

Abstract

Li-rich rocksalt oxides are promising candidates as high-energy density cathode materials for next-generation Li-ion batteries because they present extremely diverse structures and compositions. Most reported materials in this family contain as many cations as anions, a characteristic of the ideal cubic closed-packed rocksalt composition. In this work, a new rocksalt-derived structure type is stabilized by selecting divalent Cu and pentavalent Sb cations to favor the formation of oxygen vacancies during synthesis. The structure and composition of the oxygen-deficient Li4CuSbO5.5□0.5 phase is characterized by combining X-ray and neutron diffraction, ICP-OES, XAS, and magnetometry measurements. The ordering of cations and oxygen vacancies is discussed in comparison with the related Li2CuO2□1 and Li5SbO5□1 phases. The electrochemical properties of this material are presented, with only 0.55 Li+ extracted upon oxidation, corresponding to a limited utilization of cationic and/or anionic redox, whereas more than 2 Li+ ions can be reversibly inserted upon reduction to 1 V vs Li+/Li, a large capacity attributed to a conversion reaction and the reduction of Cu2+ to Cu0. Control of the formation of oxygen vacancies in Li-rich rocksalt oxides by selecting appropriate cations and synthesis conditions affords a new route for tuning the electrochemical properties of cathode materials for Li-ion batteries. Furthermore, the development of material models of the required level of detail to predict phase diagrams and electrochemical properties, including oxygen release in Li-rich rocksalt oxides, still relies on the accurate prediction of crystal structures. Experimental identification of new accessible structure types stabilized by oxygen vacancies represents a valuable step forward in the development of predictive models.
Short abstract
Controlling the composition and synthesis conditions of Li-rich rocksalt oxides can lead to to new structure types stabilized by oxygen vacancies. The complex atomic ordering of Li4CuSbO5.5 is described and discussed in the context of other oxygen-deficient rocksalt oxides.
1. Introduction
The development of high-energy density cathode materials for Li-ion batteries has opened a large field of investigation for solid-state chemists to propose new materials and new energy storage concepts. The discovery of the reversible redox activity of oxygen anions in the so-called “Li-rich” rocksalt oxides with formula Li1+xM1–xO2 (M = transition metal) represents an important paradigm shift in that field.1,2 The possibility to extract electrons from oxygen in addition to the transition metals gives rise to a large increase in the specific capacity of these materials compared with stoichiometric LiMO2 layered oxides such as LiCoO2 or the NMC phases Li(Ni1–x–yMnxCoy)O2. However, this increase in capacity is often accompanied by several shortcomings linked to stabilization mechanisms for oxide anions upon their oxidation. Common issues range from voltage hysteresis between charge and discharge, resulting in a low-energy efficiency, to cation migration and trapping in tetrahedral sites, leading to voltage decay, and irreversible evolution of oxygen from the surface and/or the bulk of the material that causes irreversible capacity loss.1 Research efforts are therefore focused on understanding how the redox activity of the anion sublattice can be controlled and finely tuned through appropriate choice of cations, including redox active transition metals and redox inactive elements (alkali, alkaline earth, early transition metals with no d electrons or p-block elements).
The release of gaseous oxygen is a particularly important problem, as it results in irreversible capacity loss and penalizing structural modifications such as densification of the surface of materials3−5 or complete amorphization in the most extreme cases.6 Computational evaluation of the propensity for a specific composition to undergo oxygen release and oxygen vacancy formation represents a promising path to guide future experimental exploration of high-energy density cathode materials. Several research groups have already taken that direction,2,7,8 reporting different indicators to predict the release of oxygen and leading to a more general understanding of the reversibility of oxygen redox activity. Nevertheless, these indicators rely on the accurate prediction of the structure formed upon the introduction of an oxygen vacancy and would benefit from the knowledge of experimentally determined crystal structures adopted by Li-rich rocksalt oxides containing oxygen vacancies.
In this work, the formation of structures with oxygen vacancies is taken as an opportunity to discover new structure types related to the Li-rich rocksalt oxides, with formula Li4MM′O6–x□x, where M and M′ cations must be selected in order to favor the formation of oxygen vacancies during synthesis rather than during electrochemical oxidation.
Work from McCalla and co-workers on Li–Fe–Sb–O and Li–Ni–Sb–O compounds suggests that Sb-based materials are prone to oxygen release from the bulk or the surface of the material, together with cation reduction upon deep oxidation.9,10 This mechanism of reductive elimination is an extreme and irreversible manifestation of the reductive coupling mechanism observed in some cathode materials.2 To achieve this reaction at the synthesis step, we further replaced Ni/Fe with Cu, which is unlikely to oxidize beyond divalent Cu2+ under normal solid-state synthesis conditions (high temperature, ambient atmosphere). Therefore, we targeted the composition Li4CuSbO5.5□0.5 and report its structure and electrochemical properties. In this new compound, the sum of M and M′ cation oxidation states amounts to 7 instead of 8, as is usually considered to prepare stoichiometric Li4MM′O6 rocksalt oxides.
2. Results
2.1. Synthesis and Structure Determination
Li4CuSbO5.5 can be prepared by a simple ceramic method starting from Li2CO3, CuO and Sb2O3 in proportions Li:Cu:Sb = 4.4:1:1 and heating the hand-ground mixture at 1100 °C for 12 h under air (Figure 1a, orange). Heating at a lower temperature (900 °C) also leads to the formation of Li4CuSbO5.5 with broader Bragg reflections that indicate lower crystallinity and a less ordered structure (Figure 1a, green). Prolonged heating at 1100 °C (24 h) or higher temperatures results in Li2O loss and the decomposition of Li4CuSbO5.5 into the reported Li3CuSbO5 phase (Figure 1a, gray).11 The 10 mol % excess of Li2CO3 is therefore important to delay this decomposition while heating at a temperature high enough to obtain a well crystallized material. The final cation stoichiometry was confirmed by inductively coupled plasma optical emission spectroscopy (ICP-OES) to be Li4.22(9)Cu1.027(18)Sb1.000(2), with deviations from the ideal stoichiometry that can be explained by residual Li2CO3/Li2O, Li2CuO2 and Li7SbO6 impurities (<1 wt % according to Rietveld quantitative analysis).12
Figure 1.

Diffraction data on Li4CuSbO5.5. (a) XRD patterns of samples prepared at 900 °C for 12 h (green), 1100 °C for 12 h (orange), and 1100 °C for 24 h (gray). The latter corresponds to the formation of the Li2O-deficient phase Li3CuSbO5. (b) Combined Rietveld refinement of neutron and SXRD data for Li4CuSbO5.5 prepared at 1100 °C for 12 h. Experimental points are marked by red circles, simulated pattern by a black line, difference pattern by a blue line, and reflection position for the main and secondary Li4CuSbO5.5 phases is in black and blue, and for Li2CuO2 and Li7SbO6, impurities are in green and red, respectively. (c) The sample prepared at 1100 °C for 12 h can be reasonably well fitted with two triclinic phases with slightly different cell parameters, whereas the sample prepared at 900 °C for 12 h appears as a mixture of triclinic, monoclinic, and orthorhombic phases isostructural to the parent Li5SbO5 (C2/m) and Li2CuO2 (Immm) phases.
The unit cell was determined by indexing the powder X-ray diffraction (XRD) pattern of the sample prepared at 1100 °C for 12 h using synchrotron radiation. The structure can be indexed using a triclinic cell with cell parameters: a = 5.207365(8) Å, b = 5.817536(8) Å, c = 7.888147(13) Å, α = 100.58195(11)°, β = 96.93693(11)°, and γ = 106.96091(9)°. All attempts to index the pattern with a higher symmetry cell were unsuccessful. Initial positions for Sb, Cu, and O atoms were found by simulated annealing using the program FOX13 and further determined through Fourier difference maps as implemented in the FullProf suite.14 Li positions were determined from Fourier difference maps using neutron diffraction (ND) data. Overall site occupancies and atomic displacement parameters were determined by a Rietveld refinement combining synchrotron X-ray diffraction (SXRD) data with neutron diffraction data (Figure 1b). The combined refinement with neutron data confirmed the presence of oxygen vacancies in the structure. The occupancies of all oxygen sites were refined freely and were fixed at 1 when reaching values larger than unity during the refinement. Partial occupation of cation sites by Sb, Cu, and Li was refined incrementally, using electronic and nuclear density Fourier difference maps generated by the program GFourier as implemented in Fullprof. Specifically, difference Fourier maps plotted along the (101) lattice plane (slice shown at x = 0) clearly support the absence of oxygen in site 1b (Figure 2e for ND bank 1). The fit statistics of all banks improve with a global weighted value of χ2 decreasing from 45.5 without vacancy to 16.1 with a vacancy. Unsatisfactory fit of the peak shapes, with hkl-dependent asymmetries, indicate the presence of inhomogeneous cell parameters in the sample. Adding a secondary Li4CuSbO5.5 phase to the refinement with the same atomic parameters and slightly different cell parameters (a = 5.20263(3) Å, b = 5.82002(4) Å, c = 7.88000(6) Å, α = 100.4694(6)°, β = 97.0145(5)°, γ = 107.0248(6)°) improved the fit significantly (from χ2 = 26.5 with a single phase to 16.1 with two phases, see Figure 1c, top) but still did not achieve a perfect fit of the peak shapes. A distribution of phases with continuously varying cell parameters would likely give a result more representative of the reality. The secondary Li4CuSbO5.5 phase amount varies between 20 and 32 wt %, depending on the diffraction bank considered, and its volume is 0.17% smaller than that of the dominant phase. It is likely that this secondary phase corresponds to a slightly different composition or atomic ordering compared to the main one. Attempts at refining the composition of this phase led to unstable results, and we therefore decided to keep the same atomic parameters for the main and secondary phases as an approximation. Additional experiments show that cell parameters, and potentially the atomic ordering, are affected by the sample’s cooling conditions (see Methods section). Small additional peaks corresponding to Li2CuO2 and Li7SbO6 impurities were also observed in the synchrotron X-ray and neutron data and were added to the fit. The result of the combined refinement is shown in Figure 1b and Table 1, and the structure is presented in Figure 2.
Figure 2.

Structure of Li4CuSbO5.5. The atomic ordering is derived from the NaCl rocksalt structure, represented here in the cubic cell setting (a) and in the triclinic setting (b) corresponding to the structure of Li4CuSbO5.5 (c). Sodium and chlorine atoms are in yellow and gray, respectively. Lithium, copper, antimony, oxygen atoms, and oxygen vacancies are in green, blue, brown, red, and white, respectively. The presence of oxygen vacancies ordered in the (101) lattice plane (d), corresponding to the (110) lattice plane of the parent cubic structure (a), is confirmed by Fourier difference maps using synchrotron and neutron diffraction data. Nuclear density maps along the (101) lattice plane using the ND bank 1 are compared with and without the presence of oxygen in site 1b (e). Site 1b is highlighted by white arrows. (f) Perspective view of the structure, with polyhedra shown only around copper and antimony atoms in the (101) lattice planes containing the antimony atoms and oxygen vacancies.
Table 1. Structural Parameters for the Main Li4CuSbO5.5 Phase from a Combined Rietveld Refinement on Neutron and Synchrotron X-ray Diffraction Data.
| refinement parameters | ||||
|---|---|---|---|---|
| formula | Li8Cu2Sb2O11 | |||
| temperature (K) | 298 | |||
| pressure | atmospheric | |||
| source | neutron time-of-flight | synchrotron X-ray | ||
| data bank | ND, bank 1 | ND, bank 2 | ND, bank 3 | SXRD |
| angle (°)/wavelength (Å) | 168.657 | 90.248 | 29.930 | 0.82468(1) |
| d spacing range (Å) | 0.68–2.59 | 0.89–3.89 | 3.14–6.28 | 0.58–9.44 |
| TOF (μs)/2θ (°) range | 40000–125000 | 31000–135500 | 40000–80000 | 5–120 |
| TOF (μs)/2θ (°) step | 20.3307 | 48.7172 | 75.1060 | 0.004 |
| no. of reflections | 1459 | 1120 | 15 | 6082 |
| no. of refined parameters | 95 | |||
| Rp | 13.4 | 8.78 | 25.7 | 14.0 |
| Rwp | 11.9 | 6.89 | 17.2 | 16.7 |
| Rexp | 5.35 | 1.96 | 9.42 | 3.39 |
| χ2 | 4.92 | 12.4 | 3.34 | 24.3 |
| ρmin/max residuals (fm·Å–3/e–·Å–3) | [−0.004/+0.005] | [−0.003/+0.002] | [−0.0002/+0.0003] | [−0.9/+1.5] |
| Phases | Estimated Mass Fraction (%) | |||
| Li4CuSbO5.5, main | 79.3 | 66.2 | 94.0 | 76.5 |
| Li4CuSbO5.5, secondary | 20.0 | 32.3 | – | 22.1 |
| Li2CuO2 | 0.6 | 0.8 | 0.9 | 1.1 |
| Li7SbO6 | 0.1 | 0.7 | 5.1 | 0.3 |
| structure
parameters for Li4CuSbO5.5 | |||||||
|---|---|---|---|---|---|---|---|
| space group | Z | density (g·cm–3) | formula weight (g·mol–1) | ||||
| P1 | 1 | 4.459 | 602.13 | ||||
| a (Å) | b (Å) | c (Å) | α (°) | β (°) | γ (°) | volume (Å3) | |
| main | 5.207365(8) | 5.817536(8) | 7.888147(13) | 100.58195(11) | 96.93693(11) | 106.96091(9) | 220.7860(6) |
| secondary | 5.20263(3) | 5.82002(4) | 7.88000(6) | 100.4694(6) | 97.0145(5) | 107.0248(6) | 220.413(3) |
| atom | site | x | y | z | occupancy | Biso (Å2) |
|---|---|---|---|---|---|---|
| O1 | 2i | 0.4547(10) | 0.4047(9) | 0.1628(7) | 1 | 0.66(10) |
| O2 | 2i | 0.5547(9) | 0.0990(9) | 0.8389(6) | 1 | 0.59(10) |
| O3 | 2i | 0.0067(10) | 0.1693(9) | 0.6879(7) | 1 | 0.65(11) |
| O4 | 2i | 0.0050(10) | 0.3225(8) | 0.3134(6) | 1 | 0.35(9) |
| O5 | 2i | 0.4768(10) | 0.2336(9) | 0.4789(6) | 0.959(8) | 0.78(11) |
| O6 | 1g | 0 | 0 | 0 | 0.906(6) | 0.28(15) |
| vacancy | 1b | 0 | 0.5 | 0 | 0 | – |
| Sb8 | 2i | 0.23390(15) | 0.11964(15) | 0.24415(10) | 1 | 0.351(4) |
| Cu9/Li9 | 2i | 0.2395(4) | 0.6223(4) | 0.2526(3) | 0.633/0.367(2) | 0.35(3) |
| Cu10/Li10 | 2i | 0.7483(8) | 0.0411(7) | 0.4149(5) | 0.249/0.751(2) | 0.38(6) |
| Cu11/Li11 | 2i | 0.2691(19) | 0.8018(17) | 0.9062(13) | 0.061/0.939(2) | 0.76(19) |
| Li12 | 2i | 0.669(3) | 0.691(2) | 0.1043(19) | 1 | 2.1(3) |
| Li13 | 2i | 0.749(3) | 0.528(3) | 0.4070(19) | 1 | 1.3(3) |
2.2. Structure Description
The structure of Li4CuSbO5.5 is derived from the cubic rocksalt structure of NaCl (Figure 2a–c). The unit cell parameters of the triclinic cell are related to those of the cubic cell by the following relation:
 |
The symmetry lowering from a cubic to a triclinic space group is a consequence of the complex atomic ordering established in Li4CuSbO5.5. This specific ordering of cations and oxygen vacancies is the result of a balance between competing electrostatic interactions and electronic structure effects. The main features of this arrangement can be most simply understood by focusing on the atomic ordering along the (101) lattice plane (Figure 2d), which corresponds to the (110) lattice plane of the parent cubic structure (Figure 2a). This plane contains the antimony site (Sb8), the main copper site (Cu9/Li9), oxygen sites (O5 and O6), and the vacant oxygen site (site 1b). The cation ordering is closely related to those of the parent compounds Li2CuO2 (also written as Li4Cu2O4□2) and Li5SbO5 (or Li4LiSbO5□1), as shown in Figure 3. All are derived from the cubic rocksalt structure, but the atomic ordering depends on the cationic nature and the content of oxygen vacancies. This can be better understood by writing their formula as Li4(MM′)O6–x□x: Li4(CuCu)O4□2, Li4(LiSb)O5□1 and Li4(CuSb)O5.5□0.5 with decreasing vacancy content, respectively. The number of oxygen vacancies x is directly related to the sum of oxidation states m and m′ of the cations M and M′ (x = (8 – m – m′)/2).
Figure 3.

Comparison of rocksalt-derived Li4(MM′)O6–x□x crystal structures with oxygen vacancies of Li2CuO2 (left), Li5SbO5 (middle), and Li4CuSbO5.5 (right). Oxygen atoms and oxygen vacancies are represented in red and white, respectively. Copper, antimony, and lithium atoms are in blue, brown, and green, respectively. Polyhedra are shown for M and M′ sites only, including the position of the oxygen vacancies to facilitate the comparison between structures. The top view (a, d, g) represents the (MM′) layers along the (110) lattice plane of the parent cubic structure that clearly displays the ordering of oxygen vacancies with M and M′ atoms. The center view (b, e, h) shows the arrangement of the (MM′) layers separated by Li atoms along the edge-sharing direction, and the bottom view (c, f, i) shows the same arrangement along the corner-sharing direction of M and M′ polyhedra.
In the three structures, M and M′ sites are assembled in one plane (Figure 3a,d,g), corresponding to the equivalent (110) lattice plane in the parent cubic rocksalt structure, the (001) plane in the orthorhombic Immm cell of Li2CuO2, the (100) plane in the monoclinic C2/m cell of Li5SbO5, and the (101) plane in the triclinic cell of Li4CuSbO5.5. The M and M′ sites share edges along one direction ([100] for Li2CuO2, [001] for Li5SbO5 and [010] for Li4CuSbO5.5) and corners in the perpendicular direction ([010] for Li2CuO2 and Li5SbO5, [212] for Li4CuSbO5.5). Layers of Li atoms separate each (MM′) layer from the next one (Figure 3b,c,e,f,h,i). The edge-sharing oxygen sites are all fully occupied, whereas the corner-sharing oxygen sites may or may not be occupied depending on the two cations present in the two adjacent M and M′ sites. In the case of Li4(CuCu)O4□2, all M and M′ sites are occupied by Cu2+, so that the oxygen corner-sharing sites remain empty, resulting in a square planar coordination for all Cu atoms (Figure 3a,b). Cu sites are therefore connected only by edges, forming ribbons along the [100] direction (Figure 3a). The electronic configuration of Cu2+ favors strong Jahn–Teller distortion of its coordination environment, making this square planar environment relatively stable. In Li4(LiSb)O5□1, the high valence of Sb5+ cations prevents oxygen vacancies from forming in their immediate surrounding, preserving the octahedral coordination environment. Instead, oxygen vacancies are found between Li+ ions, which are, therefore, sitting in a square planar environment. Sb sites form chains of corner-shared octahedra along the [010] direction, and each chain is separated from the next one by square planar Li sites in the [001] direction (Figure 3d). Turning back to Li4(CuSb)O5.5□0.5, we now understand that oxygen vacancies will preferentially be found in corner-shared sites between two Cu2+ cations, rather than at a vertex of the octahedral Sb5+ cation site. This is possible by ordering Cu2+ and Sb5+ cations in the (101) plane to alternate between Cu and Sb along the edge-sharing [010] direction −(Cu–Sb–Cu–Sb)– and between pairs of Cu and Sb along the corner shared [212] direction −(Cu–Cu–Sb–Sb)– (Figure 3g). Oxygen vacancies are therefore found between two Cu2+ cations, each sitting in opposingly oriented square pyramidal sites. The Cu–O distance at the apex of the pyramid (d(Cu9–O5) = 2.288(5) Å) is significantly longer than the Cu–O equatorial distances forming the base of the pyramid (1.975(6)–2.045(7) Å) due to the strong Jahn–Teller distortion discussed above (Figure S1 and Table S1 in SI). The Sb5+ cations reside in pairs of octahedral sites connected by a corner. Generally, the repulsion between highly charged cations in rocksalt oxides is the main driving force for cation ordering, leading to completely isolated MO6 octahedra to minimize electrostatic energy. However, the Sb2O11 dimers present in Li4(CuSb)O5.5 indicate that cationic repulsion comes only second to spreading oxygen vacancies in the structure. The SbO6 octahedra show little deviation from a perfectly regular octahedral environment except for a slight displacement of Sb5+ cations away from each other (d(Sb8–O6) = 2.0307(8) Å > d(Sb8–O5) = 1.991(5) Å). This displacement is likely the result of the minimization of electrostatic energy between the two highly charged Sb5+ cations. It also helps to mitigate the deviation from an ideal bond valence sum (BVS) of two for the corner-sharing oxygen (O6) at the apex of the two Sb octahedra. This oxygen site is coordinated to two Sb5+ and four Li+ cations, strongly deviating from Pauling’s rule of electroneutrality. However, it conserves a reasonable BVS of 2.028(6) thanks to relatively large bond lengths with the coordinating cations (Table S1 in SI), making it the oxygen atom with the largest octahedral volume in the structure. Finally, it can be noted that Cu and Li occupy both 6 and 5-coordinate sites with some cation mixing of Cu and Li in site 9 (36.7(2)% Li, see Table 1), site 10 (24.9(2)% Cu), and site 11 (6.1(2)% Cu), suggesting that there is little energetic difference between Li and Cu occupying the different sites. Indeed, site 10 is also significantly distorted with long apical distances (2.247(6) and 2.265(6) Å) which is favorable for Jahn–Teller Cu2+ ions (Table S1 in SI). The energies of several phases with different Cu/Li orderings were studied via density-functional theory (DFT) calculation, confirming that phases with Cu in sites 9 and 10 are indeed very close in energy of formation (within 0.25 eV/formula unit, see Figure S2 in SI). The structure refinement also suggests about 4.1(8)% and 9.4(6)% of oxygen vacancies in sites 5 and 6, respectively. This deviation from the stoichiometric composition and imperfect chemical ordering could point to inhomogeneities in composition and local structure in the sample, which would explain the asymmetric peak shapes associated with a distribution of cell parameters in the material.
Given the similarity between the structures of Li2CuO2, Li5SbO5, and Li4CuSbO5.5, one could expect to find a group/subgroup relationship between the three structures. This is straightforward between Li2CuO2 and Li5SbO5, with the latter belonging to a direct subgroup of the former through splitting of the Li, Cu, and O positions. The group–subgroup relationships of Li4CuSbO5.5 with Li5SbO5 and Li2CuO2 is implied by the corresponding Bragg reflections that could match the structures of Li5SbO5 (C2/m) and Li2CuO2 (Immm), observed when preparing Li4CuSbO5.5 at 900 °C instead of 1100 °C (Figure 1c). The refined cell volumes of these phases do not match with the parent Li5SbO5 and Li2CuO2 phases (Table S2 in SI), but instead with those of Li4CuSbO5.5 prepared at 1100 °C. Here, the group–subgroup relationship does not seem to be direct and its study would require using a large supercell of Li5SbO5 or Li2CuO2 given the more complex order found in Li4CuSbO5.5. Observing higher symmetry monoclinic and orthorhombic structures at lower temperature could point to an imperfect ordering of cations in this sample. The difference in local structure between the two samples prepared at 900 and 1100 °C was studied using two local structure probe techniques, namely X-ray pair distribution function (XPDF) and extended X-ray absorption fine structure (EXAFS). Interestingly, the local atomic arrangements investigated using XPDF (Figure S3a) show almost no difference below a 20 Å radial distance for the two samples prepared at different temperatures. This suggests that Sb, the element which scatters the strongest with X-rays, is ordered at intermediate ranges in the 900 °C sample. Clear deviations beyond 20 Å are consistent with the different average long-range structures observed from the Bragg data, which could correspond to the higher symmetry structures of Li5SbO5 and Li2CuO2. The Cu K-edge EXAFS data (Figure S3b) present largely similar local environments for Cu2+ cations with an excellent overlap of the two sample’s signals up to R = 6 Å. These two local structural probes clearly show that the local chemical environments for both materials are essentially identical, indicating that the square pyramidal configuration of Cu is strongly favored, as would be expected for this chemistry. The deviation at higher R in the EXAFS data would indicate that the long-range Cu ordering is different in Li4CuSbO5.5 when prepared at 900 and 1100 °C. Altogether, these data suggest that the ordering of cations and oxygen vacancies is incomplete at 900 °C, leading to different long-range average structural models. Higher temperature helps to overcome kinetic barriers related to structural defects and to complete the long-range ordering of the structure.
2.3. Cu and Sb Oxidation States
Confirmation of Cu and Sb oxidation states was obtained through X-ray absorption near-edge spectroscopy (XANES) and magnetometry measurements. Measurement at the Cu K-edge (Figure 4a) was compared to reference materials with different formal oxidation states (CuI2O, CuIIO, NaCuIIIO2) and to the reported Li3CuSbO5 phase,11 which contains Cu2+ and Sb5+ in comparable coordination environments. The tendency for Cu to adopt strongly distorted coordination environments clearly appears on the Cu K-edge data, with intense pre-edge features that overlap with the edge itself. Nevertheless, there is a good match between the edge position of Li4CuSbO5.5 and CuO, and even more so with Li3CuSbO5 which shows a very similar edge shape as can be expected from the comparable environments in both materials. Li4CuSbO5.5 shows an additional pre-edge feature at 8984 eV that can be interpreted as the presence of Cu2+ in square pyramidal environment and is not observed in Li3CuSbO5 for which all Cu occupies distorted octahedral environments. Measurements at the Sb K-edge (Figure 4b) and Sb L1-edge (inset), which can better resolve mixed oxidation states, unambiguously confirm a +5 valence for antimony.
Figure 4.

X-ray absorption near-edge spectra of Li4CuSbO5.5 and selected reference materials, measured at the Cu K-edge (a), Sb K-edge (b), and Sb L1-edge (b, inset).
Initial magnetization measurement of Li4CuSbO5.5 in an applied field of 100 Oe between 2 and 300 K shows a paramagnetic behavior which was fitted with a Curie–Weiss function modified with a temperature-independent paramagnetic contribution between 50 and 300 K. An effective magnetic moment of 1.313(3) μB per Cu atom was obtained, which is lower than the expected spin-only value for Cu2+ (μS.O. = 1.73 μB for S = 1/2). Isothermal field-dependent magnetization curves measured between −70 and 70 kOe at 2, 10, 50, and 300 K (Figure 5a) show a deviation from linearity below 50 K that suggests the presence of impurities or additional magnetic interactions in the material. X-ray and neutron diffraction revealed a small amount of Li2CuO2 impurity (<1 wt %) which can affect the response of the material to the magnetic field. Even a small mole fraction of ferromagnetic impurities can affect the measurement while remaining undetectable by diffraction methods. To suppress the contribution of such impurities that saturate below 4 T, the magnetic susceptibility data measured at 60 kOe and 40 kOe were subtracted, and the data measured below 50 K were discarded (Figure 5b). This way, an effective moment of 1.571(4) μB per Cu atom is found, which is closer to the expected value for Cu2+.
Figure 5.

Magnetization measurements on Li4CuSbO5.5. (a) Field-dependent magnetization loops measured between 2 and 300 K. (b) Temperature-dependent magnetization curve obtained from the difference between 60 kOe and 40 kOe field-cooled measurements. Only the data above 50 K are used due to the nonlinearity of the 2 and 10 K isotherms between 40 and 60 kOe. The data are also plotted as the inverse of the magnetization after subtracting the temperature-independent paramagnetic contribution χ0.
By combining XAS and magnetization measurements, we can confirm the presence of divalent Cu in the sample, which is consistent with the detection of oxygen vacancies by neutron diffraction. The presence of defects and/or composition variations in the material may affect the magnetic susceptibility of the material. Further studies of the magnetic properties of Li4CuSbO5.5 could bring more detailed insight into these aspects.
2.4. Electrochemical Properties
The properties of Li4CuSbO5.5 as a positive electrode material in Li cells were studied at a rate of C/20 (1 Li+ exchanged in 20 h). Upon oxidation to 5 V vs Li+/Li, a charge capacity of 50 mAh/g is obtained with several features at 3.4, 4.3, and 4.9 V (Figure 6a and Figure S4a) and a reversible discharge capacity of only 20 mAh/g with a cutoff at 1.5 V vs Li+/Li. The theoretical capacity for the extraction of one Li+ ion corresponds to 90 mAh/g, suggesting that about 0.55 Li+ ions are extracted from the material and 0.22 are reinserted. Further cycling in this voltage range improves only slightly the capacity up to 35 mAh/g reversible capacity after 15 cycles (Figure S4b). The high-voltage plateau at 4.9 V vs Li+/Li is only observed on the first cycle and is reminiscent of the activation plateau of Li-rich layered oxides corresponding to irreversible oxygen oxidation. In addition to the presence of native oxygen vacancies in the pristine structure, it is possible that additional oxygen is released upon high-voltage electrochemical oxidation instead of, or concomitantly, with the oxidation of Cu2+ to Cu3+. In any case, this situation is not favorable for reversible insertion reactions.
Figure 6.

Electrochemical properties of Li4CuSbO5.5 as a cathode material in Li cells. A small capacity is obtained upon oxidation to 5 V vs Li+/Li (a), whereas about 2.5 Li+ ions can be reversibly exchanged upon reduction to 1 V vs Li+/Li with a large voltage hysteresis between charge and discharge (b). The first and second cycles for each cell are shown in darker and lighter colors, respectively. An asterisk marks a momentary interruption of the cycling.
Remarkably, when the material is discharged to 1 V vs Li+/Li, a large capacity reaching 222 mAh/g is obtained with a long plateau at 1.2 V (Figure 6b and Figure S4d). The next charge and discharge give a capacity of 237 and 226 mAh/g, respectively, with, however, a large voltage hysteresis (3.5 V) between discharge and charge. From the second cycle, the low voltage plateau on reduction is now found at 1.75 V instead of 1.2 V vs Li+/Li. A reversible capacity between 220 and 270 mAh/g was maintained for 20 cycles before observing a rapid capacity drop (Figure S4e).
To shed further light on the structural evolution of the material upon (de)lithiation, XRD data were obtained in situ while first charging the material to 5 V, then discharging to 1 V and charging again to 5 V (Figure S5). The capacities obtained for each step are lower than those measured in coin cells (46, 171, and 148 mAh/g, respectively), which can be explained by the larger amount of material used and higher polarization in the in situ cell, but they are representative enough to observe the structural evolution of the material. Very little change is observed upon the initial charge to 5 V, which is not unexpected given the small amount of Li (∼0.51) removed from the structure. Deinsertion of lithium, which is a weak X-ray scatterer, does not make an observable difference to the intensity of Bragg peaks using a lab X-ray source. However, we should be able to measure changes in lattice parameters or the creation of additional oxygen vacancies. This is not the case here. The following discharge to 1 V is marked by a strong reduction in peak intensity of all Bragg reflections corresponding to Li4CuSbO5.5. No new peaks were observed to replace them; however, a general increase of the background contribution can be noted, which suggests the formation of an amorphous phase rather than a new crystalline phase. This transition is incomplete, with some peaks from the pristine phase still observed at the end of discharge. This can be understood from the lower capacity obtained in the in situ cell (171 mAh/g) compared to the coin cell (222 mAh/g) for which the lower polarization lets the reaction proceed to completion. The second charge step shows only weak variations in the remaining peak intensity and positions as well as the background intensity. The amorphization reaction is irreversible, and the structure of Li4CuSbO5.5 is not recovered upon oxidation. The large capacity obtained is therefore likely to come from a conversion reaction that eventually leads to the cycling of an amorphous composite, thus explaining the large voltage hysteresis between discharge and charge.
3. Discussion
Li4CuSbO5.5 was prepared to explore the possible incorporation of oxygen vacancies in a rocksalt oxide. Before discussing this result, it is interesting to comment on the electrochemical properties of this material in light of other published work. First, we attributed the large reversible capacity on reduction to a conversion reaction that leads to an amorphous phase. It should be noted that the capacity of this reduction process (222 mAh/g) corresponds approximately to the reaction of 2.5 Li+ ions with Li4CuSbO5.5. This is slightly more than the capacity expected for the full reduction of Cu2+ to Cu0 (180 mAh/g), but consistent with such a reaction. Several reports have mentioned the possibility for Li+ to displace copper from a structure and form Cu nanoparticles.15−18 In a related example, Larcher et al. reported that the trirutile CuSb2O6 structure forms an amorphous Li2Sb2O6 phase and Cu nanoparticles upon electrochemical reduction vs metallic Li.15 It is possible that Li4CuSbO5.5 follows a similar reaction pathway:
| 1 |
The composition “Li6SbO5.5” does not correspond to a reported crystalline structure, and most likely forms Li5SbO5, Li7SbO6, and/or Li2O instead, although we cannot exclude an unknown composition for an amorphous phase. This reaction pathway could explain the short cycle life of the cell, as Li-rich pentavalent Sb phases may form an electronically insulating matrix that eventually prevents copper nanoparticles from reacting reversibly.
Second, the capacity obtained upon oxidation between 4 and 5 V vs Li+/Li may be explained by the Cu2+/Cu3+ redox couple or oxidation of oxygen. Multiple advanced characterization techniques would be required to fully pin down the charge compensation mechanism in this case, while maintaining a large degree of uncertainty given the small capacity associated with this process. Instead, we can probe the possibility for oxygen evolution by evaluating the Gibbs energy of eq 2, using first principle methods, with the known experimental structures for Li4CuSbO5.5, Li3CuSbO5,11 and Li2O:
| 2 |
This value can be combined with the Gibbs energy of formation of Li2O at 298 K (ΔG ≈ −610 kJ·mol–1)19 to obtain the energy of eq 3, which corresponds to the deintercalation of 1 mole of lithium from Li4CuSbO5.5, oxygen release, and plating of one mole of lithium at the anode:
| 3 |
We can therefore obtain an estimate of the average cell voltage for eq 3:
This result shows that oxygen evolution is not limited by thermodynamics, as the phase Li3CuSbO5 is known to exist, and we pushed the cell voltage well beyond 3.39 V. However, no signs of Li3CuSbO5 were observed by in situ XRD, which suggests that this reaction may be kinetically limited. Indeed, this reaction requires good oxygen diffusion and a large reorganization of cation and anion lattices, moving from the rocksalt-derived structure of Li4CuSbO5.5 with oxygen vacancies (density of 4.5 g·cm–3) to the close-packed rocksalt structure of Li3CuSbO5 (density of 4.9 g·cm–3).
It is interesting to note that another phase is compositionally related to the two discussed above: the quantum-spin liquid LiCuSbO4,20 which can be obtained by removing another equivalent of Li2O from Li3CuSbO5 (see Figure 7). This is an example of phase dimensional reduction21 for which the connectivity of SbO6 octahedra decreases with increasing content of Li2O in the structure, moving from a bidimensional framework of SbO6 octahedra connected by edges and corners in LiCuSbO4 to isolated Sb2O10 dimers sharing an edge in Li3CuSbO5 and isolated Sb2O11 dimers sharing a corner only in Li4CuSbO5.5 (Figure S6 in SI). Beyond their study of intercalation materials, these compounds could present interesting magnetic properties controlled by the progressive dilution and different orderings of the S = 1/2 Cu2+ ions.
Figure 7.

Li2O-CuO-Sb2O5 phase diagram. Li4CuSbO5.5 is indicated by a red marker and known phases by black markers. The black dashed line highlights the relationship between Li4CuSbO5.5, Li3CuSbO5, and LiCuSbO4, the red dashed line corresponds to the compositions for which the formation of oxygen vacancies is expected, including the Li3.5Zn1.5SbO5.75 (i) composition,22 and the gray dashed line corresponds to the ideal rocksalt stoichiometries with the same number of cations and anions. Representative examples of disordered rocksalt structures with divalent MII and pentavalent M′V cations are also indicated (ii and iii from refs (23 and 24)), showing compositions very close to the region of oxygen vacancy formation.
Focusing now on the existence of Li-rich rocksalt oxides with ordered
oxygen vacancies, several materials have been reported in the literature,
although not always identified as rocksalt-derived structures. Identifying
such structures from an electronic database remains challenging given
that we are searching for a “missing” structural feature
(e.g., vacancy); therefore, some structures may have been unintentionally
excluded from our search. Examples of materials with only one cation
in addition to lithium and ordered oxygen vacancies are Li2CuO2, Li2PdO2, LiCu2O2, Li3CuO3, Li3AuO3, LiCu3O3, Li3Cu2O4, Li5AuO4, Li5SbO5, Li5BiO5, Li6TeO6, Li6Zr2O7, and Li4Cu4O4 (Table S3 in SI).25−35 Li2NiO2, which is structurally related to
Li2CuO2, could also enter this list but is more
often described as a layered rocksalt containing excess Li in tetrahedral
sites.36 The ordering of oxygen vacancies
in these structures directly depends on the proportion of unoccupied
sites in the oxygen sublattice and, most of the time, allows to maximize
the distance between vacancies. Examples of vacancy ordering patterns
for some of these structures are shown in Figure S7 in SI. Most structures can be described by a single hexagonal
anion sublattice plane stacked with a given periodicity along the
[111] direction of the corresponding cubic rocksalt cell. However,
Li4CuSbO5.5 requires two different alternating
sublattice planes, one with a close-packed array of oxygen atoms and
another with only 5 out of 6 oxygen sites occupied. Among the structures
cited above, only Li6Zr2O7 also requires
two different sublattices planes.35 The
smallest distance between two vacancies, normalized by the average
rocksalt a lattice parameter, is presented in Figure 8 as a function of
the proportion of vacancy in the anionic sublattice. For structures
with a concentration of oxygen vacancies larger than 1/6, the shortest
distance between two vacancies corresponds to the shortest distance
between two anionic sites in a rocksalt lattice (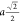 , corresponding to the vector
, corresponding to the vector 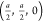 in the equivalent cubic rocksalt lattice).
For structures with a concentration of oxygen vacancies smaller than
1/6, the shortest distance between two vacancies is
in the equivalent cubic rocksalt lattice).
For structures with a concentration of oxygen vacancies smaller than
1/6, the shortest distance between two vacancies is 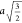 (corresponding to the vector
(corresponding to the vector  in the equivalent cubic rocksalt lattice).
For a proportion of oxygen vacancy of exactly 1/6, an intermediate
situation is observed: The distance between oxygen vacancies in Li5SbO5 and Li5BiO5 corresponds
to the a lattice parameter in the cubic parent. Li4CuSbO5.5, with 1 out of 12 oxygen sites in the
unit cell being vacant, is the rocksalt structure with the smallest
amount of ordered oxygen vacancy we were able to identify. It is noteworthy
that Sr8Fe8O23, derived from the
perovskite structure,37 maintains ordered
oxygen vacancies with as little as 1 in 24 sites unoccupied. In that
case, vacancies are separated by a distance of 2 × a, corresponding to the (2a, 0, 0) vector in the
equivalent perovskite lattice. Sr4Fe4O11 and Sr2Fe2O5, the n = 2 and 4 members of the oxygen-deficient SrnFenO3n–1 series,37 follow a similar trend and
can be presented in Figure 8 for comparison, as the lattice parameter a and vectors discussed above are also relevant in the cubic perovskite
lattice. Given this comparison, it cannot be excluded that Li-rich
rocksalt oxides with more complex compositions could be prepared with
a lower concentration of ordered oxygen vacancies than in Li4CuSbO5.5. In that regard, it should be noted that Greaves
and Katib22 reported the preparation of
Li3.5Zn1.5SbO5.75□0.25 and Li3.5Zn1.5BiO5.75□0.25, that is, 1 in 24 oxygen sites unoccupied, with, however,
oxygen vacancies statistically distributed over all oxygen positions.
This result shows that (i) other divalent MII/pentavalent
M′V cation couples may be used to prepare Li-rich
rocksalt oxides with oxygen vacancies and (ii) the amount of oxygen
vacancies in Li3+2xMII2–2xM′VO6–x□x may vary between
Li3MII2M′VO6 (x = 0) and Li5M′VO5□1 (x = 1)
(Figure 7). This large
chemical versatility is further supported by an early report from
Brixner38 of oxygen-deficient cubic rocksalt
phases based on Nb/Ta pentavalent cations and Mn/Fe/Co/Ni/Cu/Zn divalent
cations. Altogether, it appears that the presence of oxygen vacancies
in Li-rich rocksalt oxides is not limited to oxygen release induced
by electrochemical delithiation but may already exist in the pristine
state of materials after their synthesis to balance ionic charges
and achieve charge neutrality. Synthesis conditions (temperature,
partial pressure of oxygen) that favor the formation of trivalent
cations tend to give the well-known layered honeycomb Li5ReO6-type structure, as is the case for Cr, Al, and Ga
compounds.39 However, by using reducing
conditions to stabilize divalent cations, we can expect the formation
of oxygen vacancies with other transition metals (Mn, Fe, Co, Ni)
beyond Cu and Zn-based compounds for which normal atmospheric conditions
are sufficient. Two-step synthesis procedures, starting with the reaction
of precursors at high temperature, followed by a second annealing
step under oxidizing or reducing conditions, have been successful
in the preparation of double perovskites with controlled oxygen vacancy
content (e.g., Sr2CoSbO6–x,Ca2MnNbO6–x).40−42 Transposing this approach to the preparation of Li-rich rocksalt
oxides could lead to a better understanding of defect formation in
this structural family. Reciprocally, the discovery of Li-rich rocksalt
oxides Li4MM′O6–x can be used to prepare functional double perovskite A2MM′O6–x (A = alkaline-earth)
by simple metathesis solid-state routes using ACl2 salts.43,44 This approach enables the formation of double perovskite with new
cation ordering patterns at reduced temperatures (700–900 °C)
compared to conventional synthesis routes (>1000 °C).
in the equivalent cubic rocksalt lattice).
For a proportion of oxygen vacancy of exactly 1/6, an intermediate
situation is observed: The distance between oxygen vacancies in Li5SbO5 and Li5BiO5 corresponds
to the a lattice parameter in the cubic parent. Li4CuSbO5.5, with 1 out of 12 oxygen sites in the
unit cell being vacant, is the rocksalt structure with the smallest
amount of ordered oxygen vacancy we were able to identify. It is noteworthy
that Sr8Fe8O23, derived from the
perovskite structure,37 maintains ordered
oxygen vacancies with as little as 1 in 24 sites unoccupied. In that
case, vacancies are separated by a distance of 2 × a, corresponding to the (2a, 0, 0) vector in the
equivalent perovskite lattice. Sr4Fe4O11 and Sr2Fe2O5, the n = 2 and 4 members of the oxygen-deficient SrnFenO3n–1 series,37 follow a similar trend and
can be presented in Figure 8 for comparison, as the lattice parameter a and vectors discussed above are also relevant in the cubic perovskite
lattice. Given this comparison, it cannot be excluded that Li-rich
rocksalt oxides with more complex compositions could be prepared with
a lower concentration of ordered oxygen vacancies than in Li4CuSbO5.5. In that regard, it should be noted that Greaves
and Katib22 reported the preparation of
Li3.5Zn1.5SbO5.75□0.25 and Li3.5Zn1.5BiO5.75□0.25, that is, 1 in 24 oxygen sites unoccupied, with, however,
oxygen vacancies statistically distributed over all oxygen positions.
This result shows that (i) other divalent MII/pentavalent
M′V cation couples may be used to prepare Li-rich
rocksalt oxides with oxygen vacancies and (ii) the amount of oxygen
vacancies in Li3+2xMII2–2xM′VO6–x□x may vary between
Li3MII2M′VO6 (x = 0) and Li5M′VO5□1 (x = 1)
(Figure 7). This large
chemical versatility is further supported by an early report from
Brixner38 of oxygen-deficient cubic rocksalt
phases based on Nb/Ta pentavalent cations and Mn/Fe/Co/Ni/Cu/Zn divalent
cations. Altogether, it appears that the presence of oxygen vacancies
in Li-rich rocksalt oxides is not limited to oxygen release induced
by electrochemical delithiation but may already exist in the pristine
state of materials after their synthesis to balance ionic charges
and achieve charge neutrality. Synthesis conditions (temperature,
partial pressure of oxygen) that favor the formation of trivalent
cations tend to give the well-known layered honeycomb Li5ReO6-type structure, as is the case for Cr, Al, and Ga
compounds.39 However, by using reducing
conditions to stabilize divalent cations, we can expect the formation
of oxygen vacancies with other transition metals (Mn, Fe, Co, Ni)
beyond Cu and Zn-based compounds for which normal atmospheric conditions
are sufficient. Two-step synthesis procedures, starting with the reaction
of precursors at high temperature, followed by a second annealing
step under oxidizing or reducing conditions, have been successful
in the preparation of double perovskites with controlled oxygen vacancy
content (e.g., Sr2CoSbO6–x,Ca2MnNbO6–x).40−42 Transposing this approach to the preparation of Li-rich rocksalt
oxides could lead to a better understanding of defect formation in
this structural family. Reciprocally, the discovery of Li-rich rocksalt
oxides Li4MM′O6–x can be used to prepare functional double perovskite A2MM′O6–x (A = alkaline-earth)
by simple metathesis solid-state routes using ACl2 salts.43,44 This approach enables the formation of double perovskite with new
cation ordering patterns at reduced temperatures (700–900 °C)
compared to conventional synthesis routes (>1000 °C).
Figure 8.

Shortest distances between vacancies in various structures with ordered oxygen sublattices. The distance is normalized by the average lattice parameter of the equivalent cubic NaCl or perovskite cells. Dashed lines indicate specific anion–anion distances in the equivalent cubic structures, with the corresponding vectors indicated on the right-hand side of the graph. The detailed values for each structure are presented in Table S3 in SI.
Formation of oxygen vacancies is not the only mechanism to balance ionic charges in Li-based rocksalt structures. Another known mechanism is the formation of cation vacancies (lithium and/or transition metals) as studied for materials such as Li(Ni1/6□1/6Mn2/3)O245 and phases derived from the Li4FeSbO6 composition.46 These examples pointed out that cation vacancies in a rocksalt structure are found when an overstoichiometry of anions compared to metal atoms is required to ensure electroneutrality. This is more likely to be achieved in relatively oxidizing conditions, in opposition with the formation of structures with oxygen vacancies.
Overall, a better understanding of the formation of oxygen vacancies at the synthesis step in Li-rich rocksalt oxides could greatly benefit the search for advanced cathode materials for Li-ion batteries. This is particularly true concerning the recent focus on disordered rocksalt oxides, made of divalent cations and d0 cations such as Nb5+, Sb5+, Ta5+ to increase the Li content.23,24 The cationic disorder in those compounds is statistically expected to yield oxygen sites with a higher redox activity, but it could also result in sites containing oxygen vacancies given that their average composition is close to the oxygen-vacancy structure domain (Figure 7). Integrating the possibility for the presence of oxygen vacancies in theoretical models and structural analysis for this family of material may help to shed new light on their complex charge compensation mechanisms.
4. Conclusion
We report the synthesis and crystal structure of a new Li4CuSbO5.5□0.5 phase containing ordered oxygen vacancies, which are confirmed by the combined Rietveld refinement of synchrotron and neutron powder diffraction data and indirectly from spectroscopic evidence for divalent copper. Only one in 12 oxygen sites is unoccupied in the lattice, which represents the lowest concentration of ordered oxygen vacancies reported in a Li-rich rocksalt oxide. The cation and oxygen sites order to maximize the distance between vacancies, requiring two inequivalent hexagonal sublattice planes to fully describe the vacancy order. Cationic repulsion between highly charged cations comes only second to the ordering of oxygen vacancies. The electrochemical properties of this material as a cathode material in a Li cell present interesting features, despite performances which are not competitive with other compositions. More importantly, this work offers a new insight into the crystal chemistry of Li-rich rocksalt oxides, namely the possible presence of oxygen vacancies in as-prepared materials, which can have a strong effect on the understanding of the charge compensation mechanism in high-energy density cathode materials.
5. Methods
5.1. Synthesis
Li4CuSbO5.5 is prepared from Li2CO3 (Sigma-Aldrich, 99.99% trace metals basis), CuO (Sigma-Aldrich, 99.99% trace metals basis), and Sb2O3 (Sigma-Aldrich, 99.99% trace metals basis). All precursors are kept in a drying oven at 200 °C before use. The precursors in the proportions Li:Cu:Sb = 4.4:1:1 are either hand ground or ball-milled using a planetary mill with the same outcome. They are then fired in air at 1100 °C for 12 h (1 °C/min heating ramp, cooling inside the furnace turned off). The excess of Li2CO3 used plays a role in stabilizing Li4CuSbO5.5 as longer heating at 1100 °C will result in Li2O loss from the compound to form Li3CuSbO5 according to the reaction Li4CuSbO5.5 → Li3CuSbO5 + 1/2 Li2O. Rapidly quenching the sample from 1100 °C between stainless steel plates or slowly cooling (0.1 °C/min) it from 850 °C to room temperature leads to small variations in cell parameters of the sample (see Table S2 in SI). The low-temperature sample was prepared with the same procedure, using a firing temperature of 900 °C instead of 1100 °C. For structural characterization by synchrotron X-ray and neutron diffraction, a sample was prepared using enriched 7Li2CO3 (99% 7Li atom, Sigma-Aldrich) to reduce the absorption of neutrons due to 6Li in the sample. The material was manipulated in air but kept in an Ar-filled glovebox during storage to prevent building a Li2CO3 layer at the surface with prolonged contact with ambient air.
5.2. Diffraction
Routine analysis of phase purity and lattice parameters were performed on a Panalytical diffractometer with a monochromatic Co source (Kα1, λ = 1.78901 Å) in Bragg–Brentano geometry with sample rotation. SXRD was performed at the I11 beamline at Diamond Light Source (Oxfordshire, UK), with an incident wavelength of 0.82468(1) Å using five multianalyzer crystal detectors. The samples were sealed in Ø = 0.3 mm glass capillaries and spun during measurement. Time-of-flight (ToF) neutron powder diffraction data were collected on the HRPD instrument at ISIS neutron source (Oxfordshire, UK). Samples were sealed in Ø = 6 mm vanadium cylindrical cans in an argon-filled glovebox. Indexing was performed using the DICVOL method as implemented in the Fullprof suite.14 A simulated annealing method was used to obtain initial atomic positions with the FOX program.13 The structural model was completed and refined by the Rietveld method47,48 and using Fourier difference maps as implemented in the Fullprof suite.14 In situ XRD was performed using an electrochemical cell equipped with a Be window (250 μm thick) and an Al current collector (3 μm thick) on a Rigaku SmartLab diffractometer with a 9 kW rotating anode providing a parallel beam of Mo Kα1 radiation (λKα1 = 0.709032 Å). X-ray total scattering measurements were done at the I15-1 (XPDF) beamline at Diamond Light Source (Oxfordshire, UK). Samples were loaded into quartz capillaries with a 1 mm inside diameter and measured while spinning. Data were collected using a PerkinElmer XRD1611 CP3 area detector with an active area of 409.6 × 409.6 mm2 with a Q range of 36 Å–1. Pair distribution functions were calculated using GudrunX using the appropriate composition, background data, instrument corrections, and a Q max of 30 Å–1.
5.3. Electrochemical Characterization
Electrochemical characterization was performed in two-electrode Swagelok cells and 2025-type coin cells. The positive electrode consisted of a laminated mixture of active material (Li4CuSbO5.5), conductive carbon (C65 from Timcal), and binder (polytetrafluoroethylene, PTFE dried from a 60% aqueous suspension from Sigma-Aldrich) in proportions 85:10:5 in weight. For ex situ characterization by XRD, the active material was simply mixed with 10 wt % C65 conductive carbon and used as a powder. Active material loadings were typically between 5 and 10 mg. Metallic Li was used as an anode, LP30 (1 M LiPF6 in EC:DMC 1:1) was used for the electrolyte, and Whatman GF/D borosilicate glass fiber membranes dried under vacuum at 300 °C for 24 h as the separator. All parts were assembled in an Ar-filled glovebox. Galvanostatic cycling was performed at a C/20 rate (defined as 1 Li+ exchanged in 20 h, considering the chemical formula Li4CuSbO5.5) between 1 and 5 V vs Li+/Li. After cycling, samples for ex situ characterization were recovered inside the glovebox, washed three times in anhydrous DMC, and dried under vacuum.
5.4. X-ray Absorption Spectroscopy
Pellets of sample diluted with cellulose were prepared with an optimized density for X-ray absorption measurements at the Cu K-edge, Sb K-edge, and Sb L1-edge. X-ray absorption spectra were measured at the B18 beamline at Diamond Light Source (Oxfordshire, UK) in transmission mode. The spectra were calibrated by fixing at 8979/30491/4966 eV, the maximum of the derivative of Cu/Sb/Ti metal foil references placed after the samples for the corresponding edges, respectively, and normalized with the Athena software.49 Fourier transform of the EXAFS Cu K-edge data was done using a sine window function from 3.8 to 12.7 Å–1 (k-range).
5.5. Magnetization Measurement
About 30–40 mg of freshly prepared sample in the form of a powder was sealed in a high purity quartz tube (Suprasil Medium Wall EPR Tubes, Goss Scientific). Magnetic measurements were carried out using a commercial superconducting quantum interference device magnetometer MPMS3 (Quantum Design, USA). The contribution of the quartz tube to the magnetization was confirmed to be negligible prior to measuring the sample. Zero-field cooled and field cooled measurements were first performed at 100 Oe from 2 to 300 K, followed by field-cooled measurements at 40 kOe and 60 kOe. Finally, magnetic field-dependent magnetization M(H) loops were measured between −70 and 70 kOe at 2, 10, 50, and 300 K.
5.6. Computational Methods
All calculations were conducted with DFT as implemented in VASP-5.4.450 with PAW pseudopotentials.51 The atomic and vacancy positions in the structures with mixed site occupancies, Li4CuSbO5.5 and Li3CuSbO5 were determined by, first, optimizing the geometries of possible atomic configurations in 2 × 2 × 1 supercell structures (obtained with Supercell program52 from experimentally obtained atomic positions in a unit cell), the interatomic forces are reduced below 10–3 eV/Å, and then comparing their total energies. The lowest energies were used in eqs 2 and 3. Calculations were performed with a 700 eV kinetic energy cutoff for plane waves, 5 × 5 × 5 k-points sampling, and LDSA53 approach to account for strongly correlated 3d electrons in Cu, with Hubbard U = 9.79 eV and J = 2.5.54
Acknowledgments
We thank EPSRC for funding under EP/N004884. T.W.S. thanks EPSRC for funding under EP/R011753/1. This work was supported by the Faraday Institution project “CATMAT – Next Generation Li-Ion Cathode Materials” (grant number FIRG016). We thank Diamond Light Source for access to beamlines I11 (proposal CY23666), I15-1 (proposal CY23167), and B18, Dr. Sarah Day, Dr. Dean Keeble, Prof. Alan Chadwick, and Dr. Giannantonio Cibin for assistance on the beamlines. We thank ISIS Neutron and Muon Source for access to HRPD and Dr. Dominic Fortes for running the measurements. We acknowledge Dr. Konstantin Luzyanin and Mr. Stephen Moss (University of Liverpool) for ICP-OES measurements.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.1c02882.
Details on the structure of Li4CuSbO5.5 obtained by Rietveld refinement and used for DFT calculation. PDF and EXAFS data on low- and high-temperature samples of Li4CuSbO5.5. Capacity retention upon long-term electrochemical cycling. In situ XRD data during cycling. Comparison of structures with ordered oxygen vacancies (PDF)
Accession Codes
CCDC 2108517 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- Assat G.; Tarascon J.-M. Fundamental Understanding and Practical Challenges of Anionic Redox Activity in Li-Ion Batteries. Nat. Energy 2018, 3 (5), 373–386. 10.1038/s41560-018-0097-0. [DOI] [Google Scholar]
- Ben Yahia M.; Vergnet J.; Saubanère M.; Doublet M.-L. Unified Picture of Anionic Redox in Li/Na-Ion Batteries. Nat. Mater. 2019, 18 (5), 496–502. 10.1038/s41563-019-0318-3. [DOI] [PubMed] [Google Scholar]
- Armstrong A. R.; Holzapfel M.; Novák P.; Johnson C. S.; Kang S.-H.; Thackeray M. M.; Bruce P. G. Demonstrating Oxygen Loss and Associated Structural Reorganization in the Lithium Battery Cathode Li[Ni0.2Li0.2Mn0.6]O2. J. Am. Chem. Soc. 2006, 128 (26), 8694–8698. 10.1021/ja062027+. [DOI] [PubMed] [Google Scholar]
- Tran N.; Croguennec L.; Ménétrier M.; Weill F.; Biensan Ph; Jordy C.; Delmas C. Mechanisms Associated with the “Plateau” Observed at High Voltage for the Overlithiated Li1.12(Ni0.425Mn0.425Co0.15)0.88O2 System. Chem. Mater. 2008, 20 (15), 4815–4825. 10.1021/cm070435m. [DOI] [Google Scholar]
- Wu Y.; Ma C.; Yang J.; Li Z.; Allard L. F.; Liang C.; Chi M. Probing the Initiation of Voltage Decay in Li-Rich Layered Cathode Materials at the Atomic Scale. J. Mater. Chem. A 2015, 3 (10), 5385–5391. 10.1039/C4TA06856D. [DOI] [Google Scholar]
- Perez A. J.; Jacquet Q.; Batuk D.; Iadecola A.; Saubanère M.; Rousse G.; Larcher D.; Vezin H.; Doublet M.-L.; Tarascon J.-M. Approaching the Limits of Cationic and Anionic Electrochemical Activity with the Li-Rich Layered Rocksalt Li3IrO4. Nat. Energy 2017, 2 (12), 954. 10.1038/s41560-017-0042-7. [DOI] [Google Scholar]
- Xie Y.; Saubanère M.; Doublet M.-L. Requirements for Reversible Extra-Capacity in Li-Rich Layered Oxides for Li-Ion Batteries. Energy Environ. Sci. 2017, 10 (1), 266–274. 10.1039/C6EE02328B. [DOI] [Google Scholar]
- Kim S.; Aykol M.; Hegde V. I.; Lu Z.; Kirklin S.; Croy J. R.; Thackeray M. M.; Wolverton C. Material Design of High-Capacity Li-Rich Layered-Oxide Electrodes: Li2MnO3 and Beyond. Energy Environ. Sci. 2017, 10 (10), 2201–2211. 10.1039/C7EE01782K. [DOI] [Google Scholar]
- McCalla E.; Rowe A. W.; Brown C. R.; Hacquebard L. R. P.; Dahn J. R. How Phase Transformations during Cooling Affect Li-Mn-Ni-O Positive Electrodes in Lithium Ion Batteries. J. Electrochem. Soc. 2013, 160 (8), A1134–A1138. 10.1149/2.047308jes. [DOI] [Google Scholar]
- Ting M.; Burigana M.; Zhang L.; Finfrock Y. Z.; Trabesinger S.; Jonderian A.; McCalla E. Impact of Nickel Substitution into Model Li-Rich Oxide Cathode Materials for Li-Ion Batteries. Chem. Mater. 2020, 32 (2), 849–857. 10.1021/acs.chemmater.9b04446. [DOI] [Google Scholar]
- Trujillo T. S.; Bernès S.; Castellanos R. M. A. Crystal Structure of Li3CuSbO5. J. Solid State Chem. 2001, 156 (2), 321–324. 10.1006/jssc.2000.9001. [DOI] [Google Scholar]
- Hill R. J.; Howard C. J. Quantitative Phase Analysis from Neutron Powder Diffraction Data Using the Rietveld Method. J. Appl. Crystallogr. 1987, 20 (6), 467–474. 10.1107/S0021889887086199. [DOI] [Google Scholar]
- Favre-Nicolin V.; C̆erný R. FOX, `free Objects for Crystallography’: A Modular Approach to Ab Initio Structure Determination from Powder Diffraction. J. Appl. Crystallogr. 2002, 35 (6), 734–743. 10.1107/S0021889802015236. [DOI] [Google Scholar]
- Rodríguez-Carvajal J. Recent Advances in Magnetic Structure Determination by Neutron Powder Diffraction. Phys. B 1993, 192 (1–2), 55–69. 10.1016/0921-4526(93)90108-I. [DOI] [Google Scholar]
- Larcher D.; Prakash A. S.; Laffont L.; Womes M.; Jumas J. C.; Olivier-Fourcade J.; Hedge M. S.; Tarascon J.-M. Reactivity of Antimony Oxides and MSb2O6 (M = Cu, Ni, Co), Trirutile-Type Phases with Metallic Lithium. J. Electrochem. Soc. 2006, 153 (9), A1778–A1787. 10.1149/1.2219711. [DOI] [Google Scholar]
- Strauss F.; Rousse G.; Corte D. A. D.; Hassine M. B.; Saubanère M.; Tang M.; Vezin H.; Courty M.; Dominko R.; Tarascon J.-M. Electrochemical Activity and High Ionic Conductivity of Lithium Copper Pyroborate Li6CuB4O10. Phys. Chem. Chem. Phys. 2016, 18 (22), 14960–14969. 10.1039/C6CP01581F. [DOI] [PubMed] [Google Scholar]
- Sun M.; Rousse G.; Abakumov A. M.; Saubanère M.; Doublet M.-L.; Rodríguez-Carvajal J.; Van Tendeloo G.; Tarascon J.-M. Li2Cu2O(SO4)2: A Possible Electrode for Sustainable Li-Based Batteries Showing a 4.7 V Redox Activity vs Li+/Li0. Chem. Mater. 2015, 27 (8), 3077–3087. 10.1021/acs.chemmater.5b00588. [DOI] [Google Scholar]
- Morcrette M.; Rozier P.; Dupont L.; Mugnier E.; Sannier L.; Galy J.; Tarascon J.-M. A Reversible Copper Extrusion-Insertion Electrode for Rechargeable Li Batteries. Nat. Mater. 2003, 2 (11), 755–761. 10.1038/nmat1002. [DOI] [PubMed] [Google Scholar]
- Chase M. W.NIST-JANAF Themochemical Tables, 4th ed.; American Institute of Physics: College Park, MD, 1998; pp 1–1951. [Google Scholar]
- Dutton S. E.; Kumar M.; Mourigal M.; Soos Z. G.; Wen J.-J.; Broholm C. L.; Andersen N. H.; Huang Q.; Zbiri M.; Toft-Petersen R.; Cava R. J. Quantum Spin Liquid in Frustrated One-Dimensional LiCuSbO4. Phys. Rev. Lett. 2012, 108 (18), 187206. 10.1103/PhysRevLett.108.187206. [DOI] [PubMed] [Google Scholar]
- Tulsky E. G.; Long J. R. Dimensional Reduction: A Practical Formalism for Manipulating Solid Structures. Chem. Mater. 2001, 13 (4), 1149–1166. 10.1021/cm0007858. [DOI] [Google Scholar]
- Greaves C.; Katib S. M. A. The Structural Chemistry of Li3Zn2MO6 (M = Sb, Bi) and Related Phases. Mater. Res. Bull. 1990, 25 (9), 1175–1182. 10.1016/0025-5408(90)90148-U. [DOI] [Google Scholar]
- Yabuuchi N.; Takeuchi M.; Nakayama M.; Shiiba H.; Ogawa M.; Nakayama K.; Ohta T.; Endo D.; Ozaki T.; Inamasu T.; Sato K.; Komaba S. High-Capacity Electrode Materials for Rechargeable Lithium Batteries: Li3NbO4-Based System with Cation-Disordered Rocksalt Structure. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (25), 7650–7655. 10.1073/pnas.1504901112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twu N.; Li X.; Urban A.; Balasubramanian M.; Lee J.; Liu L.; Ceder G. Designing New Lithium-Excess Cathode Materials from Percolation Theory: Nanohighways in LixNi2–4x/3Sbx/3O2. Nano Lett. 2015, 15 (1), 596–602. 10.1021/nl5040754. [DOI] [PubMed] [Google Scholar]
- Migeon H.-N.; Zanne M.; Gleitzer C.; Courtois A. Préparation et Étude de LiCuO. J. Solid State Chem. 1976, 16 (3–4), 325–330. 10.1016/0022-4596(76)90048-7. [DOI] [Google Scholar]
- Hoffmann R.; Hoppe R.; Schäfer W. Neutronenbeugung an Li2CuO2. Z. Anorg. Allg. Chem. 1989, 578 (1), 18–26. 10.1002/zaac.19895780103. [DOI] [Google Scholar]
- Wolf R.; Hoppe R. Notiz über Li2PdO2. Z. Anorg. Allg. Chem. 1986, 536 (5), 77–80. 10.1002/zaac.19865360509. [DOI] [Google Scholar]
- Hibble S. J.; Köhler J.; Simon A.; Paider S. LiCu2O2 and LiCu3O3: New Mixed Valent Copper Oxides. J. Solid State Chem. 1990, 88 (2), 534–542. 10.1016/0022-4596(90)90251-R. [DOI] [Google Scholar]
- Berger R.; Önnerud T.; Tellgren R. Structure Refinements of LiCu2O2 and LiCu3O3 from Neutron Powder Diffraction Data. J. Alloys Compd. 1992, 184 (2), 315–322. 10.1016/0925-8388(92)90505-4. [DOI] [Google Scholar]
- Wasel-Nielen H.-D.; Hoppe R. Zur Kristallstruktur von Li3AuO3, Li5AuO4, KAuO2 und RbAuO2. Z. Anorg. Allg. Chem. 1970, 375 (1), 43–54. 10.1002/zaac.19703750107. [DOI] [Google Scholar]
- Migeon H.-N.; Courtois A.; Zanne M.; Gleitzer C.; Aubry J. Préparation et Propriétés d’un Oxyde de Lithium-Cuivre(III): Li3CuO3. Rev. Chim. Miner. 1975, 12 (3), 203–209. [Google Scholar]
- Berger R.; Önnerud P.; Laligant Y.; Le Bail A. The Structure of Li3Cu2O4, a Compound with Formal Mixed Valence. J. Alloys Compd. 1993, 190 (2), 295–299. 10.1016/0925-8388(93)90415-J. [DOI] [Google Scholar]
- Greaves C.; Katib S. M. A. The Structures of Li5BiO5 and Li5SbO5 from Powder Neutron Diffraction. Mater. Res. Bull. 1989, 24 (8), 973–980. 10.1016/0025-5408(89)90181-5. [DOI] [Google Scholar]
- Hauck J.; Hirschberg A. Über Eine Hochdruckmodifikation Des Li6TeO6. Z. Naturforsch., B: J. Chem. Sci. 1969, 24 (12), 1656–1656. 10.1515/znb-1969-1234. [DOI] [Google Scholar]
- Abrahams I.; Lightfoot P.; Bruce P. G. Li6Zr2O7, a New Anion Vacancy Ccp Based Structure, Determined by Ab Initio Powder Diffraction Methods. J. Solid State Chem. 1993, 104 (2), 397–403. 10.1006/jssc.1993.1175. [DOI] [Google Scholar]
- Dahn J. R.; von Sacken U.; Michal C. A. Structure and Electrochemistry of Li1±yNiO2 and a New Li2NiO2 Phase with the Ni(OH)2 Structure. Solid State Ionics 1990, 44 (1), 87–97. 10.1016/0167-2738(90)90049-W. [DOI] [Google Scholar]
- Hodges J. P.; Short S.; Jorgensen J. D.; Xiong X.; Dabrowski B.; Mini S. M.; Kimball C. W. Evolution of Oxygen-Vacancy Ordered Crystal Structures in the Perovskite Series SrnFenO3n-1 (N = 2, 4, 8, and ∞), and the Relationship to Electronic and Magnetic Properties. J. Solid State Chem. 2000, 151 (2), 190–209. 10.1006/jssc.1999.8640. [DOI] [Google Scholar]
- Brixner L. H. Preparation, Structure and Electrical Properties of Some Substituted Lithium-Oxo-Metallates. J. Inorg. Nucl. Chem. 1960, 16 (1), 162–163. 10.1016/0022-1902(60)80105-4. [DOI] [Google Scholar]
- Kumar V.; Bhardwaj N.; Tomar N.; Thakral V.; Uma S. Novel Lithium-Containing Honeycomb Structures. Inorg. Chem. 2012, 51 (20), 10471–10473. 10.1021/ic301125n. [DOI] [PubMed] [Google Scholar]
- Primo-Martín V.; Jansen M. Synthesis, Structure, and Physical Properties of Cobalt Perovskites: Sr3CoSb2O9 and Sr2CoSbO6-δ. J. Solid State Chem. 2001, 157 (1), 76–85. 10.1006/jssc.2000.9041. [DOI] [Google Scholar]
- Vasala S.; Karppinen M. A2B′B″O6 Perovskites: A Review. Prog. Solid State Chem. 2015, 43 (1), 1–36. 10.1016/j.progsolidstchem.2014.08.001. [DOI] [Google Scholar]
- Kruth A.; Tabuchi M.; Guth U.; West A. R. Synthesis, Structure, Electrical and Magnetic Properties of the New Non-Stoichiometric Perovskite Phase, Ca2MnNbOγ. J. Mater. Chem. 1998, 8 (11), 2515–2520. 10.1039/a804865g. [DOI] [Google Scholar]
- Mandal T. K.; Gopalakrishnan J. From Rocksalt to Perovskite: A Metathesis Route for the Synthesis of Perovskite Oxides of Current Interest. J. Mater. Chem. 2004, 14 (8), 1273–1280. 10.1039/b315263d. [DOI] [Google Scholar]
- Mandal T. K.; Gopalakrishnan J. New Route to Ordered Double Perovskites: Synthesis of Rock Salt Oxides, Li4MWO6, and Their Transformation to Sr2MWO6 (M = Mg, Mn, Fe, Ni) via Metathesis. Chem. Mater. 2005, 17 (9), 2310–2316. 10.1021/cm050064e. [DOI] [Google Scholar]
- McCalla E.; Rowe A. W.; Camardese J.; Dahn J. R. The Role of Metal Site Vacancies in Promoting Li-Mn-Ni-O Layered Solid Solutions. Chem. Mater. 2013, 25 (13), 2716–2721. 10.1021/cm401461m. [DOI] [Google Scholar]
- McCalla E.; Abakumov A.; Rousse G.; Reynaud M.; Sougrati M. T.; Budic B.; Mahmoud A.; Dominko R.; Van Tendeloo G.; Hermann R. P.; Tarascon J.-M. Novel Complex Stacking of Fully-Ordered Transition Metal Layers in Li4FeSbO6 Materials. Chem. Mater. 2015, 27 (5), 1699–1708. 10.1021/cm504500a. [DOI] [Google Scholar]
- Rietveld H. M. Line Profiles of Neutron Powder-Diffraction Peaks for Structure Refinement. Acta Crystallogr. 1967, 22 (1), 151–152. 10.1107/S0365110X67000234. [DOI] [Google Scholar]
- Rietveld H. M. A Profile Refinement Method for Nuclear and Magnetic Structures. J. Appl. Crystallogr. 1969, 2 (2), 65–71. 10.1107/S0021889869006558. [DOI] [Google Scholar]
- Ravel B.; Newville M. Athena, Artemis, Hephaestus: Data Analysis for X-Ray Absorption Spectroscopy Using IFEFFIT. J. Synchrotron Radiat. 2005, 12 (4), 537–541. 10.1107/S0909049505012719. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Hafner J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B: Condens. Matter Mater. Phys. 1993, 47 (1), 558–561. 10.1103/PhysRevB.47.558. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Joubert D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B: Condens. Matter Mater. Phys. 1999, 59 (3), 1758–1775. 10.1103/PhysRevB.59.1758. [DOI] [Google Scholar]
- Okhotnikov K.; Charpentier T.; Cadars S. Supercell Program: A Combinatorial Structure-Generation Approach for the Local-Level Modeling of Atomic Substitutions and Partial Occupancies in Crystals. J. Cheminf. 2016, 8 (1), 17. 10.1186/s13321-016-0129-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrbach A.; Hafner J.; Kresse G. Electronic Correlation Effects in Transition-Metal Sulfides. J. Phys.: Condens. Matter 2003, 15 (6), 979–996. 10.1088/0953-8984/15/6/325. [DOI] [Google Scholar]
- Isseroff L. Y.; Carter E. A. Importance of Reference Hamiltonians Containing Exact Exchange for Accurate One-Shot GW Calculations of Cu2O. Phys. Rev. B: Condens. Matter Mater. Phys. 2012, 85 (23), 235142. 10.1103/PhysRevB.85.235142. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.