

Summary
Presynaptic homeostatic plasticity (PHP) stabilizes synaptic transmission by counteracting impaired neurotransmitter receptor function through neurotransmitter release potentiation. PHP is thought to be triggered by impaired receptor function and to involve a stereotypic signaling pathway. However, here we demonstrate that different receptor perturbations that similarly reduce synaptic transmission result in different responses at the Drosophila neuromuscular junction. While receptor inhibition by the glutamate receptor (GluR) antagonist γ-D-glutamylglycine (γDGG) is not compensated by PHP, the GluR inhibitors Philanthotoxin-433 (PhTx) and Gyki-53655 (Gyki) induce compensatory PHP. Intriguingly, PHP triggered by PhTx and Gyki involve separable signaling pathways, including inhibition of distinct GluR subtypes, differential modulation of the active-zone scaffold Bruchpilot, and short-term plasticity. Moreover, while PHP upon Gyki treatment does not require genes promoting PhTx-induced PHP, it involves presynaptic protein kinase D. Thus, synapses not only respond differentially to similar activity impairments, but achieve homeostatic compensation via distinct mechanisms, highlighting the diversity of homeostatic signaling.
Keywords: synaptic transmission, neurotransmitter release, glutamate receptors, homeostatic plasticity, Protein Kinase D, Drosophila neuromuscular junction
Graphical abstract
Highlights
-
•
Presynaptic homeostatic plasticity (PHP) is induced and reversed within seconds
-
•
Glutamate receptor inhibition per se is not sufficient for PHP induction
-
•
Different glutamate receptor antagonists trigger distinct molecular PHP pathways
Synapses counteract neurotransmitter receptor perturbations by homeostatic neurotransmitter release potentiation. Homeostatic compensation is thought to be triggered by impaired receptor function, and to involve a stereotypic signaling pathway. Nair et al. here show that different glutamate receptor antagonists trigger distinct homeostatic signaling pathways at the Drosophila neuromuscular junction.
Introduction
A variety of homeostatic signaling systems stabilize neural function by counteracting diverse perturbations (Delvendahl et al., 2019; Frank et al., 2006; Ibata et al., 2008; Keck et al., 2013; Petersen et al., 1997; Teichert et al., 2017; Turrigiano et al., 1998). Chemical synapses compensate for neural activity perturbations through homeostatic regulation of neurotransmitter release (Frank et al., 2006; Petersen et al., 1997) or neurotransmitter receptors (Turrigiano et al., 1998). There are also emerging links between homeostatic synaptic plasticity and neural disease, such as autism spectrum disorders, schizophrenia, or amyotrophic lateral sclerosis (Genç et al., 2020; Perry et al., 2017; Tatavarty et al., 2020).
Presynaptic homeostatic plasticity (PHP) is a major form of homeostatic plasticity that is characterized by homeostatic upregulation of presynaptic release in response to reduced postsynaptic neurotransmitter receptor activity (Delvendahl and Müller, 2019). PHP has been observed at diverse synapses in different species (Delvendahl et al., 2019; Frank et al., 2006; Wang et al., 2018), implying evolutionary conservation. Experimentally, PHP is induced by pharmacological or genetic neurotransmitter receptor impairment (Frank et al., 2006; Petersen et al., 1997). Correspondingly, reduced postsynaptic excitation caused by receptor impairment, as quantified by a reduction in quantal size (q), is thought to trigger homeostatic signaling. In this regard, Ca2+ flux through postsynaptic receptors has been hypothesized to be a major synaptic variable sensed by the signaling system promoting PHP (Frank et al., 2006; Ouanounou et al., 2016; Wang et al., 2016). In agreement, postsynaptic signaling by Ca2+/calmodulin-dependent protein kinase (CaMKII) and the Ca2+-binding protein Peflin has been implicated in PHP at the Drosophila neuromuscular junction (NMJ) (Haghighi et al., 2003; Kikuma et al., 2019; Li et al., 2018). However, PHP can be induced in the absence of extracellular Ca2+ at the Drosophila NMJ (Goel et al., 2017). Moreover, there is some evidence that ion flux through nicotinic acetylcholine receptors is dispensable for PHP at the mouse NMJ (Wang et al., 2018). It is thus currently unclear whether PHP is triggered by reduced Ca2+ influx or synaptic function resulting from receptor inhibition.
The molecular mechanisms underlying PHP are best understood at the Drosophila NMJ. At this synapse, PHP can be rapidly expressed within minutes after pharmacological glutamate receptor (GluR) inhibition by the GluR antagonist Philanthotoxin-433 (PhTx) (Frank et al., 2006). Similarly, PHP is induced and chronically sustained upon genetic loss of the GluRIIA subunit (Petersen et al., 1997). Electrophysiology-based genetic screens at the Drosophila NMJ have implicated several genes in PHP, including the schizophrenia-susceptibility gene dysbindin (dysb) (Dickman and Davis, 2009), or the gene encoding the active zone protein rab3-interacting molecule (RIM) (Müller et al., 2012). Many of the identified genes are required for both rapid and chronic PHP expression upon pharmacological or genetic receptor impairment, implying that a stereotypic PHP signaling pathway is triggered by acute and chronic receptor perturbation. However, recent work revealed that some genes that promote chronic PHP expression are dispensable for acute PHP expression upon pharmacological receptor impairment (Böhme et al., 2019; James et al., 2019). It was concluded that different PHP phases (rapid versus chronic) involve different molecular mechanisms. An alternative yet less explored hypothesis is that PHP is promoted by distinct molecular pathways depending on how the receptors have been perturbed, e.g., genetic ablation versus pharmacological inhibition, or receptor inhibition by different antagonists.
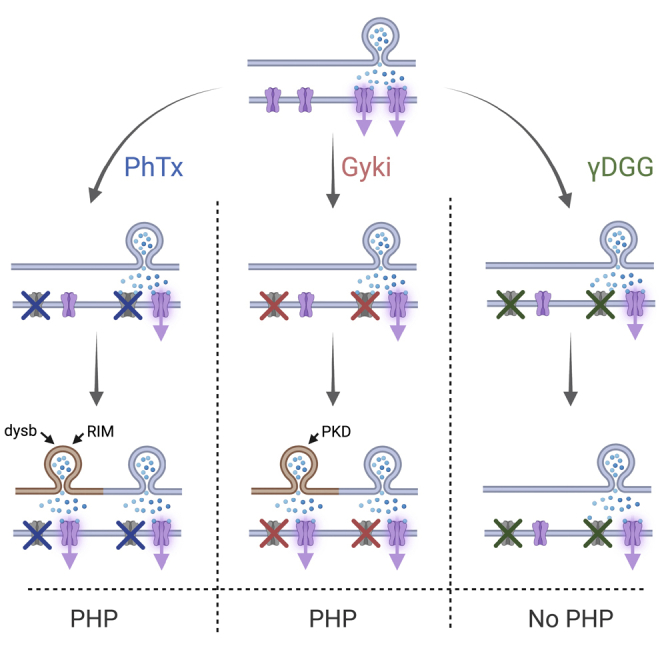
Here, we tested whether perturbing receptors with different antagonists could activate distinct PHP pathways at the Drosophila NMJ. We observed that two GluR antagonists, PhTx and Gyki-53655 (Gyki), induced PHP, whereas the GluR antagonist γ-D-glutamylglycine (γDGG) did not. Interestingly, PHP induced by PhTx and Gyki exhibited differences in the regulation of active-zone structure, short-term plasticity, and the underlying signaling molecules. While PhTx-induced PHP relied on the known PHP genes dysb and RIM, Gyki-induced PHP was promoted by presynaptic protein kinase D (PKD). Moreover, Gyki triggered PHP in six mutants previously shown to disrupt PHP upon PhTx treatment. Together, our data suggest that distinct molecular mechanisms mediate PHP in response to GluR inhibition by different antagonists, and that GluR inhibition per se is not sufficient for PHP expression
Results
GluR perturbation does not induce PHP per se
To examine whether different receptor perturbations induce distinct PHP pathways, we investigated rapid PHP at the Drosophila larval NMJ after GluR perturbation using three different antagonists: PhTx, γDGG, and Gyki. Although the mechanisms of antagonism of these three inhibitors have not been examined in Drosophila, all of them inhibit Drosophila GluRs: while PhTx and γDGG were previously shown to block GluRs at the Drosophila NMJ (Frank et al., 2006; Miśkiewicz et al., 2011; Pawlu et al., 2004), Gyki had not been tested on Drosophila GluRs. We observed that Gyki treatment reduced the amplitude of miniature excitatory postsynaptic potentials (mEPSPs) in a dose-dependent manner (Figure S1B). Drosophila GluRs contain several conserved Gyki interaction amino acids identified for rat GluA2 (Figure S1A) (Yelshanskaya et al., 2016). Together, these results are consistent with the idea that Gyki blocks Drosophila GluRs.
We next assayed PHP using either PhTx, γDGG, or Gyki (Figure S1C). PhTx reduced the median mEPSP amplitude by ∼40% (Figure 1A), indicating robust postsynaptic receptor inhibition. By contrast, there was no significant reduction in the amplitude of action potential (AP)-evoked excitatory postsynaptic potentials (EPSPs) upon PhTx treatment (Figures 1A and S1D). The disproportionate decrease in mEPSP and EPSP amplitude translates into a significant increase in quantal content (QC = EPSP/mEPSP) (Figure 1A), suggesting enhanced neurotransmitter release (Frank et al., 2006). The quantal content value of the majority of PhTx-treated NMJs falls within the boundary of the expected quantal content range to restore EPSP amplitudes within ±20% of the median amplitude of untreated EPSPs (Figure 1B). These data suggest that PhTx-dependent GluR inhibition induces PHP, in line with earlier work (Delvendahl and Müller, 2019).
Figure 1.

Different glutamate receptor perturbations produce different presynaptic homeostatic plasticity (PHP) responses
(A) Representative traces (mEPSPs and EPSPs), mEPSP amplitudes, EPSP amplitudes, and quantal content (QC) from saline (Ctrl)- or PhTx-treated NMJs. n = 25 (Ctrl) versus 23 (PhTx).
(B) QC and median mEPSP amplitude of individual Ctrl- and PhTx-treated cells along with group median and median absolute deviation as error bars.
(C) Representative traces (mEPSPs and EPSPs), mEPSP amplitudes, EPSP amplitudes, and QC from Ctrl- or γDGG-treated NMJs. n = 22 (Ctrl) versus 22 (γDGG).
(D) QC and median mEPSP amplitude of individual Ctrl- and γDGG-treated cells along with group median and median absolute deviation as error bars.
(E) Representative traces (mEPSPs and EPSPs), mEPSP amplitudes, EPSP amplitudes, and QC from Ctrl- or Gyki-treated NMJs. n = 23 (Ctrl) versus 22 (Gyki).
(F) QC and median mEPSP amplitude of individual Ctrl- and Gyki-treated cells along with group median and median absolute deviation as error bars. The solid and dashed lines in (B), (D), and (F) represent the expected QC to restore EPSP amplitudes to the untreated median EPSP amplitude (±20%). Extracellular calcium concentration ([Ca2+]e) = 0.3 mM. The whiskers of the box plots extend to either 1.5 times the inter-quartile range or to the farthest data point, whichever is closest to the respective quartile. Individual data points are shown as gray dots. p, p value; r, effect size; ns: p ≥ 0.05; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
γDGG also reduced the median mEPSP amplitude by ∼40% (Figure 1C). Unlike PhTx, however, EPSP amplitudes dropped proportional to mEPSP amplitudes upon γDGG treatment (Figures 1C and S1E). Consequently, there was no increase in quantal content (Figure 1C), and the quantal content value of the majority of γDGG-treated cells did not reach the expected range to restore EPSPs to baseline control amplitudes (Figure 1D). These observations indicate that the γDGG-induced decrease in mEPSP amplitude is not compensated by PHP.
Treating NMJs with Gyki also significantly decreased mEPSP amplitudes, but not EPSP amplitudes, translating into a significant increase in quantal content (Figures 1E and S1F). The quantal content value of the majority of Gyki-treated NMJs was close to the expected quantal content to restore baseline EPSPs (Figure 1F), implying PHP expression upon Gyki treatment. Since PhTx, γDGG, and Gyki reduced mEPSP amplitude by a similar magnitude (Figures 1A, 1C, and 1E), but only PhTx and Gyki resulted in a homeostatic increase in quantal content, these results indicate that GluR inhibition per se is not sufficient to produce PHP.
Rapid induction and reversal of Gyki-induced PHP
Given that γDGG application did not result in PHP, we focused the rest of our analysis on Gyki and PhTx. Since Gyki has not been used for PHP induction at the Drosophila NMJ, we characterized PHP after Gyki treatment.
First, we studied the dynamics of PHP expression in response to Gyki treatment. PhTx induces PHP within 10 min after antagonist treatment (Frank et al., 2006). However, little is known about the PHP time course at the Drosophila NMJ, because PhTx blocks GluRs in an activity-dependent fashion (Frank et al., 2006). Moreover, PhTx only induces PHP when applied to “semi-intact,” i.e., not fully dissectedlarval preparations, which are less amenable to electrophysiological recordings, thereby complicating the analysis of PHP dynamics (Frank et al., 2006). In contrast to PhTx, we revealed robust PHP expression upon Gyki treatment of fully dissected preparations (Figure S2A), allowing to investigate PHP dynamics. To probe the dynamics of PHP induction, we quantified mEPSP amplitude, EPSP amplitude, and quantal content before and after Gyki application every 35 s in the same preparation (Figure 2A). As expected, mEPSP amplitudes decreased over time after Gyki application (Figures 2A, 2B, and S2B). However, EPSP amplitudes remained largely unchanged (Figures 2A, 2B, and S2B). Consequently, quantal content increased without a measurable delay with regard to mEPSP amplitude reduction (Figures 2A, 2B, and S2B). Considering the temporal resolution of 35 s, this implies that EPSP amplitudes are restored continuously as the receptors are being inhibited, indicating a low latency of PHP signaling.
Figure 2.

Rapid induction and reversal of Gyki-induced PHP
(A) Left: Normalized median mEPSP amplitude, EPSP amplitude, and QC of a representative cell as a function of time before and after Gyki treatment. Values are normalized to respective mean baseline values before Gyki application. Right: Representative mEPSP and EPSP traces at specified time points. [Ca2+]e = 0.3 mM.
(B) Mean normalized mEPSP amplitude, EPSP amplitude, and QC as a function of time before and after Gyki treatment. The shaded region represents SEM around the mean. Values for individual cells are normalized to the respective mean baseline values before Gyki application. n = 15.
(C) Representative traces (mEPSPs and EPSPs), mEPSP amplitudes, EPSP amplitudes, and QC of NMJs after Ctrl or Gyki washout. n = 23 (Ctrl wash) versus 22 (Gyki wash). [Ca2+]e = 0.3 mM.
(D) Left: Normalized median mEPSP amplitude, EPSP amplitude, and QC of a representative cell as a function of time before and after Gyki wash. Values are normalized to respective mean baseline values before Gyki wash. Right: Representative mEPSP and EPSP traces at specified time points. [Ca2+]e = 0.3 mM.
(E) Mean normalized mEPSP amplitude, EPSP amplitude, and QC as a function of time before and after Gyki wash. The shaded region represents SEM. Values for individual cells are normalized to the respective mean baseline values before Gyki wash. n = 5.
(F) Representative EPSC traces from a Ctrl- and Gyki-treated NMJ measured at different [Ca2+]e.
(G) EPSC amplitude at different [Ca2+]e for Ctrl- and Gyki-treated NMJs. n = 16, 16, 51, 27 (Ctrl) versus 14, 14, 53, 23 (Gyki); Hill’s coefficient = 3.6 (Ctrl) versus 3.3 (Gyki). Error bars represent SEM.
(H) Inverse of squared coefficient of variation (CV−2) of EPSC amplitudes recorded at [Ca2+]e = 0.3 mM and estimated quantal size (q) for Ctrl- and Gyki-treated NMJs. n = 10 (Ctrl) versus 8 (Gyki).
(I) EPSC amplitude spread as a function of [Ca2+]e and relation between EPSC variance and mean for a sample variance-mean analysis, along with estimated q and functional release sites (N) for Ctrl- and Gyki-treated NMJs. n = 10 (Ctrl) versus 8 (Gyki). The error bars represent standard error of the estimator. The whiskers of the box plots extend to either 1.5 times the inter-quartile range or to the farthest data point, whichever is closest to the respective quartile. Individual data points are shown as gray dots. p, p value; r, effect size; ns: p ≥ 0.05; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
We then tested whether Gyki-induced PHP can be reversed upon antagonist washout. NMJs were either incubated with Gyki or saline for 15 min, followed by a wash and saline incubation for 15 min. mEPSP amplitudes were comparable between Gyki-treated and saline-treated NMJs (Figure 2C), demonstrating that Gyki reversibly antagonizes Drosophila GluRs. Both groups had comparable EPSP amplitudes and quantal content (Figure 2C), indicating that PHP reversed upon Gyki removal within 15 min. We next explored PHP reversibility dynamics at NMJs that were preincubated with Gyki for 15 min. mEPSP, EPSP amplitudes, and quantal content were quantified every 35 s before and during Gyki washout (Figure 2D). As expected, mEPSP amplitude increased over time after Gyki washout (Figures 2D, 2E, and S2C). While the evolution of EPSP amplitude over time was relatively variable between cells (Figure S2C), it remained largely unchanged on average (Figure 2E). Correspondingly, quantal content decreased without any delay compared with the increase in mEPSP amplitude (Figures 2E and S2C), indicating that quantal content was continuously updated as q increased during Gyki washout. Together with the dynamics of PHP induction, these results indicate that PHP at the Drosophila NMJ is a highly dynamic and a low-latency process.
Spontaneous and AP-evoked release may involve different subsets of active zones (Melom et al., 2013; Peled et al., 2014). The observation of similar EPSP amplitudes in the absence and presence of Gyki could, in principle, be due to a lack of receptor inhibition at synapses that are predominantly activated by AP-evoked release. We therefore next aimed at providing evidence that Gyki also acts on GluRs that are activated by evoked release and that Gyki indeed potentiates presynaptic release. To this end, we analyzed the amplitudes of AP-evoked excitatory postsynaptic currents (EPSCs) using two-electrode voltage clamp (TEVC) at different extracellular Ca2+ concentrations (Figure 2F). EPSC amplitudes recorded in the absence and presence of Gyki treatment were similar at different Ca2+ concentrations (Figures 2F and 2G), suggesting that Gyki does not change the Ca2+ sensitivity and Ca2+ cooperativity of release, similar to PhTx (Frank et al., 2006). At Gyki-treated NMJs, we observed a significantly increased squared inverse coefficient of variation (CV−2) of EPSC amplitudes recorded under conditions of low release probability (pr) at 0.3 mM Ca2+ (Figure 2H), indicating an increase in quantal content (McLachlan, 1978). Furthermore, q, estimated as the ratio between mean EPSC amplitude and CV−2, was significantly lower for Gyki-treated NMJs, implying postsynaptic receptor inhibition (Figure 2H). Next, we performed EPSC amplitude variance-mean analysis (Clements and Silver, 2000; Saviane and Silver, 2007) to independently estimate the quantal parameters q and functional release site number (N) (Figure 2I). Upon Gyki treatment, there was a significant decrease in q and a significant increase N, suggesting Gyki-dependent GluR inhibition and presynaptic release potentiation (Figure 2I). Together, these results independently verify that Gyki treatment impairs GluR function and enhances neurotransmitter release, without major changes in the Ca2+ sensitivity or Ca2+ cooperativity of release.
Differential GluR subtype specificity of Gyki and PhTx
Drosophila NMJs harbor two types of GluR complexes, either containing a GluRIIA or a GluRIIB subunit (Marrus et al., 2004). We next tested which receptor complexes are inhibited by Gyki and compared them to PhTx, which is considered to predominantly inhibit GluRIIA-containing receptor complexes (Frank et al., 2006). We recorded mEPSPs from NMJs mutant for either GluRIIA (GluRIIASP16; Petersen et al., 1997) or GluRIIB (GluRIIBSP5; Muttathukunnel et al., 2021) in the presence and absence of either inhibitor. Whereas mEPSP amplitudes recorded from GluRIIA mutant NMJs were largely unaffected by PhTx (Figure 3A), they significantly decreased in the presence of Gyki (Figure 3A). mEPSP amplitudes were strongly reduced by both PhTx and Gyki application in GluRIIBSP5 mutants (Figure 3B). Although miniature synaptic transmission cannot be assessed in GluRIIA-GluRIIB double mutants due to lethality (Marrus et al., 2004; Qin et al., 2005), these results suggest that Gyki inhibits both GluRIIA- and GluRIIB-containing receptor complexes, whereas PhTx predominantly acts on GluRIIA-containing receptors, as previously reported (Frank et al., 2006). Thus, we conclude that Gyki and PhTx may differentially inhibit Drosophila GluR types. The differential action on GluR types presents a possibility that Gyki and PhTx may induce PHP via distinct pathways.
Figure 3.

PhTx and Gyki exhibit differential effects on GluRIIA- and GluRIIB-containing receptors
(A) Representative mEPSP traces and mEPSP amplitude quantification for PhTx-untreated or -treated and Gyki-untreated or -treated GluRIIASP16 mutant NMJs. n = 12 (−PhTx) versus 11 (+PhTx); n = 14 (−Gyki) versus 14 (+Gyki).
(B) Representative mEPSP traces and mEPSP amplitude quantification for PhTx-untreated or -treated and Gyki-untreated or -treated GluRIIBSP5 mutant NMJs. n = 13 (−PhTx) versus 10 (+PhTx); n = 14 (−Gyki) versus 12 (+Gyki). The whiskers of the box plots extend to either 1.5 times the inter-quartile range or to the farthest data point, whichever is closest to the respective quartile. Individual data points are shown as gray dots. p, p value; r, effect size; ns: p ≥ 0.05; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Gyki and PhTx increase RRP size with different effects on short-term plasticity
PhTx induces PHP by increasing readily releasable pool (RRP) size without apparent effects on short-term dynamics (Ortega et al., 2018; Weyhersmüller et al., 2011). We therefore next carried out a comparative analysis of these synaptic parameters during PhTx- and Gyki-induced PHP using TEVC (1.0 mM extracellular Ca2+). As expected, both PhTx and Gyki strongly reduced miniature (m)EPSC amplitudes (Figures 4A and 4B [left]), while EPSC amplitudes remained largely unchanged (Figures 4A and 4B [middle]), resulting in an increase in quantal content (Figure 4B, right). Moreover, PhTx and Gyki significantly accelerated mEPSC decay kinetics (Figure 4C, left) and EPSC decay kinetics (Figure 4C, middle). Interestingly, while Gyki treatment similarly accelerated mEPSC and EPSC decay kinetics (Figure 4C, right), the PhTx-induced acceleration of mEPSC kinetics was more pronounced than the one of EPSC kinetics (Figure 4C, right). These data indicate that PhTx, but not Gyki, may potentiate neurotransmitter release during the EPSC decay phase or that the two antagonists differentially affect GluR properties during evoked transmission.
Figure 4.

Gyki and PhTx increase RRP size, but with differences in short-term plasticity
(A–C) Representative traces (mEPSCs and EPSCs) (A); mEPSP amplitudes, EPSC amplitudes, and QC (B); and mEPSP decay t-half (t50), EPSP t50, and ratio of EPSC t50 and mEPSP t50 (C) for PhTx-untreated or -treated and Gyki-untreated or -treated NMJs.
(D and E) Representative EPSC trains (60 stimuli at 60 Hz frequency), mEPSC, and linear fitting of last 10 cumulative EPSCs (D) and first two EPSCs of the train (E) from PhTx-untreated or -treated and Gyki-untreated or-treated NMJs.
(F and G) RRP size (RRPCum. EPSC) (F) and release probability (ptrain) (G) estimated from cumulative EPSCs of PhTx-untreated or -treated and Gyki-untreated or -treated NMJs.
(H and I) Depression time constant of normalized EPSC amplitudes (τw) (H) and paired-pulse ratio (EPSC2/EPSC1) (I) for PhTx-untreated or -treated and Gyki-untreated or -treated NMJs. For all figure panels, n = 16 (−PhTx) versus 15 (+PhTx); n = 15 (−Gyki) versus 18 (+Gyki). [Ca2+]e = 1.0 mM. The whiskers of the box plots extend to either 1.5 times the inter-quartile range or to the farthest data point, whichever is closest to the respective quartile. Individual data points are shown as gray dots. p, p value; r, effect size; ns: p ≥ 0.05; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
We next probed RRP-size modulation during PHP induction by Gyki and compared it to PhTx by back-extrapolating the steady state of cumulative EPSC amplitudes during repetitive stimulation (Figure 4D) (Schneggenburger et al., 1999). RRP size (RRPCum. EPSC) significantly increased after Gyki and PhTx treatment (Figure 4F). A similar increase in RRP size upon Gyki and PhTx application was obtained with the Elmquist-Quastel method (RRPEQ; Figure S3A) (Elmqvist and Quastel, 1965). Hence, Gyki, similar to PhTx, potentiates RRP size.
Intriguingly, pr, as assessed by dividing the first EPSC amplitude of a train by the cumulative EPSC amplitude (ptrain; Delvendahl et al., 2019) was significantly reduced after Gyki, but not PhTx, treatment (Figures 4G and S3B), indicating that Gyki- and PhTx-induced PHP may differentially affect pr. Consistently, we revealed a significantly slower time course (τw) of synaptic short-term depression during train stimulation upon Gyki, but not PhTx, treatment (Figures 4H and S3C), indicative of a pr decrease after Gyki application. Moreover, Gyki increased the paired-pulse ratio (PPR) of EPSC amplitudes (EPSC2/EPSC1; 60 Hz; Figure 4E) compared with untreated controls (Figures 4E and 4I). The Gyki-induced PPR increase (by 36%) was more pronounced than the PPR change upon PhTx treatment (17%; Figures 4E, right, and 4I). Together, these results show that both Gyki and PhTx treatment increase RRP size with different effects on pr and short-term plasticity, indicating the Gyki-induced PHP may involve different mechanisms compared to PhTx-induced PHP.
PhTx, but not Gyki, upregulates Brp abundance
PhTx application at the Drosophila NMJ results in an increase in immunofluorescence intensity of several active-zone proteins, including the scaffolding protein Bruchpilot (Brp), suggesting increased Brp abundance and/or reorganization (Böhme et al., 2019; Mrestani et al., 2021; Gratz et al., 2019; Weyhersmüller et al., 2011). We therefore examined whether Gyki-induced PHP also involves Brp modulation. After PhTx treatment, we detected a significant increase in Brp-fluorescence intensity (Figure 5B), consistent with previous reports (Böhme et al., 2019; Weyhersmüller et al., 2011). On the other hand, we did not observe an increase in Brp-fluorescence intensity after Gyki treatment (Figure 5B). Hence, although both PhTx and Gyki inhibit GluRs and induce PHP, only PhTx leads to an increase in Brp abundance, further supporting the idea that different receptor perturbations may trigger different downstream pathways.
Figure 5.

PhTx and Gyki exhibit differences in Brp regulation
(A) Representative confocal images (maximum intensity projection) of anti-Brp-stained NMJs, treated with Ctrl, PhTx, or Gyki. Scale bar, 1 μm.
(B) Quantified mean intensities for Ctrl-, PhTx-, or Gyki-treated NMJs. Number of NMJs, n = 15 (Ctrl), 10 (PhTx), and 15 (Gyki). Displayed statistical significance level obtained from Dunn’s post hoc comparison with Ctrl. Multi-comparison p value is ∼0.003. The whiskers of the box plots extend to either 1.5 times the inter-quartile range or to the farthest data point, whichever is closest to the respective quartile. Individual data points are shown as gray dots. ns: p ≥ 0.05; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Gyki induces PHP at mutant NMJs with impaired PhTx-induced PHP
Electrophysiology-based genetic screens in Drosophila have identified several genes involved in PhTx-induced PHP (Delvendahl and Müller, 2019). Assaying the roles of these genes on Gyki-induced PHP could reveal whether PHP induced by Gyki and PhTx involve different molecular mechanisms.
We first tested PHP in a rim null mutant, which was shown to have disrupted PhTx-induced PHP (rimΔ103) (Müller et al., 2012). PhTx application significantly reduced mEPSP and EPSP amplitudes at rimΔ103 mutant NMJs (Figures 6A and 6B). As the median mEPSP and EPSP amplitudes were reduced by a similar fraction upon PhTx treatment in rimΔ103 mutants (Figure 6C), there was no significant increase in quantal content (Figures 6B, right, and 6D, left), suggesting PHP impairment. Although Gyki treatment significantly decreased mEPSP and EPSP amplitudes in rimΔ103 (Figures 6A and 6B), the relative decrease in mEPSP amplitude was more pronounced than the relative EPSP amplitude reduction (Figure 6C). Correspondingly, Gyki significantly enhanced quantal content in rimΔ103 mutants (Figures 6B, right, and 6D, right), with the majority of Gyki-treated NMJs reaching the quantal content range required to restore the median baseline EPSP amplitude (Figure 6D, right). Thus, while PhTx-induced homeostatic quantal content potentiation requires rim (Müller et al., 2012), Gyki enhances quantal content in rim mutants, suggesting separable molecular PHP pathways.
Figure 6.

Gyki induces PHP at mutant NMJs with impaired PhTx-induced PHP
(A and B) Representative traces (mEPSPs and EPSPs) (A) and mEPSP amplitudes, EPSP amplitudes, and QC (B) for PhTx-untreated or -treated and Gyki-untreated or -treated rim mutant NMJs.
(C) Comparison of median mEPSP amplitude (open circles with solid line) and EPSP amplitude (closed circles with dashed line) without or with either treatment.
(D) QC and mEPSP amplitude of individual Ctrl and treated cells along with the median and median absolute deviation as error bars. n = 29 (−PhTx) versus 28 (+PhTx); n = 30 (−Gyki) versus 33 (+Gyki).
(E and F) Representative traces (mEPSPs and EPSPs) (E) and mEPSP amplitudes, EPSP amplitudes, and QC (F) for PhTx-untreated or -treated and Gyki-untreated or treated dysb mutant NMJs.
(G) Comparison of median mEPSP amplitude (open circles with solid line) and EPSP amplitude (closed circles with dashed line) without or with either treatment.
(H) QC and mEPSP amplitude of individual Ctrl and treated cells along with the median and median absolute deviation as error bars. n = 26 (−PhTx) versus 24 (+PhTx); n = 28 (−Gyki) versus 24 (+Gyki). The solid and dashed lines in (D) and (H) represent the expected QC to restore EPSP amplitudes to the untreated median EPSP amplitude (±20%). [Ca2+]e = 0.4 mM. The whiskers of the box plots extend to either 1.5 times the inter-quartile range or to the farthest data point, whichever is closest to the respective quartile. Individual data points are shown as gray dots. p, p value; r, effect size; ns: p ≥ 0.05; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Next, we probed PHP after loss of dysb, another gene implicated in PhTx-induced PHP (Dickman and Davis, 2009; Wentzel et al., 2018). Both PhTx and Gyki treatment significantly reduced mEPSP amplitudes in dysb1 mutants (Figures 6E and 6F). EPSP amplitudes were significantly decreased upon PhTx application (Figure 6F), to a similar extent as mEPSP amplitudes (Figure 6G). On the other hand, EPSP amplitudes were largely unchanged after Gyki treatment (Figures 6F and 6G). Consequently, quantal content was unchanged after PhTx application, but significantly increased after Gyki treatment (Figures 6F and 6H), thereby restoring EPSP amplitudes to baseline levels (Figure 6H). These data provide genetic evidence that dysb is required for PhTx-induced PHP, but dispensable for Gyki-induced PHP.
In addition, we assayed Gyki-induced PHP in four additional mutants that were previously shown to disrupt PhTx-induced PHP (Figure S4). We revealed a significant increase in quantal content upon Gyki application in previously published mutant lines of kainate-type ionotropic glutamate receptor subunit 1D (Kiragasi et al., 2017), pickpocket11 (Younger et al., 2013), Drosophila multiplexin (dmp) (Wang et al., 2014), and insomniac (Kikuma et al., 2019) (Figures S4A–S4D), indicating intact Gyki-induced PHP in all four cases. Thus, six independent experiments provide evidence that PhTx- and Gyki-induced PHP involve distinct molecular mechanisms.
Presynaptic PKD promotes Gyki-induced PHP, but not PhTx-induced, PHP
We next carried out a small electrophysiology-based genetic screen to identify molecular players involved in Gyki-induced PHP (Figure 7A). To this end, we assayed Gyki-induced PHP after genetic perturbation (presynaptic/postsynaptic RNAi expression or validated mutants) of genes encoding for second messenger signaling elements and previously characterized PHP-related genes. While most transgenic lines displayed a significant increase in quantal content upon Gyki application that restored EPSP amplitudes toward baseline levels (within 20% of the median baseline EPSP; Figure 7A), Gyki treatment did not enhance quantal content in 8 transgenic lines, which represent candidate PHP genes. One of the identified genotypes was an RNAi targeting Drosophila Protein Kinase D (PKD) presynaptically (D42-Gal4>UAS-PKDRNAi; Figure 7A, red circle), a gene not studied in the context of synaptic transmission in Drosophila so far. This candidate gene was selected for further analysis.
Figure 7.

Presynaptic PKD promotes Gyki-induced PHP-induced but not PhTx-induced PHP
(A) Summary of the screen to identify genes involved in Gyki-induced PHP. Each genotype is represented by a dot representing the relation between ratio of median QC with or without Gyki treatment and ratio of median mEPSP amplitude with or without Gyki treatment. The solid line and dashed lines below and above the solid line represent functions 1/x, 0.8/x, and 1.2/x, respectively. Genotypes falling within the upper and lower dashed lines have similar EPSPs (±20%) with or without Gyki treatment. Genotypes below the lower dashed line have EPSP reduction of more than 20% after Gyki treatment. Red dot is presynaptic PKD knockdown.
(B) Expression of fluorescently tagged endogenous PKD (stained with anti-GFP) and the neuronal membrane marker HRP. PKD fluorescence (green) overlaps with HRP (red) in the merged image. In addition, PKD expression was seen in other non-neuronal compartments, such as trachea. Scale bar, 10 μm.
(C) Representative traces (mEPSPs and EPSPs), mEPSP amplitudes, EPSP amplitudes, and QC for Ctrl- or Gyki-treated NMJs with presynaptic PKD knockdown.
(D) QC and mEPSP amplitude of individual Ctrl and treated cells along with the median and median absolute deviation as error bars. n = 40 (Ctrl) versus 37 (Gyki).
(E) Representative traces (mEPSPs and EPSPs), mEPSP amplitudes, EPSP amplitudes, and QC for Ctrl- or Gyki-treated hypomorphic PKDH mutant NMJs.
(F) QC and mEPSP amplitude of individual Ctrl and treated cells along with the median and median absolute deviation as error bars. n = 48 (Ctrl) versus 55 (Gyki).
(G) Representative traces (mEPSPs and EPSPs), mEPSP amplitudes, EPSP amplitudes, and QC for Ctrl or Gyki-treated NMJs expressing wild-type PKD presynaptically in the PKDH mutant background.
(H) QC and mEPSP amplitude of individual Ctrl and treated cells along with the median and median absolute deviation as error bars. n = 21 (Ctrl) versus 21 (Gyki).
(I) Representative traces (mEPSPs and EPSPs), mEPSP amplitudes, EPSP amplitudes, and QC for Ctrl- or PhTx-treated hypomorphic PKDH mutant NMJs.
(J) QC and mEPSP amplitude of individual Ctrl and treated cells along with the median and median absolute deviation as error bars. n = 32 (Ctrl) versus 41 (Gyki). The solid and dashed lines in (D), (F), (H), and (J) represent the expected QC to restore EPSP amplitudes to the untreated median EPSP amplitude (±20%). [Ca2+]e = 0.3 mM. The whiskers of the box plots extend to either 1.5 times the inter-quartile range or to the farthest data point, whichever is closest to the respective quartile. Individual data points are shown as gray dots. p, p value; r, effect size; ns: p ≥ 0.05; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Although PKD shows a broad expression pattern in Drosophila (Maier et al., 2006), its presence at the NMJ had not been investigated. Therefore, we analyzed PKD expression at the larval body wall of third-instar larvae using fluorescently tagged endogenous PKD (PKDGFP) generated from an intronic Minos-mediated integration cassette (MiMIC) insertion in the PKD gene (Nagarkar-Jaiswal et al., 2015). PKD fluorescence was present in various compartments, including motor neurons, muscles, and trachea (Figure 7B). It was particularly enriched in close proximity to the neural membrane marker horseradish peroxidase (HRP), demonstrating PKD expression in neurons and the synaptic compartments.
We next aimed at verifying a role for PKD in Gyki-induced PHP and at revealing in which synaptic compartment PKD acts during PHP using genetic analysis. Gyki application significantly reduced mEPSP and EPSP amplitudes by a similar degree at NMJs expressing PKDRNAi presynaptically (Figure 7C). Consequently, there was no significant increase in median quantal content upon Gyki treatment after presynaptic PKDRNAi expression (Figure 7C), with the majority of NMJs failing to reach quantal content values required to restore EPSPs to median baseline levels (Figure 7D). By contrast, Gyki potentiated quantal content in the genetic controls (UAS-PKDRNAi/+; Figure S5A), suggesting PHP expression. Furthermore, NMJs expressing PKDRNAi postsynaptically (Mef2-Gal4>UAS-PKDRNAi) also exhibited an increase in quantal content after Gyki treatment (Figure S5B). Together, these results indicate a presynaptic role for PKD in Gyki-induced PHP. Gyki-induced PHP was not blocked after presynaptic PKDRNAi expression at elevated extracellular Ca2+ (Figure S5C), similar to genes that predominantly promote PhTx-induced PHP at low extracellular Ca2+ concentration (Genç et al., 2017; Hauswirth et al., 2018; Kiragasi et al., 2017).
Next, we independently verified a role of PKD in Gyki-induced PHP using a previously characterized hypomorphic allele (PKDH) (Ashe et al., 2018). Gyki treatment of PKDH NMJs resulted in a similar fractional reduction in mEPSP and EPSP amplitudes (Figure 7E), translating into no significant changes in quantal content after Gyki treatment (Figures 7E and 7F). By contrast, isogenic controls (ywMiMIC) exhibited a significant increase in quantal content upon Gyki treatment (Figure S5D). We then tested whether the PHP impairment in PKDH mutants could be rescued by synaptic compartment-specific expression of a wild-type PKD transgene (UAS-PKDWT) (Maier et al., 2007) in the PKDH mutant background. Gyki treatment of PKDH NMJs expressing PKD presynaptically (OK371-Gal4>UAS-PKDWT; PKDH) resulted in a significant decrease in mEPSP amplitudes (Figure 7G). However, unlike in the case of PKDH (Figure 7E) and the rescue control (UAS-PKDWT; PKDH (No Gal4); Figure S5E), there was no reduction in EPSP amplitude, and thus a significant increase in quantal content, indicating robust PHP expression (Figures 7G and 7H). By contrast, Gyki application did not result in a significant quantal content increase after muscle-specific PKD expression in PKDH mutants (Mef2-Gal4>UAS-PKDWT; PKDH) (Figure S5F). Thus, presynaptic, but not postsynaptic, PKD expression restores PHP in the PKDH mutant background, supporting our conclusion of a presynaptic function of PKD during Gyki-induced PHP.
In principle, the PHP defect in PKD mutants may arise from impaired NMJ development or baseline synaptic transmission. We therefore next investigated possible effects of presynaptic PKDRNAi expression on NMJ morphology and synaptic transmission (Figure S6). The total NMJ surface area, as quantified by the neuronal membrane marker HRP, and the number of active zones, as quantified by the number of Brp puncta, were not affected after presynaptic PKDRNAi expression (Figure S6A). Moreover, we did not detect apparent differences in miniature and evoked synaptic transmission between presynaptic PKDRNAi-expressing NMJs and controls (Figure S6B). Furthermore, RRP size (Figure S6C), release probability (Figure S6C), synaptic depression kinetics (Figure S6D), and PPRs were similar between presynaptic PKDRNAi-expressing NMJs and controls (Figure S6E). We conclude that presynaptic PKD plays no major role in the regulation of gross NMJ morphology and baseline synaptic transmission and that the PHP defect is unlikely a secondary consequence of major changes in NMJ development or impaired baseline synaptic transmission.
Finally, we investigated whether PKD is also involved in PhTx-induced PHP by probing synaptic responses of PKDH mutant NMJs after PhTx application (Figure 7I). PhTx strongly reduced mEPSP amplitudes in PKDH mutants with only a small effect on EPSP amplitudes, translating into a significant increase in quantal content (Figures 7I and 7J). Hence, PhTx treatment in PKDH mutants results in an increase in quantal content, suggesting that PKD is not required for PhTx-induced PHP expression. This differential requirement of PKD for Gyki- and PhTx-induced PHP further supports the notion that Gyki and PhTx induce PHP via distinct molecular mechanisms.
Discussion
We here demonstrate that neurotransmitter receptor impairment by different GluR antagonists (PhTx and Gyki) induces PHP via distinct mechanisms, including differential inhibition of GluR subtypes, differential modulation of an active zone scaffold, and short-term plasticity during PHP. Importantly, we provide genetic evidence that separable molecular mechanisms promote PHP in response to Gyki and PhTx treatment. On the other hand, the GluR antagonist γDGG did not produce PHP, despite a robust inhibition of postsynaptic receptors. Together, our data suggest that distinct molecular mechanisms mediate PHP in response to GluR inhibition by different antagonists and that GluR inhibition per se is not sufficient for PHP expression.
Our results contrast with prevalent models of homeostatic synaptic plasticity, which rest on the assumption that synaptic activity changes induce homeostatic signaling (Frank et al., 2006; Turrigiano, 2008). In the case of PHP, the magnitude of the homeostatic increase in presynaptic release scales with the decrease in the amplitude of postsynaptic miniature events (Frank et al., 2006). Based on these data, it was proposed that reduced ion flux through the receptors triggers a homeostatic signaling cascade in the postsynaptic cell, which is relayed to the presynaptic compartment where it adjusts release. A prediction that directly follows is that any receptor perturbation with similar effects on the amplitude of synaptic miniature events produces a homeostatic response via similar underlying mechanisms. We observed that two GluR antagonists, PhTx and Gyki, induced PHP, while one antagonist, γDGG, did not. All three perturbations similarly decreased q, indicating a similar reduction of ion flux through the receptors. Although we cannot exclude the possibility that γDGG may have directly blocked PHP expression, our results suggest that ion flux is unlikely the sole signal responsible for PHP induction at the Drosophila NMJ. This agrees with recent observations at the Drosophila and mouse NMJ indicating that Ca2+ flux through the receptor is likely dispensable for PHP induction (Goel et al., 2017; Wang et al., 2018). How could pharmacological receptor perturbation induce PHP independent of ion flux through the receptor? One intriguing possibility is that perturbation-specific conformational changes of the receptor may be involved in PHP signaling (Goel et al., 2017; Wang et al., 2018). Conformational changes of kainate-, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPA)-, and N-Methyl-d-aspartate (NMDA)-type GluRs at mammalian synapses are known to signal independent of ion flux in the context of synaptic plasticity, possibly through metabotropic signaling (Dai et al., 2021; Valbuena and Lerma, 2016). It is also known that different antagonists stabilize different conformational states of AMPA receptors (Balannik et al., 2005; Twomey et al., 2018). Thus, it is conceivable that distinct antagonists may trigger different PHP signaling pathways, depending on the different signaling partners that could be recruited by different conformational states. This hypothesis requires further investigation in relation to molecular signaling underlying PHP induction.
Although PhTx and Gyki robustly induced PHP through an increase in RRP size, we observed several differences between PhTx- and Gyki-induced PHP. First, PhTx and Gyki differentially affected GluR subtypes. Whereas PhTx mainly inhibits GluRIIA-containing receptors, Gyki reduced mEPSP amplitude at either GluRIIA or GluRIIB mutant NMJs. Since mEPSPs at the Drosophila NMJs are mediated by GluRIIA- and GluRIIB-containing receptors (Marrus et al., 2004), these results indicate that Gyki may act on both types of receptor complexes. An intriguing possibility to be tested in the future is that PhTx-dependent receptor inhibition may trigger PHP by affecting a signaling module associated with GluRIIA-containing receptor complexes, whereas Gyki-induced PHP may be triggered by mechanisms beyond GluRIIA-dependent signaling. Second, while Gyki similarly accelerated the decay of mEPSCs and EPSCs, the EPSC decay was slower than that of mEPSCs upon PhTx application. The mismatch between the mEPSC and EPSC decay kinetics after PhTx treatment may either result from presynaptic changes, such as the recruitment of synaptic vesicles with a low pr during the EPSC decay phase (Wentzel et al., 2018), or different effects on GluR desensitization, saturation, or diffusion during evoked release (Takahashi et al., 1995). Regardless, as this mismatch was not observed after Gyki treatment, it either reflects a difference in antagonist action or in Gyki- and PhTx-induced PHP. Third, Gyki, but not PhTx, resulted in altered short-term plasticity, indicative of a decrease in pr or increased GluR desensitization and/or saturation. Fourth, while PhTx increased Brp-fluorescence intensity, indicating elevated Brp abundance (Weyhersmüller et al., 2011), Gyki did not. Although we cannot exclude Brp modulation at a different time point with regard to Gyki application, this observation suggests differential regulation of this core active zone protein during PhTx- and Gyki-induced PHP. Fifth, we revealed that Gyki application induced PHP in six mutants that were previously shown to block PHP upon PhTx treatment. Sixth, we identified a gene (PKD) that is required for Gyki-induced PHP, but not PhTx-induced, PHP. Thus, although we cannot exclude the possibility of partially overlapping molecular mechanisms, these data demonstrate separable molecular pathways between Gyki- and PhTx-induced PHP.
Diverging homeostatic signaling has been observed in the context of acute versus chronic receptor perturbations. At the Drosophila NMJ, mutants with intact acute PHP in response to PhTx application displayed impaired chronic PHP upon GluRIICRNAi expression (James et al., 2019). However, it is difficult to separate whether these differences emerge because of differences in the nature or the timescale of receptor perturbation. Since our experiments focus on acute perturbations, our results support the idea that distinct homeostatic signaling pathways can be triggered depending on the specific way receptors have been perturbed. PhTx, γDGG, and Gyki inhibit AMPA receptors via different modes of antagonism. While PhTx is an activity-dependent pore blocker (Jackson et al., 2011), γDGG acts as a low-affinity competitive antagonist (Liu et al., 1999) and Gyki is an allosteric inhibitor (Balannik et al., 2005). PhTx and γDGG inhibit Drosophila GluRs with characteristics similar to AMPA receptor inhibition (Frank et al., 2006; Miśkiewicz et al., 2011; Pawlu et al., 2004). Similarly, the conserved Gyki-interacting residues between rat and Drosophila GluRs highlight the possibility that Gyki could also act similarly on Drosophila and mammalian GluRs (Figure S1A). Thus, PhTx, γDGG, and Gyki may inhibit Drosophila GluRs via different modes of antagonism, which may be involved in triggering distinct homeostatic signaling pathways. Differential homeostatic signaling has also been observed in the context of firing rate homeostasis, where molecular responses depend on whether the protein or the conductance of a sodium channel protein is eliminated (Kulik et al., 2019). Thus, distinct molecular signaling mechanisms that are specific to the nature, rather than the functional effects of a perturbation, may be a general theme in the context of neuronal homeostasis.
We also implicated presynaptic PKD signaling in synaptic homeostasis. Although PKD has been linked to various intracellular processes, such as vesicle sorting (Baron and Malhotra, 2002), endocytosis (Ellwanger and Hausser, 2013), and the regulation of the actin cytoskeleton (Olayioye et al., 2013), its synaptic function is less explored. Recently, PKD was associated with synaptic plasticity (Oueslati Morales et al., 2020). Our data clearly demonstrate that presynaptic PKD is required for Gyki-induced PHP, but not for PhTx-induced PHP, and establish that separable molecular pathways govern Gyki- and PhTx-induced PHP. However, PKD was required for PHP only at low extracellular Ca2+ concentration, similar to a number of genes supporting PhTx-induced PHP (Genç et al., 2017; Hauswirth et al., 2018; Kiragasi et al., 2017). This indicates that PKD signaling may contribute to the robustness of PHP under conditions of low pr. It is worth highlighting that the PHP defect observed upon presynaptic PKD knockdown and in PKD hypomorphs may not be fully penetrant, likely because of the genetic nature of the perturbation, and/or genetic compensation by other members of the Protein Kinase C/Calmodulin-Dependent Kinase family, as previously described (Maier et al., 2019). This genetic compensation is particularly pronounced in PKD null mutants (Maier et al., 2019), which precluded the analysis after complete loss of PKD function.
Unlike PhTx (Frank et al., 2006), Gyki induced PHP in the fully dissected NMJ preparation, which is more amenable to electrophysiology and imaging approaches, and thus allowed probing the dynamics of PHP induction. It is currently unclear why PhTx-induced PHP cannot be observed in the full preparation. One hypothesis is that the muscles/synapses may be stretched in the full preparation, which could lead to a disruption of signaling domains relevant for PhTx-induced PHP (Frank et al., 2006). A second possibility could be the potential disruption of neuro-glial signaling that is important for PhTx-induced PHP in the full preparation (Wang et al., 2020). The fact that Gyki results in PHP after full dissection demonstrates that PHP induction is not limited to the “semi-intact” preparation per se. It will be interesting to investigate whether different molecular pathways promoting PHP in response to Gyki and PhTx treatment may be differentially affected by the type of the preparation. In this regard, Gkyi induced PHP in dmp mutants, which were shown to disrupt PhTx-dependent PHP and neuro-glia signaling (Wang et al., 2020).
Gyki application to the fully dissected preparation revealed very rapid, low-latency PHP induction kinetics, which maintained EPSP amplitudes constant despite Gyki-induced mEPSP amplitude reduction. However, due to our sampling interval of 35 s, we cannot rule out shorter latencies. In addition, the exact latency of PHP could not be estimated because of the relatively slow kinetics of mEPSP reduction in our experiments. The reversible GluR block by Gyki also revealed rapid, low-latency PHP reversal after Gyki washout, consistent with previous observations at mouse cerebellar mossy fiber boutons (Delvendahl et al., 2019) or at mouse NMJs (Wang et al., 2016). Our results imply continuous, bidirectional PHP signaling that compensates for receptor impairment within ∼35 s, similar to observations at the mouse NMJ (Wang et al., 2016). It will be interesting to explore the molecular mechanisms underlying the induction of distinct PHP pathways and whether distinct molecular pathways also rapidly stabilize synaptic efficacy at other synapses.
Limitations of the study
Although our results indicate that PHP signaling depends on specific receptor perturbations, the mechanisms of antagonism of the used antagonists have not been studied for Drosophila GluRs. Moreover, we can neither exclude that the lack of PHP upon γDGG treatment may result from off-target inhibition of PHP signaling, nor that Gyki acts independent of GluRs to induce PHP. However, given Gyki’s known mechanism of allosteric antagonism of mammalian AMPARs (Yelshanskaya et al., 2016), and its potent reduction of mEPSP amplitudes at the Drosophila NMJ (Figure S1A), we consider this possibility unlikely. Furthermore, given the evolutionary conservation of Gyki-interacting residues in Drosophila GluR subunits (Figure S1A), it is likely that Gyki acts on all Drosophila GluR subunits. However, we cannot test the effect of Gyki on individual receptor subunits, because GluRIIC–E subunits are essential for receptor formation (Han et al., 2015; Marrus et al., 2004; Qin et al., 2005). We used homozygous Drosophila lines harboring mutations in genes required for PhTx-induced PHP to test whether PhTx and Gyki activate different downstream signaling pathways. Our analysis did not systematically investigate the effects of potential off-site mutations that may have accumulated over time in these fly lines. However, most strains were kept over a balancer chromosome, and Gyki-induced PHP proceeded normally in six mutants that were previously shown to disrupt PhTx-induced PHP (Figure 6; Figure S4), suggesting that a major contribution of off-target mutations is unlikely.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Brp | DSHB | Cat# nc82, RRID:AB_2314866 |
| Rabbit polyclonal anti-Discs large | Jan Pielage lab | NA |
| Mouse anti-GFP | Molecular Probes | Cat# mAb 3E6, RRID:AB_2313858 |
| Goat Alexa-677 conjugated anti-HRP | Jackson ImmunoResearch | Cat# 123-605-021, RRID:AB_2338967 |
| Alexa 488 conjugated anti-mouse | Molecular Probes | Cat# A-11029, RRID:AB_138404 |
| Alexa-555 conjugated anti-rabbit | Molecular Probes | Cat# A-21429, RRID:AB_2535850 |
| Atto594 conjugated anti-mouse | Sigma-Aldrich | Cat# 76085, RRID:AB_1137653 |
| Chemicals, peptides, and recombinant proteins | ||
| Dulbecco’s Phosphate Buffered Saline | Sigma-Aldrich | D8537 |
| Triton X-100 | Sigma-Aldrich | X100 |
| ProLong Gold Antifade | Thermo Fisher Scientific | P36930 |
| ethanol | Merck | CAS# 64-17-5 |
| KCl | Sigma-Aldrich | 746436 |
| NaCl | Sigma-Aldrich | 71380 |
| Na-HEPES | Sigma-Aldrich | H7006 |
| HEPES | Sigma-Aldrich | H3375 |
| Trehalose | Sigma-Aldrich | T9531 |
| Sucrose | Sigma-Aldrich | 84100 |
| MgCl2 | Fluka Analytical | 63020 |
| CaCl2 | Fluka Analytical | 21114 |
| Philanthotoxin-433 | Santa Cruz Biotechnology | Cat # sc-255421 |
| γDGG | Tocris | Cat # 0112 |
| Gyki-53655 | Tocris | Cat # 2555 |
| Experimental models: Organisms/strains | ||
| D. melanogaster: Control: w1118 | Bloomington Drosophila Stock Center | BDSC: 3605; FlyBase: FBal0018186 |
| D. melanogaster: D42-Gal4: w∗; P{GawB}D42 | Bloomington Drosophila Stock Center | BDSC: 8816; FlyBase: FBti0002759 |
| D. melanogaster: OK371-Gal4: w1118; P{GawB}OK371 | Bloomington Drosophila Stock Center | BDSC: 26160; FlyBase: FBal0243489 |
| D. melanogaster: MHC-Gal4: w1118; P{GawB}MHC | Graeme Davis lab | FlyBase: FBal0256907 |
| D. melanogaster: Mef2-Gal4: w1118; P{GawB}Mef2 | Monica Zwicky lab | FlyBase: FBal0288220 |
| D. melanogaster: dysb1: w1118; PBac{RB}Dysbe01028/TM6B, Tb1 | Dion Dickman lab | BDSC: 17918; FlyBase: FBal0161105 |
| D. melanogaster: rimΔ103: w1118; rimΔ103/TM6B, Tb1 | Graeme Davis lab | FlyBase: FBal0161105 |
| D. melanogaster: GluRIIASP16: w∗; GluRIIASP16/CyO | Graeme Davis lab | FlyBase: FBal0085982 |
| D. melanogaster: GluRIIBSP5: w∗; GluRIIBSP5 | Dion Dickman lab | NA; Muttathukunnel et al., 2021 |
| D. melanogaster: KaiR1D2: y1w67c23; Mi{ET1}KaiR1DMB01010 | Bloomington Drosophila Stock Center | BDSC: 22962; FlyBase: FBst0022962 |
| D. melanogaster: ppk11Mi: w1118; Mi{ET1}ppk11MB02012 | Bloomington Drosophila Stock Center | BDSC: 23781; FlyBase: FBst0023781 |
| D. melanogaster: dmpΔC: w∗; dmpΔC | Graeme Davis lab | NA; Wang et al., 2014 |
| D. melanogaster: inckk3: w1118; inckk3 | Dion Dickman lab | NA; Kikuma et al., 2019 |
| D. melanogaster: PKDH: y1w∗; Mi{MIC}PKDMI03619 | Bloomington Drosophila Stock Center | BDSC: 37604; FlyBase: FBal0265471 |
| D. melanogaster: PKDGFP: y1w∗; Mi{PT-GFSTF.0}PKDMI09308-GFSTF.0/TM3, Sb1 Ser1 | Bloomington Drosophila Stock Center | BDSC: 60273; FlyBase: FBst0060273 |
| D. melanogaster: RNAi of PKD: y1v1; P{TRiP.HMC04179}attP2/TM3, Sb1 | Bloomington Drosophila Stock Center | BDSC: 55898; FlyBase: FBst0055898 |
| D. melanogaster: UAS-PKDWT: y1w67c23; P{UAS-PKD.GFP} | Anette Preiss lab | FlyBase: FBal0212370 |
| Software and algorithms | ||
| Fiji / ImageJ | https://fiji.sc | RRID: SCR_002285 |
| Clampex | Axon CNS, Molecular Devices | RRID: SCR_011323 |
| Leica Application Suite X | Leica Microsystems | RRID:SCR_013673 |
| NumPy | https://www.numpy.org/ | RRID:SCR_008633 |
| SciPy | https://www.scipy.org/ | RRID:SCR_008058 |
| IPython | http://ipython.org | RRID:SCR_001658 |
| Neo | http://neuralensemble.org/neo/ | RRID:SCR_000634 |
| MatPlotLib | https://matplotlib.org/ | RRID:SCR_008624 |
| scikit posthocs | https://github.com/maximtrp/scikit-posthocs | RRID:SCR_021363 |
| Biorender | http://biorender.com | RRID:SCR_018361 |
Resource availability
Lead contact
Further information and requests should be directed to and will be fulfilled by the lead contact, Martin Müller (martin.mueller@mls.uzh.ch).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Drosophila stocks were raised at 25°C on normal molasses-containing food. w1118 served as wild-type control. Details regarding genotypes and sources can be found in the Key resources table. Either the MHC-Gal4 or the Mef2-Gal4 driver line was used for muscle-specific expression and either D42-Gal4 or OK371-Gal4 was used for motor-neuron expression.
Method details
Electrophysiology
Wandering third-instar larvae were dissected in HL3 solution (5 mM KCl, 70 mM NaCl, 10 mM Na-HEPES, 5 mM HEPES, 5 mM trehalose, 115 mM sucrose, 10 mM MgCl2) with 0.3 mM CaCl2 for sharp-electrode membrane voltage recordings (unless stated otherwise), or 1.0 mM CaCl2 for two-electrode voltage clamp (TEVC) recordings. The internal organs, including brain and ventral nerve cord, were carefully removed from the body-wall with intact muscle fibers and innervating motor nerves. Sharp-electrode and TEVC recordings were performed on muscle 6 of segments 3 and 4 with sharp glass electrodes (resistance 10-25 MΩ) using an Axoclamp 900A amplifier (Molecular Devices) and a combination of HS 9A x0.1 and HS 9A x10 headstages. For TEVC recordings, muscle cells were clamped to a membrane potential of −65 mV for EPSCs and −100 mV for mEPSCs. For individual NMJs, mEPSP/Cs were recorded prior to EPSP/Cs induced by stimulating the respective hemi-segmental nerve with single APs (3 ms or 0.3 ms stimulus duration for EPSPs and EPSCs, respectively, 0.3 Hz). Note that the comparably long stimulus duration of 3 ms for eliciting EPSPs was previously used in PHP studies in the same system (Gaviño et al., 2015). A total of 50 EPSCs or 30 EPSPs were recorded to obtain the median EPSC or EPSP value, respectively, for each cell. Train responses were obtained from 5 sweeps of 60 EPSCs at 60 Hz. Variance-mean analysis was done with EPSC measurements from individual cells with varying extracellular Ca2+ concentration (0.3 mM, 0.5 mM, 1.0 mM and 1.5 or 3.0 mM; 100 EPSCs at 0.3 Hz per Ca2+ concentration).
For PHP experiments, semi-intact (dorsally dissected; with internal organs, brain and ventral nerve cord intact) larvae were incubated with GluR antagonists for ∼15 minutes. This was followed by removal of internal organs, brain and ventral nerve cord to obtain a fully-stretched preparation for electrophysiological recordings. Recordings for γDGG and Gyki were performed in the presence of the respective inhibitors. For PhTx, we used the previously described protocol in which PhTx was thoroughly washed out with HL3 after the removal of internal organs, brain and ventral nerve cord (Frank et al., 2006). Due to the irreversible binding of PhTx, a significant fraction of receptors remains inhibited after washout. The following inhibitor concentrations were used: PhTx (20 μM), Gyki (10 μM) and γDGG (5 mM). Except for experiments in Figure 1, all experiments with Gyki were performed by directly applying Gyki to the fully stretched larval preparation. For the PHP induction and reversibility time course experiments, NMJ activity was recorded as successive sweeps (inter-sweep interval = 5 s) of 30 s duration containing either one or five EPSPs. Gyki was applied or removed by serial exchange of the bath solution.
Immunohistochemistry
Third-instar larvae were dissected in HL3 and fixed for 10 minutes in 100% ice-cold ethanol followed by rinsing 3 times with PBS. Larvae were washed afterward 5x 10 minutes with PBS and 0.1% PBT on a rotating wheel. This was followed by 2 hours blocking with 3% normal goat serum in PBT, before incubating the samples with primary antibodies at 4°C overnight. Larvae were washed 5x 10 min with PBT on the following day and then incubated with secondary antibodies in blocking solution for ∼2 hours. This was followed by 5x 10 min wash with PBT before mounting on glass slides with ProLong Gold Antifade. The following primary antibodies were used: anti-Brp (1:100), anti-Discs large (1:1000), anti-GFP (1:1000) and Alexa-677 conjugated anti-HRP (1:1000). The following secondary antibodies were used: Alexa 488 conjugated anti-mouse (1:100), Alexa-555 conjugated anti-rabbit (1:100), and Atto594 conjugated anti-mouse (1:100). Images were acquired using an upright Leica Stellaris or inverted Leica SP8 laser scanning microscope (University of Zurich Center for Microscopy and Image Analysis) controlled with LASX software suite (Leica Microsystems, Germany). Images were acquired using either 63x or 100x (for Brp intensity quantification) oil immersion objectives (HC PL APO 1.40 NA Oil STED WHITE; Leica Microsystems, Germany). Emitted light was detected with two HyD detectors in photon counting mode (Leica Microsystems, Germany).
Quantification and statistical analysis
Electrophysiology Data Analysis
Electrophysiology data were analyzed using scientific python libraries, including numpy, scipy, IPython and neo (Garcia et al., 2014). mEPSP/C events were detected using a template-matching algorithm (Clements and Bekkers, 1997). Due to high noise levels of the high-gain current-injecting headstage, the recorded raw trace was filtered using a second-order Savitzky-Golay filter over 5 ms prior to mEPSC detection. Quantal content was calculated as the ratio of the median EPSP/C amplitude and the median mEPSP/C amplitude for a given cell. The expected quantal content for a given mEPSP amplitude to restore EPSP to the untreated control level was estimated as: , where q is the median mEPSP amplitude for a given NMJ, QC is quantal content as a function of mEPSP, and EPSPCtrl is the median EPSP amplitude of the untreated control group.
As the decay kinetics of NMJ currents do not follow exponential kinetics (Pawlu et al., 2004), mEPSC and EPSC decay kinetics were quantified using the time taken to reach half maximum (t-half) by fitting the decay phase to Hill’s equation: , where I(t), Imax, t50, and n are synaptic current, EPSC amplitude, t-half and steepness coefficient.
For the time course experiments, mEPSP amplitude, EPSP amplitude and quantal content were quantified in 35 s bins. The mEPSP, EPSP and quantal content timeseries were then filtered using a first-order Savitzky-Golay filter over three data points.
The readily releasable pool (RRP) size was estimated by the method of back-extrapolation of cumulative EPSC amplitudes measured during stimulus trains (Schneggenburger et al., 1999). Briefly, presynaptic nerve terminals were stimulated with 60 stimuli at 60 Hz and EPSCs were recorded. The cumulative EPSC amplitudes were then plotted as a function of stimulus number. The last 10 data points were fitted with a straight line, and this line was extrapolated to the y axis intercept. The y-intercept indicates the total response produced by the RRP. RRP size (RRPCum. EPSC) was estimated as the ratio of the y-intercept and the median mEPSC amplitude from the same cell. Release probability (ptrain) was estimated by dividing the amplitude of the first EPSC of the train by the total response produced by RRP (y-intercept). RRP size was also independently estimated by analyzing the train responses using the Elmqvist-Quastal method (Elmqvist and Quastel, 1965). Briefly, EPSC amplitudes were plotted as a function of cumulative EPSC amplitudes. The first 5 data points were fitted with a straight line, and this line was extrapolated to the x axis intercept. The x axis intercept, in this case, indicates the total response produced by the RRP. RRP size (RRPEQ) was estimated as the ratio of the x-intercept and the median mEPSC amplitude from the same cell. Release probability (ptrainEQ) was estimated by dividing the amplitude of the first EPSC of the train by the total response produced by the RRP (x-intercept).
The weighted time constant (τw) for synaptic short-term depression was estimated by fitting the normalized EPSC amplitudes during the train with a double exponential function: . τw was then estimated using the fitted double exponential function parameters as: . The depression of train EPSC amplitudes at the Drosophila NMJs do not follow single exponential decay, and could be better fitted using a double exponential, possibly due to the compound response of Is and Ib synapses, which have different short-term dynamics (Kurdyak et al., 1994).
For variance-mean analysis, EPSC amplitude variance for all Ca2+ concentrations were plotted as a function of mean EPSC amplitude for a respective Ca2+ concentration and then fitted with: , where σ2, I, q and N are EPSC amplitude variance, mean, quantal size and number of functional release sites, respectively.
For the electrophysiology-based genetic screen to identify candidates involved in Gyki-induced PHP, we scored the relationship between the ratio of quantal content with or without Gyki, and the ratio of mEPSP amplitude with or without Gyki. and , where QCCtrl, EPSPCtrl, mEPSPCtrl, QCGyki, EPSPGyki and mEPSPGyki are median quantal content, median EPSP amplitude and median mEPSP amplitude, without or with Gyki treatment, respectively. Therefore, . If Gyki treatment results in the restoration of EPSP amplitude within ± 20% of the untreated control EPSP level, then . This will follow . Therefore, we scored for every genotype, and any genotype with was considered as a candidate gene involved in Gyki-induced PHP.
Microscopy Data Analysis
Microscopy images were analyzed using custom routines written in ImageJ (version 1.51n, National Institutes of Health, USA). Brp fluorescence intensity quantification was performed as follows: First individual Brp puncta were isolated by segmenting binary threshold masks (15%-35% of the maximum intensity value), background corrected (rolling ball, radius = 1 μm) and filtered (3 × 3 median) maximum intensity projection images. Average intensity values were then calculated for each Brp puncta from raw maximum intensity projection images. HRP-positive regions-of-interest were manually selected and the area were calculated with ImageJ area measurement tool.
Statistical Analysis
Statistical tests were performed using the stats module of scipy and scikit-posthocs. Two-sided Mann-Whitney U Tests were used to perform statistical comparison, and for multiple data comparison, a Kruskal-Wallis test followed by Dunn’s post hoc comparison was performed. p values are listed in respective figures as follows; ns: p > = 0.05; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. Effects sizes (r) were quantified as the rank biserial correlation calculated from the U statistics of the Mann-Whitney U Test and are listed in respective figures. The number of samples (n) are provided in respective figure captions, where n refers to the number of NMJs. All statistical details can be found in the figures and figure captions.
Acknowledgments
We thank the Dickman lab for generating and providing the GluRIIBSP5 mutants. We are grateful to members of the Müller lab for helpful discussions and critical comments. In particular, we thank Igor Delvendahl and Olivier Urwyler for their valuable input. This research was funded by a Forschungskredit grant of the University of Zurich and an International Postdoc grant (2018-06813) by the Swedish Research Council to A.G.N., as well as a Swiss National Science Foundation Assistant Professor grant (PP00P3–15) and a European Research Council starting grant (SynDegrade-679881) to M.M.
Author contributions
M.M. and A.G.N. conceptualized and designed experiments. A.G.N. and P.M. conducted research and analyzed data. A.G.N., P.M., and M.M. interpreted data. M.M. and A.G.N. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: December 14, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.110105.
Supplemental information
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
All data needed to evaluate the conclusions in the paper are present in the paper and the Supplemental information (Table S1).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Ashe S., Malhotra V., Raghu P. Protein kinase D regulates metabolism and growth by controlling secretion of insulin like peptide. Dev. Biol. 2018;434:175–185. doi: 10.1016/j.ydbio.2017.12.008. [DOI] [PubMed] [Google Scholar]
- Balannik V., Menniti F.S., Paternain A.V., Lerma J., Stern-Bach Y. Molecular mechanism of AMPA receptor noncompetitive antagonism. Neuron. 2005;48:279–288. doi: 10.1016/j.neuron.2005.09.024. [DOI] [PubMed] [Google Scholar]
- Baron C.L., Malhotra V. Role of diacylglycerol in PKD recruitment to the TGN and protein transport to the plasma membrane. Science. 2002;295:325–328. doi: 10.1126/science.1066759. [DOI] [PubMed] [Google Scholar]
- Böhme M.A., McCarthy A.W., Grasskamp A.T., Beuschel C.B., Goel P., Jusyte M., Laber D., Huang S., Rey U., Petzoldt A.G., et al. Rapid active zone remodeling consolidates presynaptic potentiation. Nat. Commun. 2019;10:1085. doi: 10.1038/s41467-019-08977-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements J.D., Bekkers J.M. Detection of spontaneous synaptic events with an optimally scaled template. Biophys. J. 1997;73:220–229. doi: 10.1016/S0006-3495(97)78062-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements J.D., Silver R.A. Unveiling synaptic plasticity: a new graphical and analytical approach. Trends Neurosci. 2000;23:105–113. doi: 10.1016/s0166-2236(99)01520-9. [DOI] [PubMed] [Google Scholar]
- Dai J., Patzke C., Liakath-Ali K., Seigneur E., Südhof T.C. GluD1 is a signal transduction device disguised as an ionotropic receptor. Nature. 2021;595:261–265. doi: 10.1038/s41586-021-03661-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delvendahl I., Müller M. Homeostatic plasticity-a presynaptic perspective. Curr. Opin. Neurobiol. 2019;54:155–162. doi: 10.1016/j.conb.2018.10.003. [DOI] [PubMed] [Google Scholar]
- Delvendahl I., Kita K., Müller M. Rapid and sustained homeostatic control of presynaptic exocytosis at a central synapse. Proc. Natl. Acad. Sci. USA. 2019;116:23783–23789. doi: 10.1073/pnas.1909675116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickman D.K., Davis G.W. The schizophrenia susceptibility gene dysbindin controls synaptic homeostasis. Science. 2009;326:1127–1130. doi: 10.1126/science.1179685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellwanger K., Hausser A. Physiological functions of protein kinase D in vivo. IUBMB Life. 2013;65:98–107. doi: 10.1002/iub.1116. [DOI] [PubMed] [Google Scholar]
- Elmqvist D., Quastel D.M.J. A quantitative study of end-plate potentials in isolated human muscle. J. Physiol. 1965;178:505–529. doi: 10.1113/jphysiol.1965.sp007639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank C.A., Kennedy M.J., Goold C.P., Marek K.W., Davis G.W. Mechanisms underlying the rapid induction and sustained expression of synaptic homeostasis. Neuron. 2006;52:663–677. doi: 10.1016/j.neuron.2006.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia S., Guarino D., Jaillet F., Jennings T., Pröpper R., Rautenberg P.L., Rodgers C.C., Sobolev A., Wachtler T., Yger P., Davison A.P. Neo: an object model for handling electrophysiology data in multiple formats. Front. Neuroinform. 2014;8:10. doi: 10.3389/fninf.2014.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaviño M.A., Ford K.J., Archila S., Davis G.W. Homeostatic synaptic depression is achieved through a regulated decrease in presynaptic calcium channel abundance. eLife. 2015;4:1–19. doi: 10.7554/eLife.05473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genç Ö., Dickman D.K., Ma W., Tong A., Fetter R.D., Davis G.W. MCTP is an ER-resident calcium sensor that stabilizes synaptic transmission and homeostatic plasticity. eLife. 2017;6:e22904. doi: 10.7554/eLife.22904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genç Ö., An J.Y., Fetter R.D., Kulik Y., Zunino G., Sanders S.J., Davis G.W. Homeostatic plasticity fails at the intersection of autism-gene mutations and a novel class of common genetic modifiers. eLife. 2020;9:1–32. doi: 10.7554/eLife.55775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel P., Li X., Dickman D. Disparate Postsynaptic Induction Mechanisms Ultimately Converge to Drive the Retrograde Enhancement of Presynaptic Efficacy. Cell Rep. 2017;21:2339–2347. doi: 10.1016/j.celrep.2017.10.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratz S.J., Goel P., Bruckner J.J., Hernandez R.X., Khateeb K., Macleod G.T., Dickman D., O’Connor-Giles K.M. Endogenous tagging reveals differential regulation of Ca2+ channels at single active zones during presynaptic homeostatic potentiation and depression. J. Neurosci. 2019;39:2416–2429. doi: 10.1523/JNEUROSCI.3068-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghighi A.P., McCabe B.D., Fetter R.D., Palmer J.E., Hom S., Goodman C.S. Retrograde control of synaptic transmission by postsynaptic CaMKII at the Drosophila neuromuscular junction. Neuron. 2003;39:255–267. doi: 10.1016/s0896-6273(03)00427-6. [DOI] [PubMed] [Google Scholar]
- Han T.H., Dharkar P., Mayer M.L., Serpe M. Functional reconstitution of Drosophila melanogaster NMJ glutamate receptors. Proc. Natl. Acad. Sci. USA. 2015;112:6182–6187. doi: 10.1073/pnas.1500458112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauswirth A.G., Ford K.J., Wang T., Fetter R.D., Tong A., Davis G.W. A postsynaptic PI3K-cII dependent signaling controller for presynaptic homeostatic plasticity. eLife. 2018;7:e31535. doi: 10.7554/eLife.31535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibata K., Sun Q., Turrigiano G.G. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron. 2008;57:819–826. doi: 10.1016/j.neuron.2008.02.031. [DOI] [PubMed] [Google Scholar]
- Jackson A.C., Milstein A.D., Soto D., Farrant M., Cull-Candy S.G., Nicoll R.A. Probing TARP modulation of AMPA receptor conductance with polyamine toxins. J. Neurosci. 2011;31:7511–7520. doi: 10.1523/JNEUROSCI.6688-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James T.D., Zwiefelhofer D.J., Frank C.A. Maintenance of homeostatic plasticity at the Drosophila neuromuscular synapse requires continuous IP3-directed signaling. eLife. 2019;8:1–28. doi: 10.7554/eLife.39643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck T., Keller G.B., Jacobsen R.I., Eysel U.T., Bonhoeffer T., Hübener M. Synaptic scaling and homeostatic plasticity in the mouse visual cortex in vivo. Neuron. 2013;80:327–334. doi: 10.1016/j.neuron.2013.08.018. [DOI] [PubMed] [Google Scholar]
- Kikuma K., Li X., Perry S., Li Q., Goel P., Chen C., Kim D., Stavropoulos N., Dickman D. Cul3 and insomniac are required for rapid ubiquitination of postsynaptic targets and retrograde homeostatic signaling. Nat. Commun. 2019;10:2998. doi: 10.1038/s41467-019-10992-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiragasi B., Wondolowski J., Li Y., Dickman D.K. A Presynaptic Glutamate Receptor Subunit Confers Robustness to Neurotransmission and Homeostatic Potentiation. Cell Rep. 2017;19:2694–2706. doi: 10.1016/j.celrep.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulik Y., Jones R., Moughamian A.J., Whippen J., Davis G.W. Dual separable feedback systems govern firing rate homeostasis. eLife. 2019;8:1–27. doi: 10.7554/eLife.45717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurdyak P., Atwood H.L., Stewart B.A., Wu C.-F. Differential physiology and morphology of motor axons to ventral longitudinal muscles in larval Drosophila. J. Comp. Neurol. 1994;350:463–472. doi: 10.1002/cne.903500310. [DOI] [PubMed] [Google Scholar]
- Li X., Goel P., Chen C., Angajala V., Chen X., Dickman D.K. Synapse-specific and compartmentalized expression of presynaptic homeostatic potentiation. eLife. 2018;7:e34338. doi: 10.7554/eLife.34338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G., Choi S., Tsien R.W. Variability of neurotransmitter concentration and nonsaturation of postsynaptic AMPA receptors at synapses in hippocampal cultures and slices. Neuron. 1999;22:395–409. doi: 10.1016/s0896-6273(00)81099-5. [DOI] [PubMed] [Google Scholar]
- Maier D., Hausser A., Nagel A.C., Link G., Kugler S.J., Wech I., Pfizenmaier K., Preiss A. Drosophila protein kinase D is broadly expressed and a fraction localizes to the Golgi compartment. Gene Expr. Patterns. 2006;6:849–856. doi: 10.1016/j.modgep.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Maier D., Nagel A.C., Gloc H., Hausser A., Kugler S.J., Wech I., Preiss A. Protein kinase D regulates several aspects of development in Drosophila melanogaster. BMC Dev. Biol. 2007;7:74. doi: 10.1186/1471-213X-7-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier D., Nagel A.C., Kelp A., Preiss A. Protein Kinase D Is Dispensable for Development and Survival of Drosophila melanogaster. G3 (Bethesda) 2019;9:2477–2487. doi: 10.1534/g3.119.400307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrus S.B., Portman S.L., Allen M.J., Moffat K.G., DiAntonio A. Differential localization of glutamate receptor subunits at the Drosophila neuromuscular junction. J. Neurosci. 2004;24:1406–1415. doi: 10.1523/JNEUROSCI.1575-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan E.M. The statistics of transmitter release at chemical synapses. Int. Rev. Physiol. 1978;17:49–117. [PubMed] [Google Scholar]
- Melom J.E., Akbergenova Y., Gavornik J.P., Littleton J.T. Spontaneous and evoked release are independently regulated at individual active zones. J. Neurosci. 2013;33:17253–17263. doi: 10.1523/JNEUROSCI.3334-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miśkiewicz K., Jose L.E., Bento-Abreu A., Fislage M., Taes I., Kasprowicz J., Swerts J., Sigrist S., Versées W., Robberecht W., Verstreken P. ELP3 controls active zone morphology by acetylating the ELKS family member Bruchpilot. Neuron. 2011;72:776–788. doi: 10.1016/j.neuron.2011.10.010. [DOI] [PubMed] [Google Scholar]
- Mrestani A., Pauli M., Kollmannsberger P., Repp F., Kittel R.J., Eilers J., Doose S., Sauer M., Sirén A., Heckmann M., Paul M.M. Active zone compaction correlates with presynaptic homeostatic potentiation. Cell Rep. 2021;37:1–11. doi: 10.1016/j.celrep.2021.109770. [DOI] [PubMed] [Google Scholar]
- Müller M., Liu K.S.Y., Sigrist S.J., Davis G.W. RIM controls homeostatic plasticity through modulation of the readily-releasable vesicle pool. J. Neurosci. 2012;32:16574–16585. doi: 10.1523/JNEUROSCI.0981-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muttathukunnel P., Frei P., Perry S., Dickman D., Müller M. Rapid homeostatic modulation of transsynaptic nanocolumn rings. BioRxiv. 2021 doi: 10.1101/2021.06.15.448550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarkar-Jaiswal S., Lee P.T., Campbell M.E., Chen K., Anguiano-Zarate S., Gutierrez M.C., Busby T., Lin W.W., He Y., Schulze K.L., et al. A library of MiMICs allows tagging of genes and reversible, spatial and temporal knockdown of proteins in Drosophila. eLife. 2015;4:1–28. doi: 10.7554/eLife.05338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olayioye M.A., Barisic S., Hausser A. Multi-level control of actin dynamics by protein kinase D. Cell. Signal. 2013;25:1739–1747. doi: 10.1016/j.cellsig.2013.04.010. [DOI] [PubMed] [Google Scholar]
- Ortega J.M., Genç Ö., Davis G.W. Molecular mechanisms that stabilize short term synaptic plasticity during presynaptic homeostatic plasticity. eLife. 2018;7:1–28. doi: 10.7554/eLife.40385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouanounou G., Baux G., Bal T. A novel synaptic plasticity rule explains homeostasis of neuromuscular transmission. eLife. 2016;5:1–20. doi: 10.7554/eLife.12190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oueslati Morales C., Ignácz A., Bencsik N., Rátkai A.E., Lieb W., Eisler S., Schlett K., Hausser A. PKD promotes activity-dependent AMPA receptor endocytosis in hippocampal neurons. Traffic. 2020;22:454–470. doi: 10.1111/tra.12819. [DOI] [PubMed] [Google Scholar]
- Pawlu C., DiAntonio A., Heckmann M. Postfusional control of quantal current shape. Neuron. 2004;42:607–618. doi: 10.1016/s0896-6273(04)00269-7. [DOI] [PubMed] [Google Scholar]
- Peled E.S., Newman Z.L., Isacoff E.Y. Evoked and spontaneous transmission favored by distinct sets of synapses. Curr. Biol. 2014;24:484–493. doi: 10.1016/j.cub.2014.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry S., Han Y., Das A., Dickman D. Homeostatic plasticity can be induced and expressed to restore synaptic strength at neuromuscular junctions undergoing ALS-related degeneration. Hum. Mol. Genet. 2017;26:4153–4167. doi: 10.1093/hmg/ddx304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen S.A., Fetter R.D., Noordermeer J.N., Goodman C.S., DiAntonio A. Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron. 1997;19:1237–1248. doi: 10.1016/s0896-6273(00)80415-8. [DOI] [PubMed] [Google Scholar]
- Qin G., Schwarz T., Kittel R.J., Schmid A., Rasse T.M., Kappei D., Ponimaskin E., Heckmann M., Sigrist S.J. Four different subunits are essential for expressing the synaptic glutamate receptor at neuromuscular junctions of Drosophila. J. Neurosci. 2005;25:3209–3218. doi: 10.1523/JNEUROSCI.4194-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saviane C., Silver R.A. Estimation of quantal parameters with multiple-probability fluctuation analysis. Methods Mol. Biol. 2007;403:303–317. doi: 10.1007/978-1-59745-529-9_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R., Meyer A.C., Neher E. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron. 1999;23:399–409. doi: 10.1016/s0896-6273(00)80789-8. [DOI] [PubMed] [Google Scholar]
- Takahashi M., Kovalchuk Y., Attwell D. Pre- and postsynaptic determinants of EPSC waveform at cerebellar climbing fiber and parallel fiber to Purkinje cell synapses. J. Neurosci. 1995;15:5693–5702. doi: 10.1523/JNEUROSCI.15-08-05693.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatavarty V., Torrado Pacheco A., Groves Kuhnle C., Lin H., Koundinya P., Miska N.J., Hengen K.B., Wagner F.F., Van Hooser S.D., Turrigiano G.G. Autism-Associated Shank3 Is Essential for Homeostatic Compensation in Rodent V1. Neuron. 2020;106:769–777.e4. doi: 10.1016/j.neuron.2020.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichert M., Liebmann L., Hübner C.A., Bolz J. Homeostatic plasticity and synaptic scaling in the adult mouse auditory cortex. Sci. Rep. 2017;7:17423. doi: 10.1038/s41598-017-17711-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G.G. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G.G., Leslie K.R., Desai N.S., Rutherford L.C., Nelson S.B. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Twomey E.C., Yelshanskaya M.V., Vassilevski A.A., Sobolevsky A.I. Mechanisms of Channel Block in Calcium-Permeable AMPA Receptors. Neuron. 2018;99:956–968.e4. doi: 10.1016/j.neuron.2018.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valbuena S., Lerma J. Non-canonical Signaling, the Hidden Life of Ligand-Gated Ion Channels. Neuron. 2016;92:316–329. doi: 10.1016/j.neuron.2016.10.016. [DOI] [PubMed] [Google Scholar]
- Wang T., Hauswirth A.G., Tong A., Dickman D.K., Davis G.W. Endostatin is a trans-synaptic signal for homeostatic synaptic plasticity. Neuron. 2014;83:616–629. doi: 10.1016/j.neuron.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Pinter M.J., Rich M.M. Reversible Recruitment of a Homeostatic Reserve Pool of Synaptic Vesicles Underlies Rapid Homeostatic Plasticity of Quantal Content. J. Neurosci. 2016;36:828–836. doi: 10.1523/JNEUROSCI.3786-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., McIntosh J.M., Rich M.M. Muscle Nicotinic Acetylcholine Receptors May Mediate Trans-Synaptic Signaling at the Mouse Neuromuscular Junction. J. Neurosci. 2018;38:1725–1736. doi: 10.1523/JNEUROSCI.1789-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Morency D.T., Harris N., Davis G.W. Epigenetic Signaling in Glia Controls Presynaptic Homeostatic Plasticity. Neuron. 2020;105:491–505.e3. doi: 10.1016/j.neuron.2019.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentzel C., Delvendahl I., Sydlik S., Georgiev O., Müller M. Dysbindin links presynaptic proteasome function to homeostatic recruitment of low release probability vesicles. Nat. Commun. 2018;9:267. doi: 10.1038/s41467-017-02494-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyhersmüller A., Hallermann S., Wagner N., Eilers J. Rapid active zone remodeling during synaptic plasticity. J. Neurosci. 2011;31:6041–6052. doi: 10.1523/JNEUROSCI.6698-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelshanskaya M.V., Singh A.K., Sampson J.M., Narangoda C., Kurnikova M., Sobolevsky A.I. Structural Bases of Noncompetitive Inhibition of AMPA-Subtype Ionotropic Glutamate Receptors by Antiepileptic Drugs. Neuron. 2016;91:1305–1315. doi: 10.1016/j.neuron.2016.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younger M.A., Müller M., Tong A., Pym E.C., Davis G.W. A presynaptic ENaC channel drives homeostatic plasticity. Neuron. 2013;79:1183–1196. doi: 10.1016/j.neuron.2013.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
All data needed to evaluate the conclusions in the paper are present in the paper and the Supplemental information (Table S1).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.