures.41, 42 While each of these-Omics technologies has distinct advantages for directing natural products discovery, an increasing number of studies are beginning to integrate genomics and metabolomics datasets to prioritize the identification of novel, bioactive metabolites and link them to their biosynthetic pathways. Despite the accelerated production and utilization of such tools, defining the structures of genetically encoded secondary metabolites remains a central challenge.
A number of excellent review papers on the utilization of genomics and metabolomics for natural products discovery have been written in recent years, and we invite the interested reader to examine these manuscripts for additional commentary on the topic.43–45 In this review, we seek to add to this discussion by providing an outline of the current methodologies for mining genomes and metabolomes of natural product-producing organisms for targeted discovery of new chemical entities. We will highlight both genomic and metabolomic strategies for targeting relatives of known molecules, rare and novel scaffolds, as well as molecules with specific biological activities, placing particular emphasis on integrated genomics-metabolomics approaches that have found success over the last several years. We also hope to shed light on source-organism specific considerations and overarching challenges that continue to stymie discovery efforts. We hope to provide guidance to investigators seeking to use large-scale-Omics technologies to unlock the untapped biosynthetic potential of the natural world.
2. Definitions of genomics and metabolomics in the context of natural products
Genomics and metabolomics, though both relatively established fields, have only recently begun to be applied to natural products discovery. In general, ‘-Omics’ technologies such as genomics and metabolomics aim to gain a comprehensive understanding of the molecules that comprise a given organism.46 Unlike reductionist or hypothesis-driven approaches that have been historically used for natural products discovery including bioactivity- and chemical signature-guided fractionation, -Omics technologies are more hypothesis-generating, using untargeted analyses to gain a ‘birds-eye’ perspective of the organism(s) under study. It is without question that genomics and metabolomics have made an impact in a huge variety of scientific disciplines, aiding in disease diagnosis, biomarker discovery, drug toxicity studies, and more.46, 47 While the majority of these applications rely on the evaluation of primary metabolic genes and metabolites, natural products discovery relies primarily on the assessment of secondary metabolites and biosynthetic genes. In the context of this paper, genomics approaches utilize genotypic profiles of natural product-producing organisms to identify their secondary metabolite genes (and thus their overall biosynthetic potential), while metabolomics studies evaluate chemical profiles of these organisms to determine the secondary metabolite products that are actually expressed, providing insight into gene expression and the overall phenotype of the organism under study.
3. Acquisition of metabolomics and genomics datasets
In recent years, advances in molecular biology technologies have enabled an enormous increase in genome sequencing for natural product-producing organisms, particularly in bacteria and fungi.20, 43, 45 Likewise, the continued development of analytical instrumentation, including nuclear magnetic resonance (NMR) and mass spectrometry (MS), has allowed for thousands of spectra from bacteria, fungi, and plants to be mined for natural products discovery.21, 22, 25, 45 Using genome sequences, it is possible to evaluate the full biosynthetic potential for a given organism,48 while metabolic fingerprints provide insight into secondary metabolite expression signatures as a snapshot at given experimental conditions. Of course, the ability to extract meaningful information from large datasets relies heavily on their quality, which is largely influenced by the choice of data acquisition and analysis procedures. Here, we provide an overview of the most popular techniques that are employed to acquire genomic and metabolomic datasets for natural products discovery.
3.1. Obtaining sets of gene clusters from genomics datasets
Many have promised that genomics has the potential to revolutionize the field of natural products, enabling high-throughput and organized discovery of secondary metabolites and their biosynthetic pathways.40 To generate such data, scientists must sequence, assemble, and annotate genomes to identify genes likely to be involved in secondary metabolite biosynthesis.43, 45 The two essential points to consider while annotating a sequenced genome are the contiguity of assemblies as well as their accuracy, which both depend on the quality of sequencing.45 The Illumina next generation sequencing (NGS) platform provides high quality sequencing data with low error rates and low cost but has the disadvantage of providing short reads. Since this results in fragmented assemblies of small contigs, the biggest challenge is obtaining a full biosynthetic gene cluster (BGC) from these assemblies large enough to be properly picked up and annotated by BGC identification algorithms. Additionally, inaccuracy can lead to misassembles, which could result in deletion, duplication, or rearrangements that shift the true order of genes or protein domains.45 These disadvantages have been addressed by the generation of more advanced single molecule sequencing technologies such as Pacific Biosciences (PacBio) and Oxford Nanopore sequencing methods, which are capable of providing longer reads. However, these platforms have higher error rates that could lead to frameshifts that change gene annotation.
Arguably, the most important key to unlock the untapped biosynthetic potential of Nature from a genomics standpoint is the choice of computational approaches for identifying BGCs.40 These diverse algorithms, which have grown increasingly sophisticated over the years, are paramount to understand the biosynthetic potential and prioritize exploratory efforts towards uncharted chemical space. Some of the earliest tools for detecting biosynthetic genes in genome sequences are simple comparison tools such as BLAST 49 and HMMer,50 which require manual construction of query sequences using pre-defined lists of genes. More recently, a number of algorithms have been developed for the targeted detection of biosynthetic genes, including ClustScan,32 SMURF,33 CLUSEAN,51 np.searcher,52 antiSMASH,53 and PRISM.31, 54 These strategies, rather than using a user-defined list of genes as a query sequence, instead use a library of profile Hidden Markov Models (pHMMs)55 to identify genetic regions likely to encode signature biosynthetic genes. The regions before and after the identified core genes are then scanned to detect regions encoding transporters, tailoring enzymes, or transcription factors.33, 37, 56 antiSMASH has been continually curated over the last several years,39, 53, 56, 57 currently containing detection rules for more than 50 classes of BGCs.39 The antiSMASH algorithm, though originally modeled using bacterial and fungal genome sequences,53 has now been expanded to plants using modified rules developed specifically for these organisms.29
More recently, the linkage-based CO-OCCUR algorithm was developed and applied to 101 genomes from phylogenetically diverse Dothideomycetes. Using CO-OCCUR, biosynthetic genes were identified through their frequency and co-occurrence around signature biosynthetic genes, regardless of gene function.37 The authors went on to compare the CO-OCCUR pipeline to the more established SMURF33 and antiSMASH53 pipelines, illustrating that no single algorithm can identify all accessory genes of interest in regions surrounding signature biosynthetic genes. While the three algorithms all predicted approximately the same number of BGCs, the BGCs varied in their predicted content. CO-OCCUR identified 51.2% and 37.7% of the BGCs detected by SMURF and antiSMASH, respectively, while SMURF and antiSMASH detected 40.7% and 42% of the BGCs detected by CO-OCCUR (Figure 1). Using all of these algorithms together, the authors were able to get a more comprehensive picture of the BGC compositional diversity in Dothideomycetes.37 Future genome mining studies will likely also benefit from the incorporation of multiple algorithmic approaches.
Figure 1.
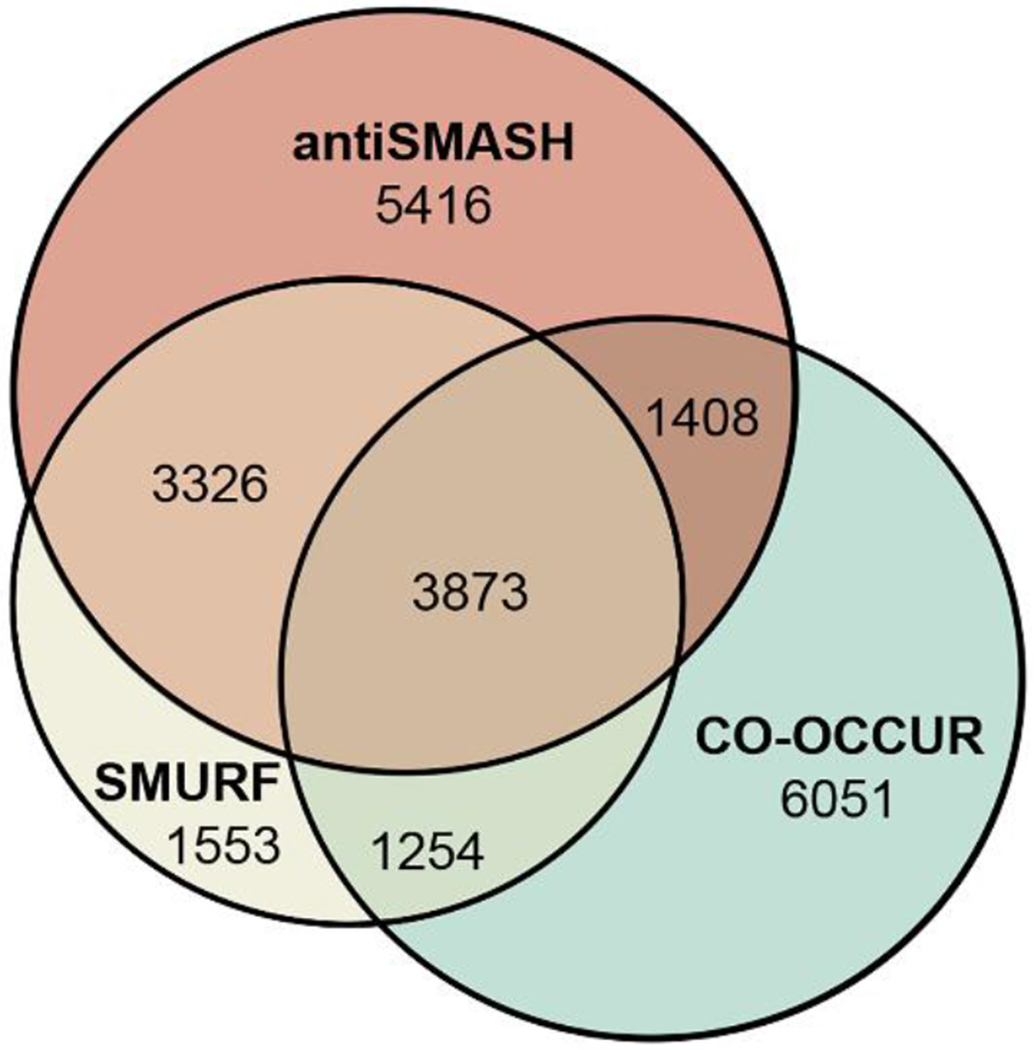
Unique and overlapping BGC genes detected from Dothideomycetes using SMURF, antiSMASH, and CO-OCCUR gene cluster detection algorithms. Figure has been reused and modified from Gluck-Thaler et al. 2020.37
No algorithm is foolproof and understanding the limitations of the available approaches will enable researchers to choose and, if desired, combine the most fitting algorithms to reach project-specific goals. For de novo BGC annotation, pHMM approaches are unmatched, but they fail to identify noncanonical biosynthetic genes and those that are unique to a lineage of organisms not used in the training dataset. Co-occurrence based detection algorithms, because they are not dependent on training datasets, are able to detect noncanonical genes more readily, but are not suited for identifying biosynthetic genes that are not evolutionarily conserved. Because of the nature of pHMM algorithms, they are not currently suited to full integration with alternative methods for BGC prediction. As such, the utilization of multiple approaches is computationally expensive and would result in multiple predictions. Researchers can then integrate this output and rank/score predictions based on the goals of the project at hand.
Notably, pattern-matching algorithms such as ClusterFinder and the more targeted antiSMASH are particularly challenged by the identification of BGCs encoding Ribosomally synthesized and Posttranslationally modified Peptides (RiPPs) because of their small size (20-30 base pairs) and lack of signature genes.45, 58 However, RiPP-specific algorithms, such as RODEO27 and DeepRIPP28 have found recent success in overcoming these challenges. Undoubtedly, continual development, invention, and integration of algorithms to identify and prioritize BGCs will enable researchers to truly take advantage of the rich biosynthetic capacity of Nature.
3.2. Detecting secondary metabolites from metabolomics datasets
While genomics data can provide insight into the biosynthetic capacity of organisms, metabolomics provides the key to truly understanding natural product biosynthetic pathways, revealing structural information of downstream secondary metabolites and their reaction intermediates.59–61 Of course, a metabolomics experiment can only identify metabolites that are expressed, so the success of the study relies on maximizing gene expression of the organism(s) under study. Some recent studies have suggested that up to 90% of secondary metabolite gene clusters remain silent under standard growth conditions.62–64 Numerous native and heterologous expression strategies to activate silent clusters have recently been developed, and we invite readers to explore some recent reviews for more information on the topic. 43, 62, 65, 66
The first requirement for any metabolomics study is choosing a method to measure the secondary metabolite production from the chemical extract of interest, and as such, the use of analytical instrumentation is paramount to the metabolomics field.25, 62 Typically, an untargeted approach is required to identify metabolites of interest within complex extracts from the hundreds or thousands of metabolites contained within the biological samples. MS remains the most popular tool for untargeted metabolomics analysis, particularly those using large sample sizes, as it is unparalleled in sensitivity.25, 62 NMR technologies, though less sensitive than those using MS, are gaining popularity, as they provide more structural information and are not biased towards ionizable compounds. As the volume of genomic sequencing data continues to grow, so do the size of metabolomics datasets, with experiments often consisting of hundreds of samples.45 To extract meaningful information from these massive datasets, metabolite dereplication is paramount.43, 67 Very often, such metabolite dereplication is achieved by comparing the chemical data with in-house or publicly available databases which can contain UV, NMR, and/or mass spectra for a variety of natural product-producing organisms. A plethora of public databases for natural products discovery have become available in the last decade, making it easier than ever to tap into the ever-growing knowledge base of natural products chemistry. An overview of these databases, as well as the conceptual and practical challenges of their curation and maintenance, is provided in a recent review.68
Numerous MS techniques are used to acquire metabolomics data, yielding a dataset comprised of metabolomic features corresponding to individual ions with mass-to-charge (m/z) ratios and other descriptive information (such as retention time, isotope patterns, and fragmentation spectra). One very popular method for mass spectrometry-based natural products discovery workflows is the use of data-dependent fragmentation methods in which the top 3-5 ions in a given scanning cycle are fragmented for enhanced structural analysis.62 Mass spectrometry is the analytical platform of choice for metabolomics analysis due to its unparalleled sensitivity, but with this sensitivity comes the challenge of identifying metabolites of interest from the thousands of detected features.25, 43, 45 One strategy to identify metabolites associated with a lab-grown sample involves labeling the media with isotopically labeled carbon and/or nitrogen so that isotope ratios can be exploited to distinguish real signals from background noise.69 Additional strategies to reduce background noise include removing features found in the solvent blank70 or that are inconsistent across injection replicates of the same samples.71 Once the data have been filtered, they must then be processed. Covington et al. recently covered the MS-based computational methods that can be used to process metabolomics datasets for natural products discovery,62 and Grim et al. have covered MS-1 and MS/MS-based networking strategies for visualizing metabolite relatedness.25 It is worth noting that MS-based analyses are often dependent on how well metabolites fragment, and some compound classes do not ionize as well as others (if they ionize at all). While methods can be developed to improve ionization of metabolites under study and maximize the number of molecular classes covered in a given experiment, mass spectrometry nonetheless remains a biased detection method and cannot give absolute numbers or quantification of metabolites within a sample.
While MS is unparalleled in sensitivity, NMR is unparalleled in its ability to provide unbiased structural information.43, 72 Currently applied most often to simplified mixtures or purified compounds, new NMR-based tools for analyzing large numbers of complex samples are being developed to streamline the natural products discovery workflow. For example, MADByTE (Metabolomics And Dereplication By Two-dimensional Experiments) enables comparative analysis of complex NMR spectra from large datasets. By identifying common features between samples using both heteronuclear and homonuclear experiments, MADByTE has the potential to identify high-priority samples and generate detailed structural information about the molecular scaffolds present. MADByTE is freely available to the natural products community at https://www.madbyte.org/ and represents the first open-source tool capable of analyzing complex natural products mixtures and grouping them by shared spin system features.73 Similarly, the machine learning tool “SMART 2.0” (Small Molecule Accurate Recognition Technology) has been developed for NMR-based mixture analysis and natural product discovery.72 While numerous raw data repositories exist for MS data, the same cannot be said for NMR data repositories. Typically publicly available NMR spectra are already processed, leading to the inability to utilize computational tools to reprocess or analyze the data.74 The NMR Raw Data Initiative continues to urge the scientific community to advance open-sharing mechanisms for raw NMR data, which will lead to improved reproducibility, transparency, and integrity of natural product research as a whole.74, 75
4. Analysis and prioritization of metabolomics and genomics datasets
4.1. Genomics-driven natural products discovery: from genes to molecules
The conceptual knowledge of BGCs and biosynthetic enzymes together with the upsurge in the number of available genome sequences has fostered the development of powerful bioinformatic tools to identify and classify BGCs within a genome, which endorsed genome mining as a strategy for natural products discovery.76 Bioinformatic tools can analyse BGCs and predict structures of the produced secondary metabolites, so far as possible, allowing researchers to dereplicate known clusters and prioritize specific targets for discovery.43, 45 However, the pace at which BGCs are identified is considerably faster than their verification or characterization of their encoded compounds. There are currently more than 400,000 bioinformatically predicted BGCs according to the atlas of biosynthetic gene clusters IMG-ABC, but less than 1% of those clusters has been experimentally verified.77
Since experimental verification of all identified BGCs would consume extensive wet lab effort, cost, and time to confirm the predicted “gene-to-molecule” correlations, there is a paramount need for BGC prioritization. The selection of the prioritization criteria depends of course on the researcher’s interests. For example, BGCs could be prioritized based on characteristic structural features, novelty of biosynthetic pathways, or their likelihood of encoding molecules with bioactivities (Figure 2).
Figure 2.
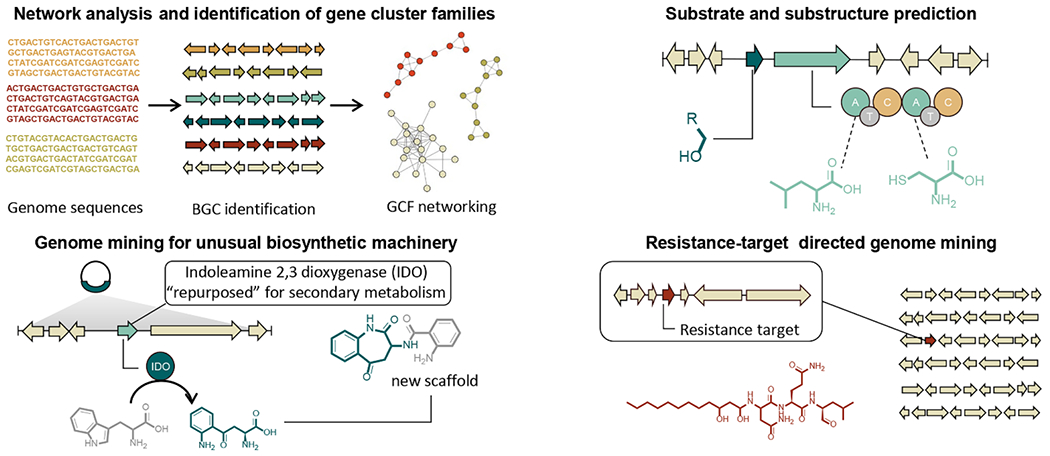
Examples of genomics-based strategies for identifying new analogs of known chemical entities, molecules with unusual scaffolds, and bioactive compounds. Once biosynthetic gene clusters have been identified, similarity networking can be used to cluster BGCs into gene cluster families (GCFs) that are most likely to encode similar molecular scaffolds. Gene clusters can also be inspected and partial structures of encoded metabolites predicted by evaluating likely substrates for key biosynthetic enzymes and identification of tailoring enzymes. BGCs that contain genes not typically involved in secondary metabolism often bind unusual substrates yielding novel chemical scaffolds. To identify bioactive molecules with a specific biological target, genomes can be mined for BGCs that contain a duplicated target that does not play a role in biosynthesis, but instead confers resistance to the producing organism.
4.1.1. Targeting relatives of known molecules
One of the powerful aspects of natural products is their huge structural diversity, even within the same compound family. This arises from biosynthetic pathways that share similar core enzymes but differ in tailoring enzymes, some incorporated monomers, or both. Indeed, in a recent study, Belknap et al.78 looked into the diversity of BGCs and their phylogenetic distribution among Streptomyces genomes, and focused on the distribution of 38 known chemotherapeutic gene clusters (CGC) encoding metabolites with antitumor activity. They noted a wide distribution of these clusters among numerous Streptomyces species and observed that the distribution of each CGC usually shares a common set of genes and pathways but differs in other cluster components, suggesting the production of different derivatives of each of the 38 known antitumor metabolites yet to be discovered.78
A powerful strategy to analyse and compare the huge number of publicly available BGCs is to sort and group similar clusters together to identify known and novel clusters. Indeed, several groups have attempted to group BGCs into gene cluster families (GCFs) based on their sequence similarity, where clusters within the same GCF are expected to produce highly related natural products (Figure 2).79–81 Thus, relatives of a natural product with a known BGC could be identified by targeting other BGCs belonging to the same GCF. In order to automate this clustering in a user-friendly manner, BiG-SCAPE and CORASON were generated.82 BiG-SCAPE can automatically generate sequence similarity networks, assemble GCFs, and display them in an interactive manner, where CORASON can construct BGC phylogenies enabling comparison and exploration of BGCs from different organisms. Importantly, BiG-SCAPE measures the similarity between complete and fragmented gene clusters using a glocal alignment mode where it starts with the longest common subcluster between a BGC pair and extends this alignment using a match/mismatch penalty system. This computational tool uses a Jaccard Index, adjacency index, and a domain sequence similarity index to calculate domain content similarity, synteny conservation, and sequence identity between protein domains encoded by the input BGCs. Sequence similarity networks are then generated by applying a user-determined cut-off value to the calculated distance matrix. Lower cut-offs group BGCs producing similar compounds more efficiently, while higher cut-offs provide a broader perspective on related GCFs and natural products.
The pairwise comparison strategy used in BiG-SCAPE limits the capacity of this sensitive tool to the analysis and clustering of tens of thousands of BGCs.83 To expand the GCF networking capabilities to more than a million BGCs within this same computational time, BiG-SLICE has been recently developed which employs BGC vectorization and a near-linear clustering algorithm to generate networks at a much faster pace.83 Medema et al. improved the efficiency of BiG-SLICE by collecting ~1.2 million publicly available microbial genomes and metagenome assemblies and clustering them into 29,955 GCFs within 10 days.83, 84 Furthermore, they made this wealth of information and collection of GCFs available for exploration through a new user-friendly database called BiG-FAM.84 BiG-FAM integrates the BiG-SLICE platform and provides an atlas of microbial secondary metabolic diversity and biosynthetic potential. Advantageously, the clustering methodology used in BiG-SLICE enables linear BGC-to-GCF matching and allows BiG-FAM users to submit a specific BGC directly from antiSMASH for analysis through an online query submission feature in BiG-FAM. The query BGC is placed in the global map of precalculated GCFs, which enables the user to easily explore the global distribution of this BGC among taxa and facilitates the identification of clusters producing similar compounds.
4.1.2. Targeting rare/novel scaffolds
While BiG-SCAPE, BiG-SLICE and BiG-FAM can instantly identify BGCs for structurally related natural products within large genomic datasets, they can also facilitate the identification of novel biosynthetic pathways, which would simply stand out as GCFs with no reference BGCs from the known biosynthetic gene cluster databases, such as the MIBiG repository for experimentally characterized biosynthetic gene clusters and the antiSMASH-DB and IMG-ABC databases for computationally predicted BGCs.85 These novel GCFs and their BGC components could then be prioritized for further detailed analysis to predict and assess the novelty of the produced scaffolds.
Algorithms based on pHMMs are unparalleled in their ability to detect gene clusters of known biosynthetic classes both quickly and with high precision.36, 37, 43 This enables researchers to obtain a birds-eye view of the BGC repertoire of a given organism from a genome sequence, and has proven particularly useful for researchers looking for gene clusters of a known biosynthetic class and for large-scale genome annotation applications.40 However, because these algorithms have been trained using known gene cluster data, and often using a limited set of organisms,33 they cannot detect unknown classes of gene clusters.45 Additionally, they may not identify BGCs that are unique to a particular lineage of organisms not included in the training set. As such, new approaches for identifying gene clusters from unknown classes are being developed, as they are likely to biosynthesize novel chemical scaffolds.40, 86
ClusterFinder, for example, uses a strategy built upon the same concept as pHMM-driven algorithms to identify new classes of BGCs, but instead of looking for specific signature genes, genomic regions rich in enzyme families common to secondary metabolism are identified using the Pfam database.79 ClusterFinder is capable of identifying new BGC classes because tailoring enzymes, regulatory elements, and transporters are involved in secondary metabolite biosynthesis, regardless of BGC class.87, 88 In addition to focusing on core and accessory genes that play roles in secondary metabolism, some researchers have employed algorithms to mine genomes for paralogues of primary metabolic enzymes. For example, the EvoMining approach searches genomes for duplicated primary metabolic genes that have undergone significant sequence divergence that may have been repurposed for secondary metabolism.89 Since this approach does not rely only on sequence similarity searches of previously identified biosynthetic enzymes, it can identify repurposed enzyme families which potentially catalyse new biosynthetic conversions. This was demonstrated by the discovery of a family of BGCs for arseno-organic metabolites for the first time, produced by model actinomycetes Streptomyces coelicolor A390 and Streptomyces lividans.89 Researchers have also demonstrated the promise of targeting BGCs containing genes that typically play a role in primary metabolism, as they may have been neofunctionalized for biosynthesis, resulting in the production of interesting molecular scaffolds. For example, a gene cluster encoding a repurposed indoleamine 2,3-dioxygenase (IDO) was targeted for metabolite discovery in Aspergillus terreus, yielding the novel metabolite terreazepine, which contains an unusual chemical scaffold resulting from the cyclization of the IDO-supplied kynurenine (Figure 2).91
Furthermore, algorithms based on function-agnostic criteria are gaining popularity for genome mining.92 For example, the MIPS-CG algorithm compares genetic sequences of two genomes and identifies BGCs by searching for small syntenic regions within otherwise non-syntenic blocks of genetic code. This motif-independent approach allowed for the detection of gene clusters that did not contain core enzymes, including the kojic acid and oxylipin gene clusters.30 Additional methods which go beyond sequence similarity approaches have also proved effective in detecting BGCs for novel and unusual natural products. For instance, the new RiPP genome mining algorithm decRiPPter uses a Support Vector Machine (SVM) classifier combined with pan-genomic analysis that predicts RiPP precursor genes located within clusters encoding multiple enzymes and distributed within the accessory genomes of a genus. Using this tool, 42 new candidate RiPP families were identified that would have otherwise been missed. Further analysis led to the identification of a new class of lantipeptides names “class V”.93
4.1.3. Targeting molecules with specific bioactivity
A particularly promising strategy for the discovery of bioactive molecules is mining microbial genomes for specific features likely to be linked to biological and pharmacological properties. Often, a toxic metabolite producer develops self-resistance against its own toxin to avoid suicide.94 Genes related to such self-resistance are often co-localized with the bioactive natural product’s BGC, which sets the foundation for the resistance gene-guided discovery approach.95
Resistance can be acquired through different mechanisms, including efflux pumps, chemical modification, compound sequestration, target modification, and target duplication.96 In the case of target duplication, the genome of a toxin producer will harbor a second copy of an essential gene in close proximity to or within the toxin’s BGC. Such duplicated genes establish resistance in the producer by either providing excess target or by including a slight variation that produces a target with greater tolerance against the produced toxin.97 Accordingly, mining for duplicated house-keeping genes is a good strategy for identifying molecules with a target mechanism of action (Figure 2). This target-directed genome mining approach was first proven effective by Moore’s group, where a group of fatty acid synthase inhibitors including thiolactomycin and their associated BGCs was discovered through identifying duplicated fatty acid synthase genes in Salinospora strains.98 Recent studies have also shown that bacteria producing proteasome inhibitors acquire self-resistance through gene duplication, where a gene putatively encoding a resistance β-proteosome subunit was observed within the BGCs encoding the proteasome inhibitors salinosporamide A, epoxomicin and eponemycin.99, 100 The BGC producing the fungal proteasome inhibitor fellutamide B also encloses a proteosome subunit-encoding gene, providing further validation for this mining strategy to discover natural proteosome inhibitors.101 Another example is the BGC encoding fumagillin, a methionine aminotransferase inhibitor, which contains both type I and type II methionine aminopeptidase genes in the gene cluster.102, 103 Similarly, the herbicide sesquiterpenoid aspeterric acid and its associated biosynthetic genes have been discovered through scanning sequenced fungal genomes for duplicates of the dihydroxyacid dehydratase (DHAD) gene, a key enzyme in the biosynthesis of branched chain amino acids in plants.104
Based on known resistance mechanisms and the promising potential of this target-directed genome mining strategy, the antibiotic resistant target seeker (ARTS) was developed in 2017.105 It is a user-friendly web tool that can automatically detect resistance genes in genomes. ARTS identifies BGCs using antiSMASH and locates resistance genes based on three criteria: gene duplication, colocalization within a BGC and evidence of horizontal gene transfer.105 ARTS initially included actinobacterial genomes only, but it has been recently updated to version 2.0, which allows the analysis of the entire kingdom of bacteria as well as metagenomic data.93 In addition, ARTS also applies the BiG-SCAPE algorithm to all the detected BGCs and provides gene cluster sequence similarity networks, which allows users to detect similar BGCs from multiple sources.
Beside the general resistance strategies mentioned above, some known resistance mechanisms related to a specific mechanisms of action can also be targeted. For example, topoisomerase-targeting pentapeptide repeat proteins (TTPRP) are known to be involved in a defense mechanism against topoisomerase inhibitors. A search for TTPRP encoding genes in myxobacterium Pyxidicoccus fallax An d4 revealed a gene in close proximity to an unknown type II PKS BGC. The metabolites produced by this cluster were identified as the new polyketide topoisomerase inhibitors pyxidicyclines A and B, which include an intriguing nitrogen-containing tetracene quinone scaffold. 106 Another notable example for a specific resistance mechanism is the conserved vanHAX operon, which is known to confer resistance to glycopeptide antibiotics by mutating cell wall precursors through conversion of the canonical D-Ala-D-Ala terminal to D-Ala-D-Lac. The Wright group used this specific resistance to design a screen for the discovery of glycopeptide antibiotics. By combining this strategy with a phylogeny-based screening filter for biosynthetic genes, they were able to discover the new antibiotic pekiskomycin, which encloses an unusual peptide scaffold.107
While this approach shows promise for identifying molecules with known mechanisms of action, molecules with a new modes of action will probably be missed by using known resistance strategies for genome mining. To overcome this disadvantage, Culp et al. used the phylogeny of biosynthetic genes to search for novel glycopeptides most likely to possess new bioactivities or modes of action. For this, they targeted divergent glycopeptide BGCs that lacked a known resistance determinant. This effort resulted in the discovery of a new functional class of glycopeptide antibiotics, including the new compound corbomycin, that work via a new mode of action in which they inhibit peptidoglycan remodelling.108
4.1.4. Source organism considerations
Although the clustering of biosynthetic genes was once thought to be a phenomenon unique to bacteria and fungi, several recent studies have shown that plant specialized metabolic gene clustering does exist.109, 110 However, BGC identification in plants obeys different strategies than those seen in bacteria and fungi. First, plant biosynthetic enzymes and pathways are unique from those in bacteria and fungi. Second, biosynthetic pathways in plants are often split across several BGCs and not fully clustered as typically seen with biosynthetic pathways in bacteria and fungi. Third, in some cases, biosynthetic enzymes produced by a set of clustered genes do not constitute a biosynthetic pathway and instead catalyze unrelated biosynthetic steps.29, 111 Finally, biosynthetic enzymes encoded by clustered genes are not always co-expressed.111 Notably, a recent definition was proposed for plant BGCs as genomic loci which enclose genes encoding at least three functionally different biosynthetic enzymes.109, 110, 112
To allow for the automated detection of plant BGCs, two similar computational tools plantiSMASH29 and phytoClust113 were independently developed. Both tools were built following the general implementation of antiSMASH but with the application of different rules to address the aforementioned differences between plant BGCs and those in bacteria and fungi. For example, plantiSMASH does not identify core biosynthetic genes, but instead identifies all genes predicted to produce biosynthetic enzymes, where candidate biosynthetic enzymes are identified based on pHMMs for known plant enzyme families, and enzyme subclasses are identified based on sequence-based clustering using CD-HIT algorithm.29 Additionally, since coexpression analysis is a powerful tool for the identification of gene sets that are scattered in the chromosome but are functionally related, plantiSMASH and phytoClust allow for the integration of transcriptomic analysis datasets through their integrated co-expression modules. Finally, plantiSMASH enables comparative genomic analysis through a plant-specific version of ClusterBlast.29
Despite the common BGC features that bacteria and fungi share, fungal specialized metabolism has some distinctive differences.35 First, fungal BGCs are often composed of fewer genes that are sometimes split over multiple loci. For this, algorithms such as MIDDAS-M114 utilize co-expression correlation to identify functionally related genes that compose a biosynthetic pathway. Second, unlike the multimodular assemblies found in bacteria, many fungal NRPS and PKS machineries act in an iterative manner which complicates structural predictions. Third, fungal genes enclose introns which could complicate gene structural annotation and sometimes challenge the precise implementation of bioinformatic technologies. Furthermore, some genes could have a bidirectional orientation, where two genes can share a cis-regulatory region,35 which makes gene prediction more challenging. All these differences must be taken into consideration by bioinformatic tools to improve fungal BGC detection.
4.1.5. Lingering challenges
Although there have been considerable advancements in developing bioinformatic tools that facilitate charting the biosynthesis of secondary metabolites in sequenced genomes, significant challenges remain. The very first challenge is linked to the input used for genome mining, where the quality of genome sequencing and annotation will influence the outcome of the analysis. A compromise must be obtained between short reads with lower cost and error rates resulting in fragmented genomes, and longer reads which offer better contiguity of assemblies but are prone to more errors.115–117
Additionally, it is particularly challenging to obtain contiguous BGC sequences from metagenome assemblies.45 Advanced assembly tools such as BiosyntheticSPAdes118 address this concern to a great extent. However, as proliferative as metagenomes might be as sources of novel secondary metabolite BGCs, all detected clusters remain putative and must be validated. Also, the presence of unculturable microorganisms as well as cryptic clusters that are resistant to activation through several manipulations, including direct cloning and heterologous expression, represents a cluster hypothesis that must be validated experimentally. If these predictions cannot be translated into specialized chemicals, they would defy the ultimate goal of the genome-driven discovery approach and encumber the natural product discovery pipeline.119
Other lingering challenges include predicting structures for rare or unusual monomers using genome mining tools, specifically from less-studies organisms, where accurate enzyme function prediction is difficult and training data are limited. Also, the RiPP family of natural products remains challenging since their high structural diversity arises from diverse enzyme families that are not conserved across all RiPP pathways, and therefore they lack a universal genetic marker for their detection.120
4.2. Metabolomics-driven natural products discovery: from molecules to genes
Genomic tools have greatly enhanced the ability of the natural products community to identify biosynthetic gene clusters of interest for secondary metabolite discovery. However, structure elucidation remains a significant challenge in natural products discovery, and several partial solutions using a variety of analytical and statistical approaches are beginning to emerge. These approaches can be used alone or in combination to prioritize discovery efforts towards derivatives of known molecules, novel chemical scaffolds, and biologically active compounds, enhancing our understanding of natural product chemical space. A schematic overview of some basic metabolomics-based strategies for prioritizing compounds for natural products discovery can be found in Figure 3.
Figure 3.
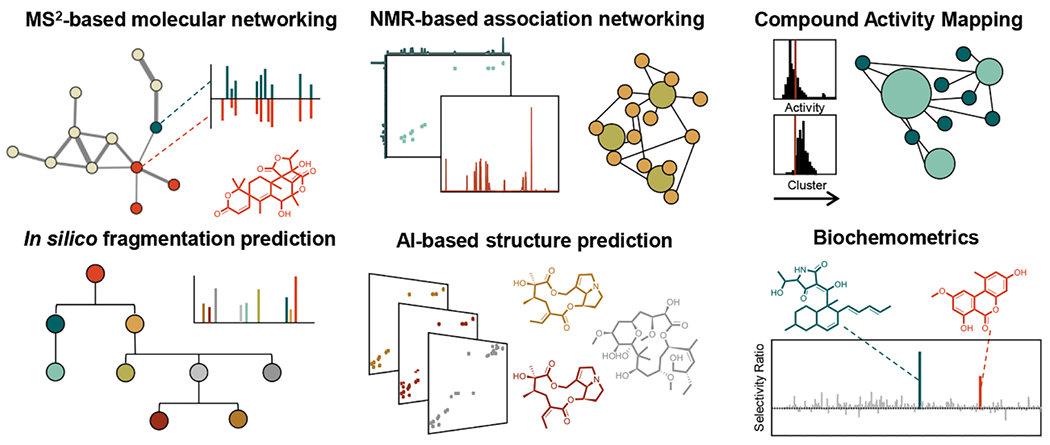
Mass spectrometry, nuclear magnetic resonance, and bioactivity guided metabolomics strategies for targeting analogs of known molecules, new chemical scaffolds, and bioactive molecules from complex natural products extracts.
4.2.1. Targeting relatives of known molecules
To facilitate compound dereplication and identification of novel analogues of important secondary metabolites, it may be fair to say that no tool is more popular than molecular networking. Molecular networking, based upon the premise that metabolites with similar molecular structures will yield similar fragmentation (MS/MS) spectra, groups structurally related classes of molecules based on similarities between their MS/MS spectra.25, 45, 62, 121 Using this information, researchers can visualize relative mass differences between product ions that share a common backbone structure but possess different modifications including sugars, methyl patterns, and amino acids.25 In 2012, the Global Natural Product Social (GNPS) molecular networking platform was made publicly available, providing scientists around the world with a streamlined molecular networking pipeline and an ever-growing database of reference spectra.122 Currently, GNPS contains spectra for only about 3% of known natural products45 but the database is continually growing, strengthening the ability of researchers to ground their unknown datasets with reference spectra that provide chemical information.
Recently, the GNPS platform has expanded to include “feature-based” molecular networking, which not only incorporates isotope patterns, retention times, and fragmentation patterns, but can also include ion mobility separation data, facilitating improved spectral annotation and the differentiation of isomers which could previously not be resolved.123 METLIN, a structurally diverse database containing over 850,000 molecules, can be applied not only for the identification of unknowns using fragment ion similarity searching, but has also recently expanded to include neutral loss searching, which can reveal unmistakable similarity between molecules that appear unrelated using MS/MS data alone.124
The majority of spectral networking is achieved by using a cosine similarity score which groups spectra based on peak m/z matches. While the cosine score has been widely adopted and successfully used by researchers in the natural products field, cosine-based methods often fail to group similar molecules that have multiple local chemical modifications.125 To address this problem, a new similarity scoring metric, Spec2Vec, was recently proposed. Spec2Vec, inspired the natural language processing algorithm Word2Vec,126 evaluates co-occurrences across large spectral datasets to learn fragmental relationships between peaks.125 When compared to cosine similarity scores and modified cosine similarity scores, Spec2Vec showed overall better correlations to Tanimoto-based structural similarity, achieves better library matching, and requires less computational power.125 GNPS has begun integration of Spec2Vec into their pipeline, and users can currently calculate Spec2Vec scores for positive mode data using pre-existing training sets.125
Despite the obvious strengths of molecular networking, network annotations are still largely performed manually given the limited size of currently available spectral libraries. To overcome this bottleneck, computational methods for producing theoretical fragmentation patterns have been developed.62, 127, 128 Such in-silico MS/MS databases can be coupled to molecular networking to further facilitate the identification of known natural product derivatives.129 The MetWork server, for example, works to expand the annotation of molecular networks by generating putative structures of molecules contained within a network that are grounded with a reference spectrum.127 Another web server for in silico fragmentation prediction is the machine learning-based probabilistic model CFM-ID, which can annotate spectral peaks for a known chemical structure, predict spectra for a given chemical structure, and predict chemical structures for a given metabolite spectrum.130 Additionally, SIRIUS 4 has been developed, which integrates high-resolution isotope patterns with in silico fragmentation trees generated with CSI:FingerID131 to generate predictions and ranking for putative molecular structures.132 Recently, Qemistree, a new tree-guided exploration approach integrating both SIRIUS and CSI:FingerID, was developed for computing and representing chemical features detected in MS/MS untargeted metabolomics studies. Qemistree is available to the metabolomics community through GNPS, enabling users to generate both molecular networks and chemical trees if so desired.133 Because most (though not all) of these tools rely on databases of experimental spectra, continued expansion and curation of public databases by the natural products community will be paramount to improve structural predictions using these tools.
Recently, a number of algorithms have been developed to identify specific classes of compounds from untargeted MS data.134–136 For example, the MeHaloCoA algorithm was recently developed to identify halogenated compounds, which often contain bioactive properties.137 MeHaloCoA was successfully used to discovery to halogenated compounds, griseophenone I and chlorogriseofulvin, which have antiproliferative properties.136 The most well-established tools for systematically linking structures to mass fragmentation patterns are typically dedicated to identifying peptidic natural products. For example, NRPquest was developed to identify and sequence peptidic natural products from MS/MS spectra, even if they incorporate non-proteinogenic amino acids or have been structurally modified during biosynthesis.135 The DEREPLICATOR algorithm compares mass fragmentation patterns to theoretical spectra using the in silico fragmentation rules defined by NRPquest for identifying peptide scaffolds. This algorithm was recently expanded to DEREPLICATOR+, which extends annotation to polyketides, flavonoids, and terpenes.134
The aforementioned mass spectrometry-based tools for visualizing compound relatedness and annotating structural features have prompted dramatic changes in natural products discovery efforts. However, while these tools can provide some structural information, unambiguous characterization still requires confirmation by NMR.138, 139 Typically applied to purified compounds, structural characterization by NMR is often stymied by the time-consuming nature of both compound purification and structure elucidation itself. Recently, SMART, a deep convolutional neural network (CNN) was trained using HSQC spectra found in the Journal of Natural Products. The training set was then used to analyze new spectra and accelerate the structural characterization of lipopeptide molecules.74, 140 More recently, SMART 2.0 was developed by using over 25,000 HSQC spectra from natural products in the JEOL database and ~28,000 spectra computed using the ACD Laboratories predictor of mostly marine natural products as a training set to guide mixture analysis from a marine cyanobacterial extract. Using a combination of SMART 2.0, molecular networking, and cytotoxicity screening, researchers were able to rapidly annotate a new chimeric macrolide, symplocolide A, annotate known mixture constituents, and identify several new derivatives thereof. SMART 2.0 very accurately and efficiently predicts structural types from NMR spectra, enabling researchers to have a clear starting point for full compound characterization. SMART 2.0 takes less than 30 minutes from data acquisition to structure prediction, and is an extremely promising tool for accelerating natural products discovery efforts.72
4.2.2. Targeting rare/novel scaffolds
While identifying derivatives of known natural products is of great interest both for understanding structure-activity relationships of bioactive molecules and gaining insight into evolutionary strategies of compound diversification, the ability to identify novel molecular scaffolds promises an even greater impact. Although the natural products community has made major strides in developing metabolomics tools for annotating known molecular classes from complex mixtures, the development of tools for discovering entirely new scaffolds is only in its infancy. Further development of such tools could represent a paradigm shift in natural products discovery efforts, enabling researchers to prioritize unusual molecular scaffolds from Nature at an accelerated pace.
Many researchers have begun incorporating multivariate statistical analyses into their natural products discovery workflow, such as partial least squares (PLS) or principal component analyses (PCA), which can help to simplify complex metabolomics datasets and prioritize samples for future study.62 PCA/PLS scores plots enable similarity/dissimilarity visualization of samples based on feature variances to determine not only which samples are most metabolically unique, but also which metabolic features are responsible for this uniqueness. While these analyses do not provide additional structural information about metabolites contained within the evaluated samples, they do have the ability to prioritize metabolically diverse strains for future study, increasing the likelihood of novel natural product discovery.141, 142
Although in silico fragmentation tools have primarily been utilized for dereplication of datasets and identification of structural analogues of known scaffolds, they do have the added benefit of assessing the chemical novelty of identified features. These tools have great promise for aiding researchers in the prioritization of novel scaffolds, with tools like SIRIUS 4 achieving identification of more than 70% of features in complex metabolomics datasets.132 Another key emerging strategy for molecular annotation and dereplication (and with it identification of putative novel scaffolds) is the MS2LDA workflow.143 MS2LDA extracts biochemically relevant molecular substructures, called “Mass2Motifs” using an adapted text mining algorithm in which common patterns of mass fragments and neutral losses are extracted from fragmentation spectra. Similarly, CANOPUS (class assignment and ontology prediction using mass spectrometry) uses a deep neutral network to predict nearly 2500 compound classes from fragmentation spectra, even in the absence of training data.144 MS2LDA has been expanded recently to take advantage of combinatorial in silico tools to match experimentally detected features to candidate molecule substructures as well as automated machine learning molecule classifications.145 Additionally, Mass2Motifs resulting from MS2LDA analyses can also be stored, browsed, and accessed through the open database MotifDB.145 Recently, MolNetEnhancer was developed with the goal of integrating molecular networking, MS2LDA substructure discovery, and in silico annotation tools.146 In addition to helping with compound dereplication and mixture annotation, these tools can highlight compound families that are not reliably connected to known compound classes, potentially highlighting novel chemistry.
As mentioned in section 3.2, NMR metabolomics methods such as SMART 2.0 and MADByTE have the potential to transform the field in terms of identifying novel scaffolds, though they have not yet met this potential. SMART 2.0, for example, upon continued development, may be capable of detecting rare structural features from crude extracts.72 MADByTE, constructs spin systems from TOCSY/COSY data and correlates them with HSQC cross peaks, ultimately producing a network of nodes comprised of related spin systems.73 Users can choose to visualize the resulting datasets as full association networks, which show all nodes generated from all samples for direct comparison, similarity networks showing nodes that shared some degree of overlap between samples, or hybrid networks, which combine the features of shared nodes across all systems.73 Although these NMR tools have not yet found widespread use, they have the potential to revolutionize natural products discovery, especially when combined with orthogonal mass spectrometric and genome mining approaches.
4.2.3. Targeting molecules with specific bioactivity
Bioactivity-guided fractionation, once the gold-standard approach for natural products discovery, has improved dramatically with advancements in chromatographic separation techniques and dereplication protocols. However, this strategy still tends to be biased towards compounds that are most easily isolated from a mixture rather than those that are most likely to be active.18, 147 As such, many researchers have begun combining chemical and biological activity data of samples using multivariate statistics to identify putative bioactive constituents early in the purification process.18, 147–155 PLS analysis is the most popular approach for integrating such datasets, and bioactive constituents can be identified from resulting data matrices using scores plots, S-plots, and selectivity ratio plots.18, 156, 157 In 2016, Kellogg et al. compared the efficacy of these analyses to identify antimicrobial constituents from both Alternaria and Pyrenochaeta fungal species. While the S-plot and the selectivity ratio plot both confidently identified the abundant macrosphelide A as bioactive from Pyrenochaeta sp., the selectivity ratio plot was superior in identifying altersetin from Alternaria sp. as the most bioactive constituent in the mixture, despite its low abundance.18 Examples of both S-plots and selectivity ratio plots are provided in Figure 4. A number of recent manuscripts have highlighted the utility of the selectivity ratio for the identification of bioactive secondary metabolites early in the fractionation process from both plants and fungi.18, 147, 148, 151, 152 Importantly, meaningful information can only be extracted from properly processed datasets, and data transformation, model simplification, and data filtering protocols can greatly impacting multivariate analyses.152
Figure 4.

Examples of an (A) S-plot and a (B) selectivity ratio plot for predicting bioactive compounds from complex mixtures. In each plot, points or bars represent individually detected features from natural product extracts. In the S-plot, points with the highest correlation and covariance to a given bioactivity are pulled to the upper righthand quadrant of the plot and can be prioritized for isolation. With the selectivity ratio plot, bioactive features are identified by their explained/residual variance.
Of course, while these multivariate approaches provide insight into which features of a complex extract are likely to possess biological activity, they do not provide structural information about the bioactive constituents themselves. To overcome this challenge, a recent study aimed to integrate both selectivity ratio predictions and molecular networking with GNPS to identify molecular classes of antimicrobial constituents from the botanical Angelica keiskei. With this strategy, a group of chalcone analogues were targeted for isolation, yielding three active constituents, including a low-abundance compound not previously known to possess antimicrobial activity.151 GNPS now offers the ability for researchers to incorporate bioactivity data directly into molecular networks in a process called “bioactive molecular networking,” where the predicted bioactivity score informs the size of individual nodes in a molecular network.153 More recently, the SeaPEPR pipeline was developed, combining phenotypic activity screening assays with metabolic fingerprinting for the identification of prioritized samples for further processing. Using 76 extracts from marine sponges, authors were able to dereplicate active compounds from crude extracts and identify a new dibrominated aplysinopsin and an hypothetical chromazonarol stereoisomer.158 Some studies have utilized metabolomics to not only identify bioactive constituents from complex mixtures, but to predict their modes of action as well. For example, Compound Activity Mapping, a combined approach integrating phenotypic screens with untargeted metabolomics, has been utilized to predict modes of action for putative active molecules early in the fractionation process. This approach was successfully used to discover the quinocinnolinomycins, a new family of natural products predicted to possess cytotoxic activity via endoplasmic reticulum stress, from a dataset containing 234 natural products extracts.154
Although the majority of approaches used to target bioactive molecules rely on mass spectral data, NMR-based strategies have recently shown preliminary success for the prioritization of putative bioactive molecules from complex mixtures. Using MADByTE, Egan et al.73 overlaid antimicrobial bioactivity data onto a pre-fractionated extract library. Using this dataset, networks were generated only containing spin systems from fractions possessing antimicrobial activity. These simplified networks contained several bioactivity “hotspots” that were prioritized for further characterization. Using NMR-guided isolation, researchers targeted a predicted bioactive component, yielding collismycin A. Bioactivity screening confirmed collismycin A’s antibacterial activity, providing evidence of the promise of MADByTE for prioritizing bioactive constituents from complex fraction libraries.73
The majority of tools currently utilized for grouping small molecules rely on the small-molecule similarity principle, which through some metric or another groups molecules by structural information alone. However, such an approach does not necessarily translate to biological activity. Indeed, molecules with similar cell-sensitivity profiles or side effects often share the same mechanism of action, despite differences in chemical structure. Recently, the Chemical Checker was released (available at https://chemicalchecker.org/), which provides processed bioactivity data on ~800,000 small molecules using data from five levels of complexity ranging from simple chemical properties to clinical outcomes.159 These multivariate data are combined into a vector format, expanding compound similarity matching to include both chemical and biological data. The Chemical Checker database not only has the potential to aid in drug discovery tasks such as target identification and library characterization, but could be capable of predicting missing bioactivity data and characterizing any molecule of interest. While this tool has not been explicitly used for natural products discovery, it has the potential to greatly facilitate existing discovery platforms for the discovery of bioactive natural products.
4.2.4. Source organism considerations
When selecting biological material for natural products discovery workflows, it is important to consider the inherent differences in chemistry between biological organisms. For example, in a recent analysis, 24,595 natural products derived from both bacterial and fungal sources were assigned chemical ontology terms and subjected to PCA.160 This analysis revealed that bacterial and fungal taxonomic groups occupy distinct regions of the resulting scores plots. This analysis revealed that fungi have twice the frequency of both lipids and heterocyclic compounds such as aromatic polyketides, while bacteria have higher prevalence of peptides. Within the fungal kingdom, different taxonomic groups also possessed distinct chemical repertoires.160 While this may not be representative of the full biosynthetic capacity of a given organism, this information can be used to prioritize certain taxonomic groups for further analysis, depending on the goals of the project. Of course, there are many taxonomic groups for which very little is known chemically, and the ability to assign spectral peaks and dereplicate datasets from understudied organisms is quite limited. For example, while numerous open access databases exist for microbial organisms such as bacteria, archaea, and fungi, there are no dedicated databases for microalgae.161
Part of the bias of the natural products field towards microbes, particularly Actinomycetes, may be due to the ease at which these organisms can be grown and scaled up in laboratory conditions. The reality is that only a small fraction of microbes are amenable to growth in laboratory settings,162 while certain organisms are typically not grown in a laboratory setting at all. When source material is collected from the field, not only do scientists constantly have to face the issue of material limitation, but source material chemistry is susceptible to changes based on location, climate conditions, and stage of life.163 Further complicating plant metabolomics projects aiming to link metabolites to gene clusters is the issue of microbial endophytes which may be the source of detected metabolites rather than the plant itself.164, 165
4.2.5. Lingering challenges
Although major strides have been made in the last decade improving our ability to interpret large metabolomics datasets for natural products discoveries, numerous challenges remain. One of the major challenges in using metabolomics to access biosynthetic dark matter is the issue of gene expression itself. It has become clear that the majority of BGCs remain silent under laboratory conditions, and undoubtedly, these cryptic clusters represent an important source of new molecules. Although many strategies have been developed to activate cryptic clusters with varying success,43, 62, 65, 66, 162 we still lack the comprehensive understanding of the complex and interconnected factors that regulate gene expression as a whole, hampering our efforts to truly access this untapped potential.
The rapid expansion of publicly accessible metabolomics databases has certainly improved our ability to dereplicate metabolomics datasets, predict compound structures, and prioritize secondary metabolites for discovery. However, biases in our existing datasets will introduce bias into our future discoveries. For example, we are now very well equipped to annotate peptidic natural products originating from NRPS-containing gene clusters due in large part to the ease at which these molecules are fragmented, and the characteristic fragmentation patterns they produce.45, 134, 135 While novel algorithms and prediction tools are being developed for other biosynthetic classes, we still lag behind in our ability to target novel scaffolds. Likely, the increased utilization and development of NMR-based metabolomics tools will improve our ability to identify and predict unusual molecular scaffolds. Additionally, continued expansion of publicly available raw data repositories, including both MS and NMR spectra, will improve in silico prediction tools for future discovery efforts.
5. Finding the needle in the haystack: integrating metabolomics and genomics for enhanced discovery
Advances in metabolomic and genomic technologies have reinvigorated secondary metabolism research, allowing researchers to explore the biosynthetic potential of plants, bacteria, and fungi at an unprecedented scale. Genome mining and molecular networking tools to predict structures of biosynthetic products have improved dramatically over the years, but these tools are insufficient particularly for understudied organisms and biosynthetic types. An increasing number of studies have begun to integrate genome sequencing data with untargeted metabolomics data, enabling improved structural prediction and compound prioritization, unearthing patterns of secondary metabolite evolution, and providing insight into genomic and chemical markers of pathogenicity.166, 167 Given the enormous impact of data processing and annotation on both metabolomics and genomics datasets, it may come as no surprise that the integration of such multi-Omics datasets comes with a fair share of challenges. Even with high quality genome sequences and metabolite profiles, establishing biologically relevant relationships between such disparate data types is no straightforward task.59 A number of approaches have been developed to correlate genetic and chemical data in order to extract meaningful information from integrated datasets, including pattern-based, weighted pattern-based (correlation based), and feature-based methods.44, 45, 168 An outline of strategies for integrating metabolomics and genomics datasets is provided in Figure 5, and selected examples of compounds that have been discovered using these approaches can be found in Figure 6.
Figure 5.

Examples of pattern-based, weighted pattern-based, and feature-based methods for integrating metabolomics and genomics datasets. (A) Pattern-based strategies utilize presence-absence matrices of gene cluster content and metabolite detection across strains in order to identify strongly overlapping gene cluster-metabolite pairs for targeted study. (B) Weighted pattern-based strategies, in addition to looking at presence-absence patterns, develop specific metrics to score metabolite gene cluster pairs. For example, fungal artificial chromosomes (FACs) can be used to heterologously express metabolites from yet uncharacterized gene clusters. To identify heterologously expressed metabolites from the thousands of host-encoded metabolites, a FAC-score was developed to quickly rank metabolites most likely to be encoded by the FAC-encoded gene cluster. (C) Feature based methods use BGC sequence data to infer structural features of encoded metabolites, enabling generation of predicted spectral profiles, and comparison to experimental data for targeted compound discovery.
Figure 6.
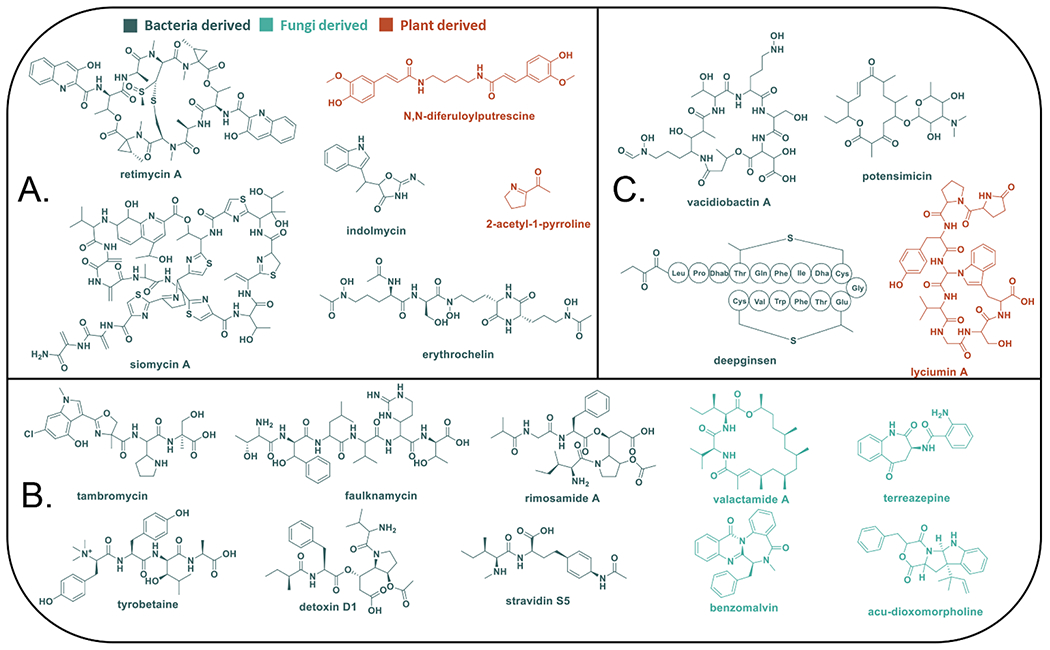
Bacterial, fungal, and plant-derived compounds discovered using (A) pattern-based, (B) weighted pattern-based, and (C) feature based strategies for integrating metabolomics and genomics datasets.
5.1. Pattern-based strategies
Because many organisms (particularly fungi and bacteria) have biosynthetic machinery clustered in their genetic code, it is possible to correlate presence/absence patterns of BGCs with the expressed secondary metabolites, enabling the linkage of BGCs to their encoded metabolites (Figure 5A).35, 45, 168 The first study to use such “pattern-based” genome mining was conducted by Duncan et al. in 2015 using 35 Salinispora strains. By combining both BGC data with molecular networking data, the group was able to overlay presence/absence matrices of molecular families and BGCs, accelerating prioritization of uncharted biosynthetic space. Using this technology, the BGC for the depsipeptide retimycin A was discovered by using the overlaid matrices to exclude all other candidate BGCs.168
A similar study was conducted on thirteen strains related to Pseudoalteromonas luteoviolacea, where untargeted metabolomics data were combined with whole-genome sequences to identify both chemical and genetic diversity between strains and to correlate BGC data with metabolites.169 Authors found that there was considerable diversity, both chemical and genetic, across these closely related strains, with 2% of chemical features and 7% of BGCs represented in all strains, and 30% of chemical features and 24% of genes unique to single strains. Using support vector machines and a genetic algorithm, the list of chemical features was filtered to prioritize 50 features that were most informative for discriminating strains, and these features were dereplicated using molecular networking.169 Using this strategy, the antibiotic indolmycin was identified as a key discriminating feature for three strains. Using pattern-based genome mining, researchers were able to identify presence/absence patterns matching the distribution of indolmycin and identify the BGC encoding its biosynthesis. Researchers also illustrated the usefulness of this approach for identifying strains as “hot spots” of biosynthetic diversity to prioritize for future studies.169 More recently, a pattern-based approached was used to assess the biosynthetic richness of the Planomonospora genus. Using 72 strains of Planomonospora sp., researchers were able to link a new salinichelin-like metabolite to the erythrochelin BGC, link a urylene-containing molecular family to its NRPS gene cluster, and manually link siomycin metabolites to a RiPP BGC. Of the 59 GCFs identified in this study, only 3 were linked to known metabolites, highlighting this genus as a rich source for future discovery.170
In plants, linking metabolites to their biosynthetic pathways is particularly challenging because the majority of biosynthetic genes are not found in clusters.59, 171 Nonetheless, secondary metabolite profiles can still be used to identify regions of gene sequences associated with particular phenotype,172–174 and genome wide association studies (GWAS) are becoming popular for identifying quantitative trait loci that can be used for breeding and genetic improvement.174–178 Combining GWAS and transcript data has proven particularly fruitful for the identification of both individual genes and gene networks that affect secondary metabolism. In a large scale study, metabolite features from 702 maize genotypes planted at different locations were evaluated, and nearly 1500 locus-trait associations were identified through metabolite guided genome-wide association mapping.174 Five representative genes from this large dataset were chosen to validate findings and determine the relevance of functional variations in candidate sequences. Using these data, the PHT locus was identified as a player in N,N-diferuloylputrescine biosynthesis, and the CCoAOMT1 locus affected production of both N-(caffeoyl-O-hexoside)-spermidine and its derivatives. With this information and metabolic profile data from transgenic rice and maize knockouts, researchers were able to reconstruct the biosynthetic pathway of phenolamides and construct a proposed biosynthetic pathway for flavonoids in maize kernel.174 Similar QTL mapping studies were used to discover the role of the TomLoxC promoter in apocarotenoid production in tomatoes, which contributes to desirable tomato flavor,176 and to provide insight into the production and accumulation of 2-acetyl-1-pyrroline in rice, a compound responsible for the aromatic fragrance in elite rice varieties.175
Pattern-based genome mining has found great success in linking secondary metabolites to their biosynthetic machinery; however, the presence of a gene cluster does not necessarily mean that the metabolite(s) it encodes will be detected or expressed, and as such, these methods may be limited. For example, a study evaluating the expression/detection of secondary metabolites from known biosynthetic gene clusters in Photorabdus and Xenorabdus spp. found that while it is relatively common for a BGC to be found without its corresponding secondary metabolites (~34% of cases), it is extremely rare for a metabolite product to be found without its corresponding BGC (<1% of cases).179 Pattern-based methods for linking metabolites to gene clusters, as such, are biased towards cases in which metabolite expression is consistent across organisms under study, and may miss linkages in which metabolite expression is inconsistent or low. Nonetheless, this strategy has laid an important foundation for the development of more nuanced methods that take these considerations into account.
5.2. Weighted Pattern-based strategies
Several strategies have been developed that consider not only presence/absence patterns between BGCs and molecules, but that also help to rank the quality of a given BGC-metabolite match by adding weighted scores to the correlations themselves. A widely used strategy in bacteria, for example, is the metabologenomics platform.80, 121, 180–182 This platform enables researchers to prioritize the most promising BGC-metabolite links from the large-scale analysis of genome sequences and chemical data using a metabologenomic score, which boosts and penalizes presence/absence patterns differently. For example, the presence of both a BGC and a metabolite feature is boosted ten times more than the absence of both a BGC and metabolite feature, while the presence of a metabolite without the BGC is penalized ten times more than the presence of a BGC without its corresponding metabolite.80 The first study using this scoring system was conducted at an unprecedented scale, correlating genome sequences from 178 actinomycetes with their corresponding mass spectrometry data. After collapsing similar BGCs and molecules into GCFs and molecular families, authors were able to experimentally validate this method by linking 27 known natural products to their known gene clusters, despite the fact that 77% of known gene clusters remained cryptic in the majority of strains under study.80 Subsequent studies have shown the promise of this platform, leading to the discovery of novel natural products including the antiproliferative tambromycin,180 the unusual hexapeptide faulknamycin,183 the rimosamides,182 and the tyrobetaines.121 In addition to identifying new molecules and their biosynthetic origins, the metabologenomics platform has found success in linking the long-orphaned detoxins182 and the antibacterial stravidins181 to their BGCs (Figure 6).
While the metabologenomics platform has shown great success for elucidating biosynthetic pathways for both known and unknown metabolites, its applications thus far remain limited to bacteria. In fungi, a unique and scalable pipeline has been developed which utilizes heterologous expression to link metabolites to their BGCs.91, 184–187 Heterologous expression offers numerous benefits that complement studies in native producers, including activation of silent gene clusters, increased production of valuable metabolites, and facile genetic manipulation of pathways under study. However, the size of fungal BGCs makes their heterologous expression challenging. To address this challenge, fungal artificial chromosomes (FACs) are produced using the AMA1 shuttle vector to capture up to 300 kb of genomic DNA, large enough to contain full-length clusters including backbone genes, tailoring enzymes, and regulatory elements. The first study using FACs resulted in the successful production of the known metabolite terezine D from its BGC, illustrating the utility of FACs as expression vectors in fungi.187 However, without a scoring metric to distinguish heterologously expressed metabolites from the thousands of host-encoded metabolites, large-scale correlative studies were not possible. To achieve this goal, the FAC-score was developed, in which metabolites found in replicates of the same FAC, but not in other FACs or the negative control, are given positive scores, while metabolites detected in multiple FACs are given negative scores (Figure 5B). Using a dataset of 56 FACs containing uncharacterized BGCs from Aspergillus terreus, A. aculeatus, and A. wentii, 17 natural products were correlated to their biosynthetic gene clusters. From this dataset, novel secondary metabolites and their biosynthetic pathways have been elucidated, including valactamide A186 and terreazepine,91 and the biosynthetic pathways of known metabolites including benzomalvins185 and acu-dioxomorpholines184 were identified (Figure 6). Though this platform has seen great success, discoveries using this approach have remained limited to the Aspergillus genus, and future studies will be required to assess the ability of this platform to expand to organisms with greater genetic distance from the A. nidulans host.
5.3. Feature-based strategies
Feature-based approaches to data integration, though perhaps the most promising of all approaches, have only been modestly pursued due to a lack of established in silico models for many biosynthetic types. There have, however, been significant advances in developing such integration strategies for natural products with well-defined building blocks, such as peptidic metabolites and glycosylated products.28, 135, 188–190 Using feature-based strategies, specific monomers can be predicted from biosynthetic gene sequences, as can specific tailoring reactions (such as glycosylation or methylation). In tandem, molecular scaffolds can be predicted from chemical datasets. Predictions from each of these datasets can then be combined to rank spectral features for prioritized study (Figure 5C). Thus far, the chemical datasets used for feature-based predictions have been mass spectral in origin, though one can imagine that the enhanced structural information provided through NMR analysis would greatly improve this part of the pipeline.
The earliest automated example of feature-based integration is NRPquest, designed to detect nonribosomal peptides and their gene clusters.135 NRPquest begins by annotating genomes using NRPSpredictor2 to identify all possible amino acid substrates for each detected adenylation domain.191 Using this information, NRPquest then constructs a database of the biosynthetic potential for nonribosomal peptides in the organism(s) under study. Additionally, NRPquest searches genome sequences for methylation and P450 domains, and accounts for corresponding methylations and cyclizations that could result from these biosynthetic players. Mass spectral data are then matched against theoretical spectra for each putative peptide in the organism’s database. Using an algorithm designed for linear, cyclic, and branched cyclic peptides,192 NRPquest then calculates the statistical significance of each peptide-spectral match and produces a molecular network containing all significant identifications.135 This approach was validated in a blind experiment using extracts from Streptomyces roseosporus, S. pristinaespiralis, Bacillus subtilis subsp. subtilis, and B. brevis, finding that many of the peptides identified by NRPquest corresponded to previous sequenced molecules. NRPquest also identified several additional unknown metabolites that likely represented novel variants of known nonribosomal peptides.135
The Genomes to Natural Products (GNP) platform is another genome-guided natural products discovery platform that can automatically predict both nonribosomal peptides and polyketides from their biosynthetic pathways. Using Hidden Markov models and BLAST databases, GNP identifies gene clusters and their associated domains, predicts substrate specificities, and uses this information to predict chemical structures. These automated predictions are then combinatorialized to construct libraries of hypothetical fragmentation patterns. These in silico predictions are then compared to experimental data to link compounds to their putative clusters.193 Using GNP, a novel NRPS-PKS cluster was identified in Acidovorax citrulli and a predicted scaffolds used to target metabolites for isolation in experimental LC-MS data. Targeted isolation efforts yielded vacidobactins A and B, novel NRPS/PKS natural products derived from this gene cluster. Additionally, a BGC predicted to encode machinery for polyketide and deoxysugar biosynthesis was identified and predicted scaffolds were used to target and isolate the encoded metabolite potensimicin.193 The GNP platform represents an integrated platform for feature-based identification of nonribosomal peptides, type I polyketides, and deoxysugar-containing natural products, and is available to the public at http://magarveylab.ca/gnp.
More recently, the DeepRiPP platform, designed to identify ribosomally synthesized and post translationally modified peptides (RiPPs) from genomic and metabolomic datasets.28 DeepRiPP recognizes several limitations of a central assumption in many other computational approaches: that biosynthetic pathways are encoded by chromosomally adjacent genes.40 As discussed previously, the presence of biosynthetic gene clusters is not always a given. Not only have there been examples of sequences for precursor peptides separated from the rest of the encoded RiPP biosynthetic machinery (such as the prochlorosin lantipeptides),194 even if the genes involved in biosynthesis are clustered, fragmented and low-quality genome assemblies may fail to resolve gene clusters,116 making it appear as if precursor peptide sequences are distant from the rest of their biosynthetic machinery. Additionally, if sequences diverge from expected sequences as a result of biosynthetic novelty, overly-targeted algorithms may fail. To overcome the limitations of this assumption, DeepRiPP first uses a deep learning approach, NLPPrecursor, to classify precursor peptides from across the entire genome (regardless of their genomic content) and to predict their cleavage patterns. The putative cleaved peptides are then integrated into the RiPP-PRISM system,195 a combinatorial tool to predict final molecular structures. A particularly clever inclusion in the DeepRiPP pipeline is the BARLEY algorithm, which uses a cheminformatic local alignment framework to prioritize loci most likely to encode novel compounds, and to identify microbial taxa enriched for structural novelty. Finally, the CLAMS algorithm integrates metabolomics data to identify target products in mass spectrometry datasets for subsequent studies. Using an impressive 10,498 extract dataset originating from 463 bacterial strains, DeepRiPP was applied to expand the landscape of novel RiPPs by a factor of six, leading to the identification of novel compounds “deepstreptin,” “deepflavo,” and “deepginsen,” whose structures were exactly as predicted by DeepRiPP, available publicly at http://deepripp.magarveylab.ca.28 Feature-based integration strategies have also guided the discovery of the lyciumins, branched cyclic RiPPs encoded in the botanical Lycium barbarum, and their biosynthetic machinery.196
Feature-based strategies, in addition to being used as a sole integrated approach, have great promise for facilitating pattern-based discovery methods. Indeed, pattern-based methods are often slowed due to the extensive manual verification required to determine which of the ranked BGC-metabolite pairs are the most promising for follow up studies. Recently, the NPLinker framework was developed that demonstrates the strength of using multiple link-scoring functions in tandem to prioritize true links. Using a standardized metabologenomics score to link BGCs to putative metabolites and combining this with the feature-based Input-Output Kernel Regression score, 197 NPLinker has the potential to significantly accelerate integrated discovery pipelines.198 Recently, the potential of this platform was illustrated in a comparative metabologenomics analysis of 25 polar bacterial strains. Using NPLinker to link BGC and molecular features using a standardized strain correlation score and a novel feature-based Rosetta score, researchers quickly linked the known metabolites ectoine and chloramphenicol to their associated BGCs.199 Despite the preliminary success of feature-based integration approaches, they are still quite limited in scope. Of course, users of feature-based tools must be aware that predictions are only as good as the algorithms used to generate them, and the genome sequences and chemical information provided. As such, mining genomes for compounds of unusual biosynthetic origin and/or with rare molecular scaffolds remains a central challenge. Continued improvements to algorithms for identifying rare and novel scaffolds and noncanonical biosynthetic machinery, particularly in understudied organisms, will be paramount for feature-based integration approaches to reach their full potential.
5.4. Validation of metabolite-gene correlations
Once a potential metabolite-gene cluster pair has been identified, their correlations must be validated. This is often done through gene deletions, in which entire biosynthetic gene clusters, or individual genes in the cluster, are knocked out of the genetic sequence via homologous recombination to confirm their linkage to the metabolite(s) of interest.43 In fungi, however, this process is somewhat challenging due to the frequent occurrence of non-homologous end-joining.200, 201 In model organisms such as Aspergillus nidulans,202 this has largely been overcome through the discovery of the nkuA gene, a major player in this pathway, the deletion of which improved gene targeting efforts to a 90% success rate.203 Additionally, the 2020 Nobel Prize-winning CRISPR-Cas9 genome editing technology stands out as a particularly promising tool for gene editing in a wide variety of organisms.204 Originally adapted from a genome editing system used by bacteria to protect against invading viruses, CRISPR-Cas9 enables the production of an RNA guide sequence designed to attach to a target DNA sequence and cut the DNA at the targeted location, following which genetic material can be added, deleted, or altered for follow up studies.205
The heterologous transfer of putative biosynthetic genes into a non-producing host organism is another valuable strategy to confirm gene cluster products. Heterologous expression of target BGCs was successfully utilized in a number of metabologenomics studies, enabling validation of BGC-metabolite linkages for the tyrobetaine, rimosamide, faulknamycin, and stravidin BGCs.121, 181–183 Similarly, GWAS findings in plants can be validated using transgenic expression approaches to scrutinize the activity of candidate sequences.174, 176 The use of heterologous expression in the FAC-MS pipeline, in addition to enabling facile linkage of BGCs to their metabolites using the FAC-score itself, also enables easy manipulation of heterologously encoded gene cluster, allowing targeted gene deletions in biosynthetic machinery to elucidate specific biosynthetic routes for metabolites and their intermediates.91, 184–187
6. Challenges and outlook
In recent years, advances in analytical instrumentation and genome sequencing capabilities have made possible the collection of unprecedented numbers of metabolomic profiles and genomes from a wide variety of organisms. A growing number of laboratories are taking advantage of these datasets, either individually or in combination, to mine the untapped chemical potential of Nature. While genome mining and metabolome mining offer unique benefits for natural product discovery, the integration of such datasets enables improved predictions, enhanced prioritization of novel biosynthetic pathways, and ultimately, targeted discovery of novel natural products. While the last decade has witnessed an inspiring growth in technologies for unifying metabolomics and genomics datasets, numerous challenges remain that prevent the potential of this strategy to be fully realized.
6.1. Source-specific challenges
The success of any natural products discovery project depends on acquisition of sufficient source material for genetic and/or chemical profiling, a task which should not be taken for granted. For example, it has been estimated that only a tiny fraction, ~1%, of bacteria on Earth can be readily cultured in laboratory settings;206 however, it should be noted that the remaining ~99% of organisms are deemed ‘unculturable’ not because they can never be cultured, but rather than we lack crucial information on their biology, and likely, the diversity of microbial organisms as a whole.207 While significant efforts in the last decade have been made to develop culturing methods for such ‘unculturable’ organisms (covered recently in 62, 206), even in lab-cultured organisms, the production of secondary metabolites is often limited, with some sources estimating a lack of expression for >90% of biosynthetic gene clusters.63, 64 Many recent studies have identified the impact of various environmental stimuli on microbial gene expression (for example, exposure to subinhibitory concentrations of toxic compounds,208, 209 heavy metals,210–212 and competing organisms,213–215 though undoubtedly more studies will be required to gain a comprehensive understanding of the myriad stimulatory methods scientists can utilize to regulate secondary metabolite gene clusters for natural product discovery.
Of course, the ability to link an expressed metabolite to its biosynthetic machinery depends on the successful identification of biosynthetic gene clusters in the producing organism. This endeavour has become routine for a wide variety of microorganisms of bacterial and fungal origin, but still lags behind in understudied organisms such as microalgae.161 In microalgae the detection of biosynthetic genes becomes even more challenging because enzymes involved in a given biosynthetic pathway are often distributed in different genomic contexts.161 Even if gene-metabolite linkages are made, advanced methods for manipulating genes are limited in marine algae,161 making it challenging to validate bioinformatic predictions. In plants, biosynthetic discoveries remain largely elusive due to the complexity of plant genomes.29, 59, 171 Not only are plant genomes considerably larger (and more expensive to sequence) than those of bacteria and fungi, but they are fraught with genetic redundancy, cellular compartmentalization, and tight genetic regulation.59 Moreover, detected metabolites in plants may not actually originate from the plants themselves, and may instead be formed by fungal endophytes (either directly or as biotransformation products of plant metabolites),164, 216 further limiting the ability of bioinformatic pipelines to identify biosynthetic pathways of interest. As we gain a greater understanding of the unique chemical space occupied by bacteria, fungi, and plants, it will likely become more straightforward to identify natural products derived from fungal endophytes within a plant extract.
6.2. Overarching challenges
The advancement of analytical instrumentation with increased sensitivity and resolution, including both MS and NMR platforms, has enabled rising numbers of laboratories to detect molecules of low abundance and gain insight into their structural features even in the most complex natural products mixtures.43 While the complexity of such extracts may vary from organism to organism, the ability for researchers to mine complex datasets, regardless of source organism, remains a central challenge and continual development of computational tools to interpret and prioritize labor-intensive wet lab studies will be crucial to maintain momentum in this field of study. To achieve this, it is critical to monitor data quality, including data filtering, pre-processing, and simplification, prior to its inclusion in bioinformatics pipelines. Poorly filtered and/or processed spectra can skew statistical analyses, ultimately preventing researchers from extracting meaningful information from chemical datasets.152 Likewise, fragmented genomes or those with poor quality gene reads, can cause mis-annotation of biosynthetic genes. While this becomes more challenging to achieve as datasets grow in size, large datasets can improve linkage predictions between metabolites and their biosynthetic genes, and a careful balance between data quality and quantity must be met to optimize workflows moving forward.
Even with the highest-quality datasets, metabolomic and genomic analyses are still subject to a number of biases. We have made great strides in recent years in predicting molecular substructures from mass spectral data, particularly for molecules formed from canonical biosynthetic gene clusters (such as nonribosomal peptides). It is these same substructures that we can predict most efficiently from genomic datasets, where algorithms are most well-established for detecting biosynthetic types and predicting the types of precursors they incorporate into the final molecular structure. While such algorithms are undoubtedly ground breaking achievements in their own right, the use of such tools lead primarily to the discovery of modular metabolites, limiting our capacity to find the “unknown unknowns” that likely exist in natural product chemical space. Indeed, the vast majority of compounds discovered using genome mining and integrated metabolomic/genomic approaches thus far have been nonribosomal peptides, polyketides, and hybrids or these biosynthetic types. Increasingly sophisticated and interdisciplinary tools will play a pivotal role in exploiting integrated datasets for the identification of unusual molecular scaffolds and biosynthetic pathways, but undoubtedly represent exciting avenues for future research.
6.3. Where do we go from here?
Genomics and metabolomics provide powerful insight into the biosynthetic capacity of living organism and offer exciting opportunities for discovery. Because metabolomics datasets can only provide insight into natural products that are actually expressed by the producing organism, the linkage of metabolites to any detected biosynthetic gene clusters, which represent a combination of transcriptionally active and inactive genes, is not optimized. One can imagine the power of combining additional-Omics datasets, particularly transcriptomics, as an intermediate strategy to underscore metabolite-gene linkages.43 Microarray data, RNA-seq, and transcriptome shotgun sequencing not only provide researchers with the opportunity to minimize false associations by focusing in on transcriptionally active genes, but also enables identification of biosynthetic pathways that are not clustered together (or split biosynthetic pathways), facilitating investigation into understudied organisms such as microalgae and plants. Additionally, the incorporation of bioactivity data into multi-omics datasets could enable targeted prioritization of metabolites with desirable biological effects, providing insight into the types of biosynthetic pathways that produce molecules privileged for bioactivity, and accelerating the discovery of novel drugs for therapeutic use.
The successful utilization of multi-omics strategies will required continued improvements to data mining algorithms, maximizing their capacity to handle the massive and ever-expanding datasets available to us today. The ability for researchers to share datasets are essential pursuits, not only to avoid redundancy in discovery efforts, but also to provide the natural products community with complementary, standardized datasets that can improve statistical predictions.43, 45, 217 An exciting recent initiative to facilitate this goal is the Paired omics Data Platform (https://pairedomicsdata.bioinformatics.nl)218 where multi-omics datasets can be stored, annotated, and shared with the goal of stimulating community use for compound discovery. Such community efforts will improve targeted discovery efforts, enable facile validation of findings, improve upon machine learning models, and cultivate the much-needed engagement of experts from interdisciplinary fields to link complex datasets and truly discover the bounty that Nature has to offer.
7. Concluding Remarks
As genomic and metabolomic pipelines become increasingly high throughput, the natural products community stands on the precipice of a new era. No longer challenged by a paucity of data, researchers instead find themselves in a data-rich environment; however, without proper tools to extract meaning from these datasets, they may instead find themselves buried by meaningless information. Data alone is not enough—data must be organized to extract information, and then to build our knowledge base, before true discoveries can be made. Although strategies to analyze such datasets are only in their infancy, they are already proving quite successful, as illustrated by the numerous examples presented above. Such successes will only be accelerated through the continued development of pattern recognition software, improved sensitivity of analytical instrumentation, community development and curation of databases, and integrated machine learning approaches for identifying targets for future discovery.
Acknowledgements
This research was supported by the National Institute of General Medical Sciences of the National Institutes of Health under grant number 5 F32 GM132679. The authors would also like to thank Michael Mullowney and Grant Nickles for their insight into paper structure and content.
Footnotes
Conflicts of interest
There are no conflicts to declare.
8. References
- 1.Newman DJ and Cragg GM, J. Nat. Prod, 2020, 83, 770–803. [DOI] [PubMed] [Google Scholar]
- 2.Petrovska BB, Pharmacogn. Rev, 2012, 6, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bernardini S, Tiezzi A, Laghezza Masci V and Ovidi E, Nat. Prod. Res, 2018, 32, 1926–1950. [DOI] [PubMed] [Google Scholar]
- 4.Cragg GM and Pezzuto JM, Med. Princ. Pract, 2016, 25 Suppl 2, 41–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roemer T, Xu D, Singh SB, Parish CA, Harris G, Wang H, Davies JE and Bills GF, Chem. Biol, 2011, 18, 148–164. [DOI] [PubMed] [Google Scholar]
- 6.Genilloud O, Current Opinion in Microbiology, 2019, 51, 81–87. [DOI] [PubMed] [Google Scholar]
- 7.Bills GF and Gloer JB, Microbiol. Spectr, 2016, 4. [DOI] [PubMed] [Google Scholar]
- 8.Pye CR, Bertin MJ, Lokey RS, Gerwick WH and Linington RG, Proc. Natl. Acad. Sci, 2017, DOI: 10.1073/pnas.1614680114, 201614680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kinghorn AD, Curr. Org. Chem, 1998, 2, 597–612. [Google Scholar]
- 10.Wall ME and Wani MC, J. Ethnopharmacol, 1996, 51, 239–253. [DOI] [PubMed] [Google Scholar]
- 11.Oberlies NH and Kroll DJ, J. Nat. Prod, 2004, 67, 129–135. [DOI] [PubMed] [Google Scholar]
- 12.Tu Y, Nat. Med, 2011, 17, 1217–1220. [DOI] [PubMed] [Google Scholar]
- 13.Noble RL, Biochem. Cell. Biol, 1990, 68, 1344–1351. [PubMed] [Google Scholar]
- 14.Weller MG, Sensors (Basel), 2012, 12, 9181–9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jensen PR, Chavarria KL, Fenical W, Moore BS and Ziemert N, J. Ind. Microbiol. Biotechnol, 2014, 41, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prince EK and Pohnert G, Anal. Bioanal. Chem, 2010, 396, 193–197. [DOI] [PubMed] [Google Scholar]
- 17.Bachmann BO, Van Lanen SG and Baltz RH, J. Ind. Microbiol. Biotechnol, 2014, 41, 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kellogg JJ, Todd DA, Egan JM, Raja HA, Oberlies NH, Kvalheim OM and Cech NB, J. Nat. Prod, 2016, 79, 376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Almeida A, Nayfach S, Boland M, Strozzi F, Beracochea M, Shi ZJ, Pollard KS, Sakharova E, Parks DH, Hugenholtz P, Segata N, Kyrpides NC and Finn RD, Nat. Biotechnol, 2020, DOI: 10.1038/s41587-020-0603-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zou Y, Xue W, Luo G, Deng Z, Qin P, Guo R, Sun H, Xia Y, Liang S, Dai Y, Wan D, Jiang R, Su L, Feng Q, Jie Z, Guo T, Xia Z, Liu C, Yu J, Lin Y, Tang S, Huo G, Xu X, Hou Y, Liu X, Wang J, Yang H, Kristiansen K, Li J, Jia H and Xiao L, Nat. Biotechnol, 2019, 37, 179–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang KB, Ernst M, van der Hooft JJJ, da Silva RR, Park J, Medema MH, Sung SH and Dorrestein PC, Plant J, 2019, 98, 1134–1144. [DOI] [PubMed] [Google Scholar]
- 22.Nguyen DD, Melnik AV, Koyama N, Lu X, Schorn M, Fang J, Aguinaldo K, Lincecum TL, Ghequire MGK, Carrion VJ, Cheng TL, Duggan BM, Malone JG, Mauchline TH, Sanchez LM, Kilpatrick AM, Raaijmakers JM, De Mot R, Moore BS, Medema MH and Dorrestein PC, Nat. Microbiol, 2016, 2, 16197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Didelot X, Bowden R, Wilson DJ, Peto TE and Crook DW, Nat. Rev. Genet, 2012, 13, 601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kersten RD and Dorrestein PC, ACS Chem. Biol, 2009, 4, 599–601. [DOI] [PubMed] [Google Scholar]
- 25.Grim CM, Luu GT and Sanchez LM, FEMS Microbiol. Lett, 2019, 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang JY, Sanchez LM, Rath CM, Liu X, Boudreau PD, Bruns N, Glukhov E, Wodtke A, de Felicio R, Fenner A, Wong WR, Linington RG, Zhang L, Debonsi HM, Gerwick WH and Dorrestein PC, J. Nat. Prod, 2013, 76, 1686–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tietz JI, Schwalen CJ, Patel PS, Maxson T, Blair PM, Tai H-C, Zakai UI and Mitchell DA, Nat. Chem. Biol, 2017, 13, 470–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Merwin NJ, Mousa WK, Dejong CA, Skinnider MA, Cannon MJ, Li H, Dial K, Gunabalasingam M, Johnston C and Magarvey NA, Proc. Natl. Acad. Sci, 2020, 117, 371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kautsar SA, Suarez Duran HG, Blin K, Osbourn A and Medema MH, Nucleic Acids Res, 2017, 45, W55–W63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takeda I, Umemura M, Koike H, Asai K and Machida M, DNA Res, 2014, 21, 447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skinnider MA, Dejong CA, Rees PN, Johnston CW, Li H, Webster AL, Wyatt MA and Magarvey NA, Nucleic Acids Res, 2015, 43, 9645–9662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Starcevic A, Zucko J, Simunkovic J, Long PF, Cullum J and Hranueli D, Nucleic Acids Res, 2008, 36, 6882–6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khaldi N, Seifuddin FT, Turner G, Haft D, Nierman WC, Wolfe KH and Fedorova ND, Fungal Genet. Biol, 2010, 47, 736–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chavali AK and Rhee SY, Brief. Bioinform, 2018, 19, 1022–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van der Lee TA and Medema MH, Fungal Genet Biol, 2016, 89, 29–36. [DOI] [PubMed] [Google Scholar]
- 36.Weber T and Kim HU, Synth. Syst. Biotechnol, 2016, 1, 69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gluck-Thaler E, Haridas S, Binder M, Grigoriev IV, Crous PW, Spatafora JW, Bushley K and Slot JC, Mol. Biol. Evol, 2020, 37, 2838–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boddy CN, J. Ind. Microbiol. Biotechnol, 2014, 41, 443–450. [DOI] [PubMed] [Google Scholar]
- 39.Blin K, Shaw S, Steinke K, Villebro R, Ziemert N, Lee SY, Medema MH and Weber T, Nucleic Acids Res, 2019, 47, W81–W87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Medema MH and Fischbach MA, Nat. Chem. Biol, 2015, 11, 639–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang M, Carver JJ, Phelan VV, Sanchez LM, Garg N, Peng Y, Nguyen DD, Watrous J, Kapono CA, Luzzatto-Knaan T, Porto C, Bouslimani A, Melnik AV, Meehan MJ, Liu W-T, Crüsemann M, Boudreau PD, Esquenazi E, Sandoval-Calderón M, Kersten RD, Pace LA, Quinn RA, Duncan KR, Hsu C-C, Floros DJ, Gavilan RG, Kleigrewe K, Northen T, Dutton RJ, Parrot D, Carlson EE, Aigle B, Michelsen CF, Jelsbak L, Sohlenkamp C, Pevzner P, Edlund A, McLean J, Piel J, Murphy BT, Gerwick L, Liaw C-C, Yang Y-L, Humpf H-U, Maansson M, Keyzers RA, Sims AC, Johnson AR, Sidebottom AM, Sedio BE, Klitgaard A, Larson CB, Boya P CA, Torres-Mendoza D, Gonzalez DJ, Silva DB, Marques LM, Demarque DP, Pociute E, O’Neill EC, Briand E, Helfrich EJN, Granatosky EA, Glukhov E, Ryffel F, Houson H, Mohimani H, Kharbush JJ, Zeng Y, Vorholt JA, Kurita KL, Charusanti P, McPhail KL, Nielsen KF, Vuong L, Elfeki M, Traxler MF, Engene N, Koyama N, Vining OB, Baric R, Silva RR, Mascuch SJ, Tomasi S, Jenkins S, Macherla V, Hoffman T, Agarwal V, Williams PG, Dai J, Neupane R, Gurr J, Rodríguez AMC, Lamsa A, Zhang C, Dorrestein K, Duggan BM, Almaliti J, Allard P-M, Phapale P, Nothias L-F, Alexandrov T, Litaudon M, Wolfender J-L, Kyle JE, Metz TO, Peryea T, Nguyen D-T, VanLeer D, Shinn P, Jadhav A, Müller R, Waters KM, Shi W, Liu X, Zhang L, Knight R, Jensen PR, Palsson BØ, Pogliano K, Linington RG, Gutiérrez M, Lopes NP, Gerwick WH, Moore BS, Dorrestein PC and Bandeira N, Nat. Biotechnol, 2016, 34, 828–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramos AEF, Evanno L, Poupon E, Champy P and Beniddir MA, Nat. Prod. Rep, 2019, 36, 960–980. [DOI] [PubMed] [Google Scholar]
- 43.Hautbergue T, Jamin EL, Debrauwer L, Puel O and Oswald IP, Nat. Prod. Rep, 2018, 35, 147–173. [DOI] [PubMed] [Google Scholar]
- 44.Soldatou S, Eldjarn GH, Huerta-Uribe A, Rogers S and Duncan KR, FEMS Microbiol. Lett, 2019, 366, fnz142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van der Hooft JJJ, Mohimani H, Bauermeister A, Dorrestein PC, Duncan KR and Medema MH, Chem. Soc. Rev, 2020, 49, 3297–3314. [DOI] [PubMed] [Google Scholar]
- 46.Horgan RP and Kenny LC, Obstet. Gynaecol, 2011, 13, 189–195. [Google Scholar]
- 47.Johnson CH, Ivanisevic J and Siuzdak G, Nat. Rev. Mol. Cell. Biol, 2016, 17, 451–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Van Lanen SG and Shen B, Curr. Op. Microbiol, 2006, 9, 252–260. [DOI] [PubMed] [Google Scholar]
- 49.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K and Madden TL, BMC Bioinformatics, 2009, 10, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eddy SR, PLoS Comput. Biol, 2011, 7, e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weber T, Rausch C, Lopez P, Hoof I, Gaykova V, Huson D and Wohlleben W, J. Biotechnol, 2009, 140, 13–17. [DOI] [PubMed] [Google Scholar]
- 52.Li MH, Ung PM, Zajkowski J, Garneau-Tsodikova S and Sherman DH, BMC Bioinformatics, 2009, 10, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, Weber T, Takano E and Breitling R, Nucleic Acids Res, 2011, 39, W339–W346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Skinnider MA, Merwin NJ, Johnston CW and Magarvey NA, Nucleic Acids Res, 2017, 45, W49–W54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eddy SR, Bioinformatics (Oxford, England), 1998, 14, 755–763. [DOI] [PubMed] [Google Scholar]
- 56.Blin K, Wolf T, Chevrette MG, Lu X, Schwalen CJ, Kautsar SA, Suarez Duran HG, de Los Santos ELC, Kim HU, Nave M, Dickschat JS, Mitchell DA, Shelest E, Breitling R, Takano E, Lee SY, Weber T and Medema MH, Nucleic Acids Res, 2017, 45, W36–w41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Blin K, Medema MH, Kazempour D, Fischbach MA, Breitling R, Takano E and Weber T, Nucleic Acids Res, 2013, 41, W204–W212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL and Clardy J, Natural Prod. Rep, 2013, 30, 108–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rai A, Saito K and Yamazaki M, Plant J, 2017, 90, 764–787. [DOI] [PubMed] [Google Scholar]
- 60.Prosser GA, Larrouy-Maumus G and de Carvalho LPS, EMBO Rep, 2014, 15, 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rai A and Saito K, Curr. Op. Biotechnol, 2016, 37, 127–134. [DOI] [PubMed] [Google Scholar]
- 62.Covington BC, McLean JA and Bachmann BO, Nat. Prod. Rep, 2017, 34, 6–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Scherlach K and Hertweck C, Org. Biomol. Chem, 2009, 7, 1753–1760. [DOI] [PubMed] [Google Scholar]
- 64.Walsh CT and Fischbach MA, J. Am. Chem. Soc, 2010, 132, 2469–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mao D, Okada BK, Wu Y, Xu F and Seyedsayamdost MR, Curr. Op. Microbiol, 2018, 45, 156–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ochi K and Hosaka T, Appl. Microbiol.Biotechnol, 2012, 97. [Google Scholar]
- 67.El-Elimat T, Figueroa M, Ehrmann BM, Cech NB, Pearce CJ and Oberlies NH, J. Nat. Prod, 2013, 76, 1709–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van Santen JA, Kautsar SA, Medema MH and Linington RG, Nat. Prod. Rep, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bueschl C, Kluger B, Neumann NKN, Doppler M, Maschietto V, Thallinger GG, Meng-Reiterer J, Krska R and Schuhmacher R, Anal. Chem, 2017, 89, 9518–9526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Berg M, Vanaerschot M, Jankevics A, Cuypers B, Breitling R and Dujardin J-C, Comp. Struct. Biotechnol. J, 2013, 4, e201301002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Caesar LK, Kvalheim OM and Cech NB, Anal. Chim. Acta, 2018, 1021, 69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Reher R, Kim HW, Zhang C, Mao HH, Wang M, Nothias L-F, Caraballo-Rodriguez AM, Glukhov E, Teke B, Leao T, Alexander KL, Duggan BM, Van Everbroeck EL, Dorrestein PC, Cottrell GW and Gerwick WH, J. Am. Chem. Soc, 2020, 142, 4114–4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Egan JM, van Santen JA, Liu DY and Linington RG, J. Nat. Prod, 2021, DOI: 10.1021/acs.jnatprod.0c01076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McAlpine JB, Chen S-N, Kutateladze A, MacMillan JB, Appendino G, Barison A, Beniddir MA, Biavatti MW, Bluml S and Boufridi A, Nat. Prod. Rep, 2019, 36, 35–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bisson J, Simmler C, Chen S-N, Friesen JB, Lankin DC, McAlpine JB and Pauli GF, Nat. Prod Rep, 2016, 33, 1028–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ziemert N, Alanjary M and Weber T, Nat. Prod. Rep, 2016, 33, 988–1005. [DOI] [PubMed] [Google Scholar]
- 77.Palaniappan K, Chen I-MA, Chu K, Ratner A, Seshadri R, Kyrpides NC, Ivanova NN and Mouncey NJ, Nucleic acids Res, 2020, 48, D422–D430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Belknap KC, Park CJ, Barth BM and Andam CP, Sci. Rep, 2020, 10, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cimermancic P, Medema MH, Claesen J, Kurita K, Brown LCW, Mavrommatis K, Pati A, Godfrey PA, Koehrsen M and Clardy J, Cell, 2014, 158, 412–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Doroghazi JR, Albright JC, Goering AW, Ju KS, Haines RR, Tchalukov KA, Labeda DP, Kelleher NL and Metcalf WW, Nat. Chem. Biol, 2014, 10, 963–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ziemert N, Lechner A, Wietz M, Millán-Aguiñaga N, Chavarria KL and Jensen PR, Proc. Natl. Acad. Sci, 2014, 111, E1130–E1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Navarro-Muñoz JC, Selem-Mojica N, Mullowney MW, Kautsar SA, Tryon JH, Parkinson EI, De Los Santos EL, Yeong M, Cruz-Morales P and Abubucker S, Nat. Chem. Biol, 2020, 16, 60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kautsar SA, Van Der Hooft JJ, De Ridder D and Medema MH, GigaScience, 2021, 10, giaa154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kautsar SA, Blin K, Shaw S, Weber T and Medema MH, Nucleic Acids Res, 2021, 49, D490–D497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Medema MH, Nat. Prod. Rep, 2021. [DOI] [PubMed] [Google Scholar]
- 86.Fischbach MA and Walsh CT, Science, 2009, 325, 1089–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pelzer S, Wohlert SE and Vente A, Ernst Schering Res. Found. Workshop, 2005, DOI: 10.1007/3-540-27055-8_11, 233–259. [DOI] [PubMed] [Google Scholar]
- 88.Weng JK and Noel JP, Cold Spring Harb. Symp. Quant. Biol, 2012, 77, 309–320. [DOI] [PubMed] [Google Scholar]
- 89.Cruz-Morales P, Kopp JF, Martínez-Guerrero C, Yáñez-Guerra LA, Selem-Mojica N, Ramos-Aboites H, Feldmann J and Barona-Gómez F, Genome Biol. Evol, 2016, 8, 1906–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kloosterman AM, Cimermancic P, Elsayed SS, Du C, Hadjithomas M, Donia MS, Fischbach MA, van Wezel GP and Medema MH, PLoS Biol, 2020, 18, e3001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Caesar LK, Robey MT, Swyers M, Islam MN, Ye R, Vagadia PP, Schiltz GE, Thomas PM, Wu CC, Kelleher NL, Keller NP and Bok JW, mBio, 2020, 11, e01691–01620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gluck-Thaler E, Vijayakumar V and Slot JC, Mol. Ecol, 2018, 27, 5120–5136. [DOI] [PubMed] [Google Scholar]
- 93.Mungan MD, Alanjary M, Blin K, Weber T, Medema MH and Ziemert N, Nucleic Acids Res, 2020, 48, W546–W552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cundliffe E and Demain AL, J. Ind. Microbiol. Biotechnol, 2010, 37, 643–672. [DOI] [PubMed] [Google Scholar]
- 95.Yan Y, Liu N and Tang Y, Nat. Prod. Rep, 2020, 37, 879–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Almabruk KH, Dinh LK and Philmus B, ACS Chem. Biol, 2018, 13, 1426–1437. [DOI] [PubMed] [Google Scholar]
- 97.Tran PN, Yen M-R, Chiang C-Y, Lin H-C and Chen P-Y, Appl. Microbiol. Biotechnol, 2019, 103, 3277–3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tang X, Li J, Millán-Aguiñaga N, Zhang JJ, O’Neill EC, Ugalde JA, Jensen PR, Mantovani SM and Moore BS, ACS Chem. Biol, 2015, 10, 2841–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kale AJ, McGlinchey RP, Lechner A and Moore BS, ACS Chem. Biol, 2011, 6, 1257–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schorn M, Zettler J, Noel JP, Dorrestein PC, Moore BS and Kaysser L, ACS Chem. Biol, 2014, 9, 301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yeh H-H, Ahuja M, Chiang Y-M, Oakley CE, Moore S, Yoon O, Hajovsky H, Bok J-W, Keller NP and Wang CC, ACS Chem. Biol, 2016, 11, 2275–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lin H-C, Chooi Y-H, Dhingra S, Xu W, Calvo AM and Tang Y, J Am. Chem. Soc, 2013, 135, 4616–4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wiemann P, Guo C-J, Palmer JM, Sekonyela R, Wang CCC and Keller NP, Proc. Natl. Acad. Sci, 2013, 110, 17065–17070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yan Y, Liu Q, Zang X, Yuan S, Bat-Erdene U, Nguyen C, Gan J, Zhou J, Jacobsen SE and Tang Y, Nature, 2018, 559, 415–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Alanjary M, Kronmiller B, Adamek M, Blin K, Weber T, Huson D, Philmus B and Ziemert N, Nucleic Acids Res, 2017, 45, W42–W48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Panter F, Krug D, Baumann S and Müller R, Chem. Sci, 2018, 9, 4898–4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Thaker MN, Wang W, Spanogiannopoulos P, Waglechner N, King AM, Medina R and Wright GD, Nature Biotechnol, 2013, 31, 922–927. [DOI] [PubMed] [Google Scholar]
- 108.Culp EJ, Waglechner N, Wang W, Fiebig-Comyn AA, Hsu Y-P, Koteva K, Sychantha D, Coombes BK, Van Nieuwenhze MS and Brun YV, Nature, 2020, 578, 582–587. [DOI] [PubMed] [Google Scholar]
- 109.Nützmann H-W and Osbourn A, Curr. Op. Biotechnol, 2014, 26, 91–99. [DOI] [PubMed] [Google Scholar]
- 110.Nützmann HW, Huang A and Osbourn A, New Phytol, 2016, 211, 771–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Witjes L, Kooke R, van der Hooft JJ, de Vos RC, Keurentjes JJ, Medema MH and Nijveen H, BMC Res. Notes, 2019, 12, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Medema MH and Osbourn A, Nat. Prod. Rep, 2016, 33, 951–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Töpfer N, Fuchs L-M and Aharoni A, Nucleic Acids Res, 2017, 45, 7049–7063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Umemura M, Koike H, Nagano N, Ishii T, Kawano J, Yamane N, Kozone I, Horimoto K, Shin-ya K and Asai K, PloS One, 2013, 8, e84028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Derakhshani H, Bernier SP, Marko VA and Surette MG, BMC Genomics, 2020, 21, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Goldstein S, Beka L, Graf J and Klassen JL, BMC Genomics, 2019, 20, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wick RR, Judd LM, Gorrie CL and Holt KE, PLoS Comp. Biol, 2017, 13, e1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Meleshko D, Mohimani H, Tracanna V, Hajirasouliha I, Medema MH, Korobeynikov A and Pevzner PA, Genome Res, 2019, 29, 1352–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lin Z, Nielsen J and Liu Z, Front. Bioeng. Biotechnol, 2020, 8, 526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kloosterman AM, Medema MH and van Wezel GP, Curr. Op. Biotechnol, 2021, 69, 60–67. [DOI] [PubMed] [Google Scholar]
- 121.Parkinson EI, Tryon JH, Goering AW, Ju K-S, McClure RA, Kemball JD, Zhukovsky S, Labeda DP, Thomson RJ, Kelleher NL and Metcalf WW, ACS Chem. Biol, 2018, 13, 1029–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yang JY, Sanchez LM, Rath CM, Liu X, Boudreau PD, Bruns N, Glukhov E, Wodtke A, De Felicio R and Fenner A, J. Nat. Prod, 2013, 76, 1686–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nothias L-F, Petras D, Schmid R, Dührkop K, Rainer J, Sarvepalli A, Protsyuk I, Ernst M, Tsugawa H and Fleischauer M, Nat. Meth, 2020, 17, 905–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Xue J, Guijas C, Benton HP, Warth B and Siuzdak G, Nat. Meth, 2020, 17, 953–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Huber F, Ridder L, Verhoeven S, Spaaks JH, Diblen F, Rogers S and van der Hooft JJJ, PLoS Comp. Biol, 2021, 17, e1008724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mikolov T, Sutskever I, Chen K, Corrado GS and Dean J, Adv. Neur. Inform. Proc. Syst 2013, 26. [Google Scholar]
- 127.Beauxis Y and Genta-Jouve G, Bioinformatics, 2019, 35, 1795–1796. [DOI] [PubMed] [Google Scholar]
- 128.Hufsky F, Scheubert K and Böcker S, Nat. Prod. Rep, 2014, 31, 807–817. [DOI] [PubMed] [Google Scholar]
- 129.Allard P-M, Péresse T, Bisson J, Gindro K, Marcourt L, Pham VC, Roussi F, Litaudon M and Wolfender J-L, Anal. Chem, 2016, 88, 3317–3323. [DOI] [PubMed] [Google Scholar]
- 130.Allen F, Pon A, Wilson M, Greiner R and Wishart D, Nucleic Acids Res, 2014, 42, W94–W99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dührkop K, Shen H, Meusel M, Rousu J and Böcker S, Proc. Natl. Acad. Sci, 2015, 112, 12580–12585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Dührkop K, Fleischauer M, Ludwig M, Aksenov AA, Melnik AV, Meusel M, Dorrestein PC, Rousu J and Böcker S, Nat. Meth, 2019, 16, 299–302. [DOI] [PubMed] [Google Scholar]
- 133.Tripathi A, Vázquez-Baeza Y, Gauglitz JM, Wang M, Dührkop K, Nothias-Esposito M, Acharya DD, Ernst M, van der Hooft JJJ, Zhu Q, McDonald D, Brejnrod AD, Gonzalez A, Handelsman J, Fleischauer M, Ludwig M, Böcker S, Nothias L-F, Knight R and Dorrestein PC, Nat. Chem. Biol, 2021, 17, 146–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mohimani H, Gurevich A, Shlemov A, Mikheenko A, Korobeynikov A, Cao L, Shcherbin E, Nothias LF, Dorrestein PC and Pevzner PA, Nat. Commun, 2018, 9, 4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mohimani H, Liu W-T, Kersten RD, Moore BS, Dorrestein PC and Pevzner PA, J. Nat. Prod, 2014, 77, 1902–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Roullier C, Guitton Y, Valery M, Amand S, Prado S, Robiou du Pont T, Grovel O and Pouchus YF, Anal. Chem, 2016, 88, 9143–9150. [DOI] [PubMed] [Google Scholar]
- 137.Gribble GW, Marine Drugs, 2015, 13, 4044–4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Schymanski EL, Jeon J, Gulde R, Fenner K, Ruff M, Singer HP and Hollender J, Environ. Sci. Technol, 2014, 48, 2097–2098. [DOI] [PubMed] [Google Scholar]
- 139.Sumner LW, Amberg A, Barrett D, Beale MH, Beger R, Daykin CA, Fan TW-M, Fiehn O, Goodacre R and Griffin JL, Metabolomics, 2007, 3, 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhang C, Idelbayev Y, Roberts N, Tao Y, Nannapaneni Y, Duggan BM, Min J, Lin EC, Gerwick EC and Cottrell GW, Sci. Rep, 2017, 7, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Genilloud O, González I, Salazar O, Martín J, Tormo JR and Vicente F, J. Ind. Microbiol. Biotechnol, 2011, 38, 375–389. [DOI] [PubMed] [Google Scholar]
- 142.Hou Y, Braun DR, Michel CR, Klassen JL, Adnani N, Wyche TP and Bugni TS, Anal. Chem, 2012, 84, 4277–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.van Der Hooft JJJ, Wandy J, Barrett MP, Burgess KE and Rogers S, Proc. Natl. Acad. Sci, 2016, 113, 13738–13743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Dührkop K, Nothias L-F, Fleischauer M, Reher R, Ludwig M, Hoffmann MA, Petras D, Gerwick WH, Rousu J and Dorrestein PC, Nat. Biotechnol, 2021, 39, 462–471. [DOI] [PubMed] [Google Scholar]
- 145.Rogers S, Ong CW, Wandy J, Ernst M, Ridder L and Van Der Hooft JJ, Faraday Discuss, 2019, 218, 284–302. [DOI] [PubMed] [Google Scholar]
- 146.Ernst M, Kang KB, Caraballo-Rodríguez AM, Nothias L-F, Wandy J, Chen C, Wang M, Rogers S, Medema MH and Dorrestein PC, Metabolites, 2019, 9, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Britton ER, Kellogg JJ, Kvalheim OM and Cech NB, J. Nat. Prod, 2018, 81, 484–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Caesar LK, Nogo S, Naphen CN and Cech NB, Anal. Chem, 2019, 91, 11297–11305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Li P, AnandhiSenthilkumar H, Wu S.-b., Liu B, Guo Z.-y., Fata JE, Kennelly EJ and Long C.-l., J. Chromatog. B, 2016, 1011, 179–195. [DOI] [PubMed] [Google Scholar]
- 150.Chan K-M, Yue GG-L, Li P, Wong EC-W, Lee JK-M, Kennelly EJ and Bik-San Lau C, J. Chromatog. A, 2017, 1487, 162–167. [DOI] [PubMed] [Google Scholar]
- 151.Caesar LK, Kellogg JJ, Kvalheim OM, Cech RA and Cech NB, Planta medica, 2018, 84, 721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Caesar LK, Kellogg JJ, Kvalheim OM and Cech NB, J. Nat. Prod, 2019, 82, 469–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Nothias L.-F. l., Nothias-Esposito M. l., da Silva R, Wang M, Protsyuk I, Zhang Z, Sarvepalli A, Leyssen P, Touboul D and Costa J, J. Nat. Prod, 2018, 81, 758–767. [DOI] [PubMed] [Google Scholar]
- 154.Kurita KL, Glassey E and Linington RG, Proc. Natl. Acad. Sci, 2015, 112, 11999–12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Inui T, Wang Y, Pro SM, Franzblau SG and Pauli GF, Fitoterapia, 2012, 83, 1218–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Rajalahti T and Kvalheim OM, Int. J. Pharm, 2011, 417, 280–290. [DOI] [PubMed] [Google Scholar]
- 157.Rajalahti T, Arneberg R, Kroksveen AC, Berle M, Myhr K-M and Kvalheim OM, Anal. Chem, 2009, 81, 2581–2590. [DOI] [PubMed] [Google Scholar]
- 158.Riyanti, Marner M, Hartwig C, Patras MA, Wodi SIM, Rieuwpassa FJ, Ijong FG, Balansa W and Schäberle TF, Marine Drugs, 2020, 18, 649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Duran-Frigola M, Pauls E, Guitart-Pla O, Bertoni M, Alcalde V, Amat D, Juan-Blanco T and Aloy P, Nat. Biotechnol, 2020, 38, 1087–1096. [DOI] [PubMed] [Google Scholar]
- 160.Robey MT, Caesar LK, Drott MT, Keller NP and Kelleher NL, Proc. Natl. Acad. Sci, 2021, 118, e2020230118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lauritano C, Ferrante MI and Rogato A, Marine drugs, 2019, 17, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lewis WH, Tahon G, Geesink P, Sousa DZ and Ettema TJG, Nat. Rev. Microbiol, 2020, DOI: 10.1038/s41579-020-00458-8. [DOI] [PubMed] [Google Scholar]
- 163.Sampaio BL, Edrada-Ebel R and Da Costa FB, Sci. Rep, 2016, 6, 29265–29265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Egan JM, Kaur A, Raja HA, Kellogg JJ, Oberlies NH and Cech NB, Phytochem. Lett, 2016, 17, 219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Tawfike AF, Tate R, Abbott G, Young L, Viegelmann C, Schumacher M, Diederich M and Edrada-Ebel R, Chem. Biodivers, 2017, 14. [DOI] [PubMed] [Google Scholar]
- 166.Scossa F, Benina M, Alseekh S, Zhang Y and Fernie AR, Planta medica, 2018, 84, 855–873. [DOI] [PubMed] [Google Scholar]
- 167.Steenwyk JL, Mead ME, Knowles SL, Raja HA, Roberts CD, Bader O, Houbraken J, Goldman GH, Oberlies NH and Rokas A, Genetics, 2020, 216, 481–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Duncan KR, Crüsemann M, Lechner A, Sarkar A, Li J, Ziemert N, Wang M, Bandeira N, Moore BS, Dorrestein PC and Jensen PR, Chem. Biol, 2015, 22, 460–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Maansson M, Vynne NG, Klitgaard A, Nybo JL, Melchiorsen J, Nguyen DD, Sanchez LM, Ziemert N, Dorrestein PC, Andersen MR and Gram L, mSystems, 2016, 1, e00028–00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zdouc MM, Iorio M, Maffioli SI, Crüsemann M, Donadio S and Sosio M, J. Nat. Prod, 2021, 84, 204–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zhao N, Wang G, Norris A, Chen X and Chen F, Crit. Rev. Plant Sci, 2013, 32, 369–382. [Google Scholar]
- 172.Riedelsheimer C, Czedik-Eysenberg A, Grieder C, Lisec J, Technow F, Sulpice R, Altmann T, Stitt M, Willmitzer L and Melchinger AE, Nat. Genet, 2012, 44, 217–220. [DOI] [PubMed] [Google Scholar]
- 173.Meyer RC, Steinfath M, Lisec J, Becher M, Witucka-Wall H, Törjék O, Fiehn O, Eckardt Ä, Willmitzer L, Selbig J and Altmann T, Proc. Natl. Acad. Sci, 2007, 104, 4759–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wen W, Li D, Li X, Gao Y, Li W, Li H, Liu J, Liu H, Chen W, Luo J and Yan J, Nat. Comm, 2014, 5, 3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Daygon VD, Calingacion M, Forster LC, Voss JJD, Schwartz BD, Ovenden B, Alonso DE, McCouch SR, Garson MJ and Fitzgerald MA, Sci. Rep, 2017, 7, 8767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Gao L, Gonda I, Sun H, Ma Q, Bao K, Tieman DM, Burzynski-Chang EA, Fish TL, Stromberg KA, Sacks GL, Thannhauser TW, Foolad MR, Diez MJ, Blanca J, Canizares J, Xu Y, van der Knaap E, Huang S, Klee HJ, Giovannoni JJ and Fei Z, Nat. Genet, 2019, 51, 1044–1051. [DOI] [PubMed] [Google Scholar]
- 177.Riedelsheimer C, Lisec J, Czedik-Eysenberg A, Sulpice R, Flis A, Grieder C, Altmann T, Stitt M, Willmitzer L and Melchinger AE, Proc. Natl. Acad. Sci, 2012, 109, 8872–8877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Chan EKF, Rowe HC, Corwin JA, Joseph B and Kliebenstein DJ, PLoS Biol, 2011, 9, e1001125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Tobias NJ, Wolff H, Djahanschiri B, Grundmann F, Kronenwerth M, Shi YM, Simonyi S, Grün P, Shapiro-Ilan D, Pidot SJ, Stinear TP, Ebersberger I and Bode HB, Nat. Microbiol, 2017, 2, 1676–1685. [DOI] [PubMed] [Google Scholar]
- 180.Goering AW, McClure RA, Doroghazi JR, Albright JC, Haverland NA, Zhang Y, Ju K-S, Thomson RJ, Metcalf WW and Kelleher NL, ACS Cent. Sci, 2016, 2, 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Montaser R and Kelleher NL, ACS Chem. Biol, 2020, 15, 1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.McClure RA, Goering AW, Ju K-S, Baccile JA, Schroeder FC, Metcalf WW, Thomson RJ and Kelleher NL, ACS Chem. Biol, 2016, 11, 3452–3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Tryon JH, Rote JC, Chen L, Robey MT, Vega MM, Phua WC, Metcalf WW, Ju K-S, Kelleher NL and Thomson RJ, ACS Chem. Biol, 2020, DOI: 10.1021/acschembio.0c00663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Robey MT, Ye R, Bok JW, Clevenger KD, Islam MN, Chen C, Gupta R, Swyers M, Wu E, Gao P, Thomas PM, Wu CC, Keller NP and Kelleher NL, ACS Chem. Biol, 2018, 13, 1142–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Clevenger KD, Ye R, Bok JW, Thomas PM, Islam MN, Miley GP, Robey MT, Chen C, Yang K, Swyers M, Wu E, Gao P, Wu CC, Keller NP and Kelleher NL, Biochemistry, 2018, 57, 3237–3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Clevenger KD, Bok JW, Ye R, Miley GP, Verdan MH, Velk T, Chen C, Yang K, Robey MT, Gao P, Lamprecht M, Thomas PM, Islam MN, Palmer JM, Wu CC, Keller NP and Kelleher NL, Nat. Chem. Biol, 2017, 13, 895–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Bok JW, Ye R, Clevenger KD, Mead D, Wagner M, Krerowicz A, Albright JC, Goering AW, Thomas PM, Kelleher NL, Keller NP and Wu CC, BMC Genomics, 2015, 16, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Cao L, Gurevich A, Alexander KL, Naman CB, Leão T, Glukhov E, Luzzatto-Knaan T, Vargas F, Quinn R, Bouslimani A, Nothias LF, Singh NK, Sanders JG, Benitez RAS, Thompson LR, Hamid M-N, Morton JT, Mikheenko A, Shlemov A, Korobeynikov A, Friedberg I, Knight R, Venkateswaran K, Gerwick WH, Gerwick L, Dorrestein PC, Pevzner PA and Mohimani H, Cell. Syst, 2019, 9, 600–608.e604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Kersten RD, Yang YL, Xu Y, Cimermancic P, Nam SJ, Fenical W, Fischbach MA, Moore BS and Dorrestein PC, Nat. Chem. Biol, 2011, 7, 794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kersten RD, Ziemert N, Gonzalez DJ, Duggan BM, Nizet V, Dorrestein PC and Moore BS, Proc. Natl. Acad. Sci, 2013, 110, E4407–4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Röttig M, Medema MH, Blin K, Weber T, Rausch C and Kohlbacher O, Nucleic Acids Res, 2011, 39, W362–W367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Mohimani H, Kim S and Pevzner PA, J. Proteome Res, 2013, 12, 1560–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Johnston CW, Skinnider MA, Wyatt MA, Li X, Ranieri MRM, Yang L, Zechel DL, Ma B and Magarvey NA, Nat. Comm, 2015, 6, 8421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Li B, Sher D, Kelly L, Shi Y, Huang K, Knerr PJ, Joewono I, Rusch D, Chisholm SW and van der Donk WA, Proc. Natl. Acad. Sci, 2010, 107, 10430–10435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Skinnider MA, Johnston CW, Edgar RE, Dejong CA, Merwin NJ, Rees PN and Magarvey NA, Proc. Natl. Acad. Sci, 2016, 113, E6343–e6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Kersten RD and Weng J-K, Proc. Natl. Acad. Sci, 2018, 115, E10961–E10969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Brouard C, Shen H, Dührkop K, d’Alché-Buc F, Böcker S and Rousu J, Bioinformatics (Oxford, England), 2016, 32, i28–i36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Hjörleifsson Eldjárn G, Ramsay A, van der Hooft JJJ, Duncan KR, Soldatou S, Rousu J, Daly R, Wandy J and Rogers S, PLoS Comp. Biol, 2021, 17, e1008920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Soldatou S, Eldjárn GH, Ramsay A, van der Hooft JJJ, Hughes AH, Rogers S and Duncan KR, Marine Drugs, 2021, 19, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Ninomiya Y, Suzuki K, Ishii C and Inoue H, Proc. Natl. Acad. Sci, 2004, 101, 12248–12253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Krappmann S, Sasse C and Braus GH, Eukaryot. Cell, 2006, 5, 212–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Caesar LK, Kelleher NL and Keller NP, Fungal Genet. Biol, 2020, 103477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Nayak T, Szewczyk E, Oakley CE, Osmani A, Ukil L, Murray SL, Hynes MJ, Osmani SA and Oakley BR, Genetics, 2006, 172, 1557–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Cohen J, Science, 2020, 370, 271–272. [DOI] [PubMed] [Google Scholar]
- 205.Hsu PD, Lander ES and Zhang F, Cell, 2014, 157, 1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Vartoukian SR, Palmer RM and Wade WG, FEMS Microbiol. Lett, 2010, 309, 1–7. [DOI] [PubMed] [Google Scholar]
- 207.Stewart EJ, J. Bacteriol, 2012, 194, 4151–4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Imai Y, Sato S, Tanaka Y, Ochi K and Hosaka T, Appl. Environ. Microbiol, 2015, 81, 3869–3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Wang W, Ji J, Li X, Wang J, Li S, Pan G, Fan K and Yang K, Proc. Natl. Acad. Sci, 2014, 111, 5688–5693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Haferburg G and Kothe E, J. Basic Microbiol, 2007, 47, 453–467. [DOI] [PubMed] [Google Scholar]
- 211.Kawai K, Wang G, Okamoto S and Ochi K, FEMS Microbiol. Lett, 2007, 274, 311–315. [DOI] [PubMed] [Google Scholar]
- 212.Tanaka Y, Hosaka T and Ochi K, J. Antibiot, 2010, 63, 477–481. [DOI] [PubMed] [Google Scholar]
- 213.Abdelmohsen UR, Grkovic T, Balasubramanian S, Kamel MS, Quinn RJ and Hentschel U, Biotechnol. Adv, 2015, 33, 798–811. [DOI] [PubMed] [Google Scholar]
- 214.Derewacz DK, Covington BC, McLean JA and Bachmann BO, ACS Chem. Biol, 2015, 10, 1998–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Knowles SL, Raja HA, Wright AJ, Lee AML, Caesar LK, Cech NB, Mead ME, Steenwyk JL, Ries LN and Goldman GH, Front. Microbiol, 2019, 10, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Gupta S, Chaturvedi P, Kulkarni M and van Staden J, Biotechnol. Adv, 2019, 39, 107462. [DOI] [PubMed] [Google Scholar]
- 217.Meyer V, Andersen MR, Brakhage AA, Braus GH, Caddick MX, Cairns TC, de Vries RP, Haarmann T, Hansen K, Hertz-Fowler C, Krappmann S, Mortensen UH, Peñalva MA, Ram AFJ and Head RM, Fungal Biol. Biotechnol, 2016, 3, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Schorn MA, Verhoeven S, Ridder L, Huber F, Acharya DD, Aksenov AA, Aleti G, Moghaddam JA, Aron AT, Aziz S, Bauermeister A, Bauman KD, Baunach M, Beemelmanns C, Beman JM, Berlanga-Clavero MV, Blacutt AA, Bode HB, Boullie A, Brejnrod A, Bugni TS, Calteau A, Cao L, Carrión VJ, Castelo-Branco R, Chanana S, Chase AB, Chevrette MG, Costa-Lotufo LV, Crawford JM, Currie CR, Cuypers B, Dang T, de Rond T, Demko AM, Dittmann E, Du C, Drozd C, Dujardin J-C, Dutton RJ, Edlund A, Fewer DP, Garg N, Gauglitz JM, Gentry EC, Gerwick L, Glukhov E, Gross H, Gugger M, Guillén Matus DG, Helfrich EJN, Hempel B-F, Hur J-S, Iorio M, Jensen PR, Kang KB, Kaysser L, Kelleher NL, Kim CS, Kim KH, Koester I, König GM, Leao T, Lee SR, Lee Y-Y, Li X, Little JC, Maloney KN, Männle D, Martin H C, McAvoy AC, Metcalf WW, Mohimani H, Molina-Santiago C, Moore BS, Mullowney MW, Muskat M, Nothias L-F, O’Neill EC, Parkinson EI, Petras D, Piel J, Pierce EC, Pires K, Reher R, Romero D, Roper MC, Rust M, Saad H, Saenz C, Sanchez LM, Sørensen SJ, Sosio M, Süssmuth RD, Sweeney D, Tahlan K, Thomson RJ, Tobias NJ, Trindade-Silva AE, van Wezel GP, Wang M, Weldon KC, Zhang F, Ziemert N, Duncan KR, Crüsemann M, Rogers S, Dorrestein PC, Medema MH and van der Hooft JJJ, Nat. Chem. Biol, 2021, 17, 363–368. [DOI] [PMC free article] [PubMed] [Google Scholar]