

Abstract
Single-cell RNA sequencing data is characterized by a large number of zero counts, yet there is growing evidence that these zeros reflect biological variation rather than technical artifacts. We propose to use binarized expression profiles to identify the effects of biological variation in single-cell RNA sequencing data. Using 16 publicly available and simulated datasets, we show that a binarized representation of single-cell expression data accurately represents biological variation and reveals the relative abundance of transcripts more robustly than counts.
INTRODUCTION
Single cell RNA sequencing (scRNAseq) data are highly sparse, and the common belief is that the zero values are primarily caused by technical artifacts (often referred to as dropouts). Although more zeros are observed in scRNAseq data than expected, these can largely be explained by biological rather than technical factors (1). Also, the amount of zeros in scRNAseq is in line with distributional models of molecule sampling counts (2,3). These distributional models show that a zero observation is not simply a missing-value, as a missing-value would provide no information. On the contrary, a zero observation for a gene reveals that the respective gene is unlikely to be highly expressed (3). Methods that utilize zero observations for feature selection (4,5) and cell type clustering (6) have recently been developed and perform better or comparable with methods relying on the continuous expression values of highly variable genes. For instance, Qiu (6) binarized scRNAseq count data, where each zero remains zero and every non-zero value was assigned a one. With this binary representation, cell type clusters were identified based on co-occurrence of transcripts. Yet, it is not clear whether differences in the number of zeros for a gene also reflect differences across distinct biological cell populations. Therefore, we investigated whether biological differences across cell population can be identified using Binary Differential Analysis (BDA) rather than the commonly used differential expression analysis (DEA). Instead of relying on changes in the expression value of genes across cell populations, which can be sparse and are subject to pre-processing steps, we analyzed the binary expression patterns across biological distinct cell populations, i.e. are there more (or less) zeros for a gene in condition A compared to condition B. Taken together the main contribution of our work is that we show that the binarization of gene expression is biologically relevant and can be used to test for differences between a wide variety of groupings, and that this holds across different datasets, as well as different single-cell protocols.
MATERIALS AND METHODS
Single-cell RNA-seq datasets
In total, 16 scRNAseq dataset (14 human and 2 mouse) were used to investigate the utility and biological relevance of binarized expression profiles of genes (Table 1). All datasets had pre-annotated cell types and conditions. From the corresponding references, un-normalized count matrices were acquired, and only annotated cells were kept for further analysis. For each dataset, we extracted the annotated cell type, patient ID, and to which of contrasting cell population the cell belonged from the included meta data. This was slightly different for the aging mouse atlases and cancer atlas. For the aging mouse atlases (7), instead of annotated cell types we retrieved the tissue names. For the cancer atlas (8), the contrasting cell populations were defined by cell type, so we retrieved the tissue and the cancer-type for each cell. Each dataset was separately pre-processed. For BDA, the count matrices were transformed to a binary representation, where each zero remain zero and every non-zero value was assigned a one. For the DEA, each count matrix was log-normalized using Seurat 3.2.2 (9), such that  , where
, where 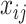 and
and 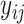 are the raw and normalized values for every gene
are the raw and normalized values for every gene  in every cell
in every cell 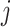 , respectively. This normalizes the feature expression measurements for each cell by the total expression, multiplies this by a scale factor (10 000 by default), and log-transforms the result. The cancer atlas was already normalized, as it was a merger of multiple datasets.
, respectively. This normalizes the feature expression measurements for each cell by the total expression, multiplies this by a scale factor (10 000 by default), and log-transforms the result. The cancer atlas was already normalized, as it was a merger of multiple datasets.
Table 1.
Single cell datasets included in this study.
| Dataset | No. of unique individuals | No. of cells | No. of genes | Contrasting subpopulation defined by: | Description | Protocol | Reference |
|---|---|---|---|---|---|---|---|
| Alzheimer’s Disease (AD) | 14 | 13 214 | 10 850 | Control versus AD | Entorhinal cortex | 10x Chromium | (21) |
| Major Depressive Disorder (MDD) | 34 | 78 886 | 30 062 | Control versus MDD | Prefrontal cortex | 10x Chromium | (31) |
| Type 2 Diabetes (T2D) | 10 | 3514 | 26 271 | Control versus T2D | Pancreas | Smart-seq2 | (32) |
| Coronavirus Disease 2019 (COVID19) | 13 | 44 721 | 26 361 | Control versus COVID19 | PBMCs | Seq-Well | (33) |
| Lung adenocarcinoma (LUAD, Lung) | 22 | 88 144 | 29 634 | Normal tissue versus cancerous tissue | Lung | 10x Chromium | (34) |
| Lung adenocarcinoma (LUAD, Lymph node) | 17 | 54 577 | 29 634 | Normal tissue versus cancerous tissue | Lymph node | 10x Chromium | (34) |
| Four cancers (T-cells) | 14 | 132 549 | 22 815 | Normal tissue versus cancerous tissue | Colon, Endo, Lung, Renal | 10x Chromium | (35) |
| Aging Mouse Atlas FACS | 14 | 74 157 | 22 966 | 3m versus 24m | Aging mouse | Smart-seq2 | (7) |
| Aging Mouse Atlas Droplet | 11 | 83 262 | 20 138 | 3m versus 24m | Aging mouse | 10x Chromium | (7) |
| Allen Brain Atlas (Medialis temporalis Gyrus) | 8 | 14 689 | 48 304 | Inhibitory neurons versus excitatory neurons | Medialis temporalis Gyrus | Smart-Seq v4 | (36) |
| Colorectal Cancer | 23 | 63 502 | 27 946 | Normal tissue versus cancerous tissue | Colon CRC cells | 10x Chromium | (37) |
| Cortex Neurons | 5 | 9451 | 28 985 | Inhibitory neurons versus excitatory neurons | Cortex | 10x Chromium | (25) |
| Cortex Oligodendrocytes | 5 | 307 | 28 985 | OPC versus ODC | Cortex | 10x chromium | (25) |
| Substantia Nigra | 7 | 4711 | 28 985 | OPC versus ODC | Substantia Nigra | 10x chromium | (25) |
| Cancer Atlas (1) | 171 | 33 346 | 50 705 | Regulatory T cells versus T helper cells | Cancer atlas | Multiple | (8) |
| Cancer Atlas (2) | 162 | 30 105 | 48 942 | Naive-memory CD4 T cells versus Transitional memory CD4 T cells | Cancer atlas | Multiple | (8) |
Statistical analysis
P-values were corrected for multiple tests with the Benjamini–Hochberg procedure and significance was assumed at an adjusted P-value of PFDR ≤ 0.05. Spearman’s rank correlation coefficient and the associated P-values were calculated using the cor.test function in R v4.0.2.
Differential expression analysis
DEA was performed using the Wilcoxon Rank Sum test using the FindMarkers function in Seurat 3.2.2 (9). Note that the Wilcoxon Rank Sum test from Seurat takes into account zero measurements and handles them as ties between contrasting cell populations. Genes coding for ribosomal proteins were excluded and we only tested genes that were expressed in at least 10% of the cells in either of the respective groups of interest. This is the default option in the FindMarkers function and speeds up testing by ignoring infrequently expressed genes. P-values were corrected for multiple tests.
Binary differential analysis (BDA)
As the sampling process of biomolecules is the main cause for generating zeros, as illustrated by Svensson (2) and Sarkar and Stephens (3). The probability of measuring a gene is dependent on the relative abundance; more abundant genes are less likely to result in a zero observation. Extrapolating this to a population of cells, the number of zeros for a gene is representative of the abundance within the respective cell population, and differences in the number of zeros between two groups of cells are representative of differential abundance. Lastly, we assume that within a single-cell experiment zeros induced by stochastic processes are not confounded by the groupings. In other words, a stochastically induced zero is equally likely to happen in either cell population, as such, in this setting can be ignored.
Implementation
In the main analyses, to statistically test for significant differences of zero observations between pre-defined groups in scRNAseq data, we used a logistic regression (BDA-LR). Specifically, the glm(family = ‘binomial’) function in R v4.0.2, with the binarized expression pattern of the genes as outcome variables and the grouping (i.e. healthy versus diseased) as predictor variable. We have used logistic regression because it allows to add covariates to correct for potential confounding factors. Moreover, predictor variables as well as covariates can be continuous, allowing for complex study designs. All genes that were tested with DEA were also tested with BDA. The resulting association P-values were corrected for multiple tests (see Statistical analysis). In addition to logistic regression, we used the Chi-squared test (BDA-chisq), the Fisher’s exact test (BDA-fisher) and binary Pearson's correlation (BDA-Phi) on the simulated data. The Chi-squared test and the Fisher’s exact tests were performed with the chisq.test() and fisher.test() R functions, respectively. These tests were performed for each gene on the contingency table representing the binarized gene expression against the pre-defined groupings. The binary Pearson’s correlation was calculated between each binarized gene and the pre-defined groupings and performed with the cor.test() R function, where one group was defined as 0 and the other group as 1. In a binary setting the outcome statistic of Pearson’s correlation is called Phi (φ).
BDA–DEA comparison
For the comparison between BDA and DEA, we investigated agreement and disagreement between detected genes and the linear association between the logOR and logFC. Agreement was calculated by the Jaccard index, i.e. number of genes that both tests commonly detected, divided by the total number of genes that were detected. Agreement was calculated on the combination of all datasets and for each individual dataset. The disagreement was investigated by means of inspecting characteristics of BDGs-only and DEGs-only. BDGs-only were defined as genes that were detected (PFDR ≤ 0.05) by BDA and were not detected (PFDR > 0.05) by DEA. Conversely, DEGs-only were defined as genes that were detected (PFDR ≤ 0.05) by DEA and were not detected (PFDR > 0.05) by BDA. The Spearman’s rank correlation coefficients between the logOR and logFC were calculated with the estimates of all tested genes of the respective datasets. The scale differences for every dataset, between logOR and logFC, were calculated with a linear model on the estimates of all tested genes of the respective datasets, using the lm function in R v4.0.2. The logOR was specified as outcome variable and the logFC as predictor variable. The resulting slopes were interpreted as scale differences between the logOR and logFC
Simulation
Data were simulated with muscat 1.2.1 (10). The provided PBMC dataset (11) was used as reference. In total, 100 simulated datasets were generated with varying sample sizes (1000 cells, 2000 cells, 5000 cells and 10 000 cells), 25 simulations per sample size. For each simulation 1000 genes were generated of which 25% were differently expressed between two groups of equal size. For all tests we calculated the False Positive Rate (FPR), Positive Predictive Value (PVV) and accuracy (F1-score) per simulation. Performance was evaluated of 12 DEA methods. Eight methods implemented in Seurat (wilcox, bimod, t, negbinom, poisson, LR, MAST (12), DESeq2 (13)), 4 additional methods (DEsingle (14), BPSC (15), monocle (16), limmaVoom (17)) and 4 BDA methods (logistic regression, chi squared test, Fisher’s exact test and binary Pearson’s correlation). For the runtime benchmark, each run of the simulation of every tests was also timed with proc.time()function in R. Tests requiring >20 min computational time on one simulated dataset were excluded.
Validation with existing bulk RNA-seq data
The AD bulk RNA-seq datasets were acquired from Gemma (18). The first dataset from Friedman et al. (19) consisted of 33 controls (CT) and 84 samples from individuals diagnosed with Alzheimer’s Disease (AD) collected from the fusiform gyrus. This dataset was reprocessed by Gemma and no batch effects were present. For the differential expression analysis in bulk we used the Wilcoxon Rank Sum test from limma v3.44.3 (20). In total, 2228 genes were tested for differential expression, as these genes were also included in the scRNAseq AD dataset (21) analysis. The second dataset from Hokama et al. (22) consisted of 47 controls and 32 AD samples. The samples originated from the frontal cortex (NCT = 18, NAD = 15), temporal cortex (NCT = 19, NAD = 10) and hippocampal formation (NCT = 10, NAD = 7). The data was reprocessed and batch corrected by Gemma. For the differential expression analysis no distinction was made between brain regions. In total, 2001 genes were tested for differential expression. All resulting association P-values were corrected for multiple tests. For validation, the significant BDGs and DEGs from the scRNAseq AD dataset analyses were compared with the significantly differentially expressed genes from the bulk analyses. Venn diagrams were plotted with ggVennDiagram(v0.3) (23). Correlations were calculated between the logOR and logFC of the single-cell analysis with the bulk logFC.
RESULTS
BDA competitive with Wilcoxon Rank Sum test
As proof of concept, we performed BDA with a simple logistic regression on binarized expression profiles from 16 scRNAseq datasets (662 825 cells in total, Table 1). We compared the results of BDA-LR with those of differently expressed genes (DEGs) detected using the commonly used Wilcoxon Rank Sum test, which is also top ranked for single cell analyses (9,24). We tested each gene using both BDA-LR and DEA for differences between conditions (6 datasets), cell types (6 datasets) and normal versus cancerous tissues (4 datasets, Figure 1A). Across all datasets, a total of 96 275 significant genes (PFDR ≤ 0.05) were identified with either BDA-LR (92 381 genes) or DEA (91 521 genes). Of these, 87 627 were identified by both tests, resulting in a Jaccard index of 0.91. This high degree of agreement is also reflected in each individual dataset (median = 0.92, minimum = 0.76, and maximum = 0.99). We did not use a log fold-change (logFC) or log odds-ratio (logOR) threshold, as for each dataset and comparison different thresholds are appropriate. In all datasets, the logFC and logOR were significantly (spearman) correlated (median (ρ) = 0.90, minimum (ρ) = 0.49 and maximum (ρ) = 0.98, P ≤ 5 × 10–100). The three datasets with the lowest correlation coefficient between logOR and logFC (ρ ≤ 0.62) were datasets generated using the Smart-seq protocol (Table 1). Across the datasets, we observed an average increase of 1.80 in logOR (median = 1.70, Q1 = 1.59, Q3 = 2.10), for every increase in logFC (see Figure 1B for the cancer atlas (2) dataset (8)). The high degree of agreement of detected genes shows that BDA-LR performs on par with the Wilcoxon Rank Sum test, and the strong correlation of the logFC and logOR across all datasets shows that the results can be interpreted in a similar way.
Figure 1.

(A) Heatmap of the dataset characteristics, general overview of the results of both BDA-LR and DEA per dataset, and a comparison of the results. The rows represent the datasets, the first column shows the cell populations that were used as contrast for testing. (B) Plot of the logOR and logFC of the Cancer Atlas (2) dataset. The x-axis represent the logFCs of each tested gene, and the y-axis represent the logORs for the same genes. The blue lines shows the linear association between the logFC and logOR. The Spearman’s rank correlation coefficient (ρ) is also shown in the plot. (C) Barplots of the F-score of four BDA methods and 12 DEA methods on simulated data. Numbers above the barplots show the number of cells that were generated within the simulation. Height of bar defines the median value from 25 simulations, error bars are the first and third quartile. (D) Two density plots of MTUS2 from cortex neuron dataset. The top plot shows the density of MTUS2 in inhibitory neurons and the bottom plot shows the density of MTUS2 in excitatory neurons. (E) Two density plots of Gnb2I1 from the aging mouse atlas droplet dataset. The top plot shows the density of Gnb2I1 in 3-month-old mice and the bottom plot shows the density of Gnb2I1 in 24-month-old mice. Both (D and E) are supported with fraction of zeros and variance of each cell population.
BDA among the best performing tests on simulated data
To compare the performance of binary methods with methods relying on counts in a controlled manner, we simulated scRNAseq data with muscat (10) using the provided dataset as reference (11). We generated scRNAseq data with varying number of cells and 25% of differentially expressed genes. With 1000 and 2000 simulated cells, DEsingle (14) performed the best as the F1-score (Figure 1C) and positive predictive value (PPV, Supplementary Figure S1) were the highest and the false positive rate (FPR, Supplementary Figure S2) was the lowest. However, this performance comes at a cost in terms of considerably required computational time (Supplementary Figure S3a). For that reason, we excluded DEsingle when simulating 5000 and 10 000 cells (running time >20 min). All binary-based methods performed consistently good, with 1000 and 2000 cells ranking tightly together. BDA-fisher and BDA-Phi had decreased relative performance with 5000 and 10 000 cells, while the performances of BDA-chisq and BDA-LR were also among the best with 5000 and 10 000 cells. Taken together, this shows that differences in the frequency of zeros between groups can represent biological variation and can most accurately be detected with BDA-chisq and BDA-LR in a time efficient manner.
Differences in test outcomes explained by differences in variance between contrasting cell populations
Despite the observed association between mean expression and number of zeros, which has been previously described (2), and similar performance of the two tests, there were 4754 and 3894 genes uniquely identified using BDA and DEA, respectively, across all datasets. To better understand these differences, we highlighted two extreme exemplar cases that were not significant differentially expressed (PFDR ≥ 0.05), while they were binary differential genes (BDGs, PFDR ≤ 5.27 × 10–115). In the cortex dataset (25), MTUS2 had significantly less zeros in excitatory neurons (logOR = 1.30, PFDR|BDA = 5.27 × 10–115, Figure 1D) compared to inhibitory neurons, while the expression levels were not significantly different (logFC = -1.70 × 10–3, PFDR|DEA = 5.70 × 10–2), implying additional high ranked expressions for every additional zero. In the aging mouse atlas droplet dataset (7), Gnb2l1 had significantly less zeros in the 3-month-old mice (logOR = -0.67, PFDR|BDA = 1.81 × 10–122, Figure 1E) compared to the 24-month-old mice, while again the expression levels were not significantly different (logFC = 3.15 × 10–3, PFDR|DEA = 6.97 × 10–1). These examples show that differences in variance between contrasting cell populations can interfere with the association between observed zeros and mean expression, resulting in disparities between BDA and DEA. Of note, most BDGs-only and DEGs-only had small differences in P-values between the two tests i.e. a borderline significant difference in frequency of zeros while not having a significant difference in median expression (Supplementary Figure S4). The mean PFDR for the 4754 genes uniquely identified using BDA was 9.57 × 10–3, while the mean PFDR of the same genes using DEA was 3.18 × 10–1. As for the 3894 genes uniquely identified using DEA the mean PFDR of BDA was 3.45× 10–1 and mean PFDR of DEA was 8.56 × 10–3.
Binary differential genes are not driven by technological or biological process
To exclude that the differentially behaving genes between BDA and DEA associate with a specific technological or biological process, we investigated whether there were genes repeatedly detected by a one of the two methods. In most cases, genes that were identified as BDG-only (or DEG-only) were found within a single dataset (Supplementary Figure S5a, Supplementary Figure S5b), suggesting the absence of a driving process for them.
Binary differential genes validated with bulk RNA sequencing data
To provide additional insight that differences in zero observations are indeed biologically relevant and represent differential abundance we compared the results of the Alzheimer’s disease (AD) dataset (21) (entorhinal cortex) with DEA analysis performed on a bulk RNAseq AD dataset (19), an approach followed by others (26). The bulk RNAseq datatset was comprised of samples from the fusiform gyrus. For genes measured in both, the scRNAseq dataset and the bulk dataset (N = 2177), the majority of BDG-only (59.4%) were also differentially expressed in bulk (Figure 2A). The logOR of the single cell analysis was also significantly correlated with the logFC of the bulk analysis (ρ = 0.39, P = 9.70 × 10–79, Figure 2B). Similarly, in a second dataset (22), 65.9% of the BDG-only genes were differentially expressed in bulk samples from the frontal cortex, temporal cortex and hippocampal formation (Supplementary Figure S6). Given that the differences in zero observations for genes between the tested groups (expressed in logOR) highly correlates with the differences in median expression in bulk RNAseq data (expressed in logFC), and that the majority of BDG-only were still detected in bulk, further emphasizes that binarized scRNAseq expression data can be used to detect differentially abundant genes.
Figure 2.

(A) Venn diagram of genes detected (PFDR ≤ 0.05) in a bulk AD dataset (Friedman et al.), in the single cell AD dataset with BDA-LR (BDGs) and with DEA (DEGs). Each section shows the number and percentage of genes belonging to that section. (B) Plot of the logFC from the AD bulk dataset (x-axis) and the logOR from the single cell AD dataset (y-axis). The red line represents the linear association between the bulk logFC and logOR. The Spearman's rank correlation coefficient (ρ) and corresponding association P-value are also shown. Outlier genes (n = 9, B) were removed from the plots.
Binarization with a threshold of one most appropriate for BDA
To test the binarization scheme, we performed a BDA on the AD dataset for binary profiles generated with different thresholds for binarization (thresholds ranging from one to ten counts). Naturally, for every increase in the threshold, the number of genes with zero measurements across all cells increased, resulting in a decreasing number of tested and significant genes (Supplementary Figure S7a,b). With higher thresholds, we found a decrease in correlation of the logORs from BDA with the logFC from DEA (Supplementary Figure S7c). These results show that the default binarization scheme where zeros remain zero and every non-zero value is assigned a one, is indeed appropriate.
DISCUSSION
Altogether, our results show that binarized expression patterns across cell populations represent biological variation and can be used as measure of relative abundance of transcripts. Across 16 datasets and a variety of contrasting cell populations (disease versus healthy, cell types and cancer status), BDA detected biologically relevant genes that were missed by DEA. While the performance of BDA and DEA on real data is largely comparable, with a known ground truth, BDA performed better than DEA on simulated data. Additionally, BDA benefits reproducibility and is more robust than DEA, since the only pre-processing step required for BDA is the binarization of counts. In contrast, DEA requires normalization and transformation of counts, where an analyst can choose from an excess of equally valid methods (27). Performing BDA on datasets generated using the Smart-seq protocol should be approached with more caution: although the agreement of detected genes between BDA and DEA was high, we observed the lowest correlation between the logOR and logFC for these datasets.
With six of the sixteen datasets we performed the differential analyses between cell types that were based on clusters that were determined with the expression data itself, opposed to a case–control setting. We should note that this is a circular analysis (double dipping) and that the resulting P-values in these comparisons are thus not guaranteed to be controlled for false discoveries. This is, however, still common practice in single-cell differential analyses, as this setup is used to identify cell type markers. For the other ten dataset, the results are not compromised statistically as the case–control definitions are not based on the single-cell data itself.
In our main approach to test for BDA, we have used logistic regression. A logistic regressor for differential expression has been used before (12,28,29). These previous applications, however, use continuous expression values of genes as input, while we propose to use the binary expression value. As for MAST (12), a logistic regression on binarized expression values is implemented to take into account the zeros (expressed versus not expressed) and is combined in a hurdle model with a linear Gaussian model for the continuous values. In contrast to the previously described methods, we show that the frequencies of zeros alone are sufficient to capture biological variation and to identify differential expression of genes between biologically distinct groups in single-cell data.
A commonly used term for observed zeros in single-cell data is dropouts. As zeros in single-cell data can largely be explained by distributional models of molecule sampling counts (2,3), the use of the term dropout can be misleading, as indicated by Sarkar and Stephens (3). This work contributes to clarifying the origin of zeros in single-cell RNAseq data, by showing that the frequency of zeros can actually be used to identify biological differences.
Performing BDA is normalization-free, time efficient and an accurate alternative for DEA for which we see three potential use cases. First, BDA could be performed in isolation as a fast and accurate alternative to DEA. For different use cases, different BDA tests can be used. For more complex study designs BDA-LR could be used as it allows to adjust for covariates, allowing to take into account biological replicates, which decreases false discoveries (30). More straightforward designs could be performed with BDA-chisq. Second, BDA could be performed in addition to DEA to identify more genes. Finally, BDA could be used to validate pre-processing, normalization and DEA as a big discrepancy between the BDGs and DEGs could indicate an aberration in the DEA results.
DATA AVAILABILITY
The datasets used and prepared for this study can be downloaded from Zenodo (http://doi.org/10.5281/zenodo.4487320). The results are also made available in an interactive shiny dashboard: http://insyprojects.ewi.tudelft.nl:5000/BinaryDifferentialAnalysis/. The scripts, functions and source data for the figures are available at the Github repository: https://github.com/gbouland/binary-differential-analysis, including two vignettes describing a BDA starting from a Seurat object and a raw count matrix. BDA is also implemented in an R-package and is available at: https://github.com/gbouland/BDA.
Supplementary Material
ACKNOWLEDGEMENTS
Author Contributions: G.A.B., A.M. and M.J.T.R. conceived the study and designed the experiments. G.A.B. performed all experiments and drafted the manuscript. G.A.B., A.M. and M.J.T.R. reviewed and approved the manuscript.
Contributor Information
Gerard A Bouland, Delft Bioinformatics Lab, Delft University of Technology, Delft 2628 XE, The Netherlands; Department of Human Genetics, Leiden University Medical Center, Leiden 2333ZC, The Netherlands.
Ahmed Mahfouz, Delft Bioinformatics Lab, Delft University of Technology, Delft 2628 XE, The Netherlands; Department of Human Genetics, Leiden University Medical Center, Leiden 2333ZC, The Netherlands; Leiden Computational Biology Center, Leiden University Medical Center, Leiden 2333ZC, The Netherlands.
Marcel J T Reinders, Delft Bioinformatics Lab, Delft University of Technology, Delft 2628 XE, The Netherlands; Department of Human Genetics, Leiden University Medical Center, Leiden 2333ZC, The Netherlands; Leiden Computational Biology Center, Leiden University Medical Center, Leiden 2333ZC, The Netherlands.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
This research received funding by an NWO Gravitation project: BRAINSCAPES: A Roadmap from Neurogenetics to Neurobiology (NWO: 024.004.012).
Conflict of interest statement. None declared.
REFERENCES
- 1. Choi K., Chen Y., Skelly D.A., Churchill G.A.. Bayesian model selection reveals biological origins of zero inflation in single-cell transcriptomics. Genome Biol. 2020; 21:183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Svensson V. Droplet scRNA-seq is not zero-inflated. Nat. Biotechnol. 2020; 38:147–150. [DOI] [PubMed] [Google Scholar]
- 3. Sarkar A., Stephens M.. Separating measurement and expression models clarifies confusion in single-cell RNA sequencing analysis. Nat. Genet. 2021 536. 2021; 53:770–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Andrews T.S., Hemberg M., Birol I.. M3Drop: Dropout-based feature selection for scRNASeq. Bioinformatics. 2019; 35:2865–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li R., Quon G.. ScBFA: modeling detection patterns to mitigate technical noise in large-scale single-cell genomics data. Genome Biol. 2019; 20:193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Qiu P. Embracing the dropouts in single-cell RNA-seq analysis. Nat. Commun. 2020; 11:1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Almanzar N., Antony J., Baghel A.S., Bakerman I., Bansal I., Barres B.A., Beachy P.A., Berdnik D., Bilen B., Brownfield D.et al.. A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature. 2020; 583:590–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nieto P., Elosua-Bayes M., Trincado J.L., Marchese D., Massoni-Badosa R., Salvany M., Henriques A., Mereu E., Moutinho C., Ruiz S.et al.. A single-cell tumor immune atlas for precision oncology. Genome Res. 2021; 31:1913–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., Hao Y., Stoeckius M., Smibert P., Satija R.. Comprehensive integration of single-cell data. Cell. 2019; 177:1888–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Crowell H.L., Soneson C., Germain P.L., Calini D., Collin L., Raposo C., Malhotra D., Robinson M.D.. muscat detects subpopulation-specific state transitions from multi-sample multi-condition single-cell transcriptomics data. Nat. Commun. 2020; 11:6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kang H.M., Subramaniam M., Targ S., Nguyen M., Maliskova L., McCarthy E., Wan E., Wong S., Byrnes L., Lanata C.M.et al.. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol. 2018; 36:89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Finak G., McDavid A., Yajima M., Deng J., Gersuk V., Shalek A.K., Slichter C.K., Miller H.W., McElrath M.J., Prlic M.et al.. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015 161. 2015; 16:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014 1512. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miao Z., Deng K., Wang X., Zhang X.. DEsingle for detecting three types of differential expression in single-cell RNA-seq data. Bioinformatics. 2018; 34:3223–3224. [DOI] [PubMed] [Google Scholar]
- 15. Vu T.N., Wills Q.F., Kalari K.R., Niu N., Wang L., Rantalainen M., Pawitan Y.. Beta-Poisson model for single-cell RNA-seq data analyses. Bioinformatics. 2016; 32:2128–2135. [DOI] [PubMed] [Google Scholar]
- 16. Trapnell C., Cacchiarelli D., Grimsby J., Pokharel P., Li S., Morse M., Lennon N.J., Livak K.J., Mikkelsen T.S., Rinn J.L.. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014 324. 2014; 32:381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Law C.W., Chen Y., Shi W., Smyth G.K.. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014 152. 2014; 15:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zoubarev A., Hamer K.M., Keshav K.D., McCarthy E.L., Santos J.R.C., Van Rossum T., McDonald C., Hall A., Wan X., Lim R.et al.. Gemma: a resource for the reuse, sharing and meta-analysis of expression profiling data. Bioinformatics. 2012; 28:2272–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Friedman B.A., Srinivasan K., Ayalon G., Meilandt W.J., Lin H., Huntley M.A., Cao Y., Lee S.H., Haddick P.C.G., Ngu H.et al.. Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of alzheimer's disease not evident in mouse models. Cell Rep. 2018; 22:832–847. [DOI] [PubMed] [Google Scholar]
- 20. Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K.. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Grubman A., Chew G., Ouyang J.F., Sun G., Choo X.Y., McLean C., Simmons R.K., Buckberry S., Vargas-Landin D.B., Poppe D.et al.. A single-cell atlas of entorhinal cortex from individuals with alzheimer's disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 2019; 22:2087–2097. [DOI] [PubMed] [Google Scholar]
- 22. Hokama M., Oka S., Leon J., Ninomiya T., Honda H., Sasaki K., Iwaki T., Ohara T., Sasaki T., LaFerla F.M.et al.. Altered expression of diabetes-related genes in alzheimer's disease brains: the hisayama study. Cereb. Cortex. 2014; 24:2476–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gao C.-H. ggVennDiagram: A ‘ggplot2’ Implement of Venn Diagram. 2019; [Google Scholar]
- 24. Soneson C., Robinson M.D.. Bias, robustness and scalability in single-cell differential expression analysis. Nat. Methods. 2018; 15:255–261. [DOI] [PubMed] [Google Scholar]
- 25. Agarwal D., Sandor C., Volpato V., Caffrey T.M., Monzón-Sandoval J., Bowden R., Alegre-Abarrategui J., Wade-Martins R., Webber C.. A single-cell atlas of the human substantia nigra reveals cell-specific pathways associated with neurological disorders. Nat. Commun. 2020; 11:4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mathys H., Davila-Velderrain J., Peng Z., Gao F., Mohammadi S., Young J.Z., Menon M., He L., Abdurrob F., Jiang X.et al.. Single-cell transcriptomic analysis of alzheimer's disease. Nature. 2019; 570:332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lytal N., Ran D., An L.. Normalization methods on single-cell RNA-seq data: an empirical survey. Front. Genet. 2020; 11:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Satija R., Farrell J.A., Gennert D., Schier A.F., Regev A.. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015 335. 2015; 33:495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ntranos V., Yi L., Melsted P., Pachter L.. A discriminative learning approach to differential expression analysis for single-cell RNA-seq. Nat. Methods 2019 162. 2019; 16:163–166. [DOI] [PubMed] [Google Scholar]
- 30. Squair J.W., Gautier M., Kathe C., Anderson M.A., James N.D., Hutson T.H., Hudelle R., Qaiser T., Matson K.J.E., Barraud Q.et al.. Confronting false discoveries in single-cell differential expression. Nat. Commun. 2021 121. 2021; 12:5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nagy C., Maitra M., Tanti A., Suderman M., Théroux J.F., Davoli M.A., Perlman K., Yerko V., Wang Y.C., Tripathy S.J.et al.. Single-nucleus transcriptomics of the prefrontal cortex in major depressive disorder implicates oligodendrocyte precursor cells and excitatory neurons. Nat. Neurosci. 2020; 23:771–781. [DOI] [PubMed] [Google Scholar]
- 32. Segerstolpe Å., Palasantza A., Eliasson P., Andersson E.M., Andréasson A.C., Sun X., Picelli S., Sabirsh A., Clausen M., Bjursell M.K.et al.. Single-Cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab. 2016; 24:593–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wilk A.J., Rustagi A., Zhao N.Q., Roque J., Martínez-Colón G.J., McKechnie J.L., Ivison G.T., Ranganath T., Vergara R., Hollis T.et al.. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020; 26:1070–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim N., Kim H.K., Lee K., Hong Y., Cho J.H., Choi J.W., Lee J. Il, Suh Y.L., Ku B.M., Eum H.H.et al.. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat. Commun. 2020; 11:2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wu T.D., Madireddi S., de Almeida P.E., Banchereau R., Chen Y.J.J., Chitre A.S., Chiang E.Y., Iftikhar H., O’Gorman W.E., Au-Yeung A.et al.. Peripheral t cell expansion predicts tumour infiltration and clinical response. Nature. 2020; 579:274–278. [DOI] [PubMed] [Google Scholar]
- 36. Hodge R.D., Bakken T.E., Miller J.A., Smith K.A., Barkan E.R., Graybuck L.T., Close J.L., Long B., Johansen N., Penn O.et al.. Conserved cell types with divergent features in human versus mouse cortex. Nature. 2019; 573:61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee H.O., Hong Y., Etlioglu H.E., Cho Y.B., Pomella V., Van den Bosch B., Vanhecke J., Verbandt S., Hong H., Min J.W.et al.. Lineage-dependent gene expression programs influence the immune landscape of colorectal cancer. Nat. Genet. 2020; 52:594–603. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and prepared for this study can be downloaded from Zenodo (http://doi.org/10.5281/zenodo.4487320). The results are also made available in an interactive shiny dashboard: http://insyprojects.ewi.tudelft.nl:5000/BinaryDifferentialAnalysis/. The scripts, functions and source data for the figures are available at the Github repository: https://github.com/gbouland/binary-differential-analysis, including two vignettes describing a BDA starting from a Seurat object and a raw count matrix. BDA is also implemented in an R-package and is available at: https://github.com/gbouland/BDA.