

Abstract
CRISPR/Cas9-mediated therapeutic gene editing is a promising technology for durable treatment of incurable monogenic diseases such as myotonic dystrophies. Gene-editing approaches have been recently applied to in vitro and in vivo models of myotonic dystrophy type 1 (DM1) to delete the pathogenic CTG-repeat expansion located in the 3′ untranslated region of the DMPK gene. In DM1-patient-derived cells removal of the expanded repeats induced beneficial effects on major hallmarks of the disease with reduction in DMPK transcript-containing ribonuclear foci and reversal of aberrant splicing patterns. Here, we set out to excise the triplet expansion in a time-restricted and cell-specific fashion to minimize the potential occurrence of unintended events in off-target genomic loci and select for the target cell type. To this aim, we employed either a ubiquitous promoter-driven or a muscle-specific promoter-driven Cas9 nuclease and tetracycline repressor-based guide RNAs. A dual-vector approach was used to deliver the CRISPR/Cas9 components into DM1 patient-derived cells and in skeletal muscle of a DM1 mouse model. In this way, we obtained efficient and inducible gene editing both in proliferating cells and differentiated post-mitotic myocytes in vitro as well as in skeletal muscle tissue in vivo.
Keywords: myotonic dystrophy, gene therapy, CRISPR/Cas9, skeletal muscle, DMSXL mouse model, DM1, CTG repeats, gene editing
Graphical abstract
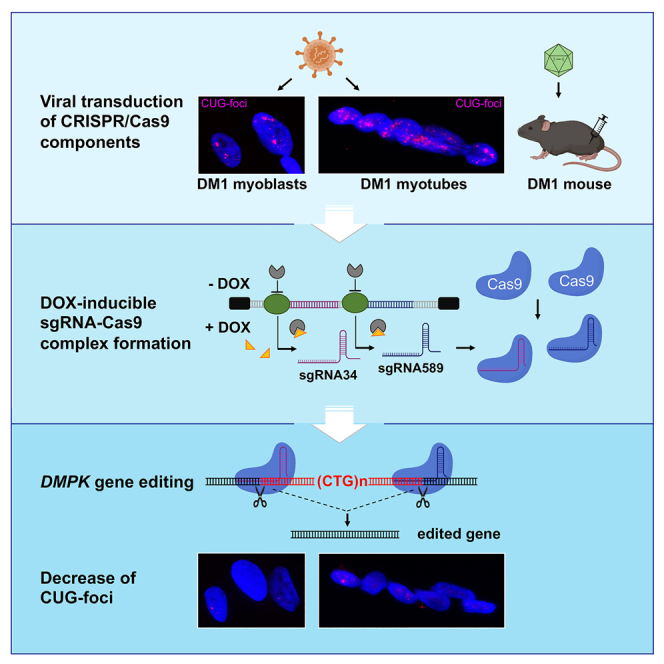
This paper describes a gene-editing approach designed to remove the pathologic CTG expansion mutation that causes myotonic dystrophy type 1. By using muscle-specific and drug-inducible expression of the CRISPR/Cas9 complex, effective deletion of the CTG expansion was obtained in myogenic cells and in mouse skeletal muscle.
Introduction
Myotonic dystrophy type 1 (DM1) is one of the most common dominant neuromuscular disorders that results in multisystemic symptoms, including myotonia, muscle wasting, cardiac conduction defects, insulin resistance, cataracts, and cognitive dysfunction.1 The underlying cause of DM1 is a toxic gain-of-function of an expanded (CTG)n repeat in the 3′ untranslated region (UTR) of the DM1 protein kinase (DMPK) gene on chromosome 19.2, 3, 4 Several pathogenic mechanisms likely contribute to disease in DM1.1, 2, 3, 4 At the DNA level, the hairpin-like structures of the repeats can induce chromatin changes, such as CpG methylation, resulting in haploinsufficiency of DMPK and neighboring genes, or cause replication fork stalling during DNA replication, leading to cell stress.5, 6, 7 Substantial experimental evidence, however, recognizes a toxic function of expanded DMPK transcripts carrying CUG repeats that are retained in cell nuclei and sequester members of the muscle-blind-like (MBNL) family of RNA-binding proteins through their hairpin-like structures. These repeated RNA-MBNL aggregates form characteristic nuclear foci in cells and tissues from DM1 patients. In addition, mutant DMPK transcripts stabilize CELF1 (CUGBP Elav-like family member 1), altering the balance of MBNL and CELF1 protein levels. This leads to aberrant alternative splicing and alternative polyadenylation of many transcripts as well as abnormal microRNA processing.8, 9, 10, 11, 12, 13
Both in vitro and in vivo models of DM1 have greatly contributed to clarifying the pathogenetic mechanisms of the disease. Cells derived from DM1 patients have been established in culture and shown to reproduce molecular alterations typical of DM1.14, 15, 16 These in vitro models have been very useful in discovering crucial molecules and cellular pathways involved in the pathology and for testing therapeutic strategies. For in vivo studies, transgenic mouse models expressing synthetic CTG repeats or mutant DMPK genes have been generated and found to mimic some aspects of the human disease.17, 18, 19, 20 Both in vitro and in vivo models have been used to test therapeutic molecules, including antisense synthetic oligonucleotides or pharmacologic compounds such as small molecules or peptides that target the mutant transcripts.21, 22, 23, 24, 25, 26 Although successful in improving the phenotype in DM1 mouse models, these approaches require repeated administration of the therapeutic molecule, target only some aspects of the multisystemic disorder, and do not eliminate the disease-causing mutation. In addition, their application in DM1 patients has obtained scarce success so far.27
More recently, gene-editing technology, including the clustered regularly interspaced short palindromic repeats (CRISPR)-associated protein 9 (Cas9) system, has been exploited.16,28, 29, 30, 31 The system utilizes the cleaving capacity of the Streptococcus pyogenes Cas9 (SpCas9) endonuclease, which can be directed to specific DNA or RNA sequences in virtually any genome by engineered single guide RNAs (sgRNAs).32,33 CRISPRs and Cas9, first identified as part of the bacterial immune system to play a role in viral defense,34,35 were then adapted for the rapid editing of eukaryotic genomes.36,37 The Cas9 endonuclease requires sgRNA-mediated conformational activation to facilitate DNA binding and cleavage.38 The resultant sites of DNA cleavage are usually repaired by non-homologous end-joining (NHEJ), but other repair mechanisms may also be recruited at the double-strand breaks (DSBs).39,40
Therapeutic approaches using CRISPR-mediated RNA targeting have been tried both in vitro and in vivo.41, 42, 43 Direct targeting of mutant RNA, however, does not correct expanded DNA-mediated transcriptional effects, such as silencing of neighboring genes,5,6 and requires persistent expression of the CRISPR elements in order to maintain suppression of the expanded transcripts.
CRISPR/Cas9 technology can be exploited for excision of DM1 expansion, due to the capability of Cas9 to elicit multiple DNA breaks simultaneously. Indeed, we and others have recently applied the CRISPR/Cas9 gene-editing system in ad hoc generated cell models from DM1 patients and have succeeded in removing pathogenetic CTG expansions permanently, resulting in phenotypic reversion of edited cells.16,29, 30, 31,44,45 Although DSB repair mechanisms are known to be error-prone pathways causing insertions and deletions (indels), in the case of DM1 this is unlikely to be damaging, since editing involves the non-coding region of the DMPK gene, and protein production is not significantly affected.16 For the evaluation of gene therapy approaches the DMSXL transgenic mouse model is particularly suitable, since mice carry a human DMPK gene with >1,000 CTG repeats from a DM1 patient, and homozygous animals exhibit a diseased phenotype that reproduces many aspects of human pathology.44,46, 47, 48, 49 Intramuscular injection of recombinant adeno-associated virus (AAV) vectors expressing CRISPR/SaCas9 components in these mice decreased the number of pathological RNA foci in myonuclei.44
Despite the advantages offered by CRISPR/Cas9-mediated gene editing, safety and efficacy need to be evaluated and refined. In regard of a gene therapy application in DM1 patients, a major challenge is that the genetic defect should be corrected in non-dividing cells, i.e., satellite cells, and in post-mitotic cells such as skeletal muscle fibers, cardiomyocytes, and neurons, which are crucial target cells in muscular dystrophies. In addition, one concern with the use of this strategy to modify the human genome is the specificity of these genome-editing tools. Although no off-target effects were observed in initial studies of genome editing,16,29,30 it is possible that certain sgRNAs produce significant off-target cuts on sequences related to those selected for editing the gene of interest and can potentially generate adverse effects.50 Another potential event to avoid is an immune response to the Cas9 protein that has been observed in certain circumstances.42,51 It is necessary, therefore, to develop methods allowing one to control and limit temporally Cas9 activity in order to minimize these undesirable effects. Most current in vivo CRISPR/Cas9 tools do not control the induction of Cas9 or sgRNAs via external stimuli. A drug-inducible CRISPR/Cas9 system would be ideal for this purpose and would be especially useful when combined with tissue-specific expression of Cas9 or sgRNAs, allowing for full spatiotemporal control.
To obtain such flexible editing tools, we sought to improve upon existing CRISPR/Cas9 components and make the system (1) efficient, (2) applicable to both proliferating cells and post-mitotic differentiated myotubes, and (3) temporally controllable. To this end, we engineered a lentiviral vector system that allows for efficient transduction of cells and subsequent inducible expression of sgRNAs with concomitant constitutive expression of Cas9, and validated the efficacy and specificity of this approach in DM1-patient-derived cell lines in vitro. In addition, the feasibility of the inducible editing approach was tested in the skeletal muscle of DMSXL mice.
Results
Characterization of inducible CRISPR/Cas9 editing of CTG repeats in DM1-patient-derived myogenic cell lines
Different options were evaluated to design efficient, specific, and adjustable CRISPR/Cas9 gene editing of the CTG repeats in the DMPK gene of DM1-patient-derived cells. Four different lentiviral constructs were employed (Figure S1): (a) pLenti-EF1a-SpCas9-EGFP, a lentiviral vector expressing the SpCas9 nuclease and fluorescent EGFP protein from EF1a constitutive promoter; (b) pLenti-CK8-eSpCas9-EGFP, a lentiviral vector expressing eSpCas9(1.1) and fluorescent EGFP protein from muscle-specific CK8 promoter;52,53 (c) pLenti-EF1a-eSpCas9-EGFP, a lentiviral vector expressing eSpCas9(1.1) and fluorescent EGFP protein from EF1a constitutive promoter; (d) pLenti-H1-Tet-sgRNA-mCherry, a lentiviral vector containing two tetracycline (TetO) inducible sgRNA cassettes and also expressing Tet Repressor (TetR) and fluorescent mCherry protein under UbC constitutive promoter. The sgRNAs sg34 and sg589 cloned in this construct were designed to target DMPK 3′ UTR sequences upstream and downstream the CTG repeats and were previously shown to induce efficient deletions of the repeat region in myogenic cells derived from a DM1 patient carrying >1,000 repeats.16 Two lentiviruses with the same constitutive EF1a promoter but driving two slightly different Cas9s were prepared to correlate the editing outcomes of nucleases described to have different specificities: compared with SpCas9, the enhanced eSpCas9(1.1) was shown to produce fewer off-target effects.52
The different lentiviral particles were used to infect myoblast-convertible immortalized fibroblasts derived from DM1 patient skin fibroblasts as previously described.16 Three viral combinations, one containing lentiviruses a + d, hereafter named EF1-Crispr, one containing lentiviruses b + d, hereafter named CK8-Crispr, and one containing lentiviruses c + d, hereafter named EF1-Crispr(1.1), were used to infect DM1 fibroblasts at a multiplicity of infection of 5 in order to ensure double infection of most cells. Infected cells were then sorted to select those co-expressing higher levels of GFP and mCherry fluorescent proteins. Fibroblasts were maintained in growth medium (GM) or induced to differentiate into myotubes by activation of estrogen-inducible MYOD1 in differentiation medium (DM). To set up the timing conditions for optimal gene-editing efficiency, we studied the expression of sgRNAs and Cas9 protein. To determine effective inducibility and de-inducibility of the sgRNAs, we analyzed their expression by qPCR following doxycycline (DOX) addition (1 μg/mL). As shown in Figure 1A, sg34 and sg589 expression was increased in both GM and DM after 7 days of induction, although sg34 showed a higher fold increase compared with sg589. Induction of both sg34 and sg589 was stronger in DM. Next, we studied the expression of sgRNAs at different time points after removal of DOX from the medium. Infected cells were treated with DOX in GM for 4 days and then cultured in either the presence or absence of DOX in GM for 4 and 7 days. Analysis of sg34 and sg589 RNAs showed a clear reduction of expression of up to 70% after DOX removal (Figure 1B). These results show that expression of both sgRNAs can be induced by DOX addition and reduced effectively after DOX removal, allowing a time-controlled regulation of CRISPR/Cas9 complex assembly on-target sequences. Expression of Cas9 protein was detectable in cells infected with either the EF1-Crispr or CK8-Crispr in both GM and DM, but Cas9 expression driven by muscle-specific CK8 promoter was higher in DM and increased during differentiation time; expression of TetR was comparable in all infected cells (Figure 1C). Although CK8 promoter was reported to be expressed exclusively in post-mitotic cardiac and skeletal muscle cells in vivo,53 it showed a basal activity also in GM in the adopted cell culture system in vitro prior to differentiation. Finally, we analyzed the DOX-dependent occurrence of gene editing in EF1-Crispr-, CK8-Crispr-, and EF1-Crispr(1.1)-transduced fibroblasts. Cells were treated with DOX for 3 or 7 days in both GM and DM, and genomic DNA was extracted and analyzed by PCR using primers binding upstream and downstream of the CTG expansion to amplify the CTG-deleted genomic fragment and unedited wild-type allele (Figure 2A). As expected, gene editing was detectable only in DOX-treated cells, confirming the inducibility of CRISPR/Cas9 complex activity (Figures 2A andS2A). To estimate the efficiency of CTG-repeat deletion, we grew EF1-Crispr-transduced fibroblasts in DOX-containing GM for 7 days and then plated them as single cells to allow for clonal expansion. Twenty-five percent of the clones analyzed (14 out of 56) had undergone CTG-repeat deletion in either wild-type (36%) or mutated (43%) alleles, or in both alleles (21%), showing a high editing efficiency. For a more precise evaluation of editing efficiency, genomic DNAs from myogenic cells transduced with EF1-Crispr in GM/DM and CK8-Crispr in DM were analyzed with droplet digital PCR (ddPCR) (Figure 2B). Investigation of the region around sg589, to quantify the DNA remaining unedited following DOX treatment compared with total DMPK gene copies, showed an editing efficiency ranging from about 5% in EF1-Crispr in DM to 12% in CK8-Crispr in DM, and up to 14% in EF1-GM. To obtain insight into the different possible editing events, we investigated deletions and inversions. The analysis of CTG-repeat deletions (by double cut and repair) revealed efficiency from about 1% to 3%. To detect possible inversion events due to CTG-repeat excision and then reinsertion in the reverse orientation, we performed qPCR analyses using as positive control a previously described cell clone derived from DM1 edited cells carrying a repeat inversion in the mutated allele (clone C12).16 The results revealed that inversions occurred with a frequency of 0.6%–3.5% in EF1-Crispr in GM/DM and in CK8-Crispr in DM (Figure 2C). Collectively, these data indicate that, in addition to the expected CTG-repeat deletions, other editing events occur at on-target regions, such as inversions, possibly big deletions (encompassing the PCR-primer binding sites), and other undetected events. The sum of the total editing outcomes is close to the efficiency estimated by ribonuclear foci analysis (see below).
Figure 1.

Characterization of Cas9 and sgRNA expression following DOX addition and removal in transduced DM1 cells
(A) qRT-PCR of sg34 and sg589 RNAs expressed in EF1-Crispr and CK8-Crispr myoblasts, in growing medium (GM) and differentiation medium (DM) for 7 days with or without doxycycline (DOX). Expression of sgRNAs was normalized on rpL23 mRNA and expressed relative to the levels measured in untreated cells set as 1 (mean ± SEM). n = 3; ∗∗p < 0.01; ∗∗∗p < 0.001. (B) qRT-PCR of sg34 and sg589 RNAs from myoblasts infected with EF1-Crispr in GM after treatment with DOX for 4 days, followed by either DOX removal for 4 and 7 days or left in DOX medium for the same time. Expression of sgRNAs was normalized on rpL23 mRNA and shown relative to the levels measured after DOX treatment for 4 days set as 1 (mean ± SEM). n = 3; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. (C) Representative western blot analysis of Cas9 protein (Cas9), driven by either CK8 or EF1 promoter, and of the differentiation marker MYOG (MYOG) in proliferating myoblasts (GM) and differentiated myoblasts in DM for 2, 4 and 7 days, transduced with CK8-Crispr or EF1-Crispr (top panel). Tet repressor (TetR) was analyzed in a separate experiment (bottom panel). p38 is shown as loading control. NI, non-infected.
Figure 2.

DOX-inducible CTG-repeat deletions and inversions in DM1-patient-derived myogenic cells
EF1-Crispr- or CK8-Crispr-transduced cells were treated with DOX or left untreated for 3 days (A) or 7 days (B, C) in growing medium (GM) and differentiation medium (DM). The scheme in each panel shows the positions of the primers used for PCR amplification, relative to the sgRNA cutting sites. Black scissors indicate effective cuts (A and B), gray scissors indicate ineffective cut due to inversion of the target sequences (C). (A) Genomic DNA was extracted and analyzed by PCR using DMPK primers binding upstream (upF) and downstream (dwR) of CTG expansion, as shown in the diagram. The black triangle indicates the expected CTG-deleted products from both wild-type and mutated alleles, the red triangle indicates undeleted wild-type allele-derived amplicons. (B) Editing efficiency was analyzed by performing ddPCR with genomic DNAs of EF1-Crispr in GM (n = 3) and DM (n = 3) and CK8-Crispr in DM (n = 3). Primers binding near the editing site upstream and downstream of either (1) the sg589 site (dd589F, dd589R) or (2) the CTG expansion (ddDelF, ddDelR) were used together with reference primers annealing >7,000 bp upstream from editing site (ddPCR.Ref.DMPK F, ddPCR.Ref.DMPK R). Obtained concentrations (copies/μL) of replicates were averaged, and differences due to editing events were expressed in percentage relative to reference (mean ± SEM). (C) qPCR analysis of CTG-repeat inversions was performed in EF1-Crispr in GM and DM and CK8-Crispr in DM. Each sample represents a pool of genomic DNAs derived from three independent experiments. Three different qPCR reactions were performed on the same pools using primers DMPK up R (upR) and DMPK dw1 R (dw1R). The percentage of inversions is calculated compared with a DM1-derived cell clone (clone C12) where the mutated allele is inverted (mean ± SEM).
DOX-inducible decrease of nuclear foci and recovery of normal splicing in proliferating and differentiated DM1 cells
The effect of CRISPR/Cas9 gene editing was first evaluated on reduction/disappearance of ribonuclear inclusions containing CUG repeats (nuclear foci), a hallmark of DM1 cell nuclei, through fluorescent in situ hybridization (FISH) with a fluorescent (CAG)6CA probe. Representative images of nuclear foci in untreated and DOX-treated proliferating (GM) and differentiated (DM) cells are shown in Figure 3A. To ensure that at the time of editing induction cells had withdrawn from the cell cycle and initiated differentiation, we measured cell proliferation 2 days after the shift to DM (D0) by 16 h labeling with bromodeoxyuridine (BrdU), and parallel cultures were treated with DOX or left untreated for 7 days (D7). At D0 most cells were not proliferating (less than 3% BrdU-positive nuclei) and post-mitotic, since they started to express myogenin (MYOG) (data not shown). Myotubes were then fixed and processed for FISH and for MYOG staining. In both GM and DM the number of foci-negative nuclei increased significantly from 8-fold to more than 30-fold following DOX treatment for 7 days (Figure 3B). In proliferating fibroblasts infected with EF1-Crispr the number of foci per nucleus decreased in DOX medium, paralleled by an increase in the percentage of foci-negative nuclei (Figure 3C). Foci number was analyzed also in cells infected with EF1-Crispr and CK8-Crispr and induced to differentiate into myotubes. In cells infected with EF1-Crispr, both total nuclei and MYOG-positive nuclei were analyzed (Figures 3D and 3E), while in cells infected with CK8-Crispr only MYOG-positive nuclei were counted (Figure 3F), Cas9 being expressed at higher levels in differentiated cells (Figure 1C). In both cases the percentage of MYOG-positive nuclei was around 60% and was not affected by DOX treatment (EF1-Crispr: 57.4 ± 3.3 −DOX; 57.2 ± 3.1 +DOX; CK8-Crispr: 62.1 ± 3.3 −DOX; 62.7 ± 2.1 +DOX). In DM, a 4-fold decrease in the number of total (Figure 3D) or MYOG-positive (Figures 3E and 3F) nuclei containing >20 foci was detected, indicating that after induction of gene editing the number of foci in myotubes was progressively reduced. Similar results were obtained in cells infected with EF1-Crispr(1.1) (Figure S2B). It should be noted that in myotubes the number of foci per nucleus is usually much higher than in fibroblasts or undifferentiated cells, due to the increased transcription and stability of DMPK transcript in differentiated cells54 and to the lack of cell proliferation that contributes to foci dilution in dividing cells. Likely for this reason, the number of MYOG-positive and foci-negative nuclei is lower compared with total nuclei (Figures 3B and S2B). In summary, the number of foci-free nuclei in edited cells (Figures 3B–3D) increases from 0.4%–3.7% to 13.2%–28.7% of the entire cell population. These data, together with those obtained from the analysis of individual clones and from the ddPCR analysis, indicate that the total editing efficiency can be estimated at around 15%–20%.
Figure 3.

Reduction of nuclear foci following DOX-induced gene editing in DM1-derived proliferating and differentiated cells
Cells were infected with the indicated lentiviral combinations and cultured in proliferation medium (GM) or differentiation medium (DM) for 7 days (D7) with or without doxycycline (DOX), then fixed and processed for RNA FISH analysis using a fluorescent (CAG)6CA probe and MYOG staining (only for differentiated cells). (A) Representative images of untreated and DOX-treated EF1-Crispr-transduced cells in GM and DM, stained as indicated, are shown. White arrows highlight edited nuclei without foci. Scale bar, 10 μm. Note that treated myotubes contain both foci-negative and foci-positive nuclei. (B) Histograms show the increase of both total (left panel) and MYOG-positive (right panel) foci-free nuclei following DOX treatment. (C and D) Histograms show quantitation of (C) total nuclei containing no foci, ≤5 foci, and >5 foci in growing cells (GM) and (D) no foci, ≤5 foci, between 6 and 20 foci, and >20 foci in differentiated cells (DM) transduced with EF1-Crispr following DOX treatment. (E and F) Histograms show quantitation of MYOG-positive nuclei containing no foci, ≤5 foci, between 6 and 20 foci, and >20 foci in differentiated cells (DM) transduced with (E) EF1-Crispr or (F) CK8-Crispr following DOX treatment. At least 300 nuclei were counted for each condition (mean ± SEM). n = 3–4. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
One important point is to verify whether CRISPR/Cas9-mediated editing affects DMPK transcript production. In the same experimental conditions as described for foci analysis, expression of DMPK mRNA was analyzed in EF1-Crispr and CK8-Crispr by RT-PCR using primers binding upstream and downstream of the CTG repeats in DOX-treated and untreated myoblasts/myotubes. Both amplicons corresponding to unedited wild-type transcripts and edited transcripts from wild-type and mutated deleted alleles were easily detectable, implying that deleted alleles are efficiently expressed and transcripts accumulated (Figure S3A). Since the mutated DMPK mRNA cannot be easily amplified by RT-PCR, northern blot analysis in EF1-Crispr and CK8-Crispr cells following DOX induction was performed. In agreement with the decrease of ribonuclear foci, a reduced accumulation of the CUG-repeated DMPK transcripts was observed (Figure S3B).
Finally, alternative splicing of sarcoplasmic/ER calcium ATPase 1 (SERCA1) and insulin receptor (INSR) transcripts, previously found to be defective for exon inclusion in DM1 myogenic cells,16 was analyzed by standard RT-PCR in EF1-Crispr- and CK8-Crispr-infected DM1 cells and compared with cells derived from unaffected individuals (CT) and with parental uninfected DM1 cells (DM1, Figure 4A). The analysis was carried out following DOX-mediated induction of CRISPR/Cas9 gene editing for 1 day in GM and 1 day in DM, since at this early differentiation time the accumulation of the alternatively spliced isoforms shows the most pronounced difference. Since the increase of the normally spliced SERCA1 and INSR isoforms cannot be accurately quantitated in agarose gels following standard PCR (Figure 4A), qRT-PCR was performed using primer pairs specific for the spliced and the unspliced isoforms. Indeed, both SERCA1 and INSR normal transcript forms, containing exon 22 and exon 11, respectively, showed an increase in EF1-Crispr-, CK8-Crispr-, and EF1-Crispr(1.1)-infected cells following DOX induction (Figures 4B, 4C, and S2C). This small but significant increase in normal splicing is fully compatible with the estimated editing efficiency of 15%–20%. Altogether the experiments described demonstrate that the use of inducible sgRNA expression allows for a tight control of CTG-repeat removal, followed by reduction of ribonuclear foci in proliferating and differentiated cells and improvement of normal splicing. In addition, SpCas9 and eSpCas9(1.1) driven by either EF1a or CK8 promoter gave very similar results.
Figure 4.

Improvement of SERCA and INSR normal splicing after induction of gene editing
SERCA1 and INSR transcripts were analyzed by standard RT-PCR (A) or qRT-PCR (B and C) in EF1-Crispr- and CK8-Crispr-infected DM1 cells, as indicated, and following doxycycline (DOX) induction for 1 day in GM and 1 day in DM. (A) Both spliced and not spliced transcript forms were amplified using the primers hSERCA1 ex22F+R for SERCA1 transcripts and the primers hINSR ex11F+R for INSR transcripts (see diagram on the right). The splicing patterns of SERCA1 and INSR are shown compared with cells derived from unaffected control (CT) and to DM1 parental cells not expressing CRISPR/Cas9 (DM1). (B and C) qRT-PCR of SERCA1 and INSR using primers specific for each isoform normalized to the total amount of SERCA1 and INSR transcripts, respectively (Table S4). Histogram represents the percentage of exon inclusion and exclusion for SERCA1 and INSR transcripts in the absence or presence of DOX, compared with the control cells. For each transcript the percentage of increase of exon inclusion following DOX administration is shown in the table (mean ± SEM, n = 3).
Accuracy and specificity of DOX-inducible gene editing
A crucial point in a CRISPR/Cas9 editing approach is to assess the occurrence of events in undesired loci (off-targets) and the precision in the generation of the desired editing events (on-targets). For this purpose, genomic DNA, extracted from DM1-patient-derived myogenic cells infected with EF1-Crispr and CK8-Crispr and treated with DOX for 7 days as described above, was analyzed by amplicon deep sequencing.
Off-target analysis
To identify potential off-targets for sg34 and sg589, we interrogated four commonly used prediction algorithms (CRISPRoff webserver [v1.1],55 COSMID,56 CRISTA,57 and Cas-OFFinder58). The resulting lists for each sgRNA were intersected and ranked decreasingly by the number of overlaps. Specifically, 50 and 35 predicted off-targets for sg34 and sg589, respectively, were found to be in common for at least two tools (Table S1). Among the tested top 20 of each intersection, 16 and 19 potential off-target sites for sg34 and sg589, respectively (highlighted in Table S1), were amplified by PCR in three independent editing experiments using proliferating EF1-Crispr cells and differentiated CK8-Crispr cells as well as in the matched parental population that served as control. These amplicons were then processed into libraries and paired-end sequencing was performed. The median coverage of the samples was over 6,000×. The obtained reads were analyzed by software designed to identify simple nucleotide variation (SNV) and small indels and their frequencies59 (Table S2). These detailed investigations revealed the absence of indels within the region of any of the tested putative off-targets (Table S2). Only one predicted off-target (ND669, located in the intron of uncharacterized LOC107985242) showed an SNV when compared with genomic reference sequence GRCh38.p13. However, since this variation was also identified in the matched control population, it is attributable to pre-existing genomic variability rather than sequence changes caused by CRISPR/Cas9 treatment. The observed lack of off-targets at the analyzed regions suggests that limiting the expression of the sgRNAs could be functional in preventing off-target events, independently of the Cas9 employed.
On-target analysis
To determine the nature and relative abundance of on-target indels, first we performed PCR amplifications using primer pairs specifically designed to investigate three possible outcomes of editing (Table S3). For the analysis of the desired simultaneous cut of both sgRNAs (double cut), resulting in the removal of the expansion mutation, we used primers located upstream of sg34 and downstream of sg589. Here, to avoid PCR amplification of unedited wild-type alleles, all samples were pre-digested with specific restriction enzymes cutting within the region of the expected deletion. To investigate the possible occurrence of single-cut events caused by either of the sgRNAs, we used primers flanking the target site for sg34 or sg589. All primer pairs carried unique molecular identifiers (UMIs),60 created by adding eight random nucleotides to the 5′ end of the forward primer. In this way, each DNA molecule was labeled with a unique sequence before amplification by PCR, allowing the discrimination between alleles arising from the same genomic locus and sequencing reads produced by PCR. Amplicons from edited populations of EF1-Crispr in GM (n = 3) and DM (n = 2), and CK8-Crispr in DM (n = 2), were used for library preparation and sequencing. For estimation of editing accuracy and the characterization of sequence variations due to CRISPR/Cas9 activity, we took advantage of algorithms specifically designed to analyze genome-editing outcome and to identify and remove read duplicates produced by PCR (“deduplication”).60,61 To investigate the impact of duplicate formation on the outcome of the sequence variation analysis, we evaluated EF1-Crispr populations in detail before and after removal of identified read duplicates (Figures S4 and S5). We found that the percentage of the modified reads as well as fraction of observed insertions, deletions, and substitutions showed only minor or no changes before and after deduplication (Figures S4A and S4B). This was observed for double-cut (Figure S4B) as well as for possible single-cut editing events of both sgRNAs (Figures S5A and S5B). Median coverage after deduplication was more than 40,000×. Since we confirmed that duplicates did not cause artifacts in the downstream sequence analysis, for all subsequent results non-deduplicated data were used. As expected, all EF1-Crispr (in GM and DM) and CK8-Crispr cell populations showed the highest indel frequency in close proximity to the expected cleavage position of Cas9 (i.e. ∼3–4 nt upstream of the protospacer adjacent motif [PAM] sequence). This was observed in the double-cut as well as the single-cut libraries for sg34 and sg589 (Figure 5). Substitutions were found only rarely, in between 0.3% and 6.4% of the reads, always near to the predicted cleavage site (Figures 5, S4 and S5; Table 1). Overall, the proliferating EF1-Crispr populations showed a higher percentage of read modification compared with both of the differentiated CK8- and EF1-Crispr populations. This could be observed in all three possible editing events (Table 1). On average, 93% of all reads derived from double-cut editing by EF1-Crispr infection in GM showed changes compared with the reference sequence, which is the sequence predicted after perfect rejoining following double cut. Instead, 70% of the reads were changed due to double-cut editing in differentiated cell populations by infection of either EF1-Crispr (70%) or CK8-Crispr (72%) (Table 1). Interestingly, analysis of the double-cut editing events revealed a difference in the distribution and size of deletions and insertions in EF1-Crispr in GM (Figure 5A) compared with EF1-Crispr and CK8-Crispr in DM (Figures 5B and 5C). The greatest portion of sequence variations in EF1-Crispr in GM was attributed to deletions (>75% of identified modifications) (Table 1), reaching up to 170–180 bp in length. While in CK8-Crispr the most common variation was due to insertions (>85% of identified modifications), the much rarer deletions (10% or less) (Table 1) reached about 100 bp length. Interestingly, also in EF1-Crispr in DM the most common observed variation was due to insertions, although not as distinct as in CK8-Crispr (>55% of identified modifications). In EF1-Crispr in DM, deletions were more frequent than in CK8-Crispr (at least 25%) and showed maximum length of 55 bp (Table 1). The most common insertion found by double-cut analysis performed for EF1-Crispr in GM, EF1-Crispr in DM, and CK8-Crispr was the nucleotide “T” (93%). This nucleotide is the fourth nucleotide upstream of the PAM sequence in the sg34 target sequence. Furthermore, we observed this preference also in single-cut sg34 libraries of all three Crispr populations, EF1 in GM, EF1 in DM, and CK8 in DM. Here, the most common modification was an insertion (>50%, >70%, and >70%, respectively); more than 80% of these insertions were caused by an additional T (Figure 5 and Table 1). Overall, analysis of sg34 single-cut libraries indicated indels for 54% of sequenced reads in EF1-Crispr grown in GM and much lower levels for both EF1-Crispr and CK8-Crispr in DM. Single-cut modifications caused by sg589 were less frequent compared with sg34 in all analyzed libraries. Nevertheless, also in this case EF1-Crispr cells growing in GM displayed the highest level of modifications compared with the differentiated cells in DM (Figure 5 and Table 1).
Figure 5.

Amplicon sequencing analysis reveals higher editing accuracy in differentiated cells than in proliferating cells
Genomic DNA was extracted from DM1 patient-derived proliferating cells in GM infected with (A) EF1-Crispr (n = 3) and differentiated cells in DM infected with (B) EF1-Crispr (n = 3) or (C) CK8-Crispr (n = 2). The precision of CRISPR/Cas9 activity was analyzed by amplicon sequencing of three possible editing events. For the double cut (simultaneous cut of both sgRNAs), primers located upstream of sg34 and downstream of sg589 were used. Single-cut events were examined using primers flanking the target site for sg34 or sg589 (single-cut sg34, single-cut sg589). The fraction of insertions, deletions, and substitutions resulting from this analysis was obtained by comparison with the reference sequence (Reference Amplicon, expected sequence derived from perfect rejoining at single- and double-cut sites).
Table 1.
Quantitation of deep sequencing on-target analysis of edited DM1-patient-derived myogenic cells infected with EF1-Crispr and CK8-Crispr
| Sample | On-target | Total reads (×1,000) | Deletions (%) | Insertions (%) | Substitutions (%) | Unmodified (%)a |
|---|---|---|---|---|---|---|
| EF1-Crispr.GM_a | double cut | 38.3 | 77.6 | 15.4 | 1.7 | 6.6 |
| single-cut sg34 | 108.1 | 23.3 | 32.3 | 2.4 | 43.5 | |
| single-cut sg589 | 168.6 | 11.5 | 1.8 | 0.4 | 86.5 | |
| EF1-Crispr.GM_b | double cut | 4.2 | 72.5 | 16.8 | 6.4 | 7.7 |
| single-cut sg34 | 131.6 | 22.9 | 27.8 | 1.1 | 48.5 | |
| single-cut sg589 | 160.4 | 15 | 0 | 4.1 | 81.6 | |
| EF1-Crispr.GM_c | double cut | 99.2 | 74.7 | 16.7 | 5.4 | 6.3 |
| single-cut sg34 | 237.4 | 20.1 | 29 | 2.7 | 50.6 | |
| single-cut sg589 | 225.7 | 13 | 3.7 | 4.7 | 81.1 | |
| EF1-Crispr.DM_a | double cut | 298.0 | 30.1 | 39.3 | 0.5 | 30.3 |
| single-cut sg34 | 261.1 | 4.2 | 19.6 | 1.0 | 75.5 | |
| single-cut sg589 | 322.9 | 1.2 | 0.2 | 0.5 | 98.1 | |
| EF1-Crispr.DM_b | double cut | 83.1 | 17.1 | 48.9 | 3.8 | 31.3 |
| single-cut sg34 | 308.5 | 6.5 | 19.9 | 1.1 | 72.9 | |
| single-cut sg589 | 321.0 | 1.7 | 0.1 | 0.6 | 97.6 | |
| CK8-Crispr.DM_a | double cut | 7.2 | 9.2 | 74 | 2.9 | 14.5 |
| single-cut sg34 | 128.9 | 7.2 | 28.3 | 2.2 | 64.0 | |
| single-cut sg589 | 246.2 | 0.8 | 0.6 | 0.3 | 98.3 | |
| CK8-Crispr.DM_b | double cut | 6.4 | 0 | 57.9 | 0.3 | 41.9 |
| single-cut sg34 | 113.9 | 7.5 | 23.1 | 1.5 | 68.9 | |
| single-cut sg589 | 123.7 | 0 | 2.3 | 0.3 | 97.4 |
Since one read can show more than one modification, the total of modified and unmodified reads can be higher than 100%.
In conclusion, sequence variation analysis revealed a higher precision of editing in differentiated cells infected with CK8-Crispr and EF1-Crispr than in proliferating myogenic cells infected with EF1-Crispr. In addition, no differences in on-target editing outcome were observed between SpCas9 and eSpCas9(1.1) nucleases in this cell context.
DOX-inducible DMPK gene editing in skeletal muscle of DMSXL mice
After in vitro characterization, we wanted to test whether inducible gene editing could be obtained also in vivo. To this aim, tibialis anterior (TA) muscles of DMSXL hemizygous or homozygous mice carrying one or two copies, respectively, of mutant human DMPK gene with >1,000 CTG repeats4 were transduced with AAVs of serotype 9 (AAV9) expressing the CRISPR/Cas9 components using a dual-vector approach. Two AAV9 vectors carrying muscle-specific CK8-driven eSpCas9(1.1) (AAV-CK8-eSpCas9) and inducible sgRNA cassettes (AAV-H1-Tet-sgRNA-mCherry) (Figure 6A) were injected into the TA muscles of 4- to 6-week-old DMSXL mice (1.5 × 1011 viral genomes for each AAV). Injected mice were fed with normal or DOX-containing diet for 1 or 2 weeks or DOX-containing drinking water for 1 week before sacrifice. Four weeks post injection the mice were sacrificed and TA muscles collected and analyzed for DMPK gene editing and the expression of Cas9 and sgRNAs. Since DMSXL mice carry one (hemizygous) or two (homozygous) copies of the expanded human DMPK gene and no human wild-type DMPK gene, only the CTG-free edited product can be amplified using human-specific primers in the PCR condition used (see diagram in Figure 6B), and in the case of lack of editing no product is expected. CRISPR/Cas9-mediated editing occurred only in the TAs of AAV-injected and DOX-treated hemizygous (Figure 6B, lanes 4–12) or homozygous (Figure S6, lanes +) mice, while AAV-injected TA muscles of untreated mice (Figure 6B, lanes 1–3) or contralateral TAs of DOX-treated mice (Figure S6, lanes −) were negative for editing. Cas9 and TetR proteins were easily detectable in AAV-injected muscles by western blot analysis (Figure 6D). Expression of both sgRNAs was DOX inducible (Figure 6C), and the levels of Cas9 and sgRNA expression were significantly higher in those TA muscles where editing was readily detectable (Figure 6E). These experiments confirm that inducible expression of the sgRNA components of the CRISPR/Cas9 complex results in DOX-dependent gene editing also in skeletal muscle in vivo.
Figure 6.

DOX-inducible gene editing in skeletal muscle of DMSXL mice
(A) Scheme of AAV constructs containing Cas9 and sgRNAs. ITR, inverted terminal repeat; CK8, creatine kinase 8 promoter; TetO, tetracycline operator; TetR, tetracycline repressor; UbC, ubiquitin C promoter. (B) Editing of genomic DNA extracted from TA muscles of DMSXL hemizygous mice, injected with both the AAV vectors (AAV-CK8-Crispr), and treated with DOX as indicated. Primers DMPK F2 (F2) and DMPK R2 (R2) were used for PCR amplification. In the diagram the expected outcomes in the absence (−DOX) or presence (+DOX) of DOX are indicated. The black triangle indicates the expected CTG-deleted products and the gray triangle indicates non-specific PCR products. (C) qRT-PCR showing fold induction of sg34 and sg589 RNAs in untreated or DOX-treated mice, normalized to mCherry expression; ∗∗p < 0.01, n = 5 (−DOX); n = 12 (+DOX). (D) Western blot analysis of Cas9 and TetR proteins in TA of DMSXL mice, treated as indicated. PBS-injected TA muscle is shown as negative control; p38 is shown as loading control. (E) Dot plots represent qRT-PCR expression analysis of Cas9, sg34, and sg589 RNAs in TA muscles of DMSXL mice injected with the AAV vectors and treated with DOX. RNA expression in samples either with or without CTG editing was normalized to GAPDH expression: ∗p < 0.05, n = 6 (No Editing); n = 11 (Editing).
Discussion
Cas9 nuclease has recently emerged as a flexible tool to modify or interfere with both DNA and RNA through its interaction with single-strand guide RNAs. Different strategies have been used to contrast the DMPK transcript toxic effect in DM1, such as blocking transcription of DMPK using enzymatically inactive Cas9,43 elimination of single-stranded CUG-repeated RNA using RNA-targeting Cas9,41,42 or insertion of a premature polyadenylation signal in the DMPK gene.31 However, while these approaches allow for the selective reduction of mutant DMPK mRNA, most of them have a time-limited duration and do not eliminate the effects of CTG-repeat tracts on the DMPK genomic region.5, 6, 7 Removal of the expansion mutation from the genome is the most direct application of CRISPR/Cas9 in DM1 and has the advantages of being permanent and resulting in the production of expansion-free transcripts. The success of this strategy is documented in a number of in vitro studies including ours, using different cell models such as DM1-patient-derived immortalized myoblasts and MYOD1-expressing immortalized fibroblasts, induced pluripotent stem cells (iPSCs) and iPSC-derived myogenic cells and neural stem cells, embryonic stem cells, and immortalized myoblasts from transgenic mice.16,29, 30, 31,45 In all these studies, removal of the CTG expansion was accompanied by reduction in nuclear foci and reversal of aberrant splicing patterns. Importantly, long-term analysis of DM1 myoblasts undergoing complete excision of the expanded repeat, by transcriptomic, proteomic, and morphological studies, revealed that reversion of DM1-specific features is maintained over time.62 Apparently, the CTG expansion could be removed in a variety of cell types, although with different efficiencies. No clear evidence is yet available regarding the feasibility of CTG-repeat excision in terminally differentiated cells such as myotubes and in quiescent muscle stem cells (satellite cells), which represent crucial targets for in vivo therapy. In this study, using the availability of an inducible CRISPR/Cas9 system, we were able to induce gene editing specifically in post-mitotic myocytes undergoing full differentiation and myotube formation. We found that repeat excision can indeed occur efficiently in these cells and leads to the production of expansion-free DMPK gene transcripts. In addition, we show that gene editing results in a significant decrease in the number of ribonuclear foci in myofibers and in partial recovery of normal splicing.
In view of a future therapeutic application of the CRISPR/Cas9 editing approach for DM1, it is very important to assess the efficiency in the generation of the desired on-target editing events. Equally important is to evaluate the occurrence of unintended on-target events during the excision-joining of the targeted DNA regions and off-target events in undesired loci. While short indels, which frequently occur at the joining regions,16,29,30 should not be detrimental since they take place in a non-coding region of the DMPK gene, other events such as large deletions and inversions may affect the function of DMPK and neighboring genes. To minimize such undesired events, we designed and tested tools for exerting spatiotemporal control over CRISPR/Cas9 activity. By using tissue-specific and/or drug-inducible promoters, we show here that the expression of CRISPR/Cas9 components can be time-limited and localized to specific cell types, including terminally differentiated myogenic cells. In addition, sgRNA expression can be readily induced and then returned to basal levels upon drug removal, thus halting Cas9 activity. This time restriction should reduce the occurrence of off-target effects. Indeed, no evidence of off-target activity was found in the potential off-target genomic regions analyzed by deep sequencing in both proliferating and differentiated cells for both the sgRNAs used. A more comprehensive analysis to detect possible off-target effects could be performed by whole-genome sequencing, although this approach is prone to produce a high rate of false positives due to library preparation, sequencing biases, and variant calling algorithm artifacts;63 in addition, off-target mutations being usually rare, they may not be distinguishable in frequency from naturally occurring mutations.64 Deep sequencing analysis of the on-target site of each sgRNA and of the deleted region in edited cells revealed an unexpectedly high percentage of relatively large deletions at the joining site following double sgRNA cut, but much less upon single cut. Interestingly, we observed a marked difference in the occurrence of large deletions following editing between proliferating cells and post-mitotic differentiated cells. On-target deletions following Cas9-induced DSB have been previously described in different genomic regions and within the CTG repeats and appear to be cell context dependent and correlated with the sgRNA target sequence.65, 66, 67, 68, 69 DSBs elicited by Cas9 nucleases are mainly repaired by the NHEJ and the microhomology-mediated end-joining (MMEJ) pathways, which can induce small indel mutations. In the presence of longer homologies, larger deletions via the single-strand annealing (SSA) pathway may occur. Successful and accurate editing not only depends on the efficiency of NHEJ and MMEJ repair but also on the properties of the nuclease and the design of the guide RNAs utilized. Equally important is the cell context, the chromatin state of the target sites, and the cell cycle state.70 For example, while the NHEJ, a fairly accurate repair system, is active in all cell cycle phases including the post-mitotic state, the MMEJ and SSA pathways are active only in S-G2 phases in proliferating cells.39,68,71 This may explain why we detected large deletions mostly in dividing cells in GM and only marginally in post-mitotic cells in DM. In addition, the sgRNA target sequence may play a role in induction of different DSB repair mechanisms.71,72
In our edited cells insertion of a nucleotide “T” was frequently found following both double cut and sg34 single cut. In sg34 target sequence the nucleotide “T” is the fourth nucleotide upstream of the PAM sequence. It has been shown that many target sites showed recurrent insertions of the same base, suggesting that the choice of inserted nucleotide is non-random.73 Moreover, the inserted base was frequently homologous to the nucleotide at position −4 from the PAM sequence, which is typically the nucleotide upstream of the cleavage site.74 Adenine and thymine bases located at the −4 position from a PAM predominantly generate insertion of a single homologous nucleotide.73 Luckily, recently developed software can explain and predict, to some extent, these indel events,69,72 although this can be done only for a single sgRNA cut, while the outcome of the events occurring following two simultaneous cuts is not yet predictable. Indeed, in our cell context the on-target outcome of single cuts elicited by sg34 and, to a certain extent, sg589 conforms to software prediction. Conversely, the double-cut outcome appears more variable and dependent on the cell-proliferative state.
ddPCR and qPCR analyses at the on-target site indicated that, in addition to the expected CTG deletions, other events may occur such as repeat inversions, possibly big deletions, and other undetected events. Such unintended outcomes were also previously described by other groups.16,29,65, 66, 67 These events can, at least in part, account for the difference observed between the relatively low efficiency of the expected on-target deletion and the strong reduction in the number of foci observed in edited cells, paralleled by a decrease of the CUG-repeated DMPK transcript. However, since the sgRNA target sites are present in the DMPK transcript as well as in the DMPK gene, we cannot exclude a direct effect of RNA-guided Cas9 nuclease on mutated transcript accumulation in the nuclei. Although it has been reported that SpCas9 requires a PAM-presenting DNA oligonucleotide for efficient RNA cleavage in vitro,32 SpCas9 recruited specifically to CUG repeats has been shown to affect mutant RNA stability, likely due to displacement of RNA-binding proteins.41 Concerning the role of the different Cas9 nucleases on genomic DNA, the overall distribution of the repair outcome at on-target sites was shown to be very similar between SpCas9 and eSpCas9 (1.1) used in this study.52,72,73 In our cell model, both Cas9s behave identically regarding editing activity and specificity, as well as for phenotypic reversion of edited cells. In addition, editing activity at putative off-target sites was not detected by either of the two nucleases in our DM1-patient-derived cells.
To progress toward gene therapy in DM1 patients, it is crucial to test the CRISPR/Cas9 strategy in DM1 animal models. AAV-mediated systemic delivery of CRISPR/Cas9 components into mdx mice, a model of Duchenne muscular dystrophy (DMD), was applied by several research groups to delete the mutated exons in the dystrophin gene and rescue the proper reading frame. Gene editing led to restoration of dystrophin production and improvement of muscle function.53,75, 76, 77 Concerning DM1 animal models, in a recent paper local injection of AAVs carrying CRISPR/Cas9 into TA muscle of DMSXL mice resulted in CTG-repeat deletion and decreased nuclear foci in myofibers.44 Here, we confirm and expand the feasibility of this approach by using a drug-inducible CRISPR/Cas9 system, showing that Cas9 activity for as little as 7 days is sufficient to induce repeat excision in skeletal muscle of DMSXL mice. These data support the applicability of therapeutic gene editing in vivo also for DM1. However, important differences between DMD and DM1 mouse models should be evaluated. In mdx mice, even in the presence of low levels of editing, newly produced dystrophin can spread all over the muscle fiber and contribute to increased muscle strength. For DM1 mice, expression of an effective CRISPR/Cas system in a sufficient number of nuclei to achieve CTG excision at therapeutic level could be difficult to obtain, also considering that, DM1 being a multisystemic disorder, many organs must be targeted to reach disease improvement. In addition to AAVs with optimized transducing ability, other promising non-viral systems such as nanoparticle-mediated delivery of CRISPR/Cas9 components78,79 could be used for efficient in vivo genome editing in DM1 models. At the same time, the occurrence of on-target and off-target Cas9 unintended activity will undoubtedly benefit from the use of cell/tissue target-specific Cas9 and spatiotemporal control of gene editing.
Materials and methods
Lentiviral and adeno-associated viral constructs
Viral plasmids generated for this study were produced utilizing NEBuilder HiFi DNA Assembly cloning kit (New England Biolabs, Ipswich, MA). pLenti-EF1a-SpCas9-EGFP, containing a short elongation factor 1a promoter (EF1a), was obtained from Addgene (lentiCas9-EGFP #63592). pLenti-CK8-eSpCas9-EGFP was generated by assembling the BamHI-NheI vector backbone fragment from pLenti-EF1a-SpCas9-EGFP with PCR amplicons containing muscle-specific creatine kinase (CK) minimal promoter CK8 from pAAV-CK8-Cas9 (a gift from J. Chamberlain53) and eSpCas9(1.1) (enhanced specificity Cas952) from eSpCas9(1.1) (Addgene #71814). pLenti-EF1a-eSpCas9-EGFP was generated by assembling fragment BamHI-XbaI from pLenti-EF1a-SpCas9-EGFP with PCR amplicon containing eSpCas9(1.1) from pLenti-CK8-eSpCas9-EGFP. pLenti-H1-Tet-sgRNA-mCherry was generated by assembling in vitro synthesized tetracycline (tet)-responsive derivative (tetO) of the H1 promoter upstream the two sgRNAs (GenScript, Piscataway, NJ) with the fragment PacI-PacI from FgH1tUTCherry, carrying the tetracycline repressor (TetR) and mCherry gene driven by the ubiquitin C (Ubc) promoter (Addgene #85552). AAV-CK8-eSpCas9 was obtained by replacing the EcoRV-BsmI fragment of pAAV-CK8-Cas9 with the EcoRV-BsmI fragment from eSpCa9(1.1). AAV-H1-Tet-sgRNA-mCherry was obtained by assembling the H1-Tet-sgRNA cassettes and UbC-TetR-mCherry from the pLenti-H1tetO2-sgRNAs with the KpnI-SphiI vector backbone from pAAV-CK8-Cas9.
Cell culture and lentiviral infection
DM1 patient-specific myogenic cell lines and non-edited and edited 5, B9, and C12 clones were obtained from primary dermal fibroblasts transduced with hTERT and estrogen-inducible MYOD1. Fibroblasts were derived from a DM1 donor carrying 520 CTG amplifications in the 3′ UTR of the DMPK gene, as previously described.16 Myogenic cells were propagated in Dulbecco’s modified Eagle’s medium (DMEM) without phenol red (Gibco, Thermo Fisher Scientific, Waltham, MA) supplemented with 15% fetal bovine serum (Gibco, Thermo Fisher Scientific). Differentiation to myotubes was induced by growing cells to confluency on dishes coated with 50 μg/mL Collagen I (A10483-01, Gibco, Thermo Fisher Scientific) and replacing the proliferation medium with differentiation medium consisting of DMEM without phenol red supplemented with 10 μg/mL insulin (Sigma-Aldrich, St. Louis, MO), 10 μg/mL transferrin (Gibco, Thermo Fisher Scientific), and 10−7 M β-estradiol (Sigma-Aldrich). All cells were incubated under a 5% CO2 atmosphere at 37°C. Cell lines tested negative for mycoplasma.
Lentiviral particles were produced in 293FT cell line (Thermo Fisher Scientific, Waltham, MA), as previously described.80 DM1 myogenic cells were co-infected with lentiviral particles carrying Cas9 or inducible sgRNAs at a 1:1 ratio and a multiplicity of infection of 5 in order to ensure double infection of most cells. Transduced cells co-expressing EGFP and mCherry were sorted using a BD FACSAria II cell sorter (Flow Cytometry Facility, EMBL, Monterotondo, Italy).
Animal experiments and AAV9 injection
The DMSXL mice (>90% C57BL/6 background) carried 45 kb of human genomic DNA cloned from a DM1 patient as previously described.19,46 A DMSXL mouse colony was established by crossing hemizygous male and female mice. Progeny was genotyped by PCR as described.81 Homozygous DMSXL mice show a high perinatal mortality, are underweight, and require special care after weaning.46 For optimal feeding, gel diet (DietGel76A; Clear H20, Westbrook, ME) and wet food (EMMA23 protein-enriched diet; Mucedola, Settimo Milanese, Italy) were provided on the floor of the cage. Animals were subjected to an experimental protocol approved by the Veterinary Department of the Italian Ministry of Health (no. 832/2019-PR), and experiments were conducted according to the ethical and safety rules and guidelines for the use of animals in biomedical research provided by the relevant Italian laws and European Union's directives (no. 86/609/EEC and subsequent). High-titer stocks of AAVs serotype 9 (AAV9) were produced by InnovaVector (Pozzuoli, NA, Italy). Intramuscular delivery of 1.5 × 1011 viral genomes of each vector (nuclease and targeting) was performed via longitudinal injection into TA muscles of 4-week-old male and female hemizygous mice (n = 17), and 6-week-old female homozygous mice (n = 6) while an equal volume of PBS was injected in the contralateral TA muscle. DOX was administered in the food (625 mg of DOX/kg) or in sweetened drinking water (1 g/L) starting at 2 or 3 weeks post injection. At 4 weeks post injection, TA muscles were collected and frozen in liquid nitrogen for DNA, RNA, and protein extractions.
Genomic DNA analysis
Genomic DNA was isolated from cultured cells using a Kapa Express Extract kit (Roche, Sigma-Aldrich, St. Louis, MO) or a GenElute Mammalian Genomic DNA Miniprep Kit Protocol (Sigma-Aldrich). Genome editing was evaluated by standard PCR using KAPA2G Fast HS Genotyping Mix (2X) (Roche, Sigma-Aldrich). Genomic DNA (10 ng) was amplified with DMPK upF/DMPK dwR primers (Table S4) for 35 cycles.
Genomic DNA was extracted from mouse TA muscles by overnight digestion in proteinase K buffer (100 mM Tris-HCl [pH 8.5], 5 mM EDTA [pH 8], 0.2% SDS, 200 mM NaCl, and 0.5 mg/mL proteinase K) at 55°C in a thermal mixer (Biosan, Riga, Latvia) with agitation at 1,400 rpm. Samples were then treated with 0.5 mg/mL RNAse A and extracted with phenol-chloroform. Genome editing was evaluated by standard PCR using KAPA2G Fast HS Genotyping Mix (2X) (Roche, Sigma-Aldrich) using 60 ng of genomic DNA with DMPK F2/DMPK R2 primers for 40 cycles. CTG-containing fragment inversion was evaluated by qPCR using the standard curve method. Genomic DNA from a cell clone containing CTG-repeat inversion was used to generate a standard curve for both the target gene DMPK at sg589 editing site (DMPK up R/DMPK dw1 R primers) to reveal possible inversion of the deleted fragment, and, as reference, at region >7,000 bp upstream of the editing site on the same DMPK gene (DMPK ref F/DMPK ref R primers). qPCR was performed using PowerUP SYBR Green PCR Master Mix (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA) with an Applied Biosystem 7500 Fast Real-Time PCR System. The sequences of PCR primers are shown in Table S4.
Quantification of editing efficiency ddPCR
For ddPCR we designed specific primers spanning either the double-cut region (DSB and repair by sg34 and sg589 simultaneously) or single-cut region for sg589. As reference, we used the same upstream DMPK primer pair used for qPCR (Table S4). For each assay, genomic DNA (20 ng) was processed with EvaGreen Supermix (Bio-Rad, Hercules, CA, USA) following the manufacturer's instructions. The following conditions were adopted: Hold 95°C/5 min, followed by 40 cycles (95°C/30 s, 58°C/1 min) and signal stabilization (4°C/5 min, 90°C/5 min). For each step the ramp rate was set at 2°C/s. Amplicons were then measured on a QX200 droplet reader (Bio-Rad). Analysis was performed by QuantaSoft (Bio-Rad) and the obtained concentration values (copies/μL) of replicates were averaged. The values obtained by each ddPCR reaction were expressed as differences in percentage relative to reference.
RNA FISH and immunofluorescence analysis
Cells were fixed with 2% formaldehyde and subjected to FISH using a (CAG)6CA probe labeled with Texas red at the 5′ end (IDT, Coralville, IA) in combination with immunofluorescence staining, as described previously.82 To verify the number of foci in MYOG-positive post-mitotic cells, following the last post-hybridization wash the cells were incubated in PBS containing 3% BSA for 15 min and stained sequentially with mouse monoclonal antibodies (mAb) to MYOG (F5D, a gift from G. Cossu), and with goat anti-mouse antibody conjugated Alexa Fluor 488 (Thermo Fisher Scientific). Nuclei were visualized with Hoechst 33258 dye. The samples were examined with an Olympus AX70 immunofluorescence microscope. Images were recorded on an Olympus XM10 camera and processed using the Olympus CellSens standard 1.8.1 software.
Protein analysis
For protein extraction, mouse TA muscles were disrupted by Tissue Lyser LT (Qiagen, Hilden, Germany) in RIPA buffer (140 mM NaCl, 3 mM MgCl2, 1 mM EDTA, and 15 mM HEPES [pH 7.2], also containing 0.5% sodium deoxycholate, 1% NP-40, and 0.1% SDS) supplemented with a cocktail of protease inhibitors (Roche, Sigma-Aldrich) and phosphatase inhibitors. Cultured cells were lysed on plates with the same RIPA buffer. Western blots were carried out using the following antibodies: mouse mAb to MYOG (F5D), mouse mAb to TetR (Clone 9G9) from Takara Bio (Kusatsu, Japan), and mouse mAb to Cas9 (7A9-3A3) and rabbit polyclonal antibody to p38α (C-20) from Santa Cruz Biotechnology (Santa Cruz, CA). After incubation with primary antibodies, filters were incubated with horseradish peroxidase-conjugated goat anti-mouse and anti-rabbit antibodies from Santa Cruz Biotechnology and revealed with a chemiluminescence detection system by Cyanagen (Bologna, Italy). Imaging was carried out by the ChemiDoc XRS Western Blot Imaging System using ImageLab 4.0 software (Bio-Rad).
RNA analysis
Total RNA was extracted with TRIzol reagent (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) following the manufacturer’s instructions. TA muscles were minced into small pieces and disrupted by Tissue Lyser (Qiagen), and cultured cells were lysed on plates. RNA samples were then treated with gDNA Eraser (Takara Bio) to eliminate traces of DNA, and reverse-transcription was performed with the PrimeScript RT-reagent kit (Takara Bio) using oligo(dT) and random primers. PowerUP SYBR Green PCR Master Mix (Applied Biosystems, Thermo Fisher Scientific) was used to analyze RNAs by qPCR using an Applied Biosystem 7500 Fast Real-Time PCR System. Relative expression was calculated with the standard curve method using the reference genes indicated in the figure legends.
Standard PCR of reverse-transcribed mRNAs was performed with GoTaq Flexi DNA Polymerase (Promega, Madison, WI, USA) using the specific primers indicated in the figure legends. Sequences of primer pairs used for each experiment are listed in Table S4.
For northern blot analysis, polyadenylated mRNA was isolated from myogenic cells, grown in GM, and induced to differentiate in DM for 24 h, using the GeneElute Direct mRNA Miniprep kit (Sigma-Aldrich), following the manufacturer's instructions. Northern blotting was performed according to standard procedures. One to two micrograms of polyadenylated RNA was subjected to electrophoresis in a 1.2% agarose gel under denaturing conditions. RNA was transferred to positively charged nylon membranes (Roche, Sigma-Aldrich) by capillary transfer in 20× SSC and hybridized with 5′ digoxygenin-labeled (CAG)6 LNA-oligonucleotide (Eurogentec, Liège, Belgium) and GAPDH (NM_001289745, nt 133–617), generated with DIG-High Prime (Roche, Sigma-Aldrich) according to the random primed labeling technique. The probe-target hybrids were visualized by chemiluminescent assay using the CDP-Star substrate (Roche, Sigma-Aldrich) and the ChemiDoc Imaging System (Bio-Rad).
On-target analysis
Specific primers with standard Illumina adapters for library preparation, spanning either the double-cut region (DSB and repair by sg34 and sg589 simultaneously) or single-cut region (DSB and repair only by sg34 or sg589) for each sgRNA were designed to cover the relevant genomic regions. All primer pairs comprise a UMI for deep amplicon sequencing,60 consisting of a stretch of eight random nucleotides (Table S3). For each genomic DNA sample we performed PCR reactions with all three primer pairs using a Platinum Taq DNA Polymerase kit (Invitrogen, Thermo Fisher Scientific), using 5% DMSO as additive, at the following conditions: hold 94°C/2 min, then 35 cycles (94°C/30 s, 62°C/30 s, 72°C/1 min).
Library preparation for amplicon deep sequencing
Amplicons were purified by AMPURE beads (Beckman Coulter, Brea, CA, USA), with 1:1.2 (v/v) ratio, and resuspended in 30 μL of ddH2O. Library preparation and barcoding were generated using Nextera UDI (Illumina, San Diego, CA, USA) index primers following the manufacturer's instructions. PCR amplification was performed using Kapa Hi Fi polymerase (Roche, Sigma-Aldrich) adopting the following conditions: hold 98°C/3 min, followed by 18 cycles (98°C/30 s, 55°C/30 s, 72°C/1 min) and elongation (72°C for 3 min). PCR products were then purified with AMPURE beads (Beckman Coulter) using 1:0.8 (v/v) ratio. To quantify and verify the integrity of the libraries, an HS1000 DNA Chip on a Tapestation 4100 (Agilent, Santa Clara, CA, USA) was used. All libraries were pooled in equimolar ratios, and paired-end sequencing with 2 × 250 nt read length and 30% Pfix was performed on a MiSeq 600 v3 (Illumina). In detail, all libraries for off-targets, on-target double cut, and on-target single-cut sg34 were sequenced as paired-end reads (2 × 250 nt). The amplicons produced for recognition of sg589 single-cut events, with an approximate length of 200 nt, were treated as single-end reads.
On-target mutation detection and characterization
Read sequences were trimmed to remove sequencing adapters. Paired-end sequences (2 × 250 bp) covering the whole length of on-target double-cut and single-cut sg34 fragments were merged using FLASH.83 The on-target sg589 libraries have a length of roughly 200 nt; thus, these libraries were analyzed as single reads (250 bp). AmpUMI60 was used to parse and trim the UMI adapter from each read, potential UMI errors were discarded, and duplicated reads were removed. The resulting deduplicated reads were then given as input to CRISPResso2,61 the reads were then aligned to the expected corrected sequence, and detailed characterization and analysis of the read sequences was performed.
Off-target analysis
Potential off-targets were identified by interrogating with four publicly available software tools: Cas-OFFinder (http://www.rgenome.net/cas-offinder),58 COSMID (https://crispr.bme.gatech.edu),56 CRISPR-OFF (https://rth.dk/resources/crispr/crisproff),55 and CRISTA (http://crista.tau.ac.il/findofftargets.html).57 Results of the different algorithms were intersected for each sgRNA and ranked decreasingly by number of hits. Sequence variation analysis by next-generation sequencing was performed using gene-specific primers (Table S3) comprising also standard Illumina adapters for library preparation as described for the on-target analysis. For each genomic DNA sample, PCR amplicons were generated as described for the on-target analysis, without addition of DMSO.
Off-target variation detection by amplicon deep sequencing
In brief, the obtained pair-end reads (250 bp) were trimmed to remove adapters and aligned to hg38 reference genome with the BWA-MEM algorithm.84 Subsequently, Freebayes software85 was used to identify single-nucleotide variants and small indels and their frequencies within the predicted off-target sequence. Pre-processing of reads was made using GATK best practices (Genome Analysis Toolkit gatk v3.7).59
Statistical analysis
GraphPad Prism v.4.03 software (GraphPad Software, San Diego, CA) was used for statistical analysis. Continuous variables were analyzed by Student’s t test, Welch’s t test, Mann-Whitney test, or ANOVA, as opportune. All statistical tests were two-sided, and p < 0.05 was considered as statistically significant. Continuous variables were expressed as mean ± standard error of the mean (SEM).
Acknowledgments
This study was supported by Telethon-Italy to G.F. and F.M. (no. GGP19035) and AFM-Téléthon to F.M. and G.F. (no. 23054), F.M. was also supported by the Italian Ministry of Health (“Ricerca Corrente” and RF-2019-12368521), EU-CardioRNA COST Action CA17129, and EU COVIRNA agreement #101016072. M.I., J.B., and D.B. are recipients of fellowships funded by Telethon-Italy. A.P. is supported by the Italian Ministry of Health (SG-2019-12368989). We are grateful to J. Chamberlain and G. Cossu for providing precious reagents.
Author contributions
Conceptualization, G.F. and F.M.; bioinformatics analysis, J.G.-M., C.V., B.C., and C.P.; funding acquisition, G.F. and F.M.; investigation, B.C., C.P., M.I., C.V., J.B., A.P., D.B., G.S., E.G., and S.M.; methodology, G.S., M.R., F.S., and D.L.; project administration, G.F. and F.M.; resources, G.G.; supervision, G.F. and F.M.; writing – original draft, G.F.; writing – review and editing, F.M., C.V., B.C., and C.P. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.11.024.
Contributor Information
Fabio Martelli, Email: fabio.martelli@grupposandonato.it.
Germana Falcone, Email: germana.falcone@cnr.it.
Supplemental information
s
References
- 1.Meola G., Cardani R. Myotonic dystrophies: an update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim. Biophys. Acta. 2015;1852:594–606. doi: 10.1016/j.bbadis.2014.05.019. [DOI] [PubMed] [Google Scholar]
- 2.Andre L.M., Ausems C.R.M., Wansink D.G., Wieringa B. Abnormalities in skeletal muscle myogenesis, growth, and regeneration in myotonic dystrophy. Front. Neurol. 2018;9:368. doi: 10.3389/fneur.2018.00368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sicot G., Gomes-Pereira M. RNA toxicity in human disease and animal models: from the uncovering of a new mechanism to the development of promising therapies. Biochim. Biophys. Acta. 2013;1832:1390–1409. doi: 10.1016/j.bbadis.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 4.Thomas J.D., Oliveira R., Sznajder L.J., Swanson M.S. Myotonic dystrophy and developmental regulation of RNA processing. Compr. Physiol. 2018;8:509–553. doi: 10.1002/cphy.c170002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alwazzan M., Newman E., Hamshere M.G., Brook J.D. Myotonic dystrophy is associated with a reduced level of RNA from the DMWD allele adjacent to the expanded repeat. Hum. Mol. Genet. 1999;8:1491–1497. doi: 10.1093/hmg/8.8.1491. [DOI] [PubMed] [Google Scholar]
- 6.Thornton C.A., Wymer J.P., Simmons Z., McClain C., Moxley R.T., 3rd Expansion of the myotonic dystrophy CTG repeat reduces expression of the flanking DMAHP gene. Nat. Genet. 1997;16:407–409. doi: 10.1038/ng0897-407. [DOI] [PubMed] [Google Scholar]
- 7.Brouwer J.R., Huguet A., Nicole A., Munnich A., Gourdon G. Transcriptionally repressive chromatin remodelling and CpG methylation in the presence of expanded CTG-repeats at the DM1 locus. J. Nucleic Acids. 2013;2013:567435. doi: 10.1155/2013/567435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakamori M., Sobczak K., Puwanant A., Welle S., Eichinger K., Pandya S., Dekdebrun J., Heatwole C.R., McDermott M.P., Chen T., et al. Splicing biomarkers of disease severity in myotonic dystrophy. Ann. Neurol. 2013;74:862–872. doi: 10.1002/ana.23992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang H., Mankodi A., Swanson M.S., Moxley R.T., Thornton C.A. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum. Mol. Genet. 2004;13:3079–3088. doi: 10.1093/hmg/ddh327. [DOI] [PubMed] [Google Scholar]
- 10.Konieczny P., Stepniak-Konieczna E., Sobczak K. MBNL proteins and their target RNAs, interaction and splicing regulation. Nucleic Acids Res. 2014;42:10873–10887. doi: 10.1093/nar/gku767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zu T., Gibbens B., Doty N.S., Gomes-Pereira M., Huguet A., Stone M.D., Margolis J., Peterson M., Markowski T.W., Ingram M.A., et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. U S A. 2011;108:260–265. doi: 10.1073/pnas.1013343108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Falcone G., Perfetti A., Cardinali B., Martelli F. Noncoding RNAs: emerging players in muscular dystrophies. Biomed. Res. Int. 2014;2014:503634. doi: 10.1155/2014/503634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perbellini R., Greco S., Sarra-Ferraris G., Cardani R., Capogrossi M.C., Meola G., Martelli F. Dysregulation and cellular mislocalization of specific miRNAs in myotonic dystrophy type 1. Neuromuscul. Disord. 2011;21:81–88. doi: 10.1016/j.nmd.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 14.Arandel L., Polay Espinoza M., Matloka M., Bazinet A., De Dea Diniz D., Naouar N., Rau F., Jollet A., Edom-Vovard F., Mamchaoui K., et al. Immortalized human myotonic dystrophy muscle cell lines to assess therapeutic compounds. Dis. Model Mech. 2017;10:487–497. doi: 10.1242/dmm.027367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pantic B., Borgia D., Giunco S., Malena A., Kiyono T., Salvatori S., De Rossi A., Giardina E., Sangiuolo F., Pegoraro E., et al. Reliable and versatile immortal muscle cell models from healthy and myotonic dystrophy type 1 primary human myoblasts. Exp. Cell Res. 2016;342:39–51. doi: 10.1016/j.yexcr.2016.02.013. [DOI] [PubMed] [Google Scholar]
- 16.Provenzano C., Cappella M., Valaperta R., Cardani R., Meola G., Martelli F., Cardinali B., Falcone G. CRISPR/Cas9-Mediated deletion of CTG expansions recovers normal phenotype in myogenic cells derived from myotonic dystrophy 1 patients. Mol. Ther. Nucleic Acids. 2017;9:337–348. doi: 10.1016/j.omtn.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gomes-Pereira M., Cooper T.A., Gourdon G. Myotonic dystrophy mouse models: towards rational therapy development. Trends Mol. Med. 2011;17:506–517. doi: 10.1016/j.molmed.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mankodi A., Logigian E., Callahan L., McClain C., White R., Henderson D., Krym M., Thornton C.A. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science. 2000;289:1769–1773. doi: 10.1126/science.289.5485.1769. [DOI] [PubMed] [Google Scholar]
- 19.Seznec H., Lia-Baldini A.S., Duros C., Fouquet C., Lacroix C., Hofmann-Radvanyi H., Junien C., Gourdon G. Transgenic mice carrying large human genomic sequences with expanded CTG repeat mimic closely the DM CTG repeat intergenerational and somatic instability. Hum. Mol. Genet. 2000;9:1185–1194. doi: 10.1093/hmg/9.8.1185. [DOI] [PubMed] [Google Scholar]
- 20.Wang G.S., Kearney D.L., De Biasi M., Taffet G., Cooper T.A. Elevation of RNA-binding protein CUGBP1 is an early event in an inducible heart-specific mouse model of myotonic dystrophy. J. Clin. Invest. 2007;117:2802–2811. doi: 10.1172/JCI32308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bisset D.R., Stepniak-Konieczna E.A., Zavaljevski M., Wei J., Carter G.T., Weiss M.D., Chamberlain J.R. Therapeutic impact of systemic AAV-mediated RNA interference in a mouse model of myotonic dystrophy. Hum. Mol. Genet. 2015;24:4971–4983. doi: 10.1093/hmg/ddv219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jauvin D., Chretien J., Pandey S.K., Martineau L., Revillod L., Bassez G., Lachon A., MacLeod A.R., Gourdon G., Wheeler T.M., et al. Targeting DMPK with antisense oligonucleotide improves muscle strength in myotonic dystrophy type 1 mice. Mol. Ther. Nucleic Acids. 2017;7:465–474. doi: 10.1016/j.omtn.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Konieczny P., Selma-Soriano E., Rapisarda A.S., Fernandez-Costa J.M., Perez-Alonso M., Artero R. Myotonic dystrophy: candidate small molecule therapeutics. Drug Discov. Today. 2017;22:1740–1748. doi: 10.1016/j.drudis.2017.07.011. [DOI] [PubMed] [Google Scholar]
- 24.Ondono R., Lirio A., Elvira C., Alvarez-Marimon E., Provenzano C., Cardinali B., Perez-Alonso M., Peralvarez-Marin A., Borrell J.I., Falcone G., Estrada-Tejedor R. Design of novel small molecule base-pair recognizers of toxic CUG RNA transcripts characteristics of DM1. Comput. Struct. Biotechnol. J. 2021;19:51–61. doi: 10.1016/j.csbj.2020.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wheeler T.M., Leger A.J., Pandey S.K., MacLeod A.R., Nakamori M., Cheng S.H., Wentworth B.M., Bennett C.F., Thornton C.A. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature. 2012;488:111–115. doi: 10.1038/nature11362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wojtkowiak-Szlachcic A., Taylor K., Stepniak-Konieczna E., Sznajder L.J., Mykowska A., Sroka J., Thornton C.A., Sobczak K. Short antisense-locked nucleic acids (all-LNAs) correct alternative splicing abnormalities in myotonic dystrophy. Nucleic Acids Res. 2015;43:3318–3331. doi: 10.1093/nar/gkv163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pascual-Gilabert M., Lopez-Castel A., Artero R. Myotonic dystrophy type 1 drug development: a pipeline toward the market. Drug Discov. Today. 2021 doi: 10.1016/j.drudis.2021.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raaijmakers R.H.L., Ripken L., Ausems C.R.M., Wansink D.G. CRISPR/Cas applications in myotonic dystrophy: expanding opportunities. Int. J. Mol. Sci. 2019;20 doi: 10.3390/ijms20153689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Agtmaal E.L., Andre L.M., Willemse M., Cumming S.A., van Kessel I.D.G., van den Broek W., Gourdon G., Furling D., Mouly V., Monckton D.G., et al. CRISPR/Cas9-Induced (CTGCAG)n repeat instability in the myotonic dystrophy type 1 locus: implications for therapeutic genome editing. Mol. Ther. 2017;25:24–43. doi: 10.1016/j.ymthe.2016.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dastidar S., Ardui S., Singh K., Majumdar D., Nair N., Fu Y., Reyon D., Samara E., Gerli M.F.M., Klein A.F., et al. Efficient CRISPR/Cas9-mediated editing of trinucleotide repeat expansion in myotonic dystrophy patient-derived iPS and myogenic cells. Nucleic Acids Res. 2018;46:8275–8298. doi: 10.1093/nar/gky548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y., Hao L., Wang H., Santostefano K., Thapa A., Cleary J., Li H., Guo X., Terada N., Ashizawa T., Xia G. Therapeutic genome editing for myotonic dystrophy type 1 using CRISPR/Cas9. Mol. Ther. 2018;26:2617–2630. doi: 10.1016/j.ymthe.2018.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O'Connell M.R., Oakes B.L., Sternberg S.H., East-Seletsky A., Kaplan M., Doudna J.A. Programmable RNA recognition and cleavage by CRISPR/Cas9. Nature. 2014;516:263–266. doi: 10.1038/nature13769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D.A., Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 35.Wiedenheft B., Sternberg S.H., Doudna J.A. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482:331–338. doi: 10.1038/nature10886. [DOI] [PubMed] [Google Scholar]
- 36.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jinek M., Jiang F., Taylor D.W., Sternberg S.H., Kaya E., Ma E., Anders C., Hauer M., Zhou K., Lin S., et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science. 2014;343:1247997. doi: 10.1126/science.1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang H.H.Y., Pannunzio N.R., Adachi N., Lieber M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017;18:495–506. doi: 10.1038/nrm.2017.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jasin M., Haber J.E. The democratization of gene editing: insights from site-specific cleavage and double-strand break repair. DNA Repair (Amst) 2016;44:6–16. doi: 10.1016/j.dnarep.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Batra R., Nelles D.A., Pirie E., Blue S.M., Marina R.J., Wang H., Chaim I.A., Thomas J.D., Zhang N., Nguyen V., et al. Elimination of toxic microsatellite repeat expansion RNA by RNA-targeting Cas9. Cell. 2017;170:899–912 e10. doi: 10.1016/j.cell.2017.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Batra R., Nelles D.A., Roth D.M., Krach F., Nutter C.A., Tadokoro T., Thomas J.D., Sznajder L.J., Blue S.M., Gutierrez H.L., et al. The sustained expression of Cas9 targeting toxic RNAs reverses disease phenotypes in mouse models of myotonic dystrophy type 1. Nat. Biomed. Eng. 2021;5:157–168. doi: 10.1038/s41551-020-00607-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pinto B.S., Saxena T., Oliveira R., Mendez-Gomez H.R., Cleary J.D., Denes L.T., McConnell O., Arboleda J., Xia G., Swanson M.S., Wang E.T. Impeding transcription of expanded microsatellite repeats by deactivated Cas9. Mol. Cell. 2017;68:479–490 e5. doi: 10.1016/j.molcel.2017.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lo Scrudato M., Poulard K., Sourd C., Tome S., Klein A.F., Corre G., Huguet A., Furling D., Gourdon G., Buj-Bello A. Genome editing of expanded CTG repeats within the human DMPK gene reduces nuclear RNA foci in the muscle of DM1 mice. Mol. Ther. 2019;27:1372–1388. doi: 10.1016/j.ymthe.2019.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yanovsky-Dagan S., Bnaya E., Diab M.A., Handal T., Zahdeh F., van den Broek W.J.A.A., Epsztejn-Litman S., Wansink D.G., Eiges R. Deletion of the CTG expansion in myotonic dystrophy type 1 reverses DMPK aberrant methylation in human embryonic stem cells but not affected myoblasts. bioRxiv. 2019 doi: 10.1101/631457. [DOI] [Google Scholar]
- 46.Huguet A., Medja F., Nicole A., Vignaud A., Guiraud-Dogan C., Ferry A., Decostre V., Hogrel J.Y., Metzger F., Hoeflich A., et al. Molecular, physiological, and motor performance defects in DMSXL mice carrying >1,000 CTG repeats from the human DM1 locus. PLoS Genet. 2012;8 doi: 10.1371/journal.pgen.1003043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hernandez-Hernandez O., Guiraud-Dogan C., Sicot G., Huguet A., Luilier S., Steidl E., Saenger S., Marciniak E., Obriot H., Chevarin C., et al. Myotonic dystrophy CTG expansion affects synaptic vesicle proteins, neurotransmission and mouse behaviour. Brain. 2013;136:957–970. doi: 10.1093/brain/aws367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Panaite P.A., Kuntzer T., Gourdon G., Lobrinus J.A., Barakat-Walter I. Functional and histopathological identification of the respiratory failure in a DMSXL transgenic mouse model of myotonic dystrophy. Dis. Model. Mech. 2013;6:622–631. doi: 10.1242/dmm.010512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sicot G., Servais L., Dinca D.M., Leroy A., Prigogine C., Medja F., Braz S.O., Huguet-Lachon A., Chhuon C., Nicole A., et al. Downregulation of the glial GLT1 glutamate transporter and purkinje cell dysfunction in a mouse model of myotonic dystrophy. Cell Rep. 2017;19:2718–2729. doi: 10.1016/j.celrep.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang X.H., Tee L.Y., Wang X.G., Huang Q.S., Yang S.H. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol. Ther. Nucleic Acids. 2015;4:e264. doi: 10.1038/mtna.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nelson C.E., Wu Y., Gemberling M.P., Oliver M.L., Waller M.A., Bohning J.D., Robinson-Hamm J.N., Bulaklak K., Castellanos Rivera R.M., Collier J.H., et al. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019;25:427–432. doi: 10.1038/s41591-019-0344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Slaymaker I.M., Gao L., Zetsche B., Scott D.A., Yan W.X., Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351:84–88. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bengtsson N.E., Hall J.K., Odom G.L., Phelps M.P., Andrus C.R., Hawkins R.D., Hauschka S.D., Chamberlain J.R., Chamberlain J.S. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 2017;8:14454. doi: 10.1038/ncomms14454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Davis B.M., McCurrach M.E., Taneja K.L., Singer R.H., Housman D.E. Expansion of a CUG trinucleotide repeat in the 3’ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc. Natl. Acad. Sci. U S A. 1997;94:7388–7393. doi: 10.1073/pnas.94.14.7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alkan F., Wenzel A., Anthon C., Havgaard J.H., Gorodkin J. CRISPR-Cas9 off-targeting assessment with nucleic acid duplex energy parameters. Genome Biol. 2018;19:177. doi: 10.1186/s13059-018-1534-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cradick T.J., Qiu P., Lee C.M., Fine E.J., Bao G. COSMID: a web-based tool for identifying and validating CRISPR/Cas off-target sites. Mol. Ther. Nucleic Acids. 2014;3:e214. doi: 10.1038/mtna.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abadi S., Yan W.X., Amar D., Mayrose I. A machine learning approach for predicting CRISPR-Cas9 cleavage efficiencies and patterns underlying its mechanism of action. PLoS Comput. Biol. 2017;13 doi: 10.1371/journal.pcbi.1005807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bae S., Park J., Kim J.S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clement K., Farouni R., Bauer D.E., Pinello L. AmpUMI: design and analysis of unique molecular identifiers for deep amplicon sequencing. Bioinformatics. 2018;34:i202–i210. doi: 10.1093/bioinformatics/bty264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Clement K., Rees H., Canver M.C., Gehrke J.M., Farouni R., Hsu J.Y., Cole M.A., Liu D.R., Joung J.K., Bauer D.E., Pinello L. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 2019;37:224–226. doi: 10.1038/s41587-019-0032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Andre L.M., van Cruchten R.T.P., Willemse M., Bezstarosti K., Demmers J.A.A., van Agtmaal E.L., Wansink D.G., Wieringa B. Recovery in the myogenic program of congenital myotonic dystrophy myoblasts after excision of the expanded (CTG)n repeat. Int. J. Mol. Sci. 2019;20 doi: 10.3390/ijms20225685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fang H., Wu Y., Narzisi G., O'Rawe J.A., Barron L.T., Rosenbaum J., Ronemus M., Iossifov I., Schatz M.C., Lyon G.J. Reducing INDEL calling errors in whole genome and exome sequencing data. Genome Med. 2014;6:89. doi: 10.1186/s13073-014-0089-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Iyer V., Boroviak K., Thomas M., Doe B., Riva L., Ryder E., Adams D.J. No unexpected CRISPR-Cas9 off-target activity revealed by trio sequencing of gene-edited mice. PLoS Genet. 2018;14 doi: 10.1371/journal.pgen.1007503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cullot G., Boutin J., Toutain J., Prat F., Pennamen P., Rooryck C., Teichmann M., Rousseau E., Lamrissi-Garcia I., Guyonnet-Duperat V., et al. CRISPR-Cas9 genome editing induces megabase-scale chromosomal truncations. Nat. Commun. 2019;10:1136. doi: 10.1038/s41467-019-09006-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ikeda M., Taniguchi-Ikeda M., Kato T., Shinkai Y., Tanaka S., Hagiwara H., Sasaki N., Masaki T., Matsumura K., Sonoo M., et al. Unexpected mutations by CRISPR-Cas9 CTG repeat excision in myotonic dystrophy and use of CRISPR interference as an alternative approach. Mol. Ther. Methods Clin. Dev. 2020;18:131–144. doi: 10.1016/j.omtm.2020.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kosicki M., Tomberg K., Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018;36:765–771. doi: 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mosbach V., Viterbo D., Descorps-Declere S., Poggi L., Vaysse-Zinkhofer W., Richard G.F. Resection and repair of a Cas9 double-strand break at CTG trinucleotide repeats induces local and extensive chromosomal deletions. PLoS Genet. 2020;16 doi: 10.1371/journal.pgen.1008924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thomas M., Burgio G., Adams D.J., Iyer V. Collateral damage and CRISPR genome editing. PLoS Genet. 2019;15 doi: 10.1371/journal.pgen.1007994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bennett E.P., Petersen B.L., Johansen I.E., Niu Y., Yang Z., Chamberlain C.A., Met O., Wandall H.H., Frodin M. INDEL detection, the 'Achilles heel' of precise genome editing: a survey of methods for accurate profiling of gene editing induced indels. Nucleic Acids Res. 2020;48:11958–11981. doi: 10.1093/nar/gkaa975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Newman A., Starrs L., Burgio G. Cas9 cuts and consequences; detecting, predicting, and mitigating CRISPR/Cas9 on- and off-target damage: techniques for detecting, predicting, and mitigating the on- and off-target effects of Cas9 editing. Bioessays. 2020;42 doi: 10.1002/bies.202000047. [DOI] [PubMed] [Google Scholar]
- 72.Allen F., Crepaldi L., Alsinet C., Strong A.J., Kleshchevnikov V., De Angeli P., Palenikova P., Khodak A., Kiselev V., Kosicki M., et al. Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol. 2019;37:64. doi: 10.1038/nbt.4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sledzinski P., Dabrowska M., Nowaczyk M., Olejniczak M. Paving the way towards precise and safe CRISPR genome editing. Biotechnol. Adv. 2021;49:107737. doi: 10.1016/j.biotechadv.2021.107737. [DOI] [PubMed] [Google Scholar]
- 74.Chakrabarti A.M., Henser-Brownhill T., Monserrat J., Poetsch A.R., Luscombe N.M., Scaffidi P. Target-specific precision of CRISPR-mediated genome editing. Mol. Cell. 2019;73:699–713 e6. doi: 10.1016/j.molcel.2018.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Long C., Amoasii L., Mireault A.A., McAnally J.R., Li H., Sanchez-Ortiz E., Bhattacharyya S., Shelton J.M., Bassel-Duby R., Olson E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 2016;351:400–403. doi: 10.1126/science.aad5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nelson C.E., Hakim C.H., Ousterout D.G., Thakore P.I., Moreb E.A., Castellanos Rivera R.M., Madhavan S., Pan X., Ran F.A., Yan W.X., et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351:403–407. doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tabebordbar M., Zhu K., Cheng J.K., Chew W.L., Widrick J.J., Yan W.X., Maesner C., Wu E.Y., Xiao R., Ran F.A., et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2016;351:407–411. doi: 10.1126/science.aad5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Givens B.E., Naguib Y.W., Geary S.M., Devor E.J., Salem A.K. Nanoparticle-based delivery of CRISPR/Cas9 genome-editing therapeutics. AAPS J. 2018;20:108. doi: 10.1208/s12248-018-0267-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee K., Conboy M., Park H.M., Jiang F., Kim H.J., Dewitt M.A., Mackley V.A., Chang K., Rao A., Skinner C., et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat. Biomed. Eng. 2017;1:889–901. doi: 10.1038/s41551-017-0137-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cardinali B., Cappella M., Provenzano C., Garcia-Manteiga J.M., Lazarevic D., Cittaro D., Martelli F., Falcone G. MicroRNA-222 regulates muscle alternative splicing through Rbm24 during differentiation of skeletal muscle cells. Cell Death Dis. 2016;7 doi: 10.1038/cddis.2016.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gomes-Pereira M., Foiry L., Nicole A., Huguet A., Junien C., Munnich A., Gourdon G. CTG trinucleotide repeat "big jumps": large expansions, small mice. PLoS Genet. 2007;3:e52. doi: 10.1371/journal.pgen.0030052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cardani R., Mancinelli E., Sansone V., Rotondo G., Meola G. Biomolecular identification of (CCTG)n mutation in myotonic dystrophy type 2 (DM2) by FISH on muscle biopsy. Eur. J. Histochem. 2004;48:437–442. doi: 10.4081/918. [DOI] [PubMed] [Google Scholar]
- 83.Magoc T., Salzberg S.L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 2013 13033997v2. [Google Scholar]
- 85.Garrison E., Marth G. Haplotype-based variant detection from short-read sequencing. arXiv. 2012 12073907v2. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
s