

Abstract
Recent clinical successes have intensified interest in using adeno-associated virus (AAV) vectors for therapeutic gene delivery. The liver is a key clinical target, given its critical physiological functions and involvement in a wide range of genetic diseases. Here, we report the bioengineering of a set of next-generation AAV vectors, named AAV-SYDs (where “SYD” stands for Sydney, Australia), with increased human hepato-tropism in a liver xenograft mouse model repopulated with primary human hepatocytes. We followed a two-step process that staggered directed evolution and domain-swapping approaches. Using DNA-family shuffling, we first mapped key AAV capsid regions responsible for efficient human hepatocyte transduction in vivo. Focusing on these regions, we next applied domain-swapping strategies to identify and study key capsid residues that enhance primary human hepatocyte uptake and transgene expression. Our findings underscore the potential of AAV-SYDs as liver gene therapy vectors and provide insights into the mechanism responsible for their enhanced transduction profile.
Keywords: AAV, adeno-associated vectors, bioengineering, bioengineered vectors, gene therapy, liver-tropic, recombinant vectors, preclinical liver model, xenograft model
Graphical Abstract
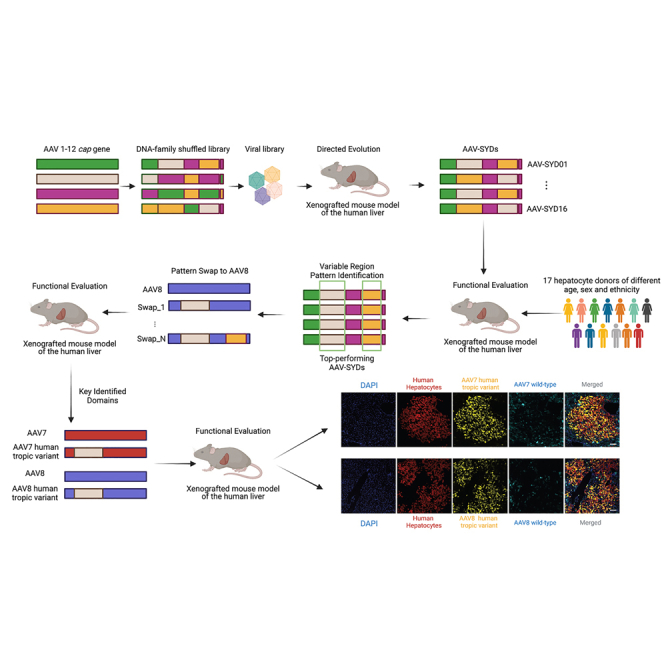
Using DNA-family shuffling and directed evolution, AAV variable regions (VRs) responsible for efficient human hepatocyte transduction in vivo were identified. Dissection of these regions via domain-swapping strategies defined key transferable domains that markedly enhanced primary human hepatocyte uptake and transgene expression of AAV7 and AAV8.
Introduction
Adeno-associated viruses (AAVs), from which AAV vector systems were derived, are putatively non-pathogenic, non-enveloped single-stranded DNA parvoviruses endemic in the human population.1 Wild-type (WT) AAV consists of a 4.7 kb genome, encoding non-structural (rep), structural (cap), assembly activating (aap), and membrane-associated accessory (maap) proteins.2,3 The AAV cap gene encodes three capsid subunits, VP1–VP3, that assemble stochastically into a T = 1 icosahedral capsid, consisting of 60 subunits.4 Diversity among AAV serotypes has been generally defined by amino acid differences within nine variable regions (VRs) found in VP3 (VR-I to VR-IX).5 These VRs are found on the capsid surface and have been associated with specific functional roles in the AAV life cycle, including receptor binding and antigenic specificity.5 Several features make AAV vectors desirable for clinical gene therapy applications, such as long-term expression in non-dividing cells, ease of manufacturability, and relatively low immunogenicity.6 To date, three capsid serotypes (AAV1, AAV2, and AAV9) have gained regulatory approval for commercial use in patients.7 Thus far, no liver-directed AAV-based therapy has obtained market approval. Since the capsid is the primary determinant of vector tropism, numerous strategies have been developed to bioengineer AAV capsids, with the ultimate goal of creating novel variants with improved targeted tropism.8
These bioengineering technologies can be broadly divided into two categories: rational design and directed evolution.8 Rational design requires knowledge of AAV capsid sequence and structure and associated phenotypic functions. Unfortunately, our understanding of these links remains limited. However, biology has the perfect algorithm for circumventing these knowledge gaps in AAV biology: evolution. By performing cycles of mutations and fitness-based selection, evolution allows organisms to continuously adapt to the ever-changing environment.9 Directed evolution mimics natural evolution by artificial selection, departing from existing proteins (AAV capsids in this case), randomly introducing mutations via various methods,8 and screening the progeny for capsids with enhanced traits,9 such as improved transduction of human hepatocytes. As in natural selection, directed evolution can only help identify the fittest variants for a given environment (selection model). Thus, it is crucial to perform such experiments in preclinical models that most closely recapitulate the intended final clinical target. When targeting the human liver, chimeric mice repopulated with primary human hepatocytes have recently gained attention for the development and testing of AAV vectors.10, 11, 12, 13 In these models, human hepatocytes proliferate in the host mouse liver scaffold and replace endogenous murine hepatocytes. This process is facilitated by a combination of genetically induced toxicity in endogenous murine hepatocytes and intrinsic immunodeficiency, as recently reviewed.14,15
While we and others have previously developed AAV vectors in murine chimeric models,10, 11, 12 here, we set out to use directed evolution as a genetic tool not only for generating vectors with improved human liver tropism but also for mapping key VRs involved in such a phenotype. With that goal, we generated a DNA-family shuffled library16 starting from twelve different prototypical AAVs (AAV1-12). We monitored the evolution of the VRs with the hypothesis that stronger pressure would be exerted toward key capsid elements required for efficient transduction of primary human hepatocytes in vivo. Specifically, we focused on regions that enhanced vector uptake into human cells in the context of competing murine hepatocytes, a phenotype that we have defined as “search-and-find”, as well as on capsid regions that enhance prompt transgene expression (functional transduction), following capsid-mediated uptake by human hepatocytes, defined here as “enter-and-express” phenotype.
Results
In vivo selection of AAV shuffled capsids in human hepatocytes leads to robust positive enrichment of VR-I (AAV2), VRs IV-V (AAV10), and VRs VI-VIII (AAV7)
To map the regions of the AAV capsid protein important for transducing human hepatocytes in vivo, we generated an AAV capsid library by shuffling DNA of the capsid genes from AAV serotypes AAV1–AAV12. We then cloned the hybrid genes into an AAV2-based replication-competent (RC) recipient construct, downstream of the rep gene open reading frame (ORF) (Figure 1A).16 The presence of fragments from all parental variants in the plasmid library was confirmed by Sanger sequencing of 24 random clones (Figure S1 and Table S1). We packaged the viral library following standard transfection protocols (Materials and Methods) and performed five rounds of iterative library selection in humanized Fah−/−/Rag2−/−/Il2rg−/− (FRG)14 mice, by injecting the library and driving replication in human hepatocytes through super-infection with WT human adenovirus 5 (Figure 1A). We found that, in this model, human adenovirus 5 transduces human hepatocytes with unequivocally higher efficacy than murine hepatocytes (Figure S2), assessed herein with an E1 deleted adenovirus 5 vector encoding for the enhanced green fluorescent protein under the control of the CMV promoter.
Figure 1.

Novel human hepatotropic AAV candidates generated by capsid shuffling and selected in humanized FRG mice
(A) Schematic representation of capsid DNA shuffled library construction and selection of hFRG mice. (B) Phylogram depicting the evolutionary relationships between the selected AAV-SYD capsid variants and the parental AAV serotypes used to construct the library. The tree is drawn to scale; branch lengths indicate the number of substitutions per site. (C) Functional comparison of 13 selected AAV-SYD variants and control AAVs in vivo in humanized FRG mouse at the levels of molecular (DNA) and functional (cDNA) transduction. DNA and cDNA levels were measured by NGS. The percentage of barcoded NGS reads that mapped to each capsid in human hepatocytes normalized to the pre-injection mix, is shown. Asterisks mark AAV-SYD vectors displaying equal or superior performance to AAV-NP59. (D) Graphic representation of the parental AAV serotype origin of the nine VRs (VR-I to VR-IX) in the indicated novel AAV-SYD variants. “|” indicates “or”; “&” indicates a recombination event on that particular VR. (E–F) Functional comparison of AAV-SYD12 and parental AAV2, AAV7, AAV8, and AAV10. The barcoded AAV mix was injected into two hFRG mice (RI = 39%). Individual capsids were identified by the corresponding barcodes (n = 5 barcodes/capsid) in the (E) DNA and at the (F) RNA (cDNA) extracted from FAC-sorted human hepatocytes. The percentage of NGS reads associated with each barcode was normalized to the pre-injection mix. Data are mean ± SD. The statistical significance of differences between the barcode means was calculated using a two-way ANOVA, and the difference between the mean of the variants and AAV-SYD12 was calculated using Sidak's multiple comparison test (∗∗∗∗p ≤ 0.0001, Data S1).
In these xenograft models, the extent of humanization can be expressed as a replacement index (RI).15 The RI refers to the degree of endogenous hepatocyte replacement with xenotransplanted primary human hepatocytes within the liver of the host mouse and can be estimated by measuring the human albumin levels in the blood.15 To enrich for capsids with enhanced tropism for human hepatocytes, or the capacity to “search-and-find” human hepatocytes, we performed the five rounds of selection in mice with a medium-low average RI of 35%. Sequencing of full-length capsid genes from n = 48 randomly chosen clones after the fifth round of selection revealed enrichment of a phylogenetically distinct subpopulation closely related to AAV7 (Figures 1B and Figures S3 and S4). We chose 16 capsid genes representative of diverse nodes across the phylogram (Figure S3), which we designated AAV-SYD01–AAV-SYD16 (where “SYD” stands for Sydney, Australia) and cloned each into a standard AAV packaging plasmid encoding for the non-structural AAV2 Rep proteins. We subsequently studied individual packaging abilities and excluded from a further evaluation three variants that produced substantially lower than average vector yields (Figure S5). We then evaluated the remaining thirteen AAV-SYD variants for the ability to molecularly and functionally transduce human hepatocytes in hFRG mice (defined here as vector uptake and transgene expression, respectively).
To enable simultaneous study of the selected AAV variants in the same animals, with the aim of minimizing the animal-to-animal variability, we independently packaged four barcoded ssAAV-LSP1-GFP-BC-WPRE-BGHpA12 expression cassettes per AAV variant. The barcodes allowed multiplexed high-throughput in vivo screening using next-generation sequencing (NGS).17 Prototypical AAV2, AAV8, AAV-LK03,10 AAV-NP59,11 and AAV-KP118 were included as controls. The barcoded equimolar mix of novel variants and control vectors was injected intravenously (total dose 1.8 × 1011 vg/animal, equal to 1× 1010 vector genomes (vg) per variant) into an hFRG mouse (RI = 21%), and the relative performance of the capsids in primary human hepatocytes and murine liver cells was evaluated one week after injection. Briefly (see Materials and Methods), we perfused the chimeric liver and isolated murine and human hepatocytes by fluorescence-activated cell sorting (FACS).19,20 We extracted DNA and RNA from the sorted cells and analyzed the barcode composition (indicative of relative vector performance) by NGS. This process is referred to throughout the manuscript as “barcoded NGS comparison”. After normalizing the number of reads to the vector composition in the pre-injection mix, we identified a cluster of eight AAV-SYD variants functionally comparable at the level of molecular transduction (DNA reads) and transgene expression (cDNA reads) in human hepatocytes to AAV-NP59, the current gold standard vector in this model11 (indicated with “∗” in Figure 1C). Most of the AAV-SYD variants, except for AAV-SYD02 and AAV-SYD14, showed reduced functional transduction (transgene expression) potency in murine liver cells (cDNA reads in Figure S6).
To investigate which features of the AAV capsid could be responsible for the ability of the eight identified variants to transduce primary human hepatocytes with high efficiency, we analyzed their sequences focusing particularly on the capsid VRs (VR-I to VR-IX).5 We hypothesized that regions subjected to strong positive selection pressure would have a common parental origin and that these regions would be important for the superior performance of AAV-SYDs in this model. In all eight cases, the VR-I was of AAV2/AAV3b origin, VR-IV and VR-V were mostly of AAV10 origin, and VR-VI, VR-VII, and VR-VIII were of AAV7 origin (Figure 1D). VR-III, which comprises only four amino acids, was identical between all AAV-SYD variants, which was expected, as these residues are common to most parental AAVs. Given the combined superior vector uptake and expression performance in human hepatocytes (Figure 1C) and VR pattern (Figure 1D), we chose AAV-SYD12 as a representative of the AAV-SYD phylogenetic family for subsequent characterization studies.
To study whether the improved human tropism observed for AAV-SYD12 was due to epistatic interactions of the VR or could be attributable to a specific contributing parental serotype, we compared the relative performance of AAV-SYD12 and that of the parental contributors of the individual VR regions: AAV2 (VR-I), AAV7 (VR-VI to VIII), and AAV10 (VR-IV and V). We included AAV8 as a positive control for functional transgene expression in murine hepatocytes.21 We prepared a barcoded equimolar mix, as described above, and tested the relative performance of each vector (1× 1010 vector genomes (vg) per tested variant) at vector uptake and functional transduction of human primary hepatocytes and murine liver cells in two hFRG mice (RI = 29% and 49%) (Figures 1E and 1F). We found AAV-SYD12 to be significantly more effective than AAV2, AAV7, AAV8, and AAV10 at the molecular and functional transduction of human hepatocytes in vivo at this particular RI (Figures 1E and 1F). Analysis of relative vector uptake and expression in the murine liver cells one week post-injection revealed that, although AAV-SYD12 entered the murine cells with similar efficiency to AAV8 and AAV10, it failed to complete the intracellular steps required for transgene expression (Figure S7).
AAV-SYD12 displays relatively superior performance over commonly used variants in FRG mice engrafted with hepatocytes of seventeen different human donors
To evaluate the relative in vivo performance of AAV-SYD12 in comparison with commonly used AAV variants, we prepared an equimolar barcoded-vector mix containing AAV-SYD12 as well as nine other variants, including the prototypical AAV2, AAV3b, AAV5, and AAV8; bioengineered AAV-LK03, AAV-NP59, AAV2-N496D,12 and AAV2-RC01 (Materials and Methods); and the naturally occurring human-liver variant, AAV-hu.Lvr02.20 We performed the barcoded NGS comparison, as described earlier, in hFRG mice engrafted with hepatocytes from seventeen different human donors of various ages, sex, and ethnicity (Figure 2A; Table S2). Given the different replacement capacities of the human donors, we injected the animals (1 × 1010 vg per AAV variant, total dose 1 × 1011 vg) at an average RI of 40% (Figure S8A). One week post-injection, we found no significant difference in GFP transgene expression nor in average vector copy number per diploid nucleus, when comparing cells from male and female human donors (Figures S8B and S8C). The amount of AAV DNA in GFP-positive human hepatocytes ranged from 247 vgs/diploid nucleus (donor 15) to 1,082.5 (donor 9) (Figure S9). The bioengineered variants AAV-NP59, AAV2-N496D, AAV2-RC01, and AAV-SYD12 and the naturally occurring AAV-hu.Lvr02 entered human hepatocytes on average more effectively than the prototypical vectors (AAV2, AAV3b, AAV5, and AAV8) and the bioengineered AAV-LK03 (Figure 2B). Mean vector uptake by AAV-hu.Lvr02 and AAV-SYD12 was significantly higher than average uptake by the other variants, including AAV-NP59 (Figures 2B and S10). Most importantly, transgene expression from AAV-SYD12 was significantly higher than expression from any of the other variants (Figures 2C and S11). Overall, while the study confirmed the relatively superior performance of the bioengineered AAV-SYD12, it also showed that the level of cellular uptake and transgene expression in human hepatocytes in this model was primarily determined by the AAV variant and less dependent on the origin of the human hepatocytes (Figures 2B and 2C).
Figure 2.

NGS-based comparison of AAV-SYD12 and relevant AAV variants in FRG mice engrafted with hepatocytes from seventeen human donors
(A) Schematic representation of the barcoded-AAV NGS comparison. Age, biological sex, and ethnicities of human donors are shown. The hepatocytes from each donor were each independently engrafted into individual FRG recipients. (B and C) Combined transduction of the barcoded AAV-mix containing the ten AAV variants in N = 32 hFRGs. Percentage of NGS reads mapped to each AAV capsid (sum of n = 5 barcodes/capsid) in human hepatocytes (B) at the DNA (vector uptake, molecular transduction) and (C) at the cDNA (expression, functional transduction) levels, normalized to the pre-injection mix, is shown. Each data point represents percentage in an independent mouse (N = 31 hFRGs analyzed for DNA and N = 32 for cDNA). Data are mean ± SD. Statistical significance among means was calculated using one-way ANOVA, and Dunnett's multiple comparison test was used to compare AAV-SYD12 with all other AAV variants (∗∗∗∗p ≤ 0.0001, n.s. p value >0.05). (D) Average percentage of mapped NGS reads per AAV capsid in FAC-sorted human hepatocytes at the DNA (N = 31 hFRGs) and cDNA (N = 32 hFRGs) levels. The EXI is defined as the quotient between average cDNA and DNA raw percentual reads per capsid.
To provide a relative measure of vector performance following vector uptake, for each variant, we calculated the vector expression index (EXI), which is an experiment and time-specific ratio of vector-driven expression (RNA/cDNA) and the average molecular transduction (DNA, vector uptake) (Figure 2D). The analysis revealed that some vectors lost relative potency at the expression level (Figure 2D) at this time point. This finding highlights differences between vector uptake and the kinetics of vector function (transgene expression). Consistent with previous reports,11,12 AAV-NP59 entered and functionally transduced human hepatocytes with high efficacy, leading to an overall relative EXI of just above 1.0. AAV8 also presented an average EXI >1, suggesting that the relatively inferior performance of this variant in human hepatocytes could be caused by suboptimal human hepatocyte uptake or by murine over human preferential transduction, rather than functional expression (Figure 2D). AAV-SYD12 was effective at uptake and at transgene expression, which led to the highest EXI among the vectors tested (Figure 2D).
Domain-swapping strategies between AAV-SYD12 and AAV8 reveal key VR-I roles in vector uptake and expression in the humanized FRG model
Next, we focused on identifying AAV capsid regions that enhance human uptake in the context of competing murine hepatocytes (“search-and-find” phenotype) and regions that enhance the kinetics of transgene expression (functional transduction) following human hepatocyte uptake (“enter-and-express” phenotype) (Figure 3A).
Figure 3.

Characterization of the “search-and-find” and “enter-and-express” AAV phenotypes through domain swapping between AAV-SYD12 and AAV8
(A) Schematic representation of functional consequences of the “search-and-find” and “enter-and-express” AAV phenotypes in humanized FRG with low and high RIs. (B) Schematic representation of AAV VRs swapped into the AAV8 capsid scaffold. VR origins are shown for reference as colored blocks. (C) In vivo comparison of AAV-SYD12, AAV8, and swapped variants in low-RI hFRGs (n = 2). (D) In vivo comparison in high-RI hFRGs (n = 2). For (C–D), percentage of NGS reads mapped to each AAV capsid (sum of n = 4 barcodes/capsid) in human hepatocytes at the DNA (vector uptake, molecular transduction) and cDNA (expression, functional transduction) levels, normalized to the pre-injection mix, is shown.
Given the observed functional differences between AAV-SYD12 and AAV8 (Figure 2), and with the intent to understand which capsid regions contribute to specific phenotypical functions, we systematically swapped the capsid gene regions encoding VR-I (AAV2 origin), VR-IV and VR-V (AAV10 origin), and VR-VI to VR-VIII (AAV7 origin) from AAV-SYD12 into the AAV8 capsid to produce seven new variants designated AAV8-Swap01-07 (Figure 3B; for specific amino acid changes, see Table S3). We subsequently generated an equimolar barcoded mix (four independent barcodes per capsid) of these variants, as outlined previously, including AAV8 and AAV-SYD12 as controls.
To map capsid regions critical for the “search-and-find” phenotype, we first injected the barcoded mix into two humanized FRGs presenting low RI (18% average) and analyzed the distribution of NGS reads in human hepatocytes one week post-injection. Theoretically, working with low RI should help in identifying variants presenting preferential human over murine transduction.
As shown in Figure 3C for AAV8-Swap05, the introduction of VR-I (AAV2 origin) and VRs-VI to VIII (AAV7 origin) was sufficient to convert AAV8 into a vector with “search-and-find” phenotype for human cells. Variants harboring only VR-I from AAV2 (AAV8-Swap01) or VR-VI to VIII from AAV7 (AAV8-Swap03) failed to display such a phenotype (Figure 3C). Addition of VRs of AAV10 origin (VR-IV and VR-V) into AAV8 (Figures 3B and 3C, AAV-Swap02) or into AAV8-Swap05 (Figures 3B and 3C, AAV8-Swap07) had no synergistic effect on vector function. Interestingly, AAV8-Swap06 (containing VRs IV and V from AAV10 and VRs VI-VIII from AAV7, but not VR-I from AAV2) displayed superior transgene expression despite lower vector uptake (Figure 3C), characteristic of the “enter-and-express” phenotype. This phenotype was also observed in the murine liver samples (Figure S12). The only difference between AAV8-Swap07 (which as Swap05 also presents a “search-and-find” phenotype) and Swap06 is that the latter retained VR-I from AAV8, suggesting a key role of this region for transgene expression when combined with VRs of AAV10 and/or VRs of AAV7 origin.
Theoretically, increasing the number of human hepatocytes in the host liver scaffold (working with higher RIs) should minimize the relative differences between vectors with and without human “search-and-find” properties, given that vectors would randomly encounter human hepatocytes more frequently. In this sense, we observed a substantial increase in relative transduction for AAV8-Swap03 when increasing the RI (Figures 3C and 3D). Nevertheless, the accelerated DNA to RNA conversion should be translatable to hFRG mice with a higher RI, given that it arises from a post-uptake step. To study this, we injected the same barcoded AAV8-Swap01-07 vector mix into two hFRG mice with an average RI of 91%. Analysis of vector performance at the cell uptake (DNA) and expression (cDNA) levels revealed that the differences in relative vector uptake by human hepatocytes were observable, although lower, in animals with high RI (Figure 3D). Importantly, AAV8-Swap06, which presented the lowest vector uptake, maintained its efficient DNA-to-RNA conversion phenotype (Figure 3D).
The “search-and-find” phenotype: AAV2's VR-I and AAV7's VR-VII/VIII enhance human hepatocytes vector uptake in the humanized FRG model
To further characterize the “search-and-find” phenotype, we generated eight additional AAV-SYD12/AAV8 swaps. Given the importance of AAV2's VR-I for this phenotype (Figure 3C), we used AAV8-Swap07 as the starting point, we kept VR-I of AAV2 origin, and we systematically reverted VRs IV to VIII back to AAV8 (Figure 4A). We generated a barcoded vector mix of the original seven swaps (AAV-Swap01 to 07) and the new swaps (AAV-Swap08 to 15) and included AAV-SYD12 and AAV8 as controls. To perform a functional analysis, we injected the mix i.v. at 1 × 1010 vg per AAV variant into two hFRG mice with low RI (average RI = 16%). NGS results at the cellular uptake (DNA) and at the functional transduction (cDNA) levels are presented in Figure 4B. Consistent with results shown previously (Figure 3C), reversion of AAV10 regions back to AAV8 had no significant effect on the “search-and-find” phenotype (Figure 4B, Swap08 and Swap09). Reversion of VR-VI was also tolerated (Figure 4B, Swap10), suggesting that VR-IV is not involved in this phenotype. In contrast, reversion of either VR-VII or VR-VIII back to AAV8 negatively affected human transduction at low RI (Figure 4B, Swap11 and Swap12). This suggests that the minimal changes required to give AAV8 the “search-and-find” phenotype are VR-I from AAV2 origin and VRs VII-VIII from AAV7 origin.
Figure 4.

The “search-and-find” phenotype is mainly driven by VR-I of AAV2 origin and AAV7's VRs VII and VIII
(A) Schematic representation of AAV VRs swapped into the AAV8 capsid scaffold. VR origins are shown for reference as colored blocks. (B) In vivo comparison of AAV-SYD12, AAV8, and swapped variants in low-RI hFRGs (n = 2). Percentage of NGS reads mapped to each AAV capsid (sum of n = 4 barcodes/capsid) in human hepatocytes at the DNA (vector uptake, molecular transduction) and cDNA (expression, functional transduction) levels, normalized to the pre-injection mix, is shown. (C) Representative immunofluorescence analysis of a humanized FRG liver transduced with AAV8-Cerulean and AAV-SYD12-Venus. Red: human GAPDH; cyan: AAV8 vector-expressed Cerulean; yellow: AAV-SYD12 vector-expressed Venus; blue: DAPI (nuclei). Scale = 100 μm. (D) Percentage of Venus-positive and Cerulean-positive human and murine hepatocytes on the same humanized FRG mouse as analyzed with FACS. (E and F) Functional comparison between AAV8-SF-Venus and AAV8-Cerulean using immunofluorescence (E) and FACS (F) analyses. (G–H) Functional comparison between AAV7-SF-Venus and AAV7-Cerulean using immunofluorescence (G) and FACS (H) analyses. (I–N) Equivalent analyses of AAV-SYD12, AAV8-SF, and AAV7-SF in mice presenting higher RIs.
At this stage, it is important to acknowledge that (i) the NGS results only give relative, not absolute, performance of capsids, and (ii) that NGS analysis provides only information on the average net transduction in the tested cells/tissue at a particular time point (one week in this case). To complement the NGS analyses presented herein, we further characterized vectors displaying the “search-and-find” phenotype by fluorescence microscopy. To compare transduction patterns in the same animal and reduce intra-animal variance, we cloned two AAV cassettes expressing the Cerulean22 or Venus23 fluorescent protein reporters under the control of the same liver-specific promoter. We validated the strategy by injecting a humanized FRG mouse with equimolar doses of both constructs packaged in AAV8 (Figure S13).
We subsequently compared the performance of AAV-SYD12 and AAV8 in a low engrafted hFRG mouse. We mixed 1 × 1011 vg of AAV8-Cerulean and AAV-SYD12-Venus (Figure S14), co-injected them i.v. into an hFRG mouse and analyzed their relative performance two weeks post-injection using immunofluorescence on liver sections (Figure 4C) and flow cytometry (Figure 4D). To do so, we tightened the left lobe of the liver prior to collagenase perfusion and perfused the rest of the liver, as described in more detail in the Materials and Methods section. As shown in Figure 4C, whereas AAV8 transduced murine cells more efficiently, AAV-SYD12 transduced human hepatocytes efficiently (see co-localization of Venus and hGAPDH, Figure 4C), even at this low RI (∼0.6%). The FACS quantification aligned well with the immunofluorescence and the previous NGS results (Figures 4D and S6).
Next, we cloned the minimal hypothesized “search-and-find” (SF) features (AAV2's VR-I + AAV7's VR-VII and VIII) into AAV8 (AAV8-SF) and compared its performance with that of parental AAV8 in an hFRG mouse (RI = 1.5%), following the same approach as described for AAV-SYD12 and AAV8. As shown in Figure 4E, the introduced changes resulted in a significantly improved in vivo targeting of human hepatocytes by AAV8-SF when compared with parental AAV8 (see co-localization of Venus and hGAPDH, Figure 4E). The increase in human targeting was also evident with FACS quantification of cells from the same liver (Figure 4F). Given the hypothesized involvement of AAV7's VR-VII and VIII in the “search-and-find” phenotype, we studied whether the incorporation of AAV2's VR-I in AAV7 would be sufficient to improve AAV7's performance in this model. Thus, we cloned VR-I from AAV2 into AAV7 (AAV7-SF) and compared its performance with parental AAV7, as described before (RI = 4.75%). As shown in Figures 4G and 4H, AAV7-SF presented a marked human specificity when compared with the parental AAV7.
Next, we investigated whether the observed search-and-find phenotype would also be retained in mice presenting higher RI. We injected the same Venus/Cerulean mixes as presented in Figures 4C–4H in three engrafted hFRG mice presenting an average RI of 84% (Table S4). These data are presented in Figures 4I–4N. As observed, AAV-SYD12, AAV8-SF, and AAV7-SF showed superior human transduction than the respective controls. In all three cases, we observed a reduction in Venus-positive human hepatocytes when increasing the RI (average of 92.65% in low; average of 66.5% in high RI). Interestingly, we also observed a marked reduction in Venus-positive murine cells (average of 33.5% in low; average of 3.5% in high RI), suggesting a marked preferential transduction of human over murine hepatocytes for AAV-SYD12, AAV8-SF, and AAV7-SF.
The “enter-and-express” phenotype: AAV8's VR-I, AAV10's VR-IV, and AAV7's VR-VII enhance transgene expression in human hepatocytes in the humanized FRG model
Given that having a better understanding of the capsid domains is critical for the “search-and-find” phenotype, the next avenue of investigation was to identify capsid regions involved in the “enter-and-express” phenotype. The latter would be characterized with a superior EXI (RNA reads/DNA reads), a characteristic that could imply either faster vector kinetics or an overall superior post-entry performance.
To further characterize the “enter-and-express” phenotype, we generated eight additional AAV-SYD12/AAV8 swaps (AAV8-Swap16-23). In this case, given the observed importance of AAV8's VR-I for the high DNA to RNA conversion (Figure 3D), we used AAV8-Swap06 as the starting point, we kept VR-I of constant AAV8 origin, and we systematically reverted VRs IV to VIII back to AAV8 (Figure 5A). We generated a barcoded vector mix, as described before, including control AAV-SYD12 and AAV8, the initial swap variants (AAV8-Swap01 through 07), and the new “enter-and-express” swaps (AAV8-Swap16-23). We compared their relative function in hFRG mice with high RI (average RI = 89%). NGS results at the uptake (DNA) and functional (cDNA) levels are presented in Figure 5B. As stated above, in this study, we focused on variants with the capacity to drive higher transgene expression at this particular time point, as indicated by higher average EXI (ratio RNA reads: DNA reads, Figure 5C). Comparing the performance of these AAV8-Swaps with that of AAV8-Swap06, the data suggested that, besides AAV8's VR-I, VR-IV (AAV10 origin) and VR-VII (AAV7 origin) were the main drivers of the “enter-and-express” phenotype. This can be inferred from the marked reduction in transgene expression of AAV8-Swap16 (reverted VR-IV, Figure 5C) and the maintenance of this phenotype for AAV8-Swap22 (reverted VRs VI and VIII, Figure 5C).
Figure 5.

The “enter-and-express” phenotype is mainly driven by VR-I of AAV8 origin, AAV10's VR-IV, and AAV7's VR-VII
(A) Schematic representation of AAV swapped used in this part of the study. VR origins are shown for reference as colored blocks. (B–C) In vivo comparison of AAV-SYD12, AAV8, and swapped variants in high-RI hFRGs (n = 2). (B) Percentage of NGS reads mapped to each AAV capsid (sum of n = 4 barcodes/capsid) in human hepatocytes at the DNA (vector uptake, molecular transduction) and cDNA (expression, functional transduction) levels, normalized to the pre-injection mix, is shown. (C) EXIs (raw cDNA read percentage/raw DNA read percentage) of the swapped variants in the same high-RI hFRGs. (D) Representative immunofluorescence analysis of a humanized FRG liver transduced with AAV8-Cerulean and AAV-SYD12-Venus. Red: human GAPDH; cyan: AAV8 vector-expressed Cerulean; yellow: AAV-SYD12 vector-expressed Venus; blue: DAPI (nuclei). Scale = 100 μm. (E) FACS analysis of the percentage of Venus-positive and Cerulean-positive human and murine hepatocytes in the humanized FRG mouse shown in (D). (F and G) Equivalent analyses as in (D and E) for AAV8-EC-Venus and AAV8-Cerulean. (H and I) Equivalent analyses as in (D and E) for AAV7-EC-Venus and AAV7-Cerulean.
Using the same approach as described above (Figures 4C and 4D), we subsequently compared the performance of AAV-SYD12 and AAV8 in a highly repopulated hFRG mouse (RI = 97.5%) (Figures 5D and 5E). We increased the vector dose to 3 × 1011 vg per capsid, as compared with 1 × 1011 vg per capsid used previously, to account for the increased percentage of human repopulation. Even at this high RI, AAV8 preferentially transduced the infrequent murine hepatocytes. On the other hand, AAV-SYD12 functionally transduced ∼50% of the human hepatocytes (Figures 5D and 5E).
Finally, we cloned the minimal hypothesized “enter-and-express” (EE) features (AAV10's VR-IV and AAV7's VR-VII) into AAV8 (AAV8-EE) and analyzed its performance in comparison with parental AAV8 (Figures 5F and 5G). We also cloned and studied the hypothesized “enter-and-express” features into AAV7 background (Figures 5H and 5I). In both cases, we observed an increase in human transduction, although the differences were less substantial than for the “search-and-find” phenotype (Figures 4E–4H). This could be linked to the fact that these variants harbored VR-I from AAV8 origin and, therefore, presented an overall lower uptake into human hepatocytes (Figure 5B). In fact, and consistent with the previous observations, both AAV8-EE and AAV7-EE retained their murine functional performance (Figures 5F–5I).
Discussion
More than a decade has elapsed since the serendipitous introduction of DNA-shuffling technologies to the world of AAV engineering by the Kay, the Samulski, and the Schaffer laboratories in 2008.16,24,25 Since then, multiple bioengineered AAV variants have been developed using directed evolution screens of AAV cap-shuffled libraries.10,11 From a bioengineering perspective, shuffled capsids present unique opportunities to study the correlation between capsid sequence/structure and vector function, since parental regions may have different functional characteristics in a shuffled context.12 In this manuscript, and similarly to the approach recently reported by Biswas and colleagues,26 we complemented rational design approaches with information gathered through directed evolution experiments. Specifically, we aimed to identify AAV capsid regions critical for in vivo transduction of human hepatocytes in a xenograft mouse model of human liver. We studied the selected pattern of VRs, enriched from a large pool of shuffled variants and, subsequently, utilized this information to perform targeted bioengineering-based improvements of other variants, exemplified herein by AAV7 and AAV8 (Figures 4 and 5). Prior to deepening the discussion, the authors want to emphasize that the work presented herein falls into the field of basic research, which aimed in this case to understand the determinants of in vivo human hepatocyte transduction in the aforementioned xenograft model.27 While these novel AAV variants have the potential to be starting points for follow-up translational studies, the future clinical significance of these variants is by no means guaranteed, since all the basic studies presented herein were carried out with reporter genes in immune-deficient mice.
Thus, once contextualized, the AAV-SYD variants present superior tropism for primary human hepatocytes in the humanized FRG model. All the selected variants, except for AAV-SYD03, were closely related to the parental WT AAV7 (Figure 1B). AAV7, along with AAV8, was isolated from a rhesus monkey,28 presents strong tropism for the murine liver,29 and more recently, has been shown to have good tropism for primary human hepatocytes in the hFRG model.30 Our results, however, indicate that AAV7 is somewhat less efficient at transducing primary human hepatocytes than AAV8 and AAV10 and significantly less efficient than AAV-SYD12, a novel variant developed in this study (Figures 1E and 1F).
It is important to highlight two limitations associated with the hFRG model, namely the lack of an adaptive immune system and the impairment of hepatic intercell crosstalk due to interspecies difference.15 A third factor that could affect vector function in hFRG mice is the source of human hepatocytes. To study this, we performed a large comparison study using animals engrafted with primary human hepatocytes from seventeen human donors of different age, sex, and ethnicity (Figure 2 and Table S2). We observed variability in the average transduction efficiency of the AAV-barcoded mix depending on human hepatocyte donor. While these differences could imply that primary hepatocytes from particular donors could be more amenable to AAV transduction, due to the limited numbers of repopulated mice per human donor and other mouse-to-mouse differences, such as RI, it is difficult to draw strong conclusions regarding differential transduction between donors. In agreement with previously published data, we did not detect a substantial influence of the sex of human hepatocytes donor on AAV transduction in this model (13 Figure S8). This suggests that factors other than the differential AAV receptor expression in hepatocytes might ultimately determine the sexual dimorphism observed in murine model.31 Recently, a study by Zou and colleagues13 using a similar mouse model, the Fah−/−/non-obese diabetic (NOD)/Rag1−/−/Il2rgnull (FNRG) model, reported substantial differences in the AAV transduction of human hepatocytes depending on the timing of vector delivery following liver injury. However, using AAV-SYD12, we did not observe significant differences in the percentage of human hepatocytes expressing the eGFP transgene when comparing the various injection protocols in humanized FRG mice with high RI (78.4%–89.7% GFP-positive cells, average RI = 69.82%) (Figures S15 and S16).
A fourth element to consider when using xenografted models for AAV capsid evaluation is the level of repopulation—the RI. Whether a low or a high RI better predicts human liver capsid performance remains to be elucidated. Others have shown that the human hepatocytes in high-RI mice present characteristics of terminally differentiated hepatocytes, whereas proliferating human hepatocyte colonies in low-RI mice exhibit an inferior degree of maturation.15 Perhaps, more therapeutically relevant is that urokinase-type plasminogen activator (uPA) transgenic mice chimeric livers with an RI higher than 70% presented signs of metabolic zonation and properly aligned cords of hepatocytes.32 This could facilitate development of portal-tropic or central-tropic AAV capsids, with the aim of targeting genetic disorders affecting those specific metabolically zonated hepatocyte populations.
The RI becomes even more important when evaluating capsids that have differential mouse versus human functional transduction abilities, such as AAV8 and AAV3b.33 Despite its inherent limitations, NGS offers unprecedented precision in studying the impact of competition between cells of the host and donor species on vector function. As suggested by Wang and colleagues,33 we observed changes in relative vector performance for AAV-SYD12 and AAV8 in mice with low and high RIs (Figure 3). However, we found AAV-SYD12 to molecularly transduce murine liver cells with equivalent efficiency to AAV8 (Figure S7A), and the main difference between both vectors appeared only evident at the transgene expression level (Figure S7B). While further studies in larger animal models will shed light on clinical predictability, we hypothesize that higher human RIs reduce the confounding effect of murine liver vector uptake. This could be relevant for variants such as AAV8-Swap03, which outperformed AAV8 in the murine liver population when studied with NGS (Figure S12), and presented a marked increase in relative vector performance at higher RIs (compare Figures 3C and 3D). Although not studied herein, this phenomena of murine over human vector uptake could also explain the limited performance of AAV5 in this model.
As in the pioneering AAV domain-swapping paper by Shen and colleagues,34 we utilized this approach to map key regions for in vivo human hepatocyte transduction. We based our designs on the information gathered on the initial directed evolution experiments (Figure 1). Given the inherent complexity of vector/cell interactions, we performed separate studies to identify regions that enhance human vector uptake (“search-and-find” phenotype) and regions that enhance vector functionality after vector uptake (DNA to RNA conversion, “enter-and-express” phenotype).
The studies that aimed at identifying the “search-and-find” regions allowed us to elucidate that the VR-I from AAV2 and VRs VII-VIII from AAV7 were sufficient to enhance the human hepatocyte transduction potential of AAV8 to levels similar to AAV-SYD12 (AAV8-Swap05, Figure 4F). On the other hand, VR-I from AAV2 was sufficient to substantially improve performance of AAV7 in this model (Figure 4I). This suggests an overall superior engagement of these vectors with AAV receptor(s) on human hepatocytes, shifting thus the natural tendency of AAV7 and AAV8 to transduce murine over human hepatocytes. Interestingly, earlier this year, we described the opposite phenomenon with AAV3b, where a single amino acid insertion in VR-I (homologous to that of AAV2) was sufficient to significantly enhance the performance of this variant in the murine liver.35 The residues in VR-I are exposed to the capsid surface at the base of the protrusions of the 3-fold axis symmetry and have been previously described to be functionally involved in AAV2's transduction and A20 and IVIG neutralization.36
On the other hand, vectors with the “enter-and-express” phenotype allow for a rapid accumulation of transgene expression, presumably arising from an overall more efficient cellular trafficking. Here, it is important to highlight that most of the NGS results presented in this study were carried out one week post-injection and thus represent a picture of the relative performance of AAV variants at that particular time point. Further studies expanding the time points will be required to evaluate relative vector performance across time. In our hands, the combination of VR-I from AAV8, VR-IV from AAV10, and VR-VII from AAV7 offered a substantial increase in DNA to RNA conversion (improved trafficking) (Figure 5). VR-IV and VR-VII are structurally adjacent and surface-exposed, forming the top and the base of the 3-fold protrusion, respectively.37
Collectively, the results presented herein provide insights into the roles of VR-I, VR-IV, and VR-VII in the in vivo performance of AAV capsids in the humanized FRG model. At the same time, the novel AAV-SYD and AAV8-swapped capsids presented herein could facilitate the start of translational studies targeting genetic diseases that are currently not amenable to gene therapy treatment due to the limited efficiency of available vectors.
Materials and methods
Shuffled AAV capsid plasmid library generation
The AAV library was generated as previously described.38 AAV variants 1 to 12 were included in the parental mix. Information on parental sequences can be found in Table S5. N = 24 individual cap genes were Sanger sequenced to confirm the library diversity (Figure S1, Table S1).
RC AAV library selection
Selection of the shuffled AAV capsid in hFRG was performed as previously described10 with minor modifications. The humanized FRG mouse of the first round of selection was infected, by intravenous tail vein administration, with 2 × 1010 vector genomes of the initial AAV library preparation. To facilitate AAV library replication, WT replication-competent human adenovirus 5 (hAd5) (8 μL, ATCC VR-5, Lot Number: 70,010,153) was administered i.v. 24 h post-AAV administration. The chimeric liver was harvested and minced 72 h post hAd5 infection. Following liver harvest, the organ was homogenized, and 0.3 g portions were snap-frozen in liquid nitrogen. Two times the volume of PBS was added to one of the liver samples, which was then subjected to three freeze-thaw cycles and further homogenization with a polypropylene pestle. The virus-containing sample was then heated at 65°C for 30 min to heat inactivate the human adenovirus 5 and spun at maximum speed in a table-top centrifuge (4°C). The virus-containing liquid supernatant was subsequently harvested, and the virus titered to confirm AAV replication. Two hundred microliters of the lysate was injected i.p. into the hFRG mouse for the subsequent round of selection.
Sanger sequencing
Sanger sequencing was performed at the Garvan Molecular Genetics facility of the Garvan Institute of Medical Research (Darlinghurst, NSW, Australia) with the primers External_Seq_F/R and internal_cap_Seq (Table S6).
Vector DNA copy number per cell
Vector copy numbers were measured via droplet-digital PCR (ddPCR, Bio-Rad, Berkeley, CA, USA) using EvaGreen supermix (Bio-Rad, catalog no. 1864034) and following the manufacturer's instructions. To detect AAV genomes, GFP primers were used (GFP-F/R), and vector genomes were normalized to human albumin copy number using primers human ALB_F/R_ddPCR (Table S6).
Mouse studies
All animal experimental procedures and care were approved by the joint Children's Medical Research Institute (CMRI) and The Children's Hospital at Westmead Animal Care and Ethics Committee. Fah−/-Rag2−/−Il2rg−/- (FRG) mice14 were bred, housed, engrafted, and monitored as recently described.12 Levels of human cell engraftment were estimated by measuring the presence of human albumin in peripheral blood, using the human albumin ELISA quantitation kit (Bethyl Laboratories, catalog no. E80-129). To evaluate the AAV transduction potential, mice were placed on 10% NTBC and were maintained in this condition until harvest. Information on sex, age, and levels of repopulation of the mice used in this study can be found in Table S4. Mice were randomly assigned to experiments and transduced via intravenous injection (lateral tail vein) with the indicated vector doses. Mice were euthanized by CO2 inhalation either 1 or 2 weeks after transduction for barcoded NGS analysis or immunohistochemistry studies, respectively. To obtain murine and human single-cell suspensions from xenografted murine livers, we followed the same collagenase perfusion procedure as recently described.20 For all experiments with the exception of that of Figure S16, cells were labeled with phycoerythrin (PE)-conjugated anti-human-HLA-ABC (clone W6/32, Invitrogen 12-9983-42; 1:20), biotin-conjugated anti-mouse-H-2Kb (clone AF6-88.5, BD Pharmigen 553,568; 1:100), and allophycocyanin (APC)-conjugated streptavidin (eBioscience 17-4317-82; 1:500). GFP-positive-labelled samples were sorted to a minimal 95% purity using a BD Influx Cell sorter. For data on Figure S16, cells were labeled with anti-mouse H-2Kb PE antibody (BD Pharmingen Catolog#553570, 1:100) HLA-ABC Biotin (eBioscience Catalog#13-9983-82, 1:100), and Streptavidin APC (eBioscience Catalog#17-4317-82, 1:500). Flow cytometry was performed in the Flow Cytometry Facility, Westmead Institute for Medical Research (WMIR), Westmead, NSW, Australia. The data were analyzed using FlowJo 7.6.1 (FlowJo LLC).
AAV transgene constructs
All vectors used in the study contain AAV2 ITR sequences. The AAV construct pLSP1-eGFP-WPRE-BGHpA, which encodes eGFP under the transcriptional control of a heterologous promoter containing one copy of the SERPINA1 (hAAT) promoter and two copies of the APOE enhancer element has been previously reported.31 The barcoded versions of this construct include a 6-mer barcode between eGFP and WPRE.20 The Venus and Cerulean versions used in Figures 4 and 5 were generated by substituting eGFP with either transgene encoding for the particular fluorophore and were kindly provided by Dr Grant Logan.
Recombinant HAdenovirus-5-eGFP vector
The initial seed of a recombinant HAd5 vector containing an enhanced green fluorescence protein under the control of the CMV promoter in the E1 deleted region was kindly offered by Dr Andrew S. Files. The recombinant vectors were rescued, propagated, and titered, as described before.39
DNA and RNA isolation and cDNA synthesis
Isolation of DNA and RNA and cDNA synthesis was performed, as described in detail before20 without modifications. Briefly, we extracted DNA using a standard phenol: chloroform protocol and RNA with the Direct-Zol kit (Zymogen Cat# R2062).
AAV vector packaging and viral production
AAV constructs were packaged into AAV capsids using HEK293 cells and a helper-virus-free system, as described previously.40 Genomes were packaged in capsid variants using packaging plasmid constructs harboring rep genes from AAV2 and a specific cap. The plasmids encoding for AAV-NP59 and AAV-KP1 were a gift from Prof. Mark Kay. AAV2-RC01 is an AAV2-peptide variant harboring the following sequence modifications 584-QGQSTTHLSPPQAA-590 (VP1 numbering; underlined 7-mer amino acid insertion). Packaging of multiple barcoded ss-LSP1-eGFP-BC-WPRE-BGHpA at increasing concentrations was achieved, as described recently.12 All vectors/libraries were purified using iodixanol gradient ultracentrifugation, as previously described.41 AAV preparations were titered using droplet-digital PCR (see AAV titration section for details).
Immunohistochemical analysis of mouse livers
Immunohistochemistry was performed, as described recently in detail12 without modifications. Briefly, mouse livers were fixed with paraformaldehyde, cryo-protected in sucrose, and frozen in O.C.T. (Tissue-Tek). Frozen liver sections (5 μm) were permeabilized in ice-cold methanol, then room temperature 0.1% Triton X-100, and then reacted with anti-human GAPDH antibody (Abcam, Cat# ab215227, Clone AF674), and DAPI (Invitrogen, D1306) at 0.08 ng/mL. For Figure S2, anti-Glutamine Synthetase (Abcam, Cat# ab73593) was used. After immunolabelling, the images were captured and analyzed on a LSM800-Airyscan microscope using ZEN Black software.
Barcode Amplification, NGS, and distribution analysis
The 150-base-pair region surrounding the 6-mer barcode was amplified with Q5 High-Fidelity DNA Polymerase (NEB, catalog no. M0491L) using BC_Fxx (barcoded to allow multiplexing of different samples) and a common BC_R primer (Table S6). NGS library preparations and sequencing using 2 × 150 paired-end configurations were performed by Genewiz (Suzhou, China) using an Illumina MiSeq instrument. A workflow was written in Snakemake (5.6)42 to process reads and count barcodes. Paired reads were merged using BBMerge and then filtered for reads of the expected length in a second pass through BBDuk, both from BBTools 38.68 (https://sourceforge.net/projects/bbmap/). The merged, filtered fastq files were passed to a Python (3.7) script that identified barcodes corresponding to AAV variants. NGS reads from the DNA and cDNA populations were normalized to the reads from the pre-injection mix.
Production of rAAV crude lysates
Production of rAAV crude lysates was performed, as described before38 with a few modifications. The cell pellet was resuspended in 600 μL of Resuspension buffer (PBS, 10 mM Tris-HCl pH 8.5, 2 mM MgCl2) and subjected to three freeze-thaw cycles. Genomic and free plasmid DNA was removed by incubating with 200 U/mL at 37°C for 1 h of Benzonase (Merck KGaA, Cat #1.101695.0002; EMD Chemicals). We added 10% sodium deoxycholate to a final concentration of 0.5% and, subsequently, added ¼ volume of 5M NaCl. The solution was then incubated at 37°C for 30 min, and cellular debris was removed by centrifugation for 30 min at 5,250 × g at 4°C. The supernatant was then used to assess vector genome yield per 15-cm HEK293T plate (Figure S4).
AAV titration
AAV titration was performed via digital droplet PCR (ddPCR, Bio-Rad, Berkeley, CA, USA) using EvaGreen supermix (Bio-Rad, catalog no. 1864034) and following the manufacturer's instructions. To detect AAV genomes on vectors, GFP primers were used (GFP-F/R). To detect AAV genomes on RC libraries, rep binding primers were used (rep2-F, rep2-R). To detect the transgene encoding for Venus and Cerulean, respective matching primers were used (Table S6).
Capsid recovery and sequence contribution analysis
Selected AAV cap genes were recovered after five rounds of iterative passage in hFRG with flanking primers (F/R-cap-recovery) and cloned directly with Gibson assembly into a standard packaging plasmid harboring rep2. To allow for visualization of parental contribution in selected capsids, the Xover tool was used (http://qpmf.rx.umaryland.edu/xover.html)43 for Figures S1 and S3.
Statistical analysis
Depending on the nature of the data, different statistical analyses detailed in the figure legends were performed. Details on tests can be found in Data S1. For all used tests, significance was represented as follows: ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001, n.s. p value >0.05).
Acknowledgments
We thank CMRI Vector and Genome Engineering Facility (VGEF) for help in vector preparation. We also thank the Cytometry Facility of the Westmead Institute for Medical Research for help with sorting of murine hepatocytes. We also would like to thank all the members of the CMRI Bioresources, with special thanks to S. Dimech.
This work was supported by project grants from the Australian National Health and Medical Research Council (NHMRC) to L.L. and I.E.A. (APP1108311, APP1156431, and APP1161583), Paediatrio Pediatric Precision Medicine Program to L.L. (PPM1 K5116/RD274) as well as funding from LogicBio Therapeutics. L.L. was also supported by research grants from the Department of Science and Higher Education of Ministry of National Defense, Republic of Poland, (“Kościuszko” k/10/8047/DNiSW/T – WIHE/3) and from the National Science Centre, Republic of Poland (OPUS 13) (UMO-2017/25/B/NZ1/02790).
Author contributions
Conceptualization: M.C.-C., M.H., L.L.; methodology: M.C.-C., E.Z., I.E.A, and L.L.; software: S.S. and L.O.W.W.; investigation: M.C.-C., R.G.N., E.Z., G.B., S.H.Y.L., A.K.A., L.H.N., and A.W.; writing—original draft: M.C.-C. and L.L.; writing—review and editing: M.C.-C., A.K.A., S.S., and L.L.; funding acquisition: I.E.A. and L.L.; visualization: M.C.-C. and M.D.; supervision: M.C.-C., A.J.T., I.E.A., and L.L.
Declaration of interests
M.C.-C., I.E.A., M.H. and L.L. are inventors on patent applications filed by Children's Medical Research Institute related to AAV capsid sequences and in vivo function of novel AAV variants. L.L. is a co-founder and a scientific advisor at LogicBio Therapeutics. M.H. is an employee of, and holds stocks in, LogicBio Therapeutics. L.L. and I.E.A. are co-founders of Exigen Biotherapeutics. L.L. has a sponsored research agreement with LogicBio Therapeutics. M.C.-C., I.E.A., and L.L. have IP licensed to biopharmaceutical companies. L.L. and I.A.E. have consulted on technologies addressed in this paper. L.L. and I.A.E. have stock and/or equity in companies with technology broadly related to this paper. A.J.T. is a cofounder and scientific consultant for Orchard Therapeutics, consultant for Rocket Pharmaceuticals, Generation bio, bluebird bio, 4Bio Capital Partners, and Sana Biotechnology. G.J.L. consults for Gyroscope Therapeutics. All other authors declare no conflicts of interest.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.11.011.
Supplemental information
References
- 1.Gao G., et al. Clades of Adeno-associated viruses are widely disseminated in human tissues. J. Virol. 2004;78:6381–6388. doi: 10.1128/JVI.78.12.6381-6388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sonntag F., et al. The assembly-activating protein promotes capsid assembly of different adeno-associated virus serotypes. J. Virol. 2011;85:12686–12697. doi: 10.1128/JVI.05359-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ogden P.J., Kelsic E.D., Sinai S., Church G.M. Comprehensive AAV capsid fitness landscape reveals a viral gene and enables machine-guided design. Science. 2019;366:1139–1143. doi: 10.1126/science.aaw2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Worner T.P., et al. Adeno-associated virus capsid assembly is divergent and stochastic. Nat. Commun. 2021;12:1642. doi: 10.1038/s41467-021-21935-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drouin L.M., Agbandje-McKenna M. Adeno-associated virus structural biology as a tool in vector development. Future Virol. 2013;8:1183–1199. doi: 10.2217/fvl.13.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu Z., Asokan A., Samulski R.J. Adeno-associated virus serotypes: vector toolkit for human gene therapy. Mol. Ther. 2006;14:316–327. doi: 10.1016/j.ymthe.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 7.Wang D., Tai P.W.L., Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019;18:358–378. doi: 10.1038/s41573-019-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li, C. & Samulski, R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 21, 255-272, doi:10.1038/s41576-019-0205-4 (2020). [DOI] [PubMed]
- 9.Arnold F.H. Directed evolution: Bringing new chemistry to life. Angew. Chem. Int. Ed. 2018;57:4143–4148. doi: 10.1002/anie.201708408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lisowski L., et al. Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature. 2014;506:382–386. doi: 10.1038/nature12875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paulk N.K., et al. Bioengineered AAV capsids with combined high human liver transduction in vivo and unique humoral seroreactivity. Mol. Ther. 2018;26:289–303. doi: 10.1016/j.ymthe.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cabanes-Creus M., et al. Attenuation of heparan sulfate proteoglycan binding enhances in vivo transduction of human primary hepatocytes with AAV2. Mol. Ther. Methods Clin. Dev. 2020;17:1139–1154. doi: 10.1016/j.omtm.2020.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zou C., et al. Experimental variables that affect human hepatocyte AAV transduction in liver chimeric mice. Mol. Ther. Methods Clin. Dev. 2020;18:189–198. doi: 10.1016/j.omtm.2020.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Azuma H., et al. Robust expansion of human hepatocytes in Fah-/-/Rag2−/−/Il2rg-/- mice. Nat. Biotechnol. 2007;25:903–910. doi: 10.1038/nbt1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sugahara G., Ishida Y., Sun J., Tateno C., Saito T. Art of making artificial liver: depicting human liver biology and diseases in mice. Semin. Liver Dis. 2020;40:189–212. doi: 10.1055/s-0040-1701444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grimm D., et al. In Vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J. Virol. 2008;82:5887–5911. doi: 10.1128/JVI.00254-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Westhaus A., et al. High-throughput in vitro, ex vivo, and in vivo screen of adeno-associated virus vectors based on physical and functional transduction. Hum. Gene Ther. 2020;31:575–589. doi: 10.1089/hum.2019.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pekrun K., et al. Using a barcoded AAV capsid library to select for clinically relevant gene therapy vectors. JCI Insight. 2019;4 doi: 10.1172/jci.insight.131610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cabanes-Creus M. Isolation of human hepatocytes by collagenase perfusion. Bio-protocol. 2020 bio-protocol.org/prep532 [Google Scholar]
- 20.Cabanes-Creus M., et al. Restoring the natural tropism of AAV2 vectors for human liver. Sci. Transl Med. 2020;12 doi: 10.1126/scitranslmed.aba3312. [DOI] [PubMed] [Google Scholar]
- 21.Cunningham S.C., Alexander I.E. AAV-mediated gene delivery to the mouse liver. Methods Mol. Biol. 2019;1937:213–219. doi: 10.1007/978-1-4939-9065-8_12. [DOI] [PubMed] [Google Scholar]
- 22.Rizzo M.A., Springer G.H., Granada B., Piston D.W. An improved cyan fluorescent protein variant useful for FRET. Nat. Biotechnol. 2004;22:445–449. doi: 10.1038/nbt945. [DOI] [PubMed] [Google Scholar]
- 23.Nagai T., et al. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 2002;20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- 24.Li W., et al. Engineering and selection of shuffled AAV genomes: a new strategy for producing targeted biological nanoparticles. Mol. Ther. 2008;16:1252–1260. doi: 10.1038/mt.2008.100. [DOI] [PubMed] [Google Scholar]
- 25.Koerber J.T., Jang J.H., Schaffer D.V. DNA shuffling of adeno-associated virus yields functionally diverse viral progeny. Mol. Ther. 2008;16:1703–1709. doi: 10.1038/mt.2008.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Biswas M., et al. Engineering and in vitro selection of a novel AAV3B variant with high hepatocyte tropism and reduced seroreactivity. Mol. Ther. Methods Clin. Dev. 2020;19:347–361. doi: 10.1016/j.omtm.2020.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Austin C.P. Translating translation. Nat. Rev. Drug Discov. 2018;17:455–456. doi: 10.1038/nrd.2018.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao G.P., et al. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. U S A. 2002;99:11854–11859. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zincarelli C., Soltys S., Rengo G., Rabinowitz J.E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 30.Shao W., et al. Superior human hepatocyte transduction with adeno-associated virus vector serotype 7. Gene Ther. 2019;26:504–514. doi: 10.1038/s41434-019-0104-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dane A.P., Cunningham S.C., Graf N.S., Alexander I.E. Sexually dimorphic patterns of episomal rAAV genome persistence in the adult mouse liver and correlation with hepatocellular proliferation. Mol. Ther. 2009;17:1548–1554. doi: 10.1038/mt.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tateno C., et al. Morphological and microarray analyses of human hepatocytes from xenogeneic host livers. Lab Invest. 2013;93:54–71. doi: 10.1038/labinvest.2012.158. [DOI] [PubMed] [Google Scholar]
- 33.Wang L., et al. Comparative study of liver gene transfer with AAV vectors based on natural and engineered AAV capsids. Mol. Ther. 2015;23:1877–1887. doi: 10.1038/mt.2015.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen X., Storm T., Kay M.A. Characterization of the relationship of AAV capsid domain swapping to liver transduction efficiency. Mol. Ther. 2007;15:1955–1962. doi: 10.1038/sj.mt.6300293. [DOI] [PubMed] [Google Scholar]
- 35.Cabanes-Creus M., et al. Single amino acid insertion allows functional transduction of murine hepatocytes with human liver tropic AAV capsids. Mol. Ther. Methods Clin. Dev. 2021;21:607–620. doi: 10.1016/j.omtm.2021.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Govindasamy L., et al. Structurally mapping the diverse phenotype of adeno-associated virus serotype 4. J. Virol. 2006;80:11556–11570. doi: 10.1128/JVI.01536-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tseng Y.S., Agbandje-McKenna M. Mapping the AAV capsid host antibody response toward the development of second generation gene delivery vectors. Front. Immunol. 2014;5:9. doi: 10.3389/fimmu.2014.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cabanes-Creus M., et al. Codon-optimization of wild-type Adeno-associated virus capsid sequences enhances DNA family shuffling while conserving functionality. Mol. Ther. Methods Clin. Dev. 2019;12:71–84. doi: 10.1016/j.omtm.2018.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim J.W., et al. A dendritic cell-targeted adenoviral vector facilitates adaptive immune response against human glioma antigen (CMV-IE) and prolongs survival in a human glioma tumor model. Neurotherapeutics. 2018;15:1127–1138. doi: 10.1007/s13311-018-0650-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xiao X., Li J., Samulski R.J. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J. Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khan I.F., Hirata R.K., Russell D.W. AAV-mediated gene targeting methods for human cells. Nat. Protoc. 2011;6:482–501. doi: 10.1038/nprot.2011.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koster J., Rahmann S. Snakemake-a scalable bioinformatics workflow engine. Bioinformatics. 2018;34:3600. doi: 10.1093/bioinformatics/bty350. [DOI] [PubMed] [Google Scholar]
- 43.Huang W., Johnston W.A., Boden M., Gillam E.M. ReX: a suite of computational tools for the design, visualization, and analysis of chimeric protein libraries. Biotechniques. 2016;60:91–94. doi: 10.2144/000114381. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.