

Abstract
Introduction:
Patients with stage II-III lung adenocarcinoma are treated with adjuvant chemotherapy (ACT) to target the premetastatic niche that persists after curative-intent resection. We hypothesized that the premetastatic niche is a scion of resected lung tumor microenvironment (TME) and that analysis of TME can stratify survival benefit from ACT.
Methods:
Using tumor and tumoral stroma from 475 treatment-naive patients with stage II-III lung adenocarcinoma, we constructed a tissue microarray and performed multiplex immunofluorescent staining for immune markers (programmed death ligand-1 [PD-L1], tumor-associated macrophages [TAMs], and myeloid-derived suppressor cells [MDSCs]), and derived myeloid-lymphoid ratio (MLR). The association between immune markers and survival was assessed using Cox models adjusted for pathologic stage.
Results:
Patients with high PD-L1 expression on TAMs or tumor cells in resected tumors had improved survival with ACT (TAMs: hazard ratio [HR], 1.79; 95% CI, 1.12–2.85; Tumor cells: HR, 3.02; 95% CI, 1.69–5.40). Among patients with high PD-L1 expression on TAMs alone or TAMs and tumor cells, ACT survival benefit is pronounced with high MLR (TAMs: HR, 3.87; 95% CI, 1.79–8.37; TAMs and tumor cells: HR, 2.19; 95% CI, 1.02–4.71) or with high stromal MDSC ratio (TAMs: HR, 2.53; 95% CI, 1.29–4.96; TAMs and tumor cells: HR, 3.21; 95% CI, 1.23–8.35). Patients with low/no PD-L1 expression on TAMs or tumor cells had no survival benefit from ACT.
Conclusions:
Our observation that PD-L1 expression on TAMs or tumor cells is associated with improved survival with adjuvant chemotherapy provides rationale for prospective investigation and developing chemoimmunotherapy strategies for lung adenocarcinoma patients.
Keywords: NSCLC, Tumor immune microenvironment, Pre-metastatic niche, MDSCs, Myeloid-Lymphoid ratio
INTRODUCTION1
Postresection recurrence of early-stage lung adenocarcinomas is a considerable clinical challenge.1,2 Adjuvant chemotherapy (ACT), administered as the standard of care for patients with resected stage II–III lung adenocarcinoma, is associated with a 5-year overall survival (OS) benefit of 4%–15%.3–5 With the exception of pathologic stage (p-stage), there are no known predictors of OS benefit from ACT.6 The beneficial effect of ACT is thought to be derived from targeting the premetastatic niche, which comprises tumor cells and associated immunosuppressive cells that persists even after resection.7,8
We and others have shown that tumor immune microenvironment (TME) factors predict recurrence in early-stage lung adenocarcinomas.9 In advanced lung adenocarcinomas, TME factors are associated with therapeutic benefit from immune checkpoint inhibitor (ICI) agents, specifically programmed death ligand-1 (PD-L1) expression on tumor-associated macrophages (TAMs) and tumor cells.10–13 Owing to the known immunomodulatory effects of chemotherapy,8 coupled with the increasing number of investigations of combination chemotherapy and ICI agents in non-small cell lung cancer (NSCLC),14,15 the TME factors associated with benefit from ACT are of high interest. The Lung Adjuvant Cisplatin Evaluation Biomarker (LACE-Bio) collaborative group investigated tumor and immune cell PD-L1 expression in early-stage NSCLC and concluded that neither tumor nor immune cell PD-L1 expression is predictive of benefit from ACT.16
Whereas tumor and/or immune cell PD-L1 expression may indicate the activity of tumor infiltrating lymphocytes, tumor immunity is a balance of effector and suppressor immune responses.17 The immunomodulatory effects of chemotherapy extend beyond the effector responses that result from the immunogenicity of cancer cell cytolysis.8 Chemotherapeutic agents decrease immunosuppressive cells such as TAMs18 and myeloid-derived superior cells (MDSCs), a type of immature myeloid cell recruited from bone marrow.19 MDSCs play a substantial role in the formation of tumor metastasis, effecting OS in murine models and clinical studies.20 In addition, immune cell ratios such as the myeloid-lymphoid ratio (MLR) in the TME and the neutrophil-lymphocyte ratio in peripheral blood have shown to be of prognostic value in solid tumors, including NSCLC.21,22 However, the interrelation between PD-L1 expression on tumor cells and TAMs and MLR and MDSC ratio, and their association with OS benefit from chemotherapy, is not clear.
We hypothesized that the premetastatic niche immune microenvironment is a scion of the resected lung adenocarcinoma TME and that analysis of PD-L1 expression on TAMs and tumor cells (metrics of effector immune response) and MLR and MDSC ratio (metrics of suppressor immune response) in the lung adenocarcinoma TME can predict OS benefit from ACT.
MATERIALS AND METHODS
Patients
This study was approved by the institutional review board at Memorial Sloan Kettering Cancer Center. From our prospectively maintained database, we identified 1211 patients who had undergone surgical resection for p-stage II–III solitary primary lung adenocarcinoma from January 2000 to December 2012. Exclusion criteria were receipt of induction therapy, lung cancer within 2 years, concurrent non–lung cancer disease progression, wedge resection, and positive surgical margin (see Figure, Supplemental Data 1, CONSORT diagram). No patients in ACT cohort received adjuvant radiation or ICI therapy.
Histologic Examination and Tissue Microarray (TMA) Construction
Tumors were classified according to the 8th edition International Association for the Study of Lung Cancer TNM classification of lung cancer.23 Histologic examination of hematoxylin and eosin (H&E)–stained tumor slides was performed by a pathologist; tumor and stromal regions were marked. Six cores from the two predominant histologic subtypes and three from predominant stromal regions marked on the H&E-stained slides were identified on the corresponding formalin-fixed, paraffin-embedded tumor blocks, and cylindrical 0.6-mm tissue cores were arrayed onto a recipient block to construct a tissue microarray (TMA) (see Figure, Supplemental Data 2A).9
Multiplex Immunofluorescence Staining
Multiplexed staining of four consecutive sections of 4-μm thickness from each TMA were stained with four panels of antibodies (see Figure, Supplemental Data 2B) using the Opal 7-plex fIHC kit (Akoya Biosciences, Marlborough, MA). Stained slides were scanned, and high-power images of individual cores were captured using the Vectra 3.0 multispectral imaging system. Quantitative assessment of cell markers was performed using inForm software version 2.2.1. Cell segmentation and phenotyping algorithms were reviewed and confirmed by the study pathologists.
Statistical Analysis
Patient characteristics were summarized as frequency (percentage) and median (interquartile range) and compared across groups using the Wilcoxon rank-sum test for continuous variables and Fisher’s exact test for categorical variables. Distributions of cell counts were compared across the four groups using the Kruskal-Wallis test.24 If results of the Kruskal-Wallis test were significant, secondary analyses were performed between pairs of groups using Dunn’s test. Dunn’s test is appropriate for groups with unequal numbers of observations. Comparisons of cell count distributions by ACT status within subsets defined by group and cell types were conducted using the Wilcoxon rank-sum test. False-discovery rate corrections were applied to address multiple testing.
The primary outcome of interest was OS (duration from surgery to death). Patients were otherwise censored at the last follow-up. OS was estimated using the Kaplan-Meier approach and compared between groups using log-rank tests. Relationships between groups and OS were quantified using Cox proportional hazards analyses, stratified by p-stage where appropriate.25 Proportionality assumptions were assessed using Schoenfeld residuals.
As a secondary endpoint, lung cancer–specific survival was assessed using a competing-risk approach from the time of surgery to the time of lung cancer death.26 Non–lung cancer deaths were considered a competing-risk event. Lung cancer cumulative incidence of death (LC-CID) was compared between groups using Gray’s test and quantified using Fine and Gray competing-risk regression. Relationships between ACT and LC-CID within each group were quantified as sub-distribution hazard ratios (SHRs) using competing-risk regression, stratified by p-stage. In the comparisons of interest between no ACT vs ACT for each time-to-event endpoint, ACT serves as the reference group, and we quantify the hazard of death without ACT compared to ACT with hazard ratios. A hazard ratio (HR) of less than 1 indicates that no ACT has a lower hazard of the event compared to ACT, while a HR greater than 1 indicates that no ACT has a greater hazard of the event compared to ACT (i.e., ACT is protective).
All analyses were two-sided; P<0.05 was considered statistically significant. Statistical analyses were conducted using Stata 15.0 (Stata Corp, College Station, TX) and R 3.6.1 (R Development Core Team, Vienna, Austria). Box and whisker plots were generated using a special version of Spotfire for quantitative pathology (TIBCO, Palo Alto, CA). Bar graphs were generated using GraphPad Prism version 9.0.2 (San Diego, CA).
RESULTS
PD-L1 Expression and Benefit from ACT
Patient demographic and clinical characteristics are listed in Table 1. Of 394 patients with stage II–III lung adenocarcinoma who underwent complete resection (R0), 211 (54%) received ACT, and 183 (46%) did not. Among patients who received ACT, 95% received platinum-based chemotherapy, 95% received ≥2 cycles of chemotherapy, and 81% completed all cycles of chemotherapy. Consistent with published data, ACT was associated with improved OS (hazard ratio [HR], 1.72; 95% CI, 1.35–2.21; P<0.001) (Figure 1A). Cell counts of lymphoid origin (CD57+ natural killer cells, CD20+ B cells, CD4+ helper T cells, and CD8+ cytotoxic T cells) and myeloid origin (MPO+ neutrophils and CD68+ and/or CD163+ TAMs) in the TME were not statistically significantly different between patients who received ACT and those who did not (Figure 1B; Figure, Supplemental Data 3). Stratification based on PD-L1 expression on tumor cells only (cutoff value ≥1%, in accordance with current clinical practice; Table, Supplemental Data 1)27 was not predictive for the overall cohort (data not shown), ACT cohort (HR, 1.79; 95% CI, 0.55–1.14; P=0.44), or no-ACT cohort (HR, 1.14; 95% CI, 0.81–1.60; P=0.20) (Figure 1C). High numbers of PD-L1+ TAMs (at or above the median) were observed in 53% of patients with low (<1%) PD-L1 expression on tumor cells and 51% of patients with high (≥1%) PD-L1 expression on tumor cells (Figure 1D).
Table 1.
Demographics and clinical characteristics
| Characteristic | Group 1 (TAM PD-L1) (N=119; 30%) |
Group 2 (tumor PD-L1) (N=82; 21%) |
Group 3 (TAM & tumor PD-L1) (N=86; 22%) |
Group 4 (No PD-L1) (N=107; 27%) |
P |
|---|---|---|---|---|---|
| Age, years | 68.9 (60.4–75.1) | 67.3 (63.0–74.0) | 66.9 (59.4–74.6) | 70.0 (63.1–75.9) | 0.3 |
| Sex | |||||
| Female | 77 (65) | 57 (70) | 50 (58) | 76 (71) | 0.3 |
| Male | 42 (35) | 25 (30) | 36 (42) | 31 (29) | |
| Smoking status | |||||
| Never | 25 (21) | 9 (11) | 9 (10) | 30 (28) | 0.013 |
| Former | 75 (63) | 60 (73) | 67 (78) | 67 (63) | |
| Current | 19 (16) | 13 (16) | 10 (12) | 10 (9.3) | |
| Pack-years | 30.0 (5.0–53.8) | 39.5 (17.0–54.0) | 34.5 (20.0–57.5) | 26.0 (0.0–52.5) | 0.13 |
| COPD (N=393) | |||||
| No | 102 (86) | 62 (76) | 66 (78) | 90 (84) | 0.2 |
| Yes | 17 (14) | 20 (24) | 19 (22) | 17 (16) | |
| Resection | |||||
| Pneumonectomy | 10 (8.4) | 5 (6.1) | 5 (5.8) | 6 (5.6) | 1 |
| Bilobectomy | 4 (3.4) | 4 (4.9) | 3 (3.5) | 4 (3.7) | |
| Lobectomy | 98 (82) | 66 (80) | 73 (85) | 89 (83) | |
| Segmentectomy | 7 (5.9) | 7 (8.5) | 5 (5.8) | 8 (7.5) | |
| FEV1 (N=389) | 85.0 (74.0–99.0) | 80.0 (70.0–97.0) | 81.0 (69.9–99.5) | 87.5 (73.0–100.0) | 0.2 |
| DLCO (N=376) | 84.0 (68.5–97.0) | 70.0 (63.5–83.2) | 76.5 (60.0–92.0) | 80.5 (67.0–90.5) | 0.008 |
| Tumor SUV max (N=334) | 7.2 (4.0–9.5) | 6.1 (4.0–9.8) | 7.3 (3.5–10.5) | 6.3 (3.1–10.1) | 0.8 |
| Tumor size, cm | 2.8 (2.2–5.1) | 2.8 (1.8–4.5) | 3.2 (2.0–4.5) | 3.0 (2.1–4.5) | 0.7 |
| Invasive tumor size, cm | 2.7 (2.0–4.5) | 2.8 (1.8–4.5) | 3.1 (2.0–4.1) | 2.7 (1.9–4.3) | 0.8 |
| Nodal status | |||||
| Negative | 24 (20) | 10 (12) | 10 (12) | 16 (15) | 0.3 |
| Positive | 95 (80) | 72 (88) | 76 (88) | 91 (85) | |
| T stage | |||||
| 1 | 38 (32) | 32 (39) | 29 (34) | 36 (34) | 0.8 |
| 2 | 41 (34) | 28 (34) | 37 (43) | 40 (37) | |
| 3 | 30 (25) | 18 (22) | 13 (15) | 23 (21) | |
| 4 | 10 (8.4) | 4 (4.9) | 7 (8.1) | 8 (7.5) | |
| p-stage (8th edition) | |||||
| IIB | 62 (52) | 47 (57) | 39 (45) | 48 (45) | 0.5 |
| IIIA | 51 (43) | 28 (34) | 40 (47) | 52 (49) | |
| IIIB | 6 (5.0) | 7 (8.5) | 7 (8.1) | 7 (6.5) | |
| LVI (N=393) | |||||
| Absent | 37 (31) | 24 (29) | 24 (28) | 26 (25) | 0.7 |
| Present | 82 (69) | 58 (71) | 62 (72) | 80 (75) | |
| Necrosis (N=381) | |||||
| No | 79 (68) | 57 (73) | 49 (59) | 85 (82) | 0.006 |
| Yes | 37 (32) | 21 (27) | 34 (41) | 19 (18) | |
| Mutation type (N=241) | |||||
| Wild type | 29 (49) | 28 (46) | 30 (64) | 37 (50) | 0.069 |
| EGFR | 10 (17) | 6 (10) | 4 (8.5) | 18 (24) | |
| KRAS | 20 (34) | 27 (44) | 13 (28) | 19 (26) | |
| STAS (N=385) | |||||
| No | 44 (38) | 27 (34) | 29 (34) | 37 (36) | 0.9 |
| Yes | 72 (62) | 52 (66) | 57 (66) | 67 (64) | |
| Histologic subtype | |||||
| Lepidic | 5 (4.2) | 2 (2.4) | 1 (1.2) | 1 (0.9) | 0.015 |
| Acinar | 46 (39) | 23 (28) | 31 (36) | 52 (49) | |
| Papillary | 17 (14) | 10 (12) | 8 (9.3) | 13 (12) | |
| Micropapillary | 17 (14) | 8 (10) | 7 (8.1) | 17 (16) | |
| Solid | 30 (25) | 37 (45) | 37 (43) | 19 (18) | |
| Invasive mucinous | 3 (2.5) | 2 (2.4) | 1 (1.2) | 5 (4.7) | |
| Colloid | 1 (0.8) | 0 (0) | 1 (1.2) | 0 (0) | |
| Adjuvant therapy | |||||
| No | 63 (53) | 33 (40) | 43 (50) | 44 (41) | 0.2 |
| Yes | 56 (47) | 49 (60) | 43 (50) | 63 (59) | |
| Immune cell counts | |||||
| CD57 | 13 (0–773) | 12 (2–125) | 16 (0–194) | 11 (0–328) | 0.092 |
| CD8 | 86 (10–806) | 84 (12–460) | 103 (10–900) | 70 (7–478) | 0.010 |
| CD20 | 115 (3–1249) | 159 (9–2076) | 168 (9–1025) | 129 (6–1449) | 0.28 |
| CD4 | 184 (13–2417) | 238 (16–1217) | 190 (5–725) | 201 (15–1106) | 0.036 |
| MPO | 85 (9–754) | 75 (9–694) | 82 (8–461) | 80 (11–596) | 0.97 |
| CD68/163 | 98 (6–627) | 84 (8–1211) | 104 (7–862) | 78 (0–495) | 0.080 |
| MLR | 0.5 (0.2–0.9) | 0.4 (0.2–0.8) | 0.4 (0.2–0.7) | 0.3 (0.2–0.6) | 0.4 |
| Median MLR | |||||
| Below | 57 (48) | 43 (52) | 43 (50) | 63 (59) | 0.4 |
| Above | 62 (52) | 39 (48) | 43 (50) | 44 (41) | |
NOTE. Data are median (interquartile range) or no. (%).
Abbreviations: COL, colloid carcinoma; COPD, chronic obstructive pulmonary disease; DLCO, diffusing capacity of the lung for carbon monoxide; FEV1, forced expiratory volume in 1 second; IMA, invasive mucinous adenocarcinoma; LVI, lymphovascular invasion; MLR, myeloid-lymphoid ratio; p-stage, pathologic stage; STAS, spread through air spaces; SUVmax, maximum standardized uptake value. The four groups based on PD-L1 expression on Tumor cells and Tumor-associated macrophages (TAMs) are Group 1 (high PD-L1 expression on TAMs), Group 2 (high PD-L1 expression on Tumor cells), Group 3 (high PD-L1 on TAMs and Tumor cells), and Group 4 (low/no PD-L1 expression on TAMs or Tumor cells).
Figure 1.
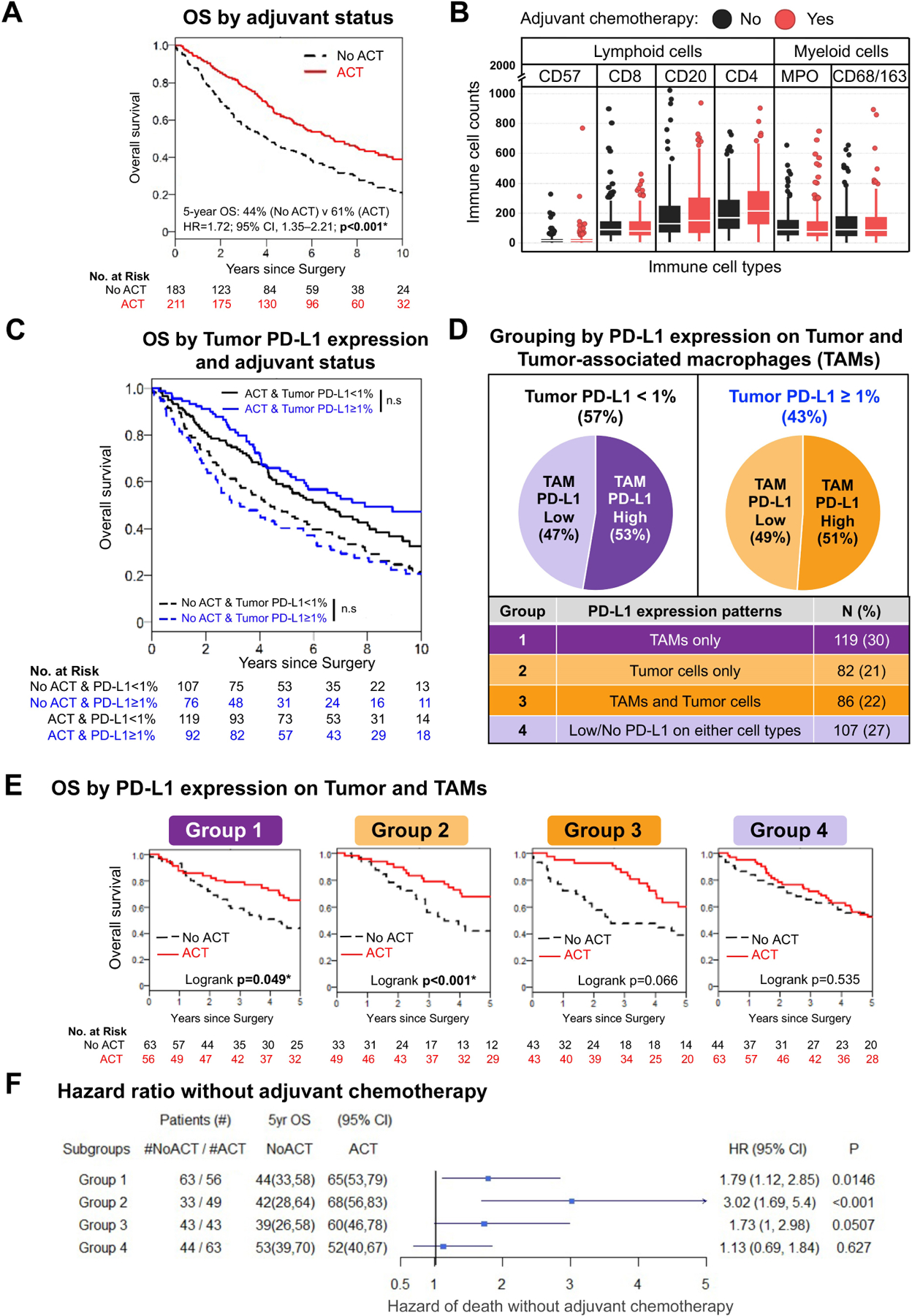
Survival benefit from adjuvant chemotherapy (ACT) by programmed death ligand-1 (PD-L1) expression on tumor-associated macrophages (TAMs) and tumor cells (Tumor cells). (A) Kaplan-Meier curves of patients stratified by ACT status demonstrating a statistically significant improvement in overall survival (OS) among patients who received ACT (5-year OS: no ACT vs ACT, 44% vs 61%; P<0.001). (B) Distribution of immune cell counts of lymphoid (CD57, CD8, CD20, CD4) or myeloid (MPO, CD68/CD163) cells, stratified by ACT (red) or no ACT (black). Each boxplot displays the median (horizontal line within the box) and interquartile range (height of box). (C) Kaplan-Meier curves of patients stratified by PD-L1 expression on Tumor cells only (cutoff ≥1%) showing no association with OS benefit in both ACT and no-ACT cohorts. (D) Pie charts and table displaying the number and percentage of patients within each of the four groups based on PD-L1 expression on Tumor cells and TAMs. (E) Kaplan-Meier curves demonstrating the association between OS and ACT in Group 1 (high PD-L1 expression on TAMs; P=0.049) and Group 2 (high PD-L1 expression on Tumor cells; P<0.001). A nonstatistically significant benefit from ACT was observed in Group 3 (high PD-L1 on TAMs and Tumor cells; P=0.066), and ACT was not associated with a survival benefit in Group 4 (low/no PD-L1 expression on TAMs or Tumor cells; P=0.535). (F) Forest plot summarizing results from pathologic stage–adjusted analyses, demonstrating an increased hazard of death among patients who did not receive ACT in Group 1 (hazard ratio [HR], 1.79; 95% CI, 1.12–2.85; P=0.015), Group 2 (HR, 3.02; 95% CI, 1.69–5.40; P<0.001), and Group 3 (HR, 1.73; 95% CI, 1.00–2.98; P=0.051). The relationship between ACT and hazard of death was not statistically significant for Group 4 (HR, 1.13; 95% CI, 0.69–1.84; P=0.627). *Statistically significant (P<0.05); n.s., not significant.
Patients were stratified into 4 groups based on PD-L1 expression on TAMs and tumor cells (Figure 1D; Table, Supplemental Data 2). Among patients with high PD-L1 expression on TAMs (Group 1; P=0.049) or tumor cells (Group 2; P<0.001), OS was statistically significantly better in those who received ACT. The results of Cox analysis, stratified by p-stage, confirmed this observation (no-ACT vs ACT, Group 1: HR, 1.79; 95% CI, 1.12–2.85; P=0.015; Group 2: HR, 3.02; 95% CI, 1.69–5.40; P<0.001). Patients with high PD-L1 expression on both Tumor cells and TAMs (Group 3) also had improved OS with ACT (P=0.066), although this association was not statistically significant. Results of analysis stratified by p-stage reflected this result (no-ACT vs ACT, Group 3: HR, 1.73; 95% CI, 1.0–2.98; P=0.051). Among patients with low/no PD-L1 expression on TAMs and tumor cells (Group 4), ACT was not associated with+ improved OS (no-ACT vs ACT, P=0.535; HR, 1.13; 95% CI, 0.69–1.84; P=0.627) (Figure 1E and F).
High MLR and Risk of Death without ACT
We next investigated the association between MLR (stratified by median, 0.39) and ACT status and OS benefit (Figure 2A and 2B). An MLR at or above the median (high myeloid content or low lymphoid content) was associated with a higher risk of death in the no-ACT cohort (HR, 1.62; 95% CI, 1.14–2.31; P=0.007) (Figure 2B) but not in the ACT cohort, which, in general, had better prognosis than the no-ACT cohort.
Figure 2.
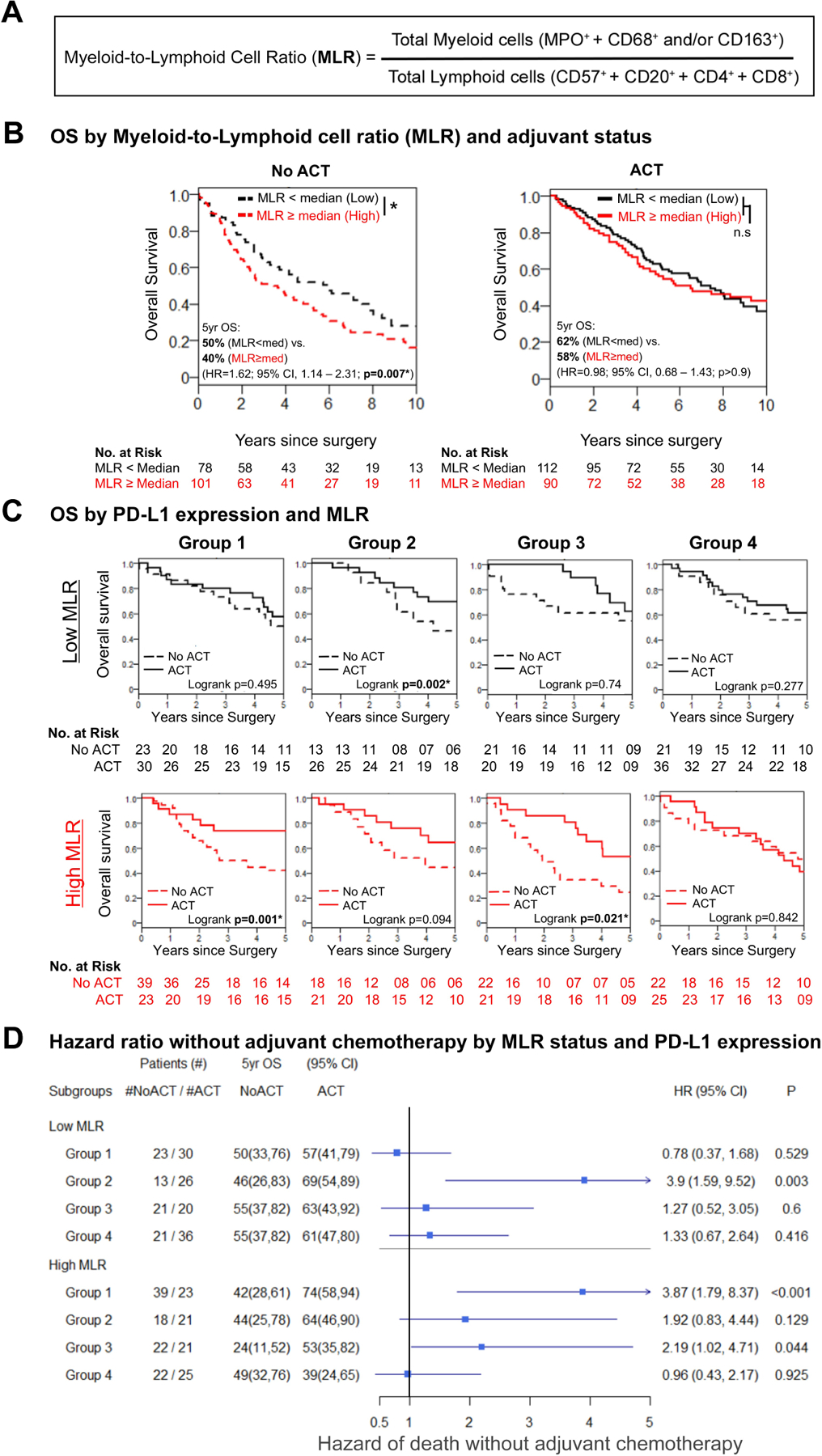
Association between survival benefit from adjuvant chemotherapy (ACT) and high programmed death ligand-1 (PD-L1) expression and high myeloid-lymphoid ratio (MLR). (A) MLR is derived from the ratio of total myeloid cells to lymphoid cells. Total myeloid cells are equal to the sum of neutrophils (MPO+ cells) and tumor-associated macrophages (CD68+ and/or CD163+ cells). Total lymphoid cells are equal to the sum of natural killer, B, helper T, and cytotoxic T cells (identified by CD57+, CD20+, CD4+, and CD8+ markers, respectively). (B) Kaplan-Meier survival estimate curves demonstrating a statistically significant association between MLR and overall survival (OS). In the no-ACT cohort, survival of patients with high MLR was statistically significantly worse (5-year OS: MLR low vs. high, 50% vs. 40%; P=0.007). Whereas survival was not significantly different in the ACT cohort by MLR status (5-year OS: MLR low vs. high, 62% vs. 58%; P>0.9), patients with high MLR benefitted more from ACT, compared with those with low MLR (5-year OS: high MLR, no ACT vs. ACT, 40% vs. 58%; low MLR, no ACT vs ACT, 50% vs. 62%). (C) Kaplan-Meier survival estimates stratified by PD-L1 expression and MLR showing an improvement in survival with ACT among patients with high PD-L1 expression on tumor-associated macrophages and high MLR (Groups 1 and 3; Group 1/high MLR, log-rank P=0.001; Group 3/high MLR, log-rank P=0.021). Patients with high PD-L1 expression on tumor cells and low MLR benefited from ACT (Group 2/low MLR, log-rank P=0.002). Patients with low/no PD-L1 expression had no improvement in OS with ACT. (D) Pathologic stage–adjusted hazard ratio (HR) plot showing an increased risk of death without ACT only among patients in Group 2 with low MLR (Group 2/low MLR, HR, 3.9; 95% CI, 1.59–9.52; P=0.003) and patients in Groups 1 and 3 with high MLR (Group 1/high MLR, HR, 3.87; 95% CI, 1.79–8.37; P<0.001; Group 3/high MLR, HR, 2.19; 95% CI, 1.02–4.71; P=0.044). Regardless of MLR status, patients in Group 4 did not have an increased risk of death without ACT. *Statistically significant (P<0.05); n.s., not significant.
Patients with high PD-L1 expression on TAMs and high MLR had an OS benefit from ACT (Group 1: P=0.001; Group 3: P=0.021); this observation was confirmed in p-stage–adjusted Cox analysis (Group 1: HR, 3.87; 95% CI, 1.79–8.37; P<0.001; Group 3: HR, 2.19; 95% CI, 1.02–4.71; P=0.044). Patients with high PD-L1 expression on tumor cells and low MLR also had an OS benefit from ACT (P=0.002), which was also confirmed in p-stage–adjusted Cox analysis (Group 2: HR, 3.90; 95% CI, 1.59–9.52; P=0.003). Patients with low/no PD-L1 expression did not benefit from ACT, regardless of MLR status (Group 4, Figure 2C and 2D).
We next investigated the association between MLR and LC-CID. Similar to the relationships observed for OS, ACT was beneficial in Group 1 patients with high MLR (P=0.027), and this association was confirmed in p-stage–adjusted competing-risk analysis (SHR, 3.23; 95% CI, 1.35–7.73; P=0.009) (Figure 3A and 3B). Similar to the patterns observed for OS, patients with high PD-L1 expression on tumor cells and low MLR had a non-statistically significant benefit with ACT (SHR, 2.32; 95% CI, 0.88–6.15; P=0.091). Regardless of MLR status, patients with low/no PD-L1 expression did not benefit from ACT (Figure 3A and 3B).
Figure 3.
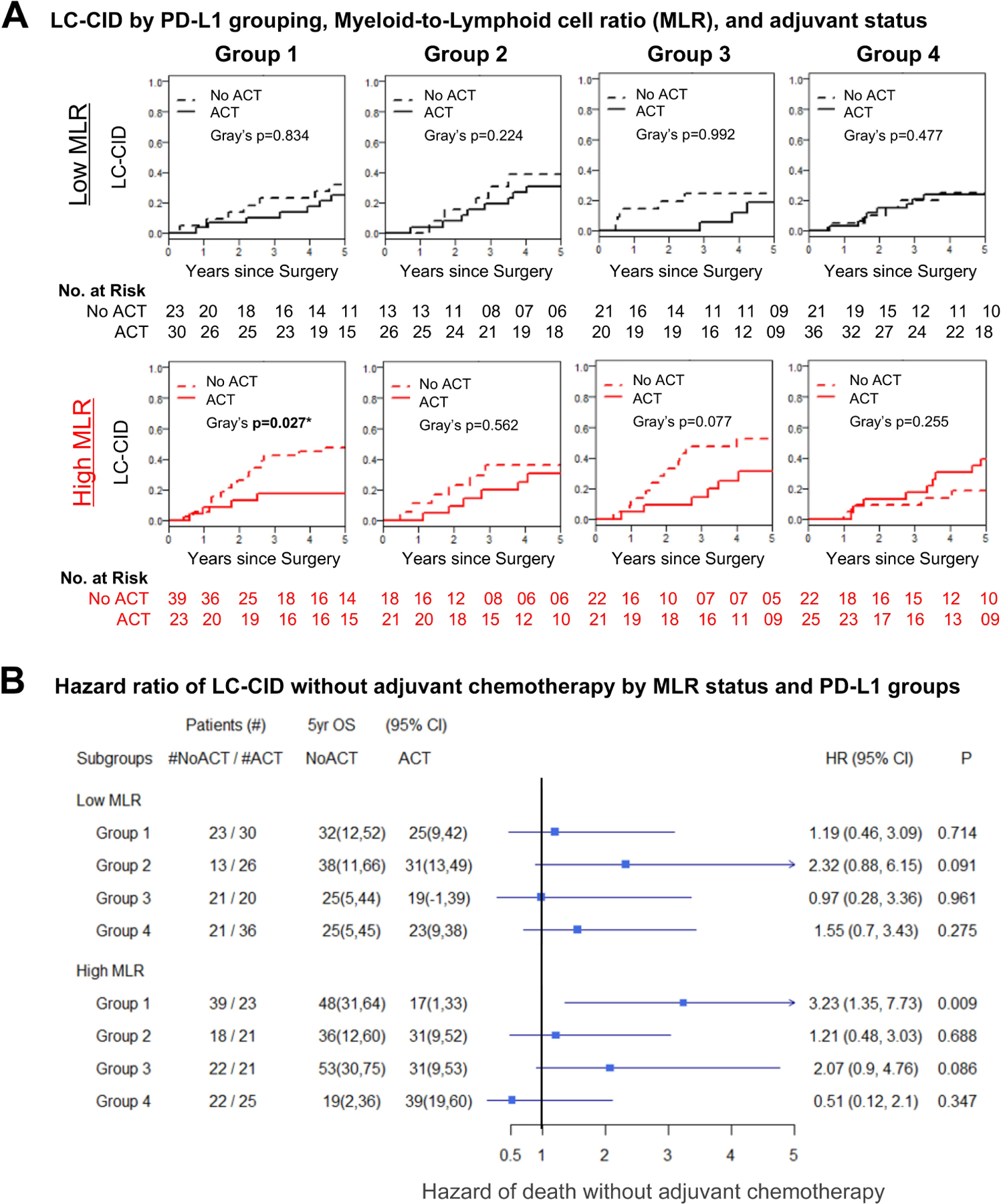
Adjuvant chemotherapy (ACT) and lung cancer cumulative incidence of death (LC-CID). (A) Kaplan-Meier curves showing significant improvement in LC-CID among patients with high programmed death ligand-1 (PD-L1) expression on tumor-associated macrophages and high myeloid-lymphoid ratio (MLR) who received ACT (Group 1/high MLR, Gray’s P=0.027). A delayed trend of LC-CID, in general, can be seen in all groups of patients who received ACT, except for patients in Group 4, regardless of MLR status. (B) Forest plot of pathologic stage–adjusted subhazard ratios (SHRs) showing a significant hazard of lung cancer–related death associated with not receiving ACT among patients in Group 1 with high MLR (Group 1/high MLR, SHR, 3.23; 95% CI, 1.35–7.73; P=0.009). A nonstatistically significant improvement in LC-CID was seen in Group 3 patients with high MLR (Group 3/high MLR, SHR, 2.07; 95% CI, 0.90–4.76; P=0.086) and in Group 2 patients with low MLR (Group 2/low MLR, SHR, 2.32; 95% CI, 0.88–6.15; P=0.091). HR, hazard ratio; OS, overall survival. *Statistically significant (P<0.05).
Stromal Infiltration of MDSCs and Benefit from ACT
With the observation that high PD-L1 expression on TAMs and high MLR was associated with better OS and LC-CID, we assessed individual immune-cell counts in all cores (see Figure, Supplemental Data 4). Statistically significant differences among the four groups were observed in CD8 (Kruskal-Wallis P=0.010) and CD4 (P=0.036) cell counts. The results of Dunn’s multiple comparison test revealed the differences in immune-cell counts were driven by individual groups, with no clear patterns across groups (see Figure, Supplemental Data 4A–C). CD68/163 counts in tumor (P=0.048) and stroma (P=0.005) differed across all groups. However, only in stroma were the differences between Groups 1 (P-adj=0.014) and 3 (P-adj=0.019) and Group 2 (see Figure, Supplemental Data 4C) statistically significant.
Given the above differences in stromal TAM counts between patients with high PD-L1 expression on TAMs (Groups 1 and 3), we attempted to identify the MDSC population that contributed to the higher MLR in the TME. We observed that a high stromal MDSC ratio, derived as the ratio of immature MDSCs (CD11b+CD33+ HLA-DR−) to monocyte-like MDSCs (CD33+ HLA-DR−) at or above the median,28–30 was associated with a benefit from ACT among patients with high PD-L1 expression on TAMs or tumor cells (Group 1: P=0.014; Group 2: P=0.003). A similar benefit was observed in Group 3, but this was not statistically significant (P=0.066) (Figure 4A). On p-stage–adjusted analyses, patients with high PD-L1 expression of any kind and high stromal MDSC ratio who did not receive ACT had a higher risk of death (Group 1: HR, 2.53; 95% CI, 1.29–4.96; P=0.007; Group 2: HR, 3.90; 95% CI, 1.57–9.68; P=0.003; Group 3: HR, 3.21; 95% CI, 1.23–8.35; P=0.017) (Figure 4A). Survival curves by tumor MDSC ratio are shown in Figure, Supplemental Data 5.
Figure 4.
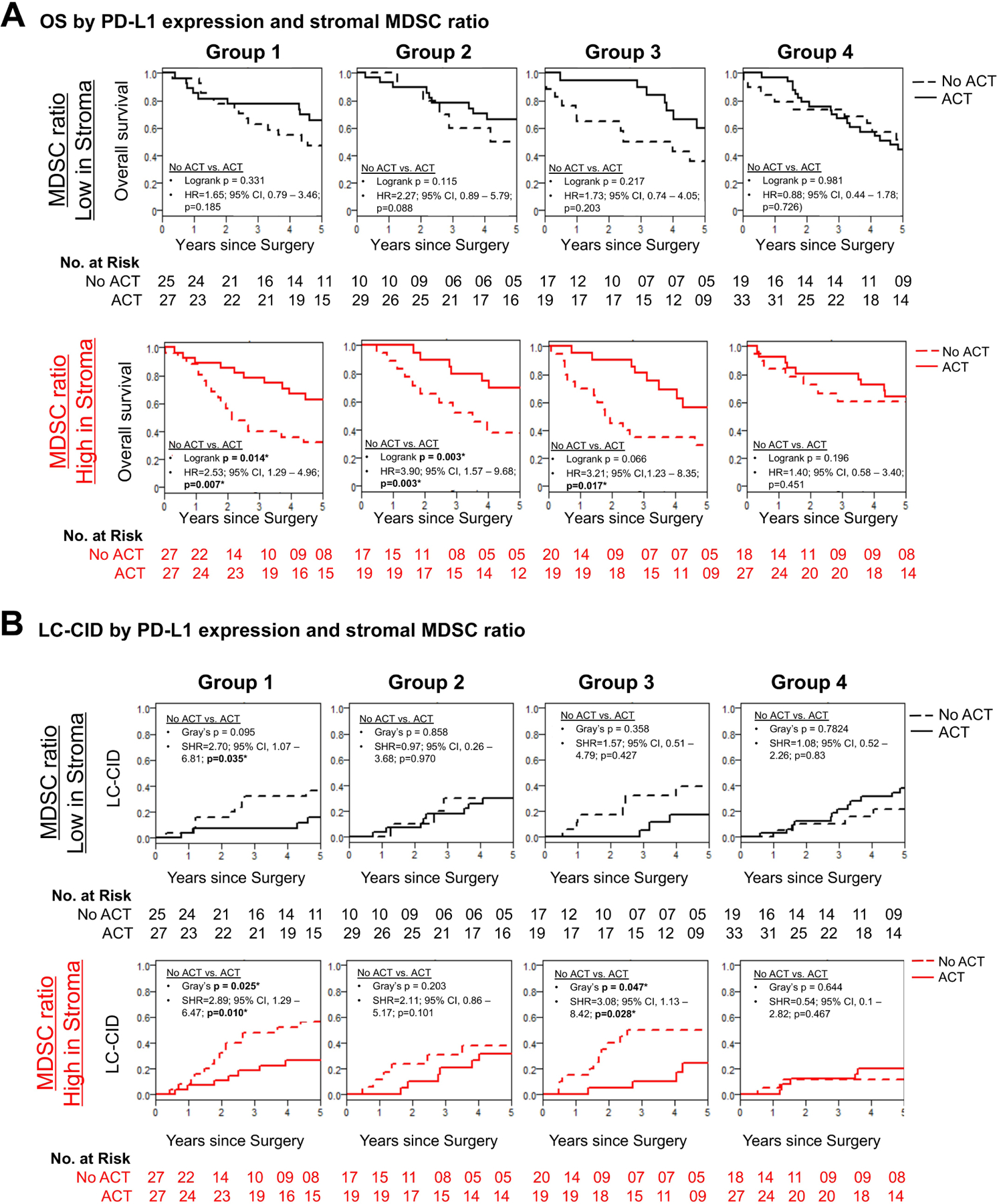
Association between survival benefit from adjuvant chemotherapy (ACT) and high stromal myeloid-derived suppressor cell (MDSC) ratio in patients with high programmed death ligand-1 (PD-L1) expression on tumor-associated macrophages (TAMs) and tumor cells (Tumor cells). (A) Kaplan-Meier survival estimate of patients across PD-L1 groups stratified by low and high stromal MDSC ratio. Patients with high stromal MDSC ratio and high PD-L1 expression on TAMs or Tumor cells benefited from ACT (Group 1/high stromal MDSC ratio, log-rank P=0.014; Group 2/high stromal MDSC ratio, log-rank P=0.03). A similar trend was observed in patients with PD-L1 expression on both Tumor cells and TAMs, irrespective of stromal MDSC ratio. Pathologic stage–adjusted hazard ratio (HR) plot showing an increased risk of death associated with the absence of ACT among patients with high PD-L1 expression on TAMs and/or Tumor cells and high MDSC ratio in Groups 1, 2, and 3 (Group 1/high stromal MDSC ratio, log-rank P=0.007; Group 2/high stromal MDSC ratio, log-rank P=0.003; Group 3/high stromal MDSC ratio, log-rank P=0.017). For patients in Group 4, regardless of stromal MDSC ratio, absence of ACT was not associated with a statistically significantly higher risk of death. (B) Kaplan-Meier survival plots of patients with high PD-L1 expression on TAMs and high stromal MDSC ratio demonstrating a statistically significant benefit from ACT (Group 1/high stromal MDSC ratio, Gray’s P=0.025; Group 3/high stromal MDSC ratio, Gray’s P=0.047). Pathologic stage–adjusted Cox proportional subhazard ratio analyses demonstrate an increased risk of lung cancer cumulative incidence of death (LC-CID) without ACT in patients with high PD-L1 expression on TAMs and high stromal MDSC ratio (Group 1/high stromal MDSC ratio, Gray’s P=0.01; Group 3/high stromal MDSC ratio, Gray’s P=0.028). No increased risk of lung cancer–related death was observed in patients with high PD-L1 expression on TAMs or Tumor cells and low stromal MDSC ratio. In contrast, in patients with low/no PD-L1 expression, lack of ACT was not associated with an increased risk of lung cancer–related death, regardless of stromal MDSC ratio. OS, overall survival. *Statistically significant (P<0.05).
Similar to OS, patients with high PD-L1 expression on TAMs and high stromal MDSC ratio had a lower LC-CID with ACT (Group 1: P=0.025; Group 3: P=0.047) (Figure 4B). After adjustment for p-stage, patients with high PD-L1 expression on TAMs and high stromal MDSC ratio had a statistically significant lower LC-CID with ACT (Group 1: SHR, 2.89; 95% CI, 1.29–6.47; P=0.010; Group 3: SHR, 3.08; 95% CI, 1.13–8.42; P=0.028).
2-yr and 5-yr Survival
Patient stratification based on PD-L1 overexpression on TAMs and tumor cells distinguished patients with differences in 2-year OS of 55% vs 95% and 5-year OS of 20% vs 75% between those who did not and did receive ACT (Figure, Supplemental Data 6).
DISCUSSION
We previously established that effector and suppressor immune markers are independently prognostic in early-stage lung adenocarcinoma and squamous cell cancers.9,31 The present study represents a comprehensive investigation of patients with locoregionally advanced stage II–III lung adenocarcinomas, with attention to immune markers in tumor and stromal compartments and their association with the clinical outcomes of OS, LC-CID, and stage-adjusted risk ratio. Our approach to investigate the resected tumor TME as a proxy for the premetastatic niche provides insights to help guide the development of combination chemoimmunotherapeutic strategies for patients with locoregionally advanced lung adenocarcinoma.
The limited survival benefit of 4%–15%3,4 with ACT, currently recommended for all patients with stage II–II lung adenocarcinomas, is compounded by the side effects of ACT, which underscore the need for biomarkers of ACT response.32,33 Unlike in advanced NSCLC, where investigation is limited by the availability of biopsy tissue, in stage II–III lung adenocarcinomas, fully resected specimens provide sufficient tissue to study the TME. Following the establishment of PD-L1 expression on tumor cells as a marker of therapy response in advanced NSCLC, it was investigated as a marker of chemotherapy response; the results, however, were inconclusive.16,34 Recent preclinical and clinical investigations convincingly demonstrate that PD-L1 expression on immune cells, in addition to tumor cells, is necessary for response to ICI agents.12,35 More importantly, PD-L1 expression on TAMs is predictive of response to ICI therapy in NSCLC, melanoma, ovarian cancer, and triple-negative breast cancer.36–39 Our study demonstrates that patient stratification based on PD-L1 overexpression on TAMs and tumor cells can distinguish patients with a 2-year OS of 95% vs 55% and 5-year OS of 75% vs 20% between those who did and did not receive ACT. Furthermore, our observation that high PD-L1 expression on TAMs alone can be a marker for selection and stratification is intriguing and underscores the need for assessment of PD-L1 expression on TAMs, in addition to tumor cells. To further illustrate the translatability of our findings, we have demonstrated that PD-L1 expression on TAMs can be assessed using the reflex immunohistochemistry testing currently performed for assessment of PD-L1 expression on tumor cells (Figure, Supplemental Data 7). Therefore, use of PD-L1 expression on TAMs and tumor cells as markers of response to ACT is immediately and highly feasible.
The LACE-Bio study investigators analyzed PD-L1 expression on tumor cells and immune cells in patients with early-stage NSCLC and, contrary to our observations, concluded that PD-L1 expression is not predictive.16 However, there are several differences between the studies. Although both studies are retrospective in nature and use the same antibody for PD-L1 expression, in the LACE-Bio study, one-third of patients had stage I disease (none in our study), only 41% of patients had lung adenocarcinomas (100% in our study), patients were predominantly men (73% vs 35% in our study), 31% of patients underwent pneumonectomy (8.4% in our study), and 51% of patients had N0 status (20% in our study).16 Whereas the LACE-Bio study investigated PD-L1 expression on all immune cells, we assessed PD-L1 expression on TAMs only on the basis of the strong rationale provided by recently published data.11–13,16 These differences, combined with our published results that highlight the differential immune composition between lung adenocarcinoma and squamous cell cancer, underscore the need for assessment of immune markers by histologic subtype rather than by all NSCLCs combined.9,31 The high-throughput evaluation of multiple markers in both tumor and tumor-associated stroma, a key requisite for assessment of immune responses, is a strength of our study. Emerging data by investigation of human non-small cell lung cancer cells and peripheral blood monocytes demonstrate differential PD-L1 expression on immune cells (macrophages, dendritic cells, and MDSCs) influenced by specific cytokines such as IL-1a, IL-10, IL-27, and IL-32g in addition to IFN-γ, which can explain the differences observed in PD-L1 expression on TAMs alone in our study in contrast to PD-L1 on all immune cells in LACE-Bio study. In addition, the variable results among both studies could have originated from the study cohort—a homogenous cohort of adenocarcinoma in our study versus heterogenous non-small cell lung cancer in LACE-BIO study.40
As high PD-L1 expression on TAMs and high myeloid content were associated with benefit from ACT, and as these patients also had high stromal TAM counts, we suspected the involvement of factors such as MDSCs, which are immature myeloid precursors that are systemically recruited and subsequently converted to immunosuppressive TAMs after entry into the TME.28,41,42 In our analysis of the tumor and stromal compartments independently, we observed that the ratio of immature MDSCs to monocyte-like MDSCs (surrogate for MDSC infiltration) in tumoral stroma was similarly predictive as MLR, suggesting that peripherally circulating immunosuppressive TAM precursors can perhaps be mitigated by the administration of ACT.18,20,43 Importantly, the increased myeloid content appeared to be associated with PD-L1 expression on TAMs, which is indicative of IFN-γ release and immune activation, whereas PD-L1 expression on tumor cells corresponds with epigenetic dysregulation of the PD-L1 gene, tumor promoter demethylation, or gene amplification, in addition to inflammatory factors.13,36,37 With consideration of these observations combined with recent publications of chemotherapy immune modulation in patients with thoracic cancers,44,45 we postulate that the myelosuppressive effects of chemotherapy on MDSCs and other protumor immune cells may contribute to the survival benefit from ACT. Our observations are further strengthened by recent evidence that tissue-resident macrophages provide a pre-metastatic niche to early NSCLC cells.46,47
The limitations of our study include its retrospective nature and lack of an internal validation set with which to construct a predictive model. In addition, only 81% of patients who received ACT completed all four cycles of chemotherapy, as was commonly seen in ACT trials.48 Although it is thought that four cycles of ACT are required to achieve cytotoxic effects, the necessary number of cycles to achieve immunomodulatory effects is unknown.49,50 In addition, the cut-off values used for TAMs and MLR (median) in this retrospective study are exploratory, not confirmatory. Future studies will set the cut-off value using ROC curves. A prospective study designed to validate our results, along with analyses of matched peripheral blood samples to assess associations of stromal MDSC ratio and MLR with circulating MDSCs and neutrophil-lymphocyte ratio, respectively, is planned. Such a study can identify the number of cycles of ACT that are required to achieve immunomodulatory effects and can inform the development of rational regimens of combination chemoimmunotherapy. The significance of our study is even more important in induction and/or adjuvant chemoimmunotherapy wherein the chemotherapy may enhance the ICI agent efficacy by modulating the tumor immune microenvironment. Examination of the full histological slide is necessary in those studies due to expected necrosis, and to complete better pathological responses.
In conclusion, PD-L1 expression on TAMs and tumor cells is associated with benefit from ACT—in particular, in patients with high MLR or stromal MDSC infiltration in the TME. This association suggests that analysis of the resected TME can inform postresection treatment. Importantly, ACT does not appear to benefit patients without substantial PD-L1 expression on TAMs or tumor cells, regardless of MLR or MDSC ratio. This suggests that the tumor cytotoxicity and immunomodulation of ACT are not associated with improved survival in patients without immune activation. These observations from our retrospective study are to be validated in external cohorts as well as in prospective studies evaluating immune cell function in addition to immune cell marker expression.
Supplementary Material
Figure, Supplemental Data 1. CONSORT Diagram. Consort flowchart showing inclusion and exclusion criteria.
{kind=link}
Figure, Supplemental Data 2. Tissue microarray (TMA) construction, antibody information, and immunofluorescence staining. (A) Primary and secondary International Association for the Study of Lung Cancer histologic subtypes were identified, and three representative tumor sections (orange circles) were marked from each subtype. Three additional cores were then marked from areas of stromal predominance (blue circles). Marked regions were punched and arrayed into recipient blocks, which were used to create TMA slides. (B) Three consecutive sections were stained using the listed panel of antibodies, at the listed dilutions, in four different panel arrangements, each consisting of at least five markers. DAPI was used to identify the nucleus. (C) Representative images of multiplex immunofluorescent staining, showing differential programmed death ligand-1 (PD-L1) expression patterns across the PD-L1 groups created on the basis of PD-L1 expression on tumor cells and tumor-associated macrophages (TAMs). NK, natural killer.
{kind=link}
Figure, Supplemental Data 3. Cell count comparison within programmed death ligand-1 groups by adjuvant chemotherapy (ACT) status. Box and whisker plots showing median cell counts (with 95% CIs) in patients grouped by ACT status within each programmed death ligand-1 group. Results of the Wilcoxon rank-sum test revealed statistically significant differences between (A) all cores (tumor + stroma) in Group 2 (P=0.043) and Group 4 (P=0.018). Comparison of cell counts by ACT status within groups revealed statistically significant differences in (B) tumor cores of Group 2 (CD4; P=0.013) and (C) stromal cores of Group 3 (MPO; P=0.045; CD68/163; P=0.004) and Group 4 (CD20; P=0.049). *Statistically significant (P<0.05).
{kind=link}
Figure, Supplemental Data 4. Cell count comparison across programmed death ligand-1 groups. Box and whisker plots showing median cell counts (with 95% CIs) in patients grouped by programmed death ligand-1 expression patterns. Results of the Kruskal-Wallis test, to compare the distribution of cell counts across groups, were statistically significant. A post hoc analysis using Dunn’s multiple comparison test revealed differences among (A) all cores (tumor + stromal) in CD8 (Group 3 vs. Group 4, P-adj=0.008) and CD4 (Group 1 vs. Group 2, P-adj=0.035) counts, (B) among tumor cores in CD8 counts (Group 3 vs. Group 4, P-adj=0.010), and (C) among stromal cores in CD4 (Group 1 vs. Group 2, P-adj=0.041) and CD68/163 (Group 1 vs. Group 2, P-adj=0.013; Group 2 vs. Group 3, P-adj=0.019) counts. *Statistically significant (P<0.05).
{kind=link}
Figure, Supplemental Data 5. Survival estimates between adjuvant chemotherapy and no adjuvant chemotherapy by tumor myeloid-derived suppressor cell (MDSC) ratio. (A) Kaplan-Meier curves showing overall survival (OS) and (B) lung cancer cumulative incidence of death (LC-CID), stratified by programmed death ligand-1 (PD-L1) grouping and MDSC ratio in tumor cores.
{kind=link}
Figure, Supplemental Data 6. Survival benefit from adjuvant chemotherapy (ACT) by programmed death ligand-1 (PD-L1) expression on tumor-associated macrophages (TAMs) and tumor cells, myeloid-lymphoid ratio (MLR), and stromal myeloid-derived suppressor cell (MDSC) ratio at 2 years and 5 years after resection. (A) Pie chart showing the percentage of patients stratified by PD-L1 expression on TAMs and tumor cells. (B) Overall survival (OS) improved with receipt of ACT among patients with high PD-L1 expression (survival benefit at 2-years defined as the difference in 2-year OS for no ACT vs. ACT: Group 1, 12%; Group 2, 14%; Group 3, 35%). (C) The 2-year survival benefit from ACT was most pronounced in patients with high PD-L1 expression on TAMs and high MLR (Group 1, 17%; Group 2, 15%; Group 3, 37%). High stromal MDSC ratio was associated with a similar OS benefit from ACT in all patients with high PD-L1 expression (Group 1, 29%; Group 2, 20%; Group 3, 50%). (D) Trends for 5-year OS were similar to those for 2-year OS, with a survival benefit from ACT in Groups 1 and 3 (Group 1, 21%; Group 2, 26%; Group 3, 22%). OS was also better in patients with high PD-L1 expression on TAMs with high MLR and high stromal MDSC ratio (MLR: Group 1, 32%; Group 2, 20%; Group 3, 29%; MDSC ratio: Group 1, 29%; Group 2, 28%; Group 3, 35%). H, high; L, low.
{kind=link}
Figure, Supplemental Data 7. Reflex programmed death ligand-1 (PD-L1) immunohistochemistry staining. A brightfield image of lung adenocarcinoma tissue section stained for PD-L1 (clone: E1L3N), showing positive staining on tumor-associated macrophages (arrows). High-power inset, highlighting cell-surface PD-L1 expression on a cluster of tumor-associated macrophages. Scale bar = 100 um.
{kind=link}
Acknowledgments
We acknowledge excellent editorial assistance from David B. Sewell and Summer Koop of the Memorial Sloan Kettering Cancer Center Thoracic Surgery Service.
Research Support:
DJG is supported, in part, by the National Institutes of Health (T32CA009501). MB’s laboratory work is supported by a grant from the National Institutes of Health (K08 CA245206) and by the Thoracic Surgery Foundation. JMI’s research is supported by the Fiona and Stanley Druckenmiller Center for Lung Cancer Research of Memorial Sloan Kettering Cancer Center. DRJ’s laboratory work is supported by grants from the National Institutes of Health (R01 CA217169 and R01 CA240472). PSA’s laboratory work is supported by grants from the National Institutes of Health (P30 CA008748, R01 CA236615-01, and R01 CA235667), the U.S. Department of Defense (BC132124, LC160212, CA170630, CA180889, and CA200437), the Batishwa Fellowship, the Comedy vs Cancer Award, the DallePezze Foundation, the Derfner Foundation, the Esophageal Cancer Education Fund, the Geoffrey Beene Foundation, the Memorial Sloan Kettering Technology Development Fund, the Miner Fund for Mesothelioma Research, the Mr. William H. Goodwin and Alice Goodwin, the Commonwealth Foundation for Cancer Research, and the Experimental Therapeutics Center of Memorial Sloan Kettering Cancer Center. PSA’s laboratory receives research support from Atara Biotherapeutics.
Conflict of Interest Statement:
Dr. Adusumilli declares research funding from ATARA Biotherapeutics; Scientific Advisory Board Member and Consultant for ATARA Biotherapeutics, Bayer, Carisma Therapeutics, Imugene, ImmPactBio, Takeda Therapeutics; Patents, royalties and intellectual property on mesothelin-targeted CAR and other T-cell therapies licensed to ATARA Biotherapeutics, issued patent method for detection of cancer cells using virus, and pending patent applications on PD-1 dominant negative receptor, wireless pulse-oximetry device, and on an ex vivo malignant pleural effusion culture system. All other authors do not have conflicts of interest to disclose.
Memorial Sloan Kettering Cancer Center (MSK) has licensed intellectual property related to mesothelin-targeted CARs and T-cell therapies to ATARA Biotherapeutics, and has associated financial interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CRediT Statement
Daniel J. Gross: Data curation, Formal analysis, Investigation, Roles/Writing - original draft, Writing - review & editing
Navin K. Chintala: Data curation, Formal analysis, Investigation, Roles/Writing - original draft, Writing - review & editing
Raj G. Vaghjiani: Data curation, Formal analysis, Investigation, Writing - review & editing
Rachel Grosser: Data curation, Formal analysis, Investigation, Writing - review & editing
Kay See Tan: Data curation, Formal analysis, Software, Writing - review & editing,
Xiaoyu Li: Data curation, Investigation
Jennie Choe: Data curation, Investigation
Yan Li: Data curation, Investigation
Rania G. Aly: Data curation
Katsura Emoto: Data curation
Zheng Hua: Data curation
Joseph Dux: Data curation
Waseem Cheema: Data curation, Project administration, Visualization
Matthew J. Bott: Data curation, Writing - review & editing
William D. Travis: Data curation, Investigation, Methodology, Resources, Supervision, Writing - review & editing
James M. Isbell: Data curation, Writing - review & editing
Bob T. Li: Data curation, Writing - review & editing
David R. Jones: Data curation, Investigation, Resources, Writing - review & editing
Prasad S. Adusumilli: Conceptualization, Data curation, Formal analysis, Investigation, Funding acquisition, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Roles/Writing - original draft, Writing - review & editing
Abbreviations: ACT, adjuvant chemotherapy; COL, colloid carcinoma; COPD, chronic obstructive pulmonary disease; DLCO, diffusing capacity of the lung for carbon monoxide; FEV1, forced expiratory volume in 1 second; HR, hazard ratio; ICI, immune checkpoint inhibitor; IMA, invasive mucinous adenocarcinoma; LC-CID, lung cancer cumulative incidence of death; LVI, lymphovascular invasion; MDSCs, myeloid-derived suppressor cells; MLR, myeloid-lymphoid ratio; NSCLC, non-small cell lung cancer; OS, overall survival; p-stage, pathologic stage; PD-L1, programmed death ligand-1; SHRs, sub-distribution hazard ratios; STAS, spread through air spaces; SUVmax, maximum standardized uptake value; TAMs, tumor-associated macrophages; TMA, tissue microarray; TME, tumor microenvironment
References
- 1.Hung JJ, Yeh YC, Jeng WJ, et al. Predictive value of the International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society classification of lung adenocarcinoma in tumor recurrence and patient survival. J Clin Oncol. 2014;32:2357–2364. 10.1200/jco.2013.50.1049 [DOI] [PubMed] [Google Scholar]
- 2.Ujiie H, Kadota K, Chaft JE, et al. Solid predominant histologic subtype in resected stage I lung adenocarcinoma is an independent predictor of early, extrathoracic, multisite recurrence and of poor postrecurrence survival. J Clin Oncol. 2015;33:2877–2884. 10.1200/JCO.2015.60.9818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chemotherapy in non-small cell lung cancer: a meta-analysis using updated data on individual patients from 52 randomised clinical trials. Non-small Cell Lung Cancer Collaborative Group. BMJ. 1995;311:899–909. [PMC free article] [PubMed] [Google Scholar]
- 4.Arriagada R, Bergman B, Dunant A, et al. Cisplatin-based adjuvant chemotherapy in patients with completely resected non-small-cell lung cancer. N Engl J Med. 2004;350:351–360. 10.1056/NEJMoa031644 [DOI] [PubMed] [Google Scholar]
- 5.Douillard JY, Rosell R, De Lena M, et al. Adjuvant vinorelbine plus cisplatin versus observation in patients with completely resected stage IB-IIIA non-small-cell lung cancer (Adjuvant Navelbine International Trialist Association [ANITA]): a randomised controlled trial. Lancet Oncol. 2006;7:719–727. 10.1016/s1470-2045(06)70804-x [DOI] [PubMed] [Google Scholar]
- 6.National Comprehensive Cancer Centers. NCCN clinical practice guidelines in oncology (NCCN Guidelines): Non-small cell lung cancer v3.2020. https://www2.tri-kobe.org/nccn/guideline/lung/english/non_small.pdf. Accessed May 19, 2021. [DOI] [PMC free article] [PubMed]
- 7.Krebs MG, Sloane R, Priest L, et al. Evaluation and prognostic significance of circulating tumor cells in patients with non-small-cell lung cancer. J Clin Oncol. 2011;29:1556–1563. 10.1200/jco.2010.28.7045 [DOI] [PubMed] [Google Scholar]
- 8.Zheng H, Zeltsman M, Zauderer MG, et al. Chemotherapy-induced immunomodulation in non-small-cell lung cancer: a rationale for combination chemoimmunotherapy. Immunotherapy. 2017;9:913–927. 10.2217/imt-2017-0052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suzuki K, Kadota K, Sima CS, et al. Clinical impact of immune microenvironment in stage I lung adenocarcinoma: tumor interleukin-12 receptor beta2 (IL-12Rbeta2), IL-7R, and stromal FoxP3/CD3 ratio are independent predictors of recurrence. J Clin Oncol. 2013;31:490–498. 10.1200/JCO.2012.45.2052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ready N, Hellmann MD, Awad MM, et al. First-line nivolumab plus ipilimumab in advanced non-small-cell lung cancer (CheckMate 568): Outcomes by programmed death ligand 1 and tumor mutational burden as biomarkers. J Clin Oncol. 2019;37:992–1000. 10.1200/JCO.18.01042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin H, Wei S, Hurt EM, et al. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade–mediated tumor regression. J Clin Invest. 2018;128:805–815. 10.1172/JCI96113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Y, Zugazagoitia J, Ahmed FS, et al. Immune cell PD-L1 colocalizes with macrophages and is associated with outcome in PD-1 pathway blockade therapy. Clin Cancer Res. 2020;26:970–977. 10.1158/1078-0432.Ccr-19-1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kowanetz M, Zou W, Gettinger SN, et al. Differential regulation of PD-L1 expression by immune and tumor cells in NSCLC and the response to treatment with atezolizumab (anti-PD-L1). Proc Natl Acad Sci USA. 2018;115:E10119–e10126. 10.1073/pnas.1802166115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gandhi L, Rodriguez-Abreu D, Gadgeel S, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med. 2018;378:2078–2092. 10.1056/NEJMoa1801005 [DOI] [PubMed] [Google Scholar]
- 15.Paz-Ares L, Luft A, Vicente D, et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N Engl J Med. 2018;379:2040–2051. 10.1056/NEJMoa1810865 [DOI] [PubMed] [Google Scholar]
- 16.Tsao MS, Le Teuff G, Shepherd FA, et al. PD-L1 protein expression assessed by immunohistochemistry is neither prognostic nor predictive of benefit from adjuvant chemotherapy in resected non-small cell lung cancer. Ann Oncol. 2017;28:882–889. 10.1093/annonc/mdx003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Binnewies M, Roberts EW, Kersten K, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24:541–550. 10.1093/annonc/mdx003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Biasi AR, Villena-Vargas J, Adusumilli PS. Cisplatin-induced antitumor immunomodulation: a review of preclinical and clinical evidence. Clin Cancer Res. 2014;20:5384–5391. 10.1158/1078-0432.Ccr-14-1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bronte V, Brandau S, Chen S-H, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150. 10.1038/ncomms12150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma J, Xu H, Wang S. Immunosuppressive role of myeloid-derived suppressor cells and therapeutic targeting in lung cancer. J Immunol Res. 2018;2018:6319649. 10.1155/2018/6319649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu Y, Qian L, Cui J. Value of neutrophil-to-lymphocyte ratio for predicting lung cancer prognosis: A meta-analysis of 7,219 patients. Mol Clin Oncol. 2017;7:498–506. 10.3892/mco.2017.1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suzuki K, Kachala SS, Kadota K, et al. Prognostic immune markers in non-small cell lung cancer. Clin Cancer Res. 2011;17:5247–5256. 10.1158/1078-0432.CCR-10-2805 [DOI] [PubMed] [Google Scholar]
- 23.Rami-Porta R, Bolejack V, Crowley J, et al. The IASLC lung cancer staging project: Proposals for the revisions of the T descriptors in the forthcoming eighth edition of the TNM. J Thorac Oncol. 2015;10:990–1003. 10.1097/JTO.0000000000000559 [DOI] [PubMed] [Google Scholar]
- 24.Kruskal WH, Wallis WA. Use of ranks in one-criterion variance analysis. J Am Stat Assoc. 1952;47:583–621. 10.2307/2280779 [DOI] [Google Scholar]
- 25.Cox DR. Regression models and life-tables. J R Stat Soc Series B Stat Methodol. 1972;34:187–220. [Google Scholar]
- 26.Dignam JJ, Zhang Q, Kocherginsky M. The Use and Interpretation of Competing Risks Regression Models. Clin Cancer Res. 2012;18:2301–2308. 10.1158/1078-0432.ccr-11-2097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hellmann MD, Ciuleanu TE, Pluzanski A, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. 2018;378:2093–2104. 10.1056/NEJMoa1801946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tcyganov E, Mastio J, Chen E, et al. Plasticity of myeloid-derived suppressor cells in cancer. Curr Opin Immunol. 2018;51:76–82. 10.1016/j.coi.2018.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Law AMK, Valdes-Mora F, Gallego-Ortega D. Myeloid-derived suppressor cells as a therapeutic target for cancer. Cells. 2020;9:561. 10.3390/cells9030561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cassetta L, Bruderek K, Skrzeczynska-Moncznik J, et al. Differential expansion of circulating human MDSC subsets in patients with cancer, infection and inflammation. J Immunother Cancer. 2020;8:e001223. 10.1136/jitc-2020-001223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kadota K, Nitadori J, Ujiie H, et al. Prognostic impact of immune microenvironment in lung squamous cell carcinoma: Tumor-infiltrating CD10+ Neutrophil/CD20+ lymphocyte ratio as an independent prognostic factor. J Thorac Oncol. 2015;10:1301–1310. 10.1097/JTO.0000000000000617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Florea A-M, Büsselberg D. Cisplatin as an anti-tumor drug: Cellular mechanisms of activity, drug resistance and induced side effects. Cancers. 2011;3:1351–1371. 10.3390/cancers3011351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McWhinney SR, Goldberg RM, McLeod HL. Platinum neurotoxicity pharmacogenetics. Mol Cancer Ther. 2009;8:10–16. 10.1158/1535-7163.Mct-08-0840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sepesi B, Cuentas EP, Canales JR, et al. Programmed death cell ligand 1 (PD-L1) is associated with survival in stage I non-small cell lung cancer. Semin Thorac Cardiovasc Surg. 2017;29:408–415. 10.1053/j.semtcvs.2017.05.008 [DOI] [PubMed] [Google Scholar]
- 35.Tang H, Liang Y, Anders RA, et al. PD-L1 on host cells is essential for PD-L1 blockade–mediated tumor regression. J Clin Invest. 2018;128:580–588. 10.1172/JCI96061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schalper KA, Carvajal-Hausdorf D, McLaughlin J, et al. Clinical significance of PD-L1 protein expression on tumor-associated macrophages in lung cancer. J Immunother Cancer. 2015;3:415. 10.1186/2051-1426-3-s2-p415 [DOI] [Google Scholar]
- 37.Hartley GP, Chow L, Ammons DT, et al. Programmed cell death ligand 1 (PD-L1) signaling regulates macrophage proliferation and activation. Cancer Immunol Res. 2018;6:1260. 10.1158/2326-6066.CIR-17-0537 [DOI] [PubMed] [Google Scholar]
- 38.Ojalvo LS, Thompson ED, Wang T-L, et al. Tumor-associated macrophages and the tumor immune microenvironment of primary and recurrent epithelial ovarian cancer. Hum Pathol. 2018;74:135–147. 10.1016/j.humpath.2017.12.010 [DOI] [PubMed] [Google Scholar]
- 39.Ahmed FS, Gaule P, McGuire J, et al. PD-L1 protein expression on both tumor cells and macrophages are associated with response to neoadjuvant durvalumab with chemotherapy in triple-negative breast cancer. Clin Cancer Res. 2020;26:5456–5461. 10.1158/1078-0432.Ccr-20-1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen S, Crabill GA, Pritchard TS, et al. Mechanisms regulating PD-L1 expression on tumor and immune cells. J Immunother Cancer. 2019;7(1):305. 10.1186/s40425-019-0770-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Corzo CA, Condamine T, Lu L, et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 2010;207:2439–2453. 10.1084/jem.20100587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ostrand-Rosenberg S, Fenselau C. Myeloid-derived suppressor cells: Immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol. 2018;200:422–431. 10.4049/jimmunol.1701019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sevko A, Michels T, Vrohlings M, et al. Antitumor effect of paclitaxel is mediated by inhibition of myeloid-derived suppressor cells and chronic inflammation in the spontaneous melanoma model. J Immunol. 2013;190:2464–2471. 10.4049/jimmunol.1202781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dammeijer F, De Gooijer CJ, van Gulijk M, et al. Immune monitoring in mesothelioma patients identifies novel immune-modulatory functions of gemcitabine associating with clinical response. EBioMedicine. 2021;64:103160. 10.1016/j.ebiom.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Goeje PL, Poncin M, Bezemer K, et al. Induction of Peripheral Effector CD8 T-cell Proliferation by Combination of Paclitaxel, Carboplatin, and Bevacizumab in Non-small Cell Lung Cancer Patients. Clin Cancer Res. 2019;25(7):2219–2227. 10.1158/1078-0432.CCR-18-2243 [DOI] [PubMed] [Google Scholar]
- 46.Casanova-Acebes M, Dalla E, Leader AM, et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature. 2021. Epub ahead of print. 10.1038/s41586-021-03651-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoshida C, Kadota K, Ikeda T, et al. Tumor-associated macrophage infiltration is associated with a higher rate of tumor spread through air spaces in resected lung adenocarcinomas. Lung Cancer. 2021;158:91–96. 10.1016/j.lungcan.2021.06.009 [DOI] [PubMed] [Google Scholar]
- 48.Pignon JP, Tribodet H, Scagliotti GV, et al. Lung adjuvant cisplatin evaluation: a pooled analysis by the LACE Collaborative Group. J Clin Oncol. 2008;26:3552–3559. 10.1200/JCO.2007.13.9030 [DOI] [PubMed] [Google Scholar]
- 49.Spreafico F The heterogeneity of the interaction between cancer chemotherapeutic agents and host resistance mechanisms. Recent Results Cancer Res. 1980;75:200–206. 10.1007/978-3-642-81491-4_31 [DOI] [PubMed] [Google Scholar]
- 50.Zagozdzon R, Golab J. Immunomodulation by anticancer chemotherapy: more is not always better (review). Int J Oncol. 2001;18:417–424. 10.3892/ijo.18.2.417 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure, Supplemental Data 1. CONSORT Diagram. Consort flowchart showing inclusion and exclusion criteria.
Figure, Supplemental Data 2. Tissue microarray (TMA) construction, antibody information, and immunofluorescence staining. (A) Primary and secondary International Association for the Study of Lung Cancer histologic subtypes were identified, and three representative tumor sections (orange circles) were marked from each subtype. Three additional cores were then marked from areas of stromal predominance (blue circles). Marked regions were punched and arrayed into recipient blocks, which were used to create TMA slides. (B) Three consecutive sections were stained using the listed panel of antibodies, at the listed dilutions, in four different panel arrangements, each consisting of at least five markers. DAPI was used to identify the nucleus. (C) Representative images of multiplex immunofluorescent staining, showing differential programmed death ligand-1 (PD-L1) expression patterns across the PD-L1 groups created on the basis of PD-L1 expression on tumor cells and tumor-associated macrophages (TAMs). NK, natural killer.
Figure, Supplemental Data 3. Cell count comparison within programmed death ligand-1 groups by adjuvant chemotherapy (ACT) status. Box and whisker plots showing median cell counts (with 95% CIs) in patients grouped by ACT status within each programmed death ligand-1 group. Results of the Wilcoxon rank-sum test revealed statistically significant differences between (A) all cores (tumor + stroma) in Group 2 (P=0.043) and Group 4 (P=0.018). Comparison of cell counts by ACT status within groups revealed statistically significant differences in (B) tumor cores of Group 2 (CD4; P=0.013) and (C) stromal cores of Group 3 (MPO; P=0.045; CD68/163; P=0.004) and Group 4 (CD20; P=0.049). *Statistically significant (P<0.05).
Figure, Supplemental Data 4. Cell count comparison across programmed death ligand-1 groups. Box and whisker plots showing median cell counts (with 95% CIs) in patients grouped by programmed death ligand-1 expression patterns. Results of the Kruskal-Wallis test, to compare the distribution of cell counts across groups, were statistically significant. A post hoc analysis using Dunn’s multiple comparison test revealed differences among (A) all cores (tumor + stromal) in CD8 (Group 3 vs. Group 4, P-adj=0.008) and CD4 (Group 1 vs. Group 2, P-adj=0.035) counts, (B) among tumor cores in CD8 counts (Group 3 vs. Group 4, P-adj=0.010), and (C) among stromal cores in CD4 (Group 1 vs. Group 2, P-adj=0.041) and CD68/163 (Group 1 vs. Group 2, P-adj=0.013; Group 2 vs. Group 3, P-adj=0.019) counts. *Statistically significant (P<0.05).
Figure, Supplemental Data 5. Survival estimates between adjuvant chemotherapy and no adjuvant chemotherapy by tumor myeloid-derived suppressor cell (MDSC) ratio. (A) Kaplan-Meier curves showing overall survival (OS) and (B) lung cancer cumulative incidence of death (LC-CID), stratified by programmed death ligand-1 (PD-L1) grouping and MDSC ratio in tumor cores.
Figure, Supplemental Data 6. Survival benefit from adjuvant chemotherapy (ACT) by programmed death ligand-1 (PD-L1) expression on tumor-associated macrophages (TAMs) and tumor cells, myeloid-lymphoid ratio (MLR), and stromal myeloid-derived suppressor cell (MDSC) ratio at 2 years and 5 years after resection. (A) Pie chart showing the percentage of patients stratified by PD-L1 expression on TAMs and tumor cells. (B) Overall survival (OS) improved with receipt of ACT among patients with high PD-L1 expression (survival benefit at 2-years defined as the difference in 2-year OS for no ACT vs. ACT: Group 1, 12%; Group 2, 14%; Group 3, 35%). (C) The 2-year survival benefit from ACT was most pronounced in patients with high PD-L1 expression on TAMs and high MLR (Group 1, 17%; Group 2, 15%; Group 3, 37%). High stromal MDSC ratio was associated with a similar OS benefit from ACT in all patients with high PD-L1 expression (Group 1, 29%; Group 2, 20%; Group 3, 50%). (D) Trends for 5-year OS were similar to those for 2-year OS, with a survival benefit from ACT in Groups 1 and 3 (Group 1, 21%; Group 2, 26%; Group 3, 22%). OS was also better in patients with high PD-L1 expression on TAMs with high MLR and high stromal MDSC ratio (MLR: Group 1, 32%; Group 2, 20%; Group 3, 29%; MDSC ratio: Group 1, 29%; Group 2, 28%; Group 3, 35%). H, high; L, low.
Figure, Supplemental Data 7. Reflex programmed death ligand-1 (PD-L1) immunohistochemistry staining. A brightfield image of lung adenocarcinoma tissue section stained for PD-L1 (clone: E1L3N), showing positive staining on tumor-associated macrophages (arrows). High-power inset, highlighting cell-surface PD-L1 expression on a cluster of tumor-associated macrophages. Scale bar = 100 um.