

Abstract
Tramadol (TR) is a centrally acting analgesic drug that is used to relieve pain. The therapeutic (0.1–0.8 mg/l), toxic (1–2 mg/l) and lethal (>2 mg/l) ranges were reported for TR. The present study was designed to evaluate which doses of TR can induce liver mitochondrial toxicity. Mitochondria were isolated from the five rats’ liver and were incubated with therapeutic to lethal concentrations (1.7–600 μM) of TR. Biomarkers of oxidative stress including: reactive oxygen species (ROS), lipid peroxidation (LPO), protein carbonyl content, glutathione (GSH) content, mitochondrial function, mitochondrial membrane potential (MMP) and mitochondrial swelling were assessed. Our results showed that ROS and LPO at 100 μM and protein carbonylation at 600 μM concentrations of TR were significantly increased. GSH was decreased specifically at 600 μM concentration. Mitochondrial function, MMP and mitochondrial swelling decreased in isolated rat liver mitochondria after exposure to 100 and 300 μM, respectively. This study suggested that TR at therapeutic and toxic levels by single exposure could not induce mitochondrial toxicity. But, in lethal concentration (≥100 μM), TR induced oxidative damage and mitochondria dysfunction. This study suggested that ROS overproduction by increasing of TR concentration induced mitochondrial dysfunction and caused mitochondrial damage via Complex II and membrane permeability transition pores disorders, MMP collapse and mitochondria swelling.
Keywords: tramadol, mitochondria, oxidative stress, liver toxicity, biomarkers
Graphical Abstract
Graphical Abstract.
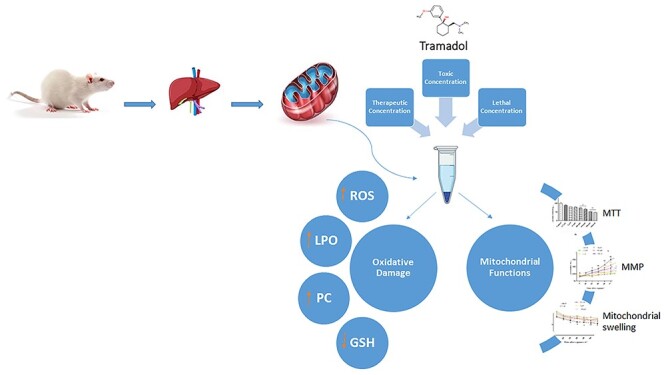
Introduction
Tramadol (TR) is a synthetic opioid-like analgesic and one of the commonly used prescription drugs for pain management [1]. It was presented as the injectable and oral forms in the pharmaceutical market. TR is extensively metabolized in the liver by O- and N-desmethylation pathways [2]. It is expected that the liver is the main target organ for TR toxicity that has been explained in the human [3–5] and animal studies [6–10]. Moreover, liver injury, such as hyperammonemia, lactic acidosis and hepatic steatosis, have been reported following TR overdoses that could attribute to mitochondrial damage [11].
The previous animal studies in mice, rats and doges have been shown that the reactive oxygen species (ROS) production and oxidative stress are the primary mechanisms of TR toxicity [10, 12, 13]. According to previous animal studies, treatment of rats with 50 mg/kg of TR for 21 days increased malondialdehyde (MDA) and protein carbonyl (PC) levels and decreased glutathione (GSH) content in the rat brain and testis tissues [14]. Also, TR administration (30 and 60 mg/kg) for 60 days resulted in a reduction in GSH content in the rat brains [15]. Moreover, chronic exposure to TR (50 mg/kg) in rats showed elevated MDA levels in homogenized brain tissues [16]. Hussein et al. reported that administration of TR at therapeutic (22.5 mg/kg) and high concentrations (30, 60 and 90 mg/kg) for 9 weeks could increase lipid peroxidation (LPO) in the rat brains [17]. Ali et al. showed that TR at therapeutic (25 mg/kg), double therapeutic (50 mg/kg) and four times therapeutic (100 mg/kg) doses for 30 days increased LPO in rats brain tissues [10]. The adverse effects of acute TR administration in rats were also reported by Faria et al. According to this study, the highest dose of TR (50 mg/kg) after 24 h enhanced the heart and lung protein oxidation [18]. In the other animal study, exposure of rats with 37.5 mg/kg (three times daily for 1 month) and 40 mg/kg (for 21 days) of TR increased LPO level and decreased GSH content in the rat liver tissue [19–21]. Sadek et al. reported that after injection of TR (15 mg/kg) for 15 days, MDA level increased and GSH content decreased in rat liver samples [22]. A similar result was reported that TR (15 mg/kg for 7 days) could increase the LPO and reduce GSH content in the liver of treated rats [23]. Hepatotoxicity with TR administration at doses of 10, 50 and 100 mg/kg was studied by Owoade et al. The results of this work showed that three experimental doses of TR after 28 days could decrease GSH content in the rat liver tissues. In another study, elevated MDA level after exposure to 50 and 100 mg/kg of TR in the rat liver samples was also reported [24]. The effect of repeated therapeutic doses of TR on liver oxidative damage was studied by Joana Barbosa et al. on the rat’s liver. In this study, rats were exposed to 10, 25 and 50 mg/kg of TR (low analgesic, intermediate and the maximum recommended daily doses, respectively) for 14 days. MDA increased only at 50 mg/kg dose of TR in the rat liver tissues and TR could not increase liver protein carbonylation [25].
TR half-life was reported as 9.24 h (in overdose), which was certainly related with higher concentrations [26]. The therapeutic doses of TR for human are 50 and 100 mg [27] and the maximum acceptable daily dose is 400 mg/day [1, 28]. Several studies showed that TR can induce toxicity at high doses (>1300 mg) [29–32]. Some studies reported that TR abuse and/or misuse is increasing among the communities [33–35]. It was stated that the maximum plasma concentration of TR following a single oral dose of 100 mg is ~0.3 mg/l [1]. According to previous studies, therapeutic blood level in human adults range (from 0.1 to 0.8 mg/l), toxic level (from 1 to 2 mg/l) and lethal concentration (>2 mg/l) were reported. Hence, it seems that TR easily can shift from the therapeutic concentrations to toxic and lethal levels [36]. Toxicological screening in post-mortem cases revealed the different TR levels in the human blood range between 0.880 to 134 mg/ml [36–38].
In the human study, administration of TR in the therapeutic range (200–400 mg/day) for management of chronic pain induced oxidative damage and enhanced serum MDA levels [6].
The previous human and animal studies have been shown that the oxidative stress can induce disruption of mitochondrial function, apoptosis and ultimately cell damage. Mitochondria, as small intracellular organelles, have a key role in free radical production [39–41]. In pathological conditions, a growing trend in hydrogen peroxide (H2O2) production was observed through the inhibition of mitochondrial electron transfer chain (ETC) complexes (I, III and IV) activity [42]. Since TR presents a positive charge at physiological pH [18], it may accumulate within negatively charged cell compartments, such as mitochondria, eventually causing mitochondrial dysfunction [12]. Mohamed et al. reported that Complexes I, III and IV activities in rat’s mitochondria were decreased after abuse of TR (progressing dose from 42 to 168 mg/kg for 30 days) via ROS formation. Therefore, TR induces the enhancement of the H2O2 formation consequences of ROS production in the mitochondria [43].
There are growing reports in different countries about the abuse, toxicity and mortality due to TR consumption. Although different animals and human studies evaluated the oxidative damage induced by acute and chronic consumption of TR in different organs, but to the best of our knowledge, the concentrations of TR that can cause toxicity along with mitochondria has not yet been determined and has not been reviewed in the isolated liver mitochondria. Hence, we designated the present study to investigate the effect of a wide range of TR concentrations under controlled conditions on oxidative stress biomarkers status and mitochondrial functions.
Experimental
Materials
TR hydrochloride (“C16H26ClNO2”) was prepared from Razak Pharmaceutical Co (Tehran, Iran). Other chemicals used in this study were prepared of analytical grade and purchased from the Merck (Darmstadt, Germany) and Sigma Aldrich Co (St. Louis, MO, USA).
Preparation of liver mitochondria
All investigation procedures were conducted according to the ethical principles and protocols that were approved by the Animal Ethics Research Committee of Mazandaran University of Medical Sciences, Sari, Iran (with the ethic number: IR.MAZUMS.REC.1399.6990). The livers of five male Wistar rats (250–300 g, mean of liver weight: 11.63 ± 1.57 g) were dissected and after that were well washed with cold isolation media (0.225 M D-mannitol, 75 mM sucrose and 0.2 mM EDTA) and then were homogenized using silent crusher M (Heidolph-Germany). The mixture was centrifuged at 1000× g for 10 min at 4°C and the supernatant was centrifuged at 10 000× g for 10 min to precipitate the mitochondria. Experiments were performed in triplicate. Depending on the type of test, rats’ liver mitochondria were suspended in their exceptional buffer solution including: Tris buffer (0.05 M Tris–HCl, 0.25 M sucrose, 20 mM KCl, 2.0 mM MgCl2 and 1.0 mM Na2HPO4, pH =7.4) for LPO, PC and GSH assay; respiration buffer (125 mM sucrose, 65 mM KCl, 10 mM HEPES, 20 mM Ca2+ and 5 mM sodium succinate) for ROS assay; mitochondrial membrane potential (MMP) buffer (68 mM mannitol, 220 mM sucrose, 10 mM KCl, 5 mM KH2PO4, 50 μM EGTA, 2 mM MgCl2 and 10 mM HEPES) and swelling buffer (125 mM sucrose, 65 mM KCl, 10 mM Hepes-KOH and 20 mM Ca2+) for MMP assay and swelling assay, respectively. All extraction procedures were performed at 4°C [44].
Mitochondrial treatment
Extracted mitochondria were placed in presence of different TR concentrations (1.7–600 μM). These concentrations have been selected based on previous human and in vitro studies [36, 45]. For evaluation of oxidative damage biomarkers in the therapeutic, toxic and lethal concentrations of TR in the mitochondria, freshly isolated mitochondria from five rats’ liver were incubated with therapeutic (0–1.7 μM), toxic (1.7–5 μM) and lethal (10, 100, 150, 300 and 600 μM) concentrations of TR [4, 10, 45]. Samples were incubated at 37°C for 1 h. After incubation time, oxidative stress biomarkers, such as ROS, LPO, PC, GSH, mitochondrial function, MMP and mitochondrial swelling, were investigated (each test was performed in triplicate).
Measurement of total protein
Samples protein concentration determined by Bradford method [46]. Briefly, mitochondrial suspension and bovine serum albumin (BSA) in different concentrations were incubated with Coomassie blue for 10 min and the absorbance was measured at 595 nm by spectrophotometry (UV-1601 PC, Shimadzu, Japan).
Mitochondrial ROS assay
Mitochondrial ROS level was estimated by using 2′,7′-dichlorofluorescein diacetate (DCFH-DA) [12]. Isolated liver mitochondria were treated with TR at 37°C for 60 min. After that, mitochondria immersed in respiration media and 5 μl of DCFH-DA (final concentration 10 μM) was added. Present suspension incubated at 37°C for 30 min. Afterward, the fluorescence intensity was measured by spectrofluorometer (JASCO, FP6200, Japan) at 480 nm excitation and 520 nm emission wavelengths.
Mitochondrial LPO assay
MDA level, as LPO index, was measured by thiobarbituric acid (TBA) as an indicator of LPO process [47]. Mitochondrial suspension, 0.2 ml phosphoric acid (85%) and 25 μl TBA were mixed and placed in a boiling Bain Marie for 30 min. After, the samples were placed in the ice bath and 0.45 ml n-butanol was added to each sample. Samples were centrifuged and absorbance was measured at 532 nm by enzyme-linked immunosorbent assay (ELISA) (Tecan Rainbow Thermo, Austria) equipment.
Mitochondrial PC assay
For assessment of PC, 200 μl of isolated mitochondria suspension and 0.5 ml of 20% (w/v) trichloroacetic acid (TCA) were added into a microtube and were kept at 4°C for 15 min. After centrifugation, the sedimentation was mixed with 0.5 ml of 0.2% 2,4-dinitrophenylhydrazine (DNPH) and was maintained at room temperature for 1 h. Then, the samples were washed with 1 ml of ethanol-ethyl acetate and were centrifuged for 10 min (recent step was repeated for three times). The ultimate precipitates were dissolved in 200 μl of 6 mol/l guanidine HCl. Measured absorbance at 365 nm wavelength by ELISA reader (Tecan, Rainbow Thermo, Austria) was indicated as carbonylated moieties [47].
Mitochondrial GSH assay
Mitochondria GSH level was measured with the Ellman method by using 5,5′-dithiobis-(2-nitrobenzoicacid) (DTNB) [44]. Mitochondrial suspension, TCA and DTNB were mixed until they developed yellowish color, and absorbance was measured at 412 nm by ELISA reader (Tecan Rainbow Thermo).
Mitochondrial complex II activity assay
Mitochondrial succinate dehydrogenase activity was evaluated by reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide dye (MTT) to formazan metabolite [44]. Formation of purple color intensity at 570 nm by ELISA reader (Tecan Rainbow Thermo) is directly related to the health and activity of this mitochondrial enzyme.
Mitochondrial MMP assay
MMP was evaluated by rhodamine 123 (Rh123) [44]. Isolated rat liver mitochondria were resuspended in the MMP media (pH = 7.2) and was incubated with Rh123 (10 μM) at 37°C for 60 min. The fluorescence was measured by spectrofluorometer (JASCO) at the 490 nm excitation and 535 nm emission wavelength at several times from 5 to 60 min of treatment (even 10 min time intervals). The fluorescence intensity is inversely related to mitochondrial membrane health.
Mitochondrial swelling assay
To evaluate the effect of different concentrations of TR on mitochondrial membrane permeability transition (MPT), fresh isolated liver mitochondria were suspended in swelling buffer (pH = 7.2). The absorbance was measured by ELISA reader (Tecan Rainbow Thermo) at 540 nm for several times from 5 to 60 min of treatment (even 10 min time intervals). Reduction in mitochondria absorbance represents an elevation in mitochondrial swelling [44].
Statistical analysis
All statistical analyses were performed using the GraphPad Prism software, version 6. Results are expressed as means ± standard error of the mean. Data were statistically analyzed by one-way ANOVA test followed by the post hoc Tukey test. P < 0.05 was considered as the statistical significance level.
Results
Figure 1 shows the effects of different concentrations of TR on ROS production. The present results showed that TR increased the ROS formation with the dose-dependent manner in the mitochondria. At concentrations range between 1.7 and 10 μM (therapeutic and toxic concentrations), no significant changes were observed in the ROS levels. By increasing of the TR concentrations from 100 μM, ROS production significantly increased at 100–600 μM of TR concentrations (100 μM: P < 0.05; 150 μM: P = 0.022; 300 μM: P = 0.004 and 600 μM: P = 0.0035) when compared with the control group. Results showed that TR concentration at 300 μM significantly increased ROS production (P < 0.01) on comparison with the 1.7 μM group. Generated of ROS at 600 μM of TR concentration was significantly enhanced when compared with 1.7 (P = 0.0037), 5 (P = 0.0049) and 10 μM (P = 0.0151).
Figure 1.
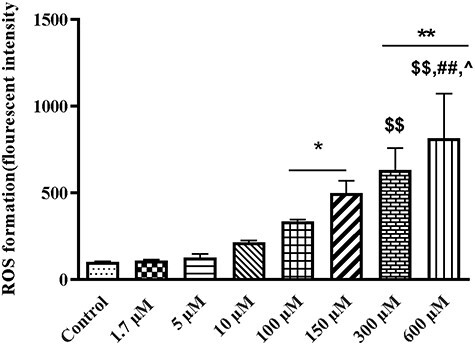
The effect of different tramadol (TR) concentrations (1.7–600 µM) on ROS formation in the rat liver isolated mitochondria. Values represented as mean ± SEM (n = 5). *(p < 0.05); **(p < 0.01): Significantly different from control group. $$(p < 0.01): Significantly different from 1.7 µM group. ##(p < 0.001): significantly different from 5 µM group. ^(p < 0.05): significantly different from 10 µM group.
LPO for the evaluation of the free radicals’ attack to the membranes lipids was assessed in this study. Measurement of MDA level reflects to membrane damage due to LPO. For evaluation of LPO, different concentrations of TR (1.7–600 μM) were added to the isolated rat liver mitochondria. After 1 h as shown in Fig. 2, mitochondrial MDA content were 5.46 ± 1.65, 8.19 ± 1.80, 12.15 ± 2.11, 15.14 ± .38, 16.49 ± .57, 18.51 ± 1.96 and 21.76 ± 2.48 μg MDA/mg protein for 1.7, 5, 10, 100, 150, 300 and 600 μM concentrations of TR, respectively. Our findings showed that LPO significantly increased (P < 0.01 and P < 0.001) from 100 to 600 μM (lethal range) when compared with the control group (4.90 ± .88 MDA/mg protein) in a concentration-dependent pattern (Fig. 2). Multiple comparisons between different concentrations of TR showed that 100 and 150 μM of TR could significantly increase (P = 0.007 and P = 0.0021, respectively) LPO in comparison with 1.7 μM. MDA content in 300 μM had significance difference with 1.7 μM (P = 0.004) and 5 μM (P = 0.044) of TR. At the highest lethal concentration (600 μM) of TR, MDA content significantly increased when compared with the 1.7 (P = 0.0003), 5 (P = 0.00045) and 10 μM (P = 0.0083) groups.
Figure 2.
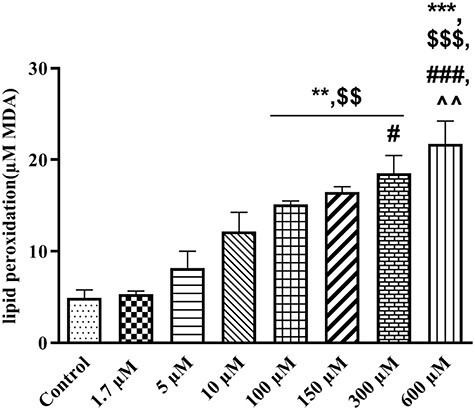
The effect of therapeutic to lethal concentrations of TR (1.7–600 µM) on lipid peroxidation in liver mitochondria. Values represented as mean ± SEM (n = 5). **(p < 0.01); ***(p < 0.001) : Significantly different compared with control mitochondria. $$(p < 0.01); $$$(p < 0.001): Significantly different compared with 1.7 µM group. #(p < 0.05); ###(p < 0.001): Significantly different compared with 5 µM group. ^^(p < 0.01): Significantly different compared with 10 µM group.
Another consequence of oxidative damage following ROS overproduction is the oxidation of proteins. We measured PC groups in the isolated rat liver mitochondria. It observed that TR only in the highest lethal concentration (600 μM) could significantly increase (P = 0.003) the production of carbonylated moieties when compared with the control group (Fig. 3). TR in 600 μM had significant difference with 1.7, 5 (P = 0.004) and 10 μM (P = 0.006) in oxidation of protein groups. Therapeutic and toxic concentrations of TR could not effect on mitochondrial PC.
Figure 3.
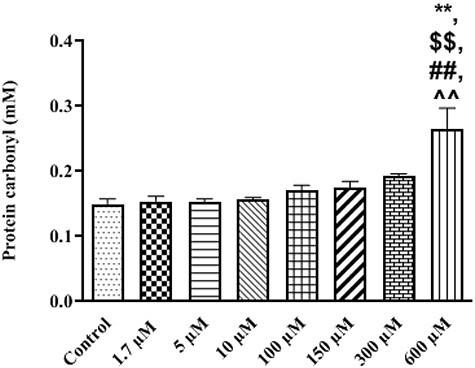
Protein carbonyl formation at different concentrations of TR (1.7–600 µM) in treated isolated mitochondria. Values represented as mean ± SEM (n = 5). **(p < 0.01): Significantly different compared with control mitochondria. $$(p < 0.01): Significantly different compared with 1.7 µM group. ##(p < 0.01): Significantly different compared with 5 µM group. ^^(p < 0.01): Significantly different compared with 10 µM group.
The effect of TR was also evaluated on the content of the non-enzymatic antioxidant system such as GSH. Mitochondrial GSH content after exposure to TR in therapeutic and toxic levels showed no significant changes. GSH level were decreased (47.02 ± .32 μg/mg protein) significantly (P = 0.01) after exposure to 600 μM (maximum lethal concentration) of TR in comparison with control group (49.28 ± .07 μg /mg protein), (Fig. 4). The maximum lethal concentration of TR could significantly decrease the GSH content in comparison with 1.7 (P = 0.0138), 5 (P = 0.014), 10, 100 and 150 μM (P < 0.05).
Figure 4.
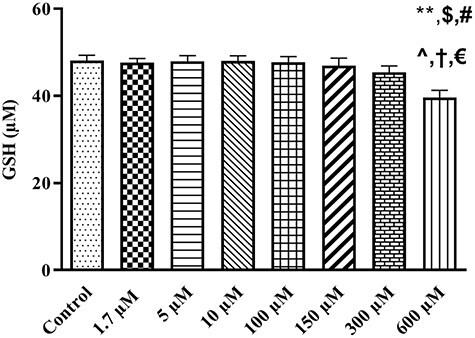
The influence of different concentrations of TR (1.7–600 µM) on GSH content in rat liver isolated mitochondria. Values represented as mean ± SEM (n = 5). *(p < 0.05): Significantly different compared with control mitochondria. $(p < 0.05): Significantly different compared with 1.7 µM group. #(p < 0.05): Significantly different compared with 5 µM group. ^(p < 0.05): Significantly different compared with 10 µM group. †(p < 0.05): Significantly different compared with 100 µM group. €(p < 0.05): Significantly different compared with 150 µM group.
The effect of the different concentrations of TR on mitochondrial succinate dehydrogenase enzyme activity was also evaluated in isolated rat liver mitochondria. Our data showed a significant decrease in the reduction of MTT to formazan metabolite with the dose-dependent manner by increasing of the TR concentration (Fig. 5). TR at 1.7 μM concentrations had no significant effect on mitochondrial complex II activity. Activity of succinate dehydrogenase enzyme was significantly inhibited at 100, 150 (P < 0.05), 300 and 600 μM (P < 0.01) concentrations of TR when compared to the control group with the dose-dependent manner. TR in 100 and 150 μM significantly (P < 0.05) inhibited Complex II activity in comparison to 1.7 μM. The 300 and 600 μM concentrations of TR showed significant difference with 1.7 (P = 0.003 and P = 0.002), 5 (P < 0.05 and P = 0.003) and 10 (P < 0.05) μM. The highest concentration of TR (600 μM) had significant difference with 100 μM (P < 0.05).
Figure 5.
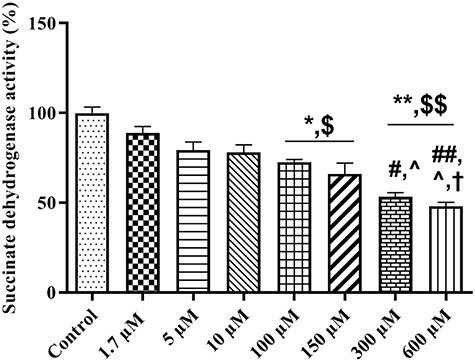
The effect of TR (1.7–600 µM) on succinate dehydrogenase enzyme activity. Mitochondrial complex II activity was measured using MTT dye. Values represented as mean ± SEM (n = 5). *(p < 0.05); **(p < 0.01): Significantly different compared with control mitochondria. $(p < 0.05): Significantly different compared with 1.7 µM group. #(p < 0.05): Significantly different compared with 5 µM group. ^(p < 0.05): Significantly different compared with 10 µM group. †(p < 0.05): Significantly different compared with 100 µM group. €(p < 0.05): Significantly different compared with 150 µM group.
As shown in Fig. 6, TR at concentrations of 100, 150, 300 and 600 μM significantly decreased MMP (P < 0.05 and P < 0.01) after 20, 40, 50 and 60 min, respectively, when compared with the control group. Our results showed that TR could induce the collapse of MMP in concentration manner- and time-dependent pattern.
Figure 6.
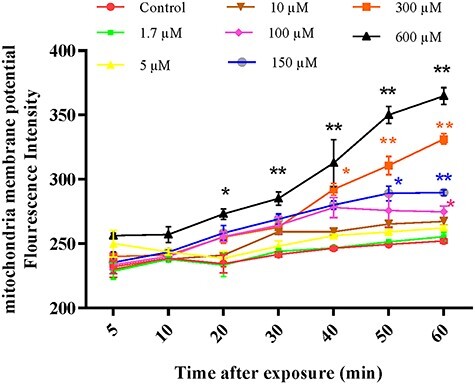
The effect of different concentrations of TR (1.7–600 µM) on the mitochondrial membrane potential (MMP) in the isolated rat liver mitochondria. MMP was measured by Rh123. Values represented as mean ± SEM (n = 5). *(p < 0.05); **(p < 0.01): Significantly different compared with control mitochondria.
Mitochondrial swelling, as an indicator of mitochondrial membrane permeability transition pore (MPTP) opening index, was assessed with absorbance changes at 540 nm. TR significantly increased (P < 0.01) mitochondrial swelling at 300 and 600 μM concentrations after 20 and 40 min, respectively (Fig. 7). TR concentration and length of time exposure are important factors for induction of swelling in isolated rat liver mitochondria.
Figure 7.
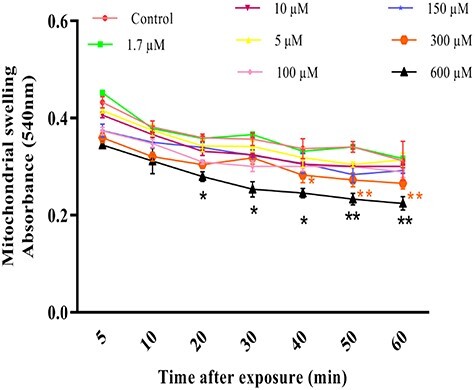
Mitochondrial swelling due to influence of different concentrations of TR (1.7–600 µ600 µM) in the isolated rat liver mitochondria. Mitochondrial swelling was measured through the determination of absorbance at 540 nm. Values represented as mean ± SEM (n = 5). *(p < 0.05); **(p < 0.01): Significantly different compared with control mitochondria.
Discussion
We designed the present study to investigate the effects of therapeutic (1.7 μM), toxic (5 μM) and lethal (10–600 μM) concentrations of TR on the mitochondrial oxidative damage and mitochondrial functions in isolated rat liver mitochondria. This study revealed that TR induces oxidative stress in isolated rat liver mitochondria at lethal concentrations. In our study, TR at 100 μM (30 mg/l) concentration, which is approximately equal to human lethal level, could induce ROS generation, LPO, inhibition of mitochondrial complex II activity and MMP collapse. Mitochondrial swelling was observed at 300 μM (90 mg/l) concentration of TR. In this study, increase of protein carbonylation and decrease of GSH content occurred at 600 μM (180 mg/l) of TR concentration.
For the accurate extrapolation of the human dose to an equivalent dose for a rat or vice versa, a method based on the body surface area was stated by the US Food and Drug Administration [48]. Based on this calculation, the used doses of 20, 40 and 60 mg/kg in rats are almost equivalent to 210.6, 421.2 and 631.8 mg for an adult human whose weight is 65 kg. Also, doses of 20, 40 and 60 mg/kg of TR in rat according to the other equation [49] could produce therapeutic (0.688 mg/l), toxic (1.375 mg/l) and lethal (2.06 mg/l) concentrations in rat blood, respectively. Abovementioned concentrations (0.688, 1.375 and 2.06 mg/l) in rat blood might be comparable to human therapeutic (0.1–0.8 mg/l), toxic (1–2 mg/l) and lethal (>2 mg/l) ranges, respectively [36].
ROS is produced during oxygen metabolism, being chemical species with one unpaired electron derived from molecular oxygen. Elevated formation of the different ROS leads to molecular damage that represented as oxidative distress. ROS can modulate a wide range of mechanisms within the biological system. They are very well known for playing both favorable and damaging roles in the human body given this “double-edged sword” characteristic of ROS [50, 51].
This study revealed that TR at therapeutic and toxic (1.7- 5 μM) concentrations couldn’t alter the ROS generation. Our data showed that ROS level could be enhanced from 100 μM of TR concentration (Fig. 1). Present findings are similar to the results of Mehdizadeh et al. who reported that consumption of TR (20 mg/kg) for 4 weeks could not effect on ROS production in rats brain mitochondria, but by increasing of doses of the TR (40 and 80 mg/kg for 4 weeks) the ROS generation in rats brain mitochondria was enhanced [52]. In another animal studies, infusion of TR (25 mg/ml) for evaluation of the seizure threshold increased ROS levels in the rat brain mitochondria [12, 47].
In our investigation, exposure of rat liver mitochondrial suspension to TR in lethal (100–600 μM) concentrations substantially increased mitochondrial LPO (Fig. 2). Previous studies carried out by Bameri et al. and Samadi et al. demonstrated that infusion of TR (25 mg/ml) could decrease seizure threshold and increase LPO in rat brain mitochondria [12, 47]. Hence, it seems that increasing the concentration of the TR increases the oxidative damage of the mitochondria by increase of LPO and elevation of MDA content. However, the important point is: When mitochondrial damage can be induced and how much TR concentrations can induce damage to the mitochondria? According to our data, mitochondrial damage can be started at a concentration of 100 μM, which can be considered equivalent to 15 times of the therapeutic concentration of TR.
Several studies identified the association between recurrent opioid administration and increased production of ROS [12, 43, 53–55]. The pathological lesions produced by chronic TR administration could be explained by the accumulation of toxic metabolites, decrease of their clearance and ability of TR for ROS generation that commonly leads to disruption and disintegration of the cell membrane and ultimately LPO [56, 57]. Previous studies have shown that chronic administration of TR even at therapeutic levels could cause mitochondrial oxidative damage [10, 12, 47]. In our study, single exposure of rat liver mitochondria to TR could only induce oxidative stress at lethal concentrations.
It is demonstrated that the protein/lipid ratio in the mitochondrial inner membrane is 3:1 [58]. So, ROS could attack to mitochondrial membrane and can induce both lipoxidative and protein oxidation damage in overproduction of ROS following TR consumption. In our study, TR at the highest concentration (600 μM), which can be equivalent to 180 mg/l of human plasma concentration, significantly increased PC groups level (Fig. 3). In confirmation of our findings, previous studies have shown that infusion of high dose of TR (25 mg/ml) could increase PC groups in rat brain mitochondria [12, 47]. Also, it was stated that administration of TR at therapeutic doses enhanced protein and lipid oxidation in liver and kidney tissues and diminished total liver antioxidant capacity [25].
GSH has a protective effect on the thiol groups of mitochondrial proteins [21]. In our study, the significant decrease in GSH levels was observed following the treatment of mitochondria with a lethal concentration (600 μM) (Fig. 4). It was similar to previous investigations that reported TR (25 mg/ml) infusion decreased GSH content in the rat brain mitochondria [12, 47].
Moreover, we investigated the activity of succinate dehydrogenase enzyme (Complex II) in rat liver mitochondria under the influence of different concentrations of TR. Toxicity assessment by measuring of the total amount of produced formazan upon MTT reduction demonstrated a considerable decrease in mitochondrial complex II activity in 100–600 μM concentrations (Fig. 5). The results of this study are compatible with previous work, which showed the TR at 50 mg/kg for 21 consecutive days diminished mitochondrial complex II activity in the testis and brain mitochondria [14]. Our results are incongruence with the Faria et al. (2016) study that showed the TR at a high concentration (600 μM) could not inhibit succinate dehydrogenase enzyme in the human SH-SY5Y neuroblastoma cells line [45]. Also, Mohamed et al. reported that TR at therapeutic (42 mg/kg) and overdose (168 mg/kg) levels could not effect on the Complex II activity in the rat brain mitochondria after 30 days of treatment [43].
Oxidative stress by inhibiting or change the mitochondrial ETC activity and opening the MPT pores could induce MMP collapse [59]. Oxidation of the mitochondrial thiol groups and structural changes in the mitochondrial permeability transition poor leads to mitochondrial dysfunction, energy failure, enhanced free radical production and ultimately mitochondrial swelling [52, 60]. Therefore, the effects of TR on MMP collapse and mitochondria swelling were investigated. Our findings showed that TR could induce MMP collapse (Fig. 6) in a concentration- and time-dependent manner. TR at the 100, 150, 300 and 600 μM after 20, 40, 50 and 60 min could induce collapse of the MMP, respectively. These findings suggest that TR at higher concentrations can effect on MMP more rapidly. The findings of the present study are similar to the Mehdyzadeh et al. results that showed TR (at 40 and 80 mg/kg) after 30 days could induce MMP collapse in the rat brain mitochondria [52]. Our study is inconsistent with an in vitro study that showed TR has not effect on MMP of the SH-SY5Y neuroblastoma cells line [45].
Mitochondrial swelling (Fig. 7) was observed at 300 and 600 μM of TR after 20 and 40 min of exposure, respectively. Mitochondrial swelling was also affected by TR in a concentration- and time-dependent pattern. These results are similar to previous studies that showed the MMP collapse and mitochondrial swelling created by the TR at 50 mg/kg in the rat brain and testis mitochondria after 3 weeks [14]. It was stated that 40 and 80 mg/kg of TR induced collapse and mitochondrial swelling in the rat brain mitochondria after 4 weeks [52]. Moreover, it demonstrated that 1 mM of TR concentration induces mitochondrial swelling in the rat neuron-like cells line [61]. The morphometric study after TR administration (50 mg/kg for 4 weeks) has shown mitochondrial swelling in the brain tissue [62].
Mohamed HM et al. stated that administration of TR could increase nitric oxide (NO) and monoamine neurotransmitter levels. NO in the redox condition can react with superoxide radicals to produce the potent oxidant peroxynitrite that attacks lipid and protein and induces lipid and protein damage and elevates MDA and PC content. Moreover, TR can decrease the activity and expression of antioxidant enzyme, apoptotic and anti-apoptotic and inflammation genes [15, 22, 63].
Mitochondria play a vital role in the cellular redox state regulation. ROS generation is also induces when mitochondrial ETC produces the electrochemical proton gradient, which leads to synthesis of ATP. It was stated that the main sources of mitochondrial ROS generation are the ubiquinone sites in Complexes I and III. The most oxygen is consumed by the mitochondrial ETC. Hence, it is a main source of ROS generation [64]. This study revealed that TR could induce changes on ETC in mitochondria and by this manner has an effect on opening the MPT pores and could induce MMP collapse and mitochondria swelling.
Moreover, intracellular calcium homeostasis is regulated by mitochondria. The MMP created by the ETC proton gradient is crucial to mitochondrial calcium uptake and efflux. MMP is changed by the mitochondrial dysfunction and subsequently diminishes its ability to implement calcium suppression and disrupts the homeostasis of calcium, which results in cell damage [64, 65]. Thus, mitochondria prepare the cellular energy, regulate cellular redox state and cellular calcium homeostasis and are necessary for cell survival. Our results obviously showed that liver mitochondrial function was disrupted by TR. It seems that TR may be able to alter the calcium homeostasis in mitochondria and affect the redox state and cause mitochondrial destruction. Hence, ROS overproduction by TR in this study induced mitochondrial dysfunction and caused mitochondrial damage possibly via stimulating and alterations of calcium homeostasis.
Previous studies have proposed two main mechanisms that are involved in TR-induced oxidative damage. The first is the effect of TR on dopaminergic system which leads to the enhancement of free radicals and the second is a decrease in the activity of the mitochondrial electron transport chain (ETC) of I, III and IV complexes with protein structure [12, 15]. In this study, the second mechanism can be considered. Because this study showed that increasing of TR concentrations induced ETC disorder and reduced the activity of Complex II.
The interaction between TR and the oxygen molecule leads to the generation of free radicals, such as superoxide, hydrogen peroxide, hydroxyl and peroxynitrite radicals. Therefore, these generated free radicals cause LPO, PC formation and reduction in the antioxidant capacity, such as GSH, induction of mitochondrial membrane damage and consequently cell death in various tissues [17, 43, 66]. Hence, it is supposed that TR could induce oxidative stress and that mitochondria are the main target of this process.
Application of isolated mitochondria in toxicological study has several advantages, such as control of the environmental conditions, monitoring of biological state and the elimination of the interfering factors [67].
Finally, according to our results, TR has oxidant effect on rat’s liver isolated mitochondria. Our findings revealed that TR, by increasing the free radicals in mitochondria, enhanced the oxidative damage via a dose-dependent manner. It can be concluded that oxidative effects of TR may be due to its change on mitochondrial ETC, activities of I, III and IV complexes, MPT pores disorders, inducing MMP collapse, mitochondria swelling and/or alterations of calcium homeostasis. However, further study is needed to understand which mechanism is most involved in TR-induced oxidative stress.
Conclusion
In conclusion, our study indicated that TR administration (from 100 to 600 μM) at the highest concentrations induced rat liver mitochondria oxidative damage and mitochondrial disruption via increasing of ROS, lipid and protein oxidation. TR showed pronounced effects on mitochondrial complex II activity, inducing of MPT pores disorders and increasing of MMP collapse and mitochondrial swelling.
Funding
Financial support for this work was provided by Mazandaran University of Medical Sciences, Sari, Iran, with reference number 6990.
Contributor Information
Leila Mohammadnejad, Department of Toxicology and Pharmacology, Faculty of Pharmacy, Mazandaran University of Medical Sciences, Sari 48157-33971, Iran.
Kambiz Soltaninejad, Department of Forensic Toxicology, Legal Medicine Research Center, Legal Medicine Organization, Tehran 48157-33971, Iran.
Mohammad Seyedabadi, Department of Toxicology and Pharmacology, Faculty of Pharmacy, Mazandaran University of Medical Sciences, Sari 48157-33971, Iran.
Seyed Khosro Ghasem Pouri, Department of Emergency Medicine, School of Medicine, Antimicrobial Resistance Research Center, Ghaem Shahr Razi Hospital, Mazandaran University of Medical Sciences, Sari 48157-33971, Iran.
Mohammad Shokrzadeh, Department of Toxicology and Pharmacology, Faculty of Pharmacy, Mazandaran University of Medical Sciences, Sari 48157-33971, Iran.
Hamidreza Mohammadi, Department of Toxicology and Pharmacology, Faculty of Pharmacy, Mazandaran University of Medical Sciences, Sari 48157-33971, Iran; Pharmaceutical Science Research Center, Hemoglobinopathy Institute, Mazandaran University of Medical Sciences, Sari 48157-33971, Iran.
Conflict of interest declaration
The authors declared no conflicts of interest.
References
- 1. Grond S, Sablotzki A. Clinical pharmacology of tramadol. Clin Pharmacokinet 2004;43:879–923. [DOI] [PubMed] [Google Scholar]
- 2. Gong L, Stamer UM, Tzvetkov MV et al. PharmGKB summary: tramadol pathway. Pharmacogenet Genomics 2014;24:374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Elmanama A, Abu Tayyem N, Esawwaf H et al. Tramadol-induced liver and kidney toxicity among abusers in Gaza Strip, Palestine. Jordan J Biol Sci 2015;8:133–7. [Google Scholar]
- 4. Mehrpour O, Sharifi M, Zamani N. Tramadol poisoning. Toxicology Studies Cells Drugs Environment 2015;101:101–26. [Google Scholar]
- 5. Nakhaee S, Hoyte C, Dart RC et al. A review on tramadol toxicity: mechanism of action, clinical presentation, and treatment. Forensic Toxicology 2021;30:1–18. [Google Scholar]
- 6. Arafa MH, Atteia HH. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6) are associated with long term tramadol treatment-induced oxidative damage and hepatotoxicity. Toxicol Appl Pharmacol 2018;346:37–44. [DOI] [PubMed] [Google Scholar]
- 7. Hussein SA, Ismail HK, Abdel Aal SA. Effect of tramadol drug on some biochemical and immunological parameters in albino male rats; evaluation of possible reversal following its withdrawal. Benha Veterinary Medical J 2017;33:418-29. [Google Scholar]
- 8. Ibrahim MA, Ibrahim HM, Mohamed AA et al. Vitamin E supplementation ameliorates the hepatotoxicity induced by tramadol: toxicological, histological and immunohistochemical study. Toxicol Mech Methods 2020;30:177-88. [DOI] [PubMed] [Google Scholar]
- 9. Sheweita SA, Almasmari AA, El-Banna SG. Tramadol-induced hepato- and nephrotoxicity in rats: role of curcumin and gallic acid as antioxidants. PLoS One 2018;13:e0202110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ali HA, Afifi M, Saber TM et al. Neurotoxic, hepatotoxic and nephrotoxic effects of tramadol administration in rats. J Mol Neurosci 2020;70:1934–42. [DOI] [PubMed] [Google Scholar]
- 11. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases, 2012. Tramadol. [Updated 2020 Nov 24]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK548235/. [Google Scholar]
- 12. Bameri B, Shaki F, Ahangar N et al. Evidence for the involvement of the dopaminergic system in seizure and oxidative damage induced by tramadol. Int J Toxicol 2018;37:164–70. [DOI] [PubMed] [Google Scholar]
- 13. Nazifi S, Tabrizi AS, Mohammadi S et al. The effect of tramadol and meloxicam, alone and in combination on oxidative stress status in dogs. Comp Clin Pathol 2019;28:1055–60. [Google Scholar]
- 14. Koohsari M, Ahangar N, Mohammadi E et al. Ameliorative effect of melatonin against reproductive toxicity of tramadol in rats via the regulation of oxidative stress, mitochondrial dysfunction, and apoptosis-related gene expression signaling pathway. Addiction Health 2020;12:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mohamed HM, Mahmoud AM. Chronic exposure to the opioid tramadol induces oxidative damage, inflammation and apoptosis, and alters cerebral monoamine neurotransmitters in rats. Biomed Pharmacother 2019;110:239–47. [DOI] [PubMed] [Google Scholar]
- 16. Ghoneim FM, Khalaf HA, Elsamanoudy AZ et al. Effect of chronic usage of tramadol on motor cerebral cortex and testicular tissues of adult male albino rats and the effect of its withdrawal: histological, immunohistochemical and biochemical study. Int J Clin Exp Pathol 2014;7:7323. [PMC free article] [PubMed] [Google Scholar]
- 17. Hussein SA, Abdel Aal SAL. Neurodegeneration and oxidative stress induced by tramadol administration in male rats: the effect of its withdrawal. Benha Vet Med J 2017;33:149–59. [Google Scholar]
- 18. Faria J, Barbosa J, Leal S et al. Effective analgesic doses of tramadol or tapentadol induce brain, lung and heart toxicity in Wistar rats. Toxicology 2017;385:38–47. [DOI] [PubMed] [Google Scholar]
- 19. Awadalla EA, Salah-Eldin A-E. Histopathological and molecular studies on tramadol mediated hepato-renal toxicity in rats. J Pharm Biol Sci 2015;10:90–102. [Google Scholar]
- 20. Elwy A, Tabl G. Effects of chronic usage of tramadol, acetaminophen and tramacet on some biochemical and immunological changes in male rats. J Drug Res Egypt 2014;35:63–71. [Google Scholar]
- 21. Ibrahim MA-L, Salah-Eldin A-E. Chronic addiction to tramadol and withdrawal effect on the spermatogenesis and testicular tissues in adult male albino rats. Pharmacology 2019;103:202–11. [DOI] [PubMed] [Google Scholar]
- 22. Sadek KM, Lebda MA, Abouzed TK et al. The molecular and biochemical insight view of lycopene in ameliorating tramadol-induced liver toxicity in a rat model: implication of oxidative stress, apoptosis, and MAPK signaling pathways. Environ Sci Pollut Res Int 2018;25:33119–30. [DOI] [PubMed] [Google Scholar]
- 23. Adikwu E, Bokolo B. Melatonin and N- acetylcysteine as remedies for tramadol-induced hepatotoxicity in albino rats. Adv Pharm Bull 2017;7:367-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Owoade A, Adetutu A, Olorunnisola O. Hematological and biochemical changes in blood, liver and kidney tissues under the effect of tramadol treatment. J Alcohol Drug Depend 2019;7:2. [Google Scholar]
- 25. Barbosa J, Faria J, Garcez F et al. Repeated administration of clinical doses of tramadol and tapentadol causes hepato-and nephrotoxic effects in Wistar rats. Pharmaceuticals 2020;13:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Khosrojerdi H, Alipour Talesh G, Danaei GH et al. Tramadol half life is dose dependent in overdose. Daru 2015;23:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Subedi M, Bajaj S, Kumar MS et al. An overview of tramadol and its usage in pain management and future perspective. Biomed Pharmacother 2019;111:443–51. [DOI] [PubMed] [Google Scholar]
- 28. Scott LJ, Perry CM. Tramadol. Drugs 2000;60:139–76. [DOI] [PubMed] [Google Scholar]
- 29. Stassinos GL, Gonzales L, Klein-Schwartz W. Characterizing the toxicity and dose-effect profile of tramadol ingestions in children. Pediatr Emerg Care 2019;35:117–20. [DOI] [PubMed] [Google Scholar]
- 30. Habibollahi P, Garjani A, Vahdati SS et al. Severe complications of tramadol overdose in Iran. Epidemiology Health 2019;41:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tashakori A, Afshari R. Tramadol overdose as a cause of serotonin syndrome: a case series. Clin Toxicol 2010;48:337–41. [DOI] [PubMed] [Google Scholar]
- 32. Doostmohammadi M, Rahimi H-R. ADME and toxicity considerations for tramadol: from basic research to clinical implications. Expert Opin Drug Metab Toxicol 2020;16:627–40. [DOI] [PubMed] [Google Scholar]
- 33. Reines SA, Goldmann B, Harnett M et al. Misuse of tramadol in the United States: an Analysis of the National Survey of Drug Use and health 2002-2017. Subst Abuse Res Treat 2020;14:1178221820930006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zabihi E, Hoseinzaadeh A, Emami M et al. Potential for Tramadol Abuse by Patients Visiting Pharmacies in Northern Iran. London, England: SAGE Publications Sage UK, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fawzi MM. Some medicolegal aspects concerning tramadol abuse: the new Middle East youth plague 2010. An Egyptian overview. Egypt J Forensic Sci 2011;1:99–102. [Google Scholar]
- 36. Clarot F, Goulle J, Vaz E et al. Fatal overdoses of tramadol: Is benzodiazepine a risk factor of lethality? Forensic Sci Int 2003;134:57–61. [DOI] [PubMed] [Google Scholar]
- 37. De Backer B, Renardy F, Denooz R et al. Quantification in postmortem blood and identification in urine of tramadol and its two main metabolites in two cases of lethal tramadol intoxication. J Anal Toxicol 2010;34:599–604. [DOI] [PubMed] [Google Scholar]
- 38. Larson SJ, Pestaner J, Prashar SK et al. Postmortem distribution of tapentadol and N-desmethyltapentadol. J Anal Toxicol 2012;36:440–3. [DOI] [PubMed] [Google Scholar]
- 39. Guo C, Sun L, Chen X et al. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen Res 2013;8:2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Krumschnabel G, Manzl C, Berger C et al. Oxidative stress, mitochondrial permeability transition, and cell death in Cu-exposed trout hepatocytes. Toxicol Appl Pharmacol 2005;209:62–73. [DOI] [PubMed] [Google Scholar]
- 41. Cui H, Kong Y, Zhang H. Oxidative stress, mitochondrial dysfunction, and aging. J Signal Transduct 2012;2012:646354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pourahmad J, Hosseini M-J. Application of isolated mitochondria in toxicological and clinical studies. Iran J Pharm Res 2012;11:703. [PMC free article] [PubMed] [Google Scholar]
- 43. Mohamed TM, Ghaffar HMA, El Husseiny RM. Effects of tramadol, clonazepam, and their combination on brain mitochondrial complexes. Toxicol Ind Health 2015;31:1325–33. [DOI] [PubMed] [Google Scholar]
- 44. Ashari S, Karami M, Shokrzadeh M et al. The implication of mitochondrial dysfunction and mitochondrial oxidative damage in di (2-ethylhexyl) phthalate induced nephrotoxicity in both in vivo and in vitro models. Toxicol Mech Methods 2020;30:1–11. [DOI] [PubMed] [Google Scholar]
- 45. Faria J, Barbosa J, Queirós O et al. Comparative study of the neurotoxicological effects of tramadol and tapentadol in SH-SY5Y cells. Toxicology 2016;359:1–10. [DOI] [PubMed] [Google Scholar]
- 46. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–54. [DOI] [PubMed] [Google Scholar]
- 47. Samadi M, Shaki F, Bameri B et al. Caffeine attenuates seizure and brain mitochondrial disruption induced by tramadol: the role of adenosinergic pathway. Drug Chem Toxicol 2019;44:1–7. [DOI] [PubMed] [Google Scholar]
- 48. Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J 2008;22:659-61. [DOI] [PubMed] [Google Scholar]
- 49. Price G, Patel DA. Drug Bioavailability. In: StatPearls. Treasure Island (FL): StatPearls Publishing, 2020. PMID: 32496732. [PubMed]
- 50. Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol 2020;21(7):363-83. [DOI] [PubMed] [Google Scholar]
- 51. Mitra S, Nguyen LN, Akter M et al. Impact of ROS generated by chemical, physical, and plasma techniques on cancer attenuation. Cancers 2019;11:1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mehdizadeh H, Pourahmad J, Taghizadeh G et al. Mitochondrial impairments contribute to spatial learning and memory dysfunction induced by chronic tramadol administration in rat: protective effect of physical exercise. Prog Neuro-Psychopharmacol Biol Psychiatry 2017;79:426–33. [DOI] [PubMed] [Google Scholar]
- 53. Abdel-Zaher AO, Abdel-Rahman MS, ELwasei FM. Protective effect of Nigella sativa oil against tramadol-induced tolerance and dependence in mice: role of nitric oxide and oxidative stress. Neurotoxicology 2011;32:725–33. [DOI] [PubMed] [Google Scholar]
- 54. Atici S, Cinel I, Cinel L et al. Liver and kidney toxicity in chronic use of opioids: an experimental long term treatment model. J Biosci 2005;30:245–52. [DOI] [PubMed] [Google Scholar]
- 55. Zhang YT, Zheng QS, Pan J et al. Oxidative damage of biomolecules in mouse liver induced by morphine and protected by antioxidants. Basic Clin Pharmacol Toxicol 2004;95:53–8. [DOI] [PubMed] [Google Scholar]
- 56. Ahmed MA, Kurkar A. Effects of opioid (tramadol) treatment on testicular functions in adult male rats: the role of nitric oxide and oxidative stress. Clin Exp Pharmacol Physiol 2014;41:317–23. [DOI] [PubMed] [Google Scholar]
- 57. Popovic M, Janicijevic-Hudomal S, Kaurinovic B et al. Antioxidant effects of some drugs on immobilization stress combined with cold restraint stress. Molecules 2009;14:4505–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schenkel LC, Bakovic M. Formation and regulation of mitochondrial membranes. Int J Cell Biol 2014;2014:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Taghizadeh G, Pourahmad J, Mehdizadeh H et al. Protective effects of physical exercise on MDMA-induced cognitive and mitochondrial impairment. Free Radic Biol Med 2016;99:11–9. [DOI] [PubMed] [Google Scholar]
- 60. Krügel K, Wurm A, Pannicke T et al. Involvement of oxidative stress and mitochondrial dysfunction in the osmotic swelling of retinal glial cells from diabetic rats. Exp Eye Res 2011;92:87–93. [DOI] [PubMed] [Google Scholar]
- 61. Elyasi B, Zhaleh M, Amini K et al. Chemical characterization and suppressor potent of Juglans regia essential oil on tramadol-induced cell death. J Essent Oil-Bear Plants 2020;23:849–61. [Google Scholar]
- 62. Ragab IK, Mohamed HZ. Histological changes of the adult albino rats entorhinal cortex under the effect of tramadol administration: histological and morphometric study. Alexandria J Med 2017;53:123–33. [Google Scholar]
- 63. Diesen DL, Kuo PC. Nitric oxide and redox regulation in the liver: part I. General considerations and redox biology in hepatitis. J Surg Res 2010;162:95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang J-F. Defects of mitochondrial electron transport chain in bipolar disorder: implications for mood-stabilizing treatment. Can J Psychiatr 2007;52:753–62. [DOI] [PubMed] [Google Scholar]
- 65. Chinopoulos C, Adam-Vizi V. Calcium, mitochondria and oxidative stress in neuronal pathology: novel aspects of an enduring theme. FEBS J 2006;273:433–50. [DOI] [PubMed] [Google Scholar]
- 66. Lemarie A, Grimm S. Mutations in the heme b-binding residue of SDHC inhibit assembly of respiratory chain complex II in mammalian cells. Mitochondrion 2009;9:254–60. [DOI] [PubMed] [Google Scholar]
- 67. Vergun O, Votyakova TV, Reynolds IJ. Spontaneous changes in mitochondrial membrane potential in single isolated brain mitochondria. Biophys J 2003;85:3358–66. [DOI] [PMC free article] [PubMed] [Google Scholar]