

Abstract
Antisense gapmer oligonucleotides containing phosphoryl guanidine (PG) groups, e.g., 1,3-dimethylimidazolidin-2-imine, at three to five internucleotidic positions adjacent to the 3′ and 5′ ends were prepared via the Staudinger chemistry, which is compatible with conditions of standard automated solid-phase phosphoramidite synthesis for phosphodiester and, notably, phosphorothioate linkages, and allows one to design a variety of gapmeric structures with alternating linkages, and deoxyribose or 2′-O-methylribose backbone. PG modifications increased nuclease resistance in serum-containing medium for more than 21 days. Replacing two internucleotidic phosphates by PG groups in phosphorothioate-modified oligonucleotides did not decrease their cellular uptake in the absence of lipid carriers. Increasing the number of PG groups from two to seven per oligonucleotide reduced their ability to enter the cells in the carrier-free mode. Cationic liposomes provided similar delivery efficiency of both partially PG-modified and unmodified oligonucleotides. PG-gapmers were designed containing three to four PG groups at both wings and a central “window” of seven deoxynucleotides with either phosphodiester or phosphorothioate linkages targeted to MDR1 mRNA providing multiple drug resistance of tumor cells. Gapmers efficiently silenced MDR1 mRNA and restored the sensitivity of tumor cells to chemotherapeutics. Thus, PG-gapmers can be considered as novel, promising types of antisense oligonucleotides for targeting biologically relevant RNAs.
Keywords: antisense oligonucleotide gapmer, phosphoryl guanidine, gene silencing, intracellular accumulation, nuclease resistance, RNase H, MDR1
Graphical abstract
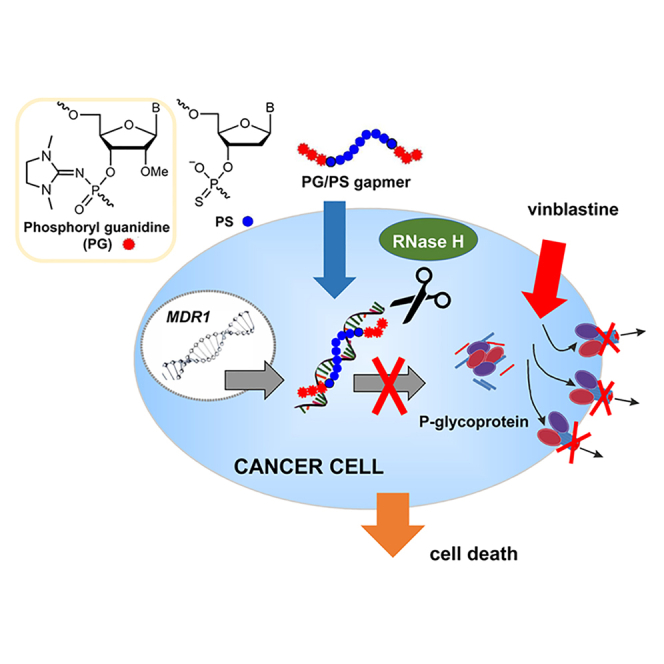
Many successful antisense oligonucleotide designs are gapmers, which contain a central section of DNA phosphorothioates flanked by RNA-like nucleotides. We describe a new type of gapmers with phosphoryl guanidine groups in the flanks, which efficiently silenced MDR1 gene expression and restored the sensitivity of drug-resistant tumor cells to conventional chemotherapy.
Introduction
In the 40 years that have elapsed since the pioneering studies of Zamecnik and Stephenson,1 concerted efforts have been focused on the development of antisense oligonucleotides (ASOs) as therapeutic medicines to combat human diseases.2,3 ASOs can function by sterically blocking mRNA translation, by mediating mRNA cleavage by the recruitment of cellular nucleases, such as RNase H,4,5 by altering pre-mRNA splicing,6 or by preventing ribosome recruitment.7 However, two main problems still limit the widespread clinical application of ASOs: the difficulty of oligonucleotide delivery to the target cells8 and rapid degradation of oligonucleotides by extra- and intracellular nucleases. While the former is a more formidable obstacle, the latter can be alleviated by chemical modification of the sugar residue9 or, more straightforwardly, the internucleotidic phosphate of synthetic oligonucleotides.10 Moreover, alteration of the internucleotidic phosphate may also help to improve cellular uptake of ASOs.11,12 Thus, efficient mRNA downregulation can be achieved by using an ASO with modified phosphate groups such as phosphorothioate (PS),13 2′-modified RNAs, such as oligo-2′-O-(2-methoxyethyl)ribonucleotides (2′-MOE)14 and bridged/locked nucleic acids (2′,4′-BNA/LNA),15,16 and by oligonucleotide analogs with a more or less radically altered sugar-phosphate backbones, such as peptide nucleic acids (PNAs)17 and phosphorodiamidate morpholino oligomers (PMOs).18
ASO development in the previous decade has firmly established the ASO concept as a straightforward approach for site-selective RNA targeting. This endeavor has culminated in the FDA approval of several ASO therapeutics with modified backbones that confer improved pharmacological properties: a 2′-MOE ASO Mipomersen (Kynamro) to treat familial hypercholesterolemia,19 and 2′-MOE ASO Nusinersen (Spinraza) and PMO Eteplirsen (Exondys 51) for the treatment of spinal muscular atrophy and Duchenne muscular dystrophy, respectively; PMOs Golodirsen (Vyondys 53) and, recently, Casimersen (Amondys 45) for DMD19, 20, 21 and 2′-MOE ASO, Inotersen (Tegsedi), for familial amyloid polyneuropathy.22
Of particular interest are the oligodeoxynucleotide analogs, which can mediate irreversible RNA inactivation in a catalytic mode by recruiting RNase H. These analogs include DNA oligonucleotides containing modification at phosphodiester bonds such PS,23 2′-deoxy-2′-fluoro-β-D-arabinonucleic acids,24 and the recently developed mesyl phosphoramidate oligodeoxynucleotides (μ-ASOs).25
These DNA analogs could be used to build gapmers: mixed backbone ASOs containing short (6–10 nucleotide [nt]) central “windows” of RNase H-competent deoxynucleotides and 3′,5′-flanking “wings” (3–5 nt) of RNase H-incompetent residues, such as 2′-OMe, 2′-MOE, or LNA, which primarily increase RNA binding but also nuclease resistance.26, 27, 28 Gapmer ASOs can efficiently silence RNAs by using the best of both mechanisms: steric blocking and RNase H-mediated RNA cleavage within the central RNase H-competent window of the heteroduplex.
In this study, we describe oligonucleotide gapmers containing the phosphoryl guanidine (PG) modification of the phosphate group that had been developed previously.29,30 Recently, PG oligonucleotides (PGOs) have shown promise as primers for PCR,31,32 antibacterial33 and antiviral34 ASOs, and G-quadruplex-forming agents.35 Introduction of a single PG group into a trinucleotide increased its stability to snake venom phosphodiesterase (SVPDE).36 PG modification of PCR primers prevented DNA multimerization during qPCR and increased discrimination between wild-type and mutated DNA.31,32 Fully substituted 2′-OMe RNA PGOs were shown to successfully target intracellular Mycobacterium smegmatis in human macrophages via selective downregulation of a bacterial gene by RNase H-independent mechanism,33 and possess potent antiviral activity against influenza A virus in Madin-Darby canine kidney (MDCK) cell culture when delivered by nanoparticles.34 G-rich deoxynucleotide PGOs can assemble into intermolecular G-quadruplexes very rapidly due to the absence of charge repulsion, and form mixed G-quartets with natural G-rich sequences presumably via strand invasion.35
Multiple drug resistance (MDR) phenotype of tumor cells provides their resistance to a wide range of chemotherapeutic agents, preventing successful cancer cell elimination.37 The MDR phenotype is often attributed to an overexpression of some members of transmembrane ATP-binding cassette proteins, referred to as the ABC transporters superfamily.38,39 The human MDR1 (ABCB1) genes, that belong to subfamily B of ABC transporters encode P-glycoprotein (Pgp), involved in active ATP-dependent transport of cytotoxic agents out of the cells.39 MDR1 mRNA is an attractive target for ASOs since its downregulation restores the sensitivity of cancer cells to chemotherapy.
The remarkable properties of PG modification prompted us to design gapmers to introduce a combination of charge-neutral PG linkages, namely, 1,3-dimethylimidazolidin-2-imine groups (Dmi), and negatively charged phosphodiester (PO) or PS groups with a mixed deoxyribo- and 2′-O-methylribonucleotide backbone to decrease the total negative charge. The ability of these oligonucleotides to penetrate cells, resist nuclease digestion in biological media, and reverse MDR1-induced drug resistance of tumor cells is reported.
Results
Design of oligonucleotides
To obtain oligonucleotides and gapmers with a decreased total negative charge, we employed a previously developed charge-neutral phosphate mimic—a PG group such as Dmi (Figure 1C). PGOs bearing exclusively PG groups at internucleotidic positions are charge neutral, similarly to PMOs, in contrast to natural oligonucleotides that have negatively charged PO groups or first-generation ASOs with PS linkages (Figures 1A and 1B, respectively).
Figure 1.

Structures of DNA phosphodiester and phosphate group mimics used in this study
(A) Phosphodiester (PO), (B) phosphorothioate (PS), and (C) a phosphoryl guanidine (PG) group, namely 1,3-dimethylimidazolidin-2-imine (Dmi), R = H or OMe.
Three sets of modified oligonucleotides were obtained: the R series was designed to assess nuclease resistance, the D series was designed to elucidate their ability to enter cells, and the M series was designed to study their gene-silencing properties (Table 1). The R and D series oligonucleotides have random sequences that contain no regions complementary to the reference sequences of known human mRNAs.
Table 1.
Structures and molecular masses of the oligonucleotides used in this study
| Name | Oligonucleotide sequence (5′→3′) | Abbreviation | Calc. [M–H]– | Found [M–H]– | Figures |
|---|---|---|---|---|---|
| Oligonucleotides for nuclease resistance study (R series)a | |||||
| R1 | agtctcgacttgctacc | deoxy/PO | 5,121.41 | 5,119.43 | S1 |
| R2 | aSgStScStScSgSaScStStSgScStSaScSc | deoxy/PS | 5,378.44 | 5,377.12 | S2 |
| R3 | a∗g∗t∗c∗t∗c∗g∗a∗c∗t∗t∗g∗c∗t∗a∗c∗c | deoxy/PG | 6,643.78 | 6,641.90 | S3 |
| R4∗ | AGUCUCGACUUGCUACC | 2′-O-Me/PO | 5,561.73 | 5,556.76 | S4 |
| R5∗ | ASGSUSCSUSCSGSASCSUSUSGSCSUSASCSC | 2′-O-Me/PS | 5,818.76 | 5,813.76 | S5 |
| R6∗ | A∗G∗U∗C∗U∗C∗G∗A∗C∗U∗U∗G∗C∗U∗A∗C∗C | 2′-O-Me/PG | 7,084.10 | 7,079.76 | S6 |
| Oligonucleotides for cellular delivery study (D series)a,b,c,d,e,g | |||||
| D1 | FAMS-ASGSUSCSUSCSGSASCSUSUSGSCSUSASCSCSUSCSAb | F-2′-OMe/PS | 7,416.50 | 7,415.00 | S7A |
| D2 | FAMS-CSUSCSCSGSASASGSASASASUSASASGSASUSCSCc | F-2′-OMe/PS | 7,174.40 | 7,172.50 | S7C |
| D3 | FAMS-GSASCSASUSCSCSASUSUSCSASASASUSGSGSUSUSUSGd | F-2′-OMe/PS | 7,840.80 | 7,839.00 | S7B |
| D4 | FAMS-GSASCSASUSCSCSASUSUSCSASASASUSGSGSUSU∗U∗Gd | F-2′-OMe/19PS/2PG | 8,002.80 | 8,001.50 | S7D |
| D5 | FAMS-ASGSUSCSUSCSGSASCSUSUSGSCSUSASCSCa | F-2′-OMe/PS | 5,945.94 | 5,944.31 | – |
| D6 | FAM-a∗g∗t∗c∗t∗c∗g∗a∗c∗t∗t∗g∗c∗t∗a∗c∗ca | F-deoxy/PG | 7,211.28 | 7,208.50 | – |
| D7 | FAM-agtctcgacttgctacca | F-deoxy/PO | 5,688.90 | 5,688.12 | – |
| D8 | FAM-cactcgcaagcaccctatcage | F-deoxy/PO | 6,887.70 | 6,887.40 | S8 |
| D9 | FAM∗-c∗a∗c∗t∗c∗g∗caagcaccctatcage | F-deoxy/7PG | 7,553.75 | 7,555.02 | S9 |
| D10 | FAM∗-c∗a∗c∗t∗c∗g∗c∗a∗agcaccctatcage | F-deoxy/9PG | 7,744.05 | 7,741.80 | S10 |
| D11 | FAM∗-c∗a∗c∗t∗c∗g∗c∗a∗a∗g∗caccctatcage | F-deoxy/11PG | 7,934.35 | 7,932.88 | S11 |
| D12 | FAM∗-c∗a∗c∗t∗c∗g∗c∗a∗a∗g∗c∗a∗ccctatcage | F-deoxy/13PG | 8,124.65 | 8,122.50 | S12 |
| D13 | FAM∗-c∗a∗c∗t∗c∗g∗c∗a∗a∗g∗c∗a∗c∗c∗ctatcage | F-deoxy/15PG | 8,314.95 | 8,310.66 | S13 |
| D14 | FAM∗-c∗a∗c∗t∗c∗g∗c∗a∗a∗g∗c∗a∗c∗c∗c∗t∗atcage | F-deoxy/17PG | 8,505.25 | 8,502.76 | S14 |
| Oligonucleotides targeted to MDR1 mRNA (M series)f | |||||
| M1 | gtccagccccatgga | deoxy/PO | 4538.05 | 4537.11 | S15 |
| M2 | GUCCAGCCCCAUGGA | 2′-OMe/PO | 4960.37 | 4969.52 | S16 |
| M3 | gStScScSaSgScScScScSaStSgSgSa | deoxy/PS | 4762.93 | 4766.72 | S17 |
| G1 | GSUSCSCSaSgScScScScSaSUSGSGSA | 2′-OMe/PS [deoxy/7PS] 2′-OMe/PS | 4975.09 | 4978.32 | S18 |
| G2 | G∗U∗C∗CagccccaU∗G∗G∗A | 2′-OMe/3PG [deoxy/7PO] 2′-OMe/3PG | 5,321.07 | 5,319.92 | S19 |
| G3 | G∗U∗C∗C∗agcccca∗U∗G∗G∗A | 2′-OMe/4PG [deoxy/7PO] 2′-OMe/4PG | 5,511.37 | 5,510.72 | S20 |
| G4 | G∗U∗C∗CSaSgScScScScSaSU∗G∗G∗A | 2′-OMe/3PG [deoxy/7PS] 2′-OMe/3PG | 5,449.59 | 5,448.32 | S21 |
Deoxyribonucleotides are in lowercase letters, 2′-OMe ribonucleotides are in capital letters; S, phosphorothioate group; ∗1,3-dimethylimidazolidin-2-imine (Dmi) group.
ON1, 5′-agtctcgacttgctacc-3′.
ON2, 5′-agucucgacuugcuaccuca-3′.
ON3, 5′-ctccgaagaaataagatcc-3′.
ON4, 5′-gacatccattcaaatggtttg-3′.
ON5, 5′-cactcgcaagcaccctatcag-3′. ON1-ON5, oligonucleotides with random sequences having no complementarity with nucleic acids in eukaryotic cells.
ON6, oligonucleotide 5′-gtccagccccatgga-3′ targeted to MDR1 mRNA.
All oligonucleotides of the D series contained a fluorescein residue (FAM) at the 5′ end.
MDR1 mRNA was selected as a target for ASOs of M series. The human gene MDR1—also known as ATP-binding cassette subfamily B member 1 (ABCB1) —belongs to subfamily B of the ABC transporters and encodes Pgp, which is involved in active ATP-dependent efflux of cytotoxic agents out of the cells. MDR1 overexpression leads to the development of a multiple drug resistance phenotype of tumor cells.39 M series ASOs were designed to target the 319–333 nt region of MDR1 mRNA and to be complementary to all four MDR1 splice variants that occur in Homo sapiens. This region of MDR1 mRNA was chosen as a target for oligonucleotides of M series. Our previously obtained results showed that ASOs targeted just to this region bound with MDR1 mRNA with high efficiency, and PO ASOs bearing a bis-pyrenyl group at the 5′ end and siRNA caused efficient silencing of MDR1 mRNA.40,41
The M series included control oligonucleotides with a uniform structure of the backbone: DNA PO (M1), 2′-OMe RNA PO (M2), and DNA PS (M3) (Table 1). Antisense gapmers G1, G2, G3, and G4 all had a 7 nt deoxynucleotide central window, either PO (G2 and G3) or PS (G1 and G4), flanked by either PS or PG 2′-OMe wings 3-4 nt in length (G2, G4, and G3, respectively, Table 1). The composition of oligonucleotides was designated as follows, using oligonucleotide G1 as an example: 2′-OMe/PS (5′-wing) [deoxy/7PS] (central window) 2′-OMe/PS (3′-wing).
These three sets of oligonucleotides allowed us to investigate in detail the impact of the PG modification on the key factors determining biological potential of ASOs, including: (1) the compatibility of the PG group with the PS linkage during automated solid-phase synthesis; (2) the stability of modified ASOs in biological media or in solutions simulating these conditions; (3) the impact of PG modifications on intracellular accumulation of modified ASOs pre-complexed with cationic liposomes or in a carrier-free mode; (4) the ability of PG-gapmers to recruit RNase H upon binding with the RNA target; and (5) the gene-silencing activity of PG-gapmers.
Synthesis of oligonucleotides
Oligonucleotides with PG groups were synthesized according to a previously developed protocol of automated solid-phase synthesis by the phosphoramidite method.30 The standard aqueous iodine oxidation of the intermediate phosphite to phosphate was replaced by the Staudinger reaction of the phosphite with commercially available Kitamura's reagent (2-azido-1,3-dimethylimidazolinium hexafluorophosphate [ADMP]).42,43 This protocol introduced the Dmi group onto the internucleotidic phosphate. One of the findings of this study is that the Staudinger reaction could be successfully carried out in the presence of PS groups in the oligonucleotide. Thus, the two phosphate modifications, PS and PG, can be introduced independently during the oxidation step of the automated solid-phase synthesis cycle without interference from each other. By using commercially available deoxy- and 2′-OMe ribonucleoside phosphoramidites and three different P(III) to P(V) conversion procedures, namely, oxidation to PO, thionation (sulfurization) to PS, and guanylation (Staudinger reaction) to PG, it was possible to obtain a variety of gapmers having both sugar (deoxy- and 2′-OMe ribonucleotide) and phosphate (PO/PS/PG) modifications on a commercial automated DNA/RNA synthesizer (Table 1). The standard sulfurizing reagent DDTT was used to introduce the PS modification.44 Standard post-synthetic processing, such as cleavage from support, protecting group removal, and reverse-phase high-performance liquid chromatography (HPLC) purification based on the “DMTr ON” mode was analogous to that of native oligonucleotides irrespective of the pattern of modification. Molecular masses of the oligonucleotides were confirmed by mass spectrometry (MS) and their integrity by electrophoretic analysis (Table 1; Figures S1–S23). MS analysis of reaction mixtures of gapmer oligonucleotides G1-G4 (see Figures S18–S21) showed that in all cases the main products correspond to desired composition and sequence. This confirms a good compatibility of both oxidation protocols with each other.
The introduction of a single Dmi group into a DNA chain does not significantly affect hybridization properties of the modified oligonucleotide with respect to either DNA or RNA. Under physiological conditions (100 mM Na+, 10 mM Mg2+), the thermal stability of the modified duplexes was only slightly lower than that of the native duplexes,35,36,45 although there were some slight fluctuations in the melting temperature (Tm) depending on the type of the template (RNA or DNA) and the position of the Dmi group within the oligonucleotide. The Tm for the duplexes formed with poly(rA) were 48°C for the oligonucleotide 5′-d(T19∗T) with the Dmi group nearest to the 3′ end, and 46.5°C for 5′-d(T11∗T9) with the Dmi group located in the middle. For the complexes of the same two oligonucleotides formed with poly(dA), the Tm were 54°C and 52.5°C, respectively; these values are similar to the Tm of the duplexes formed by unmodified dТ20 with the same templates: 48°C and 55°C, respectively.36
Stability of modified oligonucleotides in biological medium
The nuclease resistance of a modified ASO is one of the key factors underlying its biological activity.46 The stability of the R series oligonucleotides was assessed in DMEM in the presence of 50% FBS for 21 days. Nuclease resistance of PGOs R3 and R6 was compared with that of the deoxy/PO, deoxy/PS, 2′-OMe/PO, and 2′-OMe/PS oligonucleotides (R1, R2, R4, and R5, respectively). The products of degradation of ASOs R1, R2, R4, and R5 were analyzed by denaturing PAGE electrophoresis. Due to the charge-neutral character of PGOs R3 and R6, the products of their degradation were analyzed by liquid chromatography-electrospray ionization-tandem MS (LC-ESI MS/MS). Half-life times (τ½) are presented in Table S1.
As expected, oligonucleotides with PO groups—deoxy R1 and 2′-OMe R4—were the least stable, with τ½ values of 10 and 70 min, respectively. Introduction of the PS modifications both in the deoxy (R2) or 2′-OMe (R5) backbone considerably increased the stability of the oligonucleotides: R2 has a τ½ of 10 h, whereas R5 remained stable more than 24 h (Table S1). Deoxy/PG (R3) and 2′-OMe/PG (R6) PGOs both were characterized by high stability and remained intact for over 21 days (Table S1). Thus, stability of PG oligonucleotides exceeded many times the stability of 2′-OMe/PS oligonucleotide R5 (24 h). Thus, nuclease resistance of modified oligonucleotides increased in the order R2 (deoxy/PS) < R5 (2′-OMe/PS) << R3 (deoxy/PG) ≈ R6 (2′-OMe/PG). In other words, modification of internucleotidic positions in oligonucleotides by PG groups increases their stability in biological medium considerably more than that by the PS group, an outcome that correlates with recorded biological activity of PGOs.33,34
Effect of PG modification on oligonucleotide accumulation in eukaryotic cells
Accumulation of D series oligonucleotides in MDCK, KB-8-5, and A549 cells was studied under carrier-free conditions or in the presence of cationic liposomes. All oligonucleotides of the D series were labeled with fluorescein residue (FAM) at the 5′ end. The phosphate linkage introduced via the fluorescein phosphoramidite onto the 5′ end has been converted into the same modification as in the internucleotidic positions: PS for D1–D6, PO for D7 and D8, and PG for D9–D14 (Table 1).
Intracellular oligonucleotide uptake was monitored by flow cytometry. The percentage of the FAM-positive cells and the cell-associated fluorescence intensity were evaluated and are presented as uptake efficiency (UE) for carrier-free studies or transfection efficacy (TE) for liposome-mediated delivery and relative fluorescence units (RFU). The flow cytometry images used for evaluation of UE and TE are shown in Figures S25–S27.
Overall, the oligonucleotide sequence significantly affected their penetration into the cells under carrier-free conditions. Of the three different oligonucleotides tested—D1-D3 (all 2′-OMe/PS) —only D3 (21 nt long) efficiently accumulated in A549 cells: the UE of D3 reached 80% at the fluorescence intensity of 14.1 RFU, whereas D1 and D2 exhibited moderate levels of intracellular accumulation even at a 5 μM concentration (Figure 2A; Table S2). Introduction of two PG groups into D3 to produce D4 did not affect the accumulation of the 2′-OMe/PS oligonucleotide in the cells: there was no change in the values of either TE or RFU. Taking into account that the lengths of oligonucleotides differ only by one nucleotide (20, 19, and 21 nt for D1, D2, and D3, respectively) we cannot suggest that the length of the oligonucleotide accounts or impacts the uptake.
Figure 2.

Intracellular accumulation of fluorescein-labeled D series oligonucleotides in eukaryotic cells mediated by Lipofectamine 2000 or under carrier-free conditions
(A) Accumulation of D1-D4 in A549 cells in the absence of transfection agents. (B) Lipofectamine 2000-mediated delivery of D1-D4 in A549 and MDCK cells. (C) Intracellular accumulation of D5-D7 in A549 and MDCK cells under carrier-free conditions. (D) Lipofectamine 2000-mediated delivery of D5-D7 in A549 cells and MDCK cells. Under carrier-free conditions A549 cells and MDCK cells were incubated in the presence of oligonucleotides D1-D4 at concentrations of 0.1–5 μM or oligonucleotides D5-D7 at concentrations of 5 μM for 4 h. In the case of liposome-mediated delivery A549 and MDCK cells were incubated with complexes formed by oligonucleotides D5-D7 (1 μM) with Lipofectamine 2000 for 4 h. The percentage of fluorescent cells was measured by flow cytometry 4 h post-transfection. Data are presented as mean ± SEM. The data were statistically processed using Student's t test (two-tailed, unpaired); a p value of ≤0.05 was considered to indicate a significant difference; ∗∗data were statistically insignificant. All experimental points were run in triplicate for statistical analysis. C1, control, cells incubated without oligonucleotides; C2, control, cells incubated without Lipofectamine 2000; C(LF), cells incubated with Lipofectamine 2000.
Oligonucleotides D1–D4 in complexes with Lipofectamine 2000 demonstrated similar levels of accumulation in both cell lines (Figures 2B and S25). It should be noted that accumulation of D1–D4 was more efficient in A549 cells both in terms of TE and fluorescence intensity. However, because the level of accumulation under carrier-mediated conditions was sufficiently high for all the oligonucleotides, the obtained data allowed no unequivocal conclusion as to whether the introduction of two PG modifications contributed to the penetration of D4 into the cells. Oligonucleotides D1–D4 did not affect the viability of A549 cells at a concentration of 1 μM in the medium and incubation for 24 h (Figure S22).
A study of intracellular accumulation of oligonucleotides D5–D7 in A549 and KB-8-5 cells either mediated by Lipofectamine 2000 or under carrier-free conditions revealed that the completely charge-neutral PGO D6 (deoxy/PG) did not penetrate the cells in either conditions (Figures 2C, 2D, S26, and Table S2). In the absence of a transfection agent, only D5 (2′-OMe/PS) at the concentration of 1 μM moderately accumulated in both cell lines. The addition of Lipofectamine 2000 stimulated the delivery of negatively charged D5 and D7 but not that of PGO D6, which presumably was unable to form lipoplexes due to its charge-neutral character (Figures 2D, S26, and Table S2). Thus, the replacement of all internucleotidic phosphates in an oligodeoxynucleotide D6 by PG groups abolished its accumulation in the cells in the presence of cationic liposomes and at the same time did not improve its cellular uptake under carrier-free conditions compared with unmodified (PO) control D7.
The ability of oligodeoxynucleotides to enter cells was studied depending on the number of PG modifications. In the absence of a transfection agent, oligonucleotides D9–D14 as well as unmodified D8 accumulated in the cells poorly: the amount of FAM-positive cells did not exceed 20%, and fluorescence intensity reached a maximum of 1.9 RFU (Figures 3A, 3B; Table S2). Analysis of cellular uptake of oligodeoxynucleotides mediated by 2X3-DOPE showed that a gradual replacement of internucleotidic phosphates by PG groups decreased the percentage of fluorescent cells in the following order: D9 (83.4% ± 3.5%) ≈ D10 (88.9% ± 4%) ≈ D11 (80.1% ± 2.6%) > D12 (65.2% ± 5.2%) >> D13 (8.4% ± 4.2%) >> D14 (%1 ± 0.5%), whereas the control D8 (deoxy/PO) efficiently accumulated in A549 cells (98.9% ± 1%) (Figures 3B and S27; Table S2). Fluorescence intensity was also considerably decreased with the increase in the number of PG modifications. Indeed, a nearly 10-fold decline in RFU was observed when seven PG groups were introduced into the sequence (D9) compared with the unmodified control (D8): 59.5 RFU for D8 and only 4.3 RFU for D9 (Figure 3B; Table S2).
Figure 3.

Effect of the number of PG groups in oligonucleotides on their accumulation in A549 cells
(A) Under carrier-free conditions. (B) In the presence of cationic liposomes 2X3-DOPE. A549 cells were incubated with oligonucleotides D8-D14 alone at 5 μM or complexed with liposomes 2X3-DOPE at 1 μM for 4 h. The percentage of fluorescent cells was measured by flow cytometry 4 h post-transfection. Data are presented as mean ± SEM. The data were statistically processed using Student's t test (two-tailed, unpaired); a p value of ≤0.05 was considered to indicate a significant difference; ∗∗data were statistically insignificant. All experimental points were run in triplicate for statistical analysis. C1, control, cells incubated without oligonucleotides; C2, control, cells incubated without liposomes; C(L), cells incubated in the presence of liposomes 2 × 3/DOPE.
Thus, under carrier-free conditions the oligonucleotide D4 (PS/2′-OMe/2PG) penetrated into the cells with efficiency similar to D3 (2′-OMe/PS), which has the same sequence, or D5 (2′-OMe/PS), which has a different sequence. However, in the oligodeoxynucleotide series D9–D14, the increase in the number of PG modifications from 7 to 14, respectively, drastically decreased the level of their intracellular accumulation in the carrier-free mode and ability to form lipoplexes. Nevertheless, in the complexes with 2X3-DOPE, partially PG-modified oligodeoxynucleotides D9, D10, and D11 with 7, 9, and 11 PG modifications, respectively, accumulated in the cells with moderate efficiency (Figure 3B).
It should be emphasized that in D4 two PG groups are located at the 3′ end of this ASO, while in D9-D14 ASOs PG modifications were placed at their 5′ end; for D9-D14 ASOs the increase of the number of PG groups ultimately leads to a drop in the effectiveness of their penetration into cells. Considering these data, we cannot exclude the possibility that the location of PG modification (5ʹ or 3ʹ part of ASO) might play a role in intracellular accumulation of ASOs.
In the extreme cases of a completely PG-modified D6 (compare D5 and D6) and the nearly completely modified D14 (compare D8 and D14), it may be concluded that the replacement of all internucleotidic phosphates by PG groups in an oligodeoxynucleotide may entirely abolish their ability to penetrate into eukaryotic cells in the form of lipoplexes while also not improving their carrier-free uptake.
PG-containing antisense gapmers recruit RNase H
Our results indicated that the optimal number of PG modifications providing balance between nuclease resistance and cellular uptake has to be determined experimentally. A reasonable approach was to obtain gapmer oligonucleotides with 3′ and 5′ wings protected by three to four PG-modified 2′-O-methyl ribonucleotides and the central window of six to eight PO or PS deoxynucleotides (no charge-neutral PG groups in this section) to stimulate their incorporation into lipoplexes and to form a heteroduplex with RNA capable of recruiting RNase H. The M series oligonucleotides (M1-M3, G1-G4) targeting MDR1 RNA was synthesized accordingly (Table 1).
To prove that PG-gapmers targeting the 319–333 nt region of human MDR1 mRNA were indeed able to recruit RNase H, we performed experiments with a 678 nt fragment of MDR1 mRNA obtained by in vitro transcription (hereinafter referred to as MDR1 mRNA), which included the target region. Oligonucleotides M1-M3 and G1-G4 were hybridized with MDR1 mRNA followed by incubation with RNase H. RNA cleavage products were analyzed by 12% PAGE under denaturing conditions (Figure 4A). Oligo-2′-O-methylribonucleotide M2, which is unable to recruit RNase H, was used as a negative control (no hydrolysis in lane 1, Figure 4A). Both unmodified (PO) M1 and its PS counterpart M3, which formed heteroduplexes with MDR1 mRNA that recruited RNase H, were used as positive controls. A typical pattern of MDR1 mRNA cleavage in a heteroduplex with M1 and M3 by RNase H is shown in Figure 4A, lanes 2 and 3, respectively. The gapmer G1 (2′-OMe/PS[deoxy/7PS]2′-OMe/PS, Table 1) formed a heteroduplex recognized by RNase H. The cleavage pattern observed with G1 was similar to those observed for M1 and M3 (lane 4, Figure 4A). Correspondingly, the bands in lanes 5, 6, and 7 (Figure 4A) provide evidence for RNase H-mediated RNA cleavage of heteroduplexes formed by PG-gapmers G2, G3, and G4. Notably, the cleavage pattern depends not only on the nucleotide modifications within the central window, but on the number of PG modifications in the wings: the same cleavage patterns were observed for PG-gapmers G2 and G4, which both contain three PG groups at either the 5′ or 3′ ends (lanes 5 and 7, Figure 4A), but in the case of PG-gapmer G3, which contains four PG modifications in both wings, the cleavage pattern is slightly altered, possibly because additional PG linkages narrow the window recognized by RNase H (lane 6, Figure 4A).
Figure 4.

RNase H cleavage of the MDR1 RNA complexed with ASOs
(A) Autoradiograph of a 12% polyacrylamide/8 M urea gel. Lanes Im and T1, imidazole ladder and partial RNA digestion with RNase T1, respectively; C, control, RNA incubated in the absence of ASOs and RNase H. Lanes 1–5 (left panel), lanes 6 and 7 (right panel), RNA/ASO incubated with RNase H: 1, RNA/M2; 2, RNA/M1; 3, RNA/M3; 4, RNA/G1; 5, RNA/G2; 6, RNA/G3; 7, RNA/G4. MDR1 RNA (0.05 μg) was pre-complexed with 10 μM ASO at 37°C for 1 h and incubated with RNase H (1 U/mL) at 37°C for 30 min. (B) Secondary structure of the 678 nt fragment of MDR1 RNA. The ASO binding site is indicated in blue. Spontaneous cleavages at CA and UA sites of MDR1 RNA are indicated by red arrows.
It should be noted that, since MDR1 mRNA is a long transcript with pronounced secondary structure, the binding of an oligonucleotide with RNA leads to the reorganization of molecule structure. As the result the most labile linkages C-A and U-A in RNA are undergoing spontaneous cleavage after RNA unfolding, and we observed cleavage not only in the site of ASO binding provided by RNase H but also at C-A and U-A linkages due to spontaneous hydrolysis (Figure 4B).
Biological effects of M series ASOs in human KB-8-5 cells
The M series ASOs restore the sensitivity of KB-8-5 cells to vinblastine
To study the biological activity of the M series ASOs targeted to human MDR1 RNA, we used the human epidermoid carcinoma cell line KB-8-5, which exhibits high resistance to chemotherapeutics due to overexpression of Pgp encoded by MDR1 mRNA. KB-8-5 cells were transfected with ASOs pre-complexed with Lipofectamine 2000. 24 h post-transfection, vinblastine was added and the cells were incubated for 3 days. The ability of ASOs to increase the sensitivity of KB-8-5 cells to vinblastine was evaluated by real-time cell survival monitoring. PO oligonucleotides R1 and M1, PG-gapmer G2 with a PO window, and PS oligonucleotides R2, M3, and PG-gapmer G4 with a PS window were compared in two parallel experiments. The concentrations were 1 μM for PO-modified (R1, M1, and G2) and 0.1 μM for PS-modified (R2, M3, and G4) oligonucleotides. The concentration of the PS-modified oligonucleotides was lowered due to their relatively high non-specific toxicity to KB-8-5 cells observed in the presence of vinblastine. It should be mentioned that no toxic effects of gapmer oligonucleotides were observed when the cells were incubated with G2 and G4 in the absence of vinblastine (Figure S28). The full-PS-gapmer G1 was excluded from the experiments due to its especially high cytotoxicity under these conditions. The fully 2′-O-methylated oligoribonucleotide M2 was also excluded because its heteroduplex with RNA was not recognized by RNase H rendering M2 ineffective in this case. The PG-gapmer G3 containing four PG linkages in both wings did not affect the sensitivity of KB-8-3 cells to vinblastine at either 0.1 or 1 μM concentration (primary data not shown).
The control R1 with random sequences had no influence on cell viability (Figure 5A). Deoxyribonucleotide M1 at concentration 1 μM decreased 1.2-fold cell viability in the presence of vinblastine (LF versus M1 p = 0.025): cell proliferation index for M1 was 1.86 versus 2.3 for LF (Figure 5A). The gapmer G2 (2′-OMe/3PG[deoxy/7PO]2′-OMe/3PG) at the same concentration increased the sensitivity of the cells to vinblastine 2-fold (LF versus G2 p = 0.0006): cell proliferation index for G2 was 1.66 versus 2.3 for LF (Figure 5A). It should be noted that the effect of ASO M1 on cell viability differ statistically insignificantly from the effect of R1, whereas the effect of G2 was statistically significant with p = 0.008 in comparison with R1. Nevertheless, no statistically significant difference between M1 and G2 was observed.
Figure 5.

The biological effect of modified ASOs targeted to MDR1 mRNA in KB-8-5 cells
(A and B) Real-time analysis of the effect of modified ASOs on the growth rate of KB-8-5 cells in the presence of vinblastine. Cells were transfected with 1 μM of oligonucleotides R1, M1, or G2, or 0.1 μM of oligonucleotides R2, G4, or M3 pre-complexed with Lipofectamine 2000 for 4 h; LF, cells treated with Lipofectamine 2000 only. Cell viability was recorded in real time using an xCELLigence instrument for 96 h. Vinblastine was added to the cells to a concentration of 300 nM 24 h post-transfection, and the cells were cultured in an atmosphere of 5% CO2 at 37°C for 96 h. The results are shown as mean cell index ± SE. The data were statistically processed using Student's t test (two-tailed, unpaired); a p value of ≤0.05 was considered to indicate a significant difference; ∗∗data were statistically insignificant. (C and D) Analysis of the effect of modified ASOs on the level of MDR1 gene expression. Data of qPCR. KB-8-5 cells were transfected with ASOs R1, M1, G2 (0.5 μM) or R2, M3, G4 (0.1 μM) pre-complexed with Lipofectamine for 4 h and incubated for 24 and 48 h. LF, cells treated with Lipofectamine 2000 only. The expression level of MDR1 mRNA was normalized to the level of HPRT1 mRNA. (E and F) Western blot analysis of Pgp 72 h after transfection. β-Actin served as an internal control. Cells were transfected with oligonucleotides R1 (0.5 μM), M1 (0.5 μM), M3 (0.1 μM), G4 (0.1 and 0.5 μM), and G2 (0.5 μM) pre-complexed with Lipofectamine 2000 for 4 h and incubated for 72 h; LF, cells treated with Lipofectamine 2000 only. (D) The bar graph shows the semi-quantitative analysis of the western blot results for Pgp. Data of qPCR and western blot were statistically processed using one-way ANOVA with the Tukey post hoc test; p < 0.05 was considered to be statistically significant; ∗∗data were statistically insignificant. The data are presented as the mean three independent experiments with triplicate samples ±SEM.
A different pattern was observed in the case of oligonucleotides with PS linkages (Figure 5B). Both the full-PS oligodeoxynucleotide R2, with a random sequence, and the full-PS match M3 caused 50% reduction of cell survival similar to the gapmer G4 (2′-OMe/3PG[deoxy/7PS]2′-OMe/3PG) at a concentration of 0.1 μM and the differences were statistically significant compared with control cells treated with Lipofectamine: LF versus R1 p= 0.007, LF versus M3 p = 0.003 and LF versus G4 p = 0.007 (Figure 5B). However, we did not find a statistically significant difference between R2 and G4 as well as M3 and G4. The study of cytotoxicity of oligonucleotides D1-D4, including PS/2PG D4, revealed that all oligonucleotides were not toxic to the A549 cells within the concentration range of 0.1–1 μM (Figure S22). Thus, at the concentration of oligonucleotides used in this study, no decrease in cell viability due to PS-mediated toxicity was expected. However, similar values of cell survival observed in the presence of the PS ASOs R2 and M3 and the PG/PS-gapmer G4 may indicate the non-specific contribution of PS modification to the overall toxic effect on the cells. The presence of vinblastine seemed to enhance the non-specific cytotoxicity of PS oligonucleotides and mask the antisense silencing of MDR1 gene.
To compare the specific inhibition of cell viability by PG/PO-gapmer G2 and non-specific inhibition by the control random oligonucleotide R1, we calculated the G2/LF and R1/LF cell indexes. The values were 0.72 and 0.95, respectively, which indicate the specific contribution of antisense G2 to the inhibition of cell viability compared with the random oligonucleotide R1. However, in the case of the PS-modified G4 and R2, the G4/LF and R2/LF ratios were 0.75 and 0.77, respectively, which highlights the pronounced cytotoxicity of PS modification in the presence of vinblastine.
Downregulation of Pgp expression by the M series ASOs
Restoration of the sensitivity of KB-8-5 cells to vinblastine by PG-gapmers G2 and G4 can be explained by downregulation of Pgp, which mediates the drug resistance of tumor cells. The changes in the MDR1 mRNA levels under the action of ASOs were studied using qRT-PCR. For these purposes KB-8-5 cells were incubated in the presence of PO/PG- (0.5 μM) or PS/PG-modified ASOs (0.1 μM) pre-complexed with Lipofectamine at 37°C for 24 and 48 h (Figures 5C and 5D). ASOs R1 and R2 did not cause any changes in MDR mRNA level (Figures 5C and 5D). In the case of PO/PG-modified ASO G2 we observed a tendency to decrease MDR1 mRNA levels, but data were statistically insignificant (Figure 5C). The PS/PG-modified ASOs much more efficiently downregulate MDR1 mRNA in comparison with PO/PG-modified ASOs: a 30% decrease in the MDR1 mRNA level was observed for PS-modified M3 in comparison with Lipofectamine and R2 groups (Figure 5D). Gapmer G4 decreased MDR1 mRNA levels even more efficiently, reaching 50% downregulation (Figure 5D). It should be noted that gapmer G4 exhibited much higher silencing activity with respect to MDR1 mRNA in comparison with M3 (PS ASO) and this difference was statistically significant (Figure 5D).
To determine the alteration in the Pgp levels, western blot analysis was performed 72 h after KB-8-5 cells had been transfected with 0.1 or 0.5 μM of oligonucleotides R1, M1, or M3, or PG-gapmers G2, or G4 (Figures 5E and 5F). There was no statistically significant difference in the level of Pgp in tumor cells incubated with Lipofectamine 2000 and random oligonucleotide R1 at a concentration of 0.1 μM. The PO ASO M1 at 0.5 μM caused a 2-fold reduction in the Pgp level in comparison with Lipofectamine and R1 groups (Figure 5F). These data are in agreement with data on the decrease of cell viability in the presence of M1 (Figure 5A), although we did not observe a decrease in MDR1 mRNA levels. It can be assumed that the time points chosen for the analysis of both mRNA and Pgp levels are not optimal for the analysis of mRNA. It is known that the half-life of MDR1 mRNA is 4–9 h, while the half-life of Pgp is 48–72 h.47, 48, 49
Since full-PS M3 at a concentration of 0.5 μM exhibited high toxicity to KB-8-5 cells, the Pgp level for M3 at this concentration was not measured. At an M3 concentration of 0.1 μM, a reduction of the Pgp level by 1.7-fold was observed (Figure 5F). PG/PS-gapmer G4 was the most potent inhibitor of Pgp synthesis, causing a 2-fold reduction in the Pgp level at 0.1 μM and a 10-fold reduction at 0.5 μM (Figure 5F). These data are in a good agreement with the data on the decrease of MDR1 mRNA level and restoration of cell sensitivity to vinblastine observed for this gapmer (Figures 5B and 5D).
As shown above, PG/PO-gapmer G2 (1 μM) decreases the viability of KB-8-5 cells in the presence of vinblastine, which occurs due to silencing of MDR1 mRNA followed by a decrease in Pgp levels and restoration of KB-8-5 cell sensitivity to the cytotoxic drug vinblastine. Despite the restoration of cell sensitivity to vinblastine we did not detect statistically significant effect of G2 on the Pgp levels (Figure 5F), while G2 decreased the level of MDR1 RNA by 30% at the 24 h time point (Figure 5C). The reasons for this could be a 2-fold lower concentration of G2 used in these experiments, and the somewhat lower half-life of G2 in comparison with G4. Two reasons can explain the efficient Pgp silencing by G4. First, G4 is a PG/PS-gapmer, and the decrease in the number of PS linkages may reduce non-specific cytotoxicity as G4 worked efficiently at both 0.1 and 0.5 μM. Second, the experiments on downregulation of Pgp were carried out in the absence of vinblastine, which may have decreased the overall toxicity burden on the cells (compare Figures 5B and 5F).
Discussion
ASOs were first shown to inhibit protein expression at the mRNA level over four decades ago.1 To date, a number of drugs based on ASOs and small interfering RNA (siRNA) have been approved for clinical use.50,51 However, widespread development of clinically successful oligonucleotide drugs is still being slowed down by their often lower than expected therapeutic efficacy in humans.46,52, 53, 54 Therefore, the search for novel chemical modifications to improve clinical prospects of therapeutic oligonucleotides remains of great relevance.
Recently, we have reported on the promising properties of a novel class of charge-neutral DNA analogs, PGOs (Figure 1C),29 and then developed a facile way for their automated synthesis based on a solid-phase version of the Staudinger reaction with 1-azidocarbamidinium compounds,30 such as ADMP.42,43 The Staudinger chemistry that was first employed to obtain oligonucleotides with the internucleotidic phosphoramidate group in the 1970s,55 proved to be nearly as efficient as the current sulfurization chemistry for the synthesis of PS oligonucleotides,44 which enabled us to obtain a range of various PGOs including the 2′-OMe RNA analogs.33,34 In this work, we demonstrate the compatibility of Staudinger chemistry for PGOs with the standard automated phosphoramidite protocol for not only PO but, more importantly, for PS synthesis, which allows one to easily create diverse gapmer structures with alternating internucleotidic PO, PS, and PG linkages, and a mix of deoxyribose and 2′-O-methylribose and, very likely, other 2′-substituted sugars, e.g., LNA, etc. (see Figure S21 for ESI MS of the PS/PG-gapmer G4). Similar compatibility of the mesyl phosphoramidate chemistry with the PS protocol was shown recently by others.56
We describe herein the novel antisense PG-gapmers, which efficiently downregulate a target mRNA in cell culture. In this study, we demonstrate that optimal combinations of the PG modifications can be achieved when some of the PS linkages responsible for both antisense activity and some off-target toxicity were replaced by PG groups, resulting in the exceedingly stable and less-toxic PG-gapmers that retain high biological activity.
Introduction of a charge-neutral PG linkage, such as the Dmi group (Figure 1C), into an oligonucleotide affects its chemical and biological properties.57,58 Because of their chemical properties, PGOs are a close equivalent of the well-known morpholinos (PMOs), which are also charge neutral.18 Thus, PGOs, similarly to PMOs, also belong in the family of oligonucleotide analogs that act by steric block of translation via an RNase H-independent mechanism.33,59 One of the important characteristics of the PG modification is the near lack of effect on the thermal stability of PGO:RNA duplexes. The thermal stability of the duplex formed by PGO with RNA was only slightly lower than of the native DNA:RNA duplex, while the thermal stability of PG-gapmer:RNA duplexes was similar to that of the DNA:RNA duplex,36 unlike PMOs, which exhibit higher binding affinity to RNA targets.18 The unaffected thermal stability of PG-gapmer:RNA duplexes may be considered an advantage of these oligonucleotides because it provides faster dissociation of a PG-gapmer from the complex with target RNA after RNase H-mediated RNA cleavage in comparison with other RNase H-triggering ASOs.
The duplexes of completely charge-neutral PGOs with RNA were not recognized as substrates by RNase H, presumably due to the steric bulk and lack of charge of internucleotidic PG groups. Of note, these PGOs could still participate in a steric block antisense mechanism.33 Our data show that PG-gapmers containing three to four PG groups at both the 3′ and 5′ ends of the oligonucleotide and the central window of seven deoxy/PS-nucleotides were able to recruit RNase H upon binding to MDR1 mRNA. It should be noted that RNase H displays different cleavage patterns depending on the number of PG groups flanking the central window of the gapmers. The cleavage pattern of RNase H in the complex of the RNA with the G4 gapmer containing three PG groups at either end and a 7 nt PS window, as well as in the complex with the G2 gapmer, which had a 7 nt PO window, was shifted downward compared with the cleavage pattern in the complex with the G3 gapmer containing four PG groups at both ends and the same 7 nt PO window. Despite the need for RNase H to bind to the heteroduplex at a site that is no more than four nucleotides,60 the bulky PG modifications can shift the cleavage frame. However, the slight difference in the cleavage pattern of RNase H with different gapmers did not affect their antisense activity within cells as we observed a decrease of the level of Pgp under the action of the PG-gapmers followed by a decrease of tumor cell viability in the presence of vinblastine.
We showed that PGOs display much higher nuclease resistance in serum-containing medium than is usually expected for PS oligonucleotides. Furthermore, the number and position of PG groups had an impact on the nuclease resistance of partially PG-modified oligonucleotides. Our data indicate that introduction of PG groups at the ends of a gapmer oligonucleotide is more beneficial for nuclease resistance and prolonged action within cells as compared with PS groups. Earlier it was demonstrated that the introduction of a single PG group into a trinucleotide increased its resistance to SVPDE.36
One may expect that neutralization of the negative charge of an oligonucleotide may positively influence the cellular uptake in the case of PGOs. Our data showed that a fully charge-neutral deoxy-PGO penetrated eukaryotic cells no better than the unmodified counterpart. A similarly poor cellular uptake was observed for the naked PMOs and PNAs, which can be considered conceptually similar to PGO due to inherent lack of negative charge.61 However, unlike PNAs or PMOs, the PG groups could be introduced into an oligonucleotide in a gradual way, replacing the phosphates one by one to produce partially charged oligomers. For example, the introduction of just two PG linkages into an oligo-2′-OMe PS ribonucleotide did not interfere with carrier-free intracellular accumulation. The results indicate that that optimal balance between the efficiency of cellular uptake and nuclease resistance of ASOs could be achieved by introducing a limited number of charge-neutral linkages such as PG modifications. Despite the hydrophobic nature of the Dmi group, it does not significantly interfere with the packaging of partially (but not fully) PG-modified oligonucleotides into lipoplexes with cationic liposomes. Thus, even though lipoplex formulations were shown to be unsuitable for the delivery of charge-neutral oligonucleotide analogs, such as PMOs, PG-gapmers packed in liposomes efficiently penetrate into the cells.
Both deoxy- and 2′-OMe PG oligonucleotides exhibited low toxicity to eukaryotic cells: no effects on cell viability were observed at a concentration range of 1–5 μM in cell culture medium. At the same time, PS oligonucleotides displayed noticeable cytotoxicity at 1 μM. The toxicity became even more pronounced when cytotoxic compounds, such as vinblastine, were also present in cell culture medium with PS oligonucleotides. It is worth mentioning that the overall toxicity of PS modifications was significantly reduced when PG/PS-gapmer G4 was used instead of a completely PS oligonucleotide; however, the contribution of the PS modifications to the overall non-specific toxic effect on the cells and the presence of vinblastine masked specific antisense downregulation of MDR1 gene by these oligonucleotides (Figure 5). On the contrary, when vinblastine was not present in culture medium, the advantages of PG/PS-gapmers over PS oligonucleotides became apparent (Figure 5C). Specifically, the fully PS M3, which was somewhat effective at 0.1 μM, exhibited high toxicity at 0.5 μM, while PG/PS-gapmer G4 caused a 2-fold reduction of Pgp level at 0.1 μM and a 10-fold reduction at 0.5 μM. Thus, our data reveal that PG/PS-gapmer G4 is an efficient inhibitor of Pgp synthesis: it combines low toxicity and efficient gene silencing. Of note, in tumors with a MDR phenotype, the level of Pgp is increased 2–5 times compared with drug-sensitive tumors.62 As we demonstrate in this study, 2- to 5-fold downregulation of Pgp is achievable by the action of, e.g., a PG/PS-gapmer G4. Hence, the application of such ASOs could significantly increase the efficacy of applied chemotherapy.
It is interesting to compare several types of phosphate modifications, such as charge-neutral PMOs, PGOs studied in this work, which are also charge neutral, and recently developed negatively charged mesyl phosphoramidate (μ) ASOs,25,63 which are also obtained through Staudinger chemistry.64 PG and μ-modifications can be introduced at the internucleotidic position during automated phosphoramidite solid-phase assembly by Staudinger reaction with ADMP and mesyl azide, respectively (Table 2). These oligonucleotides have a number of similarities and differences. Fully or partially PG-modified ASOs are either charge neutral, similarly to PMOs, or carry a decreased negative charge, respectively, while μ-ASOs completely retain a negative charge similarly to natural oligonucleotides. PGOs, PMOs, and μ-ASOs are all able to form stable duplexes with RNA targets. Importantly, all the phosphate modifications significantly increase the nuclease resistance of ASOs, with τ1/2 > 21 days for PGOs and >7 days for μ-ASOs.
μ-ASOs, as well as PG-gapmers, efficiently recruited RNase H and caused up to 4- versus 2-fold downregulation of their RNA targets in vitro, respectively (Table 2), while PMOs and fully charge-neutral PGOs are steric blocking ASOs that work through an RNase H-independent pathway. None of the modifications facilitated cellular uptake of the ASO in a carrier-free mode (Table 2). Nonetheless, when delivered by cationic liposomes, both anti-MDR1 PG-gapmers and anti-miR-21 μ-ASOs efficiently downregulated their targets by an RNase H-dependent mechanism, which led to a decrease of tumor cell proliferation by specific miR-21 inhibition,25 significant reduction of tumor xenografts in nude mice,63 or the reversal of the MDR phenotype of MDR1-expressing cells, as shown in this work (Table 2). Because traditional cationic liposomes did not help PMO uptake, the main strategies for PMO delivery are chemical methods,65 conjugation with cell-penetrating peptides,66 or encapsulation in the so-called bubble liposomes.67 The same may be applicable to fully charge-neutral PGOs, including nanoparticle-mediated delivery.67
Table 2.
Comparison of biological properties of PMO, PGO, μ-ASOs, and PG-gapmers
| Biological properties | μ-ASO | PMO | PGO | PG-gapmers |
|---|---|---|---|---|
| Charge | negative | neutral | neutral | decreased negative |
| Type of studied RNA target | miRNA | mRNA | mRNA | mRNA |
| Thermal stability of the duplex with RNA target | slight destabilization | high stabilization | slight destabilization | not affected |
| Stability in biological media | >7 daysa | highly stable18 | >21 days | N/D |
| Recruitment of RNase H | yes | nob | no33 | yes |
| Cellular uptake in a carrier-free mode | low | somec | low | high for PS/2PG ASO |
| Liposome-mediated cellular uptake | efficient | not efficient68 | not efficient | efficient |
| Cytotoxicity | not found | low69 | low | not found for PO/PG |
| Silencing of RNA target | 2–4 times compared with untreated cells | yes | 2–4 times compared with untreated cells33 | 2 times compared with untreated cells |
The application of gapmeric ASOs for mRNA downregulation has produced notable successes in many experimental studies and clinical trials.22,70,71 These successes have opened new avenues for the therapy of various diseases, including cancer, cardiovascular disease, lung fibrosis, and neurodegenerative and neuromuscular disorders. There have been significant advances in the use of ASOs, deoxyribozymes, and siRNA for the treatment of viral infections caused by RNA viruses, such as hepatitis C virus (HCV), HIV-1, and SARS-CoV; the latter could be very relevant to the current SARS-CoV-2 pandemic.72, 73, 74 A combination of the advantages of such internucleotide modifications as PG and mesyl phosphoramidate (μ) could make it possible to design novel oligonucleotide drugs that target therapeutically important messenger or non-coding RNAs as well as viral RNAs.
Materials and methods
Oligonucleotides preparation
Automated phosphoramidite solid-phase synthesis of all the oligonucleotides was carried out on an ASM-800 DNA/RNA synthesizer (Biosset LLC, Russia). Oligonucleotides were assembled on a 0.2 μmol scale using standard commercial 2-cyanoethyl deoxynucleoside and 2′-OMe RNA phosphoramidites and modified controlled pore glass supports with the respective nucleosides attached (Glen Research, USA). Oligonucleotides with internucleotidic PG (Dmi) groups were synthesized by substituting the Staudinger reaction of polymer-supported dinucleoside 2-cyanoethyl phosphite with ADMP, for conventional iodine oxidation. This chemistry has been described previously.30
Oligonucleotides containing PS linkages were synthesized using 0.1 M 3-((dimethylaminomethylidene)amino)-3H-1,2,4-dithiazole-3-thione (DDTT) from Glen Research (USA) in pyridine–acetonitrile (3:2, v/v) according to the manufacturer's protocol.
Modified and unmodified oligonucleotides were labeled using a commercially available fluorescein modifier, 2-dimethoxytrityloxymethyl-6-(3′,6′-dipivaloylfluorescein-6-yl-carboxamido)-hexyl-1-O-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite (Glen Research, USA) according to the manufacturer's protocols.
Oligonucleotides purification and identification
Natural and modified oligonucleotides were purified as described previously.36 In brief, oligonucleotides were isolated by reverse-phased HPLC on an Agilent 1200 HPLC system (Santa Clara, USA) using a Zorbax SB-C18 5 mm column (4.6 × 150 mm) at a linear gradient of 0%–50% or 0%–90% acetonitrile in 20 mM triethylammonium acetate (pH 7.0), and a flow rate of 2 mL min−1. Fractions containing the desired product were pooled, concentrated in vacuo, dissolved in 0.1 mL deionized water, and precipitated by 1 mL 2% LiClO4 in acetone. After centrifugation at 14,500 rpm for 2 min, washing with acetone, and air drying for 20 min, oligonucleotide pellets were dissolved in deionized water and stored at −20°C. Oligonucleotide homogeneity was assessed by electrophoresis in a 20% PAAG/8 M urea gel with 0.1 M Tris-borate buffer (pH 8.3) as the running buffer. The oligonucleotide bands were visualized in the gel by staining with Stains-All (Sigma-Aldrich, USA). Molecular masses of oligonucleotides were confirmed by LC-ESI-MS/MS on an Agilent G6410A mass spectrometer in the negative ion mode. The samples were prepared by dissolving oligonucleotides in 20 mM triethylammonium acetate in 60% aq. acetonitrile at a concentration 0.1 mM. Positive ion mode was applied for PGOs. The PGOs were dissolved in 20 mM formic acid in 60% aq. acetonitrile at a concentration of 0.1 mM; the total volume was 10 μL. Analysis was carried out using 80% aq. acetonitrile as an eluent, at a flow rate of 0.1 mL/min, and with standard device settings. Molecular masses were calculated using experimental m/z values obtained for each sample.
Stability of modified oligonucleotides in biological medium
The assay was carried out as described previously.75 Oligonucleotides R1-R6 (0.3 nmol) were incubated in 140 μL DMEM supplemented with 50% FBS for 1 min to 24 h (oligonucleotides R1, R2, R4, and R5) and for 21 days (oligonucleotides R3 and R6) at 37°C. The reaction was stopped by addition of an equal volume of phenol (pH 8.0) for oligonucleotides R1 and R2 or phenol (pH 7.0) for oligonucleotides R4 and R5. The reaction was quenched by 1:1 phenol/chloroform (v/v) extraction followed by ethanol precipitation of oligonucleotides. The digestion products of oligonucleotides R1, R2, and R4 were analyzed in 20% denaturing PAAG and stained by Stains-All. The gels were photographed and analyzed using GEL PRO 4.0 software (Media Cybernetics, USA).
The described extraction procedure did not allow isolation of R3 and R6 from the serum-containing samples. Therefore, these oligonucleotides were analyzed after the reaction directly in the samples by LC-ESI-MS/MS on an Agilent G6410A mass spectrometer in the positive ion mode.
Cell cultures
The human epidermoid carcinoma KB-8-5 cell line was purchased from the Institute of Cytology RAS (St. Petersburg, Russia). KB-8-5 cells were grown in DMEM containing 10% FBS and 1% antibiotic-antimycotic solution (10 mg/mL streptomycin, 10,000 IU/mL penicillin, and 25 mg/mL amphotericin, MP Biomedicals, Germany), in the presence of 300 nM vinblastine (Sigma-Aldrich, USA) at 37°C in a humidified atmosphere with 5% CO2.
The MDCK cell line and the human bronchial epithelial carcinoma A549 cell line were obtained from the cell collection of the Institute of Chemical Biology and Fundamental Medicine SB RAS. MDCK and A549 cells were grown in DMEM containing 10% FBS and 1% antibiotic-antimycotic solution at 37°C in a humidified atmosphere with 5% CO2.
Delivery of oligonucleotides into cells under carrier-free conditions and using Lipofectamine 2000/cationic liposomes 2X3-DOPE
The UE for the D series oligonucleotides under transfectant-free conditions and the TE of oligonucleotides pre-complexed with Lipofectamine 2000 or 2X3-DOPE were evaluated in experiments with A549, MDCK, and KB-8-5 cells. Cells were seeded into 24-well plate (5 × 105 cells/well) in serum-free and antibiotic-free DMEM for 30 min before the treatment. Under carrier-free conditions the cells were incubated with oligonucleotides D1-D4 at concentrations of 0.1–5 μM and D5-D7 at concentrations of 5 μM for 4 h in a humidified atmosphere with 5% CO2.
Complexes of oligonucleotides and Lipofectamine 2000 (Thermo Fisher Scientific, USA) were prepared according to the manufacturer's protocol. Complexes of oligonucleotides and cationic liposomes 2X3-DOPE (the latter were a generous gift of Prof. M. Maslov) were formed in serum-free Opti-MEM medium (Thermo Fisher Scientific, USA) via vigorous mixing of liposome solution (1 mM) and oligonucleotides at a concentration of 1 μM; the resulting mixtures were incubated for 20 min at room temperature. ASO/Lipofectamine and ASO/2X3-DOPE lipoplexes were formed at a nitrogen-to-phosphate (N/P) ratio of 6/1 for Lipofectamine and 6/1 and 8/1 for 2X3-DOPE.
Cells were seeded into 24-well plates (5 × 105 cells/well) in serum-free and antibiotic-free DMEM for 30 min before treatment. Then the cells were incubated with ON/2X3-DOPE lipoplexes for 4 h in a humidified atmosphere with 5% CO2. The UE and TE of the fluorescein (FAM)-labeled oligonucleotides were evaluated after a 4-h transfection by flow cytometry.
Flow cytometry
Oligonucleotide delivery to the cells was assayed by flow cytometry using a FC 500 (Beckman Coulter, USA) and data were processed using CXP Analysis software (Applied Cytometry Systems, UK) as described in.76 Cells were mechanically detached from the plate using a scraper and collected by centrifugation at 1,000 rpm for 5 min. The cells were washed with phosphate-buffered saline (PBS) and fixed with 4% formaldehyde in PBS. The resulting samples were assayed by flow cytometry using a FC 500 (Beckman Coulter, USA) and data were processed using CXP Analysis software (Applied Cytometry Systems, UK).
The fluorescence intensity (FI) of individual cells was measured in RFU. All experiments were run in triplicate for statistical analysis; the standard deviation of the mean did not exceed 5%. The UE and TE were characterized by two values: a percentage of FAM-positive cells in a sample and the FI in the sample. The percentage of FAM-positive cells in the analyzed samples was calculated using the equation:
where FAM+sample (%) is the percentage of fluorescence-positive cells in the analyzed sample and FAM+control (%) is the percentage of fluorescence-positive cells in the negative control. The FI of cells was calculated using the equation:
where FIsample (RFU) is the average fluorescence of the cells in the analyzed sample and FIcontrol (RFU) is the average fluorescence of the cells in the negative control. Cells incubated in the absence of oligonucleotides and lipoplexes were used as a negative control: in these samples the percentage of fluorescence-positive cells and FI did not exceed the experimental error (1–3%). Cells incubated in the presence of Lipofectamine 2000 or 2X3-DOPE were used as a positive control.
Preparation of MDR1 RNA
MDR1 RNA was prepared using PstI-linearized plasmid pMDR670 by in vitro transcription using a TranscriptAid T7 High Yield Transcription Kit (Thermo Fisher Scientific, USA). The reaction was carried out in 20 μL of 40 mM Tris-HCl (pH 7.5) containing 6 mM MgCl2, 2 mM spermidine, 10 mM sodium chloride, 10 mM dithiothreitol (DTT), 10 mM each of nucleoside triphosphate (NTP), 1 μg PstI-linearized plasmid pMDR670, and TranscriptAid enzyme mix for 2 h at 37°C. The reaction was quenched by 1:1 phenol-chloroform (v/v) extraction followed by ethanol precipitation. After centrifugation, the RNA precipitate was rinsed twice with 80% ethanol and dissolved in water.
Dephosphorylation and RNA 5′ labeling
MDR1 RNA transcripts were dephosphorylated using FastAP thermosensitive alkaline phosphatase (Thermo Fisher Scientific, USA) according to the manufacturer's recommendations. The 120 μL reaction mixture contained 10 mM Tris-HCl (pH 8.0), 5 mM MgCl2, 100 mM KCl, 0.02% Triton X-100, 0.1 mg/mL bovine serum albumin (BSA), 2 μg MDR1 RNA transcript, and 12 U FastAP phosphatase; it was incubated at 37°C for 30 min. The reaction was quenched by 1:1 phenol-chloroform (v/v) extraction followed by ethanol precipitation.
The 5′ end labeling of dephosphorylated MDR1 RNA was carried out using [γ-32P]ATP and T4 polynucleotide kinase (Thermo Fisher Scientific, USA) according to the manufacturer's recommendations. 5′-[32P]RNA MDR1 was isolated by electrophoresis in an 8% PAAG/8 M urea gel and visualized by autoradiography on X-ray film. 5′-[32P]RNA was eluted from the gel with 300 mL of 0.3 M sodium acetate (pH 5.5) and precipitated with ethanol.
Hybridization of MDR1 RNA with oligonucleotides and cleavage by RNase H
A reaction mix containing 20 mM Tris-HCl (pH 7.8), 40 mM KCl, 8 mM MgCl2, 1 mM DTT, and 0.05 μg MDR1 RNA was incubated at 90°C for 1 min, at 0°C for 1 min, and at room temperature for 10 min. Then, oligonucleotide was added (final concentration: 10 μM) and the mixture was incubated at 37°C for 1 h. After hybridization was completed, RNase H (Termo Fisher Scientific, USA) was added at a concentration of 1 U/mL and incubated at 37°C for 30 min. The reaction was quenched with 300 μL of 0.3 M sodium acetate (pH 5.5), followed by ethanol precipitation.
RNA was collected by centrifugation and dissolved in loading buffer (6 M urea, 0.025% bromophenol blue, and 0.025% xylene cyanol). RNA cleavage products were resolved in a 12% PAAG/8 M urea gel using TBE (100 mM Tris-borate (pH 8.3), 2 mM EDTA) as a running buffer. To identify cleavage sites, an imidazole ladder77 and a G-ladder produced by partial RNA cleavage with 2 M imidazole buffer (pH 7.0) and RNase T1, respectively, were run in parallel. The gel was visualized with a Molecular Imager FX (Bio-Rad, USA).
xCELLigence real-time analysis of KB-8-5 cell proliferation in the presence of vinblastine
KB-8-5 cells were seeded in 16-well E-plates (5 × 104 cells/well) and incubated in DMEM supplemented with 10% FBS and 1% antibiotic-antimycotic solution at 37°C in a humidified atmosphere containing 5% CO2 for 24 h. The medium was then replaced with serum-free and antibiotic-free DMEM, and cells were transfected with oligonucleotides (1 μM) in complexes with 2X3-DOPE or Lipofectamine 2000—prepared as described above—at 37°C in a humidified atmosphere containing 5% CO2 for 4 h. Subsequently, the medium was replaced by DMEM supplemented with 10% of FBS and 1% antibiotic-antimycotic solution. 24h after transfection, vinblastine (Sigma-Aldrich, USA) was added to the cells at a concentration of 300 nM, and the cells were incubated at 37°C in a humidified atmosphere containing 5% CO2 for 96 h.
Cell viability was recorded in real time using an xCELLigence instrument (Agilent, USA) for 96 h. Cell proliferation experiments were run for 96 h and the cell index was monitored every 30 min for the entire experiment duration. Four replicates of each cell density were used in the cell proliferation experiment.
qRT-PCR analysis of MDR1 mRNA levels in KB-8-5 cells
Human epidermoid carcinoma cells KB-8-5 were seeded into 24-well plates at a density of 105cells/well in 500 μL of DMEM supplemented with 10% FBS in the absence of antibiotics and were maintained overnight at 37°C in a humidified atmosphere with 5% CO2. On the following day, the growth medium was replaced by 200 μL of fresh FBS-free medium containing 100 nM of ASOs R1, R2, M1, M3, G2, or G4 pre-complexed with Lipofectamine 2000 in an Opti-MEM medium according to the manufacturer's protocol and cells were incubated for 4 h under the same conditions. Then the medium was replaced with DMEM containing 10% FBS and 1% antibiotic-antimycotic solution ( and the cells were further cultivated for 48 h under standard conditions.
After 24 or 48 h, total RNA from KB-8-5 cells was extracted using TRIzol Reagent (Thermo Fisher Scientific, USA) according to the manufacturer's protocol. MDR1 gene expression level was evaluated using qRT-PCR. cDNA synthesis was performed in a total volume of 40 μL containing 1 μg of total RNA form KB-8-5 cells, RT buffer (50 mM Tris-HCl [pH 8.3], 75 mM KCl, 3 mM MgCl2), 10 mM DTT, 0.5 mM dNTPs, 100 pmol random hexa-primers, and 20 units of M-MLV reverse transcriptase. The reaction was carried out at 37°C for 60 min. The obtained cDNA was further used for qPCR. The reaction was carried out in a total volume of 20 μL using 0.5 units of Maxima Hot Start Taq DNA Polymerase (Thermo Fisher Scientific, USA), PCR Buffer (200 mM Tris-HCl [pH 8.3], 200 mM KCl, 50 mM (NH4)2SO4), 1.2 mM MgCl2, 0.2 mM dNTPs, EvaGreen (Biotium, USA), and 0.25 μM PCR forward and reverse primers for MDR1 (MDR1-F 5′-ACAGTGGAATTGGTGCTGGG-3′ and MDR1-R 5′-TAAGCTCCCCAACATCGTGC-3′) and a reference gene HPRT1 (HPRT1-F 5′-TGCTGAGGATTTGGAAAGGG-3′ and HPRT1-R 5′-ACAGAGGGCTACAATGTGATG-3′). The reaction was performed with initial preheating at 95°C for 4 min followed by 40 cycles of denaturing at 95°C for 40 s, annealing at 60°C for 30 s, and elongation at 72°C for 30 s. Relative gene expression was calculated using the standard Bio-Rad IQ5 software.
Western blotting
The level of Pgp in KB-8-5 cells was evaluated in vinblastine-free medium. Cells in the exponential growth phase were plated in 48-well plates (1 × 104 cells/well). After 24 h, the growth medium was replaced by fresh serum-free DMEM (200 μL/well). The cells were transfected with oligonucleotides (0.1–0.5 μM) using 0.8 μL Lipofectamine 2000 per well according to the manufacturer's protocol. Four hours post-transfection, FBS was added to the cells, then cells were cultured for 72 h. After the incubation, the cells were lysed in 60 μL sample buffer (Sigma-Aldrich, USA). Ten microliters of each sample were loaded on a 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel and separated at 60 mA for 1 h. The proteins were transferred to a polyvinylidene fluoride membrane (Millipore, USA) using a Criterion blotter transfer apparatus (Bio-Rad, USA). The membrane was incubated overnight in 2% nonfat dried milk in PBS to block non-specific protein binding. The membranes were incubated with monoclonal anti-Pgp and anti-β-actin (internal control) antibodies (P7965, A5441, Sigma-Aldrich, USA) at 1:1,000 and 1:3,000 dilutions, respectively, for 1 h. The membranes were then washed in PBS with 0.1% Tween 20 and subsequently incubated for 1 h with secondary rabbit anti-mouse antibodies conjugated with horseradish peroxidase (Thermo Fisher Scientific, USA). A chemiluminescent reagent kit (Abcam, USA) was used to develop protein bands, and the blot was visualized with a Versadoc 4000 MP imager (Bio-Rad, USA). The gels were analyzed using GelPro 4.0. software.
Statistical analysis
The data of flow cytometry were statistically processed using Student's t test (two-tailed, unpaired); a p value of ≤0.05 was considered to indicate a significant difference. Data of western blot analysis were statistically processed using one-way ANOVA with the Tukey post hoc test; p < 0.05 was considered to be statistically significant. All experimental points were run in triplicate for statistical analysis.
Acknowledgments
The study was initiated in the framework of Russian Government Support for Research Projects implemented under supervision of world's leading scientists (agreement no. 14.B25.31.0028 with S.A. as the leading scientist). This work was funded by the Russian Science Foundation (grant no. 19-74-30011), the Russian Foundation for Basic Research (grant nos. 18-515-57006, 18-29-09045, and 18-29-08062 to D.A.S.), and the Russian Government-funded budget project of ICBFM SB RAS no. 121031300042-1. The authors thank Ms A. Vladimirova for cell maintenance and Dr. O. Markov for the help with the analysis of flow cytometry data.
Author contributions
PGO gapmers were conceived by D.A.S. E.L.C., D.V.P., D.A.S., M.A.Z., S.A., and V.V.V. designed the study. N.L.M., A.V.F., O.A.P., and I.V.C. performed the experiments. M.S.K., D.A.S., and D.V.P. performed oligonucleotide synthesis. N.L.M., M.S.K., A.V.F., O.A.P., and I.V.C. analyzed the data. N.L.M., M.S.K., D.V.P., D.A.S., E.L.C., S.A., and M.A.Z. interpreted the data. M.A.Z., D.V.P., and V.V.V. contributed reagents and analytic tools. M.S.K., N.L.M., D.V.P., D.A.S., M.A.Z., S.A., and V.V.V. wrote and edited the manuscript. All authors read the manuscript and agreed to its contents.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.11.025.
Contributor Information
Nadezhda L. Mironova, Email: mironova@niboch.nsc.ru.
Sidney Altman, Email: sidney.altman@yale.edu.
Supplemental information
28 and Tables S1 and S2
References
- 1.Zamecnik P.C., Stephenson M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. U S A. 1978;75:280–284. doi: 10.1073/pnas.75.1.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Quemener A.M., Bachelot L., Forestier A., Donnou-Fournet E., Gilot D., Galibert M.D. The powerful world of antisense oligonucleotides: from bench to bedside. Wiley Interdiscip. Rev. RNA. 2020;11:e1594. doi: 10.1002/wrna.1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bajan S., Hutvagner G. RNA-based therapeutics: from antisense oligonucleotides to miRNAs. Cells. 2020;9:137. doi: 10.3390/cells9010137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karkare S., Bhatnagar D. Promising nucleic acid analogs and mimics: characteristic features and applications of PNA, LNA, and morpholino. Appl. Microbiol. Biotechnol. 2006;71:575–586. doi: 10.1007/s00253-006-0434-2. [DOI] [PubMed] [Google Scholar]
- 5.Pérez B., Rodríguez-Pascau L., Vilageliu L., Grinberg D., Ugarte M., Desviat L.R. Present and future of antisense therapy for splicing modulation in inherited metabolic disease. J. Inherit. Metab. Dis. 2010;33:397–403. doi: 10.1007/s10545-010-9135-1. [DOI] [PubMed] [Google Scholar]
- 6.Singh N.N., Luo D., Singh R.N. Pre-mRNA splicing modulation by antisense oligonucleotides. Methods Mol. Biol. 2018;1828:415–437. doi: 10.1007/978-1-4939-8651-4_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Opalinska J.B., Gewirtz A.M. Nucleic-acid therapeutics: basic principles and recent applications. Nat. Rev. Drug Discov. 2002;1:503–514. doi: 10.1038/nrd837. [DOI] [PubMed] [Google Scholar]
- 8.Kaczmarek J.C., Kowalski P.S., Anderson D.G. Advances in the delivery of RNA therapeutics: from concept to clinical reality. Genome Med. 2017;9:60. doi: 10.1186/s13073-017-0450-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prakash T.P. An overview of sugar-modified oligonucleotides for antisense therapeutics. Chem. Biodivers. 2011;8:1616–1641. doi: 10.1002/cbdv.201100081. [DOI] [PubMed] [Google Scholar]
- 10.Sahu N., Shilakari G., Nayak A., Kohli D. Antisense technology: a selective tool for gene expression regulation and gene targeting. Curr. Pharm. Biotechnol. 2007;8:291–304. doi: 10.2174/138920107782109985. [DOI] [PubMed] [Google Scholar]
- 11.Dirin M., Winkler J. Influence of diverse chemical modifications on the ADME characteristics and toxicology of antisense oligonucleotides. Expert Opin. Biol. Ther. 2013;13:875–888. doi: 10.1517/14712598.2013.774366. [DOI] [PubMed] [Google Scholar]
- 12.Crooke S.T., Wang S., Vickers T.A., Shen W., Liang X.H. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 2017;35:230–237. doi: 10.1038/nbt.3779. [DOI] [PubMed] [Google Scholar]
- 13.Iannitti T., Morales-Medina J., Palmieri B. Phosphorothioate oligonucleotides: effectiveness and toxicity. Curr. Drug Targets. 2014;15:663–673. doi: 10.2174/1389450115666140321100304. [DOI] [PubMed] [Google Scholar]
- 14.Koizumi M. True antisense oligonucleotides with modified nucleotides restricted in the N-conformation. Curr. Top. Med. Chem. 2007;7:661–665. doi: 10.2174/156802607780487768. [DOI] [PubMed] [Google Scholar]
- 15.Itoh M., Nakaura M., Imanishi T., Obika S. Target gene knockdown by 2′,4′-BNA/LNA antisense oligonucleotides in zebrafish. Nucleic Acid Ther. 2014;24:186–191. doi: 10.1089/nat.2013.0464. [DOI] [PubMed] [Google Scholar]
- 16.Imanishi T., Obika S., Pu L. BNAs: novel nucleic acid analogs with a bridged sugar moiety. Chem. Commun. 2002;2:1653–1659. doi: 10.1039/b201557a. [DOI] [PubMed] [Google Scholar]
- 17.Nielsen P.E. Peptide nucleic acids (PNA) in chemical biology and drug discovery. Chem. Biodivers. 2010;7:786–804. doi: 10.1002/cbdv.201000005. [DOI] [PubMed] [Google Scholar]
- 18.Summerton J., Weller D. Morpholino antisense oligomers: design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997;7:187–195. doi: 10.1089/oli.1.1997.7.187. [DOI] [PubMed] [Google Scholar]
- 19.Stein C.A., Castanotto D. FDA-approved oligonucleotide therapies in 2017. Mol. Ther. 2017;25:1069–1075. doi: 10.1016/j.ymthe.2017.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heo Y.A. Golodirsen: first approval. Drugs. 2020;80:329–333. doi: 10.1007/s40265-020-01267-2. [DOI] [PubMed] [Google Scholar]
- 21.Shirley M. Casimersen: first approval. Drugs. 2021;81:875–879. doi: 10.1007/s40265-021-01512-2. [DOI] [PubMed] [Google Scholar]
- 22.Keam S.J. Inotersen: first global approval. Drugs. 2018;78:1371–1376. doi: 10.1007/s40265-018-0968-5. [DOI] [PubMed] [Google Scholar]
- 23.Cazenave C., Stein C.A., Loreau N., Thuong N.T., Neckers L.M., Subasinghe C., Hélène C., Cohen J.S., Toulmé J.J. Comparative inhibition of rabbit globin mRNA translation by modified antisense oligodeoxynucleotides. Nucleic Acids Res. 1989;17:4255–4273. doi: 10.1093/nar/17.11.4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferrari N., Bergeron D., Tedeschi A.L., Mangos M.M., Paquet L., Renzi P.M., Damha M.J. Characterization of antisense oligonucleotides comprising 2′-deoxy-2′-fluoro-β-D-arabinonucleic acid (FANA): specificity, potency, and duration of activity. Ann. N. Y. Acad. Sci. 2006;1082:91–102. doi: 10.1196/annals.1348.032. [DOI] [PubMed] [Google Scholar]
- 25.Miroshnichenko S.K., Patutina O.A., Burakova E.A., Chelobanov B.P., Fokina A.A., Vlassov V.V., Altman S., Zenkova M.A., Stetsenko D.A. Mesyl phosphoramidate antisense oligonucleotides as an alternative to phosphorothioates with improved biochemical and biological properties. Proc. Natl. Acad. Sci. U S A. 2019;116:1229–1234. doi: 10.1073/pnas.1813376116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malchère C., Verheijen J., Van der Laan S., Bastide L., Van Boom J., Lebleu B., Robbins I. A short phosphodiester window is sufficient to direct RNase H-dependent RNA cleavage by antisense peptide nucleic acid. Antisense Nucleic Acid Drug Dev. 2000;10:463–468. doi: 10.1089/oli.1.2000.10.463. [DOI] [PubMed] [Google Scholar]
- 27.Prakash T.P., Johnston J.F., Graham M.J., Condon T.P., Manoharan M. 2′-O-[2-[N,N-Dimethylamino)oxy]ethyl]-modified oligonucleotides inhibit expression of mRNA in vitro and in vivo. Nucleic Acids Res. 2004;32:828–833. doi: 10.1093/nar/gkh220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fluiter K., Frieden M., Vreijling J., Rosenbohm C., De Wissel M.B., Christensen S.M., Koch T., Ørum H., Baas F. On the in vitro and in vivo properties of four locked nucleic acid nucleotides incorporated into an anti-H-Ras antisense oligonucleotide. ChemBioChem. 2005;6:1104–1109. doi: 10.1002/cbic.200400419. [DOI] [PubMed] [Google Scholar]
- 29.Kupryushkin M.S., Pyshnyi D.V., Stetsenko D.A. Phosphoryl guanidines: a new type of nucleic acid analogues. Acta Naturae. 2014;6:116–118. [PMC free article] [PubMed] [Google Scholar]
- 30.Stetsenko D., Kupryushkin M., Pyshnyi D. 2016. Modified Oligonucleotides and Methods for Their Synthesis. WO2016028187. [Google Scholar]
- 31.Garafutdinov R.R., Sakhabutdinova A.R., Kupryushkin M.S., Pyshnyi D.V. Prevention of DNA multimerization using phosphoryl guanidine primers during isothermal amplification with Bst exo-DNA polymerase. Biochimie. 2020;168:259–267. doi: 10.1016/j.biochi.2019.11.013. [DOI] [PubMed] [Google Scholar]
- 32.Chubarov A.S., Oscorbin I.P., Filipenko M.L., Lomzov A.A., Pyshnyi D.V. Allele-specific PCR for KRAS mutation detection using phosphoryl guanidine modified primers. Diagnostics. 2020;10:872. doi: 10.3390/diagnostics10110872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Skvortsova Y.V., Salina E.G., Burakova E.A., Bychenko O.S., Stetsenko D.A., Azhikina T.L. A new antisense phosphoryl guanidine oligo-2′-O-methylribonucleotide penetrates into intracellular mycobacteria and suppresses target gene expression. Front. Pharmacol. 2019;10:1049. doi: 10.3389/fphar.2019.01049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levina A.S., Repkova M.N., Chelobanov B.P., Bessudnova E.V., Mazurkova N.A., Stetsenko D.A., Zarytova V.F. Impact of delivery method on antiviral activity of phosphodiester, phosphorothioate, and phosphoryl guanidine oligonucleotides in MDCK cells infected with H5N1 bird flu virus. Mol. Biol. (Mosk). 2017;51:717–723. doi: 10.7868/S0026898417040139. [DOI] [PubMed] [Google Scholar]
- 35.Su Y., Edwards P.J.B., Stetsenko D.A., Filichev V.V. The importance of phosphates for DNA G-quadruplex formation: evaluation of zwitterionic G-rich oligodeoxynucleotides. ChemBioChem. 2020;21:2455–2466. doi: 10.1002/cbic.202000110. [DOI] [PubMed] [Google Scholar]
- 36.Lomzov A.A., Kupryushkin M.S., Shernyukov A.V., Nekrasov M.D., Dovydenko I.S., Stetsenko D.A., Pyshnyi D.V. Diastereomers of a mono-substituted phosphoryl guanidine trideoxyribonucleotide: isolation and properties. Biochem. Biophys. Res. Commun. 2019;513:807–811. doi: 10.1016/j.bbrc.2019.04.024. [DOI] [PubMed] [Google Scholar]
- 37.Ambudkar S.V., Dey S., Hrycyna C.A., Ramachandra M., Pastan I., Gottesman M.M. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 1999;39:361–398. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- 38.Choi C.H. ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer Cell Int. 2005;5:30. doi: 10.1186/1475-2867-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stavrovskaya A.A., Stromskaya T.P. Transport proteins of the ABC family and multidrug resistance of tumor cells. Biochem. 2008;73:592–604. doi: 10.1134/s0006297908050118. [DOI] [PubMed] [Google Scholar]
- 40.Kostenko E.V., Laktionov P.P., Vlassov V.V., Zenkova M.A. Downregulation of PGY1/MDR1 mRNA level in human KB cells by antisense oligonucleotide conjugates. RNA accessibility in vitro and intracellular antisense activity. Biochim. Biophys. Acta - Gene Struct. Expr. 2002;1576:143–147. doi: 10.1016/s0167-4781(02)00339-1. [DOI] [PubMed] [Google Scholar]
- 41.Logashenko E.B., Vladimirova A.V., Repkova M.N., Venyaminova A.G., Chernolovskaya E.L., Vlassov V.V. Silencing of MDR 1 gene in cancer cells by siRNA. Nucleosides, Nucleotides and Nucleic Acids. 2004;23:861–866. doi: 10.1081/NCN-200026032. [DOI] [PubMed] [Google Scholar]
- 42.Kitamura M., Kato S., Yano M., Tashiro N., Shiratake Y., Sando M., Okauchi T. A reagent for safe and efficient diazo-transfer to primary amines: 2-azido-1,3-dimethylimidazolinium hexafluorophosphate. Org. Biomol. Chem. 2014;12:4397–4406. doi: 10.1039/c4ob00515e. [DOI] [PubMed] [Google Scholar]
- 43.Kitamura M., Murakami K. Organic Syntheses. John Wiley & Sons, Inc.; 2016. Synthesis of 2-azido-1,3-dimethylimidazolinium hexafluorophosphate (ADMP) pp. 171–181. [Google Scholar]
- 44.Guzaev A.P. Reactivity of 3H-1,2,4-dithiazole-3-thiones and 3H-1,2-dithiole-3-thiones as sulfurizing agents for oligonucleotide synthesis. Tetrahedron Lett. 2011;52:434–437. [Google Scholar]
- 45.Golyshev V.M., Pyshnyi D.V., Lomzov A.A. Effects of phosphoryl guanidine modification of phosphate residues on the structure and hybridization of oligodeoxyribonucleotides. J. Phys. Chem. B. 2021;125:2841–2855. doi: 10.1021/acs.jpcb.0c10214. [DOI] [PubMed] [Google Scholar]
- 46.Herkt M., Thum T. Pharmacokinetics and proceedings in clinical application of nucleic acid therapeutics. Mol. Ther. 2021;29:521–539. doi: 10.1016/j.ymthe.2020.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muller C., Goubin F., Ferrandis E., Cornil-Scharwtz I., Bailly J.D., Bordier C., Benard J., Sikic B.I., Laurent G. Evidence for transcriptional control of human mdr1 gene expression by verapamil in multidrug-resistant leukemic cells. Mol. Pharmacol. 1995;47:51–56. [PubMed] [Google Scholar]
- 48.Prokipcak R.D., Raouf A., Lee C. The AU-rich 3’ untranslated region of human MDR1 mRNA is an inefficient mRNA destabilizer. Biochem. Biophys. Res. Commun. 1999;261:627–634. doi: 10.1006/bbrc.1999.1101. [DOI] [PubMed] [Google Scholar]
- 49.Richert N.D., Aldwin L., Nitecki D., Gottesman M.M., Pastan I. Stability and covalent modification of P-glycoprotein in multidrug-resistant KB cells. Biochemistry. 1988;27:7607–7613. doi: 10.1021/bi00420a006. [DOI] [PubMed] [Google Scholar]
- 50.Scharner J., Aznarez I. Clinical applications of single-stranded oligonucleotides: current landscape of approved and in-development therapeutics. Mol. Ther. 2021;29:540–554. doi: 10.1016/j.ymthe.2020.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuijper E.C., Bergsma A.J., Pijnappel W.W.M.P., Aartsma-Rus A. Opportunities and challenges for antisense oligonucleotide therapies. J. Inherit. Metab. Dis. 2021;44:72–87. doi: 10.1002/jimd.12251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou L.-Y., Qin Z., Zhu Y.-H., He Z.-Y., Xu T. Current RNA-based therapeutics in clinical trials. Curr. Gene Ther. 2019;19:172–196. doi: 10.2174/1566523219666190719100526. [DOI] [PubMed] [Google Scholar]
- 53.Smith C.I.E., Zain R. Therapeutic oligonucleotides: state of the art. Annu. Rev. Pharmacol. Toxicol. 2019;59:605–630. doi: 10.1146/annurev-pharmtox-010818-021050. [DOI] [PubMed] [Google Scholar]
- 54.Dhuri K., Bechtold C., Quijano E., Pham H., Gupta A., Vikram A., Bahal R. Antisense oligonucleotides: an emerging area in drug discovery and development. J. Clin. Med. 2020;9:2004. doi: 10.3390/jcm9062004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Letsinger R.L., Heavner G.A. Synthesis of phosphoromonoamidate diester nucleotides via the phosphite-azide coupling method. Tetrahedron Lett. 1975;16:147–150. [Google Scholar]
- 56.Anderson B.A., Freestone G.C., Low A., De-Hoyos C.L., Østergaard M.E., Migawa M.T., Fazio M., Wan W.B., Berdeja A., et al. Towards next generation antisense oligonucleotides: mesylphosphoramidate modification improves therapeutic index and duration of effect of gapmer antisense oligonucleotides. Nucleic Acids Res. 2021;49:9026–9041. doi: 10.1093/nar/gkab718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pavlova A.S., Dyudeeva E.S., Kupryushkin M.S., Amirkhanov N.V., Pyshnyi D.V., Pyshnaya I.A. SDS-PAGE procedure: application for characterization of new entirely uncharged nucleic acids analogs. Electrophoresis. 2018;39:670–674. doi: 10.1002/elps.201700415. [DOI] [PubMed] [Google Scholar]
- 58.Dyudeeva E.S., Pavlova A.S., Kupryushkin M.S., Pyshnyi D.V., Pyshnaya I.A. Problems of the synthesis of oligonucleotide derivatives in the realization of the anchimeric effect. Russ. J. Bioorg. Chem. 2021;47:505–513. [Google Scholar]
- 59.Johannes L., Lucchino M. Current challenges in delivery and cytosolic translocation of therapeutic RNAs. Nucleic Acid Ther. 2018;28:178–193. doi: 10.1089/nat.2017.0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cerritelli S.M., Crouch R.J. Ribonuclease H: the enzymes in eukaryotes. FEBS J. 2009;276:1494–1505. doi: 10.1111/j.1742-4658.2009.06908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Summerton J. Morpholino antisense oligomers: the case for an RNase H-independent structural type. Biochim. Biophys. Acta - Gene Struct. Expr. 1999;1489:141–158. doi: 10.1016/s0167-4781(99)00150-5. [DOI] [PubMed] [Google Scholar]
- 62.Mironova N., Shklyaeva O., Andreeva E., Popova N., Kaledin V., Nikolin V., Vlassov V., Zenkova M. Animal model of drug-resistant tumor progression. Ann. N. Y. Acad. Sci. 2006;1091:490–500. doi: 10.1196/annals.1378.090. [DOI] [PubMed] [Google Scholar]
- 63.Patutina O.A., Gaponova S.K., Sen’kova A.V., Savin I.A., Gladkikh D.V., Burakova E.A., Fokina A.A., Maslov M.A., Shmendel’ E.V., Wood M.J.A., et al. Mesyl phosphoramidate backbone modified antisense oligonucleotides targeting miR-21 with enhanced in vivo therapeutic potency. Proc. Natl. Acad. Sci. U S A. 2020;117:32370–32379. doi: 10.1073/pnas.2016158117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chelobanov B.P., Burakova E.A., Prokhorova D.V., Fokina A.A., Stetsenko D.A. New oligodeoxynucleotide derivatives containing N-(methanesulfonyl)-phosphoramidate (mesyl phosphoramidate) internucleotide group. Russ. J. Bioorg. Chem. 2017;43:664–668. [Google Scholar]
- 65.Brand R.M., Iversen P.L. Transdermal delivery of antisense compounds. Adv. Drug Deliv. Rev. 2000;44:51–57. doi: 10.1016/s0169-409x(00)00083-1. [DOI] [PubMed] [Google Scholar]
- 66.Lebleu B., Moulton H.M., Abes R., Ivanova G.D., Abes S., Stein D.A., Iversen P.L., Arzumanov A.A., Gait M.J. Cell penetrating peptide conjugates of steric block oligonucleotides. Adv. Drug Deliv. Rev. 2008;60:517–529. doi: 10.1016/j.addr.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Negishi Y., Ishii Y., Nirasawa K., Sasaki E., Endo-Takahashi Y., Suzuki R., Maruyama K. PMO delivery system using bubble liposomes and ultrasound exposure for Duchenne muscular dystrophy treatment. Methods Mol. Biol. 2018;1687:185–192. doi: 10.1007/978-1-4939-7374-3_13. [DOI] [PubMed] [Google Scholar]
- 68.Moulton H.M., Hase M.C., Smith K.M., Iversen P.L. HIV Tat peptide enhances cellular delivery of antisense morpholino oligomers. Antisense Nucleic Acid Drug Dev. 2003;13:31–43. doi: 10.1089/108729003764097322. [DOI] [PubMed] [Google Scholar]
- 69.Arora V., Hannah T.L., Iversen P.L., Brand R.M. Transdermal use of phosphorodiamidate morpholino oligomer AVI-4472 inhibits cytochrome P450 3A2 activity in male rats. Pharm. Res. 2002;19:1465–1470. doi: 10.1023/a:1020448430919. [DOI] [PubMed] [Google Scholar]
- 70.Shimojo M., Kasahara Y., Inoue M., Tsunoda S., Shudo Y., Kurata T., Obika S. A gapmer antisense oligonucleotide targeting SRRM4 is a novel therapeutic medicine for lung cancer. Sci. Rep. 2019;9:7618. doi: 10.1038/s41598-019-43100-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Marrosu E., Ala P., Muntoni F., Zhou H. Gapmer antisense oligonucleotides suppress the mutant allele of COL6A3 and restore functional protein in Ullrich muscular dystrophy. Mol. Ther. - Nucleic Acids. 2017;8:416–427. doi: 10.1016/j.omtn.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mutso M., Nikonov A., Pihlak A., Žusinaite E., Viru L., Selyutina A., Reintamm T., Kelve M., Saarma M., Karelson M., et al. RNA interference-guided targeting of hepatitis C virus replication with antisense locked nucleic acid-based oligonucleotides containing 8-oxo-dG modifications. PLoS ONE. 2015;10:e0128686. doi: 10.1371/journal.pone.0128686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Takahashi M., Li H., Zhou J., Chomchan P., Aishwarya V., Damha M.J., Rossi J.J. Dual mechanisms of action of self-delivering, anti-HIV-1 FANA oligonucleotides as a potential new approach to HIV therapy. Mol. Ther. - Nucleic Acids. 2019;17:615–625. doi: 10.1016/j.omtn.2019.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kumar V., Jung Y.S., Liang P.H. Anti-SARS coronavirus agents: a patent review (2008-present) Expert Opin. Ther. Pat. 2013;23:1337–1348. doi: 10.1517/13543776.2013.823159. [DOI] [PubMed] [Google Scholar]
- 75.Patutina O.A., Bazhenov M.A., Miroshnichenko S.K., Mironova N.L., Pyshnyi D.V., Vlassov V.V., Zenkova M.A. Peptide-oligonucleotide conjugates exhibiting pyrimidine-X cleavage specificity efficiently silence miRNA target acting synergistically with RNase. H. Sci. Rep. 2018;8:14990. doi: 10.1038/s41598-018-33331-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Markov O.V., Mironova N.L., Shmendel E.V., Serikov R.N., Morozova N.G., Maslov M.A., Vlassov V.V., Zenkova M.A. Multicomponent mannose-containing liposomes efficiently deliver RNA in murine immature dendritic cells and provide productive anti-tumour response in murine melanoma model. J. Control Release. 2015;213:45–56. doi: 10.1016/j.jconrel.2015.06.028. [DOI] [PubMed] [Google Scholar]
- 77.Mironova N.L., Pyshnyi D.V., Shtadler D.V., Fedorova A.A., Vlassov V.V., Zenkova M.A. RNase T1 mimicking artificial ribonuclease. Nucleic Acids Res. 2007;35:2356–2367. doi: 10.1093/nar/gkm143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
28 and Tables S1 and S2