

Abstract
Soil pH is a key controller of denitrification. We analysed the metagenomics/transcriptomics and phenomics of two soils from a long-term liming experiment, SoilN (pH 6.8) and un-limed SoilA (pH 3.8). SoilA had severely delayed N2O reduction despite early transcription of nosZ (mainly clade I), encoding N2O reductase, by diverse denitrifiers. This shows that post-transcriptionally hampered maturation of the NosZ apo-protein at low pH is a generic phenomenon. Identification of transcript reads of several accessory genes in the nos cluster indicated that enzymes for NosZ maturation were present across a range of organisms, eliminating their absence as an explanation for the failure to produce a functional enzyme. nir transcript abundances (for NO2− reductase) in SoilA suggest that low NO2− concentrations in acidic soils, often ascribed to abiotic degradation, are primarily due to biological activity. The accumulation of NO2− in neutral soil was ascribed to high nar expression (nitrate reductase). The -omics results revealed dominance of nirK over nirS in both soils while qPCR showed the opposite, demonstrating that standard primer pairs only capture a fraction of the nirK pool. qnor encoding NO reductase was strongly expressed in SoilA, implying an important role in controlling NO. Production of HONO, for which some studies claim higher, others lower, emissions from NO2− accumulating soil, was estimated to be ten times higher from SoilA than from SoilN. The study extends our understanding of denitrification-driven gas emissions and the diversity of bacteria involved and demonstrates that gene and transcript quantifications cannot always reliably predict community phenotypes.
Subject terms: Metagenomics, Soil microbiology
Introduction
During the past century human activities have accelerated the input of reactive N to the biosphere [1, 2]. This has escalated the emissions of N2O, a major greenhouse gas and contributor to ozone depletion [3–5], as well as nitric oxide (NO) and nitrous acid (HONO) which both influence chemical reactions in the troposphere, leading to formation of undesired ozone [6]. Agriculture, and especially the excessive use of synthetic N fertilisers in large parts of the world, is a major source of N2O release from terrestrial systems, primarily via denitrification and nitrification [1, 7, 8]. While CO2 emissions are predicted to decline substantially during the present century, anthropogenic N2O emissions, which have increased at an average rate of 0.6 ± 0.2 Tg N yr−1 per decade during the past 40 years, are expected to continue to increase [5] or remain nearly constant [9], unless new methods to reduce the N2O/N2 product ratio of our agroecosystems are developed [10, 11]. Some mitigation options have been identified through improved understanding of the organisms producing and reducing N2O and the environmental factors that control these emissions [12], and more are expected to emerge as we intensify our research on denitrification and denitrifying organisms in soil.
Denitrification is the stepwise reduction of nitrate (NO3−) to N2 via nitrite (NO2−), NO and N2O, and is performed by a diverse range of facultatively anaerobic organisms when O2 becomes scarce. Complete reduction of NO3− to N2 is a four-step process with each step catalysed by a different reductase [13, 14]: the NO3− reductases NarG (membrane-bound) and/or NapA (periplasmic) encoded by the genes narG and napA; the NO2− reductases NirK (copper-containing) or NirS (containing cytochrome cd1) encoded by nirK and nirS; the NO reductases cNor (cytochrome c dependent) or qNor (quinol-dependent) encoded by cnor and qnor; and the N2O reductase NosZ, of which two clades have been identified, encoded by nosZ clade I and II [15]. While many organisms have both Nar and Nap, most denitrifiers carry only one type of Nir, Nor and NosZ. The denitrification pathway is modular [16] and the absence of one or more of the reduction steps is common [17, 18]. Not only does the presence/absence of the denitrification genes affect the phenotypes of denitrifiers and the amounts of the different denitrification products that are released but gene regulation [14], in interplay with environmental factors, is a key factor as well [18–20]. Similar controls of N2O/N2 product ratios have been observed in soil communities [21]. If the production and reduction steps are not fully balanced net production of a denitrification intermediate will occur. Studies of denitrification kinetics during anoxic incubation of various denitrifying organisms always show temporal accumulation of intermediate products [18, 22], but the extent differs even between closely related strains [23]. Also, since the timing of denitrification gene transcription varies between organisms, denitrifier communities will have a succession of actively transcribing populations during an anoxic spell which will also influence levels of intermediate products [24].
Of the environmental key factors known to control denitrification perhaps the best characterised is soil pH [25, 26], which profoundly affects the accumulation of denitrification intermediates. The effects of pH on the regulation and enzymology of the four reduction steps of denitrification differ, however, and the mechanisms by which they are affected, are not well understood. While the accumulation of NO2− increases with soil pH [27, 28], there is a clear negative correlation between pH and N2O emissions [29–33]. The simple assumption that soils with high N2O emissions have fewer N2O-reducing organisms is corroborated by some studies but not by others [32, 34–36], which lends little support for a direct, causal relationship between nosZ numbers and N2O emissions. Studies of denitrifying bacteria in pure culture, extracted soil bacterial communities and in intact soils [37–39] all showed that nosZ transcription did take place under acidic conditions (pH 5.7–6.1) during periods of active denitrification but with negligible or strongly delayed N2O reduction. This suggests a post-transcriptional phenomenon, possibly interference with the maturation of the NosZ apo-protein, or with the transfer of electrons to the reductase. The quantification of nosZ transcripts in those soil communities was, however, based on PCR primers targeting only bacteria carrying nosZ clade I. Moreover, the transcripts were not sequenced, which leaves it open to speculation whether different fractions of the denitrifying bacteria transcribe nosZ at low vs high pH. This, and the fact that the post-transcriptional effect of low pH has only been tested in a few model strains, implies that it remains essentially unknown how widespread this phenomenon is among denitrifiers in soils.
The general occurrence of low NO2− concentrations in acidic soils has been attributed mainly to chemical decomposition at low pH [40]. This was challenged recently by [28], who suggested that high rates of microbial reduction of NO2− played a key role in keeping NO2− concentrations low during denitrification in acidic soil. This would also imply high rates of NO reduction since the concentration of this gas was kept low. In soil with near neutral pH, on the other hand, the rate of NO3− reduction exceeded the rate of NO2− reduction, leading to NO2− accumulation. The nir and nor gene abundance, their transcriptional activity and the organisms involved were however not investigated.
Here we aimed at resolving some of the questions raised from the above-mentioned studies about how soil pH affects the composition and activity of the denitrifier community and the accumulation/release of denitrification intermediates. We took an integrated “multi-omics” approach [41] to avoid primer biases [42] and thus to include as large a portion as possible of the microbial community, and analysed the metagenomes (MGs) and the metatranscriptomes (MTs) of two soils with significantly different pH, 3.8 (SoilA) and 6.8 (SoilN). Samples for MT analysis were taken at time intervals through anoxic incubation of soil microcosms, during which the dynamics of the denitrifier community “phenome” was followed by frequent determinations of denitrification intermediate and end products. The results from the phenomics analysis, presented in more detail in [28], showed complete denitrification in SoilN from the start of the incubation and accumulation of NO2−. SoilA, on the other hand, showed no NO2− accumulation but severely delayed N2O reduction (Fig. 1A, B). The problems with obtaining mRNA from low pH soil (pH 4) encountered earlier [38] were overcome using an optimised protocol [43], which provided nucleic acid quality suitable for both shotgun MG- and MT sequencing. The relative abundances of detected denitrification genes and transcripts were compared to results from PCR-quantification.
Fig. 1. Kinetics of NO2−, N-gases and denitrification enzyme rates.

Results for anoxic incubation of two soils with pHCaCl2 = 3.8 (SoilA) and 6.8 (SoilN), amended with NO3−. A Measured amounts of NO2−, NO, N2O and N2. Sampling for metatranscriptome analyses are indicated by red arrows (0.5 and 3 h for SoilA; 0.5, 3, 9, 12 and 27 h for SoilN). B Reduction rates for the different denitrification steps VNAR (NO3− → NO2−), VNIR (NO2− → NO), VNOR (NO → N2O) and VNOS (N2O → N2), all given as µmol N vial−1 h−1. The values were based on the measured kinetics of NO2−, NO, N2O and N2 and corrected for abiotic decomposition of NO2− as published previously by Lim et al. [28]. Abiotic NO2− decomposition was significant only in SoilA. The figure is adapted from graphs shown in [28] and based on the same dataset.
The -omics analyses allowed us to address several issues. One was to determine if the abundance of nar/nap, nir and nor genes and transcripts could explain the strong control of NO2− and NO observed in the acidic soil and which organisms were involved. In addition, the detailed kinetics data made it possible to estimate if NO2−- accumulation in neutral soil would lead to more or less HONO emission than from non-NO2− accumulating, acidic soil. Secondly, we investigated if the apparent lack of DNRA activity (dissimilatory nitrite reduction to ammonium) in these soils was due to low abundance of DNRA-related genes and transcripts. Thirdly, we clarified the complex ecophysiology of nosZ carrying bacteria to better understand their hampered N2O reduction under acidic conditions (this study and [38, 39]). To do so, we investigated to what extent the two nosZ clades were found in the MGs and MTs of two soils of differing pH; if nosZ gene transcripts originated from a few populations or represented diverse denitrifying bacteria; and if genes other than nosZ in the nos operon were transcribed in acidic soil. For the latter, we included nosR, which encodes NosR suggested to be involved in electron delivery to NosZ in organisms with nosZ cladeI; nosL, which encodes a chaperone delivering Cu to the NosZ apo-protein (both nosZ clades); and the ORF nosDFY (both nosZ clades) encoding NosD, suggested to be involved in NosZ maturation, and the ABC-transporter NosFY [44–46].
Materials and methods
Soils
Two peat soils (40–45% organic C, 2% organic N) [38] with pH 3.8 (SoilA) and 6.8 (SoilN) were sampled from a long-term field experimental site in western Norway (61°17'42”, 5°03'03”). SoilA is the original un-limed soil, and SoilN was limed in 1978 with shell sand (800 m3 ha−1; [47]). Freshly sampled soils were transported to the laboratory, sieved (4.5 mm) upon arrival, then stored in sealed plastic bags at 4 °C. All pH values were measured in 0.01 M CaCl2 (soil:CaCl2 1:5) immediately prior to further analyses.
Soil treatment
To ensure detectable transcription, 5 mg dried, powdered clover g−1 soil wet weight (ww) was mixed into the soil before incubation at 15 °C for 72 h [38]. Soil aliquots corresponding to 1.5 g soil organic C (5–8 g of soil ww depending on lime content) were placed in air-tight glass vials and sealed with butyl-rubber septa and aluminium crimps. Nitrate (as KNO3) was dissolved in autoclaved MilliQ water, which was added to reach 80% of the soil’s water holding capacity and 6.2–7.1 mM NO3− in soil moisture. Thus, at the onset of the incubation, the total amount of NO3− per vial was 37 or 26 μmol NO3− in SoilA or SoilN, respectively (see also [28]. The vials were immediately made anoxic by six cycles of gas evacuation and He filling [38], and incubated at 15 °C. Gases (CO2, O2, NO, N2O and N2) were measured in headspace every 3 h using an autosampler linked to a GC and NO analyser [48]. At each gas sampling time point, one replicate vial of each soil type was sacrificed and NO2− was extracted and concentration determined as described in [28]. A portion of soil from the same vial was snap frozen in liquid nitrogen, then stored at −80 °C until nucleic acid extraction. The soil incubation experiment was described in detail in [28], which also explains how the rates of the four steps of denitrification were calculated based on measured concentrations of NO2−, NO, N2O and N2 throughout the incubation, and corrected for abiotic decomposition of NO2− in the acid soil. The present MG and MT analyses were done on two of the three soils investigated by [28], and our presentation of the kinetics is limited to the calculated rates of the four steps of denitrification.
Nucleic acid extraction
DNA and RNA were extracted from frozen samples using the method of [43], who also tested several commercial kits for these soils without success. Briefly, 3 × 0.2 g of soil was taken at time 0 (at the start of anoxic incubation) for DNA extraction, and at selected time points (0.5–27 h) during anoxic incubation for RNA extraction. Lysis was performed with glass beads in CTAB extraction buffer and phenol-chloroform-isoamyl alcohol (25:24:1), using a FastPrep-24 instrument. After ethanol precipitation, the nucleic acids were resuspended in DEPC-treated nuclease-free water purified with the OneStep PCR Inhibitor Removal Kit (Zymo Research, Irvine, USA), then split into a fraction for DNA and one for RNA. The DNA fraction was further purified using the Genomic DNA Clean & Concentrator kit (Zymo Research), then kept at −20 °C until use. The RNA fraction was digested using the TURBO DNA-free DNase kit (Ambion, Life Technologies) according to the manufacturer’s instructions, then purified using the RNA Clean & Concentrator-5 kit (Zymo Research). Quantitative PCR (qPCR) using primers targeting the 16S rRNA gene (described below) was used to assess the presence of residual genomic DNA (gDNA) in the purified RNA fractions (defined by signal detected in the qPCR at ≤35 cycles), and only RNA fractions free of gDNA was used for further analysis. The purified and DNA-free RNA fractions were reverse transcribed using the Maxima Reverse Transcriptase with random hexamer primers (Thermo Scientific), according to the manufacturer’s instructions. Primers targeting the 16S rRNA or nosZ genes (described below) were used in qPCR to assess the quality (defined by uninhibited amplifiability) of purified DNA and reverse-transcribed cDNA.
Sequencing the metagenome (MG), metatranscriptome (MT), and 16S rRNA genes
Triplicate DNA and duplicate RNA samples were sent for metagenomic and metatranscriptomic sequencing at The Roy J. Carver Biotechnology Center (CBC)/W. M. Keck Center for Comparative and Functional Genomics at the University of Illinois at Urbana-Champaign, using HiSeq 2500 technology. All nucleic acids were shipped in a liquid nitrogen vapour dry shipper (Cryoport) and arrived within 5 days (the Cryoport Express dewar is able to maintain the temperature at −150 °C during shipment for 10 days). The RNA integrity (including confirmation of the absence of gDNA) was also independently verified by the CBC prior to sequencing the samples. The microbial community composition was analysed after sequencing of partial 16S rRNA genes using MiSeq technology (Illumina) at StarSEQ GmbH (Mainz, Germany) (2 × 300 bp paired-end sequencing with V3 chemistry). The primers used targeted the V4 region of the 16S rRNA gene, 515f and 806rB [49, 50], as detailed by the Earth Microbiome project (http://www.earthmicrobiome.org/emp-standard-protocols/16s/).
Amplicon sequence analysis of 16S rRNA genes
Processing of the sequenced 16S rRNA gene amplicons was performed by StarSEQ Gmbh, Mainz (Germany). Briefly, the sequences were demultiplexed and the adapters were trimmed locally on the MiSeq instrument with the Illumina Metagenomics 16S rRNA application, using default settings. Amplicon sequence data was processed using the Greenfield Hybrid Analysis Pipeline (GHAP) which combines amplicon clustering and classification tools from USearch [51], and RDP [52], combined with custom scripts for demultiplexing and OTU table generation [53]. Reads were subject to quality trimming with a cut-off threshold of 25 and length trimming where only reads in the length range 250–258 bp were retained. Trimmed forward and reverse reads were then merged and only successfully merged reads retained. OTU clustering was performed at a 97% sequence similarity. Representative sequences from each OTU were classified by finding their closest match in a set of reference 16S rRNA gene sequences, and by using the RDP Naïve Bayesian Classifier. The RDP 16S rRNA gene training set and the RefSeq 16S rRNA gene reference sequence collection were used for classification. NumPy, SciPy, scikit-learn and Matplotlib were used for calculation and plotting of principle component analyses (PCAs) [54–56]. Samples were rarefied to 38287 reads/sample for the purpose of determining species richness and Peilou’s evenness, calculated using USearch v11 [57].
Analysis of MG- and MT sequences
After trimming the number of reads (in millions) was between 25.9 and 31.4 in the MG samples and between 16.4 and 42.0 in the MT samples except for two samples which were lower (4.0 for one of the 12 h duplicates from SoilN and 8.5 for one of the 0.5 h duplicates from SoilA). The sequenced reads, obtained by HiSeq sequencing technology (Illumina), were quality controlled using BBDuk from the BBTools package version 35.66 (http://sourceforge.net/projects/bbmap/). For functional annotation, reads were aligned using DIAMOND with an e-value cut-off of 1 × 10−3 [58]. For analysis of reads derived from genes involved in nitrogen metabolism comparison was done against a custom dataset described in [59, 60]. The DIAMOND output was converted to m8 blast format and analysed in R. Reads must have had a matching region of >30 amino acids and an identity of >60% to be considered matching. Output of matching reads were normalised to reads per million of total reads, RPM (see below).
Reads derived from specific genes and meeting the assigned quality cutoffs were extracted from read sets using filterbyname from the BBTools suite of programs. The extracted reads were then uploaded to KBase [61] and taxonomic assignment was performed using KAIJU using default settings [62].
Statistical and quantitative analysis of meta-omic data
All reads counts were normalised for sequencing depth, generating RPM values: (number of reads)/(total reads that passed quality control) × 106. All statistical analyses and graphing were performed using in-house R scripts custom created for this purpose.
Quantitative amplification-based analysis
The genes encoding 16S rRNA and the three denitrification reductases nirK, nirS and nosZ clade I were quantified by qPCR using the primers 27F and 518R for the 16S rRNA gene [63], 517F and 1055R for the nirK gene [64], cd3aF and R3cd for the nirS gene [65] and Z-F and 1622R for the nosZ gene [66]. DNA samples were diluted to 1–10 ng of DNA per reaction. All cDNA and RNA samples (DNase-digested) were used without dilution. Each 20 µL qPCR reaction contained SYBR Premix Ex Taq II (Tli RNaseH Plus) (Takara Bio) and was run according to the manufacturer’s instructions and included 0.4 µM of each primer and 2 µL of template. The optimised qPCR cycling conditions for all primer sets were 95 °C for 5 min, 40 cycles of 95 °C for 30 s, x for 60 s, 72 °C for 30 s, 82 °C for 20 s, and a final melting curve analysis from 60 °C to 95 °C to determine the specificity of amplicons, where x = 54 °C (16S rRNA gene), or 60 °C (denitrification genes). To reduce background signals from primer dimers and unspecific PCR products, the fluorescence signal was measured during the final step of each cycle, at 82 °C. The detection limit of each qPCR run was five copies per microliter of reaction [43], which was ~4 × 102 copies g−1 soil (ww).
Results
Kinetics of denitrification intermediates depict a pH-dependent response to anoxia
The denitrification kinetics of the two soils during 45 h of anoxic incubation are shown in Fig. 1, in which the sampling occasions for MT analyses are also indicated. A more complete description of the incubation experiment is given by [28], including detailed analyses of production/reduction rates of the denitrification intermediates/end products over 70 h. The analysis included a careful mineral N budget analysis demonstrating 100% recovery of NO3−-N as N2 for the soil with pH = 6.8, which suggests negligible reduction of nitrate to ammonium in this soil. For Soil 3.8, the recovery as N-gas (N2 + N2O + NO) was lower (77%), but abiotic nitrosylation of organic material accounted for 17% [28], thus in total 94% was accounted for, leaving only 6% of the NO3−- loss that could possibly be ascribed to DNRA.
The two soils showed striking differences in their accumulation of NO2− and N2O, while they had very similar NO kinetics (Fig. 1A). The NO2− concentration in SoilA was 20–50 µM in the soil moisture during the entire anoxic incubation, except for the 36–40 h period when it reached ~100 µM. This corroborates the general notion that NO2− concentrations are low in acidic soils. In SoilN, on the other hand, NO2− steadily increased from the beginning, reaching 2–3 mM in the soil moisture at 20 h, after which levels decreased and were undetectable at the end of the sampling period. Both soils had a transient accumulation of 3–4 µmol NO vial−1, which is equivalent to 1.5–2 µM NO in the soil moisture. The soils had profoundly different N2O and N2 kinetics. SoilA accumulated N2O but little or no N2 during the first 35 h, while SoilN accumulated both N2O and N2 at similar rates from the very beginning. The gas kinetics is reflected in the calculated rates of the four steps of denitrification (Fig. 1B): In the acid soil, VNAR/NAP (NO3− → NO2−), VNIR (NO2− → NO) and VNOR (NO → N2O) were similar, while VNOS (N2O → N2) was close to zero during the first 35 h. In the soil with near neutral pH, VNAR/NAP exceeded the other enzyme rates during the first 15 h but declined rapidly to zero as NO3− was depleted. In contrast to the acidic soil, VNOS was high from the very beginning of the incubation in SoilN.
Based on the total NO2−-N (TNN = NO2− + HNO2), we calculated the concentration of undissociated HNO2 (aq) in the soil matrix using the Henderson–Hasselbalch approximation (see Supplementary material p.3), which forms an equilibrium with the gas HONO in the atmosphere, thus predicting the potential emission of HONO. Despite the high accumulation of TNN in SoilN (up to 3.6 mM), the concentration of undissociated HNO2 was ≤1.4 μM. In SoilA the concentration HNO2 was almost two orders of magnitude lower (Fig. 2).
Fig. 2. Concentrations of HNO2.

Measured total nitrite-N (TNN) and calculated HNO2 (µM in soil moisture) assuming equilibrium: [HNO2]/([HNO2] + [NO2−]) = 1/(1 + 10 pH−pKa), where pKa = 3.398.
Soil bacterial community composition differed by pH but was stable during incubation
In the 16S rRNA gene amplicon analysis >99.29% of all sequenced reads were annotated as bacterial, about 0.004% were unclassified, and the rest belonged to Archaea, which represented ≤0.60% of the reads in SoilN and ≤1.03% in SoilA. Principal component analysis of the bacterial 16S rRNA gene reads (Fig. S1A) clearly separated the reads from the two soils along PC1, which explained 94% of the total variation and showed that the variation between replicate samples was low. The most abundant classified phyla in both soils were Proteobacteria, Acidobacteria, Actinobacteria, Planctomycetes, Verrucomicrobia and Bacteroidetes. A breakdown of Proteobacteria showed that Alphaproteobacteria were most abundant in both soils, followed by Beta-, Gamma- and Deltaproteobacteria (Fig. S1B). The main differences between the soils was a larger relative abundance of Actinobacteria and Alpha- and Betaproteobacteria in SoilA than in SoilN along with a higher relative abundance of Bacteroidetes in SoilN than in SoilA. The OTU richness was higher in the SoilN samples with 2846 OTUs observed on average for the SoilN samples compared to an average of 1882 OTUs for the SoilA samples. SoilN samples were also slightly but significantly (p = 1.66 × 10−5) more even according to Peilou’s evenness measure with SoilN samples having an average Peilou’s evenness measure of 0.814 and compared to 0.796 for SoilA [57]. The microbial community profile of SoilN was stable during the 27 h incubation, suggesting that differences observed in the MT can be reasonably attributed to variations in transcription patterns, and not due to bacterial growth causing a shift in the bacterial community composition. A detailed list of OTU relative abundancies at multiple taxonomic levels is found in Table S1.
Prevalence of reads in the MG of genes encoding denitrification and DNRA reductases
Reads annotated as NAR, here defined as nap + nar reads, were twice as abundant as NIR, NOR and NOS gene reads in the MG of both soils (Fig. 3A; Table 1). Of the two types of NAR, narG reads were 6.1 ± 0.3 times more abundant than nap reads in SoilA but only 2.1 ± 0.3 times more abundant in SoilN (Table 1). Levels of NIR (nirK + nirS) were comparable in the two soils with RPM values of 36.4 ± 1.7 and 44.6 ± 1.4 for SoilA and SoilN, respectively (Table 1). nirK genes were much more abundant than nirS in both soils with a nirK/nirS ratio of about 40 in SoilA and 7 in SoilN. The NOR gene reads (cnor + qnor) were more abundant in SoilA, where RPM values were 74.9 ± 0.5 compared to 48.1 ± 0.3 for SoilN. The qnor genes dominated over cnor in both soils and were about 11 and 4 times higher in SoilA and SoilN, respectively. Total NOS reads (nosZ clade I + II) were instead somewhat higher in SoilN than in SoilA (25.8 ± 2.0 vs 18.2 ± 1.0). To summarise this for all genes, the order of gene reads from highest to lowest abundance was for SoilA narG > qnor > nirK > napA > nosZ clade I > cnor > nirS > nosZ clade II; and for SoilN narG > napA > nirK + qnor > nosZ clade II > cnor > nirS > nosZ clade I.
Fig. 3. Genetic potential and transcription of selected denitrification genes.
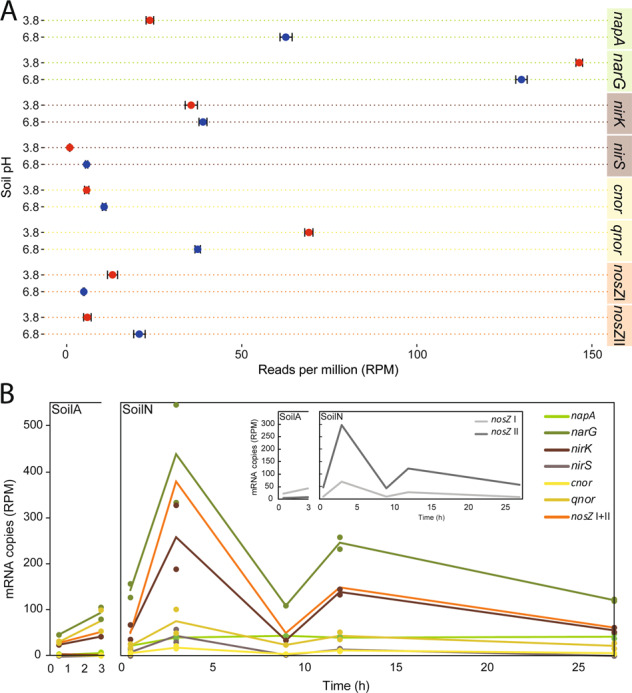
A Metagenome analysis showing a comparison of the abundance of denitrification genes in SoilA and SoilN based on number of reads annotated to the gene in question per total number of reads in the sample. Averages of triplicate soil samples taken at the start of the incubation for metagenomics analysis. Bars indicate standard deviation. B Metatranscriptome analysis showing changes in the abundance of denitrification gene transcripts during the first 3 h of incubation for SoilA and 27 h for SoilN. Averages (lines) and individual data points are shown for duplicate soil samples taken after 0.5, 3, 9, 12 and 27 h of anaerobic incubation. Reads of nirS transcripts were not detected in SoilA. The insert shows the transcriptional dynamics of the two nosZ clades only, for clarity.
Table 1.
Abundances of gene and transcript reads in the metagenome (DNA) and metatranscriptome (RNA) based on RPM values.
| Soil pH | Time | NAR | NIR | NOR | NOSa | NAR/NIR | NIR/NOR | NIR/NOS | NOR/NOS | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (h) | G | P | G/P | K | S | K/S | Q | C | Q/C | I | II | I/II | |||||
|
DNA SoilA |
0 |
146.4 (0.9) |
23.9 (1.2) |
6.13 (0.28) |
35.6 (1.72) |
0.87 (0.06) |
41.2 (4.62) |
69.2 (1.2) |
5.7 (0.6) |
12.3 (1.6) |
13.1 (1.4) |
5.9 (1.1) |
2.3 (0.7) |
4.68 (0.27) |
0.49 (0.03) |
1.64 (0.51)1) |
3.33 (0.89) |
| SoilN | 0 |
129.9 (1.6) |
62.7 (1.8) |
2.07 (0.05) |
38.9 (1.15) |
5.70 (0.36) |
6.84 (0.31) |
37.4 (0.8) |
10.7 (0.5) |
3.5 (0.2) |
4.9 (0.2) |
20.8 (1.6) |
0.24 (0.02) |
4.32 (0.12) |
0.93 (0.03) |
1.65 (0.26) |
1.78 (0.23) |
| RNA | 0.5 | 46.0 | 2.96 | 15.5 | 23.5 | 0.00 | na | 31.7 | 1.30 | 24.4 | 22.7 | 5.1 | 4.46 | 2.09 | 0.71 | 0.83 | 1.08 |
| SoilA | 0.5 | 45.1 | 1.30 | 34.7 | 23.4 | 0.00 | na | 27.3 | 0.70 | 39.0 | 1.98 | 0.84 | |||||
| 3 | 104.7 | 2.80 | 37.4 | 40.9 | 0.40 | 102.3 | 98.2 | 1.30 | 75.5 | 42.5 | 8.1 | 5.23 | 2.60 | 0.42 | 0.81 | 1.48 | |
| 3 | 79.9 | 6.60 | 12.1 | 42.1 | 0.00 | na | 52.8 | 0.10 | 528.0 | 2.05 | 0.80 | ||||||
| SoilN | 0.5 | 162.2 | 16.3 | 10.0 | 72.3 | 21.9 | 3.3 | 23.2 | 10.2 | 2.27 | 9.3 | 45.5 | 0.20 | 1.89 | 2.82 | 1.27 | 0.71 |
| 0.5 | 131.9 | 41.1 | 3.20 | 42.4 | 6.90 | 6.1 | 32.6 | 14.1 | 2.31 | 3.51 | 1.06 | ||||||
| 3 | 544.6 | 50.0 | 10.9 | 330.2 | 63.0 | 5.2 | 106.0 | 28.1 | 3.77 | 68.3 | 296.9 | 0.23 | 1.51 | 2.93 | 0.82 | 0.28 | |
| 3 | 336.1 | 41.6 | 8.10 | 192.9 | 35.8 | 5.4 | 55.8 | 22.1 | 2.52 | 1.65 | 2.94 | ||||||
| 9 | 114.5 | 49.9 | 2.30 | 40.6 | 7.96 | 5.1 | 31.0 | 10.8 | 2.87 | 8.6 | 43.1 | 0.20 | 3.38 | 1.16 | 0.89 | 0.77 | |
| 12 | 251.9 | 48.0 | 5.50 | 137.5 | 18.5 | 7.4 | 58.4 | 16.4 | 3.56 | 1.99 | 2.09 | ||||||
| 12 | 235.1 | 44.7 | 5.30 | 149.1 | 22.6 | 6.6 | 40.8 | 18.4 | 2.22 | 26.7 | 122.5 | 0.22 | 1.63 | 2.90 | 1.06 | 0.43 | |
| 27 | 127.6 | 43.8 | 2.90 | 56.5 | 7.10 | 8.0 | 21.5 | 10.6 | 2.03 | 2.69 | 1.98 | ||||||
| 27 | 124.2 | 53.1 | 2.30 | 67.4 | 7.10 | 9.5 | 34.0 | 12.6 | 2.70 | 7.7 | 56.0 | 0.14 | 2.38 | 1.60 | 1.02 | 0.58 | |
Results for anoxic incubation of SoilA (pH 3.8) and SoilN (pH 6.8), amended with NO3-. NAR = nitrate reductases, sum of narG and napA abundancies; NIR = nitrite reductases, sum of nirK and nirS abundancies; NOR = nitric oxide reductases, sum of qnor and cnor abundancies; NOS = nitrous oxide reductase; sum of clade I and clade II abundancies. DNA samples were taken just before the start of the incubation (t = 0 h, Fig. 1A). RNA samples were taken after 0.5 and 3 h incubation (SoilA) and after 0.5–27 h incubation (SoilN) (Fig. 1A). All values are in reads per million (RPM). Ratios of reads from selected genes/transcripts are also given. DNA: Average values are given for abundance of individual genes, values in parenthesis are standard deviation; n = 3. RNA: Duplicate metatranscriptome samples were analysed from each sampling time except for the 9 h sample from SoilN for which one samples was lost. Individual values for read abundances and ratios are shown except for the nosZ clades for which data from duplicate samples were pooled. Ratios including NOS are therefore calculated from average values.
na not applicable.
aValues were obtained using a manually curated database for nos clades containing sequences reported by Conthe et al. (2018). For this examination reads from duplicate samples in the MT were pooled before the analysis, thus replicates are not reported. Calculations of ratios were based on the same database as for the other denitrification genes, which was based on individual replicates. For detailed descriptions see Materials and Methods section.
Since the two soils accumulated different amounts of denitrification intermediates, we calculated ratios of MG reads representing the four steps of denitrification (Table 1), to examine if the genetic potential for production/consumption of the different intermediates was related to the net production seen in Fig. 1. The NAR/NIR ratios (based on RPM values) were similar in the two soils (4.68 ± 0.27 for SoilA and 4.32 ± 0.12 for SoilN), which, by itself, is not consistent with the higher net production of NO2− in SoilN. The NOR genes were more abundant than NIR in SoilA, with a NIR/NOR ratio of 0.49 ± 0.03 compared to 0.92 ± 0.03 in SoilN. NIR and NOR were more abundant than NOS in both soils with NIR/NOS ratios of 2.83 and 2.07 and NOR/NOS ratios of 5.81 and 2.23 in SoilA and SoilN, respectively. The two nosZ clades (I and II) differed in gene abundance in the two soils with higher values for clade I in SoilA and vice versa in SoilN (Fig. 3A). The nosZ clade I/clade II ratio was 28.1 in SoilA and 0.25 in SoilN (Table 1).
In addition to the canonical denitrification genes, we examined reads from the two DNRA-related genes nrfA and nirB. They were about three times more abundant in SoilN than in SoilA with nirB dominating over nrfA in both soils (Fig. S2). We also examined the occurrence of reads derived from the gene hmp. This gene, reported from a wide range of bacteria [67], encodes a flavohemoprotein that has a role in NO detoxification. Unlike the NOR genes, we found similar and relatively low abundances of hmp in the two soils (16 ± 0.8 and 18 ± 1.4 RPM, not shown).
Prevalence of reads in the MT of transcripts encoding denitrification and DNRA reductases
Earlier PCR-based investigations of soils from the same site as that studied here showed that denitrification genes were quickly transcribed upon onset of anoxia [24, 38]. Based on these findings, RNA samples for the present study were taken from both soils 0.5 and 3 h after the start of the experiment (red arrows, Fig. 1A) to characterise the community response to denitrifying conditions. Additional RNA samples were taken from SoilN prior to the peaks in NO2−, NO, and N2O to determine if there were new bursts of gene transcription during the incubation and, if so, if these were from already active organisms, or if different organisms became transcriptionally active at the later time points. The transcript read abundancies are shown in Fig. 3B. For clarity, the nosZ genes are shown as the sum of the two clades in the main figure, while the insert shows the clades separately. The reads in the meta-transcriptome from SoilN exhibited a strong increase between 0.5 and 3 h for almost all denitrification gene transcripts. Over the same time frame there was a smaller, yet substantial increase for several of the genes in SoilA. Two distinct spikes in denitrification gene transcription were seen at 3 and 12 h, particularly for narG, nirK, qnor and nosZ (Fig. 3B). Reads of narG represented the most common denitrification transcripts in both soils and at all time points. The overall trend in transcript read abundance in SoilN was narG > nosZ > nirK > qnor > napA > nirS > cnor, almost without exception, throughout the incubation (Table 1). The trend for SoilA was narG > qnor > nosZ > nirK > napA > cnor = nirS, the main difference compared to SoilN being that qnor was second most abundant, followed by nosZ. Similar to the gene reads in the MG, nosZ clade I transcript reads were more abundant than clade II reads in SoilA and vice versa for SoilN.
It could be argued that the ratios of transcripts representing the reductases responsible for the production and consumption of the various denitrification intermediates are more suitable to use for comparison with phenotypic data than actual RPM values (transcript read abundances and selected ratios are given in Table 1). The NAR/NIR ratios were very similar in the two soils with average values of 2.2 ± 0.3 for SoilA and 2.3 ± 0.8 for SoilN, with some variations between sampling times. The NIR/NOR ratios, on the other hand, were almost three times higher in SoilN compared to SoilA (average values including all sampling times were 2.2 ± 0.8 and 0.7 ± 0.2, respectively). The high NOR transcription in SoilA most likely contributed to keeping NO emissions low, despite known chemical reactions between NO2− and soil compounds producing NO. The NIR/NOR ratios of SoilN for the individual sampling points further showed that NOR was consistently lower than NIR throughout the 27 h incubation with NIR/NOR ratios of 1.2–2.5. Transcription of NOS (sum of clade I, clade II and ambiguous reads) was higher in SoilN than in SoilA and read numbers increased almost seven times from 56 to 380 in SoilN between 0.5 and 3 h. In SoilA the NOS transcript reads almost doubled during the same period. NIR/NOS ratios were rather similar in the two soils, around 0.8 in SoilA and 1.0 ± 0.2 in SoilN. NOR/NOS ratios were higher in SoilA than in SoilN, with an average of 1.3 ± 0.3 for the first two sampling points while, during the same time, this ratio for SoilN was 0.6 ± 0.2. These ratios reflect the higher number of NOR reads in SoilA, combined with higher reads for NOS in SoilN.
The MT was also examined with regard to the two DNRA genes nfrA and nirB, expecting low read abundance since no DNRA activity was discerned from the gas analyses. Surprisingly, these two genes were transcribed at levels comparable to the NIR and NOR genes, with sometimes high read values for nirB (Fig. S2).
Accessory genes and transcripts of the nos operon in the MG and MT
We also analysed the MG and MT with respect to some accessory genes and transcripts of the nos operon, using a manually curated database, with a focus on clade I (Fig. 4). The nosR gene is a part of the nos operon in clade I but not in nosZ clade II organisms [15, 45]. Its product is essential for N2O reduction in clade I organisms and is suggested to be involved in transcription of nosZ, and also in electron transfer to the NosZ reductase [46]. In accordance with nosZ clade I being dominant in SoilA, we found about twice as many reads derived from nosR in SoilA than in SoilN with RPM values of 16.2 ± 0.3 vs 7.0 ± 0.3, respectively. Moreover, we examined the relative abundance of the accessory genes nosL, D and Y, which are found both in nosZ clade I and clade II organisms [15, 45]. For each of these genes, the read abundance was similar in the two soils (Fig. 4), which is in accordance with the comparable abundance of NOS (nosZ clade I + II) in the two soils (12.5 RPM in SoilA and 20.8 in SoilN, Table 1). The gene reads for nosF, which is also part of the nos operon both in clade I and II [45], were 10–45 times higher than for the other accessory genes which points to uncertainties in the databases for this gene. Transcript reads of nos accessory genes were detected in both soils at all sampling occasions. The transcriptional activity increased for all these genes between 0.5 and 3 h, which could be taken as an indication that the lack of N2O reduction in SoilA was not caused by a defect in the transcriptional control mechanism. The nosR transcript abundances were comparable in the two soils.
Fig. 4. Abundance of selected genes in the nos operon and their transcription over time.
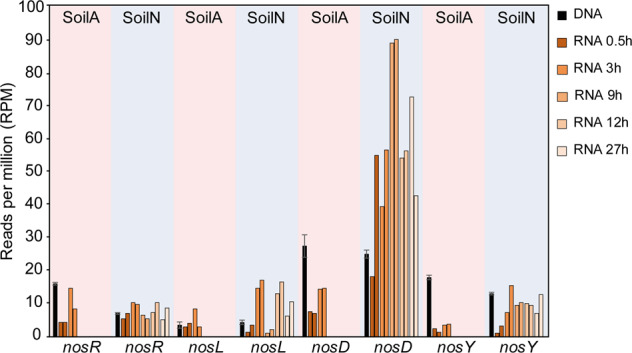
Metagenome and metatranscriptome analyses of two soils (SoilA, pH = 3.8 and SoilN, pH = 6.8). Black bars show average gene read abundances in the metagenome (n = 3; bars show sd). Colored bars show transcript read abundances after 0.5 and 3 h of incubation (SoilA) and after 0.5, 3, 9, 12 and 27 h of incubation (SoilN). Duplicate samples were analysed for each sampling point, shown as individual bars with the same color.
PCR-based quantification of functional genes and transcripts overlooked substantial parts of the community
The abundances of some of the denitrification genes and transcripts in the MGs and MTs (Fig. 3A, B; Table 1) were compared to qPCR-results in samples from the same soil incubation (Fig. 5A, B), although more time points were included in the qPCR analysis. We targeted nirK, nirS and nosZ clade I using standard primer pairs (see Materials and Methods). The comparison revealed some striking differences between -omics and PCR-based results. While the “-omics” based results showed dominance of nirK over nirS both in the MGs and MTs, the qPCR-based quantifications showed 1–2 orders of magnitude lower abundances of nirK genes and transcripts compared to nirS, except for SoilA where the difference was smaller. Comparing the nirS and nosZ (clade I) abundancies shows nearly identical abundancies of these genes in SoilN according to the MG (Fig. 3A) while qPCR showed almost ten times fewer nosZ than nirS gene copies based on qPCR (Fig. 5A). The MG- and qPCR-based results from SoilA showed better correspondence, with nosZ being more abundant than nirS. For the transcripts (Figs. 3B and 5B), nosZ was more abundant than nirS in SoilA according to the MT analysis, while the qPCR showed an opposite trend. Similarly, qPCR gave nosZ transcript levels that were 5–10 times lower than nirS in SoilN, while their levels were similar in the MT. The results indicate a critical primer bias in the amplicon-based quantification leading to severe underestimations of nirK abundance and overestimations of nirS compared to nirK and nosZ clade I.
Fig. 5. Amplification-based quantification (qPCR) of genes and transcripts.

A Boxplot showing quantified gene copy abundance g−1 soil (wet weight). Primers targeting the 16S rRNA (27F/518R), nirK (517F/1055R), nirS (cd3aF/R3cd) and nosZ clade I (nosZF/1622R) genes were used for amplification. B Quantified mRNA transcript abundance g−1 soil (wet weight) of nirK, nirS, and nosZ. Red = SoilA (pH 3.8); Blue = SoilN (pH (6.8).
Taxonomic annotation of denitrification genes and transcripts
Each gene and corresponding transcript had a unique taxonomic profile that varied by soil pH (Fig. 6). Proteobacteria, Actinobacteria and Bacteroidetes were the most abundant phyla of denitrifiers in the MG and MT in both soils. Reads assigned to these phyla were detected for most denitrification genes and transcripts with the exceptions of Bacteroidetes, which were not found among narG and nirS reads, and Actinobacteria, which were not found among the nirS, cnor and nosZ clades I and II reads. Several other phyla such as Firmicutes, Chlamydiae, Nitrospira, Spirochaetes and Verrucomicrobia were represented by reads only from one or a few genes/transcripts.
Fig. 6. Taxonomic distribution of selected denitrification genes and transcripts.

Abundance of annotated reads obtained from the metagenome (DNA), sampled at the start of incubation and from the metatranscriptome, sampled after 0.5 and 3 h anoxic incubation (SoilA) and 0.5–27 h anoxic incubation (SoilN) (for sampling points also see Fig. 1A). Averages of summed reads across replicate samples (n = 3 for metagenomes; n = 2 for metatranscriptomes). A detailed list of all read abundancies is found in Supplementary materials Table S2, which also includes read abundancies for the other denitrification genes.
The MG reads of narG, the most abundant among the denitrification genes, were dominated by Proteobacteria, mostly the classes Alpha- and Betaproteobacteria, and Actinobacteria (Table S2). Second most abundant in both soils were napA reads, which were mainly derived from Proteobacteria. Organisms belonging to these phyla also dominated the transcriptional activity for narG and napA genes. narG reads from Nitrospira were high as well in SoilN. MG reads derived from the NO2− reduction gene nirK, which was far more abundant than nirS in both soils (Table 1), were attributed to a number of phyla which were dominated by Proteobacteria, primarily Alpha- Beta- and Gamma proteobacteria, and by Actinobacteria, Bacteriodetes and Firmicutes. These same phyla also dominated the MT reads of nirK in both soils, with Proteobacteria showing particularly high transcription of this gene in SoilN at the 3 h sampling time. MG reads from nirS were predominantly from Betaproteobacteria in SoilN, which also accounted for the highest nirS transcriptional activity. Only seven classified nirS MG reads were identified in SoilA compared to 242 nirK reads (Table S2). Similar as for the nirK and nirS genes, the qnor genes dominated strongly over cnor genes in the MG, and the qnor genes belonged to several phyla while the cnor reads were mostly from Proteobacteria. The Delta- and Gammaproteobacteria had the highest number of qnor MG reads in both soils, but it was the Betaproteobacteria that dominated among the transcripts showing comparatively high and immediate transcription in SoilA. Interestingly, qnor MG and MT reads from Acidobacteria and Planctomycetes were detected in both soils at relatively high abundance. Apart from a few acidobacterial nosZ clade II reads, reads from these two phyla were not found for the other denitrification genes in the MG or MT.
The total number of nosZ MG reads was higher in SoilN than in SoilA (212 vs 152, Table S2). As expected, nosZ clade I MG and MT reads were only from Proteobacteria in both soils. nosZ clade II MG reads were more diverse, especially in SoilN, and were comprised of not only Proteobacteria, mostly Deltaproteobacteria, but also several other phyla including Bacterioidetes, which was the most highly represented group, as well as Firmicutes, Verrucomicrobia, Chloroflexi, Gemmatimonadetes and Acidobacteria among others. The transcriptional activity of nosZ clade I was dominated by Alphaproteobacteria in SoilN and by Betaproteobacteria in SoilA. The pattern was very different for transcription of nosZ clade II, which was dominated by Bacteroidetes in both soils. The transcript abundance of clade II in SoilA was low but increased between 0.5 and 3 h and included not only Bacteriodetes but also a few reads from Proteobacteria, Bacteriodetes, Chloroflexi, Gemmatimonadetes, Ignavibacteriae and Acidobacteria. Taken together, the results for the two nosZ clades show that the problem of producing functional NosZ in SoilA was common to several phyla. SoilN showed high transcriptional activity of nosZ clade II, dominated by Bacteriodetes, but transcripts were also detected from all other phyla for which nosZ clade II were registered in this soil, except Euryarchaeota.
Discussion
Liming, which has traditionally been a means to improve the fertility of acidic soils [68], is known to cause changes in microbial community composition and diversity [69, 70]. This was also seen in the present study, where the PCA of the 16S rRNA gene sequences clearly separated the two soils (Fig. S1A). The sequence analysis also revealed a slight but significant increase in diversity in the limed soil. The dominant phyla in both soils were those that generally dominate in soil studies [71] and no major differences between the soils were seen on the phylum level, or on class level for the proteobacteria, meaning that changes in the abundance of different groups had taken place at lower taxonomic levels. During the 27 h anoxic incubation, when SoilN was monitored with metatranscriptomics, the community of SoilN remained stable as judged from the 16S rRNA gene analysis (Fig. S1B), suggesting that any differences in the MT can be reasonably attributed to transcriptional regulation patterns and not caused by growth of some populations.
The accumulation of NO2− and N2O in the two soils showed contrasting patterns, with low NO2− and high N2O levels in SoilA and vice versa for SoilN, while the NO accumulation was more similar in these two soils, as presented in [28]. In the present study we investigated if the contrasting denitrification phenotypes of the two soils could be predicted from the abundance and transcription of the denitrification genes. The lower abundance of NOS genes and transcripts compared to NIR and NOR could, theoretically, be the cause of the low N2O reduction in SoilA, but it is unlikely to explain the nearly complete lack of N2O reduction in SoilA during the first 35 h of incubation (Fig. 1A). This severely delayed N2O reduction in acidic soil corroborates other studies of soils from the same site and is in line with the growing evidence for a strong negative correlation between soil pH and N2O/(N2O + N2) product ratios [12, 33], suggested to be due to impaired maturation of the NosZ enzyme under acidic conditions (pH < 6.1) [37–39]. The present study detected transcripts from both nosZ clades in SoilA, which suggests that the problem of producing functional NosZ under low pH conditions applies to both clades. Moreover, the taxonomic analysis showed that this problem is general to a diverse range of bacteria (Fig. 6), which adds new knowledge to earlier qPCR-based investigations in which taxonomy was not addressed [38, 39]. Although MTs in SoilA were only analysed from the first 3 h of the incubation, the gas kinetics suggest that functional NosZ was not produced until after 25 h (Fig. 1B). In a natural situation the N2O produced in this time period would be emitted to the atmosphere. It can only be speculated why functional NosZ started to be produced after prolonged incubation. One reason could be that successful maturation took place somewhat stochastically, slowly building up a functional pool of NosZ. Another possibility is that populations of organisms such as the Rhodanobacter strains described by Lycus et al. [18] increased. These bacteria were shown to reduce N2O under acidic conditions, but not at neutral pH [18, 72].
Another important insight provided in the present study is the analysis of nos accessory genes and transcripts (Fig. 4) encoding enzymes involved in maturation of, and electron transfer to, NosZ. To our knowledge, such analyses have hitherto not been reported from soils. It is important to note, though, that since little is known about the protein function and phylogenetic characterisation of these proteins the datasets used in this analysis are likely incomplete. Genes encoding NosR, which is involved in electron transfer to NosZ clade I [46] and NosL, a Cu chaperone that delivers Cu+ to the NosZ apo-protein of both clades [45], are of particular interest since the presence and transcription of these genes may hold a clue to understanding the severely delayed NosZ function in SoilA. The abundance of nosR gene reads (RPM values) in the MG of SoilA was almost twice as high in SoilN, which is in agreement with the higher abundance of nosZ clade I in SoilA. The abundance of nosL was instead similar in the two soils, reflecting that this gene is part of the nos operon both in clade I and clade II organisms. The abundance of gene reads encoding the nosDYF cluster were also similar in the two soils. The function of the enzymes encoded by this gene cluster, which is part of the nos operon both in clade I and II organisms, is not completely elucidated. The Y and F genes are thought to encode an ATP-binding cassette (ABC) transporter, while NosD has also been suggested to be involved maturation of the CuZ site [45, 73, 74]. Taken together, gene reads for all the accessory nos genes analysed in this study were detected in both soils, which was not unexpected.
Transcript reads encoding all accessory nos genes of clade I were detected in SoilA, which could be taken to indicate that the organisms in this soil had the tools in place for NosZ function, including nosZ transcriptional activation and electron transfer to NosZ (by NosR) and CuZ site maturation (by NosL). To conclude, there were no obvious issues with the genetic potential or the transcriptional activity of the nos operon which could explain the delayed N2O reduction in SoilA.
Net production of denitrification intermediates will take place as soon as the reduction rate of one of the denitrification steps surpasses that of the following step. The NO3− reduction rate (VNAR) in SoilN grossly exceeded the NO2− reduction rate (VNIR), as long as nitrate was present (Fig. 1B and [28]). The MT analysis showed higher transcription of NAR (napA + narG) than of NIR (nirK + nirS) (Fig. 3B) with NAR/NIR ratios between 1.6 and 3.4 (Table 1). The ratio was similarly high for SoilA, however (2.0 and 2.3 for the two time points measured in this soil), despite the near-absence of NO2− accumulation in this soil. Thus, no direct link was found between the transcript ratio NAR/NIR and NO2− accumulation or the VNAR/VNIR ratio, as affected by pH. In theory, abiotic NO2− decomposition could be the primary reason for the marginal transient accumulation of NO2− in SoilA, but this was refuted by the careful analyses of Lim et al. [28], who reached the conclusion that enzymatic NO2− reduction was the major sink for NO2− in Soil 3.8. One explanation for the marginal NO2− accumulation in SoilA could be that NO2− reductase has a low pH optimum, as shown in a study by Abraham et al. [75] where measured NirK activity in vitro was approximate four times higher at pH 4.2 than at pH 7. Thus, SoilA could have a much lower VNAR/VNIR ratio than SoilN, despite the nearly equal NAR/NIR-ratio for the two soils. The results demonstrate that transcript numbers or ratios of transcript numbers are poor predictors of metabolic activities in soils. Even more evident is the discrepancy between gene numbers and activity. The NAR/NIR-ratio in the metagenome was almost identical in the two soils (Table 1), despite the substantial difference in NO2− accumulation. The understanding of how denitrifiers control NO2− levels is far from complete, and different phenotypes have been described which are probably all present in complex soil microbial communities [18]. Some organisms perform complete denitrification of NO3− to N2 with little or no accumulation of intermediates, while others show complete inhibition of nir transcription until available NO3− to NO2− before further reduction takes place [23], and yet others reduce about half of the provided NO3− to NO2− before further reduction [76].
One reason that the NAR genes were by far more abundant than the genes for the other denitrification steps (Fig. 3A) could be that these genes are also carried by organisms performing DNRA [77]. Organisms with only NAR but lacking genes both for DNRA and denitrification are, however, also common [19], and in a study of bacterial isolates from the same field site as in the present study, 18 and 19% of the isolates from acidic and neutral soil, respectively, performed only NO3− reduction to NO2− without any further reduction [18]. The present study showed a strong dominance of narG over napA both in the MGs and in the MTs (Table 1; Fig. 3), despite the common occurrence of bacteria with napA + narG or with only napA among hitherto studied DNRA- and denitrifying organisms [78]. Still, napA was 2.6 times more abundant in the metagenome of SoilN compared to SoilA (Fig. 3A), in line with the significant positive relationship between napA and pH reported by [59], but the reason for this apparent pH dependence on the abundance of napA is not clear.
According to the MG analysis the abundance of NIR genes was similar in the two soils, with strong dominance of nirK over nirS, especially in SoilA where nirS genes were almost absent (Table 1; Fig. 3A;). This contradicts the general conception that nirS is more abundant in most environments, which is derived from primer-based studies [38, 79, 80]. However, while nirS is mainly found in Proteobactera, nirK is spread among taxonomically diverse groups, many of them being non-proteobacterial denitrifiers [16, 17, 81], which was also found in the present study (Fig. 6). Transcripts of nirK dominated over nirS also in the MT (Fig. 3B), and represented a diverse range of phyla including, in addition to Proteobacteria, also Actinobacteria, Bacteroidetes and Firmicutes (Fig. 6). This higher occurrence of nirK is in accordance with the results from a non-primerbased study of another agricultural soil by [80]. The occurrence of a taxonomically diverse group of denitrifiers actively transcribing nirK probably reflects that this gene is easily transferred horizontally and expressed in new hosts since it does not require accessory genes to produce a functional enzyme, in contrast to nirS, which is part of a multi-gene operon [81]. This, and other evolutionary mechanisms, are a likely explanation to the reported incongruences between nirK and 16S rRNA gene phylogenies [16, 82]. Recently, Nadeau et al. [59] pointed out that denitrification is a non-essential trait for microbes, and that genes are gained and lost by individual members of the denitrifying community depending on the conditions. It is likely that this applies in particular to the functional genes nirK and qnor, which are probably more easily transferred horizontally than the other denitrification genes, and it can be speculated that they comprise a pool of genes that circulate between organisms depending on their needs.
The complete recovery of all added NO3− as N2 in SoilN (Fig. 1A) indicated minimal or no conversion of NO2− to NH4+ and thus that the NO2− produced from NAR activity was not used by DNRA organisms. This is surprising, taking into account the relatively high abundance of reads from nrfA and nirB genes and transcripts (Fig. S2). Likewise, DNRA was probably insignificant in SoilA, since 94% of the NO3− loss in this soil could be accounted for by N-gas + nitrosylation (see [28]). In theory, full recovery of NO3− reduction as N-gas could be obtained in a system with equal rates of DNRA and anammox. However, members of Brocardiales, which comprises the anammox-Planctomycetes [83], were scarce in the 16S rRNA gene analysis (<0.008% of 16S rRNA gene reads in all SoilA samples), which lends little support to this hypothesis.
The net production of NO was similar in the two soils despite the substantial abiotic reduction of NO2− that contributed to NO emissions from SoilA [28], in addition to the enzymatic NO2− reduction taking place in both soils. The strong control of NO in SoilA, seen from the calculated VNOR activity (Fig. 1B), is in agreement with the 2.5 times increase in qnor transcript abundance between 0.5 and 3 h (Fig. 2B) and NIR/NOR ratios <1 (Table 1). In line with this, the gene abundance of qnor was also high in SoilA, almost two times its abundance in SoilN (Fig. 3A). This corroborates the metagenome results by Roco et al. [60] from acidic soils showing dominance of qnor over all other denitrification genes, suggesting that low pH selects for this gene. Interestingly, cnor apparently played a minor role in controlling NO, especially in SoilA where gene abundance was close to 0 and transcripts were almost undetectable (Fig. 3). The taxonomic analysis of qnor genes and transcripts (Fig. 6) demonstrated that this gene, similarly as nirK, is widely distributed over different phyla, with the largest representation in the Proteobacteria, Actinobacteria, Bacteroidetes, Chlamydiae, Acidobacteria and Planctomycetes. qNor is the product of a single structural gene, norB, existing alone or in a small operon [84], and it is conceivable that this gene/gene cluster is more readily transferred horizontally than the bigger cnor operon. Furthermore, recent evidence suggests that qNor is electrogenic [85], as opposed to cNor, and it can therefore be speculated that it provides an extra, energetic advantage to its host organism. Taken together, it is not unreasonable to conclude that, between the two functionally redundant NO reductases related to denitrification, it is qNor that plays the major role in controlling NO concentrations under denitrifying conditions, at least in these soils. A primer-based study would most likely not have detected this since current primers for nor mainly target proteobacteria, which severely biases the results, especially for qnor [42].
Nitrous acid (HNO2) in its gaseous form HONO plays a key role in atmospheric reactions by being a precursor for hydroxyl (OH) radicals, which in turn take part in the formation of undesired O3 in the troposphere. Emissions of HONO and their connection to biological N-transformations have gained increasing interest during the past years. It has only recently been shown that biological processes producing NO2− in soils contribute substantially to HONO emissions [6, 86, 87]. However, while Su et al. [86] stated that the low NO2− concentrations in acidic soils are consistent with loss of HNO2 as HONO, Oswald et al. [6] instead showed that soils with neutral and slightly alkaline pH generally emitted more HONO than acidic soils. Our results show more than ten times higher HONO production from SoilA compared to SoilN (Fig. 2), but we also show that the major portion of the NO2− is not reduced and released as HONO, but instead reduced to NO and subsequently to N2O through denitrification.
Conclusions
Despite the vast number of denitrification studies, -omics based analyses are hitherto scarce. The present study added several pieces of information to the current understanding of soil denitrifier communities and how pH affects their activity, which organisms are involved, and their control and accumulation of denitrification intermediates. The metatranscriptomic results suggest that NO3− reduction was dominated by NarG activity, with lesser contributions from NapA. The NO3− reduction was apparently connected to denitrification and not to DNRA activity since N gas+ nitrosation accounted for ~100% of the consumption of NO3−. This is surprising and indicates a minor role of DNRA in N-transformations in these soils, despite DNRA-related gene and transcript reads (nrfA and nirB) being abundant.
The concentration of NO2− in soil moisture was about two orders of magnitude higher in SoilN than in SoilA, while the NAR/NIR transcript read ratios were almost identical in the two soils. This implies that NO2− concentrations in SoilA were controlled by a combination of biological activity and chemical degradation, which supports the experimental evidence from nitrite kinetics and modelling by Lim et al. [28]. It is conceivable that denitrifying organisms in acidic environments tune their genetic regulation to avoid high levels of NO2−, which at low pH will occur mainly as toxic HNO2, and at the same time avoid high NO concentrations by maintaining high transcription of NOR, mainly qnor, which was apparently selected for over cnor by low pH. Our finding that HONO emissions from SoilA were ten times higher than from SoilN is noteworthy since, while it supports the results by Su et al. [86] it contradicts other published results which indicate that neutral HONO emissions are mainly from neutral soils [6]. More experimental evidence is however required to reach a better understanding of environmental conditions affecting the release of HONO from soils.
The analyses of the MG and MT demonstrated that diverse microorganisms transcribed nosZ genes from both clade I and II in the acidic soil, but this was not followed by any detectable Nos function until >30 h after the start of the incubation. The results did not reveal any obvious lack of genes or transcripts of nos accessory genes involved in NosZ maturation. Although not exhaustive, this information provides a new building block toward an understanding of the impaired N2O reduction under acidic conditions.
The discrepancies between qPCR and -omics based estimates of genes and transcripts provides strong evidence that primers commonly used in denitrification studies only capture a fraction of the community that carries these genes. The problem likely arises since genes such as nirK occur in diverse of microbes, while genes such as nirS are restricted to a narrower group of denitrifiers making the nirS primer sets better able to bind to the targeted gene sequences. This leads to an overestimation of nirS relative to nirK genes and transcripts in the qPCR-results. Similarly, the qPCR-results showed lower abundance of nos (clade I) in SoilA than in SoilN, while it was three times more abundant in the metagenome of SoilA compared to SoilN. These examples illustrate that primer-based results for denitrification genes may lead to erroneous conclusions and must be interpreted with caution.
Supplementary information
Acknowledgements
SV was financed by the project PASUSI within the ERA-NET Co-fund Horizon2020 program Leap-Agri, under the EC Grant agreement No. 727715 and the Research Council of Norway projects No. 290488 and No. 286888. NL obtained a PhD grant from the Norwegian University of Life Sciences.
Data availability
The 16S rRNA gene amplicon sequences are available as MG and MT FASTQ files on European Nucleotide Archive (ENA) under project PRJEB41993.
Competing interests
The authors declare no competing interests.
Footnotes
The original online version of this article was revised due to an error in Table 1.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
12/17/2021
A Correction to this paper has been published: 10.1038/s41396-021-01100-y
Contributor Information
Åsa Frostegård, Email: asa.frostegard@nmbu.no.
James P. Shapleigh, Email: jps2@cornell.edu
Supplementary information
The online version contains supplementary material available at 10.1038/s41396-021-01045-2.
References
- 1.Canfield DE, Glazer AN, Falkowski PG. The evolution and future of earth’s nitrogen cycle. Science. 2010;330:192–6. doi: 10.1126/science.1186120. [DOI] [PubMed] [Google Scholar]
- 2.Stevens CJ. Nitrogen in the environment. Science. 2019;363:578–80. doi: 10.1126/science.aav8215. [DOI] [PubMed] [Google Scholar]
- 3.Tian H, Lu C, Ciais P, Michalak AM, Canadell JG, Saikawa E, et al. The terrestrial biosphere as a net source of greenhouse gases to the atmosphere. Nature. 2016;531:225–8. doi: 10.1038/nature16946. [DOI] [PubMed] [Google Scholar]
- 4.Thompson RL, Lassaletta L, Patra PK, Wilson C, Wells KC, Gressent A, et al. Acceleration of global N2O emissions seen from two decades of atmospheric inversion. Nat Clim Change. 2019;9:993–8. [Google Scholar]
- 5.Tian H, Xu R, Canadell JG, Thompson RL, Winiwarter W, Suntharalingam P, et al. A comprehensive quantification of global nitrous oxide sources and sinks. Nature. 2020;586:248–56. doi: 10.1038/s41586-020-2780-0. [DOI] [PubMed] [Google Scholar]
- 6.Oswald R, Behrendt T, Ermel M, Wu D, Su H, Cheng Y, et al. HONO emissions from soil bacteria as a major source of atmospheric reactive nitrogen. Science. 2013;341:1233–5. doi: 10.1126/science.1242266. [DOI] [PubMed] [Google Scholar]
- 7.Thomson AJ, Giannopoulos G, Pretty J, Baggs EM, Richardson DJ. Biological sources and sinks of nitrous oxide and strategies to mitigate emissions. Philos Trans R Soc B Biol Sci. 2012;367:1157–68. doi: 10.1098/rstb.2011.0415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Butterbach-Bahl K, Baggs EM, Dannenmann M, Kiese R, Zechmeister-Boltenstern S. Nitrous oxide emissions from soils: how well do we understand the processes and their controls? Philos Trans R Soc B Biol Sci. 2013;368:20130122. doi: 10.1098/rstb.2013.0122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rogelj J, Shindell D, Jiang K, Fifita S, Forster P, Ginzburg V, et al. Mitigation pathways compatible with 1.5 °C in the context of sustainable development. In, Masson-Delmotte V, Zhai P, Pörtner HO, Roberts D, Skea J, Shukla PR, et al., editors. Global Warming of 1.5 °C. An IPCC Special Report on the impacts of global warming of 1.5 °C above pre-industrial levels and related global greenhouse gas emission pathways, in the context of strengthening the global response to the threat ofclimate change, sustainable development, and efforts to eradicate poverty. 2018. https://www.ipcc.ch/site/assets/uploads/sites/2/2019/02/SR15_Chapter2_Low_Res.pdf.
- 10.Schlesinger WH. On the fate of anthropogenic nitrogen. Proc Natl Acad Sci USA. 2008;106:203–8. doi: 10.1073/pnas.0810193105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davidson EA, Kanter D. Inventories and scenarios of nitrous oxide emissions. Environ Res Lett. 2014;9:105012. [Google Scholar]
- 12.Bakken LR, Frostegård Å. Sources and sinks for N2O, can microbiologist help to mitigate N2O emissions? Environ Microbiol. 2017;19:4801–5. doi: 10.1111/1462-2920.13978. [DOI] [PubMed] [Google Scholar]
- 13.Zumft WG. Cell biology and molecular basis of denitrification. Microbiol Mol Biol Rev. 1997;61:533–616. doi: 10.1128/mmbr.61.4.533-616.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spiro S. Nitrous oxide production and consumption: regulation of gene expression by gassensitive transcription factors. Philos Trans R Soc B Biol Sci. 2012;367:1213–25. doi: 10.1098/rstb.2011.0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hallin S, Philippot L, Löffler FE, Sanford RA, Jones CM. Genomics and Ecology of Novel N2O-Reducing Microorganisms. Trends Microbiol. 2018;26:43–55. doi: 10.1016/j.tim.2017.07.003. [DOI] [PubMed] [Google Scholar]
- 16.Shapleigh JP. Denitrifying prokaryotes. In: Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F, editors. The prokaryotes: Prokaryotic Physiology and Biochemistry. Berlin Heidelberg: Springer-Verlag; 2013. p. 405–25.
- 17.Graf DRH, Jones CM, Hallin S. Intergenomic comparisons highlight modularity of the denitrification pathway and underpin the importance of community structure for N2O emissions. PLoS ONE. 2014;9:1–20. doi: 10.1371/journal.pone.0114118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lycus P, Bøthun KL, Bergaust L, Shapleigh JP, Bakken LR, Frostegård Å. Phenotypic and genotypic richness of denitrifiers revealed by a novel isolation strategy. ISME J. 2017;11:2219–32. doi: 10.1038/ismej.2017.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roco CA, Bergaust LL, Bakken LR, Yavitt JB, Shapleigh JP. Modularity of nitrogen-oxide reducing soil bacteria: linking phenotype to genotype. Environ Microbiol. 2017;19:2507–19. doi: 10.1111/1462-2920.13250. [DOI] [PubMed] [Google Scholar]
- 20.Lycus P, Soriano-Laguna MJ, Kjos M, Richardson DJ, Gates AJ, Milligan DA, et al. A bet-hedging strategy for denitrifying bacteria curtails their release of N2O. Proc Natl Acad Sci USA. 2018;115:11820–5. doi: 10.1073/pnas.1805000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Highton MP, Bakken LR, Dörsch P, Wakelin S, de Klein CAM, Molstad L, et al. Soil N2O emission potential falls along a denitrification phenotype gradient linked to differences in microbiome, rainfall and carbon availability. Soil Biol Biochem. 2020;150:108004. [Google Scholar]
- 22.Bergaust L, Bakken LR, Frostegård Å. Denitrification regulatory phenotype, a new term for the characterization of denitrifying bacteria. Biochem Soc Trans. 2011;39:207–12. doi: 10.1042/BST0390207. [DOI] [PubMed] [Google Scholar]
- 23.Liu B, Mao Y, Bergaust L, Bakken LR, Frostegård Å. Strains in the genus Thauera exhibit remarkably different denitrification regulatory phenotypes. Environ Microbiol. 2013;15:2816–28. doi: 10.1111/1462-2920.12142. [DOI] [PubMed] [Google Scholar]
- 24.Liu B, Zhang X, Bakken LR, Snipen L, Frostegård Å. Rapid succession of actively transcribing denitrifier populations in agricultural soil during an anoxic spell. Front Microbiol. 2019;9:3208. doi: 10.3389/fmicb.2018.03208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nömmik H. Investigations on denitrification in soil. Acta Agric Scand. 1956;6:195–228. [Google Scholar]
- 26.Wallenstein MD, Myrold DD, Firestone M, Voytek M. Environmental controls on denitrifying communities and denitrification rates: Insights from molecular methods. Ecol Appl. 2006;16:2143–52. doi: 10.1890/1051-0761(2006)016[2143:ecodca]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 27.Shen QR, Ran W, Cao ZH. Mechanisms of nitrite accumulation occurring in soil nitrification. Chemosphere. 2003;50:747–53. doi: 10.1016/s0045-6535(02)00215-1. [DOI] [PubMed] [Google Scholar]
- 28.Lim NYN, Frostegård Å, Bakken LR. Nitrite kinetics during anoxia: the role of abiotic reactions versus microbial reduction. Soil Biol Biochem. 2018;119:203–09. [Google Scholar]
- 29.Wijler J, Delwiche CC. Investigations on the denitrifying process in soil. Plant Soil. 1954;5:155–69. [Google Scholar]
- 30.Bakken LR, Bergaust L, Liu B, Frostegård Å. Regulation of denitrification at the cellular level: a clue to the understanding of N2O emissions from soils. Philos Trans R Soc B Biol Sci. 2012;367:1226–34. doi: 10.1098/rstb.2011.0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Russenes AL, Korsaeth A, Bakken LR, Dörsch P. Spatial variation in soil pH controls off-season N2O emission in an agricultural soil. Soil Biol Biochem. 2016;99:36–46. [Google Scholar]
- 32.Samad MDS, Biswas A, Bakken LR, Clough TJ, De Klein CAM, Richards KG, et al. Phylogenetic and functional potential links pH and N2O emissions in pasture soils. Sci Rep. 2016;6:1–10. doi: 10.1038/srep35990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y, Guo J, Vogt RD, Mulder J, Wang J, Zhang X. Soil pH as the chief modifier for regional nitrous oxide emissions: new evidence and implications for global estimates and mitigation. Glob Chang Biol. 2018;24:617–26. doi: 10.1111/gcb.13966. [DOI] [PubMed] [Google Scholar]
- 34.Duan YF, Hallin S, Jones CM, Priemé A, Labouriau R, Petersen SO. Catch crop residues stimulate N2O emissions during spring, without affecting the genetic potential for nitrite and N2O reduction. Front Microbiol. 2018;9:2629. doi: 10.3389/fmicb.2018.02629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Domeignoz-Horta LA, Putz M, Spor A, Bru D, Breuil MC, Hallin S, et al. Non-denitrifying nitrous oxide-reducing bacteria—An effective N2O sink in soil. Soil Biol Biochem. 2016;103:376–9. [Google Scholar]
- 36.Giles ME, Daniell TJ, Baggs EM. Compound driven differences in N2 and N2O emission from soil; the role of substrate use efficiency and the microbial community. Soil Biol Biochem. 2017;106:90–8. [Google Scholar]
- 37.Bergaust L, Mao Y, Bakken LR, Frostegård Å. Denitrification response patterns during the transition to anoxic respiration and posttranscriptional effects of suboptimal pH on nitrogen oxide reductase in Paracoccus denitrificans. Appl Environ Microbiol. 2010;76:6387–96. doi: 10.1128/AEM.00608-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu B, Mørkved PT, Frostegård Å, Bakken LR. Denitrification gene pools, transcription and kinetics of NO, N2O and N2 production as affected by soil pH. FEMS Microbiol Ecol. 2010;72:407–17. doi: 10.1111/j.1574-6941.2010.00856.x. [DOI] [PubMed] [Google Scholar]
- 39.Liu B, Frostegård Å, Bakken LR. Impaired reduction of N2O to N2 in acid soils is due to a posttranscriptional interference with the expression of nosZ. mBio. 2014;5:e01383–14. doi: 10.1128/mBio.01383-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Cleemput O, Samater AH. Nitrite in soils: accumulation and role in the formation of gaseous N compounds. Fertilizer Res. 1995;45:81–9. [Google Scholar]
- 41.Jansson JK, Baker ES. A multi-omic future for microbiome studies. Nat Microbiol. 2016;1:16049. doi: 10.1038/nmicrobiol.2016.49. [DOI] [PubMed] [Google Scholar]
- 42.Ma Y, Zilles JL, Kent AD. An evaluation of primers for detecting denitrifiers via their functional genes. Environ Microbiol. 2019;21:1196–210. doi: 10.1111/1462-2920.14555. [DOI] [PubMed] [Google Scholar]
- 43.Lim NYN, Roco CA, Frostegård Å. Transparent DNA/RNA co-extraction workflow protocol suitable for inhibitor-rich environmental samples that focuses on complete DNA removal for transcriptomic analyses. Front Microbiol. 2016;7:1588. doi: 10.3389/fmicb.2016.01588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bennett SP, Soriano-Laguna MJ, Bradley JM, Svistunenko DA, Richardson DJ, Gates AJ, et al. NosL is a dedicated copper chaperone for assembly of the CuZ center of nitrous oxide reductase. Chem Sci. 2019;10:4985–93. doi: 10.1039/c9sc01053j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hein S, Simon J. Bacterial nitrous oxide respiration: electron transport chains and copper transfer reactions. Adv Micro Physiol. 2019;75:137–75. doi: 10.1016/bs.ampbs.2019.07.001. [DOI] [PubMed] [Google Scholar]
- 46.Zhang L, Wüst A, Prasser B, Müller C, Einsle O. Functional assembly of nitrous oxide reductase provides insights into copper site maturation. Proc Natl Acad Sci USA. 2019;116:12822–7. doi: 10.1073/pnas.1903819116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sognnes LS, Fystro G, Øpstad SL, Arstein A, Børresen T. Effects of adding moraine soil or shell sand into peat soil on physical properties and grass yield in western Norway. Acta Agric Scand Sect B Soil Plant Sci. 2006;56:161–70. [Google Scholar]
- 48.Molstad L, Dörsch P, Bakken LR. Robotized incubation system for monitoring gases (O2, NO, N2O N2) in denitrifying cultures. J Microbiol Methods. 2007;71:202–11. doi: 10.1016/j.mimet.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 49.Apprill A, Mcnally S, Parsons R, Weber L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat Micro Ecol. 2015;75:129–37. [Google Scholar]
- 50.Parada AE, Needham DM, Fuhrman JA. Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ Microbiol. 2016;18:1403–14. doi: 10.1111/1462-2920.13023. [DOI] [PubMed] [Google Scholar]
- 51.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–61. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 52.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–7. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Greenfield P. Greenfield Hybrid Analysis Pipeline (GHAP). v1. CSIRO. Software Collection. 2017. 10.4225/08/59f98560eba25.
- 54.Van Der Walt S, Colbert SC, Varoquaux G. The NumPy array: a structure for efficient numerical computation. Comput Sci Eng. 2011;13:22–30. [Google Scholar]
- 55.Hunter JD. Matplotlib: a 2D graphics environment. Comput Sci Eng. 2007;9:90–5. [Google Scholar]
- 56.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, et al. Scikit-learn: machine learning in Python. J Mach Learn Res. 2011;12:2825–30. [Google Scholar]
- 57.Pielou EC. The measurement of diversity in different types of biological collections. J Theor Biol. 1966;13:131–44. [Google Scholar]
- 58.Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2014;12:59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 59.Nadeau SA, Roco CA, Debenport SJ, Anderson TR, Hofmeister KL, Walter MT, et al. Metagenomic analysis reveals distinct patterns of denitrification gene abundance across soil moisture, nitrate gradients. Environ Microbiol. 2019;21:1255–66. doi: 10.1111/1462-2920.14587. [DOI] [PubMed] [Google Scholar]
- 60.Roco CA, Dörsch P, Booth JG, Pepe-ranney C, Groffman PM, Fahey TJ, et al. Using metagenomics to reveal landscape scale patterns of denitrifiers in a montane forest ecosystem. Soil Biol Biochem. 2019;138:107585. [Google Scholar]
- 61.Arkin AP, Cottingham RW, Henry CS, Harris NL, Stevens RL, Maslov S, et al. KBase: the United States department of energy systems biology knowledgebase. Nat Biotechnol. 2018;36:566–9. doi: 10.1038/nbt.4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Menzel P, Ng KL, Krogh A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat Commun. 2016;7:11257. doi: 10.1038/ncomms11257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Muyzer G, De Waal EC, Uitterlinden AG. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol. 1993;59:695–700. doi: 10.1128/aem.59.3.695-700.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen Z, Luo X, Hu R, Wu M, Wu J, Wei W. Impact of Long-Term Fertilization on the Composition of Denitrifier Communities Based on Nitrite Reductase Analyses in a Paddy Soil. Micro Ecol. 2010;60:850–61. doi: 10.1007/s00248-010-9700-z. [DOI] [PubMed] [Google Scholar]
- 65.Hallin S, Lindgren PE. PCR detection of genes encoding nitrite reductase in denitrifying bacteria. Appl Environ Microbiol. 1999;65:1652–57. doi: 10.1128/aem.65.4.1652-1657.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Throbäck IN, Enwall K, Jarvis Å, Hallin S. Reassessing PCR primers targeting nirS, nirK and nosZ genes for community surveys of denitrifying bacteria with DGGE. FEMS Microbiol Ecol. 2004;49:401–17. doi: 10.1016/j.femsec.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 67.Bowman LAH, McLean S, Poole RK, Fukuto JM. The diversity of microbial responses to nitric oxide and agents of nitrosative stress. Close cousins but not identical twins. Adv Micro Physiol. 2011;59:135–219. doi: 10.1016/B978-0-12-387661-4.00006-9. [DOI] [PubMed] [Google Scholar]
- 68.Goulding KWT. Soil acidification and the importance of liming agricultural soils with particular reference to the United Kingdom. Soil Use Manag. 2016;32:390–9. doi: 10.1111/sum.12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Frostegård Å, Bååth E, Tunlid A. Shifts in the structure of soil microbial communities in limed forests as revealed by phospholipid fatty acid analysis. Soil Biol Biochem. 1993;25:723–30. [Google Scholar]
- 70.Rousk J, Bååth E, Brookes PC, Lauber CL, Lozupone C, Caporaso JG, et al. Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J. 2010;4:1340–51. doi: 10.1038/ismej.2010.58. [DOI] [PubMed] [Google Scholar]
- 71.Fierer N. Embracing the unknown: disentangling the complexities of the soil microbiome. Nat Rev Microbiol. 2017;15:579–90. doi: 10.1038/nrmicro.2017.87. [DOI] [PubMed] [Google Scholar]
- 72.Van den Heuvel RN, Van Der Biezen E, Jetten MSM, Hefting MM, Kartal B. Denitrification at pH 4 by a soil-derived Rhodanobacter-dominated community. Environ Microbiol. 2010;12:3264–71. doi: 10.1111/j.1462-2920.2010.02301.x. [DOI] [PubMed] [Google Scholar]
- 73.Honisch U, Zumft WG. Operon structure and regulation of the nos gene region of Pseudomonas stutzeri, encoding an ABC-Type ATPase for maturation of nitrous oxide reductase. J Bacteriol. 2003;185:1895–902. doi: 10.1128/JB.185.6.1895-1902.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zumft WG, Kroneck PMH. Respiratory transformation of nitrous oxide (N2O) to dinitrogen by Bacteria and Archaea. Adv Micro Physiol. 2007;52:107–227. doi: 10.1016/S0065-2911(06)52003-X. [DOI] [PubMed] [Google Scholar]
- 75.Abraham ZH, Smith BE, Howes BD, Lowe DJ, Eady RR. pH-dependence for binding a single nitrite ion to each type-2 copper centre in the coppercontaining nitrite reductase of Alcaligenes xylosoxidans. Biochem J. 1997;324:511–6. [DOI] [PMC free article] [PubMed]
- 76.Mania D, Woliy K, Degefu T, Frostegård Å. A common mechanism for efficient N2O reduction in diverse isolates of nodule-forming bradyrhizobia. Environ Microbiol. 2020;22:17–31. doi: 10.1111/1462-2920.14731. [DOI] [PubMed] [Google Scholar]
- 77.Kraft B, Strous M, Tegetmeyer HE. Microbial nitrate respiration - Genes, enzymes and environmental distribution. J Biotechnol. 2011;155:104–17. doi: 10.1016/j.jbiotec.2010.12.025. [DOI] [PubMed] [Google Scholar]
- 78.Simon J, Klotz MG. Diversity and evolution of bioenergetic systems involved in microbial nitrogen compound transformations. Biochim Biophys Acta—Bioenerg. 2013;1827:114–35. doi: 10.1016/j.bbabio.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 79.Palmer K, Biasi C, Horn MA. Contrasting denitrifier communities relate to contrasting N2O emission patterns from acidic peat soils in arctic tundra. ISME J. 2012;6:1058–77. doi: 10.1038/ismej.2011.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Coyotzi S, Doxey AC, Clark ID, Lapen DR, Van Cappellen P, Neufeld JD. Agricultural soil denitrifiers possess extensive nitrite reductase gene diversity. Environ Microbiol. 2017;19:1189–208. doi: 10.1111/1462-2920.13643. [DOI] [PubMed] [Google Scholar]
- 81.Decleyre H, Heylen K, Bjorn T, Willems A. Highly diverse nirK genes comprise two major clades that harbour ammonium-producing denitrifiers. BMC Genom. 2016;17:155. doi: 10.1186/s12864-016-2465-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jones CM, Stres B, Rosenquist M, Hallin S. Phylogenetic analysis of nitrite, nitric oxide, and nitrous oxide respiratory enzymes reveal a complex evolutionary history for denitrification. Mol Biol Evol. 2008;25:1955–66. doi: 10.1093/molbev/msn146. [DOI] [PubMed] [Google Scholar]
- 83.Kartal B, De Almeida NM, Maalcke WJ, Op den Camp HJM, Jetten MSM, Keltjens JT. How to make a living from anaerobic ammonium oxidation. FEMS Microbiol Rev. 2013;37:428–61. doi: 10.1111/1574-6976.12014. [DOI] [PubMed] [Google Scholar]
- 84.Strube K, De Vries S, Cramm R. Formation of a dinitrosyl iron complex by NorA, a nitric oxide-binding di-iron protein from Ralstonia eutropha H16. J Biol Chem. 2007;282:20292–300. doi: 10.1074/jbc.M702003200. [DOI] [PubMed] [Google Scholar]
- 85.Gonska N, Young D, Yuki R, Okamoto T, Hisano T, Antonyuk S, et al. Characterization of the quinol-dependent nitric oxide reductase from the pathogen Neisseria meningitidis, an electrogenic enzyme. Sci Rep. 2018;8:1–13. doi: 10.1038/s41598-018-21804-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Su H, Cheng Y, Oswald R, Behrendt T, Trebs I, Meixner FX, et al. Soil nitrite as a source of atmospheric HONO and OH radicals. Science. 2011;333:1616–8. doi: 10.1126/science.1207687. [DOI] [PubMed] [Google Scholar]
- 87.Wu D, Horn MA, Behrendt T, Müller S, Li J, Cole JA, et al. Soil HONO emissions at high moisture content are driven by microbial nitrate reduction to nitrite: tackling the HONO puzzle. ISME J. 2019;13:1688–99. doi: 10.1038/s41396-019-0379-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The 16S rRNA gene amplicon sequences are available as MG and MT FASTQ files on European Nucleotide Archive (ENA) under project PRJEB41993.