

Graphical abstract
Keywords: Beta-barrel, CDR, Hypervariable loop, Lipocalin, Protein engineering, Protein scaffold
Highlights
-
•
The lipocalins exhibit four structurally variable loops at one end of a β-barrel.
-
•
Binding sites for diverse ligands occur in the natural lipocalin family members.
-
•
Loop reshaping via combinatorial protein design leads to novel ligand specificities.
-
•
Many crystal structures of Anticalins derived from the Lcn2 scaffold are available.
-
•
Graphical analysis reveals high structural plasticity of the lipocalin loop region.
Abstract
Anticalins are generated via combinatorial protein design on the basis of the lipocalin protein scaffold and constitute a novel class of small and robust engineered binding proteins that offer prospects for applications in medical therapy as well as in vivo diagnostics as an alternative to antibodies. The lipocalins are natural binding proteins with diverse ligand specificities which share a simple architecture with a central eight-stranded antiparallel β-barrel and an α-helix attached to its side. At the open end of the β-barrel, four structurally variable loops connect the β-strands in a pair-wise manner and, together, shape the ligand pocket. Using targeted random mutagenesis in combination with molecular selection techniques, this loop region can be reshaped to generate pockets for the tight binding of various ligands ranging from small molecules over peptides to proteins. While such Anticalin proteins can be derived from different natural lipocalins, the human lipocalin 2 (Lcn2) scaffold proved particularly successful for the design of binding proteins with novel specificities and, over the years, more than 20 crystal structures of Lcn2-based Anticalins have been elucidated. In this graphical structural biology review we illustrate the conformational variability that emerged in the loop region of these functionally diverse artificial binding proteins in comparison with the natural scaffold. Our present analysis provides picturesque evidence of the high structural plasticity around the binding site of the lipocalins which explains the proven tolerance toward excessive mutagenesis, thus demonstrating remarkable resemblance to the complementarity-determining region of antibodies (immunoglobulins).
Introduction
The lipocalins are a family of evolutionarily related proteins that are found ubiquitously in many phyla of life where they are involved in the transport, storage or scavenging of vitamins, hormones and metabolites (Åkerström et al., 2006, Diez-Hermano et al., 2021, Flower, 1996). Despite high sequence diversity – with only a few conserved residues throughout the family – the lipocalins share a highly conserved common fold which is dominated by the central β-barrel backed by an α-helix and an extended strand. The β-barrel is formed by eight antiparallel β-strands which are arranged in a circular manner around a central axis. Closed by short loops and a hydrophobic core of densely packed aromatic side chains at one end, the β-barrel is open to the solvent at the other end, where four loop segments connect each pair of β-strands and, thus, create a pocket to accommodate a ligand (Skerra, 2000). While the β-barrel with the attached α-helix is strictly conserved in the lipocalin fold, the set of four loops is structurally highly variable in terms of length, amino acid sequence and backbone conformation, which explains the broad spectrum of natural ligand specificities that range from vitamin A to FeIII-siderophore complexes (Schiefner and Skerra, 2015).
This bipartite protein architecture prompted efforts to reshape the ligand-binding site of natural lipocalins via combinatorial protein design to generate proteins with novel binding functions, so-called Anticalins (Beste et al., 1999, Richter et al., 2014, Skerra, 2001). This was accomplished by preparing genetic libraries encoding lipocalin variants with random mutations targeted at specific positions within the loop regions and applying powerful selection techniques such as phage display and, more recently, bacterial surface display (Gebauer and Skerra, 2012, Richter et al., 2014). X-ray crystallographic analyses of the first Anticalin examples – with specificities towards fluorescein and digoxigenin, respectively, compared with biliverdin as a natural ligand – revealed considerable changes in the loop conformations of the bilin-binding protein (BBP), a structurally well characterized lipocalin from a butterfly that was initially employed as a scaffold.
Hence, a picture emerged revealing features of the lipocalins similar to immunoglobulins (Igs). Both protein classes comprise a highly conserved framework that supports a structurally variable loop region (known as hypervariable loops or complementarity-determining region (CDR) in the case of Igs) which confers the specific antigen or ligand binding activity (Skerra, 2003). However, there is one crucial biological difference: whereas the mammalian immune system is capable of constantly generating antibodies with new antigen specificities via somatic gene recombination and hypermutation, the lipocalins are genetically fixed in a species, thus comprising an inherited spectrum of ligand-binding activities. In humans, for example, there are not more than a dozen distinct lipocalins – plus some isotypes – all of which have been structurally characterized (Schiefner and Skerra, 2015).
With a maturing Anticalin technology, the focus was directed at medical applications to provide a viable alternative to antibodies, a well-established and most successful class of biopharmaceuticals today (Strohl and Strohl, 2012). Compared with Igs, with their large size (∼1500 residues), a complicated quaternary structure and complex disulfide bridge and glycosylation patterns, the small and robust lipocalin proteins simply comprise a single polypeptide chain of approximately 180 amino acid residues. This offers several benefits, such as much easier biochemical manipulation and recombinant production as well as the facile construction of fusion proteins incorporating additional functions (Deuschle et al., 2021, Richter et al., 2014). To minimize undesired immunogenicity upon administration to patients, human lipocalins were chosen as appropriate scaffolds for protein engineering in this context, in particular the human lipocalin 1 (Lcn1), also known as tear lipocalin, and the human lipocalin 2 (Lcn2), also known as neutrophil gelatinase-associated lipocalin or siderocalin (Schiefner and Skerra, 2015). Several promising Anticalin drug candidates exhibiting specificities towards different medically relevant target proteins, mainly in the areas of oncology and inflammatory diseases, were generated in this manner and are currently subject to clinical studies at various stages (Deuschle et al., 2021, Rothe and Skerra, 2018).
The human Lcn2 has emerged as a particularly fruitful scaffold to yield Anticalin proteins directed against many different targets (Table 1). In this case, the design of the Lcn2-based random library underwent iterative improvement (Gebauer et al., 2013), taking into account X-ray structural data of early Anticalin representatives (Kim et al., 2009, Schönfeld et al., 2009) in the light of theoretical considerations on the efficient physical sampling of an astronomically large sequence space (Richter et al., 2014, Skerra, 2003). Over the years, more than 20 crystal structures of Lcn2-based Anticalins with specificities from small molecules, over peptides with varying lengths, up to macromolecular protein targets have been elucidated (Table 1). Many of these Anticalins were raised against disease-related molecular targets. Notably, all of these Lcn2-based Anticalin proteins share exactly the same lengths for all four loops, since the library design always was limited to amino acid exchanges, without insertions or deletions (Gebauer et al., 2013). Consequently, the three-dimensional structures of these Anticalins can be compared in a straightforward manner, both mutually and versus the natural Lcn2 scaffold, after superposition via the conserved β-barrel (Skerra, 2000). Here, we present a graphical review of these Lcn2-based Anticalins, which offers interesting insights into their structural biology.
Table 1.
Engineered lipocalins with novel ligand specificities used for the graphical analysis.
| Ligand specificity | Anticalin | Ligand type | PDB ID |
|---|---|---|---|
| Y-DTPA | Tb7.N9 | Hapten | 3DSZ |
| Y-DTPA | C26 | Hapten | 4IAW |
| Y-DTPA | CL31 | Hapten | 4IAX |
| Catacalin-TSA | C3A5 | Hapten | 6Z2C |
| Colchicine | Δ4-D6.2(M69Q) | Hapten | 5NKN |
| Colchicine (apo) | D6.2 | Hapten | 6Z6Z |
| Petrobactin | Δ4M2 | Hapten | 6GR0 |
| Petrobactin (apo) | Δ4M2 | Hapten | 6GQZ |
| Aβ peptide | H1GA | Peptide | 4MVL |
| Aβ peptide | US7 | Peptide | 4MVI |
| Hepcidin | Ac3 (I24) | Peptide | 4QAE |
| CTLA-4 ectodomain | PRS-010#3 (O10) | Protein | 3BX7 |
| Fn ED-B | N7A | Protein | 4GH7 |
| Fn ED-B | N7E | Protein | 5 N47 |
| Fn ED-B | N9B | Protein | 5 N48 |
| huCD98hcED | P3D11 | Protein | 6S8V |
| muCD98hcED | C1B12 | Protein | 6SUA |
| Wild-type Lcn2 | UniProt ID P80188 | Siderophore | 1L6M |
Graphical review
Basis of our structural comparison was a set of 17 Anticalins with crystal structures deposited at the Protein Data Bank (PDB), Research Collaboratory for Structural Bioinformatics (Rutgers University, New Brunswick, NJ), showing resolutions of 1.40–3.00 Å, complemented with the one of human wild-type Lcn2 (PDB ID: 1L6M) (Goetz et al., 2002) (see Table 1). Only those Anticalin structures whose coordinate sets showed a continuously modelled polypeptide chain were considered for this analysis. An amino acid sequence alignment prepared with ANTICALIgN software (Jarasch et al., 2016) is shown in Fig. 1. Side chain exchanges resulting from different random library designs (Gebauer et al., 2013), followed by selection against various molecular targets, are clustered within the four structurally variable loop regions of the lipocalin scaffold (Skerra, 2000).
Fig. 1.

Amino acid sequence alignment of 17 engineered lipocalins (Anticalins; see Table 1) with the natural Lcn2 scaffold (PDB ID: 1L6M; UniProt ID: P80188; mature sequence) shown at the top. The 4 structurally variable loop regions (#1–#4; residues 38–51, 70–76, 95–105 and 125–132) are highlighted. The eight β-strands of the barrel are labeled underneath (A–H). Note that the amino acid exchange Q28H is fixed in all Anticalins (for cloning purposes) while the free Cys residue present at position 87 of the natural protein (with its reactive thiol group) was substituted by Ser throughout.
For the purpose of comparison, a comprehensive set of 18 published crystal structures of the unmutated human Lcn2 protein, co-crystallized with different ligands and/or in different crystal forms (Clifton et al., 2019), was prepared. To this end, in total 22 coordinate sets deposited at the PDB with 100 % sequence identity to PDB ID: 1L6M (Goetz et al., 2002), which was also included as reference in the group of the Anticalin proteins described above, were considered; of those, the four entries with the lowest resolutions were omitted, resulting in a collection of 18 X-ray coordinate sets showing 2.04–2.55 Å resolution.
Manipulation of both the Anticalin and the wtLcn2 coordinate sets as well as three-dimensional graphics were prepared with UCSF Chimera 1.15 (http://www.cgl.ucsf.edu/chimera) (Pettersen et al., 2004). Generally, the polypeptide chain with identifier A was chosen for the analysis. Alternate side chain conformations, if deposited, were neglected and some incomplete side chains in the wild-type Lcn2 crystal structures (mostly surface-exposed Lys) were reconstituted with the most plausible rotamer using PyMOL 2.3.3 (Schrödinger, New York, NY). First, the 18 polypeptide chains for each group of proteins were structurally superimposed via the Cα positions of those 58 residues (positions 28–37, 52–58, 63–69, 77–84, 91–94, 106–113, 118–124, 133–139 in the mature wtLcn2 polypeptide) which are structurally conserved throughout the lipocalins (Skerra, 2000) using the wtLcn2 (PDB ID: 1L6M) as template. Then, for each of the residues defined in all crystal structures (sequence positions 7–176) the arithmetic mean position of the 18 Cα atoms was calculated, together with the sample standard deviation of the distances of these 18 Cα positions to the corresponding mean coordinate. These values reflect the local structural deviations of the main chain for the engineered as well as the natural lipocalins.
In the next step, for each residue side chain (i.e. all non-hydrogen atom positions beyond Cα) the unweighted center of mass was calculated. In the case of the amino acid glycine, which lacks a side chain, the Cα position was used. Again, the arithmetic mean position of all 18 side chain centers of mass was calculated, together with the sample standard deviation of the distances of these individual positions to the mean coordinate. Furthermore, for each amino acid position in the polypeptide chain the three-dimensional covariance matrix was calculated for these 18 side chain centers of mass using the function 'numpy.cov()' as implemented in Chimera (see https://numpy.org). These data reflect both the local structural side chain deviations for the engineered as well as the natural lipocalins and their chemical diversity (with side chains differing in size) in the case of the Anticalin group (Fig. 2).
Fig. 2.
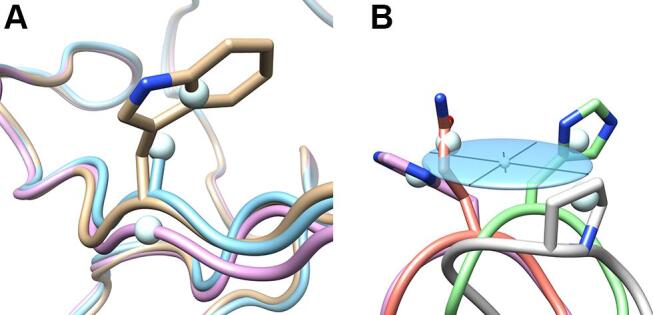
Exemplary illustration of how the side chain mass centers were calculated and visualized. A centroid is defined as the unweighted, arithmetic mean position of all non-hydrogen positions of a residue excluding its backbone atoms (Cα, N, O and C) – except for glycine, where the Cα position was used to represent the (missing) side chain. Anisotropic displacement ellipsoids were generated for illustration of the spatial side chain variance on the basis of these centroids, thus representing both conformational variations and differences in size as a result of mutagenesis. (A) Centroids calculated for tryptophan, alanine and glycine (residue position 81 in PDB IDs: 3BX7, 4GH7 and 6S8V). The centroid for alanine corresponds to Cβ (i.e., the methyl group), for glycine it is Cα. (B) Side chain variance visualized as an ellipsoid (light blue) together with its principal axes, here calculated from the covariance matrix for four side chain centroids (residue position 73 in PDB IDs: 3BX7, 4GH7, 6S8V and 6Z2C).
For both groups of proteins a coordinate set was created comprising the artificial coordinates of the mean Cα atom positions as Cα trace of the polypeptide chain A, together with the sample standard deviations stored as instance attribute 'bfactor' for the class 'Atom', thus replacing the data type normally representing the crystallographic temperature B-factors. In addition, a chain X was appended with the side chain center coordinates as well as the corresponding covariance matrices, this time replacing the anisotropic temperature B-factors (PDB, 2008). These data sets were visualized with Chimera by displaying the mean Cα positions as spheres and drawing lines to the corresponding side chain center positions (Fig. 3). On top of that, the Cα standard deviations were visualized using tubes with varying proportional radii for the polypeptide backbone, and the three-dimensional covariances of the side chains were displayed as ellipsoids using the built-in function 'aniso' of Chimera to visualize anisotropic temperature B-factors.
Fig. 3.


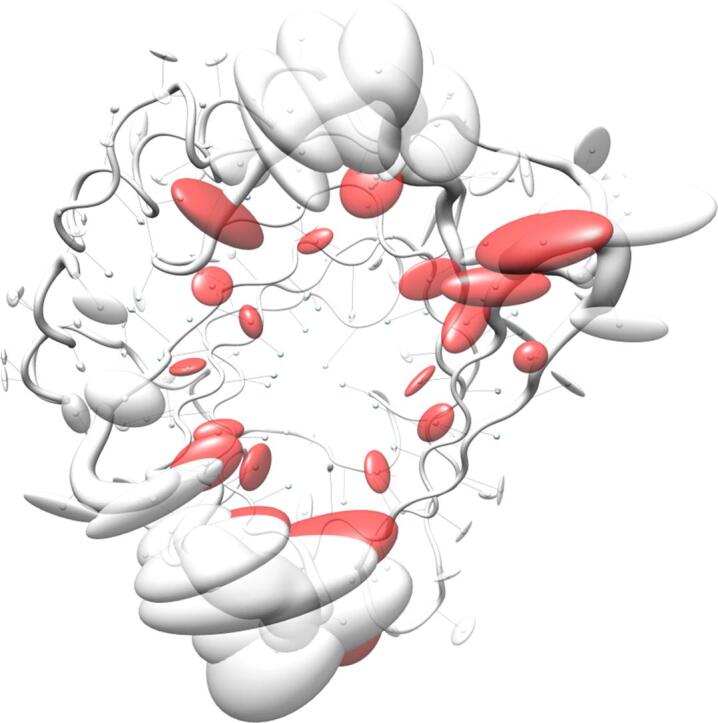
Visualization of backbone and side chain variance for engineered versus natural lipocalins in three-dimensional space. The backbone variance is defined as the standard deviation of the Cα distances to their mean positions calculated for a set of 18 representative protein structures in both groups. The corresponding tube has a proportional radius scaled from 0.1 Å to 2 Å. Note that the tube varies in radius only while the calculated standard deviations include both radial as well as axial displacements. In contrast, the spatial variance of the side chains is depicted as translucent ellipsoids (after deriving the side chain mass centers as explained in Fig. 2). (A) Tube representation for the wtLcn2 scaffold, which shows low variance for the backbone despite of its crystallization with different ligands and in different crystal forms. (B) Tube representation for Anticalins, which exhibit high variance for the backbone, apparently due to the mutagenesis in the loop region (see Fig. 1). The intensity of the blue color illustrates the number of different amino acids at a position observed within the data set (from 0, for unmutated residues, up to 10, for fully randomized positions; see Fig. 1). (C) Tube representation with side chain ellipsoids for the natural lipocalin scaffold from (A), again indicating only minor positional variations for a few side chains. (D) Tube representation with side chain ellipsoids for the Anticalins from (B). In (C) and (D), vectors are drawn from the mean Cα position to the center of each corresponding side chain ellipsoid. To this end, cardinal path calculation without smoothing was applied in Chimera to follow the guide atoms (mean Cα positions) exactly. For the ellipsoids a scale factor of 1.0 was applied.
In another representation, the distances in space from the individual Cα atoms of each of the 18 engineered or the natural lipocalins to the corresponding mean Cα position and, similarly, the corresponding deviations of the side chain center positions, were plotted against the lipocalin amino acid sequence positions using Gnuplot 5.0 (http://www.gnuplot.info) (Fig. 4). In these graphs, the standard deviations calculated for each group of 18 proteins were included for reference. These plots reveal remarkable structural variance both for the polypeptide main chain and the side chains within the group of the engineered Anticalins. Interestingly, at both levels, i.e. main chain and side chain, this results in four pronounced peak areas which coincide with those structurally variable loops that were previously defined based on a structural comparison of different natural members of the lipocalin protein family with known three-dimensional structures (Skerra, 2000) – notably, lipocalins originating from different species and recognizing diverse ligands. Thus, the engineered Anticalins with their spectrum of novel ligand specificities, here exclusively based on human Lcn2, appear like a new class of natural lipocalins.
Fig. 4.


Structural variability of main chain (Cα) and side chain center positions in a goup of 18 engineered lipocalins (Anticalins; see Table 1) versus the wtLcn2 scaffold (as structurally elucidated in 18 different crystallographic environments; see text). The distances between the individual Cα or side chain center positions and their arithmetic mean positions (for the corresponding group of 18 structures) are plotted for each Anticalin/lipocalin against the residue number in the polypeptide chain. The sample standard deviation for the 18 plotted traces (black) as well as the trace for wtLcn2 (green) are highlighted as thick lines. The four structurally variable loop regions (38–51, 70–76, 95–105, 125–132) are marked as light grey horizontal bars underneath the x-axis. (A) Cα distances and (C) side chain center distances for wtLcn2. (B) Cα distances and (D) side chain center distances for the Anticalins.
Interestingly, the precise location of the mutated side chains in the individual Anticalins, which are scattered across these four loops, does not seem to locally modulate the structural variance; rather, each loop region conformationally responds as a whole to the varying set of amino acid exchanges that were introduced versus wtLcn2. The deviation in individual Anticalins from the mean residue position can be as large as 16.8 Å for the main chain (Cα) and 19.2 Å for the side chains centers. This contrasts with a similar plot for the natural Lcn2 which was crystallized with various ligands and in different crystal forms and space groups, where the corresponding deviations are much smaller, with ≤ 1.0 and ≤ 3.6 Å, respectively. This supports the notion of conformational flexibility and structural plasticity as two different phenomena in the case of engineered as well as natural lipocalin proteins (Skerra, 2000, Skerra, 2003). In fact, for the natural Lcn2 structural flexibility is very low – both in the conserved β-barrel part and in the loop regions that shape its ligand pocket – regardless of the changing environment of the protein in different crystal forms or if differing ligands are bound.
On the other hand, if a certain number of amino acid side chains in this protein scaffold are replaced, such as in the Anticalins investigated here, the entire loop region gets reshaped, thus creating binding pockets for ligands diverse in size and shape (Deuschle et al., 2021). Interestingly, however, the (hypothetical) mean Cα trace of the group of Anticalins still closely resembles the one of wtLcn2 (as seen for the low deviation of the thick green line representing wtLcn2 in Fig. 4B and 4D), which means that the natural lipocalin still constitutes a consensus structure for the ensemble of its mutated versions, i.e. the Anticalins. This feature provides a biophysical link to the recognition of consensus sequences representing the energetic minimum of the Ig fold (Steipe et al., 1994) as well as of other protein classes whose members share high homology (Sternke et al., 2020).
The pronounced structural variability among the Anticalins is even better illustrated when looking at a superposition of the engineered versus the natural lipocalin structures in three-dimensional space (Fig. 3). Here, the backbone deviations, represented by a tube with varying radius, substantially coincide with the structural deviations of the side chains as depicted by ellipsoids. Notably, while for randomized positions these ellipsoids both depict changes in side chain size and in orientation, conformational changes alone give rise to similarly extended ellipsoids in neighboring positions that harbour conserved residues (cf. Fig. 1). Thus, while merely about half of all possible residues within the loop region of the wtLcn2 scaffold were actually randomized in the different Anticalin libraries described so far (Fig. 5) – in order to allow efficient physical sampling of the resulting sequence space as explained elsewhere (Richter et al., 2014) – many more residues within the loop region structurally respond to the mutagenesis. This results in fully reshaped ligand pockets, as noted before (Schönfeld et al., 2009), and explains why the engineered lipocalins can efficiently bind diverse ligands representing different classes, such as proteins, peptides and small molecules (Deuschle et al., 2021, Richter et al., 2014).
Fig. 5.

Illustration of mutated amino acid positions in different Lcn2-based random libraries using similar graphical visualization as in Fig. 3D. (A) First-generation library designed to select Anticalins specific for protein targets (Schönfeld et al., 2009). (B) Second-generation library designed to select Anticalins specific for small-molecule targets (Kim et al., 2009). (C) Third-generation library designed to select Anticalins against protein and peptide antigens as well as hapten targets building upon structural analyses of Anticalins successfully selected from the first two libraries (Gebauer et al., 2013).
Of course, it would be interesting to see to which extent this structural deviation is attributable to the effect of ligand binding. In fact, a case of induced fit had been described for an Anticalin selected against the immunological checkpoint receptor CTLA-4 (Schönfeld et al., 2009) where loops #3 and #4 changed their conformations considerably when comparing the crystal structure of the unbound Anticalin with the one of the ligand complex, and loop #3 even appeared structurally disordered in the absence of CTLA-4. On the other hand, in the case of Anticalins selected against the hapten-type ligand Y-DTPA, the crystal structures of two related mutants solved in the presence or absence of the ligand revealed more subtle effects, with relevant conformational changes only visible for loop #1 (Kim et al., 2009). Crystallographic analysis of an Anticalin engineered to tightly complex and scavenge the siderophore petrobactin revealed even fewer differences upon ligand binding (Dauner et al., 2018). However, so far the number of Anticalins that have been structurally characterized both in the ligand-bound and the free state is still too small to allow a dedicated comparison of the kind presented here.
In conclusion, the general feature of a loop region with pronounced structural plasticity, including high tolerance towards side chain replacements, as noted early on (Skerra, 2000), distinguishes the lipocalin scaffold from other alternative binding proteins that are derived from proteins with more rigid secondary structural elements, for example Affibodies and DARPins (Gebauer and Skerra, 2019). Taken together, the structural plasticity of their binding sites, as illustrated in this graphical review, renders engineered lipocalins functionally more similar to Igs, or antibodies, regarding the ability to specifically recognize and tightly bind vastly diverse antigens.
Declaration of Competing Interest
A.S. is cofounder and shareholder of Pieris Pharmaceuticals, Inc. All other authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper. Anticalin® is a registered trademark of Pieris Pharmaceuticals GmbH.
References
- Åkerström B., Borregaard N., Flower D.A., Salier J.-S. Landes Bioscience; Georgetown, Texas: 2006. Lipocalins. [Google Scholar]
- Beste G., Schmidt F.S., Stibora T., Skerra A. Small antibody-like proteins with prescribed ligand specificities derived from the lipocalin fold. PNAS. 1999;96(5):1898–1903. doi: 10.1073/pnas.96.5.1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifton M.C., Rupert P.B., Hoette T.M., Raymond K.N., Abergel R.J., Strong R.K. Parsing the functional specificity of Siderocalin/Lipocalin 2/NGAL for siderophores and related small-molecule ligands. J. Struct. Biol. X. 2019;2:100008. doi: 10.1016/j.yjsbx.2019.100008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauner M., Eichinger A., Lücking G., Scherer S., Skerra A. Reprogramming human siderocalin to neutralize petrobactin, the essential iron scavenger of Anthrax bacillus. Angew. Chem. Int. Ed. Engl. 2018;57(44):14619–14623. doi: 10.1002/anie.201807442. [DOI] [PubMed] [Google Scholar]
- Deuschle F.-C., Ilyukhina E., Skerra A. Anticalin® proteins: from bench to bedside. Expert Opin. Biol. Ther. 2021;21(4):509–518. doi: 10.1080/14712598.2021.1839046. [DOI] [PubMed] [Google Scholar]
- Diez-Hermano S., Ganfornina M.D., Skerra A., Gutiérrez G., Sanchez D. An evolutionary perspective of the lipocalin protein family. Front. Physiol. 2021;12:718983. doi: 10.3389/fphys.2021.718983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flower D.R. The lipocalin protein family: structure and function. Biochem. J. 1996;318(1):1–14. doi: 10.1042/bj3180001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebauer M., Skerra A. Anticalins: small engineered binding proteins based on the lipocalin scaffold. Methods Enzymol. 2012;503:157–188. doi: 10.1016/B978-0-12-396962-0.00007-0. [DOI] [PubMed] [Google Scholar]
- Gebauer M., Skerra A. Engineering of binding functions into proteins. Curr. Opin. Biotechnol. 2019;60:230–241. doi: 10.1016/j.copbio.2019.05.007. [DOI] [PubMed] [Google Scholar]
- Gebauer M., Schiefner A., Matschiner G., Skerra A. Combinatorial design of an Anticalin directed against the extra-domain B for the specific targeting of oncofetal fibronectin. J. Mol. Biol. 2013;425(4):780–802. doi: 10.1016/j.jmb.2012.12.004. [DOI] [PubMed] [Google Scholar]
- Goetz D.H., Holmes M.A., Borregaard N., Bluhm M.E., Raymond K.N., Strong R.K. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol. Cell. 2002;10(5):1033–1043. doi: 10.1016/s1097-2765(02)00708-6. [DOI] [PubMed] [Google Scholar]
- Jarasch A., Kopp M., Eggenstein E., Richter A., Gebauer M., Skerra A. ANTICALIgN: visualizing, editing and analyzing combined nucleotide and amino acid sequence alignments for combinatorial protein engineering. Protein Eng. Des. Sel. 2016;29(7):263–270. doi: 10.1093/protein/gzw016. [DOI] [PubMed] [Google Scholar]
- Kim H.J., Eichinger A., Skerra A. High-affinity recognition of lanthanide(III) chelate complexes by a reprogrammed human lipocalin 2. J. Am. Chem. Soc. 2009;131(10):3565–3576. doi: 10.1021/ja806857r. [DOI] [PubMed] [Google Scholar]
- PDB, 2008. Protein Data Bank Contents Guide: Atomic Coordinate Entry Format Description Version 3.20 – Document Published by the wwPDB.
- Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Richter A., Eggenstein E., Skerra A. Anticalins: exploiting a non-Ig scaffold with hypervariable loops for the engineering of binding proteins. FEBS Lett. 2014;588(2):213–218. doi: 10.1016/j.febslet.2013.11.006. [DOI] [PubMed] [Google Scholar]
- Rothe C., Skerra A. Anticalin® proteins as therapeutic agents in human diseases. BioDrugs. 2018;32(3):233–243. doi: 10.1007/s40259-018-0278-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiefner A., Skerra A. The menagerie of human lipocalins: a natural protein scaffold for molecular recognition of physiological compounds. Accounts Chem Res. 2015;48(4):976–985. doi: 10.1021/ar5003973. [DOI] [PubMed] [Google Scholar]
- Schönfeld D., Matschiner G., Chatwell L., Trentmann S., Gille H., Hülsmeyer M., Brown N., Kaye P.M., Schlehuber S., Hohlbaum A.M., Skerra A. An engineered lipocalin specific for CTLA-4 reveals a combining site with structural and conformational features similar to antibodies. Proc. Natl. Acad. Sci. USA. 2009;106(20):8198–8203. doi: 10.1073/pnas.0813399106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skerra A. Lipocalins as a scaffold. Biochim. Biophys. Acta. 2000;1482(1-2):337–350. doi: 10.1016/s0167-4838(00)00145-x. [DOI] [PubMed] [Google Scholar]
- Skerra A. 'Anticalins': a new class of engineered ligand-binding proteins with antibody-like properties. J. Biotechnol. 2001;74(4):257–275. doi: 10.1016/s1389-0352(01)00020-4. [DOI] [PubMed] [Google Scholar]
- Skerra A. Imitating the humoral immune response. Curr. Opin. Chem. Biol. 2003;7(6):683–693. doi: 10.1016/j.cbpa.2003.10.012. [DOI] [PubMed] [Google Scholar]
- Steipe B., Schiller B., Plückthun A., Steinbacher S. Sequence statistics reliably predict stabilizing mutations in a protein domain. J. Mol. Biol. 1994;240(3):188–192. doi: 10.1006/jmbi.1994.1434. [DOI] [PubMed] [Google Scholar]
- Sternke M., Tripp K.W., Barrick D. The use of consensus sequence information to engineer stability and activity in proteins. Methods Enzymol. 2020;643:149–179. doi: 10.1016/bs.mie.2020.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohl W.R., Strohl L.M. Woodhead; Cambridge, UK: 2012. Therapeutic Antibody Engineering: Current and Future Advances Driving the Strongest Growth Area in the Pharmaceutical Industry. [Google Scholar]