

Abstract
Background:
Severe injury predisposes patients to trauma-induced coagulopathy (TIC), which may be subdivided by the state of fibrinolysis. Systemic hyperfibrinolysis (HF) occurs in ~25% of these patients with mortality as high as 70%. Severe injury also causes the release of numerous intracellular proteins which may affect coagulation, one of which is hemoglobin, and hemoglobin substitutes induce HF in vitro. We hypothesize that the α-globin chain of hemoglobin potentiates HF in vitro by augmenting plasmin activity.
Methods:
Proteomic analysis was completed on a pilot study of 30 injured patients prior to blood component resuscitation, stratified by their state of fibrinolysis, plus 10 healthy controls. Different concentrations of intact hemoglobin (HbA), the α- and β-globin chains, or normal saline (NS, controls) were added to whole blood and tPA-challenge thrombelastography (TEG) was used to assess the degree of fibrinolysis. Interactions with plasminogen (PLG) were evaluated using surface plasmon resonance (SPR). TPA-induced plasmin activity was evaluated in the presence of the α-globin chain.
Results:
Only the α- and β-globin chains increased in HF patients (p<.01). The α-globin chain but not HbA or the β-globin chain decreased the R-time and significantly increased Ly30 on CN-TEGs (p<0.05). The PLG and α-globin chain had interaction kinetics similar to tPA:PLG, and the α-globin chain increased tPA-induced plasmin activity.
Conclusions:
the α-globin chain caused HF in vitro by binding to PLG and augmenting plasmin activity and may represent a circulating “moonlighting” mediator released by the tissue damage and hemorrhagic shock inherent to severe injury.
Level of Evidence =
III prognostic.
Keywords: α-globin chain, injury, plasmin, plasminogen, surface plasmon resonance
Background
Fibrinolysis is a physiologic process that ensures vascular patency, and multiple processes can affect the cellular and plasma fibrinolytic activity (1). Although fibrinolysis may be protective, systemic hyperfibrinolysis can lead to death from exsanguination (2–6). Approximately 25% of severely injured patients (new injury severity score (NISS) >15) develop trauma-induced coagulopathy (TIC), which was initially attributed to impaired thrombin generation or systemic hyperfibrinolysis (HF) (3–5, 7, 8). The mechanisms driving TIC have been attributed to a number of mechanisms including altered platelet activity, endothelial dysfunction, circulating extracellular vesicles, altered thrombin generation, hypoperfusion/ischemia leading to the elevation of tissue plasminogen activator (tPA), and a reduction in serine protease inhibitors (serpins) and clotting factors by activated protein C (3–5, 7–12).
Severe injury may distort physiological clot remodeling towards the two extremes: systemic hyperfibrinolysis or fibrinolysis shutdown, with both resulting in increased mortality versus similarly injured patients with normal clot remodeling (13–16). Thrombelastography (TEG) provides not only measures of clot strength, the maximal amplitude (MA), but also the kinetics of clot formation (R-time and angle) and clot remodeling, which includes total dissolution, systemic hyperfibrinolysis, as well as total cessation of clot remodeling (fibrinolytic shutdown) (7, 15, 17–20). The lysis time 30 (Ly30) allows for segregation among patients who present with systemic HF, physiologic fibrinolysis (PF) fibrinolytic shutdown (SD), and the differences among the groups become greater with the addition of exogenous tPA to the TEG assay. These tPA-challenge TEGs are an excellent predictor of massive transfusion in the severely injured (13, 19–24). Elevated tPA levels are associated with HF through plasmin activation, and similarly, patients with congenital plasminogen activator inhibitor-1 (PAI-1) deficiency may develop HF (15, 25–27). Conversely, SD has been associated with increases in PAI-1 in patients post-operatively as well as trauma patients, the source of which is likely from platelets, endothelial cells or organ parenchyma (28–31). Thus, in the severely injured the tPA:PAI-1 balance appears to be of importance in tipping the state of fibrinolysis towards the extremes of HF or SD; however, the triggers for HF and SD remain undefined (26–31).
In short, severe injury causes the release of intracellular proteins into the circulation many of which may bind or interact with plasma proteins including PAI-1 or tPA (24, 32–34). Hemoglobin is the most abundant intracellular protein in RBCs and hemoglobin-based oxygen carriers potentiate HF in vitro in tPA-challenged TEGs from healthy volunteers (35, 36). Therefore, we hypothesize that hemoglobin or one of its subunits, namely the α-globin chain, potentiates HF in vitro by directly affecting plasmin activity and may be one of the causes of severe systemic HF. The HF group exhibits some of the highest mortality in the severely injured (5, 12, 15, 16).
Materials & Methods
Materials
Human serum albumin, hemoglobin A (HbA), tPA, human plasminogen (PLG), ColorpHast® Indicator Strips pH 6.5–10.0, and phenylmethylsulfonyl fluoride (PMSF) were obtained from Sigma Aldrich, St. Louis, MO. Recombinant human, full-length α-globin chain and β-globin chain were purchased from Abcam (Cambridge, MA) and GeneTex (Irvine, CA), respectively. Enzyme-linked immunosorbent assays (ELISAs) for plasmin:α2anti-plasmin (PAP), thrombin:anti-thrombin (TAT) complexes and a plasmin activity assay kit were purchased from BioVision (Milpitas, CA). Plasminogen-depleted plasma was obtained from GE Healthcare Life Sciences (Pittsburgh, PA).
Patients
This pilot study consisted of consecutive trauma patients (n=130) meeting criteria for the highest level of activation at Denver Health Medical Center, a Level I trauma center, from 4/2014–4/2016. These patients were assigned to the Trauma Activation Protocol approved by the Colorado Multi-Institutional Review Board (COMIRB) with a waiver of consent (32). The criteria are patients >18 years of age and traumatic injury with any of the following: (a) blunt trauma with systolic blood pressure SBP <90 mmHg, (b) mechanically unstable pelvic injury, (c) penetrating neck/torso injuries with (SBP) <90 mmHg, or (d) gunshot wounds to the neck/torso or stab wounds to the neck/torso that require endotracheal intubation (32). Patients who had traumatic brain injury (TBI) received blood products, being treated with anticoagulants, or transferred from another facility, were excluded. Patients (29/30) had blood drawn within 60 minutes of ED arrival save one whose blood was obtained 3 hours and 25 minutes post ED presentation and these samples were obtained prior to transfusion. A number of these patients were reported previously from the physiologic fibrinolysis and systemic hyperfibrinolysis groups and are reported here with permission of the corresponding author (Permission from A. Banerjee) (32). Citrated and heparinized whole blood and platelet-free plasma samples were obtained upon arrival, before transfusing any blood products, tPA-challenged TEG assays were completed as described (19, 37). Targeted proteomic analysis employing stable isotope internal standards and ELISA measurements of selected proteins from the coagulation and fibrinolytic cascades were completed (32). The groups were stratified by the Ly30 obtained from tPA-challenged TEGs prior to any further analyses: 10 consecutive injured patients with HF (Ly30 ≥50%) 10 with fibrinolysis shutdown (SD) (Ly30 <5%) 10 with physiologic fibrinolysis (PF) (5%≤ Ly30 ≤20%) and 10 healthy controls (32).
Thrombelastography
Citrated whole blood was collected from healthy volunteers after obtaining informed consent under a COMIRB protocol. No study subject had a coagulation disorder, nor were they taking any medications which affect coagulation or fibrinolysis, including aspirin or non-steroidal anti-inflammatory drugs. Both groups had a median age of 30, contained 15 subjects, and all females were <54 years of age.
TPA-challenged [75 ng/ml] TEG assays were completed using the TEG 5000 Thrombelastography Hemostasis Analyzer (Haemonetics, Niles, IL) (19, 37). All TEGs were performed on citrated native (CN) samples in the presence of increasing concentrations of human HbA, the α-globin chain, or the β-globin chain, versus normal saline (NS). All TEGs yielded the following variables: reaction time (R-time, time to clot formation), angle (rate of clot propagation), MA, maximal clot strength, and percent clot lysis 30 minutes after reaching MA (Ly30) (19, 37).
Proteomics and ELISA measurement of plasma proteins
Target proteomics was performed using multiple reaction monitoring of stable isotope peptides (QconCAT reagents) and endogenous matching peptides from a tryptic digest of plasma from the injured patients (n=30) and healthy volunteers (n=10) were completed as described previously (32, 38–40). ELISAs for PAP and TAT complexes were completed per the manufacturer’s instructions.
Surface Plasmon Resonance (SPR)
Hemoglobin A and the α- and β-globin chains were concentrated using Microcon concentrators (Millipore) in PBS (phosphate-buffered saline, buffer pH 7.35 + 0.005% Tween20, PBS) with centrifugation at 8000g for 75 min, to a final volume of 200 μL and a final concentration of 195 μM, as determined by a bicinchoninic acid (BCA) protein assay. The α- and β-globin chains were dialyzed against PBS for binding to the ligand plasminogen.
Stock solutions and concentrated samples were stored at −20°C <24hrs before analysis. Plasminogen (ligand) was coupled to a chip to determine binding characteristics (41). The analyte: tPA, HbA and α- or β-globin chains, were then flowed over the PLG ligand, and the SPR (protein: protein interaction) of the analytes to PLG were analyzed on a Biacore 3000 instrument (GE Healthcare, Piscataway, NJ, USA) in the Biophysics Core of the University of Colorado, Denver. Standard amine-coupling chemistry was used to immobilize PLG or tPA to CM5 sensor chip surfaces at 25°C in separate experiments, and kinetics experiments were completed PLG-coupled CM5 surface for four different ligands using different concentrations in duplicate (41). Buffer blanks were used to double-reference the obtained kinetic data. Raw sensogram data were processed and fit using the Scrubber software package (Version 2.0b, BioLogic Software, Campbell, Australia; http://www.biologic.com.au). The analyte was stripped from the ligand by washing with 0.5M NaCO3 after each binding reaction prior to running the next reaction (41).
Plasmin AMC generation assays
A plasmin activity kit was adapted to determine plasmin activity in solution. The fluorescence is generated when the fluorophore AMC (7-Amino-4-Methylcoumarin) is cleaved by plasmin from a small polypeptide. The assay was performed in a white flat bottom 96-well microtiter plates and included assay buffer, plasma, α-globin chain or PMSF, initiated by tPA. PBS served as the negative control, and PMSF was used to inhibit tPA activity. The reaction mixture was pH 7.35, verified by using. Plasmin activity measurements were taken following the addition of all reagents to the well and every 3 minutes (Excitation: 360 nm, Emission: 450 nm) for 18 minutes, and the calculated slopes were compared and expressed as relative light units (RLU)/min.
Statistics
Statistical analyses and graphs were completed using GraphPad Software. The data were initially analyzed with the Shapiro-Wilk test to determine distribution. The data are presented as the mean ± the standard error of the mean (SEM), if normally distributed, and as the median ± the interquartiles (25%−75%) if not. Statistical significance was calculated on normally distributed data using an independent ANOVA followed by a Bonferroni test for multiple comparisons or if non-parametric, by the Kruskal-Wallis test followed by a Dunn’s test for multiple comparisons.
Results
Patient Demographics and Clinical TEG Analysis.
Patients with HF were the most severely injured as quantified by the NISS and Glasgow coma scales (GCS) scores (Table 1). There was not a difference amongst the injured patient groups in age or body mass, although the HF patients evidenced a lower plasma pH with higher base deficits than the PF or SD patients (Table 1). Importantly there was no difference in time to blood sample collection after injury because 29/30 samples were acquired within 1 hour with the longest time to collection being 3 hours 25 minutes in a female who was stabbed and did not require any crystalloid in the field. Seventy percent of HF patients died, and the mortality was zero in the PF and SD patient groups. Although the citrated, rapid TEGs, with added tissue factor, were used for clinical decisions, because R-TEGs are faster and the samples do not clot, we employed the tPA-challenged TEGs which allow for greater separation in the Ly30 amongst patient groups (Table 1.) (20, 22, 42). The groups satisfied the CN-TEG cutoffs as well as the R-TEG cutoffs for HF, PF, and SD as previously published (data not shown).
Table 1.
Patient Demographics and Clinical Laboratory Assessment
| Physiologic Lysis (n=10) | Hyperfibrinolysis (n=10) | Shutdown (n=10) | |
|---|---|---|---|
| Median (Q1–Q3) | Median (Q1–Q3) | Median (Q1–Q3) | |
| Age (years) | 36.4 (23.4–59.6) | 32.3 (24.6–43.4) | 36.4 (30.3–44.9) |
| Men (%) | 90% | 80% | 80% |
| Blunt Mechanism (%) | 30% | 60% | 50% |
| NISS | 17.0 (9.0–22.0) | 46.5 (38.0–51.8) | 17.0 (14.5–27.0) |
| Max AIS | |||
| Head/Neck | 0.0 (0.0–0.0) | 0.0 (0.0–3.5) | 0.0 (0.0–1.8) |
| Chest | 0.0 (0.0–3.0) | 3.5 (3.0–5.0) | 0.0 (0.0–2.3) |
| Abdomen/Pelvis | 0.0 (0.0–3.0) | 2.0 (0.0–3.5) | 1.0 (0.0–3.0) |
| Extremities | 0.0 (0.0–2.0) | 1.0 (0.0–2.3) | 0.0 (0.0–2.0) |
| 1st Measurement (ED) | |||
| SBP (mm Hg) | 128.0 (99.5–132.0) | 86.0 (82.0–130.0) | 110. (100.0–138.0) |
| GCS | 15.0 (9.0–15.0) | 3.0 (3.0–3.0) | 15.0 (12.5–15.0) |
| pH | 7.3 (7.3–7.3) | 6.9 (6.8–7.2) | 7.3 (7.3–7.3) |
| Base Excess (mEg/L) | −5.0 (−7.0, −2.0) | −11.0 (−17.0, −3.0) | −8.0 (−8.8, −5.8) |
| Hematocrit (%) | 40.3 (38.9–44.0) | 31.4 (27.6–37.4) | 40.4 (38.2–41.1) |
| INR | 1.1 (1.1–1.2) | 2.0 (1.8–3.4) | 1.1 (1.1–1.2) |
| PTT | 27.5 (25.3–28.2) | 66.1 (20.6–98.9) | 25.8 (23.9–28.0) |
| Fibrinogen | No Data | 68.0 (49.6–107.0) | 143.0 (122.0–164.0) |
| % Death | 0% | 70% | 0% |
| CNTEG+tPA | |||
| R | 7.8 (6.9–8.4) | 8.3 (6.6–12.9) | 6.4 (4.5–7.7) |
| Angle | 60.7 (58.2–61.9) | 48.2 (28.0–50.1) | 65.9 (62.2–71.1) |
| MA | 60.0 (57.8–62.5) | 20.3 (14.0–31.6) | 62.0 (55.6–64.3) |
| LY30 | 7.6 (6.9–8.3) | 80.0 (73.0–88.9) | 1.1 (0.5–1.5) |
Proteomics and selected analysis of the coagulation and fibrinolytic cascades of injured patients.
Proteomic analysis of the plasma from injured patients demonstrated that all severely injured patients had increased concentrations of both α- and β-globin chains versus healthy controls (p<.01), and the HF patients were also different from SD and PF patients (p<.01) without a significant decrease in haptoglobin (Table 2). HF patients also had decreased levels of anti-thrombin and plasminogen versus healthy controls, and the SD and PF groups (p<.01) (Table 2). Importantly, the ELISA data showed that HF patients had increased TAT complexes, e.g. increased thrombin activation, and PAP complexes, e.g. increased plasmin activation, as compared to healthy controls and PF and SD patient groups (Table 2). The total tPA and tPA activation, increased tPA:PAI-1 complexes, were elevated in all trauma groups versus the healthy controls, as well as being significantly increased in the HF group versus all other groups (Table 2). Lastly, both total PAI-1 and PAI-1 activity were significantly elevated in the SD groups versus the PF and HF groups and the healthy controls (p<.05) with all three injured groups showing increased tPA activation, e.g. higher circulating levels of tPA:PAI-1 complexes, versus the healthy controls (Table 2.).
Table 2.
Selected Proteomics and enzymes from the Coagulation and Fibrinolytic Cascades
| QConCat Analysis (Median (Q1–Q3) | |||||
|---|---|---|---|---|---|
| Protein | Healthy Controls | Hyperfibrinolysis | Shutdown | Physiologic | |
| Hemoglobin, alpha subunit | 0.3 (0.2–0.7) | 4.5 (1.3–6.6)** | 1.1 (0.48–3.5)** | 0.8 (0.6–1.1)** | |
| Hemoglobin, beta subunit | 1.5 (1.2–4.2) | 9.1 (2.8–20.4)** | 4.7 (2.4–13.4) | 4.5 (2.7–7.9) | |
| Haptoglobin | 28.3(16.2–36.5) | 22.8 (9.4–30.5 | 26.1 (23.0–27.9) | 29.6 (16.3–42.4) | |
| ELISA Analysis | |||||
| Alpha 2-antiplasmin (μg/ml) | 1122.4±196.7 | 890.9±217.1 | 1367.6±251.8 | 921.2±151.5 | |
| Antithrombin III (μg/ml) | 526.5±44.0 | 253.2±44.7* | 464.8±35.3 | 382.4±47.3 | |
| Thrombin(ng/ml) | 144.4±12.1 | 133.5±13.8 | 153.5±27.3 | 134.2±15.3 | |
| Plasminogen (μg/ml) | 1127.8±188.2 | 529.6±120.8* | 995.2±228.1 | 1187.2±116.7 | |
| Plasmin anti-plasmin (ng/ml) | 0.5±0.05 | 415.7±199.0* | 4.8±3.8 | 2.8±1.0 | |
| Thrombin anti-Thrombin (ng/ml) | 4.4±0.6 | 9.0±1.5* | 6.4±0.7 | 8.5±1.4* | |
| tPA | Activity (U/ml) | 0.002±0.001 | 0.89±0.2* | 0.001±0.001 | 0.03±0.01* |
| Total (ng/ml) | 3.6±0.8 | 10.9±2.3* | 14.9±3.1* | 10.3±2.1* | |
| PAI-1 | Activity (U/ml) | 4.0±1.6 | 1.9±0.1 | 79.3±26.6* | 5.0±2.0 |
| Total (ng/ml) | 7.6±2.6 | 8.41±2.3 | 115.±35.2* | 13.0±3.8 | |
| tPA:PAI-1 (ng/ml) | 5.8±1.3 | 15.4±3.7* | 24.0±4.9* | 16.5±3.4* | |
p<0.01 from Healthy Controls;
p<0.05 from Healthy Controls
Thrombelastography.
In whole blood from healthy donors, HbA and neither the α- nor the β-globin chains affected any aspect of the CN-TEGs, including the R-time, angle, MA, or Ly30 (data not shown). To mimic the injured phenotype tPA [75ng/ml]-challenged TEGs were completed with the addition of NS (control) or serial dilutions of HbA, α-globin chain, and β-globin chain. HbA had no effect on the R-time, angle, and MA (data not shown), as well as the tPA-provoked fibrinolysis: median Ly30’s being 25% (control), 17% [41 nM], 17% [123 nM], 18% [204 nM] (p=0.48) (Fig.1). Addition of the α-globin chain decreased the R-time at 550 nM and 920 nM versus NS (Fig.1a) (p<0.05) but did not affect the angle (Fig.1b) or the MA (Fig.1c), and enhanced tPA-provoked fibrinolysis: Ly30’s of 49.7% [550 nM] and 48.5% [920 nM] versus NS (31.3%) (p<0.05) (Fig.1d). Addition of the β-globin chain significantly decreased the R-time at 920 nM alone (Fig.1a) and did not affect any other parameters of the tPA-challenged TEGs including the angle, MA, or the Ly30 (Fig.1). Tranexamic acid [1mM] significantly inhibited the tPA-induced lysis by 95±4% (n=7) as expected.
Figure 1.
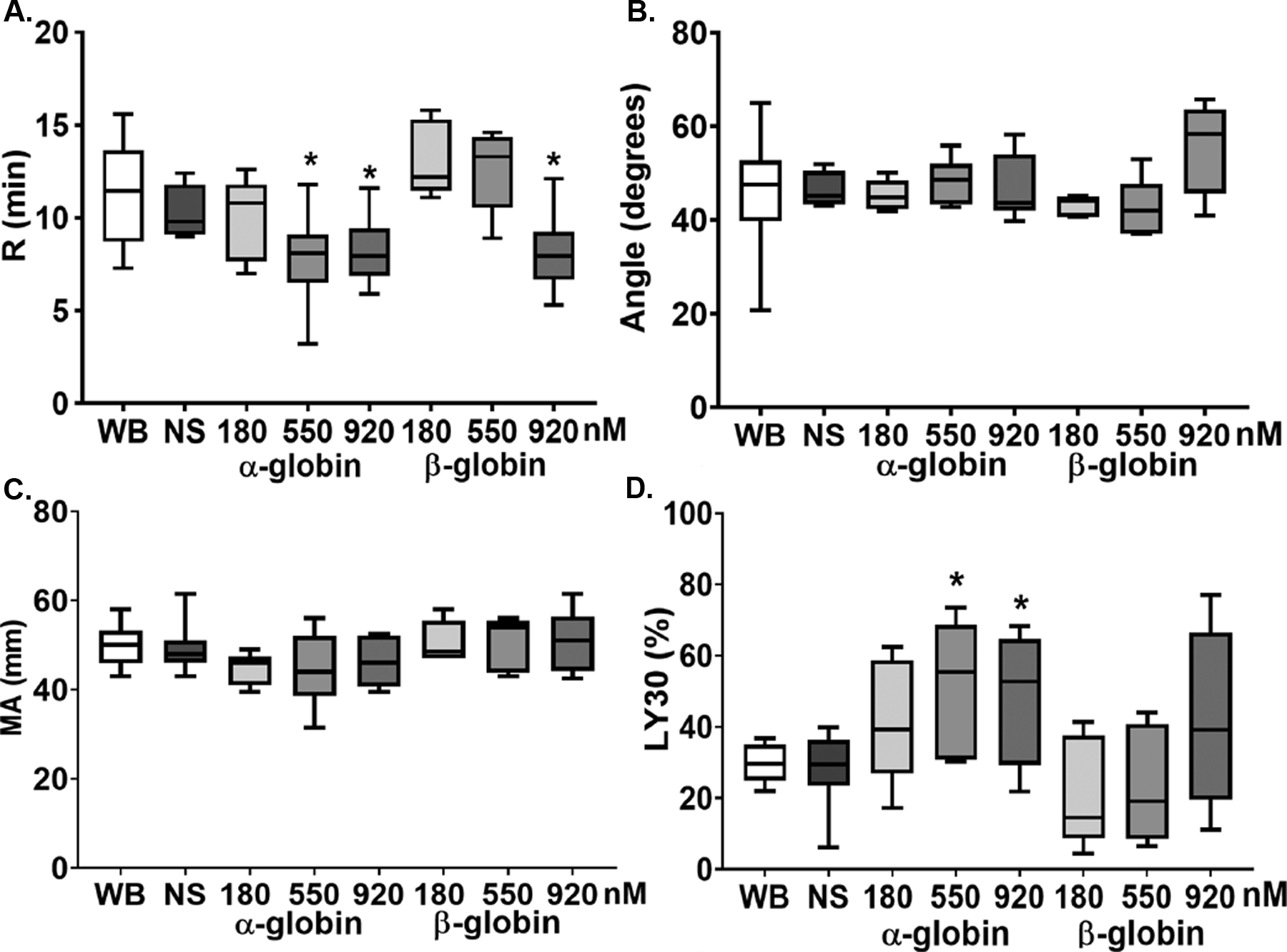
The α- and β-globin chains affect R-time and Ly30 of tPA-challenged TEGs. Whole blood was drawn from healthy controls and tPA (75 ng/ml) challenged TEGs were completed. Four TEG parameters were measured. 1a) R-time: the α-globin chain at the two highest concentrations (3μg/ml and 5 μg/ml) significantly shortened the R-time as did the highest concentration of the β-globin chain (5μg/ml). 1b) Angle: neither the a-globin chain nor the b-globin chain affected the angle in tPA-challenged TEGs at any of the concentrations employed. 1c) The maximal amplitude (MA) was unaffected by either the α-globin chain or the β-globin chain. 1d) The β-globin chain significantly increased the Ly30 in a concentration-dependent manner; whereas the b-globin chain did not affect Ly30 in the tPA-challenged TEGS (n=7, *=p<0.05 versus WB and NS).
SPR.
SPR is considered a gold standard for measuring the affinity of a biomolecular interaction between two proteins and succeeded the yeast two hybrid assay for identifying the affinity of two proteins (43). As a control, PLG was coupled to the chip and free tPA [75 ng/ml] was passed over it to measure the coupling efficiency and dissociation constant (KD) of the known enzyme: substrate pair [18]. The tPA bound with a KD of 43 nM, and with repetitions the calculated mean KD was 48±12 nM (Table 3). To ensure there was no non-specific protein:protein interaction the analyte and ligand were reversed, which yielded identical results (data not shown). Serial dilutions of α-globin chain were then passed over chips coupled to PLG yielding a KD of 93±14 nM (Table 3). HbA and the β-globin subunit demonstrated very weak molecular interactions with PLG: HbA:PLG 3.6±0.3 μM and β-globin subunit:PLG 4.0 μM, respectively. The protein: protein interactions were repeated with similar lack of any physical binding reaction between HbA or the β-globin chain and the PLG ligand bound to the chip (data not shown) (Table 3).
Table 3:
Coupling Efficiency of Ligands to Plg
| Flow over Chip | Bound to the Chip | KD (nM) |
|---|---|---|
| tPA | Plg | 48.0±12 |
| α-globin chain | Plg | 93.0 ±14 |
| β-globin chain | Plg | 4000* |
| HbA | Plg | 3600±300* |
Two replicates exhibited no physical association
Plasmin Activity assays.
The addition of tPA did not generate plasmin from plasminogen-free plasma (negative control, results not shown). When normal plasma was employed, NS served as a negative control and induced a baseline level of plasmin generation of 1,992±1,186 RLU/min (Fig.2). TPA [75 ng/ml] caused a significant (3.2±0.5-fold) increase in plasmin activity 6,374±997 RLU/min vs. NS (n=7, p<.05) (Fig.2). The addition of α-globin chain [5 μM] significantly augmented tPA-induced plasmin activity by 1.85±0.6-fold, 11,474±1,989 RLU/min (n=7, p<.05) (Fig.2). Furthermore, pre-incubation with the protease inhibitor PMSF [50 μM] significantly inhibited the tPA-induced increase in plasmin activity by 92±4% (n=7, p<.05) (Fig.2).
Figure 2.
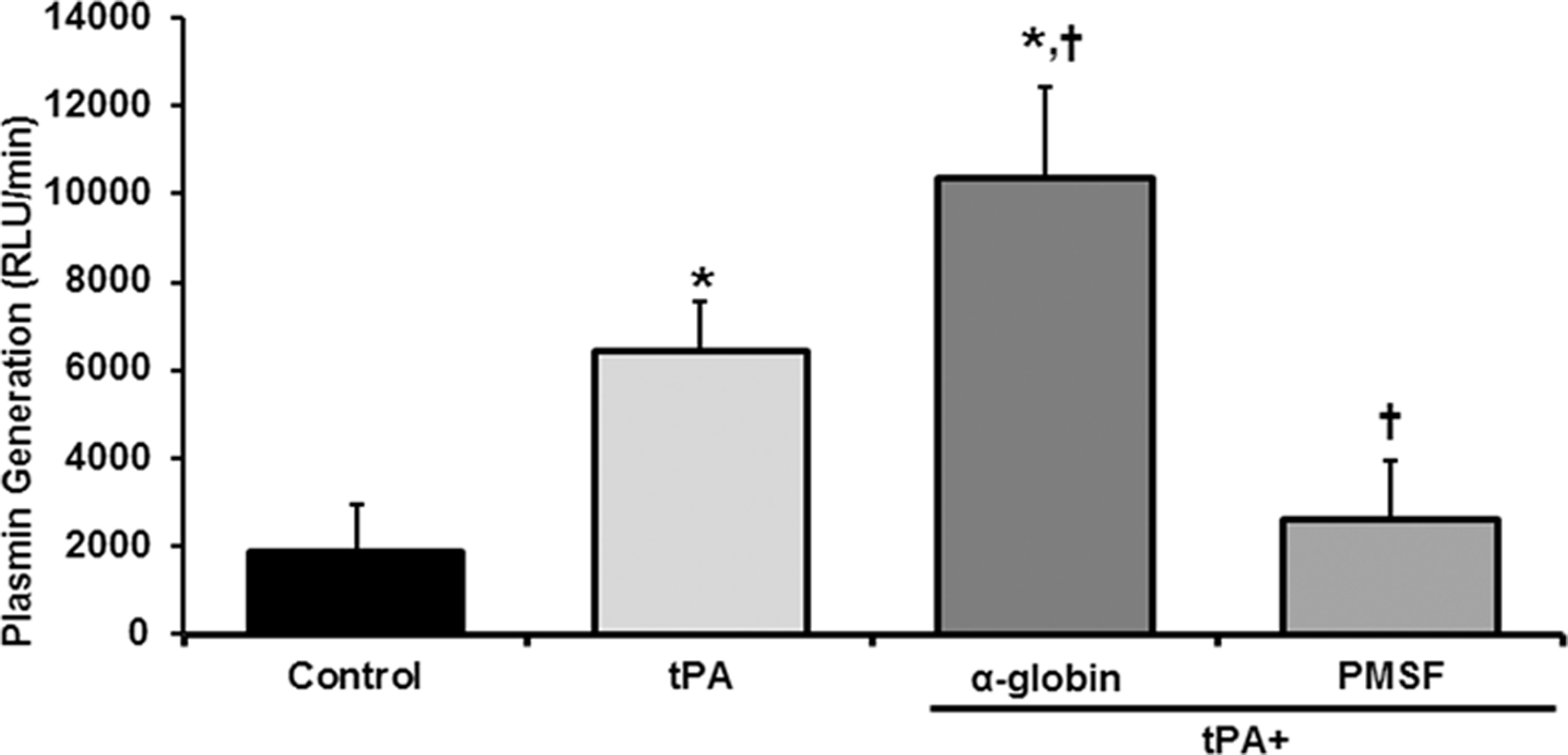
The α-globin chain augments plasmin activity. Plasmin activity is shown as a function of treatment group. The plasma alone demonstrates a baseline amount of plasmin activity. The addition of 75 ng/ml of tPA induced a significant increase over the plasma alone. The addition of 5 μg of the α-globin chain significantly increased the plasmin activity in response to tPA. In contrast the addition of the protease inhibitor PMSF [50 nM] significantly inhibited tPA-induced plasmin activity (n=7, *p<0.05 from Control, † p<0.05 from tPA).
Discussion
Following severe injury and prior to blood component resuscitation, α-globin chains accumulate in the plasma from all injured patients, regardless of TEG-defined groups, when compared to healthy controls. The patients who exhibit HF, had significantly greater amounts of the α-globin chain as compared to the SD and PF patients; whereas the β-globin chain only significantly increased in the HF patients with normal haptoglobin levels present in all groups. Similar to previous reports, the HF patients have increased thrombin-anti-thrombin and plasmin-α2-anti-plasmin complexes, decreased plasminogen and increased tPA activity versus all other groups indicating increased systemic activation of plasmin most likely by tPA (32). Both total tPA and tPA:PAI-1 complexes were increased in the injured groups versus the healthy controls; thus, the patients investigated in this report appear to be representative of injured patients who have undergone significant ischemia leading to tPA activation; however, as far as the patients with HF are concerned, the stringent criteria of using a tPA-challenged TEG selected the most severely injured patients who had significant HF hence the increased morbidity and mortality in this subset of patients (13, 15, 21, 24, 44, 45). Moreover, trauma-related hemolysis was reported decades ago and the HF group which is the most severely injured is likely evidencing such hemolysis and all of the blood samples were drawn within 60 minutes of ED arrival and may have precluded the drop in haptoglobin in these severely injured patients (46).
To evaluate traumatic injury in healthy samples tPA-challenged TEGs were completed and the injured patients were stratified by their state of fibrinolysis: HF, SD, or PF. Importantly, tPA-challenge TEGs have shortened R-times because tPA induces activation of factor V and augments the initiation of plasma-based coagulation (47). The addition of the α-globin chain significantly increased the Ly30 and decreased the R-time in tPA-challenge TEGs, while addition of the β-globin subunit only decreased the R-time at the highest concentration employed, which could be due to its ability to provide a non-specific scaffold to allow for more rapid activation of the coagulation cascade in the TEG cup. The intact hemoglobin molecule, HbA did not affect any tPA-challenge TEG parameters. To examine possible protein:protein interactions SPR experiments were completed and showed that the α-globin chain binds to PLG with a similar KD of tPA binding to PLG, a known enzyme:substrate pair (35). This binding of α-globin chain to PLG significantly augments the fibrinolytic activity of plasmin in vitro providing a possible mechanism for HF in severely injured patients by increasing and/or stabilizing the enzymatic activity of plasmin through direct binding of the α-globin chain to plasmin (Fig.3). Importantly, plasmin activation is downstream for tPA, and tPA activity was increased by the ischemia related to significant injury to the highest concentration in the HF group without increased amounts of PAI-1 seen in the SD group. Additionally, it is vital to remember that the proteomic analysis does not preserve the tetramers nor the α-β dimers of circulating free hemoglobin, therefore the data is limited to the individual chains. One would expect the presence of α-globin chains and β-globin chains in very similar concentrations if hemolysis alone were the impetus for their release into the circulation as seen in autoimmune hemolytic anemia and hemolytic crises in patients with sickle cell anemia, thalassemia, glucose-6-phosphate dehydrogenase deficiency, or hereditary spherocytosis, etc. and the monomeric α-globin chain is cleared directly by haptoglobin whereas the β-globin chain is not (48, 49). The relative increase in the α-globin chain as denoted by the 1 to 2 ratio of the α-globin chain to the β-globin chain in the HF group is curious and may be due to its release from the vascular wall in which it is present in the vascular endothelium at the myoendothelial junctions in which it functions to control vascular tone (50, 51).
Figure 3.
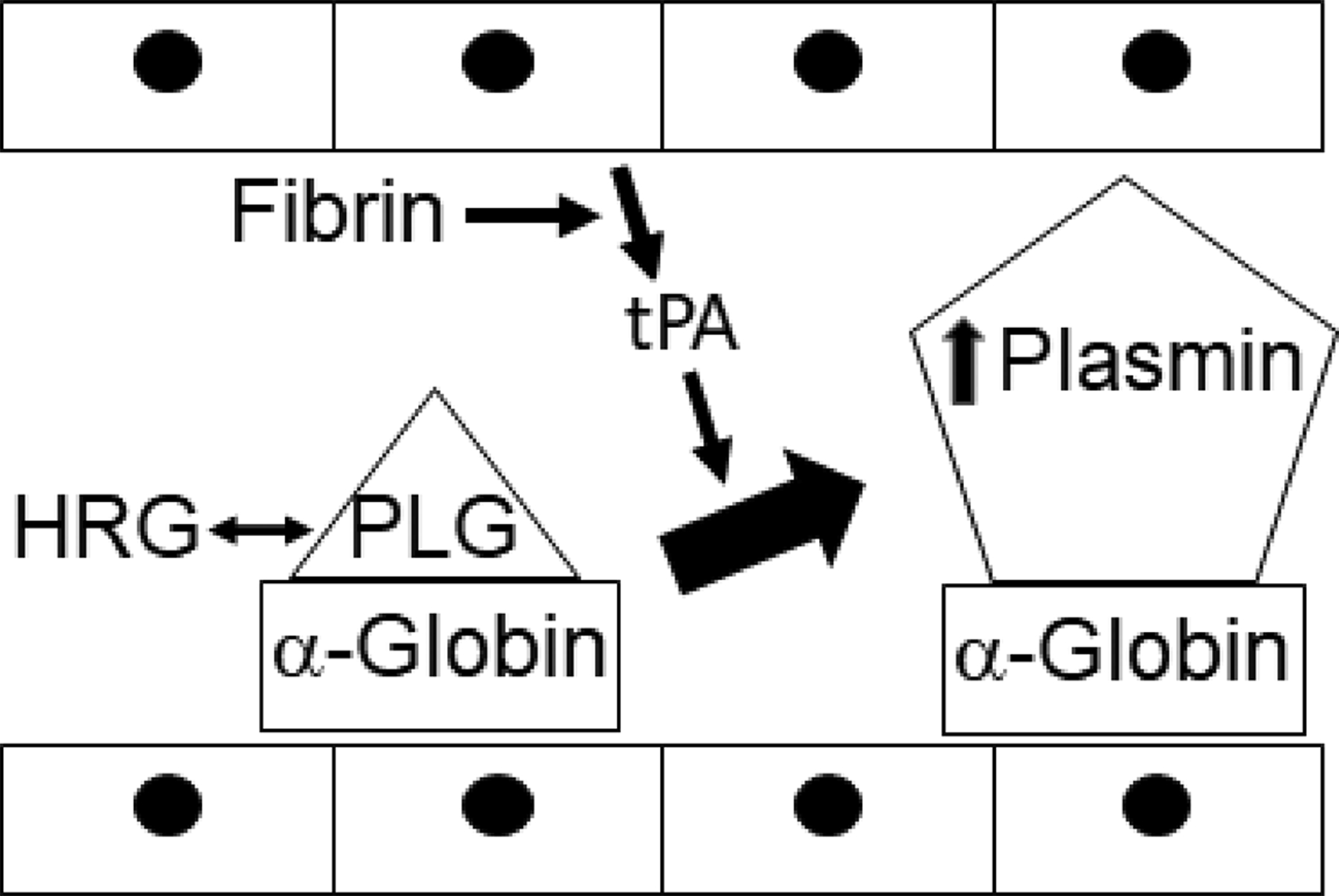
Proposed schema of the a-globin augmentation of plasmin activity. Plasminogen (PLG) circulates in a reversible complex with histidine rich glycoprotein (HRG), which is a competitive inhibitor, which can encounter the α-globin chain of hemoglobin (α-globin). In response to fibrin endothelial cells release tissue plasminogen activator (tPA) which than activated plasminogen, which is bound to the α-globin chain, and causes increased plasmin activity.
Severe injury induces the release and accumulation of intracellular proteins from damaged tissue or hemolysis into the circulation, which may alter hemostasis and may be grouped by the state of fibrinolysis: SD, HF, and physiologic (15, 23, 32, 34, 46, 52). Mortality has been reported to be greatest in the HF group, elevated in the SD group, and least in patients with PF (15). Clot stability (Ly30) appears to be crucial in TIC with systemic HF accounting for ~25% of injured patients (NISS>15) with mortality as high as 70% (4, 5, 8, 15). Thus, HF is a dangerous condition that requires better molecular description to ultimately decrease mortality in the severely injured. Importantly, these severely injured HF patients exhibit increased thrombin activity, TAT complexes, and plasmin activity, increase PAP complexes, and such increased plasmin activity is initiated by tPA with possible augmentation by a cofactor, the α-globin chain, which directly binds to plasminogen and increases plasmin activity (32). The conversion of PLG to plasmin involves a well-defined group of coagulation factors and serine proteases that may be augmented by “moonlighting” protein mediators in severely injured patients, which can be intracellular proteins released by injury and ischemia due to concomitant hemorrhagic shock (35). The α-globin chain may represent one such protein with the ability to tightly bind PLG and change the activity of plasmin following activation. Patients with significant HF as diagnosed by TEG or rotational thrombelastography (ROTEM) should receive prompt treatment with tranexamic acid and activation of a massive transfusion protocol, if warranted, with further resuscitation directed by either TEG or ROTEM.
The intact HbA protein did not affect any tPA-challenged TEG parameters and HbA, the α- and β-globin chains did not alter any of the parameters in CN-TEGs. Investigations of the PLG interactome using affinity chromatography revealed that the α-globin chain is a candidate interactor with PLG and the reported in vitro data identify a possible role for α-globin chains by binding to plasminogen and augmenting plasmin activity leading to HF in the severely injured (23, 35). The ability of the α-globin chain to augment plasmin activity may to be restricted to the severely injured in whom ischemia increases the release of tPA, which in turn may induce systemic, indiscriminate activation of plasmin resulting in weaker clots and HF (32). Importantly, in these severely injured patients the plasma haptoglobin concentrations were in the normal range; however, these haptoglobin measurements were upon arrival at the Emergency Department within minutes to hours of injury. Previous data has shown that severe injury does cause hemolysis and haptoglobin levels were decreased on the day of injury with normalization by day 3, and our most severely injured patients in the HF groups had α- and β-globin chains in their plasma (46). In this study the exact timing of the blood draws with regard to injury or ED arrival was not documented such that the normal haptoglobin levels seen here may preclude the described drop in haptoglobin, reported by others, and even in the face of massive hemolysis it may take hours to deplete the haptoglobin reserves (46). Moreover, it is unlikely that the observed increases in α-globin chain and β-globin chain were due to phlebotomy-related hemolysis because all groups were drawn under a stringently controlled protocol with identical equipment and procedures and yet only the HF and SD groups had increased α-globin and β-globin chains.
This pilot study has several limitations, including small sample size, the study patients are from a single Level I trauma center at moderate altitude, and the samples collected are from only the initial time point. Even moderate altitude may affect wound healing, platelet biology and coagulation so that these studies should be repeated at lower elevations (53–56). However, it is the initial TEG that is employed for therapeutic decisions, which may include the administration tranexamic acid, activation of trauma transfusion protocols, etc. Surprisingly, none of the SD patients died and one may expect some mortality in this group especially compared to those with PL (13, 15, 21, 24, 44, 45). In addition, the HF patients had higher NISS and increased bases excesses compared to the other two groups of trauma patients. Despite these limitations the interaction of the α-globin chain with plasmin and the resulting role in fibrinolysis needs to be verified in larger studies of the severely injured.
The importance of the α-globin chain in the hemoglobin molecule has been well documented; however, it has novel functions in other tissues (35, 50, 51). The α-globin chain is present in the vessel wall in the myoendothelial junctions and is a mediator of vascular tone in the resistance arteries (50, 51). It is also found in the central nervous system and in other cells so that its presence in the plasma may also be due to injury rather than hemolysis alone, though patients with TBI were excluded (57). In these severely injured patients the serum haptoglobin concentrations were in the normal range, which may argue against widespread hemolysis being the source of the high plasma levels of the α-globin chain; however, the reported data comes from the initial plasma draw from these injured patients and may precede a decrease in haptoglobin which has been reported in the severely injured (46). Given the altered physiology and circulation of the severely injured, more work is required to delineate the source of the α- and β-globin chains and to characterize the function of the α-globin chain as part of the PLG interactome and its role in the severely injured, especially with respect to TIC.
Funding
This work was supported by Vitalant Research Institute, Denver, CO, grants P50-GM049222, 1RM1GM131968-02, and T32-GM008315 from NIGMS, NIH and grant W81XWH-12-2008 from the Department of Defense.
Footnotes
Conflict of Interest
None of the authors have an actual or potential conflict of interest with any of the reported data or methodologies used within this manuscript.
References
- 1.Sherry S, Fletcher AP, Alkjaersig N. Fibrinolysis and fibrinolytic activity in man. Physiol Rev. 1959;39(2):343–82. [DOI] [PubMed] [Google Scholar]
- 2.Brohi K, Cohen MJ, Ganter MT, Matthay MA, Mackersie RC, Pittet JF. Acute traumatic coagulopathy: initiated by hypoperfusion: modulated through the protein C pathway? Ann Surg. 2007;245(5):812–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brohi K, Cohen MJ, Ganter MT, Schultz MJ, Levi M, Mackersie RC, Pittet JF. Acute coagulopathy of trauma: hypoperfusion induces systemic anticoagulation and hyperfibrinolysis. J Trauma. 2008;64(5):1211–7. [DOI] [PubMed] [Google Scholar]
- 4.Cotton BA, Harvin JA, Kostousouv V, Minei KM, Radwan ZA, Schochl H, Wade CE, Holcomb JB, Matijevic N. Hyperfibrinolysis at admission is an uncommon but highly lethal event associated with shock and prehospital fluid administration. J Trauma Acute Care Surg. 2012;73(2):365–70. [DOI] [PubMed] [Google Scholar]
- 5.Ives C, Inaba K, Branco BC, Okoye O, Schochl H, Talving P, Lam L, Shulman I, Nelson J, Demetriades D. Hyperfibrinolysis elicited via thromboelastography predicts mortality in trauma. J Am Coll Surg. 2012;215(4):496–502. [DOI] [PubMed] [Google Scholar]
- 6.Starzl TE, Marchioro TL, Vonkaulla KN, Hermann G, Brittain RS, Waddell WR. Homotransplantation of the Liver in Humans. Surg Gynecol Obstet. 1963;117:659–76. [PMC free article] [PubMed] [Google Scholar]
- 7.Brohi K, Cohen MJ, Davenport RA. Acute coagulopathy of trauma: mechanism, identification and effect. Curr Opin Crit Care. 2007;13(6):680–5. [DOI] [PubMed] [Google Scholar]
- 8.Schochl H, Frietsch T, Pavelka M, Jambor C. Hyperfibrinolysis after major trauma: differential diagnosis of lysis patterns and prognostic value of thrombelastometry. J Trauma. 2009;67(1):125–31. [DOI] [PubMed] [Google Scholar]
- 9.Bohm JK, Schafer N, Maegele M, Stumpges B, Bauerfeind U, Caspers M. Plasmatic and cell-based enhancement by microparticles originated from platelets and endothelial cells under simulated in vitro conditions of a dilutional coagulopathy. Scand J Trauma Resusc Emerg Med. 2021;29(1):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davenport RA, Brohi K. Cause of trauma-induced coagulopathy. Curr Opin Anaesthesiol. 2016;29(2):212–9. [DOI] [PubMed] [Google Scholar]
- 11.Gando S, Shiraishi A, Wada T, Yamakawa K, Fujishima S, Saitoh D, Kushimoto S, Ogura H, Abe T, Otomo Y. A multicenter prospective validation study on disseminated intravascular coagulation in trauma-induced coagulopathy. J Thromb Haemost. 2020;18(9):2232–44. [DOI] [PubMed] [Google Scholar]
- 12.Moore EE, Moore HB, Kornblith LZ, Neal MD, Hoffman M, Mutch NJ, Schochl H, Hunt BJ, Sauaia A. Trauma-induced coagulopathy. Nat Rev Dis Primers. 2021;7(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chapman MP, Moore EE, Moore HB, Gonzalez E, Gamboni F, Chandler JG, Mitra S, Ghasabyan A, Chin TL, Sauaia A, et al. Overwhelming tPA release, not PAI-1 degradation, is responsible for hyperfibrinolysis in severely injured trauma patients. J Trauma Acute Care Surg. 2016;80(1):16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gando S Hemostasis and thrombosis in trauma patients. Semin Thromb Hemost. 2015;41(1):26–34. [DOI] [PubMed] [Google Scholar]
- 15.Moore HB, Moore EE, Gonzalez E, Chapman MP, Chin TL, Silliman CC, Banerjee A, Sauaia A. Hyperfibrinolysis, physiologic fibrinolysis, and fibrinolysis shutdown: the spectrum of postinjury fibrinolysis and relevance to antifibrinolytic therapy. J Trauma Acute Care Surg. 2014;77(6):811–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moore HB, Moore EE, Morton AP, Gonzalez E, Fragoso M, Chapman MP, Dzieciatkowska M, Hansen KC, Banerjee A, Sauaia A, et al. Shock-induced systemic hyperfibrinolysis is attenuated by plasma-first resuscitation. J Trauma Acute Care Surg. 2015;79(6):897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gonzalez E, Moore EE, Moore HB, Chapman MP, Chin TL, Ghasabyan A, Wohlauer MV, Barnett CC, Bensard DD, Biffl WL, et al. Goal-directed Hemostatic Resuscitation of Trauma-induced Coagulopathy: A Pragmatic Randomized Clinical Trial Comparing a Viscoelastic Assay to Conventional Coagulation Assays. Ann Surg. 2016;263(6):1051–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kashuk JL, Moore EE, Sawyer M, Wohlauer M, Pezold M, Barnett C, Biffl WL, Burlew CC, Johnson JL, Sauaia A. Primary fibrinolysis is integral in the pathogenesis of the acute coagulopathy of trauma. Ann Surg. 2010;252(3):434–42. [DOI] [PubMed] [Google Scholar]
- 19.Moore HB, Moore EE, Chapman MP, Gonzalez E, Slaughter AL, Morton AP, D’Alessandro A, Hansen KC, Sauaia A, Banerjee A, et al. Viscoelastic measurements of platelet function, not fibrinogen function, predicts sensitivity to tissue-type plasminogen activator in trauma patients. J Thromb Haemost. 2015;13(10):1878–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moore HB, Moore EE, Chapman MP, Huebner BR, Einersen PM, Oushy S, Silliman CC, Banerjee A, Sauaia A. Viscoelastic Tissue Plasminogen Activator Challenge Predicts Massive Transfusion in 15 Minutes. J Am Coll Surg. 2017;225(1):138–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chapman MP, Moore EE, Moore HB, Gonzalez E, Morton AP, Chandler J, Fleming CD, Ghasabyan A, Silliman CC, Banerjee A, et al. The “Death Diamond”: Rapid thrombelastography identifies lethal hyperfibrinolysis. J Trauma Acute Care Surg. 2015;79(6):925–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Einersen PM, Moore EE, Chapman MP, Moore HB, Gonzalez E, Silliman CC, Banerjee A, Sauaia A. Rapid thrombelastography thresholds for goal-directed resuscitation of patients at risk for massive transfusion. J Trauma Acute Care Surg. 2017;82(1):114–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moore HB, Moore EE, Gonzalez E, Hansen KC, Dzieciatkowska M, Chapman MP, Sauaia A, West B, Banerjee A, Silliman CC. Hemolysis exacerbates hyperfibrinolysis, whereas platelolysis shuts down fibrinolysis: evolving concepts of the spectrum of fibrinolysis in response to severe injury. Shock. 2015;43(1):39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moore HB, Moore EE, Huebner BR, Dzieciatkowska M, Stettler GR, Nunns GR, Lawson PJ, Ghasabyan A, Chandler J, Banerjee A, et al. Fibrinolysis shutdown is associated with a fivefold increase in mortality in trauma patients lacking hypersensitivity to tissue plasminogen activator. J Trauma Acute Care Surg. 2017;83(6):1014–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cardenas JC, Matijevic N, Baer LA, Holcomb JB, Cotton BA, Wade CE. Elevated tissue plasminogen activator and reduced plasminogen activator inhibitor promote hyperfibrinolysis in trauma patients. Shock. 2014;41(6):514–21. [DOI] [PubMed] [Google Scholar]
- 26.Kuhli C, Luchtenberg M, Scharrer I, Hattenbach LO. Massive subhyaloidal hemorrhage associated with severe PAI-1 deficiency. Graefes Arch Clin Exp Ophthalmol. 2005;243(10):963–6. [DOI] [PubMed] [Google Scholar]
- 27.Minowa H, Takahashi Y, Tanaka T, Naganuma K, Ida S, Maki I, Yoshioka A. Four cases of bleeding diathesis in children due to congenital plasminogen activator inhibitor-1 deficiency. Haemostasis. 1999;29(5):286–91. [DOI] [PubMed] [Google Scholar]
- 28.Carter KT, Palei AC, Spradley FT, Witcher BM, Martin L, Hester RL, Kutcher ME. A rat model of orthopedic injury-induced hypercoagulability and fibrinolytic shutdown. J Trauma Acute Care Surg. 2020;89(5):926–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kassis J, Hirsh J, Podor TJ. Evidence that postoperative fibrinolytic shutdown is mediated by plasma factors that stimulate endothelial cell type I plasminogen activator inhibitor biosynthesis. Blood. 1992;80(7):1758–64. [PubMed] [Google Scholar]
- 30.Moore EE, Moore HB, Gonzalez E, Sauaia A, Banerjee A, Silliman CC. Rationale for the selective administration of tranexamic acid to inhibit fibrinolysis in the severely injured patient. Transfusion. 2016;56 Suppl 2:S110–S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Urano T, Suzuki Y, Iwaki T, Sano H, Honkura N, Castellino FJ. Recognition of Plasminogen Activator Inhibitor Type 1 as the Primary Regulator of Fibrinolysis. Curr Drug Targets. 2019;20(16):1695–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Banerjee A, Silliman CC, Moore EE, Dzieciatskowa M, Kelher M, Sauaia A, Jones K, Chapman MP, Gonzalez E, Moore HB, et al. Systemic Hyperfibrinolysis after Trauma: A Pilot study of Targeted Proteomic Analysis of Superposed Mechanisms in Patient Plasma. J Trauma Acute Care Surg. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bock A, Tucker N, Kelher MR, Khan SY, Gonzalez E, Wohlauer M, Hansen K, Dzieciatkowska M, Sauaia A, Banerjee A, et al. alpha-Enolase Causes Proinflammatory Activation of Pulmonary Microvascular Endothelial Cells and Primes Neutrophils Through Plasmin Activation of Protease-Activated Receptor 2. Shock. 2015;44(2):137–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peltz ED, Moore EE, Zurawel AA, Jordan JR, Damle SS, Redzic JS, Masuno T, Eun J, Hansen KC, Banerjee A. Proteome and system ontology of hemorrhagic shock: exploring early constitutive changes in postshock mesenteric lymph. Surgery. 20092009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hathaway WE, Goodnight SH. Disorders of Hemostasis and Thrombosis: A Clinical Guide. First ed. New York: McGraw-Hill, Inc.; 1993. 1993. [Google Scholar]
- 36.Morton AP, Moore EE, Moore HB, Gonzalez E, Chapman MP, Peltz E, Banerjee A, Silliman C. Hemoglobin-based oxygen carriers promote systemic hyperfibrinolysis that is both dependent and independent of plasmin. J Surg Res. 2017;213:166–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moore HB, Moore EE, Chin TL, Gonzalez E, Chapman MP, Walker CB, Sauaia A, Banerjee A. Activated clotting time of thrombelastography (T-ACT) predicts early postinjury blood component transfusion beyond plasma. Surgery. 2014;156(3):564–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.D’Alessandro A, Dzieciatkowska M, Hill RC, Hansen KC. Supernatant protein biomarkers of red blood cell storage hemolysis as determined through an absolute quantification proteomics technology. Transfusion. 2016;56(6):1329–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dzieciatkowska M, D’Alessandro A, Hill RC, Hansen KC. Plasma QconCATs reveal a gender-specific proteomic signature in apheresis platelet plasma supernatants. J Proteomics. 2015;120:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pratt JM, Simpson DM, Doherty MK, Rivers J, Gaskell SJ, Beynon RJ. Multiplexed absolute quantification for proteomics using concatenated signature peptides encoded by QconCAT genes. Nat Protoc. 2006;1(2):1029–43. [DOI] [PubMed] [Google Scholar]
- 41.Jason-Moller L, Murphy M, Bruno J. Overview of Biacore systems and their applications. Curr Protoc Protein Sci. 2006;Chapter 19:Unit. [DOI] [PubMed] [Google Scholar]
- 42.Moore HB, Winfield RD, Aibiki M, Neal MD. Is Coagulopathy an Appropriate Therapeutic Target During Critical Illness Such as Trauma or Sepsis? Shock. 2017;48(2):159–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rich RL, Myszka DG. Advances in surface plasmon resonance biosensor analysis. Curr Opin Biotechnol. 2000;11(1):54–61. [DOI] [PubMed] [Google Scholar]
- 44.Chapman MP, Moore EE, Ramos CR, Ghasabyan A, Harr JN, Chin TL, Stringham JR, Sauaia A, Silliman CC, Banerjee A. Fibrinolysis greater than 3% is the critical value for initiation of antifibrinolytic therapy. J Trauma Acute Care Surg. 2013;75(6):961–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moore HB, Moore EE, Liras IN, Gonzalez E, Harvin JA, Holcomb JB, Sauaia A, Cotton BA. Acute Fibrinolysis Shutdown after Injury Occurs Frequently and Increases Mortality: A Multicenter Evaluation of 2,540 Severely Injured Patients. J Am Coll Surg. 2016;222(4):347–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gando S, Tedo I. The effects of massive transfusion and haptoglobin therapy on hemolysis in trauma patients. Surg Today. 1994;24(9):785–90. [DOI] [PubMed] [Google Scholar]
- 47.Lee CD, Mann KG. Activation/inactivation of human factor V by plasmin. Blood. 1989;73(1):185–90. [PubMed] [Google Scholar]
- 48.Fatunmbi O, Abzalimov RR, Savinov SN, Gershenson A, Kaltashov IA. Interactions of Haptoglobin with Monomeric Globin Species: Insights from Molecular Modeling and Native Electrospray Ionization Mass Spectrometry. Biochemistry. 2016;55(12):1918–28. [DOI] [PubMed] [Google Scholar]
- 49.Schaer DJ, Buehler PW, Alayash AI, Belcher JD, Vercellotti GM. Hemolysis and free hemoglobin revisited: exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood. 2013;121(8):1276–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sangwung P, Zhou G, Lu Y, Liao X, Wang B, Mutchler SM, Miller M, Chance MR, Straub AC, Jain MK. Regulation of endothelial hemoglobin alpha expression by Kruppel-like factors. Vasc Med. 2017;22(5):363–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Straub AC, Butcher JT, Billaud M, Mutchler SM, Artamonov MV, Nguyen AT, Johnson T, Best AK, Miller MP, Palmer LA, et al. Hemoglobin alpha/eNOS coupling at myoendothelial junctions is required for nitric oxide scavenging during vasoconstriction. Arterioscler Thromb Vasc Biol. 2014;34(12):2594–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Peltz ED, D’Alessandro A, Moore EE, Chin T, Silliman CC, Sauaia A, Hansen KC, Banerjee A. Pathologic metabolism: An exploratory study of the plasma metabolome of critical injury. J Trauma Acute Care Surg. 2015;78(4):742–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carrera-Quintanar L, Lopez-Fuertes M, Climent V, Herranz-Lopez M, Micol V, Pons A, Sogorb F, Roche E. Oxidative damage is present in plasma and circulating neutrophils 4 weeks after a high mountain expedition. Eur J Appl Physiol. 2012;112(8):2923–32. [DOI] [PubMed] [Google Scholar]
- 54.Lehmann T, Mairbaurl H, Pleisch B, Maggiorini M, Bartsch P, Reinhart WH. Platelet count and function at high altitude and in high-altitude pulmonary edema. J Appl Physiol (1985). 2006;100(2):690–4. [DOI] [PubMed] [Google Scholar]
- 55.Singh VK, Wise SY, Fatanmi OO, Beattie LA, Ducey EJ, Seed TM. Alpha-tocopherol succinate- and AMD3100-mobilized progenitors mitigate radiation combined injury in mice. J Radiat Res. 2014;55(1):41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vince RV, Chrismas B, Midgley AW, McNaughton LR, Madden LA. Hypoxia mediated release of endothelial microparticles and increased association of S100A12 with circulating neutrophils. Oxid Med Cell Longev. 2009;2(1):2–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ohyagi Y, Yamada T, Goto I. Hemoglobin as a novel protein developmentally regulated in neurons. Brain Res. 1994;635(1–2):323–7. [DOI] [PubMed] [Google Scholar]