

Abstract
Purpose:
Increased availability of next generation sequencing has allowed for the genomic characterization of a variety of pediatric tumors, although genomic determinants of response to treatment remain largely unknown. We sought to evaluate the genomic landscape and genomic determinants of clinical outcomes in rhabdomyosarcoma (RMS).
Experimental Design:
Of 29,067 patients who underwent genomic profiling at our institution using a 468-gene oncopanel with complete records, 87 had RMS, of whom 22 were fusion-positive. The 10 most common genetic alterations were associated with locoregional control (LC), disease-free survival (DFS), and overall survival (OS). Tumor mutational burden (TMB), defined as the total number of somatic non-synonymous mutations normalized to the number of sequenced megabases, was also associated with clinical outcomes.
Results:
Median age at diagnosis was 16.4 years and median follow-up, 2.1 years. Patients with fusion-negative RMS had more genomic alterations and a higher TMB than those with fusion-positive RMS (mean number of genomic alterations, 6.0 versus 2.9, p=0.007 and mean TMB, 2.6 versus 1.0, p=0.01). Genetic alterations in TP53 were associated with worse OS (p=0.03). High TMB (defined as the top quartile, ≥2.8) was associated with worse LC (p=0.05), DFS (p=0.04), and OS (p=0.01), with significance retained on multivariable analysis after controlling for risk group, fusion status, and receipt of chemotherapy as per pediatric protocols.
Conclusion:
High TMB was associated with worse clinical outcomes in patients with RMS. With further validation, TMB and other genomic classifiers may be combined with traditional clinicopathologic risk factors to guide risk stratification and ultimately treatment decisions.
Keywords: rhabdomyosarcoma, genomics, tumor mutational burden, pediatric sarcomas, clinical outcomes
TRANSLATIONAL RELEVANCE
Genomic determinants of clinical outcomes in rhabdomyosarcoma (RMS) remain largely unknown aside from the PAX-FOXO1 fusion status. We utilized our institutional targeted high throughput sequencing effort to identify 87 patients with RMS, and correlated clinical outcomes with genomic alterations. Our results show that high tumor mutational burden (TMB) is associated with worse survival in RMS, independent of other well-known prognostic and treatment factors including risk group and fusion status. Pending further validation, TMB can be utilized along with traditional clinicopathologic risk factors to improve patient risk stratification and treatment options for RMS.
INTRODUCTION
Although survival improved dramatically for pediatric sarcomas such as rhabdomyosarcoma (RMS) with the advent of multimodality therapy, survival has now reached a therapeutic plateau.1 Additionally, for patients with metastatic and/or relapsed disease, prognosis remains suboptimal, with a heterogeneity of outcomes observed.2 For the patients who survive, late sequelae of intensive cytotoxic chemotherapy, surgery, and radiation therapy (RT) can be severe, including second cancers and long-term disabilities.
It is imperative to better understand the molecular landscape of RMS to guide both risk stratification and the development of novel therapies that may benefit specific subgroups. With growing availability of next generation sequencing, our knowledge of the genomic landscape of RMS has matured. Similar to other pediatric solid tumors, RMS tumors, especially fusion-positive tumors, harbor a low mutational burden, with relatively few actionable mutations. Genetic determinants of response in RMS remain largely unknown, though, aside from the prognostic significance of the PAX3/7-FOXO1 gene fusion3 and MYOD1 mutations.4 Our goal was to evaluate genomic determinants of clinical outcomes in RMS.
MATERIALS AND METHODS
Study Design and Patients
Patients with RMS who underwent targeted sequencing with our institutional oncopanel (MSK-IMPACT) from 1/2014 to 11/2018 were analyzed. MSK-IMPACT consists of a hybridization capture-based next-generation sequencing assay designed to test exons and certain introns within 468 genes (previous versions, 410 genes and 341 genes), with matched peripheral blood DNA used to determine germline versus somatic origin.5 Baseline clinicopathologic characteristics were extracted, including age, histology, stage, group, primary tumor location, primary treatment modality, and dates of local relapse, distant relapse, death, and last follow up. Genomic sequencing was performed with informed patient consent with approval from the Institutional Review Board (IRB) and in accordance with the U.S. Common Rule. Clinical correlates were identified retrospectively in RMS patients as approved by the IRB.
Statistical Analysis
The primary endpoint was overall survival (OS), with secondary endpoints including local control (LC) and disease-free survival (DFS). We screened for a possible association between the 10 most common genetic alterations and clinical outcomes (including LC, DFS, and OS). Genetic alterations were defined for a gene unit, (with one mutated gene counted as one genetic alternation), and included non-synonymous mutations, copy number changes, and/or gene fusions. In addition, tumor mutational burden (TMB), defined as the total number of somatic non-synonymous mutations normalized to the number of sequenced megabases, was associated with disease outcomes using a binary cutoff (with high TMB defined as the top quartile). LC, DFS, and OS were calculated from the date of diagnosis and evaluated via the Kaplan-Meier method, log-rank test, and Cox proportional hazard regression analysis. A paired t-test was used to assess the difference in number of genomic alterations between fusion-positive and fusion-negative patients as well the difference in age between TP53 wildtype versus mutant patients. The relationship between age at diagnosis and TMB was evaluated with Pearson’s correlation test. A p-value <0.05 was considered significant.
RESULTS
Patient population
Of 29,067 patients who underwent genomic profiling via MSK-IMPACT from 1/2014–11/2018, 96 (0.3%) had RMS. Nine patients had incomplete clinical records, leaving 87 patients in our analysis. Median age at diagnosis was 16.4 years (range, 0.2–74.3 years) and median follow up was 2.1 years (range, 0.2–8.4 years). See Table 1 for baseline patient and tumor characteristics.
Table 1.
Baseline patient and tumor characteristics
| Rhabdomyosarcoma (n=87) | |
|---|---|
| Age at diagnosis | |
| Median (Range) | 16.4 (0.2–74.3) |
|
| |
| Gender | |
| Male | 44 (51%) |
| Female | 43 (49%) |
|
| |
| Histology | |
| Embryonal | 40 (46%) |
| Alveolar | 25 (29%) |
| Spindle cell | 18 (21%) |
| Pleomorphic | 4 (5%) |
|
| |
| PAX3/7-FOXO1 Gene Fusion | |
| Present | 22 (25%) |
| Absent | 65 (75%) |
|
| |
| MYOD1 in spindle cell histology | |
| Mutated | 8 (44%) |
| Wild-type | 10 (56%) |
|
| |
| Primary site | |
| Parameningeal | 24 (28%) |
| Extremity | 20 (23%) |
| GU non-bladder/prostate | 18 (21%) |
| Perineal/perianal | 7 (8%) |
| GU bladder/prostate | 3 (3%) |
| Orbit | 3 (3%) |
| Head and neck | 3 (3%) |
| Other | 9 (10%) |
|
| |
| Stage | |
| 1 | 19 (22%) |
| 2 | 3 (3%) |
| 3 | 38 (44%) |
| 4 | 27 (31%) |
|
| |
| Group | |
| 1 | 11 (13%) |
| 2 | 8 (9%) |
| 3 | 41 (47%) |
| 4 | 27 (31%) |
|
| |
| Risk Group* | |
| Low | 22 (25%) |
| Intermediate | 38 (44%) |
| High | 27 (31%) |
|
| |
| Surgery for primary tumor | |
| Yes | 45 (52%) |
| No | 42 (48%) |
|
| |
| Radiation for primary tumor | |
| Yes | 70 (80%) |
| No | 17 (20%) |
|
| |
| Number of genetic alterations | |
| Fusion-positive tumors, median (range) | 2 (0–8) |
| Fusion-negative tumors, median (range) | 4 (0–25) |
|
| |
| Tumor mutational burden | |
| Fusion-positive tumors, median (range) | 0.9 (0–4.4) |
| Fusion-negative tumors, median (range) | 1.8 (0–13.8) |
Risk stratification based on current Children’s Oncology Group risk stratification
Mutational Landscape
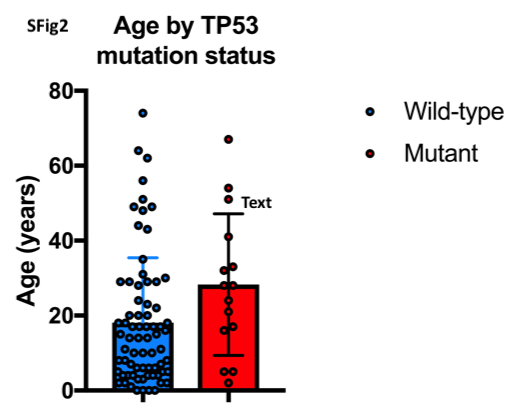
Site of tumor sequencing was from the primary tumor in 68 (78%), regional node in 6 (7%), and metastatic sites in 13 (15%). The median number of genetic alterations was 4 (range, 0–25), and median TMB was 1.1 (range, 0–13.8). Patients with fusion-negative RMS had more genomic alterations and a higher TMB than those with fusion-positive RMS (mean number of genomic alterations, 6.0 versus 2.9, p=0.007 and mean TMB, 2.6 versus 1.0, p=0.01, Figure 1). Site of tumor sequencing (primary versus metastatic) did not affect the number of genomic alterations or TMB (p=0.84 and p=0.96, respectively). Age at time of diagnosis was not associated with TMB among the entire cohort (p=0.97) nor among patients with fusion-negative disease (p=0.58). However, there was a weak positive association between age at time of diagnosis and TMB in patients with fusion-positive disease (p=0.04, Pearson coefficient = 0.44, R2 = 0.20, Supplementary Figure 1). The most common genetic alterations among the entire cohort were TP53 (17%), NF1 (13%), CDKN2A (11%), NRAS (11%), MYOD1 (10%), MDM2 (9%), BCOR (9%) CDKN2B (9%), CDK4 (8%), and GLI1 (7%). Of note, GLI1 mutational status was not profiled in 11 patients, and was altered in 7% of the entire cohort and in 8% of patients with available data. In addition, DICER1 mutations were seen in all 4 female patients with genitourinary primaries. TP53 mutations were more common in older patients (median age of 28.2 years in patients with TP53 mutations versus 14.6 years with TP53 wild-type, p=0.05, Supplementary Figure 2). Of note, all mutations in NF1 and DICER1 were somatic. One patient had a germline TP53 mutation (TMB of 0.88), and one patient had a germline mutation in APC (TMB of 0.98).
Figure 1.
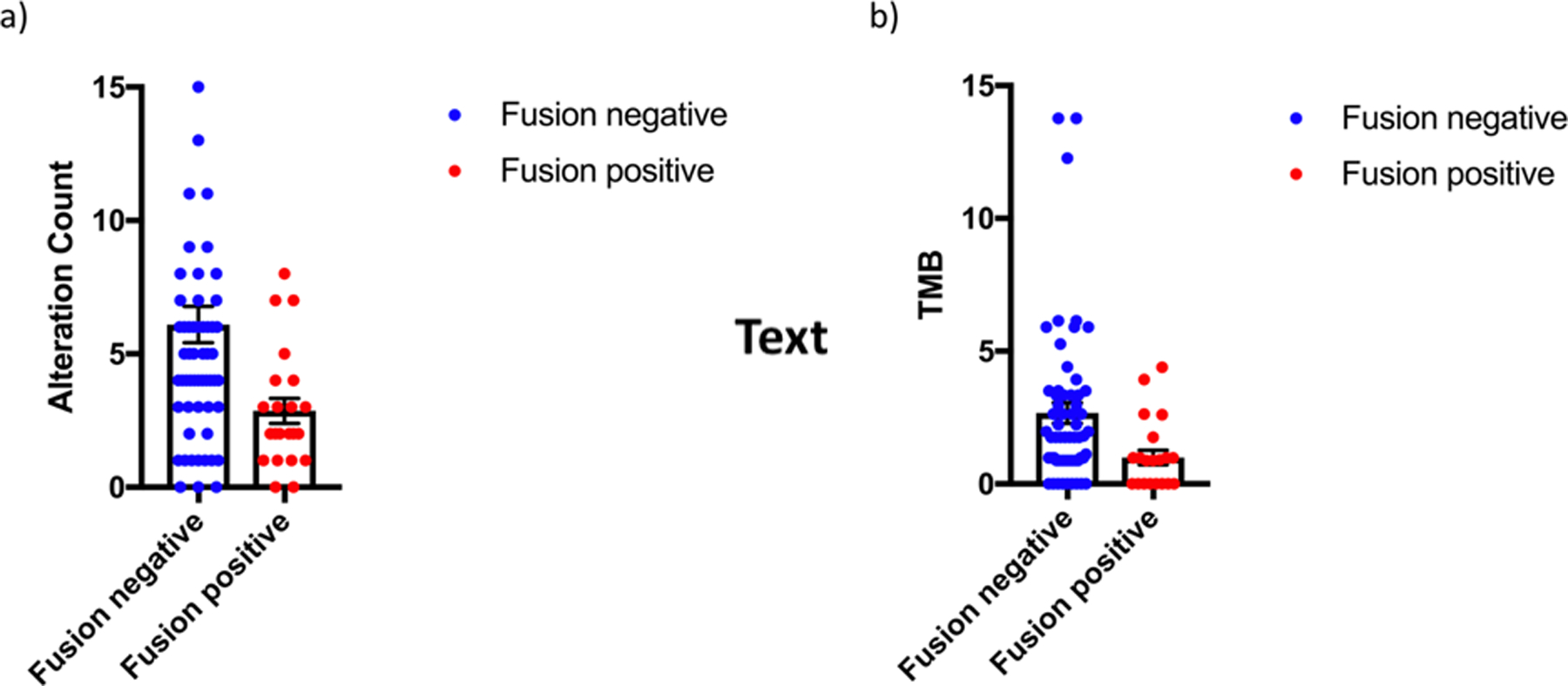
a) Number of genomic alterations in fusion-positive versus fusion-negative rhabdomyosarcoma (p=0.007) and b) tumor mutational burden (TMB) in fusion-positive versus fusion-negative rhabdomyosarcoma (p=0.01)
Patients with fusion-positive RMS were genetically distinct from those with fusion-negative RMS (see Figure 2 for oncoprints). Among patients with fusion-negative RMS, the most common mutations included TP53 (23%), NF1 (18%), MYOD1 (15%), NRAS (13%), and BCOR (13%), with the most common copy-number alterations including CDKN2A (15%) and CDKN2B (13%). In total, 18% of patients with fusion-negative tumors harbored a mutation of a RAS isoform. In fusion-positive tumors, the most common genetic alteration was CDK4 amplification (18%), with a relatively otherwise quiet genome.
Figure 2.

Oncoprints for patients with fusion-negative rhabdomyosarcoma versus fusion-positive rhabdomyosarcoma. *Denotes adjusted percentage to account for patients in which GLI1 mutational status was not assessed.
Survival Outcomes
The 2-year DFS for patients with low-, intermediate-, and high-risk disease was 66.2%, 33.7%, and 36.7%, respectively (p=0.05); and the 2-year OS was 100%, 64.2%, and 55.1% (p=0.03). DFS and OS did not differ in pediatric (age <25 years) versus adult patients (p=0.64 and p=0.19, respectively), although patients who received chemotherapy on or as per pediatric protocols had a trend towards better DFS (p=0.17) and better OS (p=0.02) on univariate analysis compared to those who did not. See Table 2 for univariate analysis of genomic factors associated with DFS and OS. There was a trend toward worse DFS in patients with PAX3/7-FOXO1 gene fusion (hazard ratio [HR] 1.7, p=0.07). Mutations in TP53 were associated with worse OS (HR 2.3, p=0.04), with a trend towards worse OS in patients with GLI1 alterations (HR 2.6, p=0.08).
Table 2.
Univariate analysis of genomic factors associated with disease-free and overall survival
| Genomic Alteration | n | DFS HR (95% CI) | P | OS HR (95% CI) | P |
|---|---|---|---|---|---|
| TP53 | 15 | 1.7 (0.9–3.3) | 0.10 | 2.3 (1.0–4.9) | 0.04 |
|
| |||||
| NF1 | 11 | 0.8 (0.3–1.8) | 0.56 | 0.5 (0.2–1.6) | 0.25 |
|
| |||||
| CDKN2A | 10 | 1.1 (0.5–2.3) | 0.90 | 1.4 (0.6–3.4) | 0.44 |
|
| |||||
| NRAS | 10 | 1.2 (0.5–2.7) | 0.73 | 1.7 (0.6–3.3) | 0.29 |
|
| |||||
| MYOD1 | 9 | 1.1 (0.5–2.3) | 0.91 | 1.4 (0.6–3.3) | 0.46 |
|
| |||||
| MDM2 | 8 | 0.4 (0.1–1.3) | 0.12 | 0.8 (0.2–2.4) | 0.63 |
|
| |||||
| BCOR | 8 | 0.4 (0.1–1.8) | 0.26 | 0.3 (0.05–2.4) | 0.27 |
|
| |||||
| CDKN2B | 8 | 0.8 (0.3–1.9) | 0.56 | 1.2 (0.5–3.1) | 0.71 |
|
| |||||
| CDK4 | 7 | 1.8 (0.8–4.2) | 0.18 | 1.9 (0.6–5.3) | 0.25 |
|
| |||||
| GLI1 | 6 | 2.3 (0.9–5.9) | 0.09 | 2.6 (0.9–7.8) | 0.08 |
|
| |||||
| PAX3/7-FOXO1 | 22 | 1.7 (1.0–2.9) | 0.07 | 1.3 (0.7–2.6) | 0.44 |
|
| |||||
| TMB ≥2.8 (3rd quartile) | |||||
| All patients | 87 | 1.9 (1.1–3.4) | 0.03 | 2.3 (1.2–4.5) | 0.01 |
| Fusion negative | 65 | 2.1 (1.1–4.2) | 0.03 | 2.4 (1.1–5.5) | 0.03 |
| Fusion positive | 22 | 5.4 (1.0–28.4) | 0.05 | 29.5 (2.6–333.3) | 0.006 |
| Low/int risk | 60 | 2.1 (1.0–4.2) | 0.04 | 2.3 (1.0–5.2) | 0.04 |
| High risk | 27 | 4.0 (1.1–15.3) | 0.04 | 6.1 (1.6–24.0) | 0.009 |
High TMB (defined as the top quartile or ≥2.8) was also associated with worse LC (HR 2.4, p=0.02), DFS (HR 1.9, p=0.03) and OS (HR 2.3, p=0.01). Specifically, the 2-year DFS and OS for patients with TMB <2.8 was 48.1% and 75.4%, respectively, compared to 24.3% and 51.6% for patients with TMB ≥2.8 (Figure 3A,B). Limiting the analysis to patients <25 years of age, DFS was 50.1% versus 29.8% for TMB <2.8 versus ≥2.8 (p=0.06), and OS was 80.3% versus 66.2% (p=0.06). The significant association between TMB and OS persisted among the subgroups of patients with fusion-positive RMS, fusion-negative RMS, localized (low/intermediate-risk) RMS, and metastatic (high-risk) RMS (Figure 3C, D, E, F and Table 2). On multivariable analysis including risk group, fusion status, and receipt of chemotherapy as per pediatric protocol, TMB ≥2.8 continued to be associated with inferior DFS (HR 2.6, p=0.005) and OS (HR, 3.5, p=0.003, Table 3). With TP53 included on multivariable analysis in addition to the above factors, TMB remained significantly associated with both DFS (HR 2.4, p=0.02) and OS (HR 3.3, p=0.01). Additionally, when TMB was treated as a continuous variable, a significant association between TMB and survival was retained (HR 1.1, p=0.04 for both DFS and OS).
Figure 3.
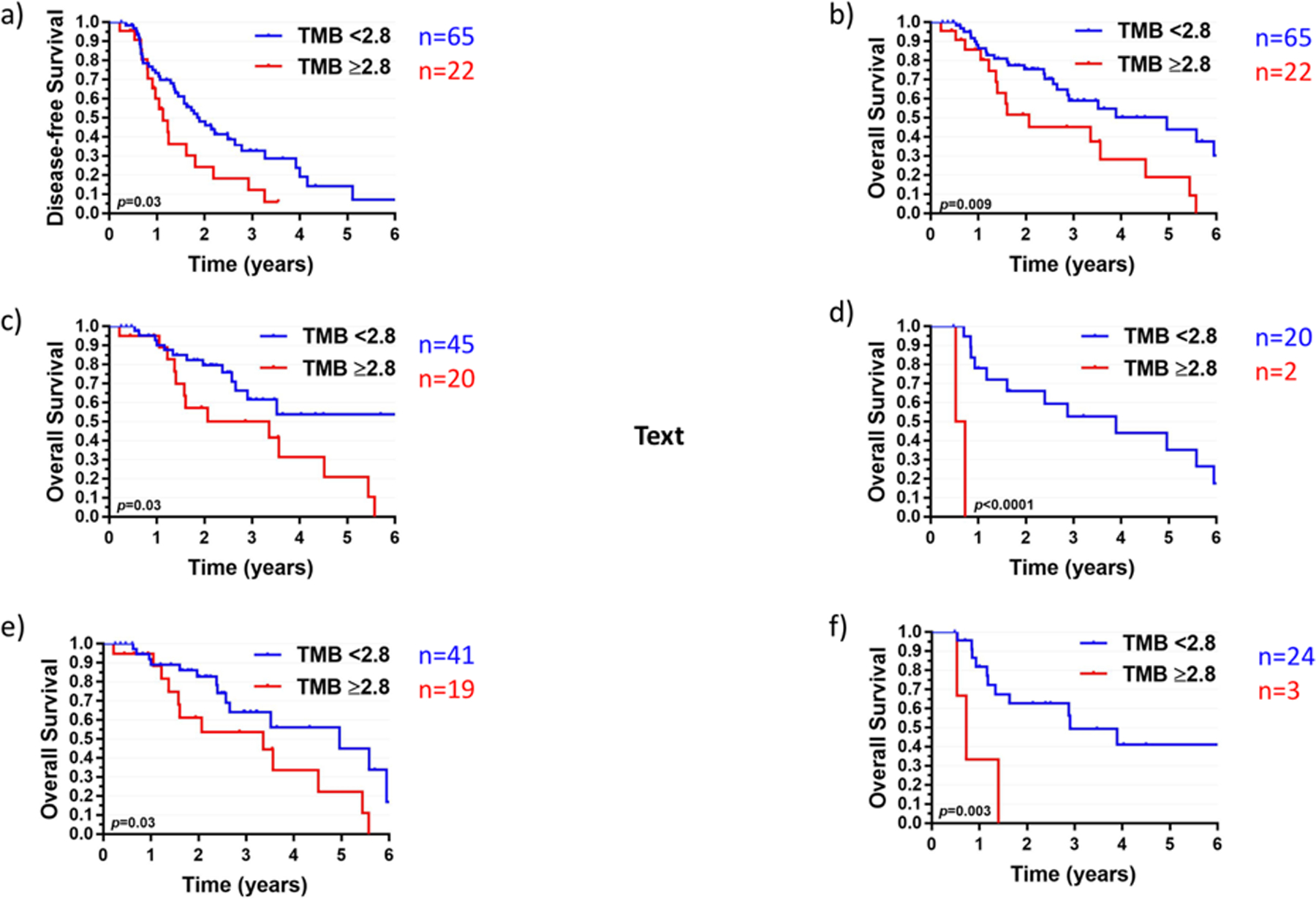
a) Disease-free survival and b) overall survival for the entire cohort stratified by tumor mutational burden (TMB) using a binary cutoff of 2.8 (third quartile). Overall survival by TMB in patients with c) fusion-negative rhabdomyosarcoma; d) fusion-positive rhabdomyosarcoma; e) localized rhabdomyosarcoma (low/intermediate-risk); and f) metastatic rhabdomyosarcoma (high-risk).
Table 3.
Multivariable analysis of factors associated with disease-free and overall survival
| Variable | DFS: HR (95% CI) | P | OS: HR (95% CI) | P |
|---|---|---|---|---|
| Risk group | ||||
| Low | 1.0 (Reference) | 1.0 (Reference) | ||
| Intermediate | 2.4 (1.1–5.5) | 0.03 | 5.4 (1.6–18.7) | 0.007 |
| High | 3.3 (1.3–8.4) | 0.01 | 6.3 (1.6–25.1) | 0.01 |
|
| ||||
| Fusion status | ||||
| Negative | 1.0 (Reference) | 1.0 (Reference) | ||
| Positive | 1.7 (0.9–3.4) | 0.12 | 1.7 (0.7–4.1) | 0.22 |
|
| ||||
| TMB ≥2.8 (Third quartile) | ||||
| No | 1.0 (Reference) | 1.0 (Reference) | ||
| Yes | 2.6 (1.3–5.1) | 0.005 | 3.5 (1.5–7.9) | 0.003 |
|
| ||||
| Receipt of chemotherapy as per pediatric protocol | ||||
| No | 1.0 (Reference) | 0.22 | 1.0 (Reference) | 0.06 |
| Yes | 0.7 (0.3–1.3) | 0.5 (0.2–1.0) | ||
DISCUSSION
This study represents the first study, to our knowledge, to evaluate the impact of TMB on clinical outcomes in RMS. Our results show that high TMB is associated with worse survival in RMS, independent of other well-known prognostic and treatment factors including risk group, fusion status, and receipt of pediatric protocol chemotherapy. Our study also confirms the distinct genomic landscapes of fusion-positive versus fusion-negative tumors, with an overall lower mutational burden in fusion-positive tumors.6 Despite this distinction, TMB remained significantly associated with survival among the subgroups of patients with either fusion-positive or fusion-negative disease. TMB has also been associated with survival in neuroblastoma,7 lung cancer,8 and pancreatic cancer.9 As it is hypothesized for neuroblastoma, it is likely that TMB in RMS serves as a surrogate marker of an aggressive biologic phenotype, irrespective of the presenting stage.
We also observed inferior survival in patients with TP53 mutations, consistent with interim findings from COG ARST14B1.10 TP53 mutations were also more common in older patients in our cohort, suggesting a possible clustering of TP53 mutations by age as has been observed for RAS isoform mutations.10 Data from our institution previously showed the aggressive biologic behavior and poor prognostic significance of MYOD1-mutant spindle and sclerosing RMS;4 approximately two-thirds of patients reported on that study underwent targeted MYOD1 mutation testing rather than genomic profiling with MSK-IMPACT, and thus were not included in our cohort. Our study also found that 18% of patients with fusion-negative disease harbored a mutation of a RAS isoform, similar to the rate of RAS-opathy (24%) reported in the large comprehensive genomic analysis of RMS.6
Limitations of our analysis include the rarity of the disease and overall low somatic mutation rate, limiting the power to evaluate the clinical significance of specific mutations. Our analysis also included adults with RMS, with an older median age than what is generally seen in COG and other cooperative group data sets; this inclusion of adults may skew the patient sample and bias the results, but also allows for a full depiction of the RMS genomic landscape across all ages, which may be missing in reports limiting their analysis to the pediatric population. In addition, although MSK-IMPACT tumor sequencing is offered routinely, there is a selection bias for an unfavorable cohort of patients that develop recurrent disease, and therefore our survival results are lower than what is typically seen on national COG trials. Our results thus warrant further validation in a larger, combined dataset. Strengths include the detailed clinical outcome analysis, beyond what is available in publicly available data, and the robustness of the relationship between TMB and clinical outcomes among subgroups of patients despite the small numbers in our cohort. Pending further validation, TMB and other genomic classifiers may be utilized in combination with traditional risk factors to refine risk stratification and guide treatments decisions for patients with RMS.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
This work was supported in part by a National Institutes of Health/National Cancer Institute Cancer Center Support Grant (P30CA008748).
Footnotes
Conflict of Interest: RMS receives loyalties related to a patent MSK has licensed for use of tumor mutational burden for the identification of patients that benefit from immunotherapy. DLC, LHW, KLP, EKS, and SLW have no relevant conflicts to disclose.
REFERENCES
- 1.Malempati S, Hawkins DS: Rhabdomyosarcoma: review of the Children’s Oncology Group (COG) Soft-Tissue Sarcoma Committee experience and rationale for current COG studies. Pediatr Blood Cancer 59:5–10, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Breneman JC, Lyden E, Pappo AS, et al. : Prognostic factors and clinical outcomes in children and adolescents with metastatic rhabdomyosarcoma--a report from the Intergroup Rhabdomyosarcoma Study IV. J Clin Oncol 21:78–84, 2003 [DOI] [PubMed] [Google Scholar]
- 3.Skapek SX, Anderson J, Barr FG, et al. : PAX-FOXO1 fusion status drives unfavorable outcome for children with rhabdomyosarcoma: a children’s oncology group report. Pediatr Blood Cancer 60:1411–7, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agaram NP, LaQuaglia MP, Alaggio R, et al. : MYOD1-mutant spindle cell and sclerosing rhabdomyosarcoma: an aggressive subtype irrespective of age. A reappraisal for molecular classification and risk stratification. Mod Pathol 32:27–36, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng DT, Prasad M, Chekaluk Y, et al. : Comprehensive detection of germline variants by MSK-IMPACT, a clinical diagnostic platform for solid tumor molecular oncology and concurrent cancer predisposition testing. BMC Med Genomics 10:33, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shern JF, Chen L, Chmielecki J, et al. : Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov 4:216–31, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hwang WL, Wolfson RL, Niemierko A, et al. : Clinical Impact of Tumor Mutational Burden in Neuroblastoma. J Natl Cancer Inst, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Owada-Ozaki Y, Muto S, Takagi H, et al. : Prognostic Impact of Tumor Mutation Burden in Patients With Completely Resected Non-Small Cell Lung Cancer: Brief Report. J Thorac Oncol 13:1217–1221, 2018 [DOI] [PubMed] [Google Scholar]
- 9.Grassi E, Durante S, Astolfi A, et al. : Mutational burden of resectable pancreatic cancer, as determined by whole transcriptome and whole exome sequencing, predicts a poor prognosis. Int J Oncol 52:1972–1980, 2018 [DOI] [PubMed] [Google Scholar]
- 10.Shern JF, Patidar R, Song Y, et al. : Targeted resequencing of pediatric rhabdomyosarcoma: report from the Children’s Oncology Group, the Children’s Cancer and Leukaemia Group, The Institute of Cancer Research UK, and the National Cancer Institute. Journal of Clinical Oncology 36:10515–10515, 2018 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.