

Abstract
Patient: Male, 10-year-old
Final Diagnosis: MOPD type II
Symptoms: Short height • psico motor delay
Medication: —
Clinical Procedure: —
Specialty: Genetics • Radiology
Objective:
Rare disease
Background:
Moyamoya syndrome is a rare cerebrovascular condition caused by blockage of the arteries of the basal ganglia. The Japanese word “moyamoya” means “a puff of smoke” which describes the appearance of the collateral compensatory vessels that develop over time. Microcephalic osteodysplastic primordial dwarfism type II (MOPD II) is a rare genetic syndrome characterized by microcephaly and short stature. In up to 25% of patients with MOPD II, there is an association with moyamoya syndrome. This report is of a Syrian boy diagnosed with moyamoya syndrome and MOPD II.
Case Report:
A 10-year-old boy was referred to our pediatric endocrinology unit for short stature (-11.1 standard deviations). Exploration of the oral cavity showed dental malposition. Laboratory tests revealed mild thrombocytosis and hypernatremia. Glucagon-based growth hormone-stimulation testing revealed pathology, with growth hormone levels peaked at 30 minutes below 1 ng/ml. No abnormalities of carbohydrate metabolism or heart function were identified. Neuropsychological assessment found moderate to severe intellectual disability. Imaging studies showed osteoporosis, bilateral coxa vara, diffuse platyspondyly without scoliosis, malrotation of the left kidney, severe microcephaly with simplified convolution pattern, and moyamoya features with secondary brain atrophy. A genetic study identified a mutation in both alleles of the pericentrin (PCNT) gene, enabling the diagnosis of microcephalic osteodysplastic primordial dwarfism type II.
Conclusions:
This case highlights the importance of identifying the cause of short stature in children and genetic syndromes that may be linked with other abnormalities. MOPDII associated with moyamoya syndrome was diagnosed by cerebrovascular imaging, which led to a multidisciplinary approach to management.
Keywords: Child, Dwarfism, Moyamoya Disease
Background
Microcephalic osteodysplastic primordial dwarfism type II (MOPDII) is rare, with only about 150 cases identified worldwide [1]. Individuals with MOPDII are among the shortest human beings [2], but there is no evidence that administration of growth hormone benefits these patients [1]. Multiple syndromes include both dwarfism and microcephaly, but very few are as severe as MOPDII [2]. The differential diagnosis should include Meier-Gorlin syndrome, ligase IV deficiency, XRCC4 deficiency, Seckel syndrome, MOPD types I and III, Schimke immune-osseous dysplasia, SOFT syndrome, SHORT syndrome, and Dubowitz syndrome. Other entities can be ruled out based on clinical features, especially cerebrovascular abnormalities, a specific feature of MOPDII that is absent in other forms of microcephalic primordial dwarfism [3,4]. Thus, in up to 25% of patients with MOPDII, there is an association with moyamoya syndrome [5]. Although we can identify some characteristic signs of a particular syndrome, the definitive diagnosis requires molecular analysis [1]. The prognosis of MOPDII depends mainly on associated cerebrovascular alterations [1].
This report is of a 10-year-old Syrian boy with short stature and microcephaly, diagnosed with moyamoya syndrome and MOPD II. Informed consent for publication of patient information and images was obtained from the patient’s parents.
Case Report
A 10-year-old boy was referred to our endocrinology unit because of his short height. Because he was a refugee from Syria, no data from his neonatal period were available. The initial physical examination noted extremely short stature (72.1 cm; −11.1 standard deviations [SD]) with signs of disharmonic growth, marked microcephaly, retrognathia, slightly dysplastic auricles, and a prominent nose with elevated nasal root and wide bridge (Figure 1). Exploration of the oral cavity revealed dental malposition and cavities. Blood pressure was normal. Sexual development (absence of pubic hair and bilateral testicular volume 1 cc) was classified as Tanner stage 1. During the examination, a nasal voice was noted. Neuropsychological assessment found moderate to severe intellectual disability.
Figure 1.

Phenotypic traits of patients with microcephalic osteodysplastic primordial dwarfism type II (MOPDII). Figures show lateral (A) and front (B) photo of the 10-year-old boy, showing characteristic phenotypic traits of (MOPDII), including a prominent nose with elevated nasal root and wide bridge.
Initial laboratory tests revealed mild thrombocytosis (616 000 platelets/µL) and hypernatremia (Na 150 mEq/L); growth hormone (GH) 0.18 ng/mL and insulin-like growth factor 1 (IGF-1) 471 ng/mL were within the normal limits. A glucagon-based GH-stimulation test revealed pathology, with GH levels peaked at 30 minutes of 0.93 ng/mL. Cortisol levels were normal. His growth rate after 6 months was 0.6 cm/year (−4.5 SD). Imaging to assess scoliosis revealed osteoporosis, bilateral coxa vara, and diffuse platyspondyly without scoliosis (Figure 2). Abdominal ultrasound showed malrotation of the left kidney. Magnetic resonance imaging (MRI) of the head showed a simplified convolution pattern, moyamoya syndrome with secondary brain atrophy, and hypoplasia of the corpus callosum and anterior commissure (Figure 3) [6,7]. The Wechsler Nonverbal Scale of Ability (WNV) revealed a nonverbal cognitive ability at the lower limit of normal results (WNV 34). In the comprehensive vocabulary test, he obtained a result equivalent to that of an 18-month-old child; he collaborated adequately in all tests. Echocardiography findings were normal. A molecular study of primitive microcephaly identified the variant c.3465-1G>A in intron 17 of the PCNT gene in homozygosis.
Figure 2.
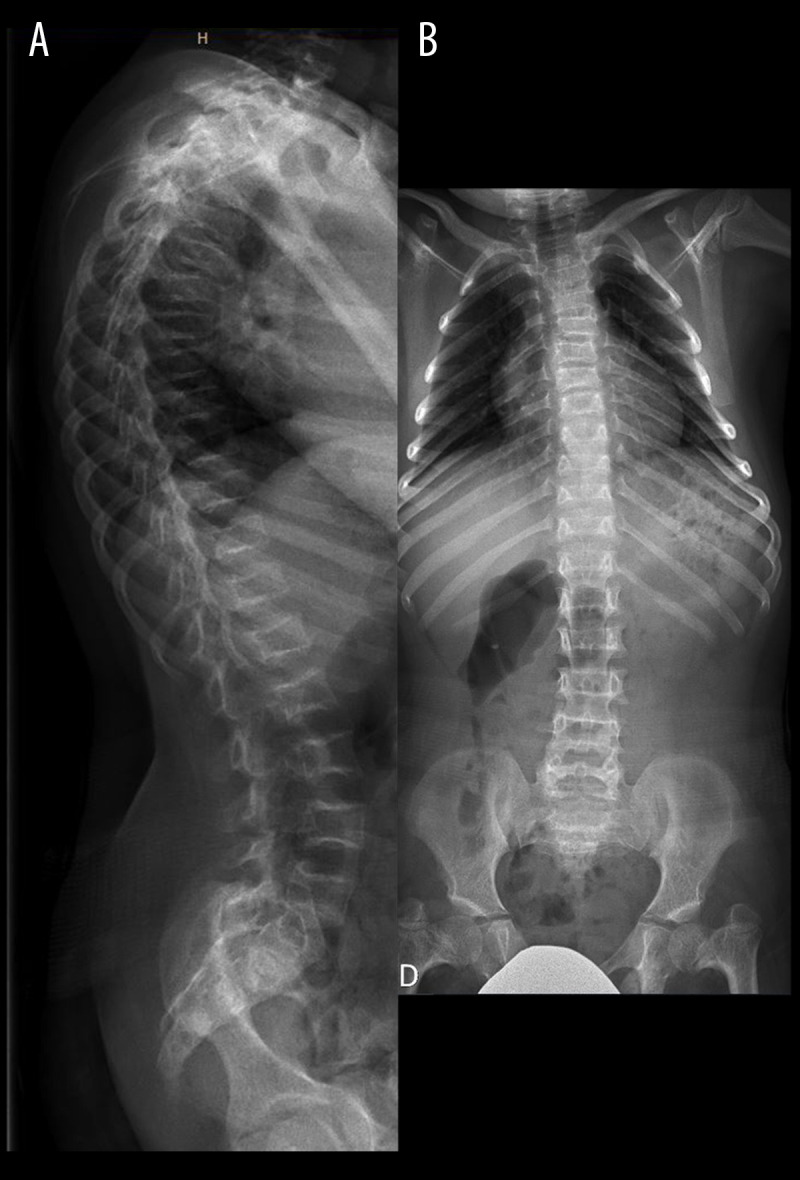
Skeletal features associated with MOPDII. Lateral (A) and antero-posterior (B) X-ray of the spine showing osteoporosis, bilateral coxa vara, and diffuse platyspondyly without scoliosis.
Figure 3.

Neurovascular abnormalities associated with MOPDII. Coronal (A), sagittal (B), and transverse (C) section of MR angiogram of the brain shows occlusion of both internal carotid arteries (marked with red arrows) and collateral supply of the anterior territory through the leptomeningeal circulation and branches of the external carotid artery, as well as inverted flow in both ophthalmic arteries. The leptomeningeal collaterals are also supplied by both posterior cerebral arteries; no clear occlusions are visible in the posterior circulation. These changes are related to moyamoya syndrome (stage IV, according to Suzuki’s classification).
The current management of the patient is based on medical follow-up by endocrinology, nephrology, and neurology. Endocrinology management consists of ruling out growth hormone deficiency, hypothyroidism, or another condition that could limit his height. We anticipate a final height below 110 cm. Nephrology evaluation involves measuring blood pressure and study of renal function to be sure it is in normal range. Neurologic management entails performing magnetic resonance imaging to confirm the possibility of moyamoya syndrome, which was diagnosed, and to assess evolution of neurovascular abnormalities and neurodevelopment.
Discussion
The PCNT gene, located at 21q22.3, encodes pericentrin, a giant coiled-coil protein that anchors regulatory and structural molecules to the centrosome. One of these structural molecules is γ-tubulin, which combines with other proteins to form the γ-tubulin ring complex, whose function is to initiate the assembly of the mitotic spindle apparatus. Thus, mutations in the PCNT gene result in disorganized mitotic spindles and chromosome mis-segregation that disrupt cell division and the cell cycle, leading to decreased cell production and increased cell death and consequently giving rise to the characteristic signs and symptoms of MOPDII [1]. PCNT’s c.3465-1G>A variant in intron 17 has been described previously and confirmed the diagnosis of MOPDII. Our patient’s phenotypic findings and complementary examinations results were similar to those found in other patients with MOPDII [1,2]. No clear correlations between genotype and pheno-type of patients with c.3465-1G>A variant have been described [8].
Suspicion of microcephalic primordial dwarfism should prompt a complete skeletal study to assess for possible abnormalities of the spine and hips. Repeated examinations are necessary to monitor the evolution of the coxa vara, which is more prevalent at younger ages, or scoliosis, which usually develops closer to puberty [1,9].
These patients require close medical follow-up, including laboratory tests starting at approximately 5 years of age to identify possible insulin resistance or diabetes, as well as routine monitoring of blood pressure and kidney function, because some cases of chronic kidney failure of uncertain etiology have been described [1].
Severe prenatal and postnatal growth retardation leads to predicted adult height below 110 cm [2], but the most prominent features of MOPDII are skeletal and craniofacial alterations. The main causes of significant morbidity and mortality in patients with MOPDII are central nervous system vascular abnormalities, particularly aneurysms and moyamoya syndrome. Moyamoya syndrome involves progressive stenosis of the terminal portion of internal carotid arteries, which results in occlusion of the vessel and formation of an anormal collateral pathways at the base of the brain [10,11]. The physiopathology of moyamoya syndrome in patients with MOPDII is still unknown, but it is believed that it may be related to the role of pericentrin in angiogenesis, which could explain why cerebrovascular abnormalities are more common in MOPDII patients than in patients with MODPI [12]. Vascular abnormalities can develop at any point in life and can cause strokes leading to disability or death. Thus, routine screening for cerebrovascular vascular anomalies is strongly recommended; the imaging technique of choice is brain MRI, including angiographic sequences, and the recommended interval between screenings is 12 to 18 months [1,13].
Multiple causes of death have been described, most related to vascular problems. The life expectancy of patients with MOPDII is currently around 30 years [1,2]. Early detection and optimal management of associated complications are the keys to improving quality of life and life expectancy in these patients.
There is no evidence to support the use of GH to treat MOPDII; to date, no statistically significant differences have been found between GH-treated and non-GH-treated patients in average SDs of height, weight, or occipitofrontal head circumference [1]. Thus, treatment usually targets complications or associated comorbidities. When moyamoya syndrome is identified, the most frequently used revascularization procedure is encephalodurosynangiosis (EDAS), in addition to endovascular coil occlusion or microsurgical clipping if aneurysms are present. These interventions aim to minimize long-term sequelae. Problems related with abnormal dentition may require minor surgical interventions and dentures or implants. Oral antidiabetic drugs should be considered in patients with insulin resistance, and antihypertensive drugs should be considered to protect kidney function in patients with hypertension [1].
Global prognosis of patients with MODPII is poor, with estimated life expectancy around 30 years. Neurovascular abnormalities in childhood deteriorate neurological prognosis and increase risk of stroke and, consequently, disabilities or death.
Follow-up by a multidisciplinary team aims to identify risks of associated comorbidities and complications, such as cerebral vasculopathy, renal and cardiac abnormalities, metabolic or hematological disorders, dental and maxillary abnormalities, and hip or spine problems.
Conclusions
This case highlights the importance of identifying the cause of short stature in children and genetic syndromes that may be associated with other abnormalities. In this case, the association between MOPDII and moyamoya syndrome was diagnosed by cerebrovascular imaging, which resulted in a multi-disciplinary approach to management.
Footnotes
Declaration of Figures’ Authenticity
All figures submitted have been created by the authors who confirm that the images are original with no duplication and have not been previously published in whole or in part.
References:
- 1.Bober MB, Jackson AP. Microcephalic osteodysplastic primordial dwarfism, type II: A clinical review. Curr Osteoporos Rep. 2017;15:61–69. doi: 10.1007/s11914-017-0348-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hall JG, Flora C, Scott CI, Jr, et al. Majewski osteodysplastic primordial dwarfism type II (MOPD II): Natural history and clinical findings. Am J Med Genet A. 2004;130A(1):55–72. doi: 10.1002/ajmg.a.30203. [DOI] [PubMed] [Google Scholar]
- 3.Bober MB, Niiler T, Duker AL, et al. Growth in individuals with Majewski osteodysplastic primordial dwarfism type II caused by pericentrin mutations. Am J Med Genet A. 2012;158A:2719–25. doi: 10.1002/ajmg.a.35447. [DOI] [PubMed] [Google Scholar]
- 4.Majewski F, Ranke M, Schinzel A. Studies of microcephalic primordial dwarfism II: The osteodysplastic type II of primordial dwarfism. Am J Med Genet. 1982;12(1):23–35. doi: 10.1002/ajmg.1320120104. [DOI] [PubMed] [Google Scholar]
- 5.Waldron JS, Hetts SW, Armstrong-Wells J, et al. Multiple intracranial aneurysms and moyamoya disease associated with microcephalic osteodysplastic primordial dwarfism type II: Surgical considerations. J Neurosurg Pediatr. 2009;4(5):439–44. doi: 10.3171/2009.6.PEDS08137. [DOI] [PubMed] [Google Scholar]
- 6.Scott RM, Smith ER. Moyamoya disease and moyamoya syndrome. N Engl J Med. 2009;360:1226–37. doi: 10.1056/NEJMra0804622. [DOI] [PubMed] [Google Scholar]
- 7.Guey S, Tournier-Lasserve E, Hervé D, Kossorotoff M. Moyamoya disease and syndromes: From genetics to clinical management. Appl Clin Genet. 2015;8:49–68. doi: 10.2147/TACG.S42772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abdel-Salam GMH, Sayed ISM, Afifi HH, et al. Microcephalic osteodys-plastic primordial dwarfism type II: Additional nine patients with implications on phenotype and genotype correlation. Am J Med Genet A. 2020;182(6):1407–20. doi: 10.1002/ajmg.a.61585. [DOI] [PubMed] [Google Scholar]
- 9.Majewski F, Goecke T. Microcephalic osteodysplastic primordial dwarfism type II: Report of three cases and review. Am J Med Genet. 1998;80:25–31. doi: 10.1002/(sici)1096-8628(19981102)80:1<25::aid-ajmg5>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 10.Fukui M. Guidelines for the diagnosis and treatment of spontaneous occlusion of the circle of Willis (‘moyamoya’ disease). Research Committee on Spontaneous Occlusion of the Circle of Willis (Moyamoya Disease) of the Ministry of Health and Welfare, Japan. Clin Neurol Neurosurg. 1997;99(Suppl. 2):S238–40. [PubMed] [Google Scholar]
- 11.Bober MB, Khan N, Kaplan J, et al. Majewski osteodysplastic primordial dwarfism type II (MOPD II): Expanding the vascular phenotype. Am J Med Genet A. 2010;152A:960–65. doi: 10.1002/ajmg.a.33252. [DOI] [PubMed] [Google Scholar]
- 12.Unal S, Alanay Y, Cetin M, et al. Striking hematological abnormalities in patients with microcephalic osteodysplastic primordial dwarfism type II (MOPD II): A potential role of pericentrin in hematopoiesis. Pediatr Blood Cancer. 2014;61:302–5. doi: 10.1002/pbc.24783. [DOI] [PubMed] [Google Scholar]
- 13.Perry LD, Robertson F, Ganesan V. Screening for cerebrovascular disease in microcephalic osteodysplastic primordial dwarfism type II (MOPD II): An evidence-based proposal. Pediatr Neurol. 2013;48(4):294–98. doi: 10.1016/j.pediatrneurol.2012.12.010. [DOI] [PubMed] [Google Scholar]