

Abstract
Increasing evidence has demonstrated that lactate and adenosine triphosphate (ATP) both play important roles in regulating abnormal metabolism in the tumor microenvironment. Herein, an O2 self‐supplying catalytic nanoagent, based on tannic acid (TA)–Fe(III) coordination complexes‐coated perfluorooctyl bromide (PFOB) nanodroplets with lactate oxidases (LOX) loading (PFOB@TA–Fe(III)–LOX, PTFL), is designed for cascade metabolic‐chemodynamic therapy (CDT) by dual‐depletion of lactate and ATP with hydroxyl •OH radicals generation. Benefiting from the catalytic property of loaded LOX and O2 self‐supplying of PFOB nanodroplets, PTFL nanoparticles (NPs) efficiently deplete tumoral lactate for down‐regulation of vascular endothelial growth factor expression and supplement the insufficient endogenous H2O2 . Simultaneously, TA–Fe(III) complexes release Fe(III) ions and TA in response to intracellular up‐regulated ATP in tumor cells followed by TA‐mediated Fe(III)/Fe(II) conversion, leading to the depletion of energy source ATP and the generation of cytotoxic •OH radicals from H2O2. Moreover, TA–Fe(III) complexes provide photoacoustic contrast as imaging guidance to enhance therapeutic accuracy. As a result, PTFL NPs efficiently accumulate in tumors for suppression of tumor growth and show evidence of anti‐angiogenesis and anti‐metastasis effects. This multifunctional nanoagent may provide new insight for targeting abnormal tumor metabolism with the combination of CDT to achieve a synergistic therapeutic effect.
Keywords: adenosine triphosphate, dual‐depletion, lactate, synergistic cancer therapy, tumor metabolism
This paper reports an O2 self‐supplying theranostic nanoagent (PFOB@TA–Fe(III)–LOX, PTFL nanoparticles) with cascade catalytic reaction for synergistic metabolic‐chemodynamic therapy by dual‐depletion of lactate and adenosine triphosphate together with the conversion of H2O2 to •OH radicals. Results demonstrate that such nanoagent efficiently accumulates at the tumor sites for anti‐angiogenesis, anti‐metastasis, and anti‐tumor growth.
![]()
1. Introduction
Regulating the tumor microenvironment (TME) has been considered as an emerging effective strategy for cancer therapy.[ 1 , 2 ] The abnormal metabolism of cancer cells gives TME unique features including low pH,[ 3 ] hypoxia,[ 4 , 5 ] redox imbalance,[ 6 , 7 ] and enzyme overexpression.[ 8 ] One feature of tumor metabolism is the Warburg effect, that is, cancer cells massively convert glucose into lactate even with adequate oxygen (O2), leading to local lactate accumulation.[ 9 , 10 ] In the past decades, lactate was regarded as a metabolic waste product of glycolysis in tumor cells, which disrupted the acid‐base balance of tumor.[ 11 ] However, increasing evidence recently has manifested that lactate accumulation leads to tumor angiogenesis, metastasis, and immune suppression to promote tumor growth and invasion.[ 11 , 12 , 13 , 14 , 15 ] Hence, lactate is believed to be an effective target to regulate abnormal metabolism in TME by blocking generation or direct depletion.[ 16 , 17 , 18 , 19 , 20 ] Lactate oxidase (LOX) is a natural bio‐enzyme for efficient lactate depletion. In the presence of O2, LOX catalyzes lactate to pyruvate and hydrogen peroxide (H2O2).[ 21 , 22 ] Moreover, compared to other metabolic substrates such as glucose,[ 23 ] lactate level is extremely high in tumors and low in normal tissues (Table S1, Supporting Information).[ 24 , 25 , 26 ] Hence, lactate depletion based on LOX has minimal side effects compared to glucose depletion based on glucose oxidase.[ 27 , 28 ] Unfortunately, there are only a few studies that have integrated LOX into nanoplatforms to regulate lactate levels for cancer therapy, and the tumor metabolic therapeutic effect of LOX‐based nanoplatforms is limited only by lactate depletion.[ 18 , 19 ] Moreover, the therapeutic effect based on LOX is notably hindered by the lack of O2 in TME. Although the by‐product H2O2, a type of reactive oxygen species (ROS), can further cause cell death, the effect is not good enough due to the relatively weak reactivity among all types of ROS.[ 29 ] Hence, additional elements are in demand to synergistically fill the gap and enhance the therapeutic efficacy, leading to a win‐win outcome.
Adenosine triphosphate (ATP) is another main biochemical component in TME to provide energy for tumor cell survival and proliferation.[ 30 , 31 ] Due to excessive glycolysis, ATP is upregulated and highly concentrated in tumor cells (1–10 × 10−3 m). Hence, ATP depletion has been considered as one promising approach to inhibit tumor growth,[ 32 , 33 ] and combining ATP depletion with lactate depletion may further enhance tumor metabolic therapy effect. Perfluorocarbon (PFC), with chemical inertness and excellent biocompatibility, is highly active in medical fields such as artificial blood,[ 34 ] organ transplantation,[ 35 ] and cancer treatment[ 36 , 37 ] due to its high O2 solubility. For cancer treatment, PFC‐based nanosystems can supply O2 to relieve the hypoxia in TME for O2‐demand therapies.[ 36 , 37 ] Chemodynamic therapy (CDT) is now widely used for its unique way to generate ROS. With the conversion from H2O2 to hydroxyl radical (•OH) by Fenton/Fenton‐like reactions, CDT can more efficiently induce cell apoptosis and necrosis.[ 32 , 37 , 38 , 39 , 41 ] However, CDT is generally limited by insufficient endogenous H2O2. It is reported that the chelate tannic acid (TA) and iron ions (TA–Fe(III)) complexes could respond to ATP and effectively induce the conversion from H2O2 to •OH.[ 33 , 42 , 43 ] Therefore, if PFC and ATP‐responsive CDT agents are integrated with LOX, a cascade catalytic route is formed. The O2‐sufficient PFC supplies O2 to promote the lactate depletion for anti‐angiogenesis/metastasis and generate H2O2. Simultaneously, the ATP‐responsive CDT agent consumes ATP and converts the produced H2O2 to cytotoxic •OH radicals without consuming additional O2 to further suppress the tumor growth.
Herein, for the first time, an O2 self‐supplying theranostic nanoagent with cascade catalytic reaction is designed for synergistic metabolic‐CDT by dual‐depletion of lactate and ATP together with the conversion of H2O2 to •OH radicals. As shown in Scheme 1 , this theranostic nanoagent consists of a perfluorooctyl bromide (PFOB) nanodroplet core coated with a LOX‐loaded TA–Fe(III) complex layer (PFOB@TA–Fe(III)–LOX, PTFL). Here, TA‐Fe(III) complexes coating can stabilize PFOB nanodroplets and also load LOX molecules. After being internalized by tumor cells, i) the O2 self‐sufficient PTFL nanoparticles (NPs) deliver O2 to relieve hypoxia. The loaded LOX catalyzes the oxidation reaction of lactate with O2, leading to lactate depletion and the generation of H2O2.[ 21 , 22 ] As a result, lactate depletion regulates abnormal metabolism in tumors and down‐regulate vascular endothelial growth factor (VEGF)/cluster of differentiation 31 (CD31) expression to promote anti‐angiogenesis.[ 14 , 15 , 18 , 19 ] ii) At the same time, TA–Fe(III) complexes exhaust intracellular ATP as an energy source and decompose to TA and Fe (III) ions. In this way, ATP was depleted to inhibit tumor growth.[ 33 ] iii) Moreover, Fe(III) ions released during ATP depletion are further reduced into Fe(II) ions by TA and subsequently catalyze H2O2 generated during lactate depletion to cytotoxic •OH, thereby enhancing CDT (Scheme 1).[ 42 , 43 ] Last, TA–Fe(III) complexes also provide photoacoustic (PA) imaging contrast as imaging guidance to further enhance the total therapeutic accuracy and efficacy.[ 44 ] This lactate/ATP dual‐depletion catalytic nanoagent could effectively suppress tumor growth and invasion by a cascade tumor metabolic‐CDT, which might provide new insight for abnormal metabolism‐targeted cancer therapy with the combination of CDT to achieve a synergistic therapeutic effect.
Scheme 1.
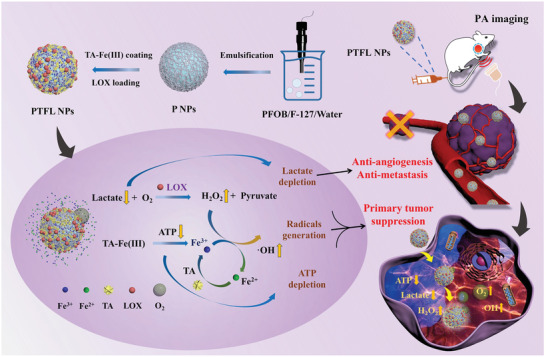
a) Schematic illustration of PTFL NPs synthesis. b,c) Schematic illustration of PTFL NPs for PA imaging‐guided synergistic and cascade metabolic‐chemodynamic therapy.
2. Results and Discussion
2.1. Design, Fabrication, and Characterization of PTFL NPs
PTFL NPs are synthesized based on the steps shown in Scheme 1a. First, PFOB in pluronic F‐127 (F‐127) aqueous solution is emulsified under probe sonication to obtain PFOB NPs (P NPs). As a biocompatible surfactant, F‐127 polymers wrap around the surface of PFOB nanodroplets and act as stabilizers. Then, iron chloride, LOX, and TA are added to form the PFOB@TA–Fe(III)–LOX (PTFL) complex. LOX, as a protein, is amphiphilic in nature, which can help F‐127 stabilize the PFOB nanodroplets, thereby attaching to the surface of the PFOB nanodroplets via hydrophobic interaction. In addition, Fe(III) ions can coordinate with the ethylene oxide of the F‐127 polymer and can also coordinate with LOX through amino acid residues on the surface of the protein, thereby acting as a bridge for loading more LOX from the solution.[ 43 , 45 ] After the addition of TA, TA forms hydrogen bonds with F‐127 and LOX, and coordinates with Fe(III) ions together to form a final complex layer (F127–Fe(III)–LOX–TA) because of the abundant dihydroxyphenyl and trihydroxyphenyl groups.[ 43 , 46 ] Finally, the formed complex layer encapsulates PFOB nanodroplets and also loads LOX into NPs. Here, PFOB NPs loaded with LOX only, TA–Fe(III) complex only, and LOX+TA–Fe(III) are abbreviated as PL NPs, PTF NPs, and PTFL NPs, respectively.
Transmission electron microscopy (TEM) image showed that the synthesized PTFL NPs had a core‐shell structure and were monodispersed in the water with an average size of 182 ± 13 nm (Figure 1a). Energy dispersive spectroscopy (EDS) elemental mapping also indicated the co‐existing of O element from F127 and TA and Fe element from coordinated iron ions in PTFL NPs (Figure S1, Supporting Information). Vis–NIR extinction spectra manifested the PTFL NPs had the extinction capability at the NIR region (Figure 1b) due to the coating of TA–Fe(III) complexes. Zeta potential measurements indicated the P NPs were −23.4 mV (Figure 1c) due to the negatively charged surface of PFOB cores.[ 47 , 48 ] The loading of LOX on P NPs weakened the zeta potential to −18.7 mV, while the formation of PTFL NPs with TA‐Fe(III) complex and LOX coating led to a zeta potential of −34.4 mV (Figure 1c). The highly negatively charged surface of PTFL NPs ensured the good water dispersity. To explore the effect of the order of adding TA or Fe(III) first together with LOX on the loading capacity of LOX, the LOX loading capacities of these two cases were measured. The results revealed that the loading capacity of LOX in the case to add TA first was ≈0.7%, and the loading capacity was ≈1% when Fe(III) was added first. These results indicated that adding Fe(III) together with LOX first to form PFOB@Fe(III)–LOX complex and then followed by adding TA for chelation with Fe(III) could increase the LOX loading capacity. This could be attributed to the weakening of electrostatic repulsion between PFOB and LOX by the positive charge of iron cations during LOX loading. The zeta potential of PTFL NPs of adding TA first (‐ 34.3 mV) was lower than that of PTFL NPs of adding Fe(III) first (−32.5 mV), indicating a higher loading of weak negatively charged LOX on PTFL NPs by adding Fe(III) first (Figure S2, Supporting Information).
Figure 1.
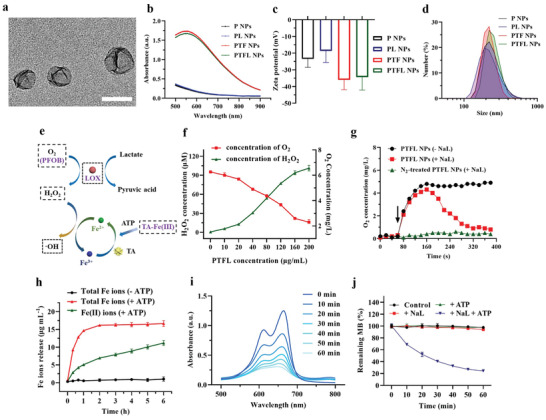
a) TEM image of PTFL NPs. Scale bar: 200 nm. b–d) Vis‐NIR extinction, Zeta potential, and DLS of P NPs, PL NPs, PTF NPs, and PTFL NPs. Data in (c) are shown as n = 3, mean ± SD. e) Schematic illustration of PTFL NPs induced H2O2 and •OH generation. f) Concentration of H2O2 and dissolved O2 generating from 15 min reaction between various concentrations of PTFL NPs and 10 mm NaL at room temperature (n = 3, mean ± SD). g) O2 concentration after adding PTFL NPs to N2‐treated solution (with/without 10 mm NaL). h). The time‐dependent concentration of total Fe ions release and the converted Fe2+ ions from PTFL NPs by 1 mg mL−1 ATP or without ATP addition, respectively. (n = 3, mean ± SD). i) Time‐dependent methylene blue (MB) (10 mm) degradation by addition of PTFL NPs (200 µg mL−1), ATP (1 mg mL−1), and NaL (10 mm). j) The ratio of remaining MB after treated with 1) 1 mg mL−1 ATP, 2) 10 mm NaL, 3) 1 mg mL−1 ATP + 10 mm NaL. MB without any treatment was set as control (n = 3, mean ± SD).
Dynamic light scattering (DLS) measurement indicated that the dynamic sizes of P NPs, PL NPs, PTF NPs, and PTFL NPs were around 260, 256, 253, and 257 nm, respectively, but the polydispersity indices of P NPs and PL NPs were larger (Figure 1d). This could be explained that the coating of TA–Fe(III) complexes stabilized PFOB cores and helped the NP well dispersed. To further confirm this, DLS sizes of P NPs, PL NPs, PTF NPs, and PTFL NPs were measured at 1, 2, 4, and 7 days after synthesis. Results showed that the sizes of P NPs and PL NPs increased to 412 and 406 nm after 7 days, whereas the size of PTF NPs and PTFL NPs showed no obvious difference (Figure S3, Supporting Information). The results demonstrated that the coating of TA–Fe(III) complexes played a stabilizing role. In addition, after dispersing in water, phosphate buffered saline (PBS) (pH = 7.4 or 5.5), or cell culture medium for 48 h, no obvious differences in the appearance and DLS sizes of PTFL NPs were observed (Figure S4, Supporting Information).
2.2. PTFL NPs Catalyzed Lactate Depletion, ATP Triggered Fe Ions Release, and •OH Generation
As mentioned above, LOX can catalyze the decomposition of lactate in the presence of O2 and generate pyruvate and H2O2 (Figure 1e). Hence, O2 and H2O2 levels were selected as indicators of the catalytic activity of PTFL NPs. H2O2 concentration was measured using an H2O2 assay kit based on an H2O2 standard curve (Figure S5, Supporting Information), while the level of O2 was determined by a portable dissolved O2 meter. Different concentrations of PTFL NPs (0, 10, 20, 40, 80, 120, 160, 200 µg mL−1) were mixed with sodium lactate (NaL) without the addition of ATP to avoid the needless release of iron ions or generation of radicals which would affect the accuracy of the detection kit. Results showed that the H2O2 concentration increased and the O2 content rapidly decreased to a low level with the increase of PTFL concentrations (Figure 1f). 200 µg mL−1 PTFL NPs could generate ≈101 µm H2O2 in 15 min. It is worth noting that when the concentration of PTFL NPs was higher than 120 µg mL−1, the increase rate of H2O2 slowed down. This could be explained by the low level of O2 that limited the rate of enzyme‐catalyzed reactions. When the PTFL NPs concentration was fixed at 200 µg mL−1 and NaL concentration was adjusted from 0 to 20 mm, the production of H2O2 showed a linear trend until the concentration of NaL exceeded 10 mm (Figure S6, Supporting Information). Figure S7, Supporting Information shows the O2 release curve of PTFL NPs in a normoxia environment for 24 h. A negligible decrease of O2 content in PTFL NPs was observed, demonstrating that PTFL NPs could contain O2 for a relatively long time in normoxia environment. As shown in Scheme 1, the natural O2‐carrying capacity of PFOB can promote the aerobic LOX enzymatic reaction. In the presence of O2‐carrying PTFL NPs and NaL (PTFL NPs +NaL), O2 content increased to 4.4 mg L−1 in the first 80 s and rapidly declined to only 0.9 mg L−1 due to the O2‐depleted LOX enzymatic reaction (Figure 1g). In the presence of only O2‐carrying PTFL NPs (PTFL NPs ‐NaL), O2 content increased to 4.9 mg L−1 in the first 80 s and then kept this high‐level O2 content due to no LOX enzymatic reaction (Figure 1g). For N2 gas‐treated PTFL NPs with NaL, O2 content was kept at a low level due to the lack of O2 supply (Figure 1g). Correspondingly, the final stable H2O2 production for the above three conditions after 380 s were also measured. As shown in Figure S8, Supporting Information, O2‐carrying PTFL NPs with NaL (PTFL NPs +NaL) showed a high H2O2 level due to the generation of H2O2 during LOX enzymatic reaction. N2 gas‐treated PTFL NPs with NaL and only O2‐carrying PTFL NPs (PTFL NPs ‐NaL) showed low H2O2 levels with no LOX enzymatic reaction due to the lack of O2 or lactate, respectively. The results indicate that the LOX enzymatic reaction is aerobic, and PTFL NPs can provide O2 to promote the LOX enzymatic reaction.
The stability of PTFL NPs in the normal environment (‐ATP) and the release of Fe ions from PTFL NPs caused by ATP (+ATP) were studied next. As shown in Figure 1e, ATP with strong chelation ability could degrade TA–Fe(III) shell to release Fe(III) ions, which were further reduced to Fe(II) ions by TA. It was reported that the ability of Fe(III) ions to catalyze H2O2 to produce •OH was far inferior to that of Fe(II) ions. Moreover, during the generation of •OH, Fe(II) ions would be re‐oxidized back to Fe(III) ions.[ 32 , 42 , 43 ] Therefore, the release of Fe(III) ions by ATP and the continuous reduction of Fe(III) ions to Fe(II) ions by TA were both essential for the subsequent production of •OH radicals. 1,10‐phenanthroline, which could coordinate with Fe(II) ions to form Fe(II)‐phenanthroline complexes with an absorption peak at 511 nm, was used to quantify the amount of Fe(II) ions (Figure S9, Supporting Information). Also, reductant hydroxylamine was used to convert Fe(III) ions to Fe(II) ions, so both Fe(III) and Fe(II) content could be quantified. As shown in Figure 1h, no Fe ions were released in the normal environment (‐ATP, black curve) and Fe ions were only released in the environment with ATP (+ATP, red curve). Moreover, most Fe ions (>85%) were released from PTFL NPs in the ATP environment within 1 h and 65% of Fe ions were reduced to Fe(II) ions at 6 h (green curve). The results demonstrated the stability of PTFL NPs in the normal physiological environment and the release of Fe ions in response to ATP. Importantly, elevated ATP expression is a common phenomenon that occurs only within the tumor cells. For in vivo experiments, PTFL NPs will not release TA–Fe(III) and LOX before accumulating in the tumor sites due to the stability in the normal physiological environment. When PTFL NPs accumulate at the tumor site and enter tumor cells, Fe(III) ions will be released in the elevated ATP environment, leading to tumor‐specific treatment.
Afterward, the generation of •OH radicals catalyzed by Fe(II) ions was evaluated by •OH caused methylene blue (MB) degradation. MB is a common oxidant indicator.[ 38 ] When treated with •OH radicals, the blue MB will fade with a significant decrease at the characteristic absorption peak at 665 nm. The declining absorption spectra of MB in Figure 1i showed that the •OH radicals were continuously generated once PTFL NPs (200 µg mL−1) were mixed with NaL (10 mm) in the presence of ATP (1 mg mL−1). Importantly, no obvious fading of MB was observed in the absence of ATP, NaL, or neither (Figure 1j), demonstrating that both ATP and NaL were essential for PTFL NPs to generate •OH radicals. In addition, ATP was significantly depleted when PTFL NPs were added into the solution containing ATP and NaL compared with the ATP and NaL solution group with no addition of PTFL NPs (Figure S10, Supporting Information). Hence, the whole process of depletion of lactate and producing •OH can be summarized as follows: (1) PTFL NPs catalyze the lactate depletion and H2O2 generation in the supplying of O2 by PFOB; (2) Fe(III) ions are released by ATP from TA‐Fe(III) complexes and reduced to Fe(II) ions by TA; (3) The reduced Fe(II) ions catalyze H2O2 to generate •OH radicals and deplete ATP, and are re‐oxidized to Fe(III) ions, which can be reduced again by TA for further •OH generation.
2.3. In Vitro Synergistic Therapy
After confirming the capacity of PTFL NPs to catalyze lactate decomposition and induce •OH radicals in response to ATP, the in vitro therapeutic effect of the nanosystem was studied using CCK‐8 assay, live/dead staining assay, and apoptosis/necrosis flow cytometry assay. 4T1 cells were first divided into 6 groups and treated with (1) Blank, (2) P NPs, (3) PL NPs, (4) PTF NPs, and (5) PTFL NPs, respectively. Herein blank means no treatment. As shown in Figure 2a, both PL NPs and PTF NPs decreased the cell viability modestly due to the lactate depletion and ATP depletion, respectively. The lowest cell viability in Group 5 manifested that the synergy of lactate depletion, ATP depletion, and CDT by employing PTFL NPs had the most anti‐cancer therapeutic effect (Fe content: 12.4 µg mL−1, LOX: 1.2 µg mL−1). Calcein‐AM/PI staining was then used to differentiate live and dead cells with green and red fluorescence, respectively. The confocal laser scanning microscopy (CLSM) results showed that 4T1 cells treated with PTFL NPs had the greatest degree of death (Figure 2b). Similar results of cell viability testing and live/dead fluorescence staining were also observed when PTFL NPs were applied for the treatment of CT26 murine colorectal carcinoma cell line (Figure S11, Supporting Information), indicating that PTFL NPs‐based therapy was applicable to different tumor types. In a contrast, MCF‐10A, a normal breast cell line, maintained high cell viability when incubated with various concentrations of PTFL NPs for 48 h, indicating a low cytotoxicity to normal cells (Figure S12, Supporting Information). Besides, flow cytometry assays were performed to evaluate the apoptosis/necrosis level of treated cells. As shown in Figure 2c, the cells treated with PTFL NPs had the highest degree of apoptosis. The total apoptosis ratio from Q2 and Q3 was 83.17%, while in P NPs, PL NPs, and PTF NPs‐treated groups, the total apoptosis ratios were 11.16%, 20.24%, and 75.1%, respectively. To determine the mechanism of PTFL NPs in causing apoptosis in 4T1 cells, western blotting of cleaved caspase‐3 protein was also performed. As shown in Figure S13, Supporting Information, PTFL NPs‐treated cells showed the highest level of cleaved caspase‐3 expression, indicating the apoptosis pathway by activation of caspases.
Figure 2.
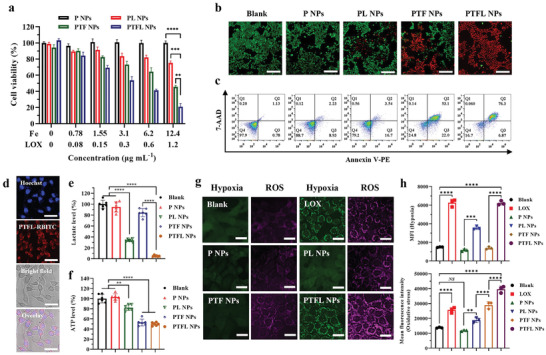
a) Cell viability of 4T1 cells after treatments with various concentrations of P NPs, PL NPs, PTF NPs, and PTFL NPs for 48 h (n = 3, mean ± SD). b) Confocal fluorescence images of calcein‐AM/PI stained 4T1 cells after various treatments (PTF NP, PTFL NPs: 72 µg mL−1). Scale bars: 200 µm. c) Apoptosis and necrosis analysis of 4T1 cells after various treatments in (b) from flow cytometry assay. d) Confocal fluorescence images of PTFL NPs endocytosis after 4 h incubation with 4T1 cells. PTFL NPs were tagged with RBITC (red). Scale bars: 50 µm. e,f) Lactate level in culture medium and intracellular ATP level of 4T1 cells after various treatments in (b) for 48 h (n = 6, mean ± SD). g) Hypoxia and ROS staining of 4T1 cells after various treatments of LOX, P NPs, PL NPs, PTF NPs, and PTFL NPs using CellROX reagent and Image‐iT Hypoxia reagent, respectively. Scale bars: 25 µm. h) Mean fluorescent intensity of both oxidative stress and hypoxia in 4T1 cells after various treatments in (g) from flow cytometry assay (n = 3, mean ± SD). Statistical significance was analyzed by one‐way ANOVA, *P < 0.05,**P < 0.01 and ****P < 0.0001, NS: nonsignificant difference.
2.4. In Vitro Cellular Uptake, Lactate/ATP Depletion, and ROS/Hypoxia Generation
As mentioned, the synergistic therapeutic effects are based on LOX‐induced lactate depletion and H2O2 generation, as well as TA–Fe(III) complexes‐induced ATP depletion and •OH radicals generation. Hence, in vitro lactate and ATP levels were measured to verify both depletions, while intracellular levels of ROS and hypoxia were investigated to confirm the •OH generation and O2 supply/consumption. In vitro cellular uptake was first evaluated by incubating PTFL NPs with 4T1 cells. As shown in Figure 2d, rhodamine B isothiocyanate (RBITC) tagged PTFL (PTFL–RBITC) NPs underwent endocytosis by 4T1 cells after 4 h incubation. It was clearly observed that PTFL–RBITC entered the cells in the cytoplasm. Subsequently, in vitro lactate and ATP levels were investigated. As shown in Figure S14, Supporting Information, in vitro lactate concentration in cell culture medium kept increasing to ≈16.9 mm after 72 h incubation time. Then 4T1 cells were treated with P NPs, PL NPs, PTF NPs, and PTFL NPs, respectively. The relative lactate level showed no obvious change for the P NPs‐treatment group, while the lactate level in the PTF NPs treatment group decreased to ≈84.5% of that in the non‐treatment group. The lactate levels of PL NPs and PTFL treatment groups significantly decreased to 33.8% and 5.1%, respectively. The difference in the effect between PL NPs‐treatment group and PTFL NPs‐treatment group was mainly due to the different loading capacities of LOX (Figure 2e). Moreover, along with the depletion of lactate, intracellular ATP was also exhausted with the release of Fe(III) ions. As shown in Figure 2f and Figure S15, Supporting Information, the intracellular ATP levels in PTF NPs treatment group and PTFL NPs treatment group were markedly decreased to 52.9% and 49.8% of that without treatment respectively, demonstrating the successful depletion of intracellular ATP. Conversely, the groups without TA–Fe(III) component (P NPs, PL NPs) showed no significant ATP suppression. Similar ATP/lactate depletion results were also observed when PTFL NPs were applied for the treatment of CT26 cell lines (Figure S16, Supporting Information), demonstrating that PTFL NPs could achieve ATP/lactate depletion for the treatment of different tumor types.
The LOX catalysis exhausted the in situ O2 content to generate local hypoxia, and the produced H2O2 was then catalyzed to •OH radicals by released iron ions, a kind of ROS. The conversion from H2O2 to •OH radicals further increase the total ROS level. Hence, intracellular hypoxia and ROS were both stained by fluorescent probes to show the change during NP treatments. Prior to the staining, the culture medium of 4T1 cells was covered with a layer of liquid paraffin to prevent the O2 exchange. As shown in Figure 2g, green color and red color represented the local hypoxia and ROS, respectively. As expected, P NPs could release the local hypoxia due to the O2‐sufficient PFOB cores. PTF NPs without LOX loading could only generate a low degree of ROS due to the insufficient intracellular H2O2. For PL NPs with LOX loading but no TA–Fe(III) coating, a certain extent of local hypoxia and ROS was generated during the LOX catalysis. Moreover, when NPs were decorated with both LOX and TA–Fe(III) coating, green and red fluorescence was enhanced obviously, which proved that PTFL NPs could produce a large amount of ROS and local hypoxia. As shown in Figure 3h, the quantitative analysis from flow cytometry assay also showed the same trend that the mean fluorescence intensity (MFI) of oxidation stress of the cells treated with PTFL NPs was 3.7 times higher than the MFI of the control group, while the MFI of the hypoxia indicator was 4.2 times higher than that of the control group, respectively.
Figure 3.
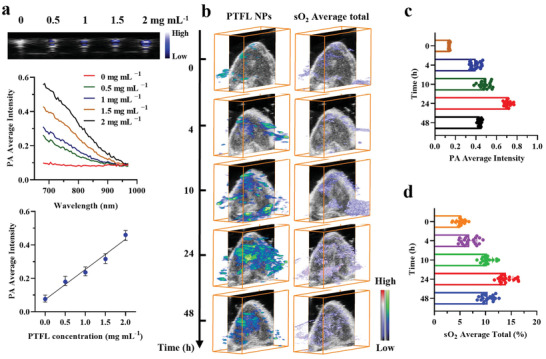
a) In vitro PA images and quantified intensities of PTFL NPs with various concentrations (n = 3, mean ± SD). b) In vivo 3D PA and sO2 tumor images at different time intervals after i.v. injection of PTFL NPs. c,d) The corresponding quantified intensities of PA and sO2 average total of tumor areas in (b) (n = 60, mean ± SD).
2.5. In Vitro and In Vivo PA Imaging Performance
The intrinsic high photothermal‐conversion capability of TA–Fe(III) complexes endowed PTFL NPs with enhanced PA contrast,[ 44 ] therefore, PA imaging of PTFL NPs was further carried out both in vitro and in vivo. First, in vitro demonstration was performed using a tissue‐mimic phantom (Figure S17, Supporting Information). As shown in Figure 3a, PA amplitude increased with the concentration of PTFL NPs. The results of broad‐spectrum PA scanning showed that the PA amplitude of PTFL NPs in the range of 680 to 970 nm gradually weakened with the increase of wavelength, which was consistent with the results of Vis–NIR extinction. Herein, the PA signal excited by 700 nm laser was selected for in vivo PA imaging. Quantitative analysis revealed that the PA amplitudes of PTFL NPs were linearly correlated with the concentrations.
Afterward, in vivo tumoral PA imaging of PTFL NPs and the subsequent changes in saturated oxygen (sO2) were carried out via the intravenous (i.v.) administration to 4T1‐bearing nude mice. To better reveal the accumulation of PTFL NPs and the corresponding sO2 level throughout the tumor area, we performed PA tomographic scanning of the tumor and subsequent 3D imaging reconstruction. Briefly, each longitudinal section of the tumor was first scanned in turn, then the PA signals and sO2 levels of the tumor in each section were analyzed. Finally, 3D reconstruction was performed on PA images of all sections to enhance the visualization and accuracy of signals throughout the tumor. As shown in Figure 3b, after i.v. administration, PTFL NPs gradually accumulated in the tumor and reached a peak after 24 h, and then weakened a little after 48 h. The corresponding sO2 level displayed the same trend. The results revealed that PTFL NPs could well accumulate and retain in tumors. It is worth noting that after 24 h of administration, the signal of PTFL NPs evenly distributed in the interior and edge of the tumor, indicating a high permeability of PTFL NPs to the tumor as well. This might be explained that PTFL NPs were in a soft “nanodroplet” state and prone to deformation to increase penetration, which was different from solid NPs.[ 49 , 50 ] The high permeability was conducive to covering the entire tumor with our nanoagents, thereby improving the overall therapeutic effect.
Generally, the O2 loading capacity of NPs follows Henry's law, that is, proportional to the exposed O2 partial pressure. The high O2 loading capacity of our PTFL NPs is due to the high O2 solubility of PFOB, which has a very high 50% v/v (i.e., 500 mLO2/LPFOB) O2 solubility under the normal atmospheric O2 partial pressure.[ 51 ] O2‐loaded PFCs‐based NPs can keep O2 encapsulated in the normoxia physiological environment for a long time due to the partial O2 pressure equilibrium and then release O2 in response to the hypoxia tumor environment due to the drop of exposed O2 partial pressure.[ 52 ] In our case, in vivo sO2 level is affected by PTFL NPs from two aspects: the O2 supply by O2 self‐sufficient PFOB nanodroplets and O2 depletion by LOX‐based catalysis. Hence, PTF NPs without LOX addition were also introduced for comparison. After the i.v. administration of PTF NPs, the sO2 level in the tumor showed a similar upward‐decreasing trend (Figure S18, Supporting Information). The difference was that the peak sO2 level in PTFL NPs‐treated tumor was only 13.9%, whereas the value was increased to 20.4% in PTF NPs‐treated tumor. Therefore, it could be concluded that the self‐carrying O2 capacity of PFOB could increase the baseline of the tumor sO2 level.
2.6. In Vivo Fluorescent (FL) Imaging and Biodistribution
IVIS spectrum imaging system was utilized for the in vivo FL imaging of PTFL NPs to study tumor accumulation and biodistribution. Prior to in vivo imaging, PTFL NPs were tagged with IR‐780 fluorescent dye (PTFL–IR780 NPs) to endow the nanoagent with FL imaging contrast. Visible‐near‐Infrared (Vis–NIR) extinction of PTFL–IR780 NPs is shown in Figure S19, Supporting Information, demonstrating the successful labeling of IR780. In addition, after 7 days of dispersion in PBS, the change in Vis–NIR extinction was found to be negligible, indicating that the labeling of IR780 was stable. The average NIR fluorescence signals of tumor sites from the IVIS images were quantified to evaluate the accumulation of PTFL–IR780 in the tumor sites. The average NIR fluorescence signals of tumor sites before the administration of PTFL‐IR780 were used as the baseline for comparison. As shown in Figure 4a,b, obvious tumor fluorescence could be seen at 2 h post‐administration, indicating that PTFL–IR780 NPs started to accumulate at the tumor site. At 24 h of administration, the average NIR fluorescence signals of tumor sites continued to increase by 5.3 times compared to that before the administration, which demonstrated the successful accumulation of NPs in the tumor sites. The average NIR fluorescence signals of tumor sites at 48 h of administration only decayed by 28% compared to that at 24 h of administration, which demonstrated the good retention of NPs in the tumor sites. This result was consistent with that of in vivo PA imaging, which assisted in proving that PTFL NPs had both excellent tumor accumulation and long‐term retention capability. At different time intervals (4 h, 10 h, 24 h, and 48 h), 4T1‐bearing nude mice were sacrificed. Major organs and tumors were excised and performed ex vivo IVIS imaging. It could be observed that PTFL NPs also accumulated in livers and lungs except for tumors (Figure 4c,d). The amount of PTFL NPs in major organs dropped to a very low level within 48 h, probably due to the clearance of TA–Fe(III) complexes and the exhalation of PFOB nanodroplets.[ 53 , 54 ]
Figure 4.
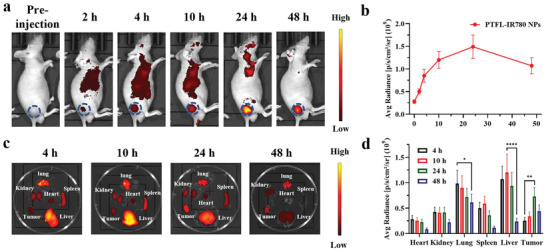
a) In vivo fluorescent images of 4T1 subcutaneous tumor‐bearing mice at various time intervals after i.v. injection of PTFL‐IR780 NPs and b) the corresponding fluorescent intensities of PTFL‐IR780 NPs at tumor sites (n = 3, mean ± SD). c) Ex vivo fluorescent images of major organs and tumors from 4T1 subcutaneous tumor‐bearing mice sacrificed at various time intervals after i.v. injection of PTFL‐IR780 NPs and d) the corresponding fluorescent intensities (n = 3, mean ± SD). Statistical significance was analyzed by one‐way ANOVA, *P < 0.05,**P < 0.01 and ****P < 0.0001, NS: nonsignificant difference.
2.7. In Vivo Synergistic Therapy
PTFL NPs have proven their excellent anti‐tumor capability in vitro, as well as their satisfactory tumor‐targeting capability in vivo. Next, the in vivo synergistic therapeutic effect was evaluated on 4T1 subcutaneous tumor‐bearing mice. Mice were randomly divided into 6 groups and treated with PBS, LOX, P NPs, PL NPs, PTF NPs, and PTFL NPs at days 0 and 4, respectively, and sacrificed at day 16 for further analysis (Figure 5a). As shown in Figure 5b, tumors in mice treated with PTFL NPs almost disappeared, whereas in mice treated with P NPs, PL NPs, and PTF NPs, tumors were only partially destroyed. Mice treated with free LOX showed no obvious tumor suppression compared to PBS treated mice because of the fast clearance and poor tumoral accumulation. The tumor growth inhibition rate in each treatment was 5.1% (free LOX), 19.5% (P NPs), 38.5% (PL NPs), 64.4% (PTF NPs), and 84.5% (PTFL NPs), respectively. No abrupt changes in body weights were found for all the groups (Figure 5c). Tumor weights and digital graphs of representative tumors after excision supported the same trend (Figure 5c,d). Hence, it can conclude that PTFL NPs suppress tumor growth to the utmost extent based on the cascade lactate/ATP depletion and synergistic CDT. Furthermore, hematoxylin and eosin (H&E) staining and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) immunostaining were performed as auxiliary proof from the pathological analysis (Figure 5f). It could be observed that the tumor treated with PTFL NPs underwent the most cell disruption and shrinkage with the largest nucleus absence in H&E staining, and had the densest fluorescence in TUNEL assay. The results revealed that PTFL NPs produced the most cell apoptosis. Whereas in tumors treated with PTF NPs, PL NPs, or P NPs, the degrees of cell apoptosis decreased in various degrees. Tumors treated with PBS or free LOX still exhibited complete cell morphology with the large nucleus and no obvious TUNEL fluorescence, indicating that the tumors were still in an uninhibited and malignant state. Besides, the biosafety of PTFL NPs was also evaluated. As shown in Figure S20, Supporting Information, no obvious histological damage was found in major organs of mice in each group, illustrating the negligible systemic toxicity as expected. All the above results demonstrate that PTFL NPs could maximally inhibit the tumor growth with negligible systemic toxicity by the cascade metabolic‐CDT based on lactate/ATP depletion and •OH radicals generation.
Figure 5.

a) Schematic illustration of in vivo therapy for 4T1 subcutaneous tumor‐bearing mice. b) Tumor volumes, c) body weights, d) tumor weights, and e) representative tumor images of 4T1 tumor‐bearing nude mice after various treatments for 16 days (n = 3, mean ± SD). Arrows indicate the time point of treatments (Days 0 and 4). f) H&E staining and TUNEL immunofluorescence staining (Green) images after various treatments. Scale bars: 100 µm. g,h) Lactate and ATP levels in 4T1 tumors with various treatments 48 h after administrations (n = 5, mean ± SD). Statistical significance was analyzed by one‐way ANOVA, *P < 0.05,**P < 0.01 and ****P < 0.0001, NS: nonsignificant difference.
2.8. In Vivo TME Regulation, Anti‐Angiogenesis, and Anti‐Metastasis Performance
Afterward, the in vivo mechanism of LOX‐induced tumor inhibition was evaluated. Tumoral lactate/ATP level, hypoxia‐related biomarker expression (HIF1‐α), angiogenesis biomarkers expression (CD31 and VEGF), as well as lung metastasis were investigated correspondingly. First, tumoral lactate/ATP concentrations were measured 48 h post‐treatment with PBS, free LOX, P NPs, PL NPs, PTF NPs, and PTFL NPs. For tumor lactate depletion, the results revealed that the tumoral lactate was almost exhausted with the depletion rate >90% in PTFL NPs‐treated group, demonstrating the excellent catalytic activity of LOX‐embedded PTFL NPs (Figure 5g). PL NPs showed a partial lactate depletion rate of ≈53.5%. Free LOX could not accumulate in tumors and P NPs/PTF NPs had no lactate depletion effect, hence the decrease of lactate level was not obvious in these three groups. For tumor ATP depletion, the results revealed that 42.4% and 45.2% of tumoral ATP was suppressed in PTF NPs and PTFL NPs‐treated groups respectively (Figure 5h). P NPs and PL NPs without TA‐Fe(III) components only showed a slight ATP decrease because of suppression of tumor growth, while free LOX with poor tumoral accumulation could not suppress ATP level. Next, the HIF1‐α (green fluorescence) as a tumor hypoxia indicator was immunostained. As shown in Figure S21, Supporting Information, the PBS/free LOX‐treated tumors showed obvious hypoxia with a high expression of HIF1‐α, whereas in P NPs/PTF NPs‐treated tumors, almost no green fluorescence was observed, indicating the relief of hypoxia due to O2 supplying by PFOB component. The PL NPs/PTFL NPs‐treated tumor showed restrained HIF1‐α expression which was attributed to the combined effect of O2 delivery by PFOB and O2 depletion by LOX.
Sequentially, two angiogenesis biomarkers (CD31 and VEGF) were immunostained to investigate the anti‐angiogenesis efficacy. As shown in the fluorescent images from Figure 6a,b and the semi‐quantitative analysis from Figure 6c, a large amount of green and red positive spots were observed in the groups treated with PBS, free LOX, and P NPs, indicating a high level of CD31 and VEGF expression. PTF NPs treatment slightly reduced both expression levels due to effective tumor suppression. PL NPs treatment distinctly decreased both expressions and for PTFL NPs treatment, areas of green and red dots dropped significantly by 67% and 86%, respectively, indicating the anti‐angiogenesis effect of lactate depletion by the loaded LOX. Next, the anti‐metastasis effect of PTFL NPs was evaluated. 4T1 cells were planted under the armpit to facilitate tumor metastasis. Mice were treated with PBS, PTF NPs, or PTFL NPs. As shown in Figure 6d, 21 days after the first administration, obvious metastasis nodules could be observed on the digital and H&E staining images of lungs in PBS‐treated group. Compared with the PBS‐treated control group, the mice treated with PTF NPs exhibited less metastasis while for the mice treated with PTFL NPs, lung metastasis nodules were almost negligible. Quantitative analysis (Figure 6e) demonstrated that PTFL NPs could prominently reduce the metastasis nodules by 89%. Hence, this nanosystem was proven to have effective anti‐angiogenesis and anti‐metastasis effects.
Figure 6.
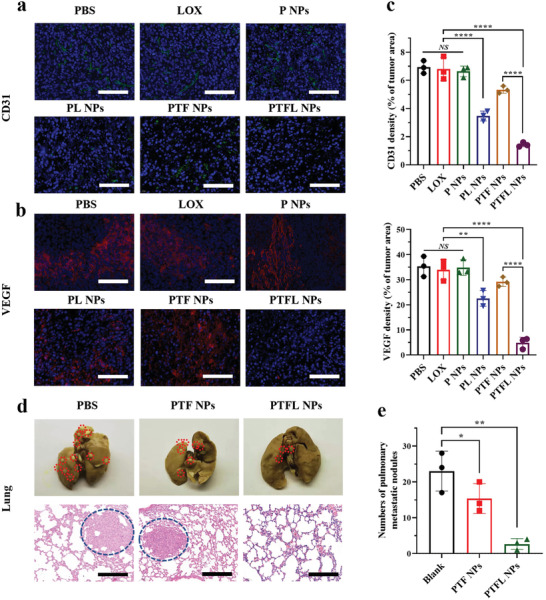
a) CD31 and b) VEGF immunostaining images and c) the corresponding quantification of CD31 and VEGF densities (n = 3, mean ± SD). Scale bars: 100 µm. d) Representative images of tumor nodules in the lungs with various treatments and e) the corresponding number of lung metastasis nodules (n = 3, mean ± SD). Scale bars: 200 µm. Statistical significance was analyzed by one‐way ANOVA, *P < 0.05,**P < 0.01 and ****P < 0.0001, NS: nonsignificant difference.
3. Conclusion
In summary, an O2 self‐supplying theranostic nanoagent (PTFL NPs) with cascade catalytic reaction was developed for synergistic metabolic‐CDT by dual‐depletion of lactate and ATP together with the conversion of H2O2 to •OH radicals. Specifically, PTFL NPs could efficiently deplete tumor lactate with the assistance of O2‐sufficient PFOB core, and generate H2O2 to supplement the insufficient endogenous H2O2. Synergistically, PTFL NPs responded to overexpressed intracellular ATP in tumor cells to deplete ATP and converted H2O2 to cytotoxic •OH radicals. In vivo studies revealed that under the guidance of PA imaging, the PTFL NPs had excellent tumor accumulation and retention for suppression of tumor growth, as well as showed the anti‐angiogenesis and anti‐metastasis effects. This unique lactate/ATP dual‐depletion cascade catalytic nanosystem with the generation of cytotoxic •OH radicals might provide new insight for synergistic metabolic‐chemodynamic tumor therapy.
4. Experimental Section
Materials
PFOB, Pluronic F‐127, Iron (III) chloride hexahydrate (FeCl3 · 6H2O), LOX from Aerococcus viridans, 2′,7′‐Dichlorofluorescin diacetate (DCFH‐DA), IR‐780 iodide, and rhodamine B isothiocyanate (RBITC) were purchased from Sigma Aldrich (USA). Methylene blue (MB) was purchased from TCI (Japan). Hydroxylamine hydrochloride, sodium lactate (NaL), and tannic acid (TA) were purchased from J&K Scientific (China). ATP and 1,10‐phenanthroline were purchased from Aladdin Reagent (China). Hydrogen peroxide (H2O2) assay kit, cell counting KIT‐8 (CCK‐8), ATP assay kit, and lactate assay kit were purchased from Solarbio (China). Live/dead cell imaging kit, image‐iT hypoxia reagent, cellROX deep red reagent, Hoechst 33342, and all cell culture reagents were purchased from ThermoFisher Scientific (USA). PE Annexin V Apoptosis Detection Kit I was purchased from BD Biosciences (USA). All reagents and chemicals were purchased commercially and without further purification.
Instrumentation and Characterization
An Ultrospec 2100 Pro spectrophotometer (Amersham Biosciences, UK) was utilized to measure the Vis–NIR extinction. A Zetasizer Nano ZS (Malvern Instruments, UK) was utilized to measure zeta potential and hydrodynamic size. A Field Emission Electron Microscope STEM (JEM‐2100F, JEOL Ltd., Japan) was utilized to observe the morphology of NPs. A Leica TCS SPE Confocal Microscope (Leica Microsystems, Germany) was utilized for in vitro fluorescence imaging. An IVIS Lumina Series III Pre‐clinical In Vivo Animal Imaging Systems (PerkinElmer, USA) was utilized for in vivo fluorescent imaging. A VEVO LAZR Imaging System (FUJIFILM VisualSonics, Canada) operating with an LZ250 transducer was utilized for PA imaging.
Synthesis of P NPs, PL NPs, PTF NPs, and PTFL NPs
For P NPs, and PTF NPs synthesis, 100 µL PFOB was first added into 10 mL ice‐cold F‐127 solution (1% w/w). After 3 min emulsification by sonication (300 W, duty cycle 50%) in an ice‐water bath, P NPs were obtained. Then FeCl3 · 6H2O was mixed at a final concentration of 0.6 mg mL−1, and the formed PFOB–Fe(III) solution kept shaking at 1400 rpm overnight. Afterward, TA was quickly added under vigorous stirring with a final concentration of 1.2 mg mL−1. The solution instantly turned from faint yellow to blue and was further centrifuged three times at 7000 rpm for 3 min to obtain PTF NPs. For PL NPs synthesis, LOX was first mixed with P NPs without the addition of FeCl3 · 6H2O and TA. After shaking overnight and washing, PL NPs were obtained. For PTFL NPs synthesis, the recipe changed that LOX was first mixed along with FeCl3 · 6H2O. After the successful emulsification of PFOB nanodroplets, the mixture of LOX and FeCl3 · 6H2O were added at a final concentration of 0.8 and 0.6 mg mL−1 respectively. After shaking overnight, the PFOB–Fe(III)–LOX solution was quickly mixed with TA to get PTFL NPs. Subsequently, the obtained PTFL NPs solution was centrifuged at 7000 rpm for 3 min and washed three times with distilled water. To evaluate the amount of loaded active LOX, H2O2 produced by LOX catalysis was measured using an H2O2 assay kit based on a standard H2O2 curve. Finally, the purified PTFL NPs were stored at 4 °C. Before all subsequent experiments, the prepared PTFL NPs were put in an aseptic chamber and exposed to a normoxia environment for 2 h. Due to the high O2 solubility of PFOB, O2 molecules then entered the NPs until the O2 saturation and O2 partial pressure balance were reached.
Catalytic Activity Measurements of PTFL NPs
The generation of H2O2 and depletion of O2 were detected to determine the catalytic activity of PTFL NPs. Various concentrations of PTFL NPs from 0 to 200 µg mL−1 were first mixed with NaL (10 mm) in PBS and incubated for 15 min. Then the concentration of H2O2 and O2 was measured by an H2O2 Assay kit and a portable dissolved O2 meter, respectively. The standard curve of H2O2 was determined according to the manufacturer's instruction. Also, the influence of NaL concentration (0, 2.5, 5, 10, 20 mm) on H2O2 generation by PTFL NPs was investigated. Last, the promotion of LOX catalytic reaction by O2 carried by PFOB core was also explored. An aqueous solution with or without NaL addition was pre‐treated with N2 to exhaust O2, and sealed with a layer of liquid paraffin to prevent air exchange. Then 200 µg mL−1 of PTFL NPs with or without N2 gas treatment were injected, and the change of O2 content was monitored throughout the process to reflect the PFOB‐assisted LOX catalysis.
Iron Ions Release and •OH Generation
Iron ions release in response to ATP was determined according to the previous study.[ 40 ] Briefly, 0.2 mg mL−1 PTFL NPs were mixed with 1 mg mL−1 ATP under continuous shaking. At different time intervals, PTFL NPs solution was centrifuged and the supernatant was divided, then added with distilled water to half and hydroxylamine hydrochloride (1 mg mL−1) to the other half for 15 min. After that, the reacted aqueous solutions were mixed with 1,10‐phenanthroline (1 mg mL−1), and the absorptions at 510 nm were measured to calculate the release amount of total iron ions and converted Fe(II) ions based on a standard curve. To investigate the •OH generation, MB degradation by •OH was measured as an indicator according to the absorption at 665 nm. Briefly, 0.2 mg PTFL, 1 mg ATP, and 10 µg MB were mixed in 1 mL NaL (10 mm) and incubated at room temperature. At different time points (0, 10, 20, 30, 40, 50, 60 min), the mixture was centrifuged and absorption of the supernatant was measured. Besides, four different combination trials were designed to prove that the MB degradation was caused by the combined effect of PTFL, lactate, and ATP. The first group contained 0.2 mg mL−1 PTFL only, while the second group contained both PTFL (0.2 mg mL−1) and ATP (1 mg mL−1). The third group consisted of PTFL (0.2 mg mL−1) and NaL (10 mm), and the last group consisted of PTFL (0.2 mg mL−1), NaL (10 mm), and ATP (1 mg mL−1). The absorption of MB at 665 nm was measured at different time intervals, which indicated the level of •OH generation in each group. Also, ATP levels were detected using an ATP assay kit.
Cell Culture
Murine mammary carcinoma 4T1 cells, murine mammary carcinoma 4T1 cells, and human non‐tumorigenic epithelial cell line MCF‐10A cells were all purchased from the American Type Culture Collection (ATCC, Manassas, VA). Murine mammary carcinoma 4T1 cells were used for in vitro cytotoxicity assay (Figure 2a), fluorescence live/dead assay (Figure 2b), flow cytometry (Figure 2c), fluorescence imaging of PTFL NPs endocytosis (Figure 2d), measurement of lactate level in culture medium and intracellular ATP level after various treatments (Figure 2e,f), hypoxia and ROS fluorescence staining (Figure 2g), fluorescent intensity analysis of both oxidative stress and hypoxia after various treatments (Figure 2h), western blot analysis of the expression levels of cleaved caspase‐3 (Figure S13, Supporting Information), measurement of lactate accumulation at different time intervals (Figure S14, Supporting Information), and intracellular ATP level treated with various concentrations of PTF NPs (Figure S15, Supporting Information). Murine colorectal carcinoma CT26 cells were used for in vitro cytotoxicity assay (Figure S11a, Supporting Information), fluorescence live/dead assay (Figure S11b, Supporting Information), and measurement of lactate level in culture medium and intracellular ATP level after various treatments (Figure S16, Supporting Information). Human non‐tumorigenic epithelial cell line MCF‐10A cells were used for in vitro cytotoxicity assay (Figure S12, Supporting Information). Both 4T1 cells and CT26 cells were cultured in RPMI 1640 supplemented with 10% heat‐inactivated fetal bovine serum (FBS), penicillin (100 units mL−1), and streptomycin (100 mg mL−1) at 37 °C in a humidified atmosphere with 5% CO2. MCF‐10A cells were cultured in complete growth medium including DMEM/F‐12 medium supplemented with horse serum (5%), epidermal growth factor (20 ng mL−1), insulin (10 µg mL−1), hydrocortisone (500 ng mL−1), penicillin/streptomycin (1 unit mL−1), and cholera toxin (0.1 µg mg−1).
Cell Viability Test
4T1 cells were seeded in 96‐well plates at a density of 5 × 103 cells per well. After 24 h, all the cell culture medium was replaced by fresh ones containing different concentrations of P NPs, PL NPs, PTF NPs, or PTFL NPs. Herein, the same concentration of P NP, PL NPs, PTF NPs, and PTFL NPs refers to the same content of PFOB. The whole co‐culture time lasted for 48 h, then the CCK‐8 assay kit was used to detect the cell viability by a microplate reader (Thermo Scientific, Varioskan LUX, USA). Besides, live & dead fluorescent imaging of treated cells was realized by calcein‐AM and PI staining. Briefly, after 24 h culture of 4T1 cells, the culture medium was replaced with fresh ones containing different NPs. 10 h later, all groups were stained by calcein‐AM and PI and further observed under a CLSM. Experiments were repeated that 4T1 cells were replaced with CT26 cells and all the conditions were kept the same.
Apoptosis & Necrosis Assay
Annexin V‐PE/7‐AAD apoptosis detection kit was used to investigate the cell apoptosis induced by P NPs, PL NPs, PTF NPs, and PTFL NPs. The co‐culture of 4T1 cells and NPs was kept for 12 h, then cells in each group were carefully harvested. The harvested cells were counted using a cell counting chamber, treated with Annexin V‐PE/7‐AAD apoptosis detection kit, and analyzed by a flow cytometer according to the manufacturer's instruction. Besides, western blot analysis was also conducted with the same treatments on 4T1 cells with GAPDH as control.
Cell Endocytosis Test
The cellular uptake of PTFL NPs was investigated by incubating PTFL NPs with 4T1 cells. To ensure PTFL NPs could be observed under CLSM, PTFL NPs were first tagged with RBITC (λ em = 580 nm). All the synthesis steps were the same except that 1 µL RBITC (20 mg mL−1) was added in PFOB emulsion together with FeCl3 · 6H2O. 4T1 in a total amount of 5 × 104 were first seeded in a 35 mm confocal dish. After 24 h, the culture medium was replaced by a fresh one containing RBITC tagged PTFL (PTFL–RBITC) NPs (120 µg mL−1). The cells were incubated for another 4 h and washed with PBS three times, then 1 µg mL−1 Hoechst 33342 was added to label the nucleus for 15 min. Subsequently, CLSM images were taken to observe the endocytosis of PTFL NPs.
Intracellular ROS and Hypoxia Detection
The generation of PTFL NPs induced oxidative stress and hypoxia was detected by Image‐iT Hypoxia Reagent (Invitrogen) and CellROX Deep Red Reagent (Invitrogen). First, 4T1 cells were seeded in 96‐well plates at a density of 5 × 103 cells per well. After 24 h, the culture medium was replaced by a fresh one containing P NP, PL NPs, PTF NPs, or PTFL NPs (120 µg mL−1) with a layer of liquid paraffin coverage to prevent the O2 exchange, then co‐cultured for another 6 h. Subsequently, 4T1 cells in each group were washed with PBS three times and stained by hypoxia and oxidative stress probes. CLSM images were taken to qualitatively analyze the intracellular level of hypoxia and oxidative stress in each group. Quantitative analysis of intracellular ROS and hypoxia was obtained by flow cytometry according to the manufacturer's instructions. The cells without any treatments were set as blank.
In Vitro Lactate Generation & Depletion
Lactate generation at different time points (12, 24, 48, 72 h) during 4T1 cells growth was evaluated by measurement of lactate level in cell supernatant using the lactate assay kit. For lactate depletion detection under various treatments, after incubation for 24 h, 4T1 cells were treated with P NP, PL NPs, PTF NPs, or PTFL NPs for another 24 h. Thereafter, the supernatants were collected to measure the lactate concentration. Experiments were repeated with CT26 cells and all the conditions were kept the same.
In Vitro ATP Depletion
4T1 cells were seeded in 96‐well plates at a density of 5 × 103 cells per well. After 24 h, the culture medium was replaced by a fresh one containing P NPs, PL NPs, PTF NPs, or PTFL NPs, and incubated for another 24 h. Thereafter, the 4T1 cells were collected, disrupted, and analyzed using an ATP assay kit for the measurement of intracellular ATP levels. Also, different concentrations of PTF NPs were used for 4T1 treatment, and the intracellular ATP levels were measured in the same manner.
Animals and Tumor Model
BALB/c nude mice (4–6 weeks old, female or male) were purchased from Centralized Animal Facilities of The Hong Kong Polytechnic University. All animal protocols were conducted under the Guidelines for Care and Use of Laboratory Animals of Department of Health of the Government of the Hong Kong Special Administrative Region. An animal ethics approval was obtained from the Animal Ethics Committee of The Hong Kong Polytechnic University (Ref No.: 20‐21/124‐BME‐R‐GRF) with an animal license issued by the Department of Health of the Government of the Hong Kong Special Administrative Region (Ref No.: (20–62) in DH/HT&A/8/2/4 Pt.2). For in vivo anti‐tumor and biodistribution study, 4T1 cells were harvested and suspended in the serum‐free medium at a density of 2 × 107 cells mL−1. Then 0.1 mL cell suspension was injected subcutaneously to the back of mice. For in vivo anti‐metastasis study, 4T1 cells were collected and injected into the right armpit of mice (2 × 107 cells mL−1, 0.1 mL).
In Vitro/In Vivo PA Imaging
In vitro demonstration of PA imaging was carried out in tissue‐mimic phantom using polyethylene (PE) tubes (0.5 mm inner diameter). Briefly, PTFL NPs with various concentrations from 0 to 2 mg mL−1 were injected into PE tubes, then all PE tubes were immersed in water for PA imaging. Both single wavelength scanning (700 nm) and spectra scanning (680–970 nm) were utilized to analyze the PA performance of PTFL NPs. For in vivo PA imaging study, after the tumor volume reached 100 mm2, 0.2 mL PTF NPs and PTFL NPs (1 mg mL−1) were intravenously (i.v.) injected into different 4T1 subcutaneous tumor‐bearing mice. At different time intervals (0, 4, 10, 24, 48 h), PA tomographic scanning was operated at 700 nm for the investigation of PTF/PTFL NPs accumulation, while oxygen saturation (sO2) scanning was operated at 750/850 nm for the analysis of tumor oxygenation level. Further quantitative data analysis and 3D reconstruction were performed on FUJIFILM VEVO software (FUJIFILM VisualSonics, Canada).
In Vivo Fluorescent Imaging and Biodistribution
IR‐780 fluorescent dye was introduced to tag PTFL NPs (PTFL–IR780) for in vivo fluorescence imaging. IR‐780 was only used for in vivo fluorescence imaging experiments and was not used for in vivo therapy experiments. All the synthesis steps were the same as PTFL NPs synthesis except that 5 µL IR‐780 (10 mg mL−1 in DMSO) was added in PFOB emulsion together with FeCl3 · 6H2O. The obtained PTFL–IR780 NPs were centrifuged to remove the unloaded IR‐780 and re‐dispersed in PBS. The successful labeling of IR780 and the stability of the labeling method were characterized by measuring the Vis–NIR extinction of the PTFL–IR780 NPs before and after dispersing in PBS for 7 days. After the tumor reached an approximate size of 100 mm3, 4T1 subcutaneous tumor‐bearing mice were i.v. injected with 0.2 mL PTFL–IR780 NPs (1 mg mL−1), then imaged under an IVIS system with an excitation wavelength of 760 nm and an emission wavelength of 845 nm at different time intervals (0, 2, 4, 10, 24, 48 h). For the biodistribution study, the PTFL‐IR780 treated mice were sacrificed at 4, 10, 24, 48 h post‐administration. The major organs (lungs, kidneys, hearts, spleens, and livers) and tumor tissues were excised and imaged ex vivo with the same excitation (760 nm) and emission (845 nm). The PerkinElmer Living Image Analysis Software was used to further conduct the region of interest analysis.
In Vivo Therapeutic Efficacy
The 4T1 subcutaneous tumor‐bearing mice were divided into 6 groups for different treatments as follows: 1) PBS, 2) LOX, 3) P NPs, 4) PL NPs, 5) PTF NPs, 6) PTFL NPs at an equivalent dose of 2.12 mg kg−1 of Fe and 0.21 mg kg−1 LOX. It should be noted that group (2) and (6) has the same amount of LOX, and for group (3), (4), (5) and (6), the amount of PFOB was the same. All the treatments were based on i.v. administration. Besides, the other treatments were performed on day 4 after the first treatments to ensure therapeutic efficacy. Tumor size and body weights were measured every 2 days. Tumor volumes were calculated as length × width2 /2. After 16 days post‐treatment, all the mice were sacrificed, and the tumors were excised and weighted.
TME Investigation
After the 4T1 subcutaneous tumor‐bearing mice were divided into 6 groups (3 mice each group) after the tumors reached an approximate size of 100 mm3. Then each group was treated with 200 µL PBS, LOX, P NPs, PL NPs, PTF NPs, and PTFL NPs. At 48 h post‐administration, all the mice were sacrificed, and tumors were collected. Subsequently, the tumors were first lysed using a Precellys Evolution tissue homogenizer (Bertin Instruments, France), then the supernatants were collected, and the concentration of lactate and ATP was measured using the lactate assay kit and ATP assay kit according to the manufacturer's instructions.
Anti‐Metastasis Evaluation
The 4T1 axillary tumor‐bearing mice were introduced to evaluate the anti‐metastasis effect. Briefly, mice were randomly assigned to 3 groups (n = 3) and treated with PBS, PTF NPs (0.2 mg), and PTFL NPs (0.2 mg) respectively at days 0, 4, and 9. 21 days after the first treatment. Mice were sacrificed, and lungs were collected and fixed by Bouin's solution. Subsequently, representative digital photographs were taken to observe the lungs and the number of pulmonary metastatic nodules were recorded. Eventually, H&E staining was used to further observe the metastasis in the lungs.
Histology and Immunofluorescence Staining
On day 16 post‐treatment, the mice in different groups were sacrificed and the major organs (lungs, kidneys, hearts, spleens, and livers) and tumor tissues were collected and fixed in 4% paraformaldehyde. First, the fixed tumor tissues were sliced and stained with H&E, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) according to the instructions. Besides, immunofluorescence staining against hypoxia‐inducible factor 1‐alpha (HIF1‐α), vascular endothelial growth factor A (VEGF‐A), cluster of differentiation 31 (CD31) biomarkers was performed to investigate the generation of tumor hypoxia, the anti‐angiogenesis, and the anti‐metastasis effects. The nuclei were stained with DAPI. All the slices were then imaged by optical microscopy or CLSM. Eventually, the major organs (lungs, kidneys, hearts, spleens, and livers) were sectioned and stained with H&E, followed by the observation under optical microscopy to confirm the biocompatibility of treated materials. Blinded histological analysis of the stained tissue slices was assessed by a trained surgeon at The First Affiliated Hospital, College of Medicine, Zhejiang University.
Statistical Analysis
Student's t‐test was utilized for single comparisons and one‐way analysis of variance (ANOVA) was utilized for multiple comparisons. All statistical analyses were performed using GraphPad Prism 8 software. Significant differences were defined as *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 while non‐significant (NS) differences were defined as p > 0.05. All data were expressed as mean ± standard deviation (SD).
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) (Grant No. 31771077), the Hong Kong Research Council Collaborative Research Grant (C5011‐19G), Innovation and Technology Fund, Guangdong‐ Hong Kong Cooperation Scheme (GHP‐039‐18GD), the Hong Kong Research Council General Research Fund (15216917), the Shenzhen‐Hong Kong‐Macao Science and Technology Plan Project (Category C, 202011033000095), and the Hong Kong Polytechnic University Internal Fund (1‐ZE1E). The authors would like to thank the facilities and technical supports from University Research Facility in Life Sciences (ULS).
Tian F., Wang S., Shi K., Zhong X., Gu Y., Fan Y., Zhang Y., Yang M., Dual‐Depletion of Intratumoral Lactate and ATP with Radicals Generation for Cascade Metabolic‐Chemodynamic Therapy. Adv. Sci. 2021, 8, 2102595. 10.1002/advs.202102595
Data Availability Statement
Research data are not shared.
References
- 1. Kv R., Liu T. I., Lu I. L., Liu C. C., Chen H. H., Lu T. Y., Chiang W. H., Chiu H. C., J. Controlled Release 2020, 328, 87. [DOI] [PubMed] [Google Scholar]
- 2. Swartz M. A., Iida N., Roberts E. W., Sangaletti S., Wong M. H., Yull F. E., Coussens L. M., DeClerck Y. A., Cancer Res. 2012, 72, 2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Boedtkjer E., Pedersen S. F., Annu. Rev. Physiol. 2020, 82, 103. [DOI] [PubMed] [Google Scholar]
- 4. Zhu W., Dong Z., Fu T., Liu J., Chen Q., Li Y., Zhu R., Xu L., Liu Z., Adv. Funct. Mater. 2016, 26, 5490. [Google Scholar]
- 5. Zhang X., Xi Z., Machuki J. O., Luo J., Yang D., Li J., Cai W., Yang Y., Zhang L., Tian J., Guo K., Yu Y., Gao F., ACS Nano 2019, 13, 5306. [DOI] [PubMed] [Google Scholar]
- 6. Gong F., Cheng L., Yang N. L., Betzer O., Feng L. Z., Zhou Q., Li Y. G., Chen R. H., Popovtzer R., Liu Z., Adv. Mater. 2019, 31, 1900730. [DOI] [PubMed] [Google Scholar]
- 7. Gao Z., Horiguchi Y., Nakai K., Matsumura A., Suzuki M., Ono K., Nagasaki Y., Biomaterials 2016, 104, 201. [DOI] [PubMed] [Google Scholar]
- 8. Chen Y., Liu X., Yuan H., Yang Z., von Roemeling C. A., Qie Y., Zhao H., Wang Y., Jiang W., Kim B. Y. S., Adv. Sci. 2019, 6, 1802070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hirschhaeuser F., Sattler U. G. A., Mueller‐Klieser W., Cancer Res. 2011, 71, 6921. [DOI] [PubMed] [Google Scholar]
- 10. Heiden M. G. V., Cantley L. C., Thompson C. B., Science 2009, 324, 1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de la Cruz‐López K. G., Castro‐Muñoz L. J., Reyes‐Hernández D. O., García‐Carrancá A., Manzo‐Merino J., Front. Oncol. 2019, 9, 1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kennedy K. M., Dewhirst M. W., Future Oncol. 2010, 6, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Colegio O. R., Chu N. Q., Szabo A. L., Chu T., Rhebergen A. M., Jairam V., Cyrus N., Brokowski C. E., Eisenbarth S. C., Phillips G. M., Cline G. W., Phillips A. J., Medzhitov R., Nature 2014, 513, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mukherjee S., Patra C. R., Nanoscale 2016, 8, 12444 [DOI] [PubMed] [Google Scholar]
- 15. Potente M., Gerhardt H., Carmeliet P., Cell 2011, 146, 873. [DOI] [PubMed] [Google Scholar]
- 16. Parks S. K., Chiche J., Pouysségur J., Nat. Rev. Cancer 2013, 13, 611. [DOI] [PubMed] [Google Scholar]
- 17. Chen Q. W., Wang J. W., Wang X. N., Fan J. X., Liu X. H., Li B., Han Z. Y., Cheng S. X., Zhang X. Z., Angew. Chem., Int. Ed. 2020, 59, 21562. [DOI] [PubMed] [Google Scholar]
- 18. Gao F., Tang Y., Liu W.‐L., Zou M.‐Z., Huang C., Liu C.‐J., Zhang X.‐Z., Adv. Mater. 2019, 31, 1904639. [DOI] [PubMed] [Google Scholar]
- 19. Tang J., Meka A. K., Theivendran S., Wang Y., Yang Y., Song H., Fu J., Ban W., Gu Z., Lei C., Li S., Yu C., Angew. Chem., Int. Ed. 2020, 59, 22054. [DOI] [PubMed] [Google Scholar]
- 20. Li K., Lin C. C., He Y., Lu L., Xu K., Tao B. L., Xia Z. Z. L., Zeng R., Mao Y. L., Luo Z., Cai K. Y., ACS Nano 2020, 14, 14164. [DOI] [PubMed] [Google Scholar]
- 21. Ghisla S., Massey V., Chemistry and Biochemistry of Flavoenzymes, CRC Press Inc., Boca Raton, FL: 1991. [Google Scholar]
- 22. Suman S., Singhal R., Sharma A. L., Malthotra B. D., Pundir C. S., Sens. Actuators, B 2005, 107, 768. [Google Scholar]
- 23. Hu X., Chao M., Wu H., Signal Transduction Targeted Ther. 2017, 2, 16047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gatenby R. A., Gillies R. J., Nat. Rev. Cancer 2004, 4, 891. [DOI] [PubMed] [Google Scholar]
- 25. Brizel D. M., Schroeder T., Scher R. L., Walenta S., Clough R. W., Dewhirst M. W., Mueller‐Klieser W., Int. J. Radiat. Oncol., Biol., Phys. 2001, 51, 349. [DOI] [PubMed] [Google Scholar]
- 26. Walenta S., Wetterling M., Lehrke M., Schwickert G., Sundfør K., Rofstad E. K., Mueller‐Klieser W., Cancer Res. 2000, 60, 916. [PubMed] [Google Scholar]
- 27. van der Vliet A., Heininger Y. M. W. J., J. Cell. Biochem. 2014, 115, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ren J., Xu Z., Kang Y., Zhang L., Zhang J., Cui H., Zhang W., Cao Y., Xue P., Biomaterials 2020, 234, 119771. [DOI] [PubMed] [Google Scholar]
- 29. Hayyan M., Hashim M. A., AlNashef I. M., Chem. Rev. 2016, 116, 3029. [DOI] [PubMed] [Google Scholar]
- 30. Di Virgilio F., Sarti A. C., Falzoni S., De Marchi E., Adinolfi E., Nat. Rev. Cancer 2018, 18, 601 [DOI] [PubMed] [Google Scholar]
- 31. Mo R., Jiang T., DiSanto R., Tai W., Gu Z., Nat. Commun. 2014, 5, 3364. [DOI] [PubMed] [Google Scholar]
- 32. Zhu H., Cao G., Fu Y., Fang C., Chu Q., Li X., Wu Y., Han G., Nano Res. 2021, 14, 222. [Google Scholar]
- 33. Luo Y., Qiao B., Zhang P., Yang C., Cao J., Yuan X., Ran H., Wang Z., Hao L., Cao Y., Ren J., Zhou Z., Theranostics 2020, 10, 6987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Riess J. G., Chem. Rev. 2001, 101, 2797. [DOI] [PubMed] [Google Scholar]
- 35. Matsumoto S., Kuroda Y., Transplantation. 2002, 74, 1804. [DOI] [PubMed] [Google Scholar]
- 36. Kee A. L. Y., Teo B. M., Ultrason. Sonochem. 2019, 56, 37. [DOI] [PubMed] [Google Scholar]
- 37. Rapoport N., Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol. 2012, 4, 492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jia T., Wang Z., Sun Q., Dong S., Xu J., Zhang F., Feng L., He F., Yang D., Yang P., Lin J., Small 2020, 16, 2001343. [DOI] [PubMed] [Google Scholar]
- 39. Xu J., Shi R., Chen G., Dong S., Yang P., Zhang Z., Niu N., Gai S., He F., Fu Y., Lin J., ACS Nano 2020, 14, 9613. [DOI] [PubMed] [Google Scholar]
- 40. Tang Z., Liu Y., He M., Bu W., Angew. Chem., Int. Ed. 2019, 58, 946. [DOI] [PubMed] [Google Scholar]
- 41. Wang X., Zhong X., Liu Z., Cheng L., Nano Today 2020, 35, 100946. [Google Scholar]
- 42. Zhang L., Wan S. S., Li C. X., Xu L., Cheng H., Zhang X. Z., Nano Lett. 2018, 18, 7609. [DOI] [PubMed] [Google Scholar]
- 43. Guo Y., Jia H. R., Zhang X., Zhang X., Sun Q., Wang S. Z., Zhao J., Wu F. G., Small 2020, 16, 2000897. [DOI] [PubMed] [Google Scholar]
- 44. Liu T., Zhang M., Liu W., Zeng X., Song X., Yang X., Zhang X., Feng J., ACS Nano 2018, 12, 3917. [DOI] [PubMed] [Google Scholar]
- 45. Hao N., Han L., Yang Y., Wang H., Webley P. A., Zhao D., Appl. Surf. Sci. 2010, 256, 5334 [Google Scholar]
- 46. Xie L. Y., Wehling R. L., Ciftci O., Zhang Y., Food Res. Int. 2017, 102, 195. [DOI] [PubMed] [Google Scholar]
- 47. Beattie J. K., Djerdjev A. M., Angew. Chem., Int. Ed. 2004, 43, 3568. [DOI] [PubMed] [Google Scholar]
- 48. McCarty L. S., Whitesides G. M., Angew. Chem., Int. Ed. 2008, 47, 2188. [DOI] [PubMed] [Google Scholar]
- 49. Hui Y., Yi X., Hou F., Wibowo D., Zhang F., Zhao D., Gao H., Zhao C. X., ACS Nano 2019, 13, 7410. [DOI] [PubMed] [Google Scholar]
- 50. Deng H., Song K., Zhang J., Deng L., Dong A., Qin Z., Chem. Commun. 2018, 54, 3014. [DOI] [PubMed] [Google Scholar]
- 51. Jaegers J., Wrobeln A., Ferenz K. B., Pfluegers Arch. 2021, 473, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cheng Y., Cheng H., Jiang C., Qiu X., Wang K., Huan W., Yuan A., Wu J., Hu Y., Nat. Commun. 2015, 6, 8785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lowe K. C., Blood Rev. 1999, 13, 171. [DOI] [PubMed] [Google Scholar]
- 54. Díaz‐López R., Tsapis N., Fattal E., Pharm. Res. 2010, 27, 1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
Research data are not shared.