

Abstract
Bromodomain containing protein 4 (BRD4) plays a critical role in controlling the expression of genes involved in development and cancer. Inactivation of BRD4 inhibits cancer growth, making it a promising anticancer drug target. The cancer stem cell population is a key driver of recurrence and metastasis in cancer patients. Here we show that cancer stem-like cells can be enriched from squamous cell carcinomas, and that these cells display an aggressive phenotype with enhanced stem cell marker expression, migration, invasion, and tumor growth. BRD4 was highly elevated in this aggressive subpopulation of cells, and its function is critical for these cancer stem cell-like properties. Moreover, BRD4 regulated ΔNp63α, a key transcription factor that is essential for epithelial stem cell function that is often overexpressed in cancers. BRD4 regulated an EZH2/STAT3 complex that led to increased ΔNp63α-mediated transcription. Targeting BRD4 in human squamous cell carcinoma reduces ΔNp63α, leading to inhibition of spheroid formation, migration, invasion and tumor growth. These studies identify a novel BRD4-regulated signaling network in a subpopulation of cancer stem-like cells elucidating a possible avenue for effective therapeutic intervention.
Keywords: BRD4, EZH2, ΔNp63α, cancer stem cells, squamous cell carcinoma
Introduction
Squamous cell carcinomas (SCC) are among the most frequently occurring solid cancers in humans, and develop in response to mutagens (1). SCC is classified by the location which they appear, frequently occurring in the skin, head and neck, oesophagus, lung and cervix, as well as more rarely in the thyroid, prostate, bladder and pancreas (2). Treatment can include surgical removal of the primary tumor, as well as radiation and chemotherapy (3). Recently, targeted therapies against EGFR have been used with modest effect (3). However, many patients experience local disease recurrence and metastatic disease following treatment (1). Increasing evidence suggests that cancer stem cells (CSCs) are to blame for recurrence, drug resistance and metastasis in multiple cancer types (4). An important goal in this context is to identify key proteins that are essential for CSC survival that can be targeted therapeutically.
Chromatin modifying proteins have been under intense focus for their potential as therapeutic targets using small molecule inhibitors. One particular family of chromatin modifying proteins under scrutiny is the Bromodomain and extra terminal (BET) domain family of proteins (5). BET proteins modulate genome processes by binding to acetylated lysine residues of both histone and non-histone proteins, including transcription factors (5). These proteins function as key regulators of transcriptional control in development and differentiation. Additionally, BET proteins are frequently critical to the deregulated transcription characteristic of transformed cells, making them ideal targets for small molecule intervention (6).
ΔNp63α is an important member of the p53 family of transcription factors that regulates epithelial cell differentiation, stem cell status and cell fate (7,8). ΔNp63α is a key controller of epithelial differentiation and maintains the undifferentiated state of the basal layer of epithelial tissues (9). The function of p63 in epithelia was first observed in p63 deficient mice, which die shortly after birth due to defects in epidermal barrier formation, revealing that ΔNp63α is essential for epidermal morphogenesis (8). Furthermore, ΔNp63α overexpression is frequently seen in human cancers, including squamous cell carcinomas (10–12). Overexpression of ΔNp63α is generally associated with poor prognosis (13). While its function and importance has been clearly demonstrated, knowledge of the mechanisms that control ΔNp63α expression is limited.
In the present study, we characterize a subpopulation of SCC cells with properties of CSCs from varying tissue types, including skin, pharynx and tongue. We define this subpopulation in three separate cell lines and show that these CSC-like cells displays elevated levels of canonical and epithelial stem cell markers. Furthermore, these cells are endowed with the features of enhanced migration, invasion and tumor growth. We show that the chromatin modifying proteins BRD4 and EZH2 are elevated in this aggressive subpopulation of cells. These chromatin modifying proteins work together to regulate the expression of a critical transcription factor, ΔNp63α, which is not only required for spheroid formation, invasion, migration and tumor growth, but is elevated in patient-derived SCC. We propose that targeting this network of CSC survival proteins offers a therapeutic strategy for preventing metastasis and recurrence.
Materials and Methods
Antibodies and Reagents
DMEM (10–013-CV), Trypsin (25–054-CI), and 6-well Ultra low attachment plates (3471) were purchased from Corning (Corning, NY). DMEM F-12 (11320–033) was purchased from Thermo Fisher (Waltham, MA). Cell lysis buffer (9803), and antibodies specific for C-MYC (5605), BRD4 (13440), STAT3 (9139), and STAT3-P (9145) were purchased from Cell Signaling Technology (Danvers, MA). Anti-EZH2 (612667) was purchased from BD Transduction Laboratories (San Jose, CA). Anti-p63 (SC-8431) was purchased from Santa Cruz (Santa Cruz, CA). Peroxidase-conjugated anti-mouse IgG and anti-rabbit IgG were obtained from GE Healthcare (Buckinghamshire, UK). Matrigel (354234) and BD Biocoat cell inserts (353097) were from BD Biosciences (San Jose, CA). DAPI (D9542) and Anti-β-actin (A5441) was purchased from Sigma (St. Louis, MO). EF.STAT3C.Ubc.GFP was a gift from Linzhao Cheng (Addgene plasmid # 24983; http://n2t.net/addgene:24983; RRID:Addgene_24983) (14). DeltaNp63alpha-FLAG was a gift from David Sidransky (Addgene plasmid # 26979; http://n2t.net/addgene:26979; RRID:Addgene_26979) (15). pcDNA3-cmyc was a gift from Wafik El-Deiry (Addgene plasmid # 16011; http://n2t.net/addgene:16011; RRID:Addgene_16011) (16).
Cell culture
Human HSC-5 cells were obtained from Sekisui Xenotech, LLC (Kansas City, KS). Human FaDu and Cal-33 cell lines were a generous gift from Leif Ellison. HSC-5, FaDu and Cal-33 cells were cultured in DMEM supplemented with 10% FBS. All cell lines tested negative for Mycoplasma contamination using Myco Alert (LT07–218) from Lonza (Basel, CH).
Lentivirus production
Lentivirus was produced using 293T cells maintained in DMEM with 1 mmol/L L-glutamine, 1 mmol/L sodium pyruvate, and 10% FBS. 293T cells were harvested and plated in 100-mm dishes at 5% confluence for 24 hours prior to transfection. Media were removed and plates were washed with Hank’s balanced Salt Solution before serum-free media were added containing 7.5 μg pPax2, 5 μg VSVG, and 5 μg sgRNA and PEI for co-transfection. After 8 hours, 10% FCS was added, and 48 hours after transfection, the medium was collected, centrifuged for 15 minutes at 1,500 rpm, sterile filtered (22 μm) and stored at −80°C in aliquots.
Creation of CRISPR-depleted cells
Wild-type cancer cells in monolayer culture were infected with 1 ml of lentivirus-containing serum-free media with PEI for 5 hours. After 5 hours, media was removed, and serum-containing media was added. After 24 h, fresh media was added containing 300 μg/ml G418. CRISPR-mediated depletion was assessed via western blot after 2 weeks of selection with G418.
Spheroid formation
Monolayer cultures were harvested with trypsin and gently pipetted to form a single-cell suspension. Trypsin was inactivated by addition of serum-containing medium and cells were collected by centrifugation at 2,000 rpm for 5 minutes. Cell pellets were resuspended in spheroid medium consisting of DMEM/F12 (1:1) containing 2% B27 serum-free supplement (17504–044, Invitrogen, Frederick, MD), 20 ng/ml EGF (AF-100–15, PeproTech, Craybury, NJ) and plated at 4×104 cells per 9.5 cm2 well in six well ultra-low attachment plates (3471, Corning, Tewksbury, MA #3471).
Immunoblotting
For immunoblot analyses, equivalent amounts of protein were electrophoresed on denaturing and reducing 10% polyacrylamide gels and transferred to nitrocellulose membrane. Membranes were blocked in 5% nonfat dry milk and then incubated with the appropriate primary (1:1000) and secondary antibody (1:5000). Secondary antibody binding was visualized using chemiluminescence detection technology.
Invasion assays
Matrigel was diluted in 0.01 Tris-HCL/0.7% NaCl, filter sterilized and 0.1 ml was used to coat individual BD BioCoat inserts (Millicell-PCF, 0.4 mm, 12 mm, PIHP01250). Cells (2.5 × 104) were plated in 100 μl growth medium supplemented with 1% FBS into the upper chambers of the inserts. The lower chambers contained growth medium supplemented with 10% FBS. After invasion, membranes were harvested and the surface of upper membranes were rinsed with PBS to remove unattached cells. Membranes were fixed in 4% paraformaldehyde, stained with 1 mg/ml DAPI, and the underside of the membrane was photographed using an inverted fluorescent microscope and cells were quantitated.
Migration assays
Cells (2 × 106) were plated in 10 cm dishes and grown as monolayer cultures in spheroid medium until confluent. A 10 ml pipette tip was used to prepare areas void of cells and the dishes were washed to remove the dislodged cells. Images were collected at 0–18 h after the scratches were made using a 10X objective, and the width of the openings was measured as a function of time as an index of cell migration potential.
Quantitative RT-PCR
Total RNA was isolated with the RNeasy Mini Kit (Qiagen) and reverse transcribed using the superscript™ III First-Strand Synthesis System (Invitrogen, Carlsbad, CA). RNA (1 mg) was used for cDNA preparation. The Power SYBR™ GREEN PCR Master Mix (ThermoFisher) was used to quantitate expression levels according to manufactures protocol. Signals were normalized to the levels of cyclophilin A mRNA to determine expression.
Tumor Xenografts
Animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC). Spheroid-derived cancer cells were dispersed with trypsin to produce single-cell suspensions, and cells were resuspended in 100 μl of spheroid medium and then mixed with Matrigel at a 1:1 ratio. The mixture, containing 1×106 cells, was injected subcutaneously into the flanks of nude mice using 26.5-gauge needles attached to 1 cc syringes. Cells, and syringes were kept on ice throughout the procedure to prevent Matrigel from solidifying. Four mice were used per data point with two tumors per mouse. For drug treatments, drugs were administered M/W/F via IP injection.
Tumor Microarrays
The tissue array of SCC samples was obtained from US Biomax (HN803f). IHC slides were stained in Discovery Ultra automatic IHC stainer (Roche) following recommended protocols. Briefly, after deparaffinization and rehydration, slides were subjected to antigen retrieval (Benchmark Ultra CC1, Roche) at 96˚C for 1hr; primary Ab incubation was performed at 37˚C for 1hr and Discovery multimer detection system (Discovery OmniMap HRP, Discovery DAB, Roche) was used to detect and amplify signals.
Statistical Analyses
All data are presented as mean values ± SEM of at least three biological replicates unless otherwise indicated. Two-tailed student t test or ANOVA (one-way or two-way) were used to determine effects of treatments. P<0.05 were considered significant.
Results
Characterization of squamous cell carcinoma cancer stem cells
Growth as non-attached multicellular spheroids can be used to enrich for cancer stem cells with enhanced tumor-forming potential (17–20). Utilizing this method, we set out to compare spheroid-derived human SCC cells to their monolayer counterparts (Fig. 1A, Supplementary Fig. 1A/B). We first examined whether cells grown as spheroids had elevated expression of epithelial and canonical stem cell markers. Monolayer- and spheroid-derived cells were analyzed via immunoblot. We found that a host of stem cell markers, including canonical stem cell markers OCT4 and SOX2 (21), as well as epithelial stem cell markers BRD4, EZH2, the mark of EZH2 activity (H3K27me3) and ΔNp63α (22–24) were increased under spheroid conditions compared to monolayer growth (Fig. 1B, Supplementary Fig. 1 A/B). Because expression of stem cell markers had previously been associated with invasion and metastasis (25), we examined whether spheroid-derived cells displayed enhanced invasion and migration compared to their monolayer counterparts. This revealed that cells grown as spheroids invaded through Matrigel-coated membranes at higher rates than monolayer-derived cells, with more cells having invaded to the high serum-containing underside of the membranes after 24 hours (Fig. 1C and Supplementary Fig. 1C/D). In line with this result, spheroid-derived cells migrated into scratch wounds faster than monolayer-derived cells (Fig. 1D, Supplementary Fig. 1E). Because CSCs are associated with the ability to seed new tumors, we assessed whether these cells were more aggressive in vivo. Therefore, we injected 1 × 106 monolayer- or spheroid-derived cells subcutaneously into the flanks of nude mice and monitored tumor growth. Spheroid-derived cells formed markedly larger, more invasive tumors than monolayer-derived cells (Fig. 1E). Additionally, these tumors maintained elevated levels of both canonical and epithelial stem cell markers (Fig. 1F), suggesting this tumorigenic phenotype was driven by the CSC population. Furthermore, tumor derived single cell suspensions resulted in increased spheroid growth compared to cells derived from monolayer tumors (Fig. 1G), suggesting these cells were maintaining stem like properties in vivo. To assess whether these CSC markers were clinically relevant, we analyzed patient-derived tumor arrays of 80 normal epithelial and SCC samples (Fig. 1H), which showed that each of these markers were elevated in SCC compared to normal control tissues. Thus, SCC spheroids are enriched for CSC-like properties and enhanced expression of CSC markers is observed in human SCC.
Figure 1.
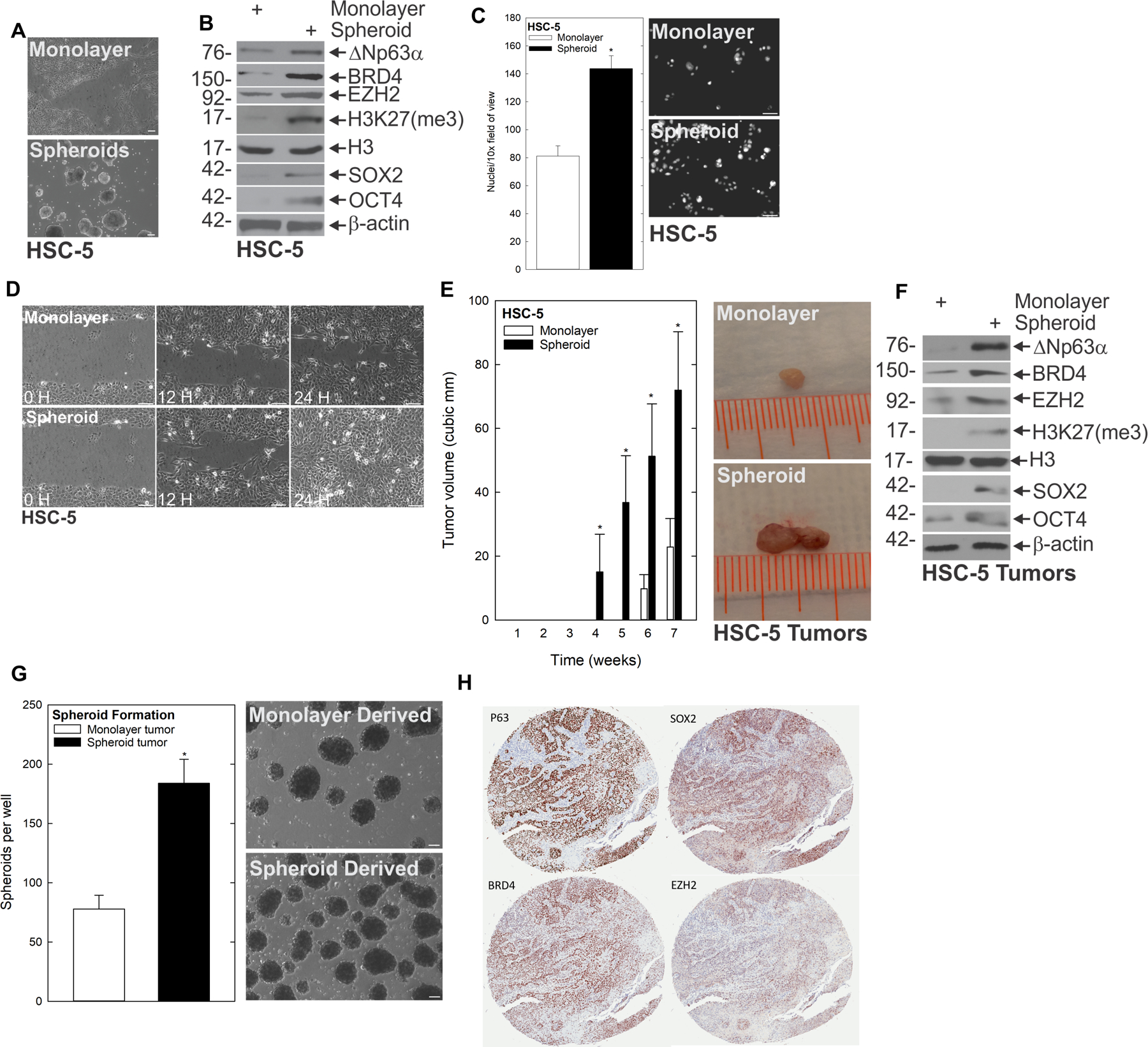
A subpopulation of SCC cells display traits of CSCs. A HSC-5 monolayer cultures maintained in growth medium were harvested and plated at 4×104 cells per 10 cm dish in monolayer cultures or in non-attached conditions as spheroids and monitored for 10 d. Bars = 200 μm B At 10 d, lysates were electrophoresed for detection of the indicated epitopes. C At 10 d, monolayer and spheroid cells were trypsinized and single-cell suspensions were seeded onto Matrigel-coated membranes in Millicell chambers for invasion assays, or D replated as monolayer cultures and allowed to reach confluence at which time they were scratched with a 10 μl pipette tip to create a wound; wound closure was monitored over time. E HSC-5 spheroid- or monolayer-derived cells were injected subcutaneously at 1×106 cells per site in nude mice and tumor growth was monitored. Tumor volume was determined by caliper measurements. The values are mean ± SEM (n=13). Asterisks indicate significant differences in tumor size between the spheroid and monolayer groups at each time point. F Protein extracts were prepared from tumors for immunoblotting to detect the indicated epitopes. G Monolayer- and spheroid-generated tumors were dissociated to create single-cell suspensions, and tumor cells were seeded for spheroid growth assays; spheroid number was monitored for 9 d. H Patient-derived SCC tumor arrays were immunostained to detect the indicated epitopes, and quantified.
BRD4 inhibition or depletion impairs the CSC phenotype
The above studies suggest that a subpopulation of cells with stem cell-like characteristics is responsible for driving an aggressive phenotype in SCC. BRD4 is involved in stem cell regulation, and is frequently overexpressed in a number of cancer types (26–28). We found that BRD4 expression was highly elevated in spheroid cultures (see Fig. 1B), suggesting a role in mediating the CSC-like phenotype. To understand the necessity of BRD4 in this context, we studied how its deficiency impacted spheroid formation, migration and invasion using CRISPR-mediated depletion. As shown in Fig. 2A, and Supplementary Fig. 2A/B, BRD4-depleted cells formed fewer spheroids than their control counterparts. Furthermore, invasion through Matrigel (Fig. 2B, Supplementary Fig. 2C/D) and migration into a scratch wound (Fig. 2C) were also impaired in BRD4-depleted cells. Using the BRD4 small molecule inhibitor JQ1, we found that treatment of pre-formed spheroids with JQ1 reduced the number of spheroids, and led to the accumulation of cell debris (Fig. 2D). We next examined the impact of JQ1 treatment prior to spheroid formation, and found that JQ1 treatment at the time of seeding (prevention conditions) resulted in a roughly 50% decrease in the number of spheroids that formed over 9 days of growth (Fig. 2E, Supplementary Fig. 2E/F). We further assessed the effect of JQ1 on invasion and migration of spheroid-derived cells, revealing that JQ1 treatment slowed the rate of both of these CSC phenotypes as well (Fig. 2F/G, Supplementary Fig. 2G/H). To further validate that these effects were due to BRD4, we used a second BRD4 inhibitor, MS436. Indeed, we found that like JQ1 treatment, inhibition of BRD4 using MS436 impaired the same CSC phenotypes; it reduced spheroid growth, invasion and migration (Fig. 2G/H/I/J/K and Supplementary Fig. 2I/J/K/L). An important goal was to assess whether BRD4 was a valid target in vivo. To determine this, we injected 2.5 × 106 spheroid-derived cells subcutaneously into the flanks of nude mice and monitored tumor growth in control and MS436-treated recipients. Tumors of MS436-treated mice were markedly smaller than controls (Fig. 2L). Additionally, single-cell suspensions of these tumors cultured under spheroid conditions indicated that MS436 treatment reduced spheroid number (Fig. 2M). These findings indicate that BRD4 is essential to SCC cancer stem cells.
Figure 2.
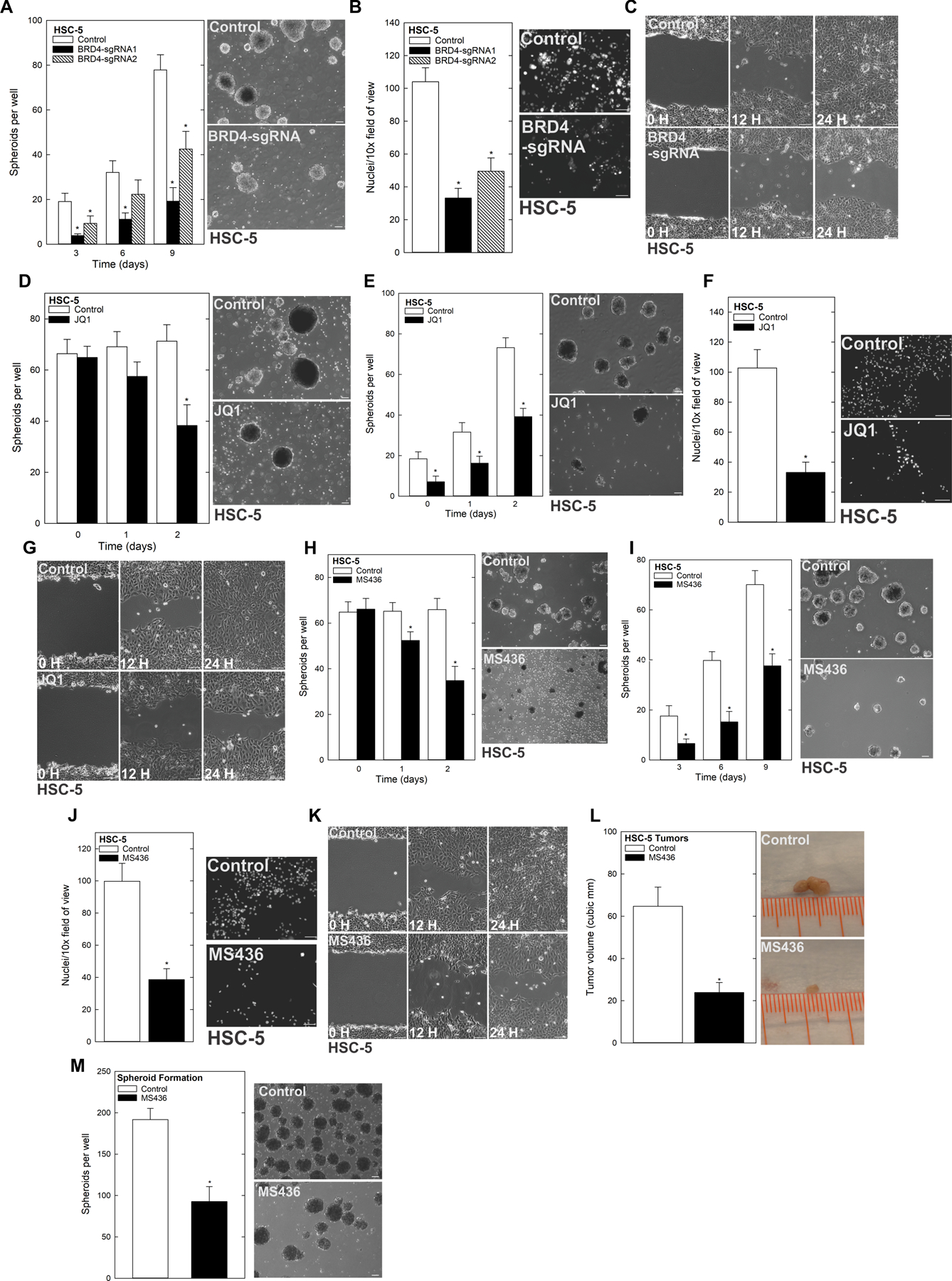
BRD4 regulates the CSC phenotype A HSC-5 Control and BRD4 CRISPR-depleted cells were seeded in spheroid growth conditions and spheroid number monitored for 9 d (left). Representative images on d 9 of growth are shown (right). B HSC-5 Control and BRD4 CRISPR depleted cells were seeded in Matrigel-coated membranes in Millicell chamber for invasion assays. C HSC-5 Control and BRD4 CRISPR-depleted cells were seeded as monolayer cultures and allowed to reach confluence before being scratched with a 10 μl pipette tip to monitor rate of scratch-wound closure. D HSC-5 cells (4×104) were plated in non-adherent six-well plates, grown for 8 d in spheroid medium, and then treated with JQ1 and spheroid numbers were counted over 48 h. The values are mean ± SEM. The asterisks indicate significant difference compared to control. Representative spheroid images following a 3 d-treatment with 0 or 2 μM JQ1 are shown. E HSC-5 cells (4×104) were plated in spheroid growth conditions and at the time of seeding, 0 or 2 μM JQ1 was added. Incubation continued for 9 d without addition of fresh JQ1 or medium, and spheroid numbers were determined at each time point (left). Representative images of day 9 spheroids are shown (right). F/G HSC-5 spheroids were trypsinized to form a single-cell suspensions and reseeded for invasion and migration assays ± JQ1. H Spheroids were grown as in panel A. On day 8, cells were treated with 4 μM MS436 and spheroid numbers were counted over 48 h. The values are mean ± SEM. The asterisks indicate significant difference compared to control. Representative spheroid images following a 3 d treatment with 0 or 4 μM MS436. I HSC-5 cells (4×104) were plated in spheroid growth conditions and at the time of seeding, 0 or 4 μM MS436 was added. Incubation continued for 9 days without addition of fresh MS436 or medium, and spheroid number was counted at each time point. Representative images of day 9 spheroids are shown. J/K HSC-5 spheroids were trypsinized to form single-cell suspensions and reseeded for invasion and migration assays ± JQ1. L HSC-5 spheroid-derived cells were injected at 2.5×106 cells per site in nude mice, and mice were treated with 0 or 50 mg/kg MS436. Tumor volume was determined using caliper measurements. The values are mean ± SEM (n=8). Asterisks indicate significant difference in tumor size between control and treated groups. Representative Control and MS436-treated tumors were harvested on week 7 and photographed. M Control and MS436 treated tumors were dissociated to create single-cell suspensions, and tumor cells were seeded for spheroid growth and spheroid numbers were counted.
To elucidate the mechanism behind the BRD4-driven aggressive phenotype of SCC CSC enrichment, we screened for potential BRD4 targets, and identified numerous changes in expression of epithelial stem cell markers. BRD4 depletion or treatment with BRD4 inhibitors JQ1 and MS436 resulted in the reduction of several key stem cell markers at the protein level, including EZH2, H3K27me3, ΔNp63α and C-MYC (Fig. 3A/B/C/D, Supplementary Fig. 2M/N/O/P). This strongly suggested that BRD4 promoted the undifferentiated state. BRD4 is associated with enhanced C-MYC expression, a known stem cell regulator (29,30). Therefore, we first focused on C-MYC to determine if the compromised stem cell marker expression caused by BRD4 depletion was being driven by C-MYC in the context of SCC. Consistent with this idea, we found that inhibition of BRD4 with MS436 treatment reduced C-MYC mRNA levels (Fig. 3E). To further investigate the role of C-MYC in BRD4-driven stemness, we performed genetic rescue experiments in which C-MYC was overexpressed in the context of BRD4 inhibition (i.e., in the presence vs. absence of JQ1), and monitored the CSC phenotype. This revealed that whereas both spheroid formation and invasion were impaired upon JQ1 treatment, this effect was mitigated in the presence of forced C-MYC expression (Fig. 3F/G). We verified this finding using a second BRD4 inhibitor, MS436, revealing that C-MYC effectively rescued the impaired spheroid formation and invasion caused by inhibiting BRD4 with this compound as well (Fig. 3H/I, Supplementary Fig. 3A/B). In addition to rescuing the biological effects of BRD4 inhibition, we found that forced C-MYC expression also mitigated MS436-induced changes in epithelial stem cell marker expression, with C-MYC effectively rescuing both EZH2 and ΔNp63α expression despite BRD4 being inhibited (Fig. 3J, Supplementary Fig. 3C/D). This evidence indicates that BRD4 is essential for CSC phenotypes and that it induces the epithelial stem cell markers EZH2 and ΔNp63α through C-MYC.
Figure 3.
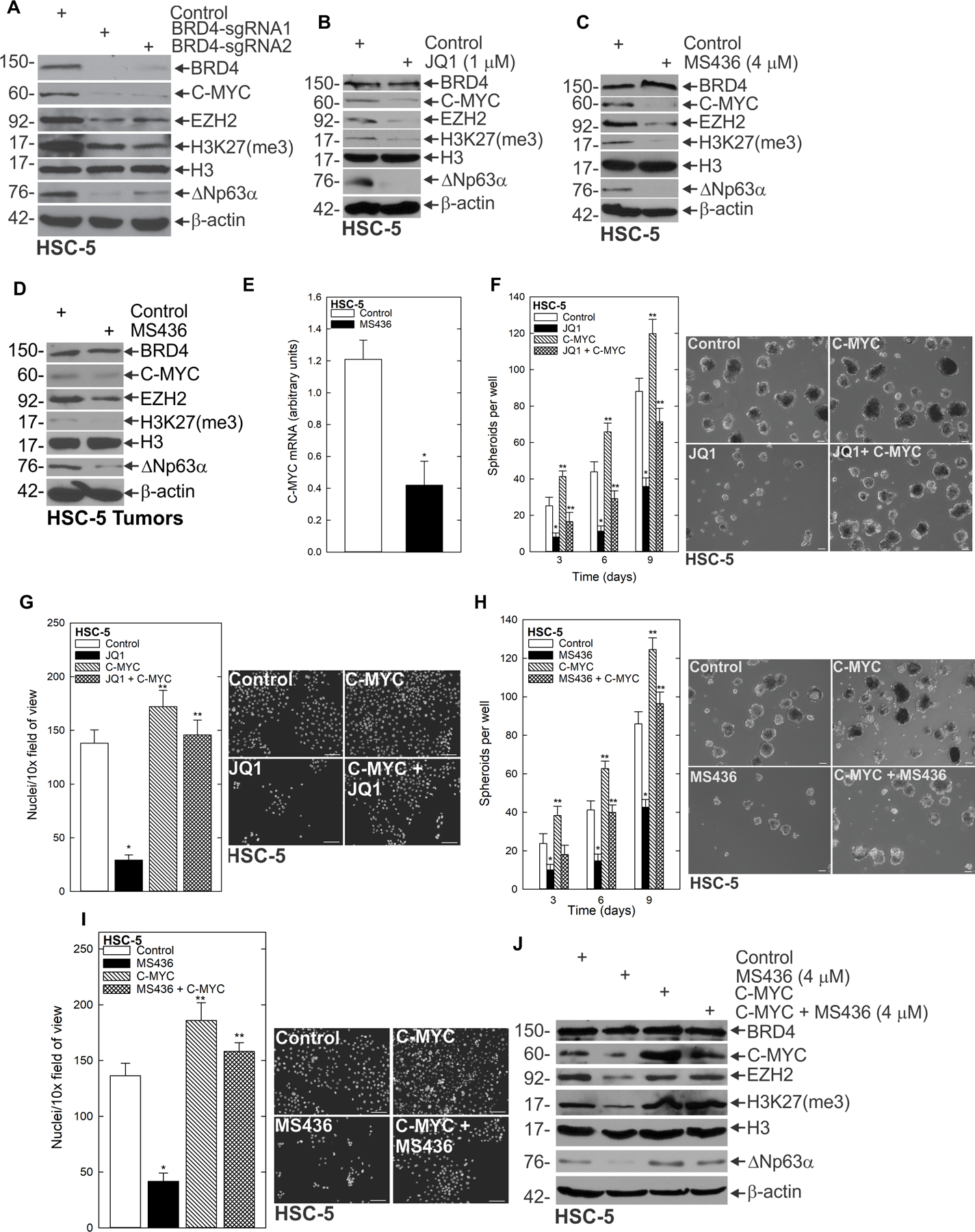
BRD4 regulates CSC markers via C-MYC A HSC-5 Control empty vector (EV) and BRD4 CRISPR-depleted cells were grown in spheroid culture and lysates were collected for immunoblotting. Spheroids were grown for 8 d then treated with JQ1 (B) or MS436 (C) for 48 h, and harvested for immunoblot. D Protein extracts were prepared from tumors for immunoblotting to detect the indicated epitopes. E Spheroids on d 8 were treated with MS436 or DMSO for 24 h and mRNA was collected for QRT-PCR. F HSC-5 cells infected with lentivirus expressing C-MYC or EV were seeded in spheroid growth conditions and treated with JQ1 or DMSO at the time of seeding. Spheroid number was monitored for 9 d. Representative spheroid images are shown from d 9 of growth. G Cells as described in F were seeded in Matrigel invasion assays ± JQ1. H/I HSC-5 cells infected with lentivirus expressing EV or C-MYC were seeded in spheroid growth conditions or for Matrigel invasion assays, treated with MS436, and monitored for spheroid formation and invasion. J Immunoblots of the indicated epitopes following 48 h of MS436 or DMSO treatment of EV and C-MYC overexpressing cells.
A non-canonical function of EZH2 regulates STAT3 activity
Understanding the mechanisms governing CSC signaling and the aggressive phenotype is a critical goal. To determine whether the compromise in EZH2 we found following BRD4 depletion was required for the CSC phenotype, we performed depletion and inhibitor studies for EZH2. Our finding that EZH2 expression was reduced when BRD4 was targeted prompted us to ask whether EZH2 loss had a similar effect as BRD4 loss in suppressing CSC phenotypes. Polycomb repressive complex 1 (PRC1) and Polycomb repressive complex 2 (PRC2) suppress gene expression via covalent modification of select histones, leading to reduced expression of tumor suppressors (31,32). EZH2 is a lysine methyltransferase and is the main catalytic subunit of the PRC2 complex. EZH2 catalyzes trimethylation of lysine 27 of histone 3 (H3K27me3), an important step in Polycomb-mediated silencing of gene expression. We assessed the impact of EZH2 depletion on the CSC phenotype, and found that CRISPR-mediated EZH2 loss compromised spheroid formation, invasion and migration (Fig. 4A/B/C, Supplementary Fig. 4A/B/C/D). Furthermore, these cellular outcomes were accompanied by reduced expression of stem cell markers (Fig. 4D). Whereas EZH2 depletion resulted in reduced ΔNp63α and compromised H3K27(me3), no appreciable changes in levels of BRD4 or C-MYC were observed, indicating that EZH2 functioned downstream of BRD4 and C-MYC, but upstream of ΔNp63α. To further validate this signaling and determine whether the effect of C-MYC on ΔNp63α expression is dependent on EZH2, we forced expression of C-MYC in BRD4 or EZH2 deficient cells and monitored for ΔNp63α expression. Expression of C-MYC in BRD4 depleted cells rescues ΔNp63α protein expression, but not in EZH2 knockout cells (Fig. 4E, Supplementary Fig. 4E/F), suggesting EZH2 is critical to C-MYC driven ΔNp63α. Having identified this BRD4-EZH2-ΔNp63α axis, we asked whether the methyltransferase activity of EZH2 was essential for the CSC phenotype. We found that inhibition of EZH2’s methyltransferase activity with GSK126—a selective, small molecule inhibitor of EZH2 that competes for the substrate-binding site of EZH2 and thereby inhibits its methyltransferase activity—reduced spheroid formation, invasion and migration (Fig. 4F/G/H, Supplementary Fig. G/H/I/J). Whereas GSK126 reduced H3K27me3 levels (hence indicating that EZH2 methyltransferase activity was being impaired) it did not alter overall levels of EZH2 (Fig. 4I). Importantly, GSK126 treatment effectively compromised ΔNp63α expression, indicating that EZH2-mediated ΔNp63α induction was dependent on the methyltransferase activity of EZH2. To assess whether EZH2 functioned to maintain ΔNp63α expression in vivo, we injected 2.5 × 106 spheroid cells per each front flank of nude mice and treated animals with 25 mg/kg body weight GSK126. Treatment with GSK126 in vivo reduced overall tumor volume, while simultaneously reducing levels of H3K27me3 and ΔNp63α (Fig. 4J, K). We found that GSK126 targeted the cell population responsible for the aggressive phenotype, as growth of dissociated tumor cells as spheroids produced significantly fewer spheroids when mice had been treated with GSK126 (Fig. 4L). These findings indicate that the methyltransferase activity of EZH2 is required for inducing the ΔNp63α–driven CSC phenotype.
Figure 4.
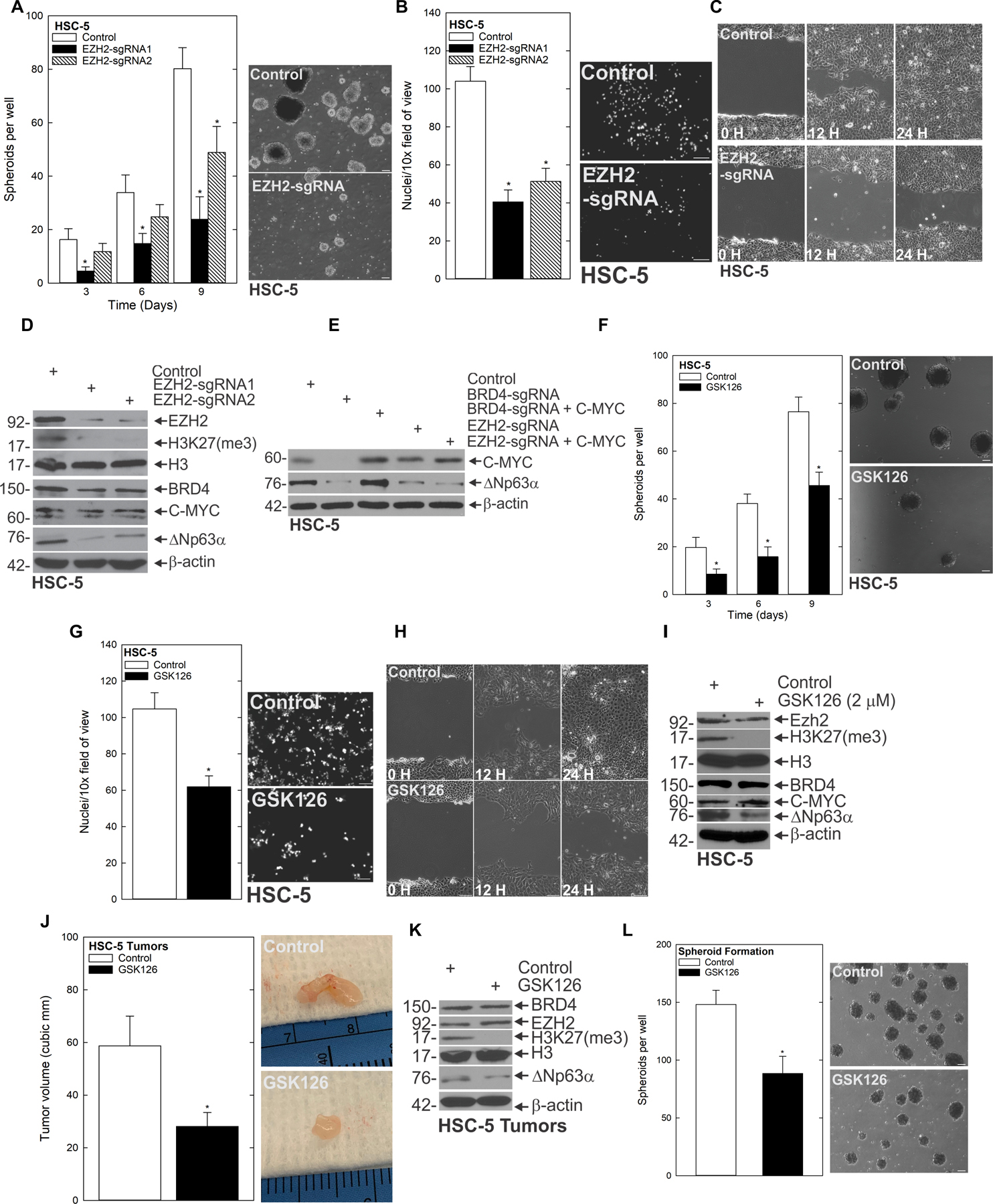
BRD4-driven EZH2 is important for the CSC phenotype A HSC-5 Control and EZH2 CRISPR-depleted cells were seeded in spheroid growth conditions and spheroid number monitored for 9 d. B/C The indicated cell lines were seeded in invasion and migration assays and monitored over time. D The indicated cell lines were grown as spheroids then harvested for immunoblot.E C-MYC plasmid was expressed in BRD4 or EZH2 CRISPR depleted cells and lysates collected for immunoblot of ΔNp63α. F HSC-5 cells (4×104) were plated in spheroid growth conditions and at the time of seeding, 0 or 2 μM GSK126 was added. Incubation continued for 9 d without addition of fresh GSK126 or medium, and spheroid number was counted at each time point G/H HSC-5 spheroid-derived cells were seeded for invasion and migration assays ± GSK126 and monitored over time. I Spheroids were grown for 8 d and then treated with DMSO or GSK126 for 48 h upon which lysates were collected for immunoblots to detect the indicated epitopes. J HSC-5 spheroid-derived cells were injected at 2.5×106 cells per site into nude mice, and mice treated with 0 or 25 mg/kg GSK126. Tumor volume was determined using caliper measurements. The values are mean ± SEM (n=12). Asterisks indicate significant difference in tumor size between control and treated groups. K Protein extracts were prepared from tumors for immunoblotting to detect the indicated epitopes. L Tumors from control and GSK126-treated mice were dissociated to create single-cell suspensions, and tumor cells were seeded to monitor spheroid growth.
EZH2 methyltransferase activity is associated with transcriptional repression and reduced expression of target genes (33). The fact that we had found reduced ΔNp63α expression in response to EZH2 depletion or inhibition suggested the possibility that EZH2 was regulating ΔNp63α in a non-canonical fashion, even though its methyltransferase activity responsible for its canonical function of placing H3K27me3 was required. To investigate how EZH2 was regulating ΔNp63α, we screened for EZH2-interacting partners that have also been linked to ΔNp63α regulation. This led us to focus on Signal Transducer and Activator of Transcription 3 (STAT3). STAT3 activation by tyrosine phosphorylation promotes cell proliferation, angiogenesis, immune evasion and metastasis (34–36). Additionally, STAT3 activity is elevated in CSC’s in a number of cancer types (37). Furthermore, STAT3 is capable of binding to the promoter of ΔNp63α, and transcriptionally activating it (37). Therefore, we hypothesized that STAT3 was part of the non-canonical mechanism by which EZH2 regulated ΔNp63α. To explore this possibility, we asked whether EZH2 was required for STAT3 expression. Depletion (Fig. 5A) and inhibition (Fig. 5B, Supplementary Fig. 4K/L) studies showed that the phosphorylated active form of STAT3 (STAT3-P) was reduced in response to both types of EZH2 perturbation. To better understand how EZH2 was activating STAT3, we pulled down EZH2 using co-immunoprecipitation; this revealed that EZH2 and STAT3 interact (Fig. 5C). Because GSK126 specifically reduced STAT3-P, we utilized co-immunoprecipitation of methyl K and STAT3 in the presence or absence of GSK126. Whereas STAT3 was methylated under control conditions, inhibition of EZH2 by GSK126 reduced STAT3 methylation (Fig. 5D). To assess whether STAT3 activity was required for the CSC phenotype, we utilized a STAT3 inhibitor, STATTIC. STATTIC is a vinyl-sulfone compound that targets the STAT3-SH2 domain, preventing its activation by phosphorylation (38). We found that STATTIC effectively targeted the CSC phenotype; it reduced spheroid maintenance and formation, as well as invasion and migration (Supplementary Fig 5A-H). Furthermore, these changes in the CSC phenotype appeared to be dependent on the ability of STAT3-P to regulate ΔNp63α expression, as treatment of preformed spheroids with STATTIC reduced ΔNp63α expression at both the transcript and protein levels (Supplementary Fig. 5I-L). To further investigate if STAT3 activity was sufficient for the EZH2-driven phenotype and ΔNp63α expression, we treated with STAT3C—a constitutively active form of STAT3—in the context of EZH2 inhibition with GSK126. This revealed that STAT3C rescued the compromised spheroid formation and invasion caused by interfering with the methyltransferase activity of EZH2 (Fig. 5E/F), and further shows that rescue of the CSC phenotype coincided with restoration of ΔNp63α expression (Fig. 5G). To further demonstrate that methylation of STAT3 by EZH2 was critical to driving the aggressive phenotype and ΔNp63α expression, we used CRISPR to deplete STAT3, then rescued with either wildtype STAT3 or STAT3 with the EZH2 methylation site mutated (K180A). Fig. 5H/I/J and Supplementary Fig. 5L-Q shows that STAT3 depletion reduces spheroid formation, invasion, and ΔNp63α expression that is rescued by wildtype STAT3, but not the methylation mutant STAT3 K180A. These findings demonstrate the importance of STAT3 methylation in driving the aggressive phenotype and ΔNp63α. Fig 5K and supplemental Fig. 5R show that when the EZH2 methylation site on STAT3 is mutated, EZH2 does not co-immunoprecipitate with STAT3. To determine if STAT3 was a potential therapeutic target to reduce ΔNp63α in vivo, tumor-bearing mice were treated with STATTIC. Tumors of STATTIC-treated mice were markedly smaller than controls (Fig. 5L), and had reduced ΔNp63α expression (Fig. 5M). Additionally, single-cell suspensions of these tumors cultured under spheroid conditions indicated that STATTIC treatment reduced spheroid number, suggesting that STATTIC was targeting the population of tumor cells with stem like properties (Fig. 5N). These findings elucidate a novel mechanism whereby EZH2 uses its non-canonical function to activate STAT3 by methylating it, serving as an oncogenic switch linking a BRD4-ΔNp63α cascade that drives the CSC population.
Figure 5.
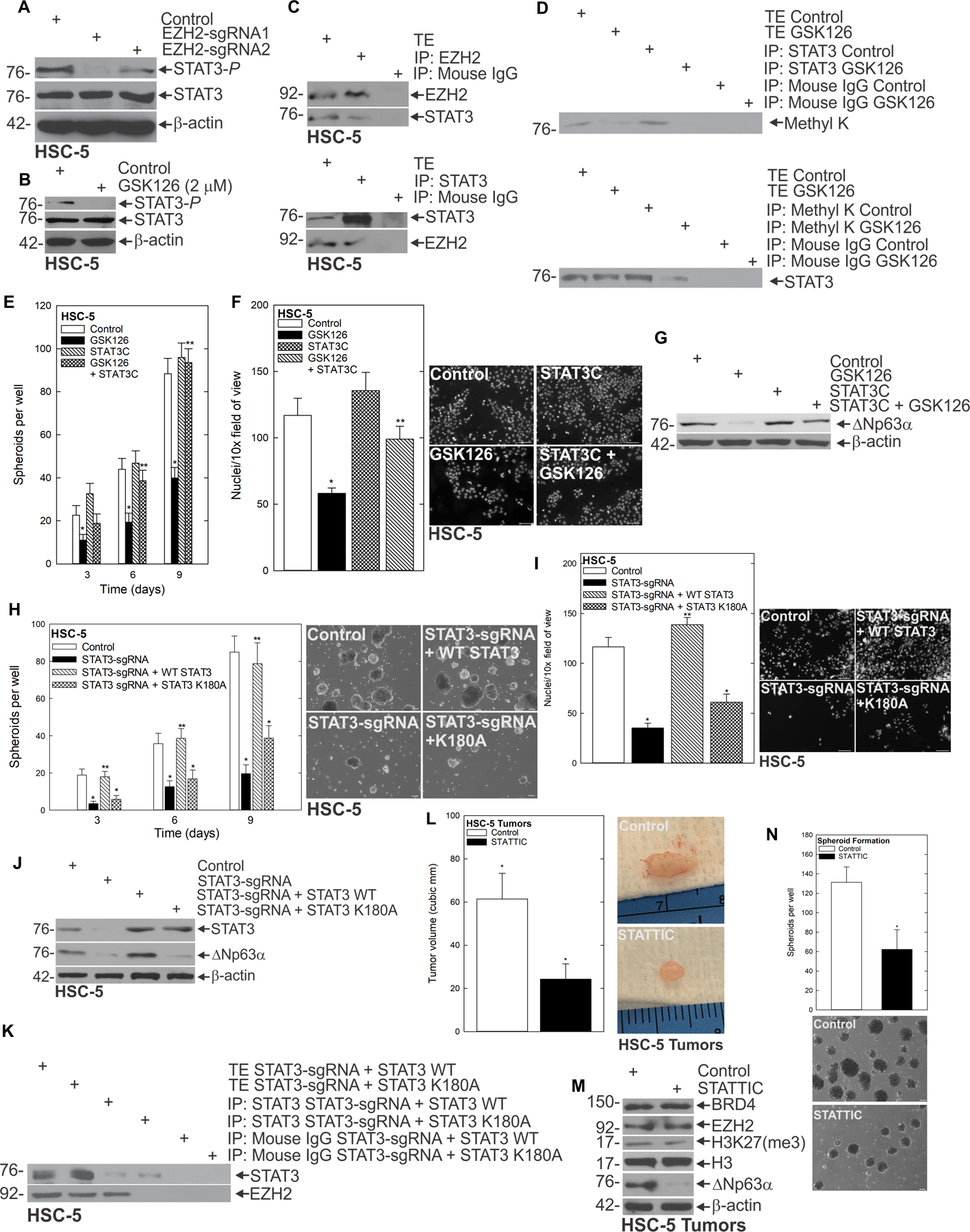
EZH2 interacts with STAT3 to regulate the CSC phenotype A Control and EZH2-sgRNA expressing cells were harvested for immunoblot and probed for the indicated epitopes. B HSC-5 spheroids were treated with GSK126 for 48 h then lysates collected for immunoblotting. C HSC-5 spheroids were harvested for protein extraction for immunoprecipitation/immunoblot. D HSC-5 spheroids were treated with GSK126 or DMSO for 48 h prior to lysates being collected for immunoprecipitation and immunoblot. HSC-5 cells expressing EV or STAT3C were treated with GSK126 and spheroid formation (E) and invasion (F) were monitored. G Protein lysates were collected for western blot of the indicated proteins. H STAT3 CRISPR depleted HSC-5 cells containing wildtype STAT3 or methylation mutant K180A STAT3 were grown as spheroids for 10 days and spheroid number counted. I STAT3 CRISPR depleted HSC-5 cells containing wildtype STAT3 or methylation mutant K180A STAT3 were seeded on a Matrigel coated membrane for invasion assays. J Protein lysates were collected from STAT3 CRISPR depleted HSC-5 cells containing wildtype STAT3 or methylation mutant K180A STAT3 and immunoblots run for the indicated epitopes. K Protein lysates were collected from STAT3 CRISPR depleted HSC-5 cells containing wildtype STAT3 or methylation mutant K180A STAT3 and Co-Immunoprecipitation done for the indicated proteins. L HSC-5 spheroid-derived cells were injected as described above and mice were treated with 0 or 25 mg/kg STATTIC. Tumor volume caliper measurements are mean ± SEM (n=8). Asterisks indicate significant difference in volume between control and treated groups Representative Control and STATTIC treated tumors were harvested on week seven and photographed. M Protein extracts were prepared from tumors for western blot to detect the indicated epitopes. N Tumors from Control and STATTIC treated mice were used to create single-cell suspensions, and cells were seeded in non-attached conditions to monitor the ability to form spheroids.
ΔNp63α is the critical regulator of the aggressive phenotype
ΔNp63α is an important member of the p53 family of transcription factors involved in epithelial cell differentiation, stem cell status and fate (39). ΔNp63α is a key bottleneck of epithelial differentiation that maintains the undifferentiated stem cell state of proliferating basal layer cells in a variety of epithelia (39). We reasoned that the BRD4-EZH2-regulated pathway was critical for the CSC phenotype because of its ability to regulate ΔNp63α expression. To test this idea, we created pan-p63-depleted cells lacking all p63 isoforms using gene editing, and found that the predominant p63 isoform in epithelial tissues and in SCC, ΔNp63α, was dramatically reduced (Fig. 6A). CSC phenotypes such as spheroid formation (Fig. 6B, Supplementary Fig. 6A/B), as well as invasion and migration (Fig. 6C/D) were dramatically reduced in pan-p63-depleted cells. To further validate ΔNp63α as the isoform responsible for the phenotype, we re-expressed ΔNp63α in pan-p63-depleted cells (Fig. 6E), and assessed its ability to rescue the effect of p63 loss. We found that whereas spheroid formation and invasion were severely compromised in cells deficient for all p63 isoforms, ΔNp63α expression alone was sufficient for restoring CSC phenotypes (Fig. 6F/G, supplementary Fig. 6C/D). To determine if ΔNp63α was capable of driving the phenotype independent of BRD4 and EZH2, we re-expressed ΔNp63α into BRD4-deficient and EZH2-deficient cells (Fig. 6H). Importantly, expression of ΔNp63α was sufficient for rescuing spheroid formation and invasion, despite the lack of BRD4 or EZH2 in these cells (Fig. 6I/J, supplementary Fig. 6E/F). These findings indicate that the BRD4-EZH2-STAT3 oncogenic network culminates with ΔNp63α to drive cancer stem cells in SCC.
Figure 6.
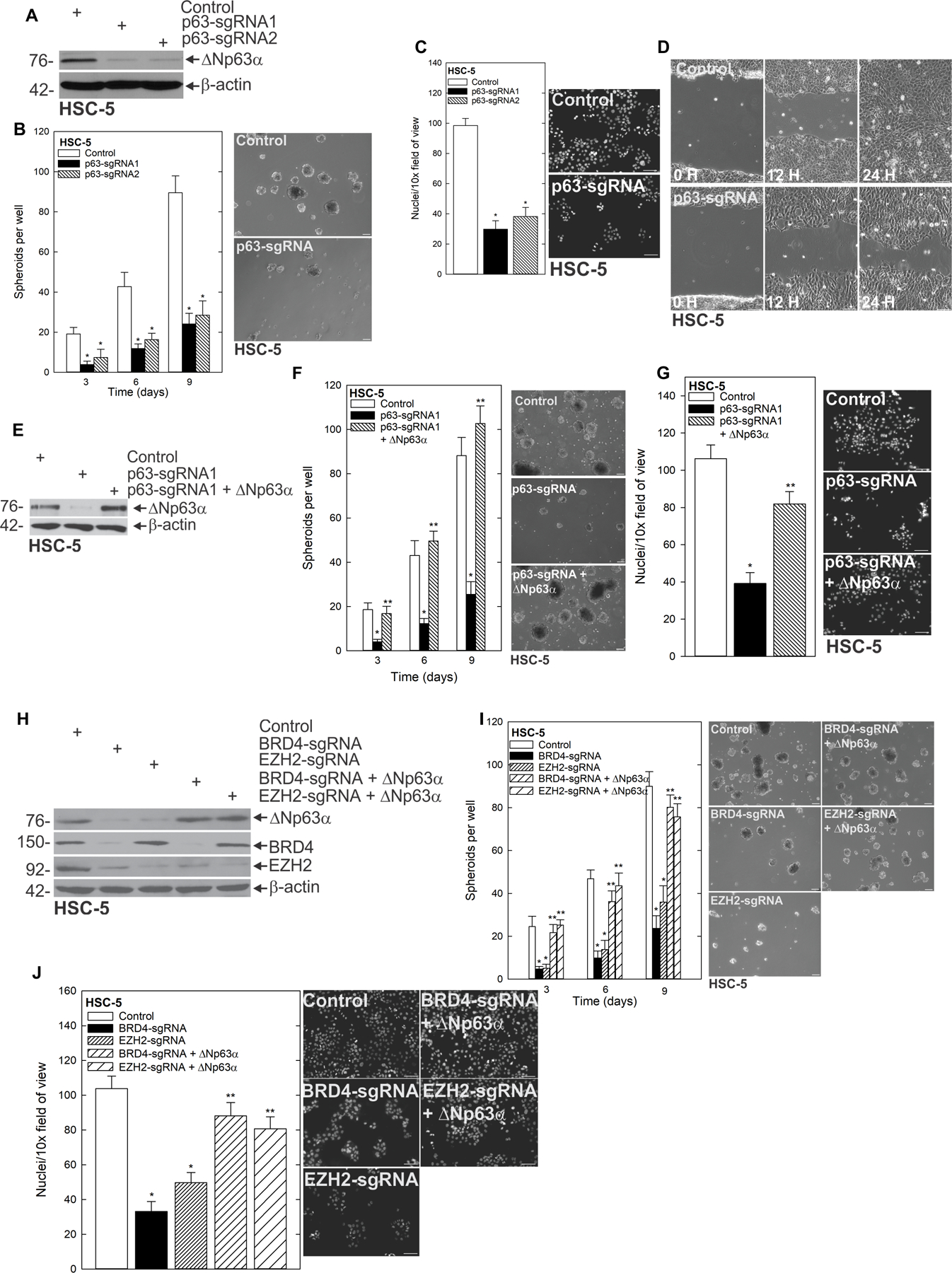
ΔNp63α is the critical component in maintaining the CSC phenotype A HSC-5 cells were treated with control (EV) or sgp63 expressing lentivirus, and harvested after selection with G418 to confirm depletion. B Control and p63 CRISPR depleted cells were seeded in spheroid growth conditions and spheroid number was counted and imaged over time. The indicated cell lines were seeded for invasion (C) and migration (D) assays, and rates of respective endpoints were monitored and imaged over time. E Control, p63-sgRNA and p63-sgRNA + ΔNp63α expressing cells were harvested for protein to confirm expression and F seeded in spheroid growth conditions and spheroid number was monitored over time. G Control, p63-sgRNA and p63-sgRNA + ΔNp63α expressing cells were seeded on membranes for an invasion assay and number of invading cells counted after 24 h. H Control, BRD4-sgRNA ± ΔNp63α and EZH2-sgRNA ± ΔNp63α expressing cells were harvested for protein and I seeded in spheroid conditions or H invasion assays and spheroid number and rate of invasion were monitored respectively. J Control, BRD4-sgRNA ± ΔNp63α and EZH2-sgRNA ± ΔNp63α expressing cells were grown as spheroids and protein lysates collected for western blot of the indicated epitopes.
Discussion
The cancer stem cell (CSC) model suggests that a fraction of cancer cells possess the ability to initiate and to sustain tumor growth, thereby contributing to tumor relapse (40). CSCs share many features with their normal stem cell counterparts, including their slow-cycling nature, and the ability to give rise to a hierarchy of daughter cells with varying degrees of differentiation (41). These CSCs undergo aberrant differentiation, creating cellular heterogeneity within the tumor (42). Furthermore, CSCs display enhanced anti-apoptotic signaling, increased membrane transporter activity, and enhanced migratory, invasive and metastatic potential (41). Identification and characterization of CSCs in various cancer types can lead to the development of therapeutic strategies that target the CSC population, thus preventing metastasis and recurrence.
An important strategy for targeting CSCs is identifying proteins that are critical for driving their aggressive phenotype. In order to do this, we screened a number of known epithelial and canonical stem cell markers in SCC CSCs compared to their bulk monolayer counterparts. We identified a subpopulation of cells (0.14%) that survive when selected under spheroid conditions. These spheroid-derived cells express elevated levels of a number of stem cell markers including BRD4, EZH2, ΔNp63α, SOX2 and OCT4. BRD4, EZH2 and ΔNp63α are expressed robustly in epithelial stem cells and are critical regulators of self-renewal and the undifferentiated state (9,23,26,43). SOX2 and OCT4 are canonical stem cell markers that are overexpressed in some cancer stem cell types including prostate, breast and epidermal squamous cell carcinoma (44,45). Overexpression of these critical stem cell markers in spheroid culture suggests that spheroids are enriched for cells with similar properties to that of embryonic stem cells and may be evidence that they are transformed. In addition to the elevated stem cell marker levels in spheroid culture, spheroid-derived cells behave more aggressively, migrating to close a scratch wound more rapidly, and invading through Matrigel at higher rates than the bulk monolayer counterparts. Spheroid-derived cells also form large and invasive tumors, further evidence of the aggressive phenotype of cells containing these CSC-like properties. We also demonstrate that stem cell marker expression is maintained in the tumors in vivo, indicating that the CSC population is enriched within the tumor. Furthermore, these markers are elevated in patient-derived SCC specimens compared to normal tissue, suggesting these targets are clinically relevant.
In these studies, we identify a BRD4-driven signaling cascade that mediates the ΔNp63α-dependent aggressive phenotype of CSCs in SCC. BRD4 is an important member of the BET domain family of proteins, characterized by two N-terminal bromodomains and an extra-terminal (ET) domain. Previous studies have shown that BRD4 binds directly to acetylated lysine residues of histones as well as those of transcription factors, and can recruit transcriptional co-regulators (46). Importantly, BRD4 is significantly overexpressed in a number of cancer types where it acts as an oncogene, including melanoma, colon, and bladder cancer (47–49).
Gene editing identified BRD4 as critical to the CSC phenotype, with BRD4 depletion impairing spheroid formation, migration and invasion. To further explore BRD4 as a potential therapeutic target of SCC CSCs, we utilized JQ1 and MS436—two small molecule inhibitors of BRD4. JQ1 competitively binds to the acetyl-lysine recognition site of BRD4’s bromodomain, displacing BRD4 from acetylated chromatin and repressing transcription of target genes (50). MS436 inhibits BRD4 transcriptional activity and has a 100-fold higher affinity for the first bromodomain of BRD4 over the second (51). Treatment with either inhibitor results in the destruction of preformed spheroids, reduces the number of spheroids forming when treated at initiation of growth, and impairs invasion and migration. Importantly, tumor growth in vivo is greatly impaired by both of these BRD4 inhibitors. This data has important translational implications, as it demonstrates that pharmacological inhibition of BRD4 can be used to target SCC CSCs.
Understanding how BRD4 regulates this phenotype was an important goal, and the changes in the CSC phenotype following perturbation of BRD4 prompted us to look at other stem cell markers elevated within the spheroid population. We show that depletion or inhibition of BRD4 causes a dramatic reduction of C-MYC expression at both the protein and the mRNA level. Additionally, other stem cell markers including EZH2 and ΔNp63α are reduced at the protein level following BRD4 depletion and inhibition. Forced expression of C-MYC rescues spheroid formation and invasion in the presence of BRD4 inhibitors, demonstrating that C-MYC is a critical mediator of the BRD4-driven aggressive CSC phenotype. Additionally, we show that EZH2 and ΔNp63α protein expression are restored by C-MYC as well. This led us to look at whether EZH2 was also an essential component of the signaling cascade regulating the aggressive phenotype.
EZH2 depletion experiments produce similar changes to the aggressive phenotype as BRD4 depletion, implicating EZH2 as a key component of the BRD4-mediated axis. GSK126 is a selective competitive inhibitor of S-adenosyl-methionine-dependent EZH2 methyltransferase activity (32). Treatment with GSK126 is associated with reduced catalytic activity as demonstrated by compromised H3K27me3 (32). An important finding is that GSK126 suppresses the aggressive phenotype, including spheroid formation and maintenance, invasion, migration and tumor growth. EZH2-depleted cells demonstrate not only a reduction in spheroid formation, migration and invasion, but reduced ΔNp63α expression. However, BRD4 and C-MYC protein levels remain unchanged, indicating that BRD4 and C-MYC are upstream of EZH2, and that EZH2 is upstream of ΔNp63α.
Immunoprecipitation studies show that EZH2 interacts with STAT3 in spheroids, and this interaction results in the methylation and subsequent activation (phosphorylation) of STAT3 via EZH2. These findings are consistent with previous reports showing that EZH2 can activate STAT3 in Glioblastoma stem cells (52). This was further shown to be dependent on methylation of K180 on STAT3 by EZH2 in glioblastoma CSCs (52). Our studies show that STAT3 is not only important for the BRD4/EZH2-driven phenotype, but that it is a critical regulator of ΔNp63α mRNA and protein. Although more studies are required to determine the exact mechanism behind increased STAT3 activation following methylation, our findings highlight EZH2 and STAT3 as potential therapeutic vulnerabilities for SCC CSCs.
The observation that ΔNp63α is reduced in each of the depletion and inhibitor studies that produce a biological response prompted us to examine whether ΔNp63α is the key driver of this phenotype. We show that pan-p63 depletion severely impairs spheroid formation, invasion and migration, and that these phenotypes are completely rescued by re-expression of the ΔNp63α isoform. Importantly, forced expression of ΔNp63α in BRD4- and EZH2-depleted cells restores the aggressive CSC phenotype as well. This demonstrates that BRD4 and EZH2 drive the CSC phenotype via regulation of ΔNp63α, and that ΔNp63α is the critical regulator of this phenotype.
The above studies indicate that ΔNp63α is a critical regulator of the aggressive phenotype of CSCs. Furthermore, two chromatin modifying proteins that are highly elevated in epithelial stem cells—BRD4 and EZH2—are essential for maintaining ΔNp63α expression. These findings highlight BRD4 and EZH2 as potential therapeutic targets in SCC to effectively reduce ΔNp63α and in turn, mitigate the CSC phenotype. This work has important therapeutic implications for preventing recurrence, drug resistance and metastasis.
Supplementary Material
Significance.
This study identifies a signaling cascade driven by BRD4 that upregulates ΔNp63α to promote cancer stem-like properties, which has potential therapeutic implications for the treatment of squamous cell carcinomas.
Acknowledgements
This work was supported by the Office of the Director, National Institutes of Health through award numbers 5P30CA045508 (Cancer Center Support Grant), CA225134 (to M.L. Fisher), CA247400 (to S.Balinth), as well as R01CA190997 and R21OD018332 (to A.A. Mills). This project was also supported through the Cold Spring Harbor Laboratory and Northwell Health Affiliation.
Footnotes
Competing Interests
The authors declare no competing interests.
References
- 1.Marur S, Forastiere AA. Head and Neck Squamous Cell Carcinoma: Update on Epidemiology, Diagnosis, and Treatment. Mayo Clin Proc 2016;91:386–96 [DOI] [PubMed] [Google Scholar]
- 2.Sánchez-Danés A, Blanpain C. Deciphering the cells of origin of squamous cell carcinomas. Nat Rev Cancer 2018;18:549–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Specenier P, Vermorken JB. Optimizing treatments for recurrent or metastatic head and neck squamous cell carcinoma. Expert Rev Anticancer Ther 2018;18:901–15 [DOI] [PubMed] [Google Scholar]
- 4.Qian X, Nie X, Wollenberg B, Sudhoff H, Kaufmann AM, Albers AE. Heterogeneity of Head and Neck Squamous Cell Carcinoma Stem Cells. Adv Exp Med Biol 2019;1139:23–40 [DOI] [PubMed] [Google Scholar]
- 5.Kumar R, Li DQ, Müller S, Knapp S. Epigenomic regulation of oncogenesis by chromatin remodeling. Oncogene 2016;35:4423–36 [DOI] [PubMed] [Google Scholar]
- 6.Qi J. Bromodomain and extraterminal domain inhibitors (BETi) for cancer therapy: chemical modulation of chromatin structure. Cold Spring Harb Perspect Biol 2014;6:a018663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo X, Mills AA. p63, cellular senescence and tumor development. Cell Cycle 2007;6:305–11 [DOI] [PubMed] [Google Scholar]
- 8.Mills AA, Zheng B, Wang XJ, Vogel H, Roop DR, Bradley A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 1999;398:708–13 [DOI] [PubMed] [Google Scholar]
- 9.Candi E, Cipollone R, Rivetti di Val Cervo P, Gonfloni S, Melino G, Knight R. p63 in epithelial development. Cell Mol Life Sci 2008;65:3126–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fisher ML, Kerr C, Adhikary G, Grun D, Xu W, Keillor JW, et al. Transglutaminase Interaction with alpha6/beta4-Integrin Stimulates YAP1-Dependent DeltaNp63alpha Stabilization and Leads to Enhanced Cancer Stem Cell Survival and Tumor Formation. Cancer Res 2016;76:7265–76 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 11.Grun D, Adhikary G, Eckert RL. NRP-1 interacts with GIPC1 and alpha6/beta4-integrins to increase YAP1/Np63alpha-dependent epidermal cancer stem cell survival. Oncogene 2018;37:4711–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keyes WM, Pecoraro M, Aranda V, Vernersson-Lindahl E, Li W, Vogel H, et al. DeltaNp63alpha is an oncogene that targets chromatin remodeler Lsh to drive skin stem cell proliferation and tumorigenesis. Cell Stem Cell 2011;8:164–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bergholz J, Xiao Z-X. Role of p63 in Development, Tumorigenesis and Cancer Progression. Cancer Microenviron 2012;5:311–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hillion J, Dhara S, Sumter TF, Mukherjee M, Di Cello F, Belton A, et al. The high-mobility group A1a/signal transducer and activator of transcription-3 axis: an achilles heel for hematopoietic malignancies? Cancer Res 2008;68:10121–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chatterjee A, Upadhyay S, Chang X, Nagpal JK, Trink B, Sidransky D. U-box-type ubiquitin E4 ligase, UFD2a attenuates cisplatin mediated degradation of DeltaNp63alpha. Cell Cycle 2008;7:1231–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ricci MS, Jin Z, Dews M, Yu D, Thomas-Tikhonenko A, Dicker DT, et al. Direct repression of FLIP expression by c-myc is a major determinant of TRAIL sensitivity. Mol Cell Biol 2004;24:8541–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adhikary G, Grun D, Kerr C, Balasubramanian S, Rorke EA, Vemuri M, et al. Identification of a population of epidermal squamous cell carcinoma cells with enhanced potential for tumor formation. PLoS One 2013;8:e84324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishiguro T, Ohata H, Sato A, Yamawaki K, Enomoto T, Okamoto K. Tumor-derived spheroids: Relevance to cancer stem cells and clinical applications. Cancer Sci 2017;108:283–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.López J, Poitevin A, Mendoza-Martínez V, Pérez-Plasencia C, García-Carrancá A. Cancer-initiating cells derived from established cervical cell lines exhibit stem-cell markers and increased radioresistance. BMC Cancer 2012;12:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mancini R, Giarnieri E, De Vitis C, Malanga D, Roscilli G, Noto A, et al. Spheres derived from lung adenocarcinoma pleural effusions: molecular characterization and tumor engraftment. PLoS One 2011;6:e21320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rizzino A, Wuebben EL. Sox2/Oct4: A delicately balanced partnership in pluripotent stem cells and embryogenesis. Biochim Biophys Acta 2016;1859:780–91 [DOI] [PubMed] [Google Scholar]
- 22.Adhikary G, Grun D, Balasubramanian S, Kerr C, Huang JM, Eckert RL. Survival of skin cancer stem cells requires the Ezh2 polycomb group protein. Carcinogenesis 2015;36:800–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ezhkova E, Pasolli HA, Parker JS, Stokes N, Su IH, Hannon G, et al. Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell 2009;136:1122–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pellegrini G, Dellambra E, Golisano O, Martinelli E, Fantozzi I, Bondanza S, et al. p63 identifies keratinocyte stem cells. Proc Natl Acad Sci U S A 2001;98:3156–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nandy SB, Lakshmanaswamy R. Cancer Stem Cells and Metastasis. Prog Mol Biol Transl Sci 2017;151:137–76 [DOI] [PubMed] [Google Scholar]
- 26.Dey A, Yang W, Gegonne A, Nishiyama A, Pan R, Yagi R, et al. BRD4 directs hematopoietic stem cell development and modulates macrophage inflammatory responses. Embo j 2019;38. [DOI] [PMC free article] [PubMed]
- 27.Liu W, Stein P, Cheng X, Yang W, Shao NY, Morrisey EE, et al. BRD4 regulates Nanog expression in mouse embryonic stem cells and preimplantation embryos. Cell Death Differ 2014;21:1950–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song H, Shi L, Xu Y, Xu T, Fan R, Cao M, et al. BRD4 promotes the stemness of gastric cancer cells via attenuating miR-216a-3p-mediated inhibition of Wnt/β-catenin signaling. Eur J Pharmacol 2019;852:189–97 [DOI] [PubMed] [Google Scholar]
- 29.Ba M, Long H, Yan Z, Wang S, Wu Y, Tu Y, et al. BRD4 promotes gastric cancer progression through the transcriptional and epigenetic regulation of c-MYC. J Cell Biochem 2018;119:973–82 [DOI] [PubMed] [Google Scholar]
- 30.Yoshida GJ. Emerging roles of Myc in stem cell biology and novel tumor therapies. J Exp Clin Cancer Res 2018;37:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gan L, Yang Y, Li Q, Feng Y, Liu T, Guo W. Epigenetic regulation of cancer progression by EZH2: from biological insights to therapeutic potential. Biomark Res 2018;6:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med 2016;22:128–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature 2011;469:343–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fan Y, Mao R, Yang J. NF-κB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell 2013;4:176–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Srivastava J, DiGiovanni J. Non-canonical Stat3 signaling in cancer. Mol Carcinog 2016;55:1889–98 [DOI] [PubMed] [Google Scholar]
- 36.Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer 2014;14:736–46 [DOI] [PubMed] [Google Scholar]
- 37.Chang JC. Cancer stem cells: Role in tumor growth, recurrence, metastasis, and treatment resistance. Medicine (Baltimore) 2016;95:S20–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol 2006;13:1235–42 [DOI] [PubMed] [Google Scholar]
- 39.Fisher ML, Balinth S, Mills AA. p63-related signaling at a glance. J Cell Sci 2020;133. [DOI] [PMC free article] [PubMed]
- 40.Dawood S, Austin L, Cristofanilli M. Cancer stem cells: implications for cancer therapy. Oncology (Williston Park) 2014;28:1101–7, 10 [PubMed] [Google Scholar]
- 41.Kuşoğlu A, Biray Avcı Ç. Cancer stem cells: A brief review of the current status. Gene 2019;681:80–5 [DOI] [PubMed] [Google Scholar]
- 42.Nassar D, Blanpain C. Cancer Stem Cells: Basic Concepts and Therapeutic Implications. Annu Rev Pathol 2016;11:47–76 [DOI] [PubMed] [Google Scholar]
- 43.Boughner JC, van Eede MC, Spring S, Yu LX, Rostampour N, Henkelman RM. P63 expression plays a role in developmental rate, embryo size, and local morphogenesis. Dev Dyn 2018;247:779–87 [DOI] [PubMed] [Google Scholar]
- 44.Liu A, Yu X, Liu S. Pluripotency transcription factors and cancer stem cells: small genes make a big difference. Chin J Cancer 2013;32:483–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Schaijik B, Davis PF, Wickremesekera AC, Tan ST, Itinteang T. Subcellular localisation of the stem cell markers OCT4, SOX2, NANOG, KLF4 and c-MYC in cancer: a review. J Clin Pathol 2018;71:88–91 [DOI] [PubMed] [Google Scholar]
- 46.Donati B, Lorenzini E, Ciarrocchi A. BRD4 and Cancer: going beyond transcriptional regulation. Mol Cancer 2018;17:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shu S, Polyak K. BET Bromodomain Proteins as Cancer Therapeutic Targets. Cold Spring Harb Symp Quant Biol 2016;81:123–9 [DOI] [PubMed] [Google Scholar]
- 48.White ME, Fenger JM, Carson WE 3rd. Emerging roles of and therapeutic strategies targeting BRD4 in cancer. Cell Immunol 2019;337:48–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu Y, Vakoc CR. Targeting Cancer Cells with BET Bromodomain Inhibitors. Cold Spring Harb Perspect Med 2017;7. [DOI] [PMC free article] [PubMed]
- 50.Shi X, Liu C, Liu B, Chen J, Wu X, Gong W. JQ1: a novel potential therapeutic target. Pharmazie 2018;73:491–3 [DOI] [PubMed] [Google Scholar]
- 51.Zhang G, Plotnikov AN, Rusinova E, Shen T, Morohashi K, Joshua J, et al. Structure-guided design of potent diazobenzene inhibitors for the BET bromodomains. J Med Chem 2013;56:9251–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N, et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013;23:839–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.