

Abstract
Bacillus Calmette-Guérin (BCG) is the most effective intravesical agent at reducing recurrence for patients with high-grade, non-muscle invasive bladder cancer. Nevertheless, response to BCG is variable and strategies to boost BCG efficacy have not materialized. Prior work demonstrated a requirement for either conventional αβ or non-conventional γδ T cells in mediating BCG treatment efficacy, yet the importance of T-cell antigen specificity for BCG’s treatment effect is unclear. Here, we provide direct evidence to show that BCG increases the number of tumor antigen-specific αβ T cells in patients with bladder cancer and protects mice from subsequent same-tumor challenge, supporting BCG induction of tumor-specific memory and protection. Adoptive T-cell transfers of antigen-specific αβ T cells into immunodeficient mice challenged with syngeneic MB49 bladder tumors showed that both tumor and BCG antigen-specific αβ T cells contributed to BCG efficacy. BCG-specific antitumor immunity, however, also required non-conventional γδ T cells. Prior work shows that mammalian target of rapamycin (mTOR) inhibitor, rapamycin, induces the proliferation and effector function of γδ T cells. Here, rapamycin increased BCG efficacy against both mouse and human bladder cancer in vivo in a γδ T cell–dependent manner. Thus, γδ T cells augment antitumor adaptive immune effects of BCG and support rapamycin as a promising approach to boost BCG efficacy in the treatment of non-muscle invasive bladder cancer.
Keywords: bladder cancer, mTOR, rapamycin, γδ T cells, immune system, BCG
INTRODUCTION
Bacillus Calmette-Guérin (BCG) treatment of bladder cancer was one of the first cancer immunotherapies to demonstrate significant benefit in a large clinical trial [1]. The first reported use of BCG in 1970 treated cutaneous melanoma [2], and in 1976, BCG treatment of bladder cancer was described [3]. BCG continues to be the standard-of-care treatment for preventing relapse and progression for patients with high-grade, non-muscle invasive bladder cancer (NMIBC) [1, 3, 4]. Nevertheless, BCG fails to completely eradicate disease in many patients [5], and there are no FDA-approved agents proven to boost BCG’s antitumor activity. Despite decades of use, the mechanisms of BCG activity are incompletely understood.
The importance of T cells in mediating BCG antitumor activity is supported by experimental models of bladder cancer showing lack of BCG efficacy in T cell–deficient mice [6] and following antibody depletion of T cells [7]. However, the role of antigen specificity in BCG’s antitumor activity is unclear. Conventional T cell–based cancer immunotherapy acts through induction of tumor-associated antigen-specific αβ T cells, which are activated by peptides derived from tumor proteins and presented by major histocompatibility (MHC) molecules on the surface of antigen-presenting cells [8, 9]. In BCG cancer immunotherapy, however, an alternative mechanism proposed is that BCG elicits tumor killing through induction of BCG antigen-specific T cells. In this regard, BCG organisms are endocytosed and processed by bladder tumor cells [10]. Then, BCG-derived peptides are bound to MHC molecules displayed on the tumor surface [10–13], and tumor destruction occurs via BCG-specific T cells [14]. Although this mechanism of antitumor activity is plausible during BCG treatment, direct evidence for BCG-specific antitumor responses in mediating BCG efficacy is lacking [15].
To address this knowledge gap, we developed an experimental model for studying antigen-specific T-cell immunity during BCG treatment of bladder cancer. We discovered that BCG mediates antitumor immunity through both tumor-specific and BCG-specific conventional αβ T cells in vivo. However, non-conventional γδ T cells were also required for antitumor activity of BCG antigen-specific T cells in our bladder cancer models. Unlike αβ T cells, recognition of antigen by γδ T cells is MHC unrestricted [16]. γδ T cells recognize a variety of ligands [17, 18], such as lipids, unprocessed tumor ligands, and small organic phosphate molecules, that are ubiquitous in nature and conserved across many biologic species, including in BCG [19, 20].
Given the contribution of γδ T cells towards BCG-mediated antigen-specific immunity, we hypothesized that activation of γδ T cells would boost BCG’s antitumor activity. Our group and others have shown that the mammalian target of rapamycin (mTOR) inhibitor, rapamycin, increases the proliferation and effector function of γδ T cells [21–24]. In patients with NMIBC, rapamycin enhances BCG-specific γδ T cells [25]. Translating γδ T-cell activity between species is complex because mouse and human γδ T cells react differently to mycobacterial antigens [26] and incompletely overlap in nomenclature and development [27]. Therefore, we studied effects of rapamycin on γδ T cells in both murine and human models. We discovered that rapamycin-mediated enhancement of γδ T-cell function improves BCG treatment efficacy in vivo against mouse and human bladder cancer pre-clinical models.
MATERIALS AND METHODS
Animals
All animal studies were conducted in accordance with, and with the approval of, an Institutional Animal Care and Use Committee (IACUC) at UT Health San Antonio. 6 – 10-week-old female wildtype (WT), Rag1 knockout (Rag1KO), TCRδ KO, female/male TCRαKO on C57BL/6 background, and female/male NSG mice were either purchased from The Jackson Laboratory, or bred under conventional (for WT) or specific pathogen free housing conditions as described and defined by IACUC protocols (20120040AR & 20150058AR). For all MB49 challenge experiments, unless in the absence of conventional αβ T cells (TCRαKO, no gender variation in results), to ensure uncompromised antitumor HY-antigen adaptive response, only females were used, including adoptive transfer studies. For immunodeficient NSG recipients (no significant gender variation), both genders were used to maximize usage of resource. Animals in each study were randomized prior to tumor challenge, and experiments were repeated independently for 2 – 3 times. Power analysis was used to estimate the minimum sample size required for each experiment or pooled experiments.
Mouse tumor model and cell lines
All cell lines were tested and confirmed free of mycoplasma by Mycoalert PLUS Detection Kit (Lonza Bioscience) during the study but were not genetically authenticated in the past year. Mouse bladder cancer cell line MB49 were generously given by Dr. Jeffrey Schlom at NIH in 2013 and maintained in complete DMEM (cDMEM-10) media (Fisher Scientific) with 10% fetal bovine serum (FBS, HyClone/Fisher Scientific), penicillin (100 IU/mL) plus streptomycin (100 μg/mL; Corning), and 2 mM of fresh L-glutamine (Corning). The last test for negative mycoplasma was in June 2018. Once thawed, MB49 cells were cultured for 2 passages (~48 h/passage) with ~70% confluency and injected subcutaneously (s.c.) into WT, Rag1KO, TCRδKO, and TCRαKO C57BL/6 mice at 0.2 × 106 cells in 100 μL of phosphate-buffered saline (PBS) per flank on both sides (lower back for initial challenge, upper back for re-challenge), on the lower back for initial MB49 challenge and upper back for tumor re-challenge at days 35 – 45. For the tumor re-challenge control, the mouse melanoma cell line B16-F10 (purchased from ATCC before 2015) was used. The last test for negative mycoplasma was in 2018. Once thawed, B16 cells were cultured for 2 passages (~48 h/passage) with ~70% confluency in complete RPMI-1640 media (Corning) with 5% FBS, penicillin (100 IU/mL) plus streptomycin (100 μg/mL), and 2 mM L-glutamine. 0.75 × 106 B16 cells in 150 μl of PBS were injected s.c. per flank in WT C57BL/6mice (upper back). Tumor volume was measured three times a week by a caliper and calculated using formula: volume = length x width x width/2 (mm3). Mice were monitored three times per week and daily whentumor volume of any single tumor was over 1500 mm3. Mice were euthanized when any tumor volume reached 2000 mm3. For immune studies, mice were euthanized for collection of tumor and tumor-draining lymph nodes (TDLNs) either within a week post-3rd BCG treatment, or whenever ~50% of tumors in control groups were over 1000 mm3.
BCG preparation
As described previously [30], live BCG.Connaught (Sanofi; for mouse) and BCG.TICE (Merck & Co; for human) were prepared and cryopreserved, with colony-forming units (CFU) determined after thawing representative vials. BCG vial(s) were thawed and pelleted at 4°C, 7000 rpm for 10 minutes prior to resuspension in PBS at the desired concentration for each treatment.
Treatment
BCG.Connaught treatment.
Starting on day (d)-5 post-MB49 tumor challenge, 1 × 106 CFU of BCG in 50 μL of PBS were injected into each s.c. tumor, weekly for 3 weeks.
CD4+ and CD8+ T-cell depletion.
200 μg of monoclonal antibody (mAb) from BioXcell, either anti-mouse CD4 (clone GK1.5), anti-mouse CD8 (clone YTS169.4), or rat IgG2b isotype mAb (clone LTF-2), were diluted in 100 μL PBS and injected intraperitoneally (i.p.) per mouse on d-1, 3, 7, and 11 post-MB49 tumor challenge.
γδ T-cell depletion.
75 μg of anti-mouse TCR γ/δ mAb (BioXcell, clone UC7–13D5) or Armenian Hamster polyclonal isotype IgG (BioXcell) were diluted in 100 μL of PBS and injected i.p. per mouse starting d-4 post-MB49 tumor challenge and every 3 – 4 days for 2 weeks.
Rapamycin.
Mice were injected i.p. with 0.075 mg/kg of rapamycin (LC laboratory, stock prepared in EtOH) or 100 μL vehicle control containing sterile 0.25% PEG-400 (Sigma-Aldrich) and 0.25% Tween-80 (Sigma-Aldrich) in water starting d-3 post-tumor challenge daily, Monday through Friday. For oral treatment, mice were placed on encapsulated rapamycin (eRapa chow; 14 parts per million/ppm; Rapamycin Holdings) or Eudragit control (Rapamycin Holdings) diet one week before tumor challenge and throughout the experiment, which contains a concentration of 14 mg/kg food and provides a ~2.24 mg of rapamycin/kg body weight/day. Dosing was justified [28, 29] and based on human to animal conversion, considering the human dose of 0.5 mg/day.
Antigen-specific T-cell adoptive transfer mouse model
As described previously [31], to prime antigen-specific T cells, female WT donor mice were immunized s.c. on top of each foot with 50 μL of Incomplete Freund’s Adjuvant (Thermo Scientific)-emulsified HY peptide (100 μg), OVA protein (100 μg) as indicated in Supplementary Table S1, or BCG.Connaught (2 × 106 CFU). On d-9, donors were sacrificed, and both inguinal and popliteal draining lymph nodes (DLNs) and spleens were harvested and placed in a 60 mm x 15 mm petri dish containing 10 mL of RPMI-1640 media. Both tissues were mashed with the back of a 10 mL syringe, and then passed through a 100-μm strainer to obtain single-cell suspension for cell counting (ViCell) prior to culture. Red blood cells (RBCs) were removed from splenocytes sterilely using hypotonic shock: loose cell pellet was resuspended in 9 mL of H2O by gentle shaking for 5 seconds, then immediately mixed with 1 mL of 10x PBS. Pooled DLNs cells or splenocytes from the same antigen group were cultured at 10 × 106 cells/mL in complete RPMI-1640 (cR-10) media with 10% FBS and 2 mM of L-glutamine plus HY peptide (10 μg/mL), OVA protein (10 μg/mL) or BCG [multiplicity of infection (MOI) of 0.1] in 6-well plates at 37°C with 5% CO2. ~50% of the supernatants were carefully removed and replaced with new media every 48 hours. After 5 days, antigen-recalled cells were harvested, re-counted, and plated at 5 × 106 cells/mL in cR-10 media containing recombinant mouse IL2 (10 ng/mL; BioLegend). ~30% of the supernatants were replaced with new media containing IL2 (10 ng/mL) every 72 hours. After 7 days, expanded DLN cells and splenocytes were pooled for magnetic enrichment by negative selection (BD, mouse T lymphocyte enrichment kit). ~5 × 106 donor T cells were injected intravenously or i.p. into TCRαKO or Rag1KO female recipients in 200 μL PBS per mouse one day prior to or after s.c. MB49 tumor challenge. After adoptive transfer, the remaining portion of donor T cells was quantified for antigen-specificity using cytokine ELISPOT assays.
Mouse antigen-specific ELISPOT assay
As described previously [24], 10,000 cells/well of magnetically enriched donor T cells were mixed with ~0.5 × 106 naïve WT female splenocytes (RBC lysed by hypotonic shock as described above) in cR-10 media containing antigen peptide (10 μg/mL; >98% purity) or protein (Supplementary Table S1): class II-restricted HY-Dby peptide, class I-restricted HY-Uty and OVA peptides, BCG-Ag85b protein, OVA protein, or BCG.Connaught (MOI 0.1), at 200 μL final volume/well on capture mAb-coated, BSA-blocked MultiScreen ImmunoSpot plates (EMD), incubated at 37°C with 5% CO2 for 24 hours. After incubation with detection mAbs and development, IFNγ-producing spot-forming cells (SFCs) were quantified using the ImmunoSpot Analyzer (S6 Micro, Cellular Technology) with ImmunoSpot Software, version 6 (Cellular Technology, RRID:SCR_011082).
Immune analysis for mouse model by flow cytometry
Tumor tissue and/or TDLNs were harvested from WT, Rag1KO, and TCRαKO C57BL/6 mice of s.c. MB49 tumor model. ≤0.5 g of tumor was minced and incubated with 3 mL of serum-free RPMI media containing collagenase IV (1.5 mg/mL; Sigma-Aldrich) and DNase I (0.25 mg/mL; Sigma-Aldrich) at 37°C with 5% CO2 for 1 hour with gentle shaking/mixing every 15 minutes. TDLNs or digested tumors were mashed with the back of a 10 mL syringe, and then passed through a 100-μm strainer to obtain single-cell suspensions for cell counting (ViCell) and flow cytometry staining using reagents listed in Supplementary Table S1. Samples and reagents were mixed thoroughly by gentle pipetting with large orifice tips prior to each incubation on 96-well U-bottom plate. Any remaining unstained portions were aliquoted and cryopreserved in freezing media freshly prepared each time by mixing cR-10 media with FBS containing 20% DMSO (Fisher Scientific) at 1:1 ratio. For surface staining, cells were resuspended in ice-cold flow buffer (2% FBS in PBS) at 1 × 106 cells/100 μL/well and incubated with 0.2 μg of mouse Fc-blocker on ice for 15 minutes, followed by incubation of fixable viability dye (FVD; 1:1000) mixed with 0.1–0.2 μg of fluorochrome-conjugated mAbs at 4°C, in the dark for 45 minutes. Samples were washed twice with up to 200 μL of flow buffer and resuspended in 150 μL of fixation buffer (2% paraformaldehyde in PBS) for at least 15 minutes or overnight at 4°C. Prior to analysis, samples were resuspended in flow buffer. For intracellular cytokine detection, single-cell suspensions were incubated with a cell activation cocktail (BioLegend) containing 0.08 μM of PMA (phorbol 12-myristate 13-acetate), 1.34 μM of ionomycin, and 5 μg/mL of Befeldin A in cR-10 at 1 × 106 cells/100 μL, 37°C for 5 hours prior to flow staining. After surface staining, cells were incubated with 100 μL of Cytofix/Cytoperm solution at 4°C for 20 minutes, followed by one wash with permeabilization buffer and incubation in 100 μL of permeabilization buffer with 0.2–0.3 μg of fluorochrome-conjugated mAbs at 4°C, in the dark for 45 minutes. Samples were washed once with up to 200 μL of permeabilization buffer and resuspended in 150 μL of flow buffer. All samples were run and analyzed on LSRII cytometer (BD) using FACSDiva software (Version 8.0.1). Gating strategies were described in the legends of each corresponding figures with flow dot-plot and/or histogram examples using tumor-infiltrating lymphocytes or TDLN cells as indicated. Each T cell subset (CD4+, CD8+, or γδ T cells) was gated under live (FVD−) CD45+CD3+ population for tumor sample analysis and gated under live (FVD−) CD3+ population for TDLN analysis if performed separately, e.g. verification of % of T cell subset after in antibody-depletion experiment. Fluorescence minus one (FMO) controls were included for the expression of any marker on any T-cell subset except for H-2Db-HY-Uty tetramer staining when an unspecific H-2Db-peptide negative control was used. Gating threshold of positive population was set to not more than 2 % in each FMO control.
Human PBMCs and tumor tissue processing and storage
Collection and use of clinical samples from bladder cancer patients were approved and abided by institutional review boards (IRB), protocol HSC2012–159H for bladder cancer repository, with informed written consent obtained prior to enrollment. The patient studies were conducted in accordance with Declaration of Helsinki ethical guidelines. Eligibility criteria for this repository included any patient with known or suspected bladder cancer. Blood, urine, lymph nodes, and tumors were collected from patients when available. Patients with available tumor tissue and blood upon enrollment, as well as with blood 3–6 months after, were selected for analyzing tumor-specific T-cell responses in vitro. For the No BCG group: two patients had muscle invasive bladder cancer and received systemic chemotherapy, and the other patient had low-grade NMIBC and did not receive BCG or chemotherapy. All three patients (No BCG) progressed and had cystectomy after 4–6 months when blood was collected again. For the BCG group: all three patients had high-grade NMIBC and were under BCG intravesical treatment, either induction phase (weekly for 6 weeks) or maintenance phase (weekly for 3 weeks). Blood was collected again when the patients returned to clinic for cystoscopy in 3–6 months. As described previously [30], peripheral blood mononuclear cells (PBMCs) were isolated from plasma-removed, non-coagulated whole blood by Ficoll-Paque (GE Healthcare) and cryopreserved in freshly prepared freezing media (as described above) at −150°C until analyzed. Surgically excised bladder tumors were transported on ice in RPMI medium containing 2% FBS and 1% penicillin-streptomycin. Tumor tissues were washed with PBS, minced, and incubated in 0.05% trypsin/EDTA (1 g of tissue per 5 mL) containing type IV collagenase (1 mg/mL; Sigma-Aldrich) and DNase I (0.25 mg/mL; Sigma-Aldrich) at 37°C, 100 rpm shaking for 1 hour. Digestion were stopped and washed with cR-10 media to obtain single-cell suspensions for cryopreservation in freshly prepared freezing media (as described above) at −150°C until analyzed.
Human tumor-specific responses in autologous patient PBMCs
Patient PBMCs were harvested at the time of tumor collection (baseline/enrollment) and at 3–6 months after. 1 × 106 cryopreserved PBMCs were thawed in cR-10 media and labeled with CFSE (Invitrogen, CellTrace), and then co-cultured with or without irradiated (25Gy, γ-irradiator) autologous tumor tissue single-cell suspensions (E:T ratio, 10:1) in cR-10 media supplemented with 10% human serum (type-AB, MP Biomedicals) at 37°C with 5% CO2 for 7 days, followed by 5 hours of re-stimulation with cell activation cocktail, flow cytometry staining using reagents listed in Supplementary Table S1 (following protocol described above), and analyzed on an LSRII cytometer using FACSDiva software (Version 8.0.1). CD4+ and CD8+ T cells were gated under FVD–CD3+ T cells. Proliferating T cells were gated as CFSElo based on medium only (without addition of irradiated autologous tumor tissue)/control sample. Gating of double-positive for IFNγ and TNFα, or IFNγ and CD107a were based on FMO control gating under CFSElo/– T cells. Total live cells were quantified before and after 7-day culture for calculating the number of proliferated/CFSElo T cells that were also positive for IFNγ and TNFα, or CD107a. Tumor responses were normalized/subtracted by media/background responses.
Establishing a patient-derived tumor cell line and model
A fresh tumor sample was processed into a single-cell suspension as described above. ~2 × 106 cells were mixed with Matrigel Matrix (Corning) at 1:1 and injected s.c. into NSG mice. Patient-derived xenografts (PDXs) were then monitored weekly for tumor growth. When tumors reached 400 mm3 in 2 months, it was resected and processed into single-cell suspensions for in vitro culture and expansion in cDMEM-10 media for approximately 2 weeks. The tumor cell line obtained (PDX257S) maintained urothelial carcinoma histology after in vivo passage and was confirmed positive for human EpCAM expression. This cell line was not tested for mycoplasma or genetically authenticated. For s.c. challenge, PDX257S cells were thawed and cultured in cDMEM-10 media for at least two passages (72 hours/passage) with ~80% confluency.
Adoptive transfer of expanded human γδ T cells from patient PBMCs
γδ T cells were expanded directly from autologous PBMCs of the PDX donor in vitro in cR-10 media with isopentenyl pyrophosphate (IPP) and IL2, with or without rapamycin (0.5 nM; LC laboratories) or live BCG.TICE (MOI 0.1) following previous protocols [22, 30]. On day 14 of culture, γδ T cells were harvested and mixed with PDX257S tumor cells at a 1:2 ratio and injected s.c. into NSG mice (1 × 106 tumor cells each site) on both flanks. Tumor growth was monitored and measured twice a week.
Immune analysis for human γδ T cells by flow cytometry
D14-expanded γδ T cells in replicates from the PDX donor PBMCs were resuspended in flow buffer at 0.5–1 × 106 cells/100 μL on 96-well U-bottom plate and either stained directly with 0.2 μg of fluorochrome-conjugated mAbs plus FVD (1:1000) at 4°C for 45 minutes in the dark after incubated with 5 μL of human Fc-blocker/TruStain (10 minutes on ice), or stimulated first with cell activation cocktail for 5 hours followed by intracellular cytokine staining using reagents listed in Supplementary Table S1 (in similar steps as described above) and analyzed on an LSRII using FACSDiva software (Version 8.0.1). Gating strategy was described in each corresponding figure legend with example of flow cytometry dot plot and/or histogram. Remaining unstained portions were cryopreserved in freshly prepared freezing media (as described above).
Statistics
Two-sided testing was performed to improve rigor by removing bias in assumptions about the direction of treatment effect. Multiple corrections were performed by Tukey’s (all pairs), Dunnett’s (comparison over control group, >one variable), or Dunn’s (comparison over control or selected groups, one variable) multiple comparison test, except for comparing mean plus SEM (when N > 100, hence Bonferroni’s multiple comparison). For analysis with one variable, the D’Agostino & Pearson omnibus normality test was used, and if p > 0.05, then a parametric t-test (two groups) or 1-way ANOVA with multiple comparison test (> two groups) was used; if p < 0.05, then a non-parametric Mann-Whitney test (two groups) or Kruskal-Wallis test with multiple comparison test (> two groups) was used. When not all groups passed normality testing: if more than half passed normality test, a parametric test was used; otherwise, a non-parametric test was used; for any group with n ≤ 7 (N too small), unless there were substantial outliers in the datasets (non-parametric distribution), it was considered to pass the normality test.
For analysis with more than one variable, interaction tests were used to identify treatment-specific outcome trajectories over time. Two-way ANOVA (two groups) or repeated measures ANOVA (> two groups) with multiple comparison test were used in tumor volume growth curves, except when two sets of pairs started from different time points (initial vs. rechallenge) or inner strain comparison occurred involving two different genotypes of mouse pairs. In those cases, repeated measures ANOVA could not be run and a two-way ANOVA was used. Differences over time in BCG versus non-BCG treated patients for tumor immunity were assessed with two-sided testing based on the group (baseline, 3–6 months) by condition (no BCG, BCG) interaction term in a linear model. Survival curves/Kaplan-Meier plots were assessed by Log-rank test. P values are two-sided and *, p < 0.05 was considered statistically significant. Statistical analyses were performed with GraphPad Prism 5–6 or Stata IC/10.1 (GraphPad Prism, RRID:SCR_002798).
RESULTS
BCG boosts bladder tumor-specific memory
To determine if BCG increased tumor-specific immunity in patients with bladder cancer, we measured the frequency of tumor antigen-specific T cells (i.e., T cells that proliferated and produced IFNγ and TNFα in response to irradiated autologous bladder tumor antigens, Supplementary Fig. S1) in peripheral blood collected from bladder cancer patients before and after intravesical BCG. At 3–6 months after BCG administration, we observed a significant increase over baseline in the number of circulating tumor-specific T cells in patients who received BCG compared to patients who did not receive BCG (Fig. 1A–C). BCG-mediated induction of tumor-specific antitumor immunity should provide long-term protection against subsequent tumor growth. To test this concept, we used MB49 tumor cells transplanted subcutaneously (s.c.) into C57BL/6 WT female mice, an established model for studying bladder cancer immunopathology [32] and immunotherapy [33], including BCG [14]. In MB49-challenged mice treated with BCG, an increase in tumor-specific immunity was observed, detected as an increase in tumor antigen-specific T cells (i.e., H-Y, male-derived tumor antigen-specific) infiltrating tumor tissue (Fig. 1D). Mice that experienced complete MB49 tumor regression after BCG treatment were protected against tumor growth during MB49 rechallenge (Fig. 1E) but were not protected against B16 melanoma tumor growth (Supplementary Fig. S2), consistent with BCG-mediated induction of tumor-specific antitumor immunity [15]. To test if BCG-induced antitumor protective immunity required αβ T cells, we repeated experiments as in Figure 1E but depleted CD4+ and/or CD8+ T cells during the MB49 rechallenge phase. Mice were still protected or partially protected against subsequent tumor challenge with depletion of CD4+ or CD8+ T cells alone but not with depletion of both (Fig. 1F), indicating that both CD4+ and CD8+ T cells contribute to BCG-mediated tumor-specific antitumor immunity in our model. Depletion of CD4+ and/or CD8+ T cells was confirmed in TDLNs at the end of study (Supplementary Fig. S3A–C). We also observed an increase in the percentage of γδ T cells in TDLNs of mice depleted of all conventional αβ T cells (Supplementary Fig. S3B, right graph).
Figure 1. BCG treatment increases tumor-specific immunity in patients and anti-tumor protection in a mouse model.
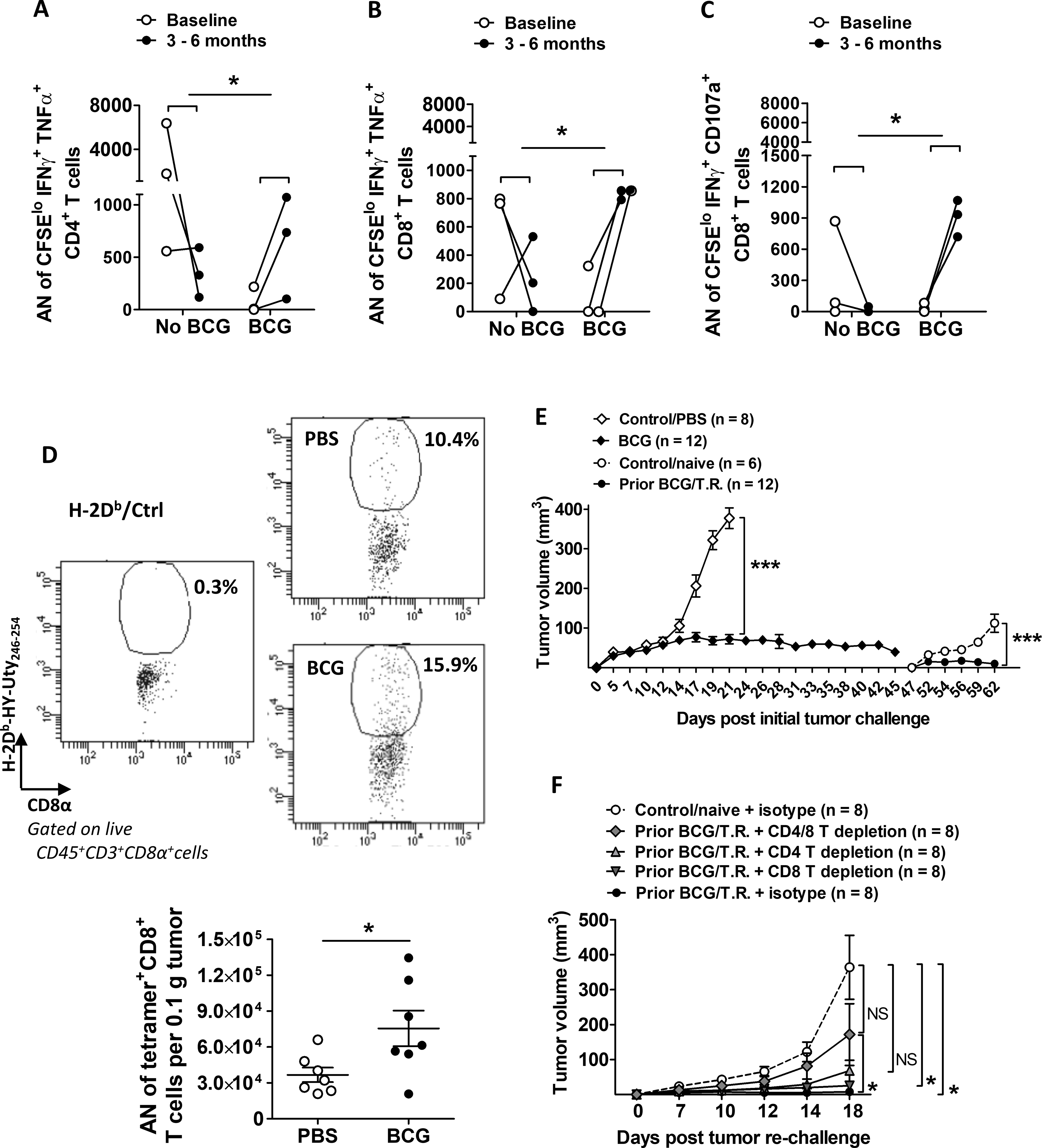
(A-C) As described in Materials and Methods, patient PBMCs harvested from baseline and at 3–6 months post BCG treatment were measured for tumor-specific responses. Paired samples indicated with lines. Shown are absolute number (AN) of (A) proliferating CD4+ or (B-C) proliferating CD8+ T cells co-expressing IFNγ and TNFα or CD107a. (n=3 patients per group). P value indicates the difference in changes between baseline and 3–6 months assessed with two-sided testing on the group (baseline, 3–6 months) by condition (no BCG, BCG) interaction term in a linear model. (D-F) WT female C57BL/6 mice were challenged with s.c. MB49 tumors and treated with BCG intratumorally weekly for 3 weeks starting on day 5. For some experiments, (D) mice were sacrificed around day 25 when tumors were harvested for immune analysis mainly for H-2Db-HY-Uty tetramer staining on CD8+ T cells (gated under FVD−CD45+CD3+CD8+ cells) using an unspecific-peptide loaded tetramer (H-2Db/Ctrl) as negative control (top panel, examples of flow dot plots for gating strategy). AN of tetramer+ CD8+ T cells per 0.1 g of tumor tissue (n=7 tumors per group) were calculated accordingly (bottom graph). Shown is representative of 3 independent experiments. Mean±SEM. Each dot represents one tumor. t-test was applied accordingly after normality test was run. (E), BCG groups (gray diamonds) compared to PBS control (open diamonds). After 45 days mice with prior BCG treatment and regressed tumors were re-challenged with s.c. MB49 cells versus a cohort of naïve mice (open circles). Shown is representative of 3 independent experiments (n=number of tumors). Two-way ANOVA for comparison of control versus treatment groups at either initial or secondary tumor challenge. Mean±SEM. (F) Used prior BCG treated/tumor regressed mice, as in (E), with or without CD4 and/or CD8 T-cell depletion or isotype antibodies. Shown is representative of 2 independent experiments (n = number of tumors). Repeated measures ANOVA with Dunnett’s multiple comparison test. Mean±SEM. T.R.=tumor regressed. *, p<0.05; ***, p<0.001; NS, not significant.
BCG promotes tumor antigen-specific T cells
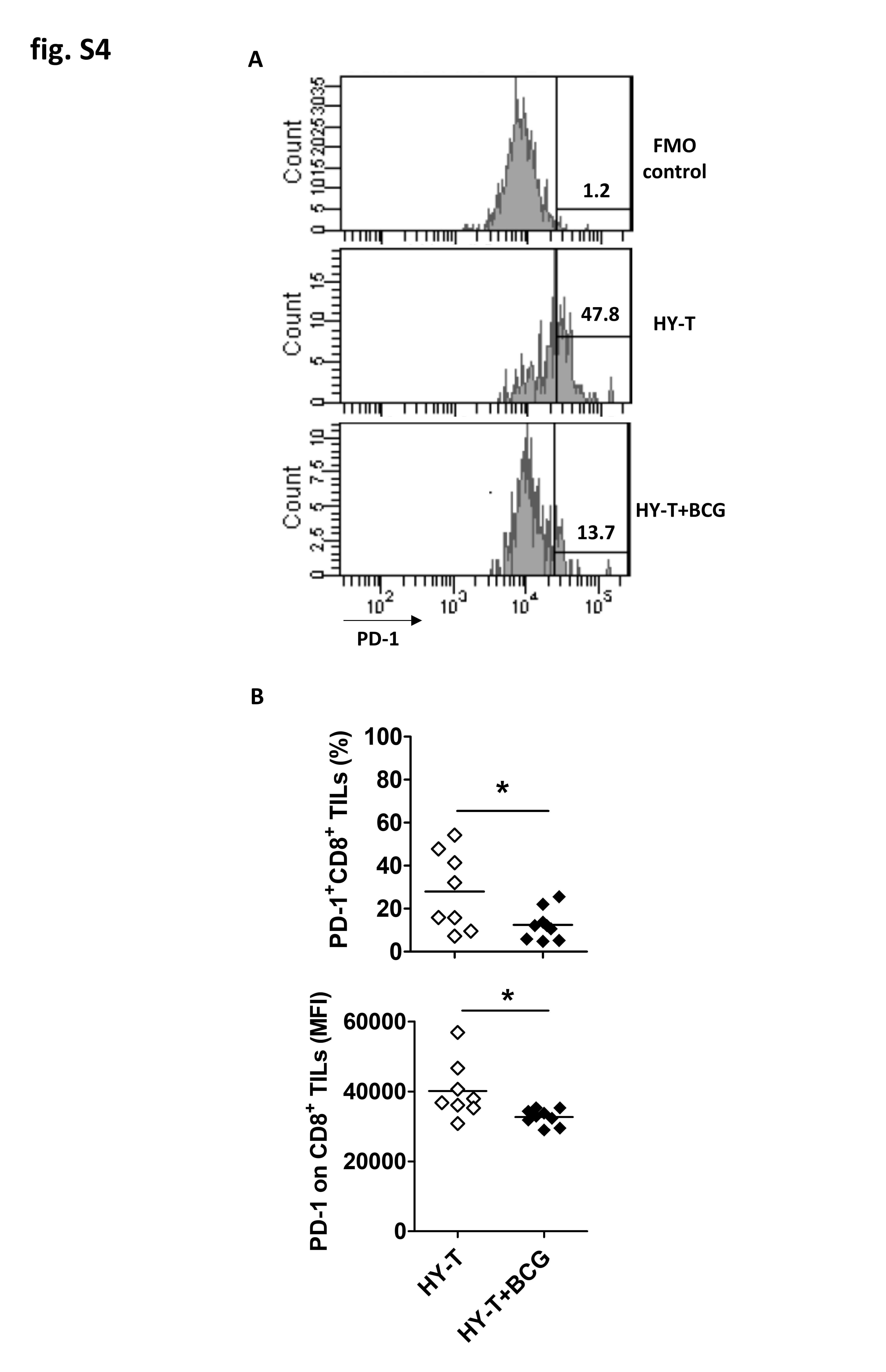
Prior studies propose that BCG treats bladder cancer through BCG-specific T cells that attack tumor cells expressing BCG antigens [10–13]. In support of this, BCG vaccination improves subsequent antitumor responses to intravesical BCG in mice [14], a strategy currently being tested in humans [30, 34]. Nevertheless, these data do not prove a BCG-specific immune mechanism explaining BCG treatment efficacy. To evaluate BCG-specific versus tumor-specific T-cell contributions toward BCG antitumor efficacy directly, we utilized a wide repertoire of antigens directly from live BCG, the male-HY antigen present on MB49 cells, and the non-tumor OVA protein to generate BCG-, tumor (HY)-, and control non-tumor OVA antigen-specific T cells, respectively, for adoptive transfer experiments (Fig. 2A). ELISPOTs confirmed high frequency and specificity of antigen-specific T cells of each type (Fig. 2B). Following adoptive transfer of T cells into Rag1KO mice, which lack all T cells, mice were challenged with s.c. MB49 and treated with intratumoral (i.t.) BCG or control (PBS). Compared to control treated tumors, tumors treated with BCG had higher numbers of BCG-specific tumor-infiltrating CD4+ T cells (Fig. 2C). BCG treatment also reduced the frequency and amount of PD-1 expressed on tumor-infiltrating tumor HY-specific CD8+ T cells (Supplementary Fig. S4). These data suggest that BCG can boost both BCG- and MB49 tumor antigen-specific T cells, by either promoting trafficking into the tumor, proliferation, or reinvigoration/reactivation inside the tumor microenvironment. BCG reduced tumor growth following adoptive transfer of tumor HY-specific T cells (Fig. 2D) but had no significant effect on tumor growth after adoptive transfer of BCG-specific (Fig. 2E) or non-tumor antigen-specific T cells (Fig. 2F). These findings indicate that BCG mediates antitumor activity, at least in part, by boosting tumor-specific T cells. However, the effect of BCG on tumor-specific T-cell control of tumor growth was modest (Fig. 2D) compared to the effects observed in WT mice (Fig. 1E, open vs. filled diamonds), and transfer of tumor-specific T cells alone did not reduce tumor growth compared with transfer of non-tumor-specific T cells (Fig. 2G), suggesting that additional immune cell subsets, lacking in Rag1KO mice, contributed to BCG-mediated antitumor immunity and/or facilitated tumor-specific T-cell efficacy.
Figure 2. BCG treatment increases tumor-infiltrating, adoptively transferred tumor- or BCG-specific T cells in MB49-bearing Rag1KO mice, but only tumor-specific T cells are protective.
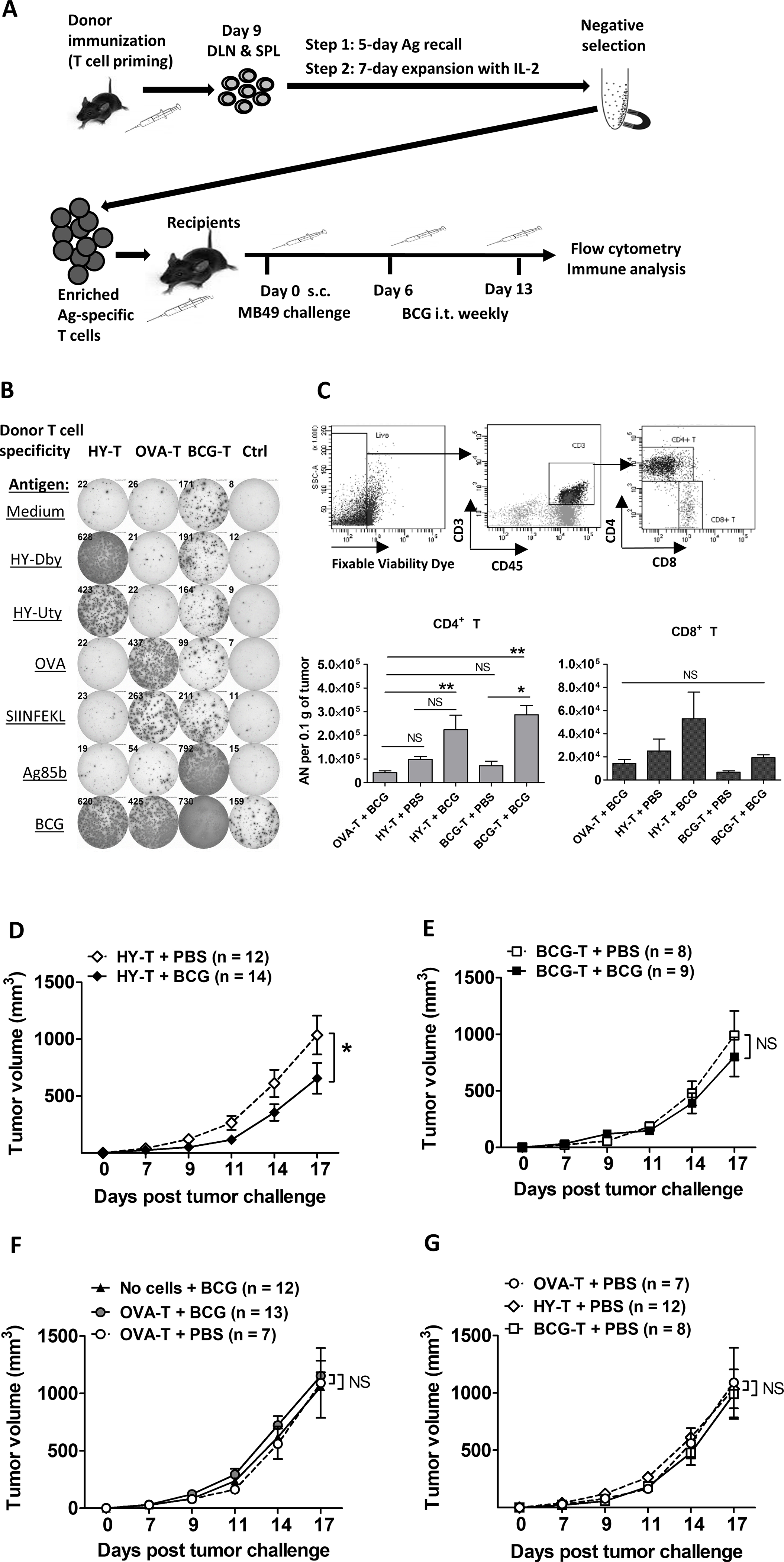
(A) Generation and expansion of antigen (Ag)-specific T cells from donor mice by vaccination and in vitro Ag re-stimulation followed by IL2 prior to adoptive transfer. After MB49 tumor challenge, BCG treatment was given intratumorally (i.t.) weekly starting day 6 in recipients. After day 17, recipients were sacrificed, and tumors and TDLNs were processed for flow cytometry analysis. DLN, draining lymph nodes; SPL, splenocytes. (B) Ag specificity of enriched donor T cells were verified and quantified on the same day of adoptive transfer by mouse IFNγ ELISPOTs. Ctrl, naive splenocytes alone as an ELISPOT control but not transferred to recipient mice. Shown is representative of 3 independent experiments. (C) Flow cytometry for CD4+ and CD8+ tumor-infiltrating T cells in Rag1KO recipients. Top, gating strategy based on TDLNs. Shown are results pooled from 2 independent experiments (n=4 – 9 tumors per group). One-way ANOVA with Tukey’s multiple comparison test was applied according to normality testing. (D-G) s.c. tumor growth in Rag1KO recipients (shown are pooled from 3 independent experiments, n=7–14 tumors per group). For 3 groups, repeated measures ANOVA with Dunnett’s multiple comparison test. For 2 groups, two-way ANOVA. Mean±SEM. *, p<0.05; **, p<0.01; NS, not significant.
γδ T cells contribute to BCG-mediated antitumor immunity in bladder cancer
We hypothesized that γδ T cells are required for CD4+ and CD8+ αβ T cell-mediated BCG antitumor effects because γδ T cells rapidly expand and develop a memory-type response during BCG infection [35], as well as provide functional help for BCG-specific αβ T cells during BCG vaccination against tuberculosis [20]. To test this hypothesis, we studied BCG treatment of MB49 tumor growth in TCRαKO mice (lack conventional αβ T cells [36, 37]) and TCRδKO mice (lack unconventional γδ T cells). MB49 tumors grew faster in TCRαKO compared to TCRδKO mice (Fig. 3A), regardless of BCG treatment, consistent with stronger antitumor protection of combined CD4+ and CD8+ αβ T cells versus γδ T cells. In the absence of γδ T cells, BCG had no discernable treatment effect (Fig. 3A, triangles), consistent with a prior report [38]. However, in the absence of αβ T cells, BCG still significantly suppressed MB49 tumor growth (Fig. 3A, circles). BCG had no direct cytotoxicity against MB49 in vitro and depleting γδ T cells with specific antibodies abolished the efficacy of BCG in TCRαKO mice, consistent with the contribution of γδ T cells towards BCG’s antitumor efficacy (Fig. 3B). To determine if γδ T cells facilitated antigen-specific immunity during BCG therapy, we adoptively transferred antigen-specific CD4+ and CD8+ T cells into TCRαKO mice, which restored BCG efficacy in reducing tumor growth using either tumor-specific (Fig. 3C) or BCG-specific T cells (Fig. 3D), but not with OVA-specific T cells (Fig. 3E). Adoptive transfer of tumor antigen-specific αβ T cells significantly reduced tumor growth compared with transfer of non-tumor specific T cells (Fig. 3F). Collectively, these data support a role for γδ T cells in mediating BCG antitumor immunity both directly (i.e., without αβ T cells) and indirectly through supporting effects for both tumor- and BCG-specific αβ T cells. These data also show that BCG mediates antitumor effects, in part, through BCG-specific T cells.
Figure 3. BCG treatment efficacy and antigen-specific immunity boost requires γδ T cells.
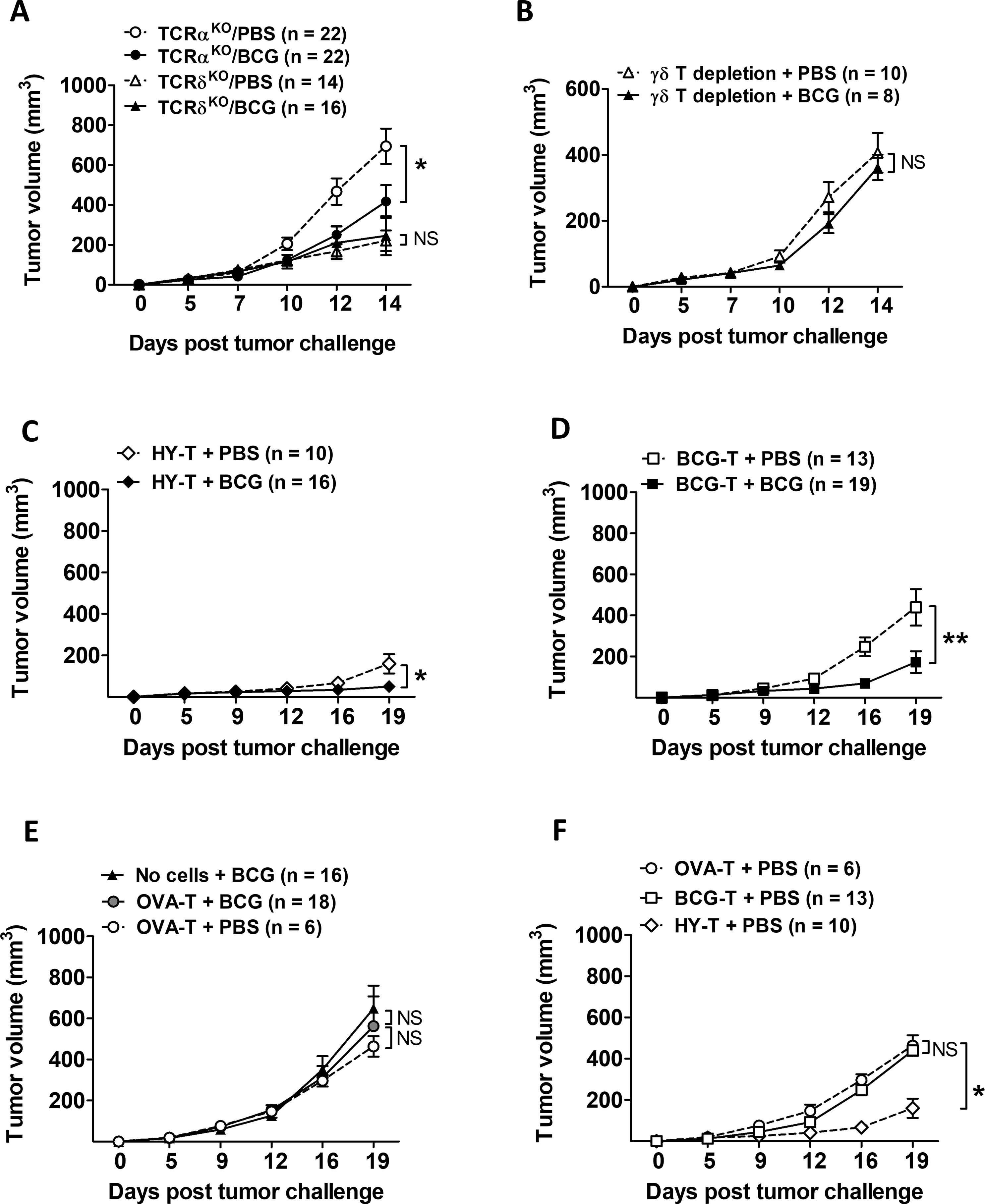
(A) TCRαKO and TCRδKO mice (pooled from 2 – 3 independent similar experiments, n=number of tumors per group), or (B) TCRαKO mice with depletion of γδ T cells (representative of 2 similar experiments, n=number of tumors per group) were challenged with s.c. MB49 cells and treated with intratumoral BCG. Two-way ANOVA. Mean±SEM. (C-F) Adoptive transfers as described in Fig. 2A but using TCRαKO mice (pooled from 3 independent experiments, n=number of tumors per group). For 3 groups, repeated measures ANOVA with Dunnett’s multiple comparison test. For 2 groups, two-way ANOVA. Mean±SEM. *, p<0.05; **, p<0.01; NS, not significant.
Rapamycin boosts γδ T-cell activity to improve BCG antitumor immunity
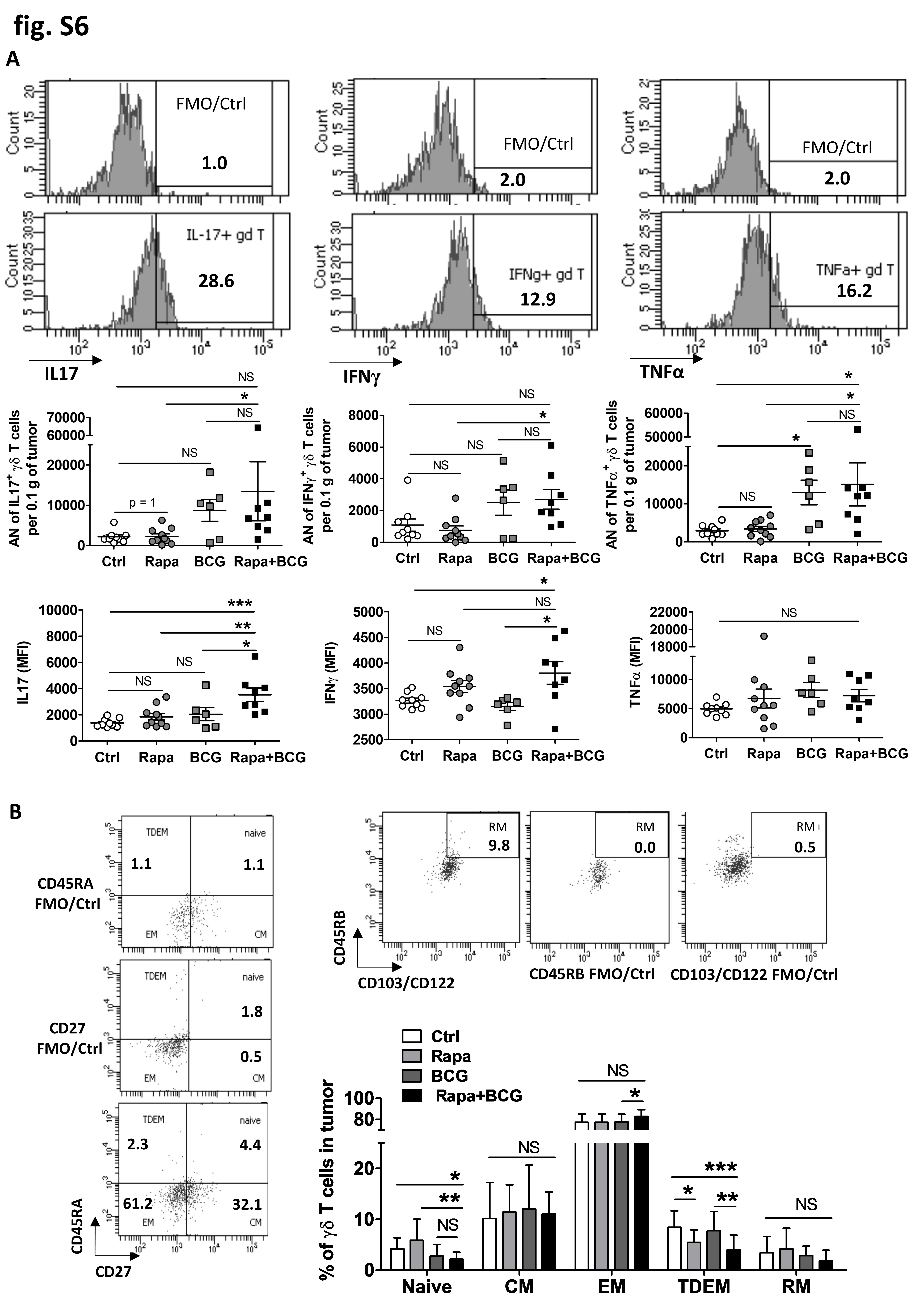
Given the role of γδ T cells in BCG-mediated antitumor responses, we questioned if improving γδ T cell function would boost BCG efficacy. The mTOR inhibitor, rapamycin, enhances γδ T cell function and antitumor immunity in mice [22] and patients with bladder cancer [25]. To test if rapamycin could augment BCG-mediated suppression of MB49 tumor growth, WT mice were challenged with s.c. MB49 and treated with ± i.t. BCG and ± intraperitoneal rapamycin. BCG alone treated MB49, but their combination further decreased tumor growth (Fig. 4A). Similarly, BCG combined with oral encapsulated rapamycin (eRapa) was effective at slowing MB49 tumor growth (Supplementary Fig. S5). To determine if the effects of the rapamycin/BCG combination therapy on MB49 tumor growth were mediated through γδ T cells, we repeated experiments in TCRαKO mice, which lack CD4+ and CD8+ T cells but retain γδ T cells. In TCRαKO mice challenged with MB49, rapamycin plus BCG significantly decreased tumor growth versus control (Fig. 4B), and this effect was abolished by γδ T-cell depletion (Fig. 4C). In TCRαKO mice, rapamycin had no discernable effect on the number of tumor-infiltrating γδ T cells (Fig. 4D–E) nor tumor-infiltrating γδ T cells producing IFNγ, TNFα, or IL17 (Supplementary Fig. S6A). However, rapamycin significantly increased surface expression of γδ TCR and activation markers CD44 and CD69 on tumor-infiltrating γδ T cells in TCRαKO mice receiving BCG (Fig. 4F–H). Rapamycin also increased production of IFNγ within γδ T cells, measured by fluorescence intensity (Supplementary Fig. S6A, middle-bottom graph). In these tumors, the majority (~80%) of tumor-infiltrating γδ T cells are effector memory (EM) cells. In mice treated with BCG, rapamycin increased the percentage of EM γδ T cells within tumors (Supplementary Fig. S6B). Conversely, combination of rapamycin and BCG decreased the percentage of both naïve and terminally differentiated effector memory (TDEM) γδ T cells in the tumor compared with control (Supplementary Fig. S6B). These data support a favorable antitumor effect of rapamycin during BCG treatment of MB49 bladder tumors, which requires EM γδ T cells.
Figure 4. Rapamycin improves γδ T-cell activity to boost BCG-mediated antitumor immunity in mice.
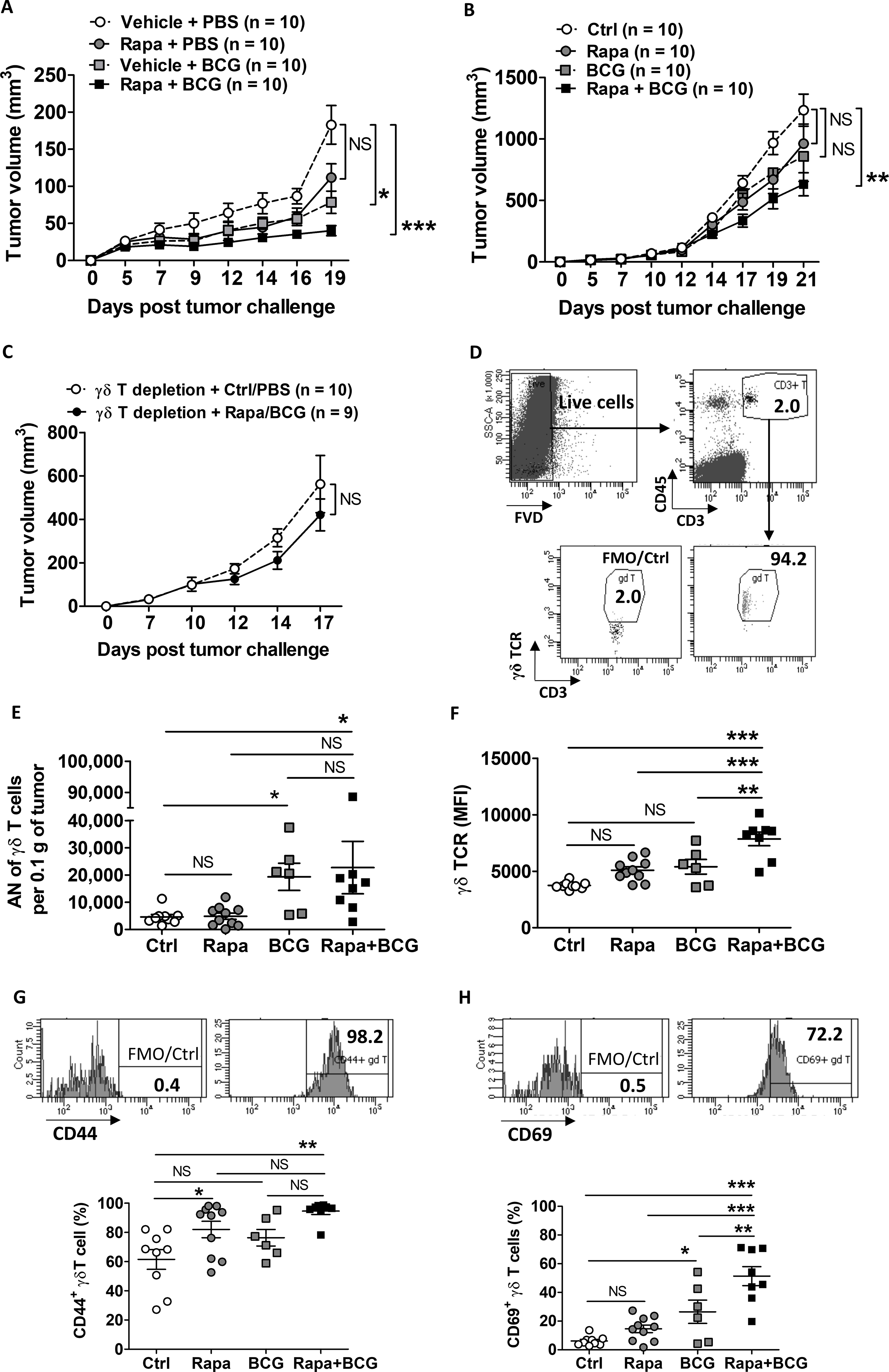
(A) WT mice challenged s.c. with MB49 and treated with rapamycin, BCG, or both starting on day 5. Shown is representative from 2 similar experiments (n=number of tumors per group). Repeated measures ANOVA with Dunnett’s multiple comparison test. Mean±SEM. (B-C) TCRαKO mice challenged s.c. with MB49 and treated with rapamycin, BCG, or with γδ T-cell depletion (representative of 2–3 independent experiments, n=number of tumors per group). (B) Repeated measures ANOVA with Dunnett’s multiple comparison test. Mean±SEM. (C) Two-way ANOVA. Mean±SEM. (D-H) TCRαKO mice treated as in (B) were sacrificed on day 18 for flow cytometry analysis of available tumors (n=6 – 10 tumors per group). Gating strategies of tumor-infiltrating γδ T cells are shown in (D) as FVD−CD45+CD3+ γδ TCR+ (top panel) and in (G-H) for cell surface expression of CD44 and CD69 with FMO control (top panels). (E) Absolute number (AN) of γδ T cells per 0.1 g of tumor tissue was calculated based on the percentage of γδ T cells out of total live tumor single-cell suspensions. (F) Mean fluorescence intensity (MFI) is shown to indicate γδ TCR expression level. (G-H) Frequency of activation marker (CD44 and CD69) expression on γδ T cells (bottom graphs). Shown are representative of 2 independent experiments. One-way ANOVA with Tukey’s or Dunn’s multiple comparison test was applied according to normality testing. Mean±SEM. *, p<0.05; **, p<0.01; ***, p<0.001; NS, not significant.
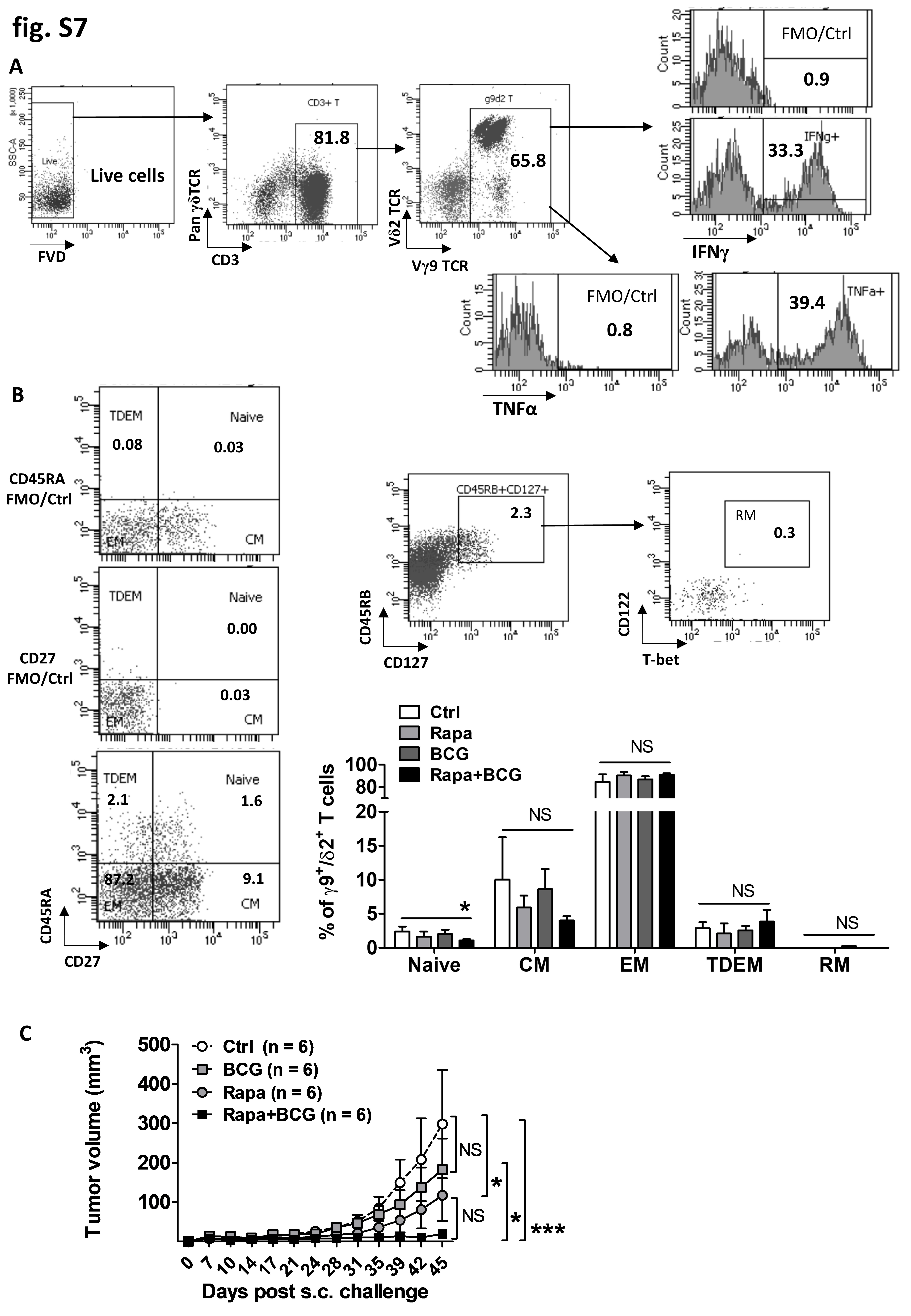
To test rapamycin’s effects on human γδ T cell-mediated antitumor activity, human peripheral blood γδ T cells (the majority were γ9+/δ2+) were expanded in vitro with IL2 and IPP as described [22] and cocultured with/without rapamycin and with/without BCG. After 14 days in culture, both rapamycin and BCG alone increased the number of expanded γ9+/δ2+ T cells, and the combination of rapamycin plus BCG yielded the highest number of γ9+/δ2+ T cells (Fig. 5A). Each agent alone and combination of rapamycin plus BCG had minimal effects on the percentage of IFNγ- or TNFα-producing γδ T cells (Fig. 5B), nonetheless, significantly increased the production of IFNγ and TNFα within γδ T cells, as estimated by mean fluorescence intensity (Fig. 5C, Supplementary Fig. S7A). Consistent with other study for in vitro-expanded γδ T cells from human PBMCs [39], the majority of γ9+/δ2+ T were EM cells (75–95%), regardless of treatment (Supplementary Fig.S7B). Rather than altering the profile/percentages of differentiated γ9+/δ2+ T cells, rapamycin significantly increased the absolute number (AN) of EM γ9+/δ2+ T cells compared to control with only IL2 and IPP in the culture. Combining rapamycin and BCG treatment yielded the lowest percentage of naïve γ9+/δ2+ T cells (Supplementary Fig. S7B) and highest numbers of EM γ9+/δ2+ T cells (Fig. 5D). To investigate rapamycin effects on γδ T cell-mediated human bladder cancer killing in vivo, cultured autologous γδ T cells were comixed in a 1:2 ratio with PDX257S human bladder cancer cells before s.c. injection into NOD/SCID/IL2Rγ-deficient mice that received no further treatment after tumor challenge as described [22]. γδ T cells cocultured with BCG did not inhibit PDX257S tumor growth, whereas rapamycin inhibited tumor growth moderately (Supplementary Fig. S7C); however, neither agent could improve survival (Fig. 5E). γδ T cells cocultured with BCG plus rapamycin significantly decreased PDX257S tumor growth and improved survival in recipient mice compared to mice receiving control γδ T cells or BCG-treated γδ T cells (Supplementary Fig. S7C, Fig. 5E). Taken together, these data support a beneficial effect of rapamycin in combination with BCG to treat human bladder cancer through γδ T cells.
Figure 5. Rapamycin improves γδ T-cell activity to boost BCG-mediated antitumor immunity against human bladder cancer cells.
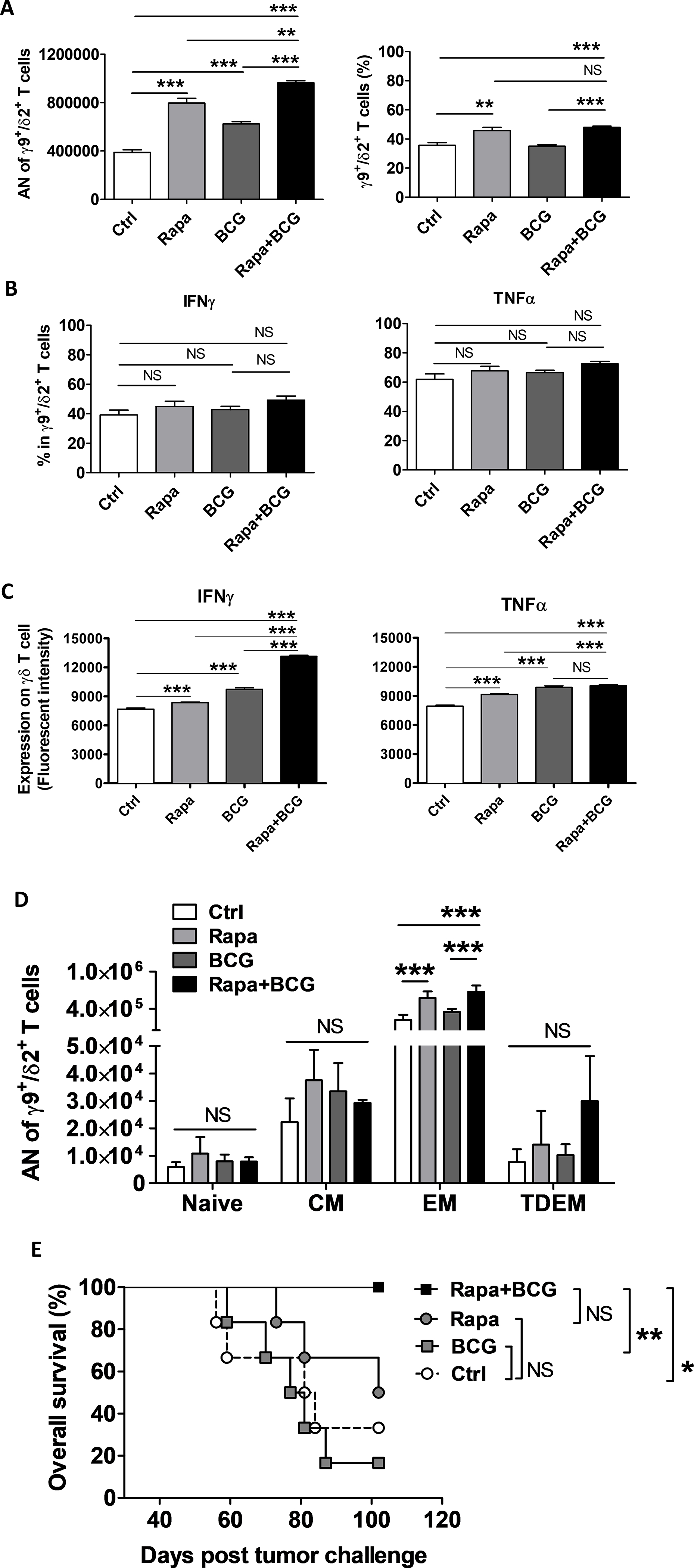
Autologous human PBMCs from the patient-derived xenograft (PDX257S) donor were expanded in vitro for γδ T cells for 14 days, and then co-cultured with rapamycin, BCG, or both in vitro, followed by quantification and flow cytometry analysis. (A) Absolution number (AN) of expanded total live CD3+ γδ T cells (mostly γ9+/δ2+ T cells) from 0.25 × 106 PBMCs and frequency. (B) Frequency of IFNγ and TNFα expression or (C) production (fluorescent intensity) in γ9+/δ2+ T cells after 5 hours of stimulation with cell activation cocktail (see Methods). (D) AN of differentiated γ9+/δ2+ T expanded from 0.25 ×106 PBMCs in d-14 culture (n=4 replicated samples per treatment group from one representative experiment). Gating strategies for differentiated γδ T cells are as follows: naïve, CD45RA+CD27+; central memory (CM), CD45RA−CD27+; effector memory (EM), CD45RA−CD27−; terminally differentiated effector memory (TDEM), CD45RA+CD27−. (A–C) Shown are representative of 2 similar experiments, in quadruplicates for each treatment condition. (A–B) One-way ANOVA with Tukey’s multiple comparison test. Mean±SEM. (C) One-way ANOVA with Bonferroni’s multiple comparison test was applied using Mean, SEM and N of each group. (D) One-way ANOVA with Tukey’s multiple comparison after normality test. Mean±SEM. (E) NSG mice were s.c. challenged with human PDX257S bladder cancer cells mixed with autologous PBMC-derived γδ T cells. Tumor volume > 1500 mm3 was used as survival cut off. Kaplan-Meier plot with log-rank test. Shown is the representative of 2 independent experiments (n=6 tumors per group). *, p<0.05; **, p<0.01; ***, p<0.001; NS, not significant.
DISCUSSION
Although the efficacy of BCG in reducing the risk of bladder cancer relapse [1] and progression [40] has been known for decades, BCG antitumor mechanisms remain incompletely defined. Most T cell-based cancer immunotherapies mediate tumor killing through tumor-specific T cells, but the role of tumor-specific immunity in BCG’s activity is not established. The first experiments to address BCG’s antitumor mechanisms used intradermal hepatocarcinoma line 10 (L-10) in guinea pigs, animals in which BCG is highly immunogenic. BCG can induce tumor immunity in weakly antigenic tumors if tumor cells are injected concurrently with live BCG [41] and shows evidence for eliciting protective antitumor immunity [42]. BCG also mediates protective antitumor immunity in MBT-2 mouse bladder tumors [43]. In both L-10 and MBT-2 tumor experiments, however, it is not clear if BCG-mediated protection against cancer growth is tumor-specific or non-specific, such as through priming or activation of T cells reactive to non-tumor peptides. Other studies have been unable to demonstrate tumor-specific immunity during BCG treatment of bladder cancer [7, 44].
Using MHC class I tetramers, we demonstrated that BCG increased tumor-antigen specific T cells in both tumor and tumor-draining lymph nodes during bladder cancer treatment. Successful treatment of bladder cancer by BCG induced protective tumor-specific immune memory against subsequent bladder tumor rechallenge. BCG was effective in treating bladder tumors in mice bearing tumor-specific or BCG-specific T cells but not in mice bearing non-tumor-specific T cells. These data support antigen-specific mechanisms underlying BCG’s antitumor activity, but with only a modest contribution towards BCG’s treatment efficacy because BCG-specific T cells can reduce tumor growth with the presence of live BCG. Although a subset of patients with bladder cancer derive long-term benefit from six weekly BCG instillations (i.e., BCG induction) [1], maintenance BCG with repeated instillations over three years is needed for long-term control [4]. BCG efficacy is contact-dependent, with little activity on distant urothelial sites [7]. These data are not consistent with BCG generating effective long-term systemic tumor-specific immunity. The improved efficacy provided by maintenance BCG could be explained by boosting poorly durable tumor-specific immunity, or by continually activating non-specific immune mechanisms.
A non-tumor-specific T-cell mechanism proposed for BCG-mediated efficacy is the activation of BCG-specific T cells that eliminate tumors expressing BCG-derived antigens, which could occur by BCG internalization into bladder tumors [45–47]. The concept of BCG-specific immunity boosting antitumor responses is supported by animal studies showing that intradermal BCG vaccination in mice boosts the antitumor activity of intravesical BCG and accelerates infiltration of T cells into the bladder in bladder cancer [14]. This concept is currently being tested in humans [34]. We found direct evidence that BCG-specific T cells can contribute to BCG-mediated antitumor activity in mice by using the full repertoire of antigen-specific T cells against live BCG, providing more supportive data compared to studies using antigen-specific T cells against a single antigen [15]. It is not clear if BCG uptake occurs equally in normal and malignant urothelium. Understanding mechanisms for BCG uptake by urothelial cells is needed and could identify approaches to enhance selective BCG uptake by transformed cells.
We validated prior findings that γδ T cells are indispensable for eliciting BCG treatment efficacy against bladder cancer in mice [38] and our previous study showing the utility of targeting γδ T cells in bladder with other immunotherapy approaches [48]. Our data support both direct and indirect antitumor effects of γδ T cells, with the latter through antigen-specific αβ T cells. The mechanisms by which γδ T cells support conventional T cells during BCG therapy is not fully clear and requires further investigation. Upon activation, γδ T cells can promote the maturation and function of dendritic cells (DCs) to enhance αβ T-cell responses [49], or can be induced to take up and process microbial and tumor soluble proteins, thereby serving as professional antigen-presenting cells [50, 51]. Activated γδ T cells are also capable of cross-priming, in which soluble proteins are taken up and processed for presentation on MHC class I molecules for CD8+ T-cell activation [52]. One report found that γδ T cells were more effective as antigen-presenting cells than DCs in inducing CD8+ T-cell proliferation, cytokine production, and target cell killing [52], but we are unaware of whether they can mediate T-cell priming. We observed an increase amount of MHC class I expression per γδ T cell during BCG therapy at later stages of tumor growth, suggesting that BCG could enhance their ability to present antigens as tumors progressed, but further work is required to understand how γδ T cells cooperate with conventional T cells to mediate antigen-specific immunity, especially in the context of combining rapamycin and BCG treatment.
The direct anti-tumor activity of γδ T cells correlates with their differentiation status, i.e. ability to migrate, proliferate, and produce effector cytokines. Central memory (CM) cells are highly proliferative with low IFNγ production, whereas EM cells are less proliferative but can produce more IFNγ [53–56]. Consistent with studies of profiling either in vitro PBMC-expanded or tumor-infiltrating γδ T cells from cancer patients [39, 57], we found that the majority of γδ T cells in mouse MB49 tumors or in vitro-expanded human γ9+/δ2+ T cells were largely EM cells. As expected, resident memory (RM) γδ T cells were infrequent among mouse tumor infiltrates (≤ 5%) and rare among in vitro-expanded human PBMCs (≤ 0.01%). In mouse tumors, rapamycin increased the percentage of EM cells during BCG therapy and decreased TDEM and naïve γδ T cells with or without the treatment of BCG, respectively, which supports favorable effects of rapamycin on γδ T-cell anti-tumor function. In contrast, for human in vitro-expanded γ9+/δ2+ T cells, rapamycin did not alter the overall differentiation status/profile of γδ T cells, except for inducing the lowest percentage of naïve γδ T cells in the presence of BCG. However, rapamycin significantly increased the AN of human EM γ9+/δ2+ T cells with or without BCG in the culture compared with control. Although these data demonstrate differences in rapamycin’s effect on murine versus human γδ T cells, in both species, rapamycin predominately acted on the EM population. Further evidence for an effect on γδ T cells effector memory function was shown by the combination of rapamycin plus BCG, which increased IFNγ production in γδ T cells in both mouse and human models.
Although mouse tumor models enable exploration of BCG anti-tumor mechanisms, interpretations here are limited by differences between mouse and human γδ T cells, which respond differently to mycobacterial antigens. Murine γδ T cells poorly recognize phosphoantigens [58, 59], whereas human BCG vaccination induces the expansion of Vγ2Vδ2 and γ9+δ2+ T cells reactive with isoprenyl pyrophosphates [20, 60, 61]. Further work is needed to understand the relevance of human γδ T cells towards response to BCG in patients.
Combining rapamycin with BCG could be useful in treating bladder cancer because mTOR inhibition improves antigen-specific immunity [62–64] and enhances BCG peptide presentation [65, 66]. In combination with anti-CLTA-4, an immune checkpoint inhibitor, rapamycin increases immune responses against EL4 lymphoma [28]. Our studies further show that rapamycin enhances MHC-independent γδ T cell-mediated BCG antitumor immunity to treat both mouse and human bladder cancer. Our previous phase I clinical trial addresses the safety and tolerability of mTOR inhibition in combination with intravesical BCG and shows that low dose rapamycin does not exacerbate BCG-related symptoms, while boosting peripheral BCG-specific and urinary γδ T cells [25]. Further work and a phase II clinical trial are underway to reveal the underlying mechanism and clinical efficacy of combination rapamycin and BCG treatment in terms of bladder tumor progression and survival of patients.
CONCLUSION
BCG treatment of bladder cancer induces a tumor-specific memory response by boosting the activity of tumor-specific T cells. BCG antigen-specific T cells also contribute to BCG’s antitumor activity, presumably by targeting BCG-derived peptides presented by MHC molecules on bladder tumors. BCG-mediated activation of conventional αβ T cells, however, requires non-conventional γδ T cells, which have innate immune features, supporting a role for both adaptive and innate immunity in BCG’s antitumor effects. In an experimental model of human bladder cancer, rapamycin improved human γδ T cell-mediated bladder cancer control. Collectively, our results identify immune mechanisms for bladder cancer treatment properties of BCG and support evaluation of rapamycin plus BCG for treating patients with non-muscle invasive bladder cancer.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
SYNOPSIS.
γδ T cells are shown to be required for boosting antitumor responses of conventional antigen-specific T cells during BCG treatment of bladder cancer. Rapamycin enhances BCG efficacy, supporting the use of the combination for treating bladder cancer.
Acknowledgments:
The work was supported by following funding: (1) 8KL2 TR000118, K23, (2) the Mays Family Cancer Center at University of Texas Health San Antonio (P30 CA054174), (3) the Roger L. And Laura D. Zeller Charitable Foundation Chair in Urologic Cancer, (4) the Max & Minnie Tomerlin Voelcker Fund, (5) CDMRP CA170270/P1P2, (6) Bladder Cancer Advocacy Network (BCAN) 2016 Young Investigator Award, (7) Research Training Award (RP170345) from the Cancer Prevention & Research Institute of Texas (8) MSTP Program (NIH T32GM113896), (9) NIH/NCATS TL1 TR002647, (10) NIA T32 AG 021890.
Declarations: The studies related to this manuscript were conducted abided by research ethics and approved IACUC or IRB guidelines for animal or clinical sample use. All authors have consent for publishing the data from the studies. Corresponding author R.S.S. discloses other roles as Consultant for FerGene and Clinical Research Support for JBL(SWOG), FKD and Decipher Biosciences. All other authors disclose no conflict of interest.
REFERENCES
- 1.Lamm DL, et al. , A randomized trial of intravesical doxorubicin and immunotherapy with bacille Calmette-Guerin for transitional-cell carcinoma of the bladder. The New England journal of medicine, 1991. 325(17): p. 1205–9. [DOI] [PubMed] [Google Scholar]
- 2.Morton D, et al. , Immunological factors which influence response to immunotherapy in malignant melanoma. Surgery, 1970. 68(1): p. 158–63; discussion 163–4. [PubMed] [Google Scholar]
- 3.Morales A, Eidinger D, and Bruce AW, Intracavitary Bacillus Calmette-Guerin in the treatment of superficial bladder tumors. J Urol, 1976. 116(2): p. 180–3. [DOI] [PubMed] [Google Scholar]
- 4.Lamm DL, et al. , Maintenance bacillus Calmette-Guerin immunotherapy for recurrent TA, T1 and carcinoma in situ transitional cell carcinoma of the bladder: a randomized Southwest Oncology Group Study. J Urol, 2000. 163(4): p. 1124–9. [PubMed] [Google Scholar]
- 5.Witjes JA, Management of BCG failures in superficial bladder cancer: a review. Eur Urol, 2006. 49(5): p. 790–7. [DOI] [PubMed] [Google Scholar]
- 6.Ratliff TL, Gillen D, and Catalona WJ, Requirement of a thymus dependent immune response for BCG-mediated antitumor activity. J Urol, 1987. 137(1): p. 155–8. [DOI] [PubMed] [Google Scholar]
- 7.Ratliff TL, et al. , T-cell subsets required for intravesical BCG immunotherapy for bladder cancer. The Journal of urology, 1993. 150(3): p. 1018–23. [DOI] [PubMed] [Google Scholar]
- 8.Mayordomo JI, et al. , Bone marrow-derived dendritic cells pulsed with synthetic tumour peptides elicit protective and therapeutic antitumour immunity. Nat Med, 1995. 1(12): p. 1297–302. [DOI] [PubMed] [Google Scholar]
- 9.Schnurr M, et al. , Tumor antigen processing and presentation depend critically on dendritic cell type and the mode of antigen delivery. Blood, 2005. 105(6): p. 2465–72. [DOI] [PubMed] [Google Scholar]
- 10.Luo Y, et al. , A novel method for monitoring Mycobacterium bovis BCG trafficking with recombinant BCG expressing green fluorescent protein. Clin Diagn Lab Immunol, 1996. 3(6): p. 761–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kavoussi LR, et al. , Fibronectin-mediated Calmette-Guerin bacillus attachment to murine bladder mucosa. Requirement for the expression of an antitumor response. The Journal of clinical investigation, 1990. 85(1): p. 62–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ratliff TL, et al. , Intravesical Bacillus Calmette-Guerin therapy for murine bladder tumors: initiation of the response by fibronectin-mediated attachment of Bacillus Calmette-Guerin. Cancer Res, 1987. 47(7): p. 1762–6. [PubMed] [Google Scholar]
- 13.Lattime EC, Gomella LG, and McCue PA, Murine bladder carcinoma cells present antigen to BCG-specific CD4+ T-cells. Cancer research, 1992. 52(15): p. 4286–90. [PubMed] [Google Scholar]
- 14.Biot C, et al. , Preexisting BCG-specific T cells improve intravesical immunotherapy for bladder cancer. Science translational medicine, 2012. 4(137): p. 137ra72. [DOI] [PubMed] [Google Scholar]
- 15.Antonelli AC, et al. , Bacterial immunotherapy for cancer induces CD4-dependent tumor-specific immunity through tumor-intrinsic interferon-gamma signaling. Proc Natl Acad Sci U S A, 2020. 117(31): p. 18627–18637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schild H, et al. , The nature of major histocompatibility complex recognition by gamma delta T cells. Cell, 1994. 76(1): p. 29–37. [DOI] [PubMed] [Google Scholar]
- 17.Tanaka Y, et al. , Natural and synthetic non-peptide antigens recognized by human gamma delta T cells. Nature, 1995. 375(6527): p. 155–8. [DOI] [PubMed] [Google Scholar]
- 18.Uldrich AP, et al. , CD1d-lipid antigen recognition by the gammadelta TCR. Nat Immunol, 2013. 14(11): p. 1137–45. [DOI] [PubMed] [Google Scholar]
- 19.Constant P, et al. , The antituberculous Mycobacterium bovis BCG vaccine is an attenuated mycobacterial producer of phosphorylated nonpeptidic antigens for human gamma delta T cells. Infect Immun, 1995. 63(12): p. 4628–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoft DF, Brown RM, and Roodman ST, Bacille Calmette-Guerin vaccination enhances human gamma delta T cell responsiveness to mycobacteria suggestive of a memory-like phenotype. J Immunol, 1998. 161(2): p. 1045–54. [PubMed] [Google Scholar]
- 21.Cao G, et al. , mTOR inhibition potentiates cytotoxicity of Vgamma4 gammadelta T cells via up-regulating NKG2D and TNF-alpha. J Leukoc Biol, 2016. 100(5): p. 1181–1189. [DOI] [PubMed] [Google Scholar]
- 22.Dao V, et al. , Immune-Stimulatory Effects of Rapamycin Are Mediated by Stimulation of Antitumor gammadelta T Cells. Cancer Res, 2016. 76(20): p. 5970–5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H and Pauza CD, Rapamycin increases the yield and effector function of human gammadelta T cells stimulated in vitro. Cancer Immunol Immunother, 2011. 60(3): p. 361–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Svatek RS, et al. , Rapamycin prevents surgery-induced immune dysfunction in patients with bladder cancer. Cancer Immunol Res, 2018: p. 466–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ji N, et al. , Rapamycin enhances BCG-specific gammadelta T cells during intravesical BCG therapy for non-muscle invasive bladder cancer: a randomized, double-blind study. J Immunother Cancer, 2021. 9(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen ZW, Immune regulation of gammadelta T cell responses in mycobacterial infections. Clin Immunol, 2005. 116(3): p. 202–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holderness J, et al. , Comparative biology of gammadelta T cell function in humans, mice, and domestic animals. Annu Rev Anim Biosci, 2013. 1: p. 99–124. [DOI] [PubMed] [Google Scholar]
- 28.Pedicord VA, et al. , Friends not foes: CTLA-4 blockade and mTOR inhibition cooperate during CD8+ T cell priming to promote memory formation and metabolic readiness. J Immunol, 2015. 194(5): p. 2089–98. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y, et al. , Biphasic Rapamycin Effects in Lymphoma and Carcinoma Treatment. Cancer Res, 2017. 77(2): p. 520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ji N, et al. , Percutaneous BCG enhances innate effector antitumor cytotoxicity during treatment of bladder cancer: a translational clinical trial. Oncoimmunology, 2019. 8(8): p. 1614857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ji N, et al. , Small molecule inhibitor of antigen binding and presentation by HLA-DR2b as a therapeutic strategy for the treatment of multiple sclerosis. J Immunol, 2013. 191(10): p. 5074–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang L, et al. , IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med, 2009. 206(7): p. 1457–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zaharoff DA, et al. , Intravesical immunotherapy of superficial bladder cancer with chitosan/interleukin-12. Cancer Res, 2009. 69(15): p. 6192–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meeks JJ, Lerner SP, and Svatek RS, Bacillus Calmette-Guerin Manufacturing and SWOG S1602 Intergroup Clinical Trial. J Urol, 2017. 197(3 Pt 1): p. 538–540. [DOI] [PubMed] [Google Scholar]
- 35.Shen Y, et al. , Adaptive immune response of Vgamma2Vdelta2+ T cells during mycobacterial infections. Science, 2002. 295(5563): p. 2255–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lindroth K, et al. , The humoral response in TCR alpha−/− mice. Can gammadelta-T cells support the humoral immune response? Scand J Immunol, 2002. 55(3): p. 256–63. [DOI] [PubMed] [Google Scholar]
- 37.Viney JL, et al. , Lymphocyte proliferation in mice congenitally deficient in T-cell receptor alpha beta + cells. Proc Natl Acad Sci U S A, 1994. 91(25): p. 11948–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takeuchi A, et al. , IL-17 production by gammadelta T cells is important for the antitumor effect of Mycobacterium bovis bacillus Calmette-Guerin treatment against bladder cancer. European journal of immunology, 2011. 41(1): p. 246–51. [DOI] [PubMed] [Google Scholar]
- 39.Ou L, et al. , Dichotomous and stable gamma delta T-cell number and function in healthy individuals. J Immunother Cancer, 2021. 9(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sylvester RJ, van der MA, and Lamm DL, Intravesical bacillus Calmette-Guerin reduces the risk of progression in patients with superficial bladder cancer: a meta-analysis of the published results of randomized clinical trials. J Urol, 2002. 168(5): p. 1964–70. [DOI] [PubMed] [Google Scholar]
- 41.Zbar B, et al. , Tumor immunity produced by the intradermal inoculation of living tumor cells and living Mycobacterium bovis (strain BCG). Science, 1970. 170(3963): p. 1217–8. [DOI] [PubMed] [Google Scholar]
- 42.Zbar B and Rapp HJ, Immunotherapy of guinea pig cancer with BCG. Cancer, 1974. 34(4 Suppl): p. suppl:1532–40. [DOI] [PubMed] [Google Scholar]
- 43.Reichert DF and Lamm DL, Long term protection in bladder cancer following intralesional immunotherapy. J Urol, 1984. 132(3): p. 570–3. [DOI] [PubMed] [Google Scholar]
- 44.Cornel EB, et al. , Antitumor effects of bacillus Calmette-Guerin in a syngeneic rat bladder tumor model system, RBT323. J Urol, 1993. 149(1): p. 179–82. [DOI] [PubMed] [Google Scholar]
- 45.Ratliff TL, et al. , Purification of a mycobacterial adhesin for fibronectin. Infect Immun, 1993. 61(5): p. 1889–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schorey JS, et al. , Characterization of the fibronectin-attachment protein of Mycobacterium avium reveals a fibronectin-binding motif conserved among mycobacteria. Mol Microbiol, 1996. 21(2): p. 321–9. [DOI] [PubMed] [Google Scholar]
- 47.Schorey JS, et al. , A Mycobacterium leprae gene encoding a fibronectin binding protein is used for efficient invasion of epithelial cells and Schwann cells. Infect Immun, 1995. 63(7): p. 2652–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reyes RM, et al. , CD122-directed interleukin-2 treatment mechanisms in bladder cancer differ from αPD-L1 and include tissue-selective γδ T cell activation. Journal for ImmunoTherapy of Cancer, 2021. 9(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ismaili J, et al. , Human gamma delta T cells induce dendritic cell maturation. Clin Immunol, 2002. 103(3 Pt 1): p. 296–302. [DOI] [PubMed] [Google Scholar]
- 50.Muto M, et al. , Myeloid molecular characteristics of human gammadelta T cells support their acquisition of tumor antigen-presenting capacity. Cancer Immunol Immunother, 2015. 64(8): p. 941–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brandes M, Willimann K, and Moser B, Professional antigen-presentation function by human gammadelta T Cells. Science, 2005. 309(5732): p. 264–8. [DOI] [PubMed] [Google Scholar]
- 52.Brandes M, et al. , Cross-presenting human gammadelta T cells induce robust CD8+ alphabeta T cell responses. Proc Natl Acad Sci U S A, 2009. 106(7): p. 2307–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dieli F, et al. , Differentiation of effector/memory Vdelta2 T cells and migratory routes in lymph nodes or inflammatory sites. J Exp Med, 2003. 198(3): p. 391–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Caccamo N, et al. , Differential requirements for antigen or homeostatic cytokines for proliferation and differentiation of human Vgamma9Vdelta2 naive, memory and effector T cell subsets. Eur J Immunol, 2005. 35(6): p. 1764–72. [DOI] [PubMed] [Google Scholar]
- 55.Dieli F, et al. , Targeting human {gamma}delta} T cells with zoledronate and interleukin-2 for immunotherapy of hormone-refractory prostate cancer. Cancer Res, 2007. 67(15): p. 7450–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meraviglia S, et al. , In vivo manipulation of Vgamma9Vdelta2 T cells with zoledronate and low-dose interleukin-2 for immunotherapy of advanced breast cancer patients. Clin Exp Immunol, 2010. 161(2): p. 290–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cordova A, et al. , Characterization of human gammadelta T lymphocytes infiltrating primary malignant melanomas. PLoS One, 2012. 7(11): p. e49878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Griffin JP, et al. , Kinetics of accumulation of gamma delta receptor-bearing T lymphocytes in mice infected with live mycobacteria. Infect Immun, 1991. 59(11): p. 4263–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dieli F, et al. , Characterization of lung gamma delta T cells following intranasal infection with Mycobacterium bovis bacillus Calmette-Guerin. J Immunol, 2003. 170(1): p. 463–9. [DOI] [PubMed] [Google Scholar]
- 60.Morita CT, Mariuzza RA, and Brenner MB, Antigen recognition by human gamma delta T cells: pattern recognition by the adaptive immune system. Springer Semin Immunopathol, 2000. 22(3): p. 191–217. [DOI] [PubMed] [Google Scholar]
- 61.Zufferey C, et al. , The contribution of non-conventional T cells and NK cells in the mycobacterial-specific IFNgamma response in Bacille Calmette-Guerin (BCG)-immunized infants. PLoS One, 2013. 8(10): p. e77334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Keating R, et al. , The kinase mTOR modulates the antibody response to provide cross-protective immunity to lethal infection with influenza virus. Nat Immunol, 2013. 14(12): p. 1266–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Beziaud L, et al. , Rapalogs Efficacy Relies on the Modulation of Antitumor T-cell Immunity. Cancer Res, 2016. 76(14): p. 4100–12. [DOI] [PubMed] [Google Scholar]
- 64.Mannick JB, et al. , mTOR inhibition improves immune function in the elderly. Sci Transl Med, 2014. 6(268): p. 268ra179. [DOI] [PubMed] [Google Scholar]
- 65.Jagannath C, et al. , Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nature medicine, 2009. 15(3): p. 267–76. [DOI] [PubMed] [Google Scholar]
- 66.Jagannath C and Bakhru P, Rapamycin-induced enhancement of vaccine efficacy in mice. Methods Mol Biol, 2012. 821: p. 295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.