

Abstract
The lack of effective treatments for autism spectrum disorder (ASD) and congenital hydrocephalus (CH) reflects the limited understanding of the biology underlying these common neurodevelopmental disorders. Although ASD and CH have been extensively studied as independent entities, recent human genomic and pre-clinical animal studies have uncovered shared molecular pathophysiology. Here, we review and discuss phenotypic, genomic, and molecular similarities between ASD and CH, and identify the PTEN-PI3K-mTOR (phosphatase and tensin homolog-phosphoinositide 3-kinase-mammalian target of rapamycin) pathway as a common underlying mechanism that holds diagnostic, prognostic, and therapeutic promise for individuals with ASD and CH.
Keywords: neurodevelopment disorders, ventriculomegaly, macrocephaly, mTOR, rapamycin
Common biological pathways in autism spectrum disorders and congenital hydrocephalus
Neurodevelopmental disorders (NDDs) include a wide range of neurological, physiological, and genetic conditions that impair normal brain development and function. While the clinical heterogeneity of NDDs reflects their multifactorial pathogenesis, the extensive symptomatic and genetic overlap among NDDs [1] suggests common underlying biological pathways. Within the intricate landscape of NDDs, autism spectrum disorders (ASDs) are a significant but poorly understood subgroup with a steadily increasing prevalence. ASDs affect one in 54 children in the United States, with a male-to-female ratio of 4.3:1 [2]. ASD is characterized by communication difficulties, restricted interests, and repetitive behaviors that often manifest before age 3 [3]. Individuals with ASD typically display deficits in socioemotional reciprocity, non-verbal communication, and social adaptation. Characteristic behaviors include motor stereotypies, repetitive speech (e.g. echolalia), and rigid adherence to behavioral, social, and environmental routine. ASD is also frequently associated with altered sensitivity to sensory stimuli, sleep dysfunction, gastrointestinal abnormalities, and motor deficits [3]. These and other social and behavioral deficits among affected individuals can vary widely in severity.
Enlargement of the brain’s cerebrospinal fluid (CSF)-filled ventricles (ventriculomegaly, see Glossary) is frequently observed in individuals with ASD [4–6], and CSF-related structural brain abnormalities such as congenital hydrocephalus (CH) are associated with ASD [7–10]. Approximately 20% of children with CH are also diagnosed with ASD – a rate 10-fold higher than the 1.9% rate in the general population [2] [11, 12]. CH has been classically attributed to decreased CSF clearance and resultant increased intracranial pressure [13]. Based on this paradigm, the mainstay of CH treatment has been neurosurgical CSF diversion [14], with high rates of complication, morbidity, and treatment failure [14, 15]. Interestingly, some CH cases, including those accompanied by comorbidities such as ASD or other NDDs [16], can be characterized by ventriculomegaly with normal or low intracerebral pressure, a critical distinction with implications for treatment [17, 18]. These observations suggest that impaired CSF dynamics alone might not drive development of ventriculomegaly in these patients. Moreover, the ventriculomegaly, macrocephaly [19, 20], and other CSF-related structural abnormalities frequently accompanying ASD suggest a degree of shared etiology between CH and ASD.
The mammalian target of rapamycin (mTOR) pathway (Figure 1) is an intracellular signaling pathway that regulates protein synthesis, cell growth, and metabolism. Emerging evidence suggests that mTOR signaling may play a central mechanistic role during neurodevelopment in both ASDs and CH [21, 22]. Although the mTOR pathway has been previously implicated in ASDs [23], recent genetic evidence supports its additional critical function in sporadic, neurosurgically treated CH [24]. Through its two major complexes, mTORC1 and mTORC2, mTOR controls cellular metabolism, proliferation, differentiation and survival [25]. Phosphatase and Tensin Homolog (PTEN) encodes a lipid and protein phosphatase that negatively regulates mTOR through PIP3 hydrolysis, preventing AKT-mediated activation of mTORC1 [25]. Importantly, de novo loss-of-function mutations in PTEN are strongly associated with both ASDs and CH [24, 26]. This intriguing link between seemingly disparate NDDs suggests the hypothesis that ASD – a disorder defined mainly by behavioral manifestations – and CH – a structural brain abnormality classically attributed to impaired CSF “plumbing” – may share, at least in part, a common targetable molecular origin.
Figure 1. The PTEN-PI3K-AKT-mTOR pathway and therapeutic targets.
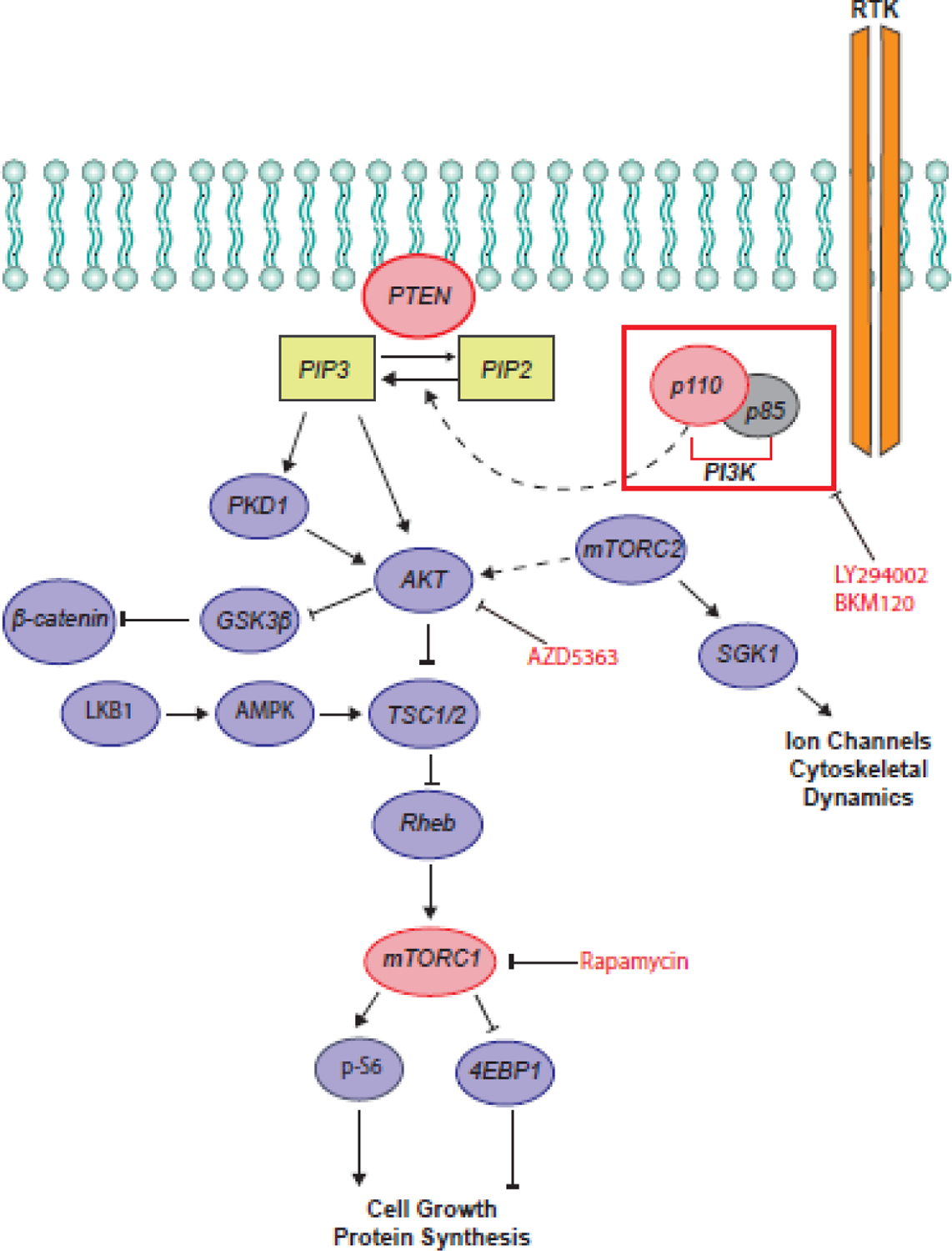
Activation of PI3K-AKT signaling can occur by binding of growth factors, hormones, or cytokines to receptor tyrosine kinases (RTK), leading to activation of phosphatidylinositol 3-phosphate kinase (PI3K). PI3K phosphorylates phosphatidylinositol-4,5-bisphosphate (PIP2), generating phosphatidylinositol 3,4,5-trisphosphate (PIP3), leading to the activation of AKT by phosphorylation at Thr308 (p-AKT). PTEN opposes the effect of PI3K by hydrolyzing PIP3 to PIP2. p-AKT deactivates glycogen synthase kinase-3 β (GSK3β), which is a negative regulator of β-catenin, and suppresses the tuberous sclerosis complex (TSC1/TSC2), a negative regulator of mTORC1. mTORC1 suppresses eukaryotic translation initiation factor 4E-binding protein (4EBP1) and activates p-S6, leading to increased protein translation. mTORC2 modulates cytoskeletal remodeling and ion channel activation and leads to phosphorylation of AKT at Ser473. Pharmacological treatment with LY294002 and BKM120 inhibits PI3K activity. AZD5363 inhibits AKT. Rapamycin inhibits mTORC1 activity. Abbreviations: AKT, protein kinase B; AMPK, AMP-activated protein kinase; mTOR, mammalian target of rapamycin; mTORC1, mammalian target of rapamycin complex 1; mTORC2, mammalian target of rapamycin complex 2; PTEN, phosphatase and tensin homolog; PKD1, polycystin 1; LKB1, liver kinase B1; SGK1, serum/glucocorticoid regulated kinase 1.
The current lack of pharmacological interventions targeting core features of ASD or CH reflects, in part, a limited understanding of the biology underlying these disorders, traditionally studied as independent entities. However, overlapping neurodevelopmental pathways and shared pathogenic elements of ASD and CH are beginning to emerge. Here, we discuss the phenotypic, genetic, and molecular similarities between ASD and CH, and identify the PTEN-PI3K (phosphoinositide 3-kinase)-mTOR pathway as a common underlying mechanism for subsets of patients with these disorders.
Phenotypic similarities between ASD and CH
Although ASD currently lacks pathognomonic neuroradiographic findings, the prevalence of structural brain abnormalities is higher in ASD compared to healthy controls, typically developing children, and children with developmental delay [27, 28]. Abnormal head sizes often described in ASD may be associated with specific genetic etiologies. Macrocephaly occurs in nearly 20% of individuals with ASD [20], and the altered ASD brain growth trajectory is characterized by overgrowth in the first few years of life, followed by a period of slowed growth [29–31]. The severity of brain structural abnormalities in children also correlates with likelihood of an ASD diagnosis. Computed tomography imaging demonstrates major structural brain abnormalities in 64% of hydrocephalic patients with ASD and 28% of those without ASD [12]. Moreover, increased extra-axial CSF spaces in neonates detected by magnetic resonance imaging (MRI) can predict future ASD diagnosis [7, 8, 10]. Such MRI detection of extra-axial CSF collections (or “benign external hydrocephalus”) analyzed by a machine learning algorithm exhibits 84% sensitivity and 60% specificity in ASD diagnosis, suggesting its use as a biomarker for a subtype of ASD [9] and reinforcing the potential of MRI as part of the diagnostic approach to ASD [32].
Gene discovery and integrative genomics implicate common pathways in ASD and CH
The complex, highly heritable but polygenic character of NDDs [33–35] has contributed to the difficulties historically encountered in their investigation. However, recent advances in human genetics are revealing new insights into the pathophysiology of these disorders. Genomic technologies such as genome-wide association studies, whole exome sequencing (WES), and whole genome sequencing have highlighted numerous disease-risk loci and afforded novel insights into ASD pathophysiology [26, 36, 37]. Recent WES analyses of families with affected offspring and unaffected parents (termed “case-parent trio design”) and of families with multiple affected individuals have similarly identified de novo and rare inherited mutations that impart large effects on CH risk [24, 38].
ASD is highly heritable, with concordance rates higher in monozygotic (50–90%) than in dizygotic twins (up to 30%), and sibling recurrence rates up to 18.7% [39–45]. The first ASD risk genes were identified in rare monogenic disorders such as Fragile X syndrome (FMR1), tuberous sclerosis complex (TSC1, TSC2), neurofibromatosis type 1 (NF1), and PTEN-associated macrocephaly [46] [47], syndromes directly or indirectly associated with dysregulated mTOR signaling (reviewed in [48]). While most inherited ASD risk is associated with common variants [49–51], recent progress in ASD gene discovery reflects identification of rare ASD risk variants. Large-scale WES and genotyping studies have revealed significant association of rare de novo variants, gene-disrupting single-nucleotide variants, and copy number variants with ASD risk [52–54, 58], leading to the identification of growing numbers of ‘high-confidence’ ASD risk genes [55, 56]. A recent analysis [26] examined rare de novo and case-control variants from almost 12,000 affected individuals, and identified 102 risk genes (FDR≤0.1) [26]. The negative regulator of mTOR signaling, PTEN, was among the top risk genes identified both by this study [26] and other WES studies [56–58].
Discovery of “high confidence” risk genes has implicated convergent pathways contributing to ASD biology, including the regulation of gene expression and neuronal communication [26, 55, 56]. Integrative genomic approaches leveraging coexpression networks to construct a spatiotemporal map of ASD risk gene expression have identified mid-fetal glutamatergic projection neurons as a point of convergence with potential disease implications [59, 60]. Moreover, ASD genes are most strongly enriched in early excitatory neurons and striatal interneurons, implicating maturing and mature neurons of both excitatory and inhibitory lineages [26]. These studies highlight the power of integrative genomics to elucidate the spatiotemporal dynamics of ASD risk genes and reveal central roles for these genes in early brain development and excitatory-inhibitory signaling.
WES has also recently illuminated the pathogenic mechanisms underlying CH. Although nearly 40% of CH cases are predicted to have genetic etiology [16], mutations in CH genes identified prior to availability of WES account for fewer than 5% of primary cases [61]. The multiple biological processes affected by these bona fide genes initially failed to suggest a unified paradigm of CH pathogenesis [62]. However, recent WES of the largest cohort of sporadic CH patients to date have revealed additional genes with significant burden of rare damaging mutations [24]. Remarkably, all of these genes are implicated in regulation of neural stem cells (NSCs), suggesting many CH cases may arise from impaired prenatal neurogliogenesis [24]. Perhaps unexpectedly, CH and ASD exhibited significant overlap of genes with rare, damaging de novo mutations, including PTEN and MTOR [24].
As with ASD genetic studies, CH-associated variants were further studied through an integrative genomics approach, using a bulk RNA-seq transcriptome characterized by WGCNA modules [63] and a single-cell [single-cell RNA sequencing (scRNA-seq)] transcriptome characterized by cell types [64] of the midgestational human brain. Of note, CH risk genes converged in a module implicated in ASD and developmental disorders, yet these genes were also found in cell types critical to fetal neurogenesis at earlier stages of differentiation than the postmitotic stages typically thought to be pertinent to autism and other developmental disorders [24]. These findings suggest that CH and ASD may arise from intersecting neurodevelopmental pathways, with disruption of early NSCs effecting a more severe structural phenotype as seen in CH. Taken together, WES and associated integrative genomics studies of ASD and CH reveal a surprising degree of convergence, and highlight PTEN-PI3K-mTOR signaling as one potentially shared pathway.
PTEN mutations and ASD
Prior to its association with ASD, PTEN was first identified as a tumor suppressor gene based on mutations present in cancer specimens [65, 66] and inherited cancer syndromes [67–69]. PTEN mutations cause Cowden syndrome, an autosomal dominant disorder characterized by macrocephaly, benign hamartomas, and an increased risk of breast, thyroid, and other cancers in adults. PTEN mutations also cause Bannayan-Riley-Ruvalcaba syndrome, a childhood-onset autosomal dominant disorder similarly characterized by macrocephaly and hamartomas, along with developmental delay, lipomas, and penile freckling [67, 68]. These disorders are collectively considered PTEN hamartoma tumor syndromes (PHTSs), reflecting their shared genetic etiology and similar clinical features [69]. Interestingly, while recent WES studies have confirmed PTEN as a high-confidence ASD risk gene [26], the association of PTEN with ASD was first established by earlier targeted sequencing of the PTEN gene in individuals with ASD and macrocephaly [70–73]. It was hypothesized [70] that PTEN mutations might be associated with co-occurrence of ASD and macrocephaly, based on reports of ASD-like behaviors in individuals with PHTS [74–76] in whom macrocephaly is common. Multiple studies have since documented PTEN mutations in at least 5–10% (up to 22%) of individuals with ASD accompanied by macrocephaly [70–73, 77–79].
Macrocephaly is the strongest clinical feature associated with PTEN mutations, and individuals with ASD and PTEN mutations typically have “extreme” macrocephaly (>2.5–3 standard deviations above the mean) [70, 73, 77, 78]. For this reason, PTEN mutation screening in children with ASD and severe macrocephaly (>2.5 standard deviations) is recommended as part of clinical genetic diagnostic evaluation [80]. While brain imaging in children with ASD, macrocephaly, and PTEN mutations may not show major findings, abnormalities on brain MRI have included ventriculomegaly, polymicrogyria, dilation of perivascular spaces, and periventricular and other white matter abnormalities [70, 72–74, 79, 81]. PTEN mutations have also been associated with seizures [82]. In addition, a WES study of 21 children with ASD, developmental delay, and macrocephaly identified in 10 of the 21 children (largely de novo) mutations in PTEN and other mTOR pathway-associated genes (MTOR, PIK3CA, and PPP2R5D) [81]. Nearly 47.6% of this cohort carried mutations in the PI3K-mTOR-AKT pathway and 19% had mutations in PTEN [81]. All patients with these mutations exhibited megalencephaly, half demonstrated polymicrogyria and white matter abnormalities, and one-third developed ventriculomegaly [81].
Pleiotropy of PTEN mutations
The spectrum of neurodevelopmental outcomes associated with PTEN mutations (Figure 2, Table 1, and Table S1 in the supplemental information online) poses a challenge to establishing genotype-phenotype correlations for PTEN. Children with ASD, macrocephaly, and PTEN mutations often lack hamartomas or other features of PHTS, and the association of these mutations with long-term cancer risk is unclear, although clinical surveillance for cancer in these individuals and family members carrying PTEN mutations is recommended [70, 71, 73]. However, reports have identified families in which a PTEN variant associated with Cowden syndrome or Bannayan-Riley-Ruvalcaba syndrome in a parent is identified in a child with ASD [71, 73, 75, 76, 83]. Conversely, the same variants initially identified in individuals with ASD and macrocephaly have also been found in Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome [73]. This suggests that, with the exception of macrocephaly, PTEN variant expressivity can be influenced by other factors such as genetic background.
Figure 2. PTEN mutations in autism spectrum disorder (ASD) and congenital hydrocephalus (CH).
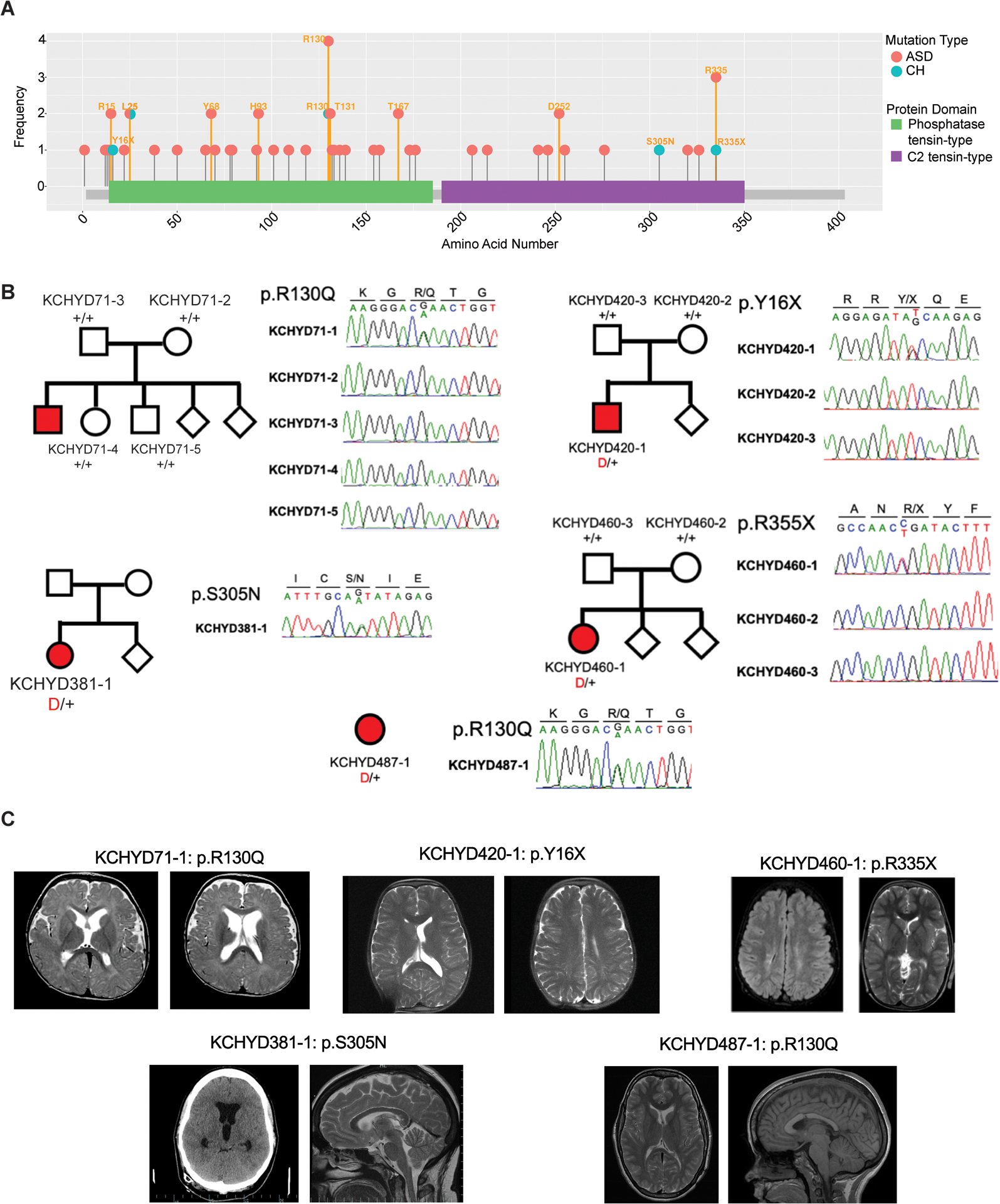
(A) PTEN mutations identified in individuals with ASD (pink) and CH (blue) are shown in relation to PTEN functional domains. This figure was generated using the MutPlot program [138] to map the missense and truncating mutations identified in our literature search (Table S1 in the supplemental information online) onto the human PTEN protein domains reported in UniProt [139]. Splice site mutations are not shown, but are listed in Table S1 in the supplemental information online. Mutation frequency at each residue (based on Table S1 in the supplemental information online) is shown on the y axis. Additional information about clinical features associated with mutations can be found in Table S1 in the supplemental information online. (B) Family pedigrees and Sanger sequencing electropherograms of individuals who underwent neurosurgical treatment for sporadic CH and harbor PTEN mutations, corresponding to reported mutations from Table 1. (B) Adapted, with permission, from [24]. Abbreviation: PTEN, phosphatase and tensin homolog. (C) Representative T1 or T2-weighted axial and sagittal brain MRIs or head CT images of neurosurgically treated CH patients harboring indicated PTEN mutations, adapted, with permission, from [24].
Table 1.
Neurosurgical treatments, cortical malformations, and developmental sequalae in PTEN-CH patients (modified from [24]) with permission.
| Subject | Treatment | Aqueductal stenosis | Cavum septum pellucidum | Cerebellar tonsillar ectopia | Corpus callosum abnormalities | Intracranialcyst | Megalencephaly | Polymicrogyria | Septal agenesis | White matter signal abnormality | White matter volume loss | Craniofacial abnormality | Developmental delay | Epilepsy | Hernia | Macrocephaly | Skeletal abnormalities |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| KCHYD71-1 | ETV | + | − | + | + | − | − | + | − | + | + | − | − | − | − | + | − |
| KCHYD420-1 | Shunted | − | − | + | − | − | − | + | − | − | + | − | + | − | + | + | − |
| KCHYD460-1 | Shunted>ETV> Shunted | − | + | + | − | − | − | − | − | + | + | + | + | − | + | + | + |
| KCHYD381-1 | Shunted | + | − | + | − | − | − | − | − | + | − | − | + | − | − | + | − |
| KCHYD487-1 | Shunted | + | − | + | + | − | − | − | − | + | + | − | + | − | − | + | − |
ETV: Endoscopic third ventriculostomy
KCHYD71-1 also exhibits mildly rotated thalami, prominence of the subarachnoid spaces overlying the cerebral convexities, small ill-defined enhancing focus in the left centrum semiovale adjacent to a medullary vein may represent a capillary telangiectasia or developmental venous anomaly
KCHYD420-1 also exhibits dilated perivascular spaces and pineal cyst
KCHYD460-1 also exhibits asymmetric caudate nuclei, signal abnormality in the globus pallidus, prominence of the extra-axial CSF spaces overlying the left cerebral convexity most pronounced at the parietal region near the vertex
KCHYD381-1 also exhibits pineal cyst
KCHYD487-1 also exhibits right lateral intraventricular cyst
Evidence indeed suggests that PTEN variant function might correlate with clinical presentation. For example, over 50% of cancer-associated PTEN variants result in protein truncation, and most cancer-associated missense variants lead to complete loss of phosphatase activity [84]. These missense variants might have dominant negative effects, resulting in more severe cancer phenotypes in animal models [85, 86]. By contrast, the majority of ASD-associated variants are missense [83]. Functional assessment in a yeast-based assay found that ASD-associated variants result in partial loss of function (LoF) compared to complete LoF of PHTS-associated variants [87]. Another study found that ASD-associated PTEN variants, though unstable when transduced in glioblastoma cells and in mouse hippocampal neurons, retained catalytic activity, whereas PHTS variants led to complete LoF in glioblastoma cells [88]. Altered ubiquitination and defective nuclear-cytoplasmic shuttling may also cause PTEN LoF [89–92]; reviewed in [25]. For example, some ASD-associated PTEN variants result in nuclear exclusion of PTEN and increased soma size when introduced into mouse dentate gyrus in vivo [91]. Restoring nuclear PTEN expression and lipid phosphatase activity rescued soma size and phospho S6 (p-S6) levels [91]. These findings suggest that mutations may affect PTEN function by altering its subcellular localization as well as its stability and activity [91].
Recent studies have developed computational approaches to assess genotype-phenotype correlation for PTEN variants [93]. One study found 106 missense and nonsense variants disrupted PTEN function in five different model systems via multiple mechanisms [94], including induced instability, loss of catalytic or substrate-binding activity, and dominant negative effects. Whereas unstable variants were highly correlated across different assays, other functional effects were less consistent, and genotype-phenotype correlations could not be assigned, reflecting clinical overlap among these variants [94]. PTEN pleiotropy might represent an avenue for exploring common mechanisms across disorders, despite this complexity. For example, missense mutations at PTEN R130 have been associated with ASD, CH, PHTS, and somatic cancers [24, 94] (Figure 2), suggesting common biology.
While integrative genomics implicates overlap in CH- and ASD-associated pathways (Figure 3), the number of CH versus ASD-associated PTEN variants identified to date is more limited (Figure 2, Table S2), complicating genotype-phenotype correlations. Identifying additional CH-associated PTEN variants is likely to provide greater leverage for differentiating variant function and revealing unique and shared downstream pathways as potential targets. As WES studies of CH and ASD have implicated altered neural proliferation and differentiation during early and mid-fetal development, both processes directly affected by PTEN and downstream mTOR signaling, these pathways may represent a convergent “hub” predisposing to a range of NDDs.
Figure 3. Integrative genomics leads to a convergent neuroscience approach to autism spectrum disorder (ASD) and congenital hydrocephalus (CH).
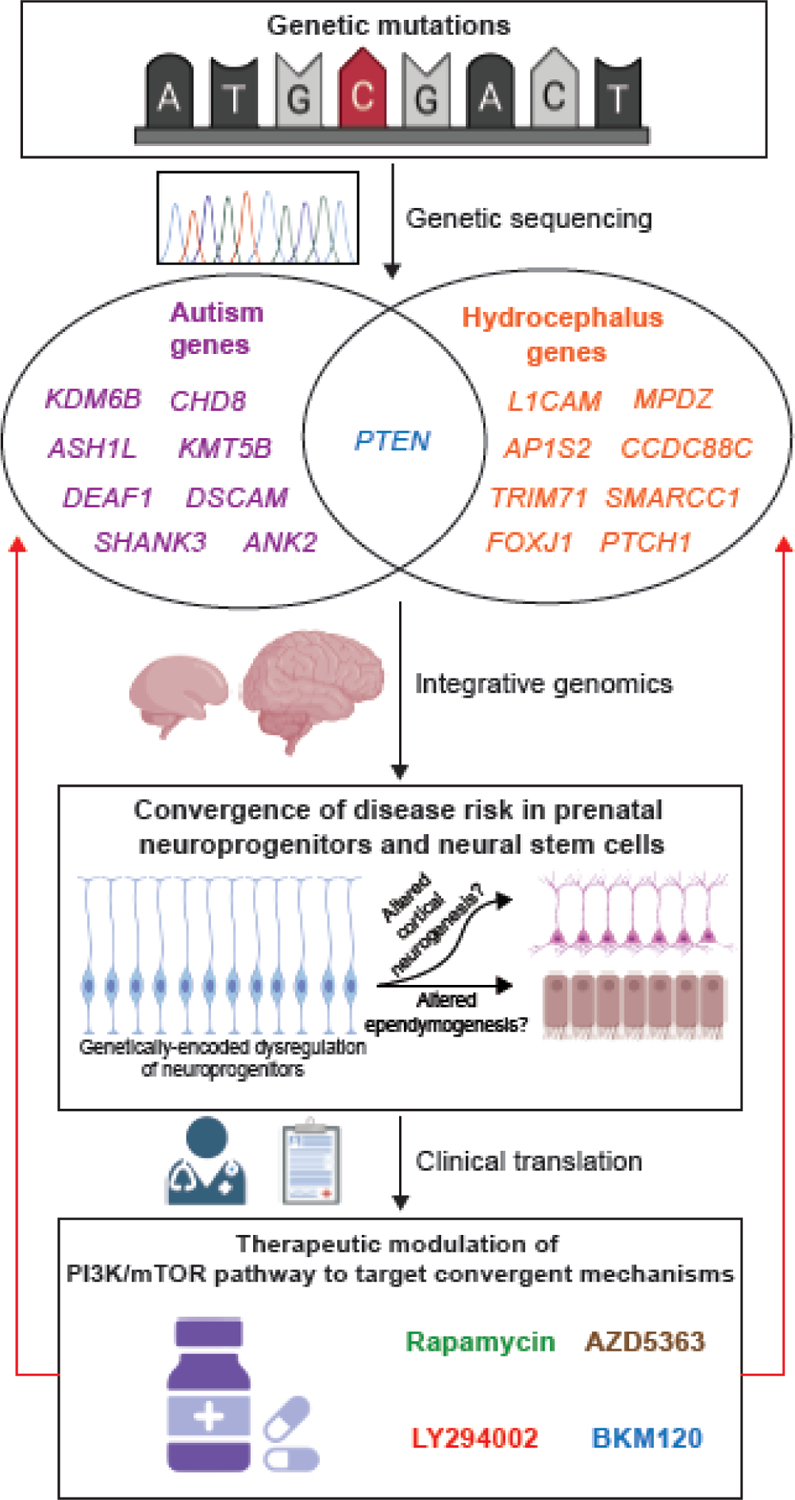
A. Mutation detection in ASD and CH patients using “case-parent trio design” whole exome sequencing approaches have identified de novo mutations that carry a high risk burden. The autism genes listed in the figure, selected from [26], were identified as ASD predominant with family-wise error rate of 0.05 or less, and the hydrocephalus genes are high-confidence genes selected from [24], with PTEN being the only gene shared between the two groups. B. Integrative genomic analyses reveal that spatial and temporal co-expression networks shared between ASD and CH candidate genes converge upon neuroprogenitors and neural stem cells in the embryonic brain. Ongoing basic science studies are examining altered cortical neurogenesis and altered ependymogenesis, as well as ASD and CH pathogenesis at the molecular, cellular, and circuit levels. C. Therapeutic modulation of the PI3K/mTOR pathway can inform the development of future potential treatments of ASD and CH in individuals carrying mutations in key genes within this pathway. Figure created using Biorender.com. Abbreviations: mTOR, mammalian target of rapamycin; PI3K, phosphoinositide 3-kinase; PTEN, phosphatase and tensin homolog.
Animal models for studying PTEN function
Animal models have provided important insights into the role of the PTEN-PI3K-AKT-mTOR pathway in the pathogenesis of ASD and CH. The PTEN gene is evolutionarily conserved across several species, including zebrafish, mice, and humans, and Pten LoF animal models have been instrumental in elucidating the effects of LoF mutations on basic neurodevelopmental mechanisms (see Table S2 in the supplemental information online).
Pten mouse models exhibit macrocephaly and ASD-like behavioral phenotypes
Because homozygous Pten LoF causes embryonic lethality in mice at about embryonic day 9.5 [95, 96], conditional Pten LoF mouse models have been developed to investigate spatial and temporal functions of PTEN in the brain. The first autism-associated Pten LoF mouse model was generated via homozygous Pten knockout in postmitotic, mature neurons (Nse-Cre × Ptenflx/flx; Nse-Pten). Nse-Pten mice exhibit macrocephaly and brain overgrowth due to neuronal hypertrophy and progressive enlargement of the cerebral cortex and dentate gyrus, as well as increased dendritic arborizations [97]. Hypertrophic neurons in the dentate gyrus have increased levels of p-Akt, p-S6, and p-GSK3ß, indicating activation of the AKT and mTOR pathways [97]. Nse-Pten mice also develop “ASD-like” behavioral phenotypes, including deficits in social interaction, impaired learning, and altered sensory processing [97]. Morphological and behavioral phenotypes in Pten mouse models have been recently reviewed [25, 99, 100].
Additional mouse models have recapitulated macrocephaly, regional brain size alterations and/or neuronal hypertrophy, as well as behavioral deficits resulting from haploinsufficient or cell-type specific loss of Pten [98, 101–106]. Interestingly, these studies highlight the complex interplay of signaling pathways downstream of PTEN. For example, haploinsufficient Pten+/− mice show brain overgrowth at birth due to neuronal hyperplasia, as well as an increase in markers of ß-catenin activity in the cortex, while adult Pten+/− mice show an increase in glia [103]. Genetic suppression of ß-catenin, not mTOR, reversed overgrowth in adult cortex, suggesting that Wnt/ß-catenin signaling downstream of PTEN might modulate brain overgrowth [103]. By contrast, genetic suppression of mTOR rescued social deficits in Pten+/− female mice as well as hypertrophy of cortical layer V neurons, implicating mTOR signaling in controlling these phenotypes [104]. PTEN LoF has also been shown to lead to decreased apoptosis and abnormal neuronal migration, and may not lead to increased proliferation of some progenitor populations [102, 107, 108], suggesting that mechanisms other than increased proliferation might also contribute to increased brain size.
Pten mouse models display seizures, excitatory-inhibitory imbalance, and synaptic alterations
Several Pten mouse models develop seizures [97, 98, 101, 102], analogous to the comorbidity of epilepsy in humans with ASD [97], and possibly reflecting a shared imbalance between cortical excitation and inhibition in ASD and epilepsy [109]. For example, epilepsy was observed in mice lacking Pten expression in astrocytes, 50–80% of cortical neurons, and in 80–90% of hippocampal pyramidal and granule neurons (Gfap-Cre × Ptenflx/flx; Gfap-Pten) [110]. Mouse models have also revealed a central role of PTEN in synaptic structure and function. For example, Nse-Pten mutants display thickened and ectopic dendrites, increased spine density, and hypertrophic axon tracts [97]. Gfap-Pten mice exhibit altered synaptic structures in both the cortex and cerebellum [101], including increased numbers of immature dendritic spines and synapses and increased pre-synaptic terminal size [101]. Synaptic plasticity is also disrupted in these mice [101].
Imbalance between excitatory and inhibitory neurons and altered synaptic transmission has also been characterized in mouse Pten LoF studies of excitatory neurons (Nse-Cre, CaMKIIα-Cre), cortical interneuron progenitors (Nkx2.1-Cre, DlxI12b-Cre and SST-IRES-Cre), and astrocytes (Gfap-Cre) [97, 101, 102, 104, 111, 112]. For example, Pten LoF in inhibitory neurons and precursors (Nkx2.1-Cre, DlxI12b-Cre) resulted in an increased ratio of parvalbumin/somatostatin (PV/SST) interneurons, likely reflecting increased apoptosis of SST neurons. Increased inhibitory postsynaptic current (IPSC) frequency in layer II/III excitatory neurons was also observed in P30 Nkx2.1-Cre+;Ptenflx/flx mice [102]. Screening of five ASD-associated PTEN variants using an in vivo complementation assay revealed these alleles as hypomorphic in their inability to rescue either increased soma size or PV-to-SST ratio, without dominant negative effects [102].
Myelination deficits
Several studies in Pten mouse models have identified myelination defects which may contribute to circuit and behavioral dysfunction [101, 113–115]. For example, a nuclear-excluded Pten mouse model (Ptenm3m4/m3m4) displayed abnormalities in oligodendrocyte development and morphology with disrupted myelination [114]. Brain MRIs of adult Pten haploinsufficient mice identified abnormal scaling affecting multiple brain regions, with prominent white matter enlargement [106]. Cortical cultures from these mice showed increased glial cell proliferation [106]. Interestingly, gene expression profiles of three ASD mouse models, including PTEN (Ptenm3m4/m3m4), MECP2 (Rett Syndrome), and TCF4 (Pitt-Hopkins syndrome), found enrichment of myelination-related genes among the shared differentially expressed genes [115], suggesting altered myelination as another point of convergence among ASD-associated genes.
Pten mouse models display congenital ventriculomegaly
Pten plays a major regulatory role in the cellular and molecular processes governing NSC proliferation and differentiation into mature neurons, ependymal cells, and oligodendrocytes. This suggests the hypothesis that Pten LoF in NSCs may not only produce structural and functional deficits in the development of the cortical circuits underlying ASDs and epilepsy, but also underpin structural and functional deficits during development of the proximal ciliated ventricular epithelium. Consistent with this, Pten mutant mice lacking Pten expression in all cortical layers, cerebellum, and hippocampus, without diminished astrocyte expression (NGfap-Pten), develop not only epilepsy, but also progressive ventriculomegaly resulting in early fatality [98, 116]. In addition, homozygous Pten knockout in NSCs (Nestin-Pten) results in macrocephaly and ventriculomegaly due to increased proliferation, decreased apoptosis, and increased NSC size [108, 117].
While few studies examined CSF dynamics directly in Pten mutant mice, an intriguing hypothesis to explain both ventriculomegaly and megalencephaly is the overaccumulation of CSF, as CSF and its hydrostatic pressure promote neural progenitor proliferation and brain enlargement [118, 119]. Interestingly, Pten expression has been localized to the apical membrane of the choroid plexus [120], within close proximity to ion transporters and water channels, which regulate the production of CSF (recently reviewed in [121]). Disrupted Pten protein phosphatase activity led to increased levels of phosphorylated Dishevelled, a key component of WNT signaling, which prevented the formation of both primary and multicilia in ependymal cells, which are thought to be responsible for the circulation of CSF [122]. Thus, developmental pleiotropy of Pten LoF mouse models in ASD, epilepsy, and CH may be related to abnormal development of ependymal cells, choroid plexus epithelial cells, and neural progenitors, all deriving from a common progenitor pool within prenatal germinal niches [123, 124], leading to functional deficits in both assembly of cortical circuits and in the proximal ventricular zone/subventricular zone.
Zebrafish models of PTEN
The transparency and rapid embryonic neurodevelopment of zebrafish offer unparalleled ability to visualize early developmental phenotypes. The duplicated teleost genome has given rise to two zebrafish PTEN orthologs, ptena and ptenb, encoding Ptena and Ptenb proteins (which are 88% and 86% identical to the human protein, respectively) [125]. Both paralogs are broadly expressed during early somitogenesis and in the central nervous system by 24 h post fertilization (hpf) [125]. Homozygous LoF of either allele is viable and exhibits no obvious morphological phenotype, but LoF of both ptena and ptenb results in embryonic lethality at 5 days post fertilization (dpf) [126]. Homozygous double mutants display an apparent increase in head size at 4 dpf [126]. While their development is grossly normal until 48 hpf, double mutants display severe developmental abnormalities by 4 dpf, partially reversible by administration of a PI3K inhibitor from 2 dpf, implicating PI3K pathway hyperactivation [126]. Homozygous double mutants also exhibit increased cell proliferation and decreased apoptosis. At 7 months, ptenb null mutants exhibit eye tumors with increased p-Akt expression [126]. These studies demonstrate overlapping cellular and molecular phenotypes in zebrafish and mouse models, consistent with evolutionary conservation of PTEN-PI3K-AKT signaling.
Pharmacological rescue in PTEN mouse models
There is evidence from preclinical models that pharmacological inhibition of mTOR signaling can reverse abnormal phenotypes. For example, treatment of Nse-Pten mice with the mTORC1 inhibitor, rapamycin, reversed social deficits, seizures, and structural abnormalities, including macrocephaly, neuronal hypertrophy, and spine density [127]. Similarly, rapamycin decreased seizures and rescued neuronal hypertrophy in mice lacking Pten in all cortical layers, cerebellum, and hippocampus using a modified Gfap-Cre (Gfap-cre x Ptenflx/flx; NGfap-Pten) [128]. In addition, early treatment of Pten+/− mice with a pharmacological inhibitor of the mTOR component, S6K1, reversed social deficits present in female mice as well as neuronal hypertrophy in cortical layer V [104]. Further, this treatment rescued increased activity and connectivity in the medial prefrontal cortex and basolateral amygdala when administered during development, but not in adulthood, suggesting there are critical periods during which mTORC1 modulation is important for the establishment of these circuits [104]. Preclinical investigations in mice have also demonstrated rapamycin efficacy in preventing ventriculomegaly [129].
Targeting the mTOR pathway in ASD and CH
No FDA-approved drugs are currently available to treat core symptoms of ASD or to attenuate ventriculomegaly in CH, but active drug discovery efforts are ongoing. At present, the only FDA-approved pharmacological treatments for ASD are atypical antipsychotics, risperidone and aripiprazole, indicated for aggression and irritability, but not for the core deficits [130]. Given evidence from preclinical studies, a number of clinical studies have investigated targeting the mTOR pathway in NDDs. Most progress to date has been in TSC, where studies have demonstrated benefit for mTOR inhibitors in treating TSC-associated seizures (reviewed in [131]). Specifically, everolimus is FDA-approved for adjunctive treatment of complex partial seizures in children (>2 years old) and adults with TSC [132]. Sirolimus is also effective in treating seizures in children with TSC [133].
At present, clinicaltrials.gov includes studies investigating pharmacological treatment targeting the mTOR pathway for ASD-associated behaviors in TSC and neurocognition in PHTS. NCT02991807I is testing mTOR inhibitor everolimus in individuals with PTEN mutations to assess safety and possible improvements in neurocognition and behavior. The completed study NCT01730209II tested everolimus in children and adolescents aged 4–17 years with TSC, yet found no improvement in intelligence quotient or ASD behaviors [134]. Another double-blind randomized, placebo-controlled trial (NCT01289912III) also found no significant improvement in neurocognition or behavior in children and adolescents with TSC following 6 months of everolimus treatment [135]. The lack of mTOR inhibitor efficacy in treating ASD-associated behaviors might reflect the need for earlier treatment, in view of the importance of critical periods of plasticity during neurodevelopment [131, 134, 135]. In addition, early seizure treatment in children with TSC may be important for improving neurocognitive and behavioral outcomes [135], a finding supported by a recent preclinical study showing that everolimus improves social behaviors in Tsc2+/− rats but not after early status epilepticus [136]. Interestingly, a recent retrospective study showed that both everolimus and sirolimus attenuated ventriculomegaly in six of 13 TSC patients diagnosed with obstructive hydrocephalus secondary to subependymal giant cell astrocytoma [137]. Because TSC1/2 is downstream of PTEN, it is possible that targeting different components of the PI3K-AKT-mTOR pathway will be important for treating specific clinical features of these disorders.
Concluding Remarks and Future Perspectives
Emerging evidence from genetics and integrative genomics strongly implicates PTEN and mTOR signaling in both ASD and CH, suggesting these disorders might share similar pathophysiological mechanisms (Figure 3). While CH has been classically attributed to abnormal CSF “plumbing,” and ASD is primarily characterized by behavioral symptoms without pathognomonic structural brain abnormalities, convergence on a common pathway has the potential to shape our conceptualization of the two disorders. For example, this reconceptualization supports characterizing CH as an NDD, similar to ASD, and suggests that children with CH may benefit from early behavioral screening and intervention, while structural imaging might be relevant for a subset of children with ASD at risk for ventriculomegaly. Moreover, the identification of PTEN-PI3K-mTOR as a central pathway has the potential not only to shed light on novel mechanisms underlying both disorders but also to present new avenues for developing novel therapeutics.
Understanding the precise mechanisms by which PTEN LoF leads to a range of neurodevelopmental phenotypes represents a critical next step (see Outstanding Questions). For example, it will be important to investigate in animal models the neural cell types, developmental time points, and molecular components downstream of PTEN that are most relevant for the development of specific phenotypes. The considerable pleiotropy of PTEN also represents a central challenge. Additional preclinical studies aimed at differentiating mechanisms by which specific PTEN variants cause diverse phenotypes are needed to elucidate how PTEN LoF predisposes to a range of clinical disorders and identify improved therapeutic targets. At the same time, gaining insight into a common pathway underlying ASD and CH holds important scientific and clinical implications for diagnosis and treatment of both disorders and identifies a targetable pathway that might lead to new avenues for pharmacological development.
Supplementary Material
Outstanding Questions Box.
How does PTEN disruption in specific neural cell types and brain regions predispose to the circuit and behavioral deficits seen in ASD?
Which period of development is most critical for PTEN function and does this period differ in distinct NDDs?
What is the mechanism underlying the pleiotropy of PTEN mutations? To what extent is the type of mutation (i.e., missense, nonsense, splice site) or its function (i.e., LoF, dominant negative) associated with specific clinical features?
How can the pleiotropy of PTEN mutations be leveraged to identify specific therapeutic targets for PTEN-related disorders?
There is evidence that PI3K-Akt-mTOR might represent a common pathway across NDDs. What are the mechanisms by which disruption of this common pathway leads to seemingly disparate clinical presentations of CH and/or ASD?
How can cellular and animal model systems be leveraged for screening the functionality of PTEN variants?
How can animal models of PTEN mutation help uncover translationally relevant therapeutic targets for future investigation in NDDs?
Highlights.
Structural brain abnormalities, such as enlargement of the brain’s ventricles, occur at an increased frequency in individuals with autism spectrum disorder (ASD). Conversely, rates of ASD are increased in congenital hydrocephalus (CH) compared to the general population. These observations raise the question of whether the two disorders might share common neurodevelopmental mechanisms.
Mutations in PTEN are associated with a range of neurodevelopmental disorders, including ASD and CH.
Integrative genomics highlights PTEN-PI3K-mTOR as one potentially shared pathway between ASD and CH.
Mouse models of PTEN loss of function exhibit macrocephaly, social deficits, seizures, synaptic alterations, myelination defects, and enlarged brain ventricles.
Preclinical studies in mouse models suggest that targeting the mTOR pathway can reverse structural and behavioral abnormalities associated with PTEN loss of function.
The PTEN-PI3K-mTOR pathway may represent a potential pharmacological target for further investigation in ASD and CH.
Acknowledgements.
KTK is supported by the NIH (NRCDP K12 228168, 1RO1NS109358, and R01 NS111029-01A1); the Hydrocephalus Association, March of Dimes, Simons Foundation, Swebilius Foundation, and Rudi Schulte Research Institute.
EJH is supported by NIH R01MH116002, Binational Science Foundation, Kavli Foundation, National Genetics Foundation, Simons Foundation, Spector Fund, and the Swebilius Foundation.
TD is supported by NIH Medical Scientist Training Program Training Grant T32GM007205 and National Institute Of Neurological Disorders And Stroke of the National Institutes of Health under Award Number F31NS115519.
MC is supported by the Interdepartmental Neuroscience Program at Yale and the NIH Training Program Grant in Genetics NIH 5T32GM007499
The content of this report is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Glossary
- De novo mutation
a mutation that is not inherited, but arose in the gametes or during early embryonic development
- Dominant negative mutation
a mutation that causes the gene product to suppress the normal function of the wild-type gene product
- Hamartoma
a benign tumor that can occur in different parts of the body (e.g. brain, skin), often associated with a genetic condition
- Integrative genomics
An analysis approach that integrates data from large-scale genetic studies with biological data sets, such as RNA-seq and protein-protein interaction networks
- Lipoma
benign tumor consisting of fatty tissue
- Loss of function (LoF) mutation
a mutation resulting in a loss of the gene product’s function
- Macrocephaly
increased head circumference, defined as over 2 standard deviations above average
- Megalencephaly
abnormal enlargement of the brain, defined as brain weight over 2.5 standard deviations above average
- Pathognomonic
a characteristic feature of a disorder that defines the diagnosis
- Pleiotropy
the production of two apparently unrelated effects by the same gene mutation, or in the clinical context, the association of mutations in a particular gene with multiple clinical outcomes. For example, PTEN is highly pleiotropic because mutations in this gene have been associated with ASD, CH, and cancer
- Polymicrogyria
abnormal brain development characterized by an increased number of small surface folds
- Ventriculomegaly
enlargement of the CSF-filled brain ventricles
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors have no conflicts of interest to report.
Search strategy and selection criteria.
We searched PubMed for articles in all year ranges with multiple combinations of search terms including, “autism spectrum disorder”, “congenital hydrocephalus”, “ventriculomegaly”, “PTEN”, “mTORC1”. There were no language exclusions and articles chosen were based on relevance to topics covered in this review.
References.
- 1.Thapar A et al. (2017) Neurodevelopmental disorders. Lancet Psychiatry 4 (4), 339–346. [DOI] [PubMed] [Google Scholar]
- 2.Maenner MJ et al. (2020) Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years - Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2016. MMWR Surveill Summ 69 (4), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Association, A.P. (2013) Diagnostic and statistical manual of mental disorders (5th ed.), American Psychiatric Publishing. [Google Scholar]
- 4.Movsas TZ et al. (2013) Autism spectrum disorder is associated with ventricular enlargement in a low birth weight population. J Pediatr 163 (1), 73–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turner AH et al. (2016) Pallidum and lateral ventricle volume enlargement in autism spectrum disorder. Psychiatry Res Neuroimaging 252, 40–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haar S et al. (2016) Anatomical Abnormalities in Autism? Cereb Cortex 26 (4), 1440–52. [DOI] [PubMed] [Google Scholar]
- 7.Shen MD (2018) Cerebrospinal fluid and the early brain development of autism. J Neurodev Disord 10 (1), 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shen MD et al. (2017) Increased Extra-axial Cerebrospinal Fluid in High-Risk Infants Who Later Develop Autism. Biol Psychiatry 82 (3), 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shen MD et al. (2018) Extra-axial cerebrospinal fluid in high-risk and normal-risk children with autism aged 2–4 years: a case-control study. Lancet Psychiatry 5 (11), 895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen MD et al. (2013) Early brain enlargement and elevated extra-axial fluid in infants who develop autism spectrum disorder. Brain 136 (Pt 9), 2825–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lindquist B et al. (2006) Behavioural problems and autism in children with hydrocephalus : a population-based study. Eur Child Adolesc Psychiatry 15 (4), 214–9. [DOI] [PubMed] [Google Scholar]
- 12.Fernell E et al. (1991) Autistic symptoms in children with infantile hydrocephalus. Acta Paediatr Scand 80 (4), 451–7. [DOI] [PubMed] [Google Scholar]
- 13.Rekate HL (2008) The definition and classification of hydrocephalus: a personal recommendation to stimulate debate. Cerebrospinal Fluid Res 5, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kahle KT et al. (2016) Hydrocephalus in children. Lancet 387 (10020), 788–99. [DOI] [PubMed] [Google Scholar]
- 15.Bondurant CP and Jimenez DF (1995) Epidemiology of cerebrospinal fluid shunting. Pediatr Neurosurg 23 (5), 254–8; discussion 259. [DOI] [PubMed] [Google Scholar]
- 16.Zhang J et al. (2006) Genetics of human hydrocephalus. J Neurol 253 (10), 1255–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fouyas IP et al. (1996) Use of intracranial pressure monitoring in the management of childhood hydrocephalus and shunt-related problems. Neurosurgery 38 (4), 726–31; discussion 731–2. [PubMed] [Google Scholar]
- 18.Whittle IR et al. (1985) Intracranial pressure changes in arrested hydrocephalus. J Neurosurg 62 (1), 77–82. [DOI] [PubMed] [Google Scholar]
- 19.Lainhart JE et al. (1997) Macrocephaly in children and adults with autism. J Am Acad Child Adolesc Psychiatry 36 (2), 282–90. [DOI] [PubMed] [Google Scholar]
- 20.Fombonne E et al. (1999) Microcephaly and macrocephaly in autism. J Autism Dev Disord 29 (2), 113–9. [DOI] [PubMed] [Google Scholar]
- 21.Bourgeron T (2009) A synaptic trek to autism. Curr Opin Neurobiol 19 (2), 231–4. [DOI] [PubMed] [Google Scholar]
- 22.Sato A (2016) mTOR, a Potential Target to Treat Autism Spectrum Disorder. CNS Neurol Disord Drug Targets 15 (5), 533–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen J et al. (2014) Dysregulation of the IGF-I/PI3K/AKT/mTOR signaling pathway in autism spectrum disorders. Int J Dev Neurosci 35, 35–41. [DOI] [PubMed] [Google Scholar]
- 24.Jin SC et al. (2020) Exome sequencing implicates genetic disruption of prenatal neuro-gliogenesis in sporadic congenital hydrocephalus. Nat Med 26 (11), 1754–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rademacher S and Eickholt BJ (2019) PTEN in Autism and Neurodevelopmental Disorders. Cold Spring Harb Perspect Med 9 (11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Satterstrom FK et al. (2020) Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 180 (3), 568–584 e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sparks BF et al. (2002) Brain structural abnormalities in young children with autism spectrum disorder. Neurology 59 (2), 184–92. [DOI] [PubMed] [Google Scholar]
- 28.Nickl-Jockschat T et al. (2012) Brain structure anomalies in autism spectrum disorder--a meta-analysis of VBM studies using anatomic likelihood estimation. Hum Brain Mapp 33 (6), 1470–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Courchesne E et al. (2003) Evidence of brain overgrowth in the first year of life in autism. JAMA 290 (3), 337–44. [DOI] [PubMed] [Google Scholar]
- 30.Redcay E and Courchesne E (2005) When is the brain enlarged in autism? A meta-analysis of all brain size reports. Biol Psychiatry 58 (1), 1–9. [DOI] [PubMed] [Google Scholar]
- 31.Schumann CM et al. (2010) Longitudinal magnetic resonance imaging study of cortical development through early childhood in autism. J Neurosci 30 (12), 4419–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dinstein I and Shelef I (2018) Anatomical brain abnormalities and early detection of autism. Lancet Psychiatry 5 (11), 857–859. [DOI] [PubMed] [Google Scholar]
- 33.Lichtenstein P et al. (2010) The genetics of autism spectrum disorders and related neuropsychiatric disorders in childhood. Am J Psychiatry 167 (11), 1357–63. [DOI] [PubMed] [Google Scholar]
- 34.Geschwind DH and Flint J (2015) Genetics and genomics of psychiatric disease. Science 349 (6255), 1489–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Polderman TJ et al. (2015) Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat Genet 47 (7), 702–9. [DOI] [PubMed] [Google Scholar]
- 36.Werling DM et al. (2018) An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nat Genet 50 (5), 727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grove J et al. (2019) Identification of common genetic risk variants for autism spectrum disorder. Nat Genet 51 (3), 431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Furey CG et al. (2018) De Novo Mutation in Genes Regulating Neural Stem Cell Fate in Human Congenital Hydrocephalus. Neuron 99 (2), 302–314 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bailey A et al. (1995) Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med 25 (1), 63–77. [DOI] [PubMed] [Google Scholar]
- 40.Constantino JN et al. (2013) Autism recurrence in half siblings: strong support for genetic mechanisms of transmission in ASD. Mol Psychiatry 18 (2), 137–8. [DOI] [PubMed] [Google Scholar]
- 41.Ozonoff S et al. (2011) Recurrence risk for autism spectrum disorders: a Baby Siblings Research Consortium study. Pediatrics 128 (3), e488–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ritvo ER et al. (1985) Concordance for the syndrome of autism in 40 pairs of afflicted twins. Am J Psychiatry 142 (1), 74–7. [DOI] [PubMed] [Google Scholar]
- 43.Rosenberg RE et al. (2009) Characteristics and concordance of autism spectrum disorders among 277 twin pairs. Arch Pediatr Adolesc Med 163 (10), 907–14. [DOI] [PubMed] [Google Scholar]
- 44.Sandin S et al. (2014) The familial risk of autism. JAMA 311 (17), 1770–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steffenburg S et al. (1989) A twin study of autism in Denmark, Finland, Iceland, Norway and Sweden. J Child Psychol Psychiatry 30 (3), 405–16. [DOI] [PubMed] [Google Scholar]
- 46.Betancur C (2011) Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res 1380, 42–77. [DOI] [PubMed] [Google Scholar]
- 47.Muhle RA et al. (2018) The Emerging Clinical Neuroscience of Autism Spectrum Disorder: A Review. JAMA Psychiatry 75 (5), 514–523. [DOI] [PubMed] [Google Scholar]
- 48.Winden KD et al. (2018) Abnormal mTOR Activation in Autism. Annu Rev Neurosci 41, 1–23. [DOI] [PubMed] [Google Scholar]
- 49.Anney R et al. (2012) Individual common variants exert weak effects on the risk for autism spectrum disorders. Hum Mol Genet 21 (21), 4781–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gaugler T et al. (2014) Most genetic risk for autism resides with common variation. Nat Genet 46 (8), 881–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klei L et al. (2012) Common genetic variants, acting additively, are a major source of risk for autism. Mol Autism 3 (1), 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sanders SJ et al. (2012) De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485 (7397), 237–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.O’Roak BJ et al. (2012) Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485 (7397), 246–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Neale BM et al. (2012) Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485 (7397), 242–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Iossifov I et al. (2014) The contribution of de novo coding mutations to autism spectrum disorder. Nature 515 (7526), 216–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.De Rubeis S et al. (2014) Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515 (7526), 209–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.O’Roak BJ et al. (2012) Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338 (6114), 1619–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sanders SJ et al. (2015) Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 87 (6), 1215–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Willsey AJ et al. (2013) Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155 (5), 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Parikshak NN et al. (2013) Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155 (5), 1008–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Adle-Biassette H et al. (2013) Neuropathological review of 138 cases genetically tested for X-linked hydrocephalus: evidence for closely related clinical entities of unknown molecular bases. Acta Neuropathol 126 (3), 427–42. [DOI] [PubMed] [Google Scholar]
- 62.Kousi M and Katsanis N (2016) The Genetic Basis of Hydrocephalus. Annu Rev Neurosci 39, 409–35. [DOI] [PubMed] [Google Scholar]
- 63.Walker RL et al. (2019) Genetic Control of Expression and Splicing in Developing Human Brain Informs Disease Mechanisms. Cell 179 (3), 750–771.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Polioudakis D et al. (2019) A Single-Cell Transcriptomic Atlas of Human Neocortical Development during Mid-gestation. Neuron 103 (5), 785–801.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li J et al. (1997) PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275 (5308), 1943–7. [DOI] [PubMed] [Google Scholar]
- 66.Steck PA et al. (1997) Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet 15 (4), 356–62. [DOI] [PubMed] [Google Scholar]
- 67.Liaw D et al. (1997) Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet 16 (1), 64–7. [DOI] [PubMed] [Google Scholar]
- 68.Marsh DJ et al. (1997) Germline mutations in PTEN are present in Bannayan-Zonana syndrome. Nat Genet 16 (4), 333–4. [DOI] [PubMed] [Google Scholar]
- 69.Marsh DJ et al. (1998) Mutation spectrum and genotype-phenotype analyses in Cowden disease and Bannayan-Zonana syndrome, two hamartoma syndromes with germline PTEN mutation. Hum Mol Genet 7 (3), 507–15. [DOI] [PubMed] [Google Scholar]
- 70.Butler MG et al. (2005) Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet 42 (4), 318–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Herman GE et al. (2007) Increasing knowledge of PTEN germline mutations: Two additional patients with autism and macrocephaly. Am J Med Genet A 143a (6), 589–93. [DOI] [PubMed] [Google Scholar]
- 72.Buxbaum JD et al. (2007) Mutation screening of the PTEN gene in patients with autism spectrum disorders and macrocephaly. Am J Med Genet B Neuropsychiatr Genet 144b (4), 484–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Varga EA et al. (2009) The prevalence of PTEN mutations in a clinical pediatric cohort with autism spectrum disorders, developmental delay, and macrocephaly. Genet Med 11 (2), 111–7. [DOI] [PubMed] [Google Scholar]
- 74.Reardon W et al. (2001) A novel germline mutation of the PTEN gene in a patient with macrocephaly, ventricular dilatation, and features of VATER association. J Med Genet 38 (12), 820–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Goffin A et al. (2001) PTEN mutation in a family with Cowden syndrome and autism. Am J Med Genet 105 (6), 521–4. [DOI] [PubMed] [Google Scholar]
- 76.Zori RT et al. (1998) Germline PTEN mutation in a family with Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome. Am J Med Genet 80 (4), 399–402. [PubMed] [Google Scholar]
- 77.McBride KL et al. (2010) Confirmation study of PTEN mutations among individuals with autism or developmental delays/mental retardation and macrocephaly. Autism Res 3 (3), 137–41. [DOI] [PubMed] [Google Scholar]
- 78.Klein S et al. (2013) Macrocephaly as a clinical indicator of genetic subtypes in autism. Autism Res 6 (1), 51–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Frazier TW et al. (2015) Molecular and phenotypic abnormalities in individuals with germline heterozygous PTEN mutations and autism. Mol Psychiatry 20 (9), 1132–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schaefer GB et al. (2013) Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions. Genet Med 15 (5), 399–407. [DOI] [PubMed] [Google Scholar]
- 81.Yeung KS et al. (2017) Identification of mutations in the PI3K-AKT-mTOR signalling pathway in patients with macrocephaly and developmental delay and/or autism. Mol Autism 8, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Marchese M et al. (2014) Autism-epilepsy phenotype with macrocephaly suggests PTEN, but not GLIALCAM, genetic screening. BMC Med Genet 15, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Leslie NR and Longy M (2016) Inherited PTEN mutations and the prediction of phenotype. Semin Cell Dev Biol 52, 30–8. [DOI] [PubMed] [Google Scholar]
- 84.Han SY et al. (2000) Functional evaluation of PTEN missense mutations using in vitro phosphoinositide phosphatase assay. Cancer Res 60 (12), 3147–51. [PubMed] [Google Scholar]
- 85.Papa A et al. (2014) Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell 157 (3), 595–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang H et al. (2010) Allele-specific tumor spectrum in pten knockin mice. Proc Natl Acad Sci U S A 107 (11), 5142–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rodríguez-Escudero I et al. (2011) A comprehensive functional analysis of PTEN mutations: implications in tumor- and autism-related syndromes. Hum Mol Genet 20 (21), 4132–42. [DOI] [PubMed] [Google Scholar]
- 88.Spinelli L et al. (2015) Functionally distinct groups of inherited PTEN mutations in autism and tumour syndromes. J Med Genet 52 (2), 128–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Trotman LC et al. (2007) Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 128 (1), 141–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mingo J et al. (2018) A pathogenic role for germline PTEN variants which accumulate into the nucleus. Eur J Hum Genet 26 (8), 1180–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fricano-Kugler CJ et al. (2018) Nuclear Excluded Autism-Associated Phosphatase and Tensin Homolog Mutations Dysregulate Neuronal Growth. Biol Psychiatry 84 (4), 265–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tilot AK et al. (2014) Germline disruption of Pten localization causes enhanced sex-dependent social motivation and increased glial production. Hum Mol Genet 23 (12), 3212–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mighell TL et al. (2020) An Integrated Deep-Mutational-Scanning Approach Provides Clinical Insights on PTEN Genotype-Phenotype Relationships. Am J Hum Genet 106 (6), 818–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Post KL et al. (2020) Multi-model functionalization of disease-associated PTEN missense mutations identifies multiple molecular mechanisms underlying protein dysfunction. Nat Commun 11 (1), 2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Di Cristofano A et al. (1998) Pten is essential for embryonic development and tumour suppression. Nat Genet 19 (4), 348–55. [DOI] [PubMed] [Google Scholar]
- 96.Suzuki A et al. (1998) High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr Biol 8 (21), 1169–78. [DOI] [PubMed] [Google Scholar]
- 97.Kwon CH et al. (2006) Pten regulates neuronal arborization and social interaction in mice. Neuron 50 (3), 377–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Backman SA et al. (2001) Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat Genet 29 (4), 396–403. [DOI] [PubMed] [Google Scholar]
- 99.Clipperton-Allen AE and Page DT (2020) Connecting Genotype with Behavioral Phenotype in Mouse Models of Autism Associated with PTEN Mutations. Cold Spring Harb Perspect Med 10 (9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Skelton PD et al. (2020) The Role of PTEN in Neurodevelopment. Mol Neuropsychiatry 5 (Suppl 1), 60–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fraser MM et al. (2008) Phosphatase and tensin homolog, deleted on chromosome 10 deficiency in brain causes defects in synaptic structure, transmission and plasticity, and myelination abnormalities. Neuroscience 151 (2), 476–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Vogt D et al. (2015) The parvalbumin/somatostatin ratio is increased in Pten mutant mice and by human PTEN ASD alleles. Cell Rep 11 (6), 944–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen Y et al. (2015) Pten Mutations Alter Brain Growth Trajectory and Allocation of Cell Types through Elevated beta-Catenin Signaling. J Neurosci 35 (28), 10252–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Huang WC et al. (2016) Hyperconnectivity of prefrontal cortex to amygdala projections in a mouse model of macrocephaly/autism syndrome. Nat Commun 7, 13421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chen CJ et al. (2019) Therapeutic inhibition of mTORC2 rescues the behavioral and neurophysiological abnormalities associated with Pten-deficiency. Nat Med 25 (11), 1684–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Clipperton-Allen AE et al. (2019) Pten haploinsufficiency disrupts scaling across brain areas during development in mice. Transl Psychiatry 9 (1), 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Marino S et al. (2002) PTEN is essential for cell migration but not for fate determination and tumourigenesis in the cerebellum. Development 129 (14), 3513–22. [DOI] [PubMed] [Google Scholar]
- 108.Groszer M et al. (2001) Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science 294 (5549), 2186–9. [DOI] [PubMed] [Google Scholar]
- 109.Ogawa S et al. (2007) A seizure-prone phenotype is associated with altered free-running rhythm in Pten mutant mice. Brain Res 1168, 112–23. [DOI] [PubMed] [Google Scholar]
- 110.Fraser MM et al. (2004) Pten loss causes hypertrophy and increased proliferation of astrocytes in vivo. Cancer Res 64 (21), 7773–9. [DOI] [PubMed] [Google Scholar]
- 111.Sperow M et al. (2012) Phosphatase and tensin homologue (PTEN) regulates synaptic plasticity independently of its effect on neuronal morphology and migration. J Physiol 590 (4), 777–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wong FK et al. (2018) Pyramidal cell regulation of interneuron survival sculpts cortical networks. Nature 557 (7707), 668–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Goebbels S et al. (2010) Elevated phosphatidylinositol 3,4,5-trisphosphate in glia triggers cell-autonomous membrane wrapping and myelination. J Neurosci 30 (26), 8953–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lee H et al. (2019) Constitutional mislocalization of Pten drives precocious maturation in oligodendrocytes and aberrant myelination in model of autism spectrum disorder. Transl Psychiatry 9 (1), 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Phan BN et al. (2020) A myelin-related transcriptomic profile is shared by Pitt-Hopkins syndrome models and human autism spectrum disorder. Nat Neurosci 23 (3), 375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kwon CH et al. (2001) Pten regulates neuronal soma size: a mouse model of Lhermitte-Duclos disease. Nat Genet 29 (4), 404–11. [DOI] [PubMed] [Google Scholar]
- 117.Groszer M et al. (2006) PTEN negatively regulates neural stem cell self-renewal by modulating G0–G1 cell cycle entry. Proc Natl Acad Sci U S A 103 (1), 111–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gato A et al. (2014) Embryonic cerebrospinal fluid in brain development: neural progenitor control. Croat Med J 55 (4), 299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Desmond ME and Jacobson AG (1977) Embryonic brain enlargement requires cerebrospinal fluid pressure. Dev Biol 57 (1), 188–98. [DOI] [PubMed] [Google Scholar]
- 120.Christensen IB et al. (2018) Choroid plexus epithelial cells express the adhesion protein P-cadherin at cell-cell contacts and syntaxin-4 in the luminal membrane domain. Am J Physiol Cell Physiol 314 (5), C519–c533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.MacAulay N (2021) Molecular mechanisms of brain water transport. Nat Rev Neurosci 22 (6), 326–344. [DOI] [PubMed] [Google Scholar]
- 122.Shnitsar I et al. (2015) PTEN regulates cilia through Dishevelled. Nat Commun 6, 8388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dani N et al. (2021) A cellular and spatial map of the choroid plexus across brain ventricles and ages. Cell 184 (11), 3056–3074.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ortiz-Álvarez G et al. (2019) Adult Neural Stem Cells and Multiciliated Ependymal Cells Share a Common Lineage Regulated by the Geminin Family Members. Neuron 102 (1), 159–172.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Croushore JA et al. (2005) Ptena and ptenb genes play distinct roles in zebrafish embryogenesis. Dev Dyn 234 (4), 911–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Faucherre A et al. (2008) Zebrafish pten genes have overlapping and non-redundant functions in tumorigenesis and embryonic development. Oncogene 27 (8), 1079–86. [DOI] [PubMed] [Google Scholar]
- 127.Zhou J et al. (2009) Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J Neurosci 29 (6), 1773–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ljungberg MC et al. (2009) Rapamycin suppresses seizures and neuronal hypertrophy in a mouse model of cortical dysplasia. Dis Model Mech 2 (7–8), 389–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Foerster P et al. (2017) mTORC1 signaling and primary cilia are required for brain ventricle morphogenesis. Development 144 (2), 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Frye RE (2018) Social Skills Deficits in Autism Spectrum Disorder: Potential Biological Origins and Progress in Developing Therapeutic Agents. CNS Drugs 32 (8), 713–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ess KC and Franz DN (2019) Everolimus for cognition/autism in children with tuberous sclerosis complex: Definitive outcomes deferred. Neurology 93 (2), 51–52. [DOI] [PubMed] [Google Scholar]
- 132.French JA et al. (2016) Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet 388 (10056), 2153–2163. [DOI] [PubMed] [Google Scholar]
- 133.Overwater IE et al. (2016) Sirolimus for epilepsy in children with tuberous sclerosis complex: A randomized controlled trial. Neurology 87 (10), 1011–8. [DOI] [PubMed] [Google Scholar]
- 134.Overwater IE et al. (2019) A randomized controlled trial with everolimus for IQ and autism in tuberous sclerosis complex. Neurology 93 (2), e200–e209. [DOI] [PubMed] [Google Scholar]
- 135.Krueger DA et al. (2017) Everolimus for treatment of tuberous sclerosis complex-associated neuropsychiatric disorders. Ann Clin Transl Neurol 4 (12), 877–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Petrasek T et al. (2021) mTOR inhibitor improves autistic-like behaviors related to Tsc2 haploinsufficiency but not following developmental status epilepticus. J Neurodev Disord 13 (1), 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Weidman DR et al. (2020) The effect of mTOR inhibition on obstructive hydrocephalus in patients with tuberous sclerosis complex (TSC) related subependymal giant cell astrocytoma (SEGA). J Neurooncol 147 (3), 731–736. [DOI] [PubMed] [Google Scholar]
- 138.Zhang W et al. (2019) Mutplot: An easy-to-use online tool for plotting complex mutation data with flexibility. PLoS One 14 (5), e0215838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Apweiler R et al. (2004) UniProt: the Universal Protein knowledgebase. Nucleic Acids Res 32 (Database issue), D115–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Resources
- I.This study is registered with ClinicalTrials.gov ( NCT0299180) https://clinicaltrials.gov/ct2/show/NCT02991807.
- II.This study is registered with ClinicalTrials.gov ( NCT01730209) https://clinicaltrials.gov/ct2/show/NCT01730209.
- III.This study is registered with ClinicalTrials.gov ( NCT01289912) https://clinicaltrials.gov/ct2/show/NCT01289912.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.