

Summary
Severe infection can dramatically alter blood production, but the mechanisms driving hematopoietic stem and progenitor cell (HSC/HSPC) loss have not been clearly defined. Using Ixodes ovatus Ehrlichia (IOE), a tick-borne pathogen that causes severe shock-like illness and bone marrow (BM) aplasia, type I and II interferons (IFNs) promoted loss of HSPCs via increased cell death and enforced quiescence. IFN-αβ were required for increased interleukin 18 (IL-18) expression during infection, correlating with ST-HSC loss. IL-18 deficiency prevented BM aplasia and increased HSC/HSPCs. IL-18R signaling was intrinsically required for ST-HSC quiescence, but not for HSPC cell death. To elucidate cell death mechanisms, MLKL- or gasdermin D-deficient mice were infected; whereas Mlkl−/− mice exhibited protected HSC/HSPCs, no such protection was observed in Gsdmd−/− mice during infection. MLKL deficiency intrinsically protected HSCs during infection and improved hematopoietic output upon recovery. These studies define MLKL and IL-18R signaling in HSC loss and suppressed hematopoietic function in shock-like infection.
Keywords: interferon, shock, IL-18, cytokines, hematopoiesis, HSC, cell death, infection, MLKL
Graphical abstract
Highlights
-
•
Type I and II IFNs regulate expression of IL-18 and IL-18R in shock-like infection
-
•
IL-18 production contributes to HSC/HSPC loss during shock-like infection
-
•
IL-18R signaling in ST-HSCs promotes infection-induced quiescence
-
•
MLKL-deficient HSCs are protected during infection
MacNamara and colleagues define a cell-autonomous role for IL-18R signaling in enforcing quiescence of ST-HSCs during severe shock-like infection. Analysis of cell death pathways revealed that MLKL was intrinsically required for the profound loss of HSCs during severe infection. IL-18R and/or MLKL targeting during acute infection may preserve hematopoietic function and improve patient outcomes.
Introduction
Hematopoietic stem and progenitor cells (HSC/HSPCs) are a rare population of cells in the bone marrow (BM) responsible for maintaining the production of blood cells throughout an organism's lifespan. Microbial infection can induce a state of demand-associated emergency hematopoiesis, characterized by increased production of myeloid cells as well as activation and expansion of the pool of HSC/HSPCs (Fuchs et al., 2019). While prolonged HSC proliferation, as seen in chronic infection, can lead to hematopoietic suppression via loss of HSC function (Baldridge et al., 2010; Pietras et al., 2016; Zhang et al., 2016a), acute demands may require HSC proliferation and differentiation to generate cells required for effective control of pathogen growth. Severe infections and trauma can induce a profound expansion in HSPCs and are often associated with hematopoietic dysfunction (Rodriguez et al., 2009; Scumpia et al., 2010). The precise mechanisms regulating HSC/HSPC function during acute shock-like illness are not well understood. We utilized an in vivo model of shock-like infection where emergency hematopoiesis is stunted to investigate how HSC/HSPC and hematopoietic suppression occurs during an acute infectious stress response.
Human monocytic ehrlichiosis (HME) is a tick-borne disease caused by an obligate intracellular bacteria, Ehrlichia chaffeensis or Ehrlichia muris-like agent (Tominello et al., 2019). Ixodes ovatus Ehrlichia (IOE) induces severe illness in mice and has been a model for understanding infection-induced shock (Sotomayor et al., 2001; Tominello et al., 2019). HME resembles septic shock, where severe complications include cardiovascular failure, liver failure, acute respiratory distress syndrome, and neurological complications, often persisting and causing long-term issues for patients (Ismail et al., 2010). In IOE infection-induced shock, type I interferons (IFNs) are essential mediators of pathology, mortality, and hematopoietic suppression (Kader et al., 2017; Smith et al., 2018; Yang et al., 2015; Zhang et al., 2014), and type IFN-I receptor (IFNAR) signals drive HSPC cell death and limit HSC proliferation (Smith et al., 2018).
HSCs exist in a quiescent state that preserves their self-renewal capacity and prevents exhaustion (Blank and Karlsson, 2015). In models of sterile inflammation, transient, acute IFN-α induced proliferation of typically quiescent HSCs in vivo (Essers et al., 2009). However, chronic IFN-αβ exposure induced by repetitive injections of polyinosinic:polycytidylic acid caused BM aplasia associated with increased IFNAR-dependent apoptosis (Pietras et al., 2014). Chronic type I IFNs were also associated with transient proliferation of HSCs and their re-entry into a quiescent state was protective against IFN-αβ-mediated apoptosis (Pietras et al., 2014). Similar to sterile inflammation, viral infection was shown to drive IFN-αβ-dependent depletion of hematopoietic progenitors revealing a key role of type I IFNs in infection-induced cytopenias (Binder et al., 1997), although the precise mechanisms driving HSPC loss were not elucidated. In the context of IOE infection we demonstrated that type I IFN-dependent hematopoietic suppression was associated with enhanced HSC quiescence and cell death (Smith et al., 2018). Similar to type I IFNs, type II IFN (IFN-γ) can induce transient activation of dormant HSCs in the context of infection (Baldridge et al., 2010; MacNamara et al., 2011). Therefore, the context and duration of IFN exposure can impact HSC/HSPC function in a variety of inflammatory and infectious conditions.
Microbial infection can drive HSC/HSPC loss via profoundly different mechanisms relative to sterile inflammation, including distinct cell death programs. Type I IFNs induce caspase-dependent apoptosis in sterile inflammation (Pietras et al., 2014). During infection with Ehrlichia and other intracellular pathogens, however, type I IFNs positively regulate both canonical and non-canonical inflammasome activation (Kader et al., 2017; Yang et al., 2015), which is associated with pyroptotic cell death and increased levels of pro-inflammatory, inflammasome-dependent cytokines, including IL-1 and IL-18. Furthermore, during IOE infection IFNAR-dependent RIPK1 activity profoundly reduced HSPCs (Smith et al., 2018). RIPK1 can drive cell death via apoptosis, pyroptosis, or necroptosis (Malireddi et al., 2020; Wallach et al., 2016), and precise cell death pathways leading to HSC/HSPC loss during IOE infection have not been identified.
IL-18 is a member of the IL-1 cytokine family and, like IL-1β, is translated as an inactive precursor and processed into an active form by caspase-1 (Dinarello et al., 2013). Elevated serum or urine concentrations of IL-18 have been linked to poor patient outcomes in clinical models of inflammatory and shock disorders (Gupta et al., 2021; Nowarski et al., 2015; Parikh et al., 2005; Sirota et al., 2013; Tao et al., 2020; Tsoukas et al., 2020), although it is unclear whether IL-18 is a correlate of severe disease or plays an active role in pathogenesis. IL-18 signals via a heterodimeric receptor, comprised of the ligand-binding α chain and the co-receptor (β chain) which together form a high-affinity complex. IL-18 biological availability is regulated both by caspases, which cleave and activate IL-18 (Wallach et al., 2016), and IL-18 binding protein (IL-18BP), which binds to free IL-18 and prevents binding to IL-18R (Dinarello et al., 2013). Expression of the α subunit of IL-18 receptor (IL-18Rα) was found on short-term HSCs (ST-HSCs), and in a model of radiation-induced injury IL-18 limited ST-HSC proliferation (Silberstein et al., 2016). IL-18 has been shown to impact immune function by enhancing IFN-γ production, although how it may impact hematopoiesis during infection has not been explored.
We found that, during acute IOE infection, type I IFNs drive pathogenesis and BM aplasia via both IL-18 signaling and mixed lineage kinase domain, like pseudokinase (MLKL). Type I IFNs were required for increased IL-18 production, and at the same time, IFN-γR was necessary for increased IL-18Rα expression on HSPCs during infection. Whereas IL-18R signaling was intrinsically required for promoting HSC quiescence during acute IOE infection, IL-18R was not directly responsible for increased cell death. Using mice lacking gasdermin D (GSDMD), the effector of pyroptosis, or MLKL, a pore-forming protein that drives necroptosis, we identify a cell-autonomous role for MLKL in HSC/HSPC loss during IOE infection. Our data reveal mechanistic insights to the precise pathways driving HSC/HSPC loss during infection that result in reduced hematopoietic activity during recovery. Targeting IL-18 and/or necroptosis may improve hematopoiesis in the context of shock-like infection and enhance recovery from severe illness.
Results
Both type I and type II IFNs contribute to HSC/HSPC loss during IOE infection
During lethal shock-like ehrlichial infection caused by IOE, profound BM aplasia and HSPC loss is observed, and we previously defined a key role for type I interferons (IFN-αβ) in this process (Pietras et al., 2014; Smith et al., 2018). Type II interferons have also been shown to activate quiescent HSCs in a context of chronic infection (Baldridge et al., 2010). As mice lacking the receptor for type I IFNs (Ifnar−/−) upregulated IFN-γ during acute IOE infection (Smith et al., 2018; Zhang et al., 2014) we next tested how IFN-γ deficiency alone or in combination with IFNAR deficiency impacted hematopoiesis during IOE-induced shock. Double knockouts for both type I and II IFNs (Ifnar−/−;Ifngr−/−) exhibited improved total BM and HSPC (Lin−, cKit+) cell numbers, relative to wild-type (WT) mice and single Ifngr−/− and Ifnar−/− mice (Figures 1A and 1B). Long-term (LT) HSCs (Lin−, cKit+, CD135−, CD150+, CD48−) were significantly reduced in WT mice compared with IOE-infected strains lacking either Ifngr−/− or Ifnar−/−, although Ifnar−/−;Ifngr−/− exhibited complete protection from LT-HSC loss (Figures S1A, and 1C). ST-HSC (Lin−, cKit+, CD135−, CD150−, CD48−) loss induced by IOE infection appeared to require type I IFNs as both Ifnar−/− and Ifnar−/−;Ifngr−/− strains showed similar ST-HSC numbers in mock- and IOE-infected conditions (Figure 1D). Ifnar−/− and Ifnar−/−;Ifngr−/− exhibited improved survival at 50% and 90%, respectively (Figure S1B), supporting a dominant role for type I IFNs in host mortality. Multipotent progenitor populations (MPPs) are more proliferative and committed to specific lineages, relative to HSCs. MPP3s showed little strain-dependent protection and were reduced significantly during infection, whereas MPP2s and MPP4s were significantly protected against infection-induced loss in the absence of IFNAR-dependent signaling (Figures S1C–S1E). Therefore, both type I and II IFNs contribute to hematopoietic suppression during IOE infection.
Figure 1.

HSC and progenitor cell loss during infection depends on both type I and II IFNs
Mice were infected with 105 copies of IOE and bone marrow (BM) was analyzed day 7 post-IOE infection (dpi).
(A) Total BM cellularity is shown for WT, Ifngr−/−, Ifnar−/−, and Ifnar−/−;Ifngr−/− mice.
(B) The absolute numbers of total hematopoietic progenitors (HSPCs; Lin− cKit+) is shown.
(C and D) (C) Long-term (LT)-HSCs and (D) short-term (ST)-HSCs of mice that were mock or IOE infected, n = 4–9 mice/group. ∗p < 0.05, ∗∗p < 0.001, ∗∗∗p < 0.0001, ∗∗∗∗p < 0.00001 (see Figure S1).
IFNs regulate HSC quiescence during infection
We previously established that IFNAR signaling was required for infection-induced quiescence of LT-HSCs, although enforced quiescence was not due to direct type I IFN signaling in HSCs (Smith et al., 2018). We next compared HSC quiescence at steady state and 7 days post-IOE infection (dpi) by staining cells for intracellular Ki-67 and DNA content to identify cell-cycle stages among phenotypic HSCs (Figure 2A). In IOE-infected Ifnar−/− and Ifnar−/−;Ifngr−/− mice we observed fewer quiescent G0 (DAPIlo and Ki-67−) LT- and ST-HSCs as compared with their WT and Ifngr−/− counterparts (Figures 2B and 2C). In addition, we noted that, in steady-state mice lacking both type I and II IFN receptors, ST-HSCs were more active, demonstrating a role for IFNs in maintaining dormancy of HSCs. To determine if type I and II IFNs impacted HSPC death differently during acute IOE infection we performed flow cytometry on BM and stained for Annexin V, a marker of regulated cell death, and 7AAD (7-aminoactinomycin) to detect dead cells (Zargarian et al., 2017). WT mice experienced a significant loss of live HSPCs after infection, and only Ifnar−/− mice showed significant protection of HSPCs from cell death compared with WT (Figure 2D).
Figure 2.

Type I and II IFNs promote HSC quiescence during infection
Mice were infected with 105 copies of IOE and BM was analyzed at 7 dpi.
(A) Representative flow cytometric staining of LT-HSCs identified in mock-infected (left panel) or IOE-infected (right panel) mice for cell-cycle metrics depicting DNA content on the x axis and Ki-67 on the y axis.
(B) The percentage of G0 LT-HSCs in WT, Ifngr−/−, Ifnar−/−, and Ifnar−/−;Ifngr−/− is shown.
(C) The percentage of G0 ST-HSCs for each genotype as in (B) comparing mock- and IOE-infected mice; n = 3–13 mice/group.
(D) Cell death breakdown of HSPCs, n = 4–7 mice/group. (∗) Denote statistic differences within a genotype and (#) denote statistic differences between genotypes.
(E and F) BM protein levels of (E) IL-1β and (F) IL-18 is shown as normalized to total protein, n = 3–8 mice/group. ∗p < 0.05, ∗∗p < 0.001, ∗∗∗p < 0.0001, ∗∗∗∗p < 0.00001 (see Figure S2).
HSCs can be activated directly by IFNs and inflammatory cytokines, including type I IFNs, have been shown to activate inflammasomes in the context of infection (Kader et al., 2017). To evaluate whether IFNs correlated with changes in inflammasome-dependent cytokines and HSC/HSPC loss during IOE infection we analyzed BM for IL-1β and IL-18. IL-1β was significantly upregulated in Ifngr−/− mice, but not in WT mice (Figure 2E). However, increased IL-18 was seen in infected WT and Ifngr−/− mice, but not in Ifnar−/− mice (Figure 2F), thus correlating with reduced ST-HSCs in WT and Ifngr−/− mice. These data suggested that IFNAR-dependent IL-18 production may promote hematopoietic suppression during IOE infection.
Partial restoration of HSCs can be achieved by blocking IL-18 receptor
Infection-induced quiescence correlated with enhanced production of IL-18, thus we next sought to determine if HSCs could directly respond to IL-18. We analyzed expression of the IL-18Rα subunit and found increased IL-18Rα expression on HSC/HSPCs of IOE-infected WT and Ifnar−/− mice, but not Ifngr−/− or Ifnar−/−;Ifngr−/− mice, demonstrating that IL-18Rα expression is dependent on IFN-γR-mediated signaling (Figures S2A–S2C). To determine the impact of blocking the IL18R-signaling axis during acute IOE infection, WT mice were infected with IOE and treated with anti-IL-18 receptor (IL-18Rα) or isotype control antibodies, on days 4 and 6, and mice were sacrificed at 7 dpi (Figure 3A). Total BM cellularity between the three groups was similar (Figure 3B) and we noted that IOE induces a similar loss of HSPCs in both the isotype and anti-IL-18Rα antibody-treated groups (Figure 3C). However, the IOE infection-induced loss of LT-HSCs and ST-HSCs was partially rescued by IL-18Rα administration (Figures 3D and 3E). Moreover, LT- and ST-HSC quiescence levels decreased with IL-18Rα blockade (Figures 3F and 3G). To assess the efficacy of anti-IL18Rα as a treatment option, we measured bacterial burden by qPCR of the IOE gene dsb and found that bacterial burden levels in the spleen of infected mice were not significantly impacted (Figure 3H), suggesting that IL-18R signaling was not required for bacterial clearance. To determine if blocking IL-18Rα during infection impacted HSPC death we performed flow cytometry on BM. Among the LT- and ST-HSC populations, the frequency of 7AAD+ cells were reduced by IL-18Rα blockade (Figures 3I and 3J). Therefore, IL-18Rα blockade partially protected HSCs from depletion during IOE infection.
Figure 3.

IL-18R blockade preserves ST-HSCs and mitigates quiescence
WT (C57BL/6) mice were inoculated with 105 copies of IOE and BM was analyzed at 7 dpi.
(A) Model depicting treatment of WT mice with either IL-18Rα blocking antibody or an isotype control.
(B–E) Absolute cellularity of (B) BM, (C) HSPCs, (D) LT-HSCs, and (E) ST-HSCs at 7 dpi, n = 3–8 mice/group.
(F and G) The frequency of quiescent (F) LT-HSCs and (G) ST-HSCs, n = 3–8 mice/group.
(H) Bacterial burden in the spleen of IOE-infected mice treated with either isotype or anti-IL-18Rα, quantified by copies of the dsb gene.
(I and J) Cell death breakdown of (I) LT-HSC and (J) ST-HSCs stained with 7AAD and Annexin V in mock- or IOE-infected WT mice treated with mock, isotype control, or anti-IL18Rα, n = 7–9 mice/group. ∗p < 0.05, ∗∗p < 0.001, ∗∗∗p < 0.0001.
IL-18 induces BM hypocellularity and HSC/HSPC loss
We next examined a genetically modified IL-18-deficient (Il18−/−) mouse model. IOE-infected Il18−/− mice had significantly increased BM cellularity, relative to IOE-infected WT mice (Figure 4A). Il18−/− mice also exhibited increased numbers of HSPCs after IOE infection, whereas a marked infection-dependent depletion of these cells was observed in WT mice (Figure 4B). LT- and ST-HSC numbers were improved in infected Il18−/− mice compared with WT mice (Figures 4C and 4D), demonstrating that IL-18 is detrimental to HSCs and MPPs (Figures S4A–S4C) during IOE infection. We next investigated if increased BM cellularity and improved HSC numbers correlated with changes to HSC quiescence/proliferation. Whereas IOE infection promoted an increase in quiescence, as determined by the percent of cells in G0 (DAPIlo and Ki-67−), among HSCs in WT mice, HSCs in Il18−/− mice exhibited very little change in G0 (Figures 4E and 4F). These data indicate that IL-18 limits HSC proliferation in the context of infection.
Figure 4.

IL-18 deficiency improves BM cellularity and protects against HSPC loss during infection
WT mice and Il18−/− mice were inoculated with 105 copies of IOE and BM was analyzed at 7 dpi.
(A and B) Total (A) BM and (B) HSPC cellularity of WT and Il18−/− mice, n = 5–9 mice/group.
(C and D) Absolute cellularity of (C) LT-HSCs and (D) ST-HSCs, n = 3–5 mice/group. (A–D) are mice from one representative experiment, repeated 3 times with similar results.
(E and F) Cell-cycle breakdown of (E) LT-HSCs and (F) ST-HSCs, n = 6–8 mice/group. ∗p < 0.05, ∗∗p < 0.001, ∗∗∗p < 0.0001, ∗∗∗∗p < 0.00001 (see Figure S3).
IOE infection is associated with robust RIPK1-dependent cell death of HSPCs (Smith et al., 2018), thus we next investigated whether IL-18 contributed to HSPC loss via cell death. WT mice had increased levels of cell death among HSPCs, LT-HSCs, and ST-HSCs, relative to Il18−/− mice (Figures 5A–5D). Therefore, IL-18 may contribute to reduced BM cellularity via both increased cell death of HSC/HSPCs and impaired proliferation. We hypothesized that IL-18 deficiency may result in HSC/HSPC protection due to reduced type I IFNs, so we measured IFN-α and IFN-β BM protein levels by ELISA. IFN-α levels did not vary significantly in either WT or Il18−/− mice (Figure S4D); however, the infection-induced increase in IFN-β was not observed in Il18−/− mice (Figure S4E). This indicated that IL-18 deficiency may confer protection via decreased type I IFN. Splenic bacterial burden of Il18−/− mice was also slightly reduced (Figure S4F), which suggested that Il18−/− mice were more resistant to IOE infection, consistent with observations in IFNAR-deficient mice (Smith et al., 2018; Zhang et al., 2014).
Figure 5.

IL-18 deficiency reduces cell death during infection
WT mice and Il18−/− mice were inoculated with 105 copies of IOE and BM was analyzed at 7 dpi.
(A) Representative flow plots of HSPCs stained for cell death markers.
(B–D) Cell death breakdown of (B) HSPCs, (C) LT-HSCs, and (D) ST-HSCs n = 6–7 mice/group. ∗p < 0.05, ∗∗p < 0.001.
Cell-autonomous impacts of IL-18R signaling in HSCs/HSPCs
To determine if IL-18 acted directly on HSC/HSPCs, we generated WT:Il18r−/− mixed BM chimeras (Figure 6A). In mixed BM chimeric mice, acute IOE infection caused an overall decrease in HSPCs although no change in the proportion of WT and Il18r−/− cells was noted (Figure 6B). LT-HSC cellularity remained stable during infection and, in contrast to our prediction, WT cells comprised a significant proportion of this population (Figure 6C). The chimerism was even among ST-HSCs before infection, although increased WT ST-HSCs were noted during infection (Figure 6D). The proportion of G0 LT-HSCs was similar among both WT and IL-18R-deficient cells (Figure 6E). However, among ST-HSCs we observed significant reduction in G0 Il18r−/− ST-HSCs, indicating a cell-autonomous role for IL-18R in restricting ST-HSC proliferation (Figure 6F). Similar levels of cell death were noted for HSPCs and ST-HSCs, among both WT or Il18r−/− donor-derived cells, whereas Il18r−/− LT-HSCs exhibited increased cell death (Figures 6G–6I). Therefore, IL-18R is not intrinsically required for HSC/HSPC cell death, but directly limits ST-HSC proliferation.
Figure 6.

IL-18R directly regulates quiescence and indirectly induces cell death
WT hosts were lethally irradiated and transplanted with an equal ratio of WT (GFP) and Il18r−/− BM cells (BMCs).
(A) Model for the generation of WT: Il18r−/− mixed chimeras. Mice were inoculated with 105 copies of IOE and BM was analyzed at 7 dpi.
(B–D) The absolute cellularity of (B) HSPCs, (C) LT- HSCs, and (D) ST-HSCs of either WT or Il18r−/− donor origin in the BM of WT recipients, n = 5–7 mice/group.
(E and F) The frequency of quiescent (E) LT-HSCs and (F) ST-HSCs in a mixed BM chimera setting.
(G–I) Cell death breakdown of (B) HSPCs, (C) LT-HSCs, and (D) ST-HSCs isolated from BM at 7 dpi in (G) HSPCs, (H) LT-HSCs, and (I) ST-HSCs, n = 5–7 mice/group.
(J and K) (J) Graphic for generation of MethoCult assays, and (K) number of colonies per plate of either WT or Il18r−/− origin that differentiated from mixed chimera BM in MethoCult media. ∗p < 0.05, ∗∗p < 0.001, ∗∗∗p < 0.0001, ∗∗∗∗p < 0.00001 (see Figure S3).
To examine the functional consequence of IL-18R deficiency on hematopoiesis we cultured BMCs from mixed BM chimeras in methocellulose media to examine colony-forming unit (CFU) potential. Hematopoietic progenitor cells will form colonies in methocellulose media, and WT colonies were distinguished from Il18r−/− colonies by GFP fluorescence (Figure 6J). In mock-infected conditions, Il18r−/− BMCs had significantly greater capacity for CFU generation (Figure 6K). After IOE infection, however, we noted a significant reduction in CFUs, although Il18r−/− cells maintained a slight advantage over their WT counterparts. The striking increase in CFU capacity among Il18r−/− cells was not due to an increase in HSCs or MPPs of this genotype (Figures S3G–S3I) supporting a functional impairment due to IL-18R signaling. Il18r deficiency resulted in reduced ST-HSC quiescence during infection, although CFU potential was greatly reduced, thus cell death may have a greater impact on HSC/HSPC functionality during infection.
IOE infection-induced hematopoietic suppression is rescued in the absence of MLKL
Type I IFNs were required for RIPK1-depedent cell death during acute IOE (Smith et al., 2018), prompting our hypothesis that necroptotic or pyroptotic cell death may play a significant role in IOE-induced hematopoietic suppression. To examine cell death pathways that promote hematopoietic suppression during IOE we utilized mice deficient in MLKL or GSDMD to target necroptotic and pyroptotic effector proteins, respectively (Wallach et al., 2016). Mlkl−/− mice showed slight protection of whole BM cellularity while Gsdmd−/− BM cellularity was reduced, similar to WT mice (Figure 7A). Mlkl−/− mice also showed increased HSPC numbers compared with WT and Gsdmd−/− mice (Figure 7B). In Mlkl−/− mice LT- and ST-HSCs were similarly protected against infection-induced loss, compared with WT and Gsdmd−/− counterparts (Figures 7C and 7D). Mlkl−/− mice also had protected MPP2-3s compared with WT mice, but they experienced a loss of MPP4s (Figures S4A–S4C). GSDMD deficiency did not confer protection to the hematopoietic compartment, suggesting that pyroptotic cell death is not a major driver of hematopoietic suppression during acute IOE infection.
Figure 7.

IOE infection induces MLKL-dependent cell death
(A and B) Total (A) BM and (B) HSPC cellularity of WT, Mlkl−/−, and Gsdmd−/− mice at 7 dpi, n = 3–7 mice/group.
(C and D) Absolute cellularity of (C) LT-HSCs and (D) ST-HSCs, n = 3–7 mice/group.
(E) Model for the generation of WT: Mlkl−/− mixed chimeras.
(F–I) The absolute cellularity of (F) total live cells, (G) HSPCs, (H) LT-HSCs, and (I) ST-HSCs of either WT or Mlkl−/− donor origin in the BM of WT recipients and overlaid percentages of each genotype, n = 4–8 mice/group.
(J) Model depicting IOE/mock infection of mice, followed by doxycycline or PBS treatment, and sacrifice at 12 dpi.
(K) Number of colonies per plate in WT, Il18r−/−, or Mlkl−/− mice after mock or IOE day 12 infection.
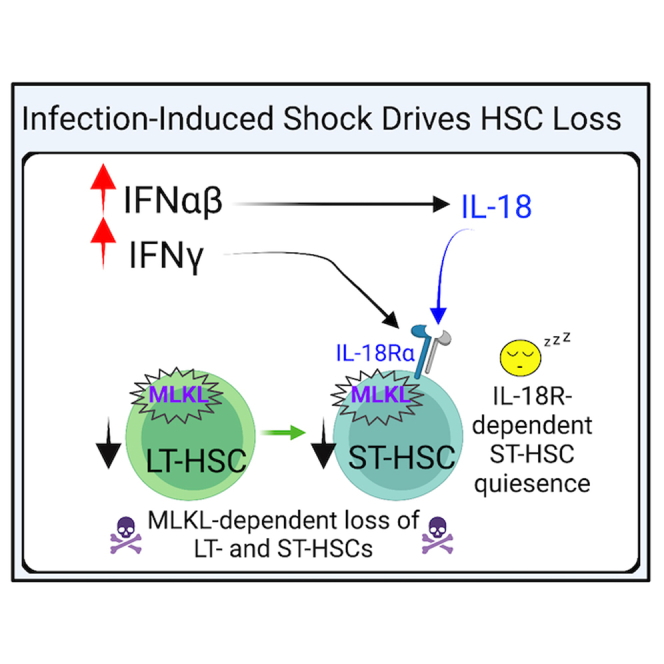
(L) Schematic of the mechanism elucidated during acute IOE wherein IFN-αβ acts directly on HSCs to induce cell death via MLKL, while also inducing IL-18, which signals on ST-HSCs to enforce quiescence. ∗p < 0.05, ∗∗p < 0.001, ∗∗∗p < 0.0001, ∗∗∗∗p < 0.00001 (see Figure S4).
We next generated mixed BM chimeras such that we could determine whether intrinsic necroptotic cell death of HSCs/HSPCs contributed to their loss during shock-like infection. WT and Mlkl−/− donor BM were transplanted into a lethally irradiated WT host and, after a 6-week reconstitution period, they were infected with IOE (Figure 7E). At 7 dpi we observed an overall dominance of Mlkl−/− cells among BM cells (Figure 7F). Mlkl−/− donor cells also comprised a majority of the HSPC pool in the mixed BM chimeras, especially after infection (Figure 7G). LT-HSCs and ST-HSCs consisted of almost exclusively MLKL-deficient cells, regardless of infection state (Figures 7H–7I), suggesting a cell-autonomous role for MLKL in limiting the HSC population. Therefore, MLKL deficiency protects HSC/HSPCs during IOE infection, and the impact of MLKL on HSC/HSPC loss is direct. Protection of HSCs/HSPCs in MLKL-deficient mice was not due to reduced type I IFNs, as IFN-α and IFN-β levels were similar in WT and Mlkl−/− mice (Figures S4D and S4E).
We next questioned whether targeting proliferation and/or cell death improved hematopoietic output after IOE infection, as we had observed with RIPK1 antagonism (Smith et al., 2018). Thus, we infected cohorts of WT, Il18r−/−, and Mlkl−/− mice and treated these mice with a course of doxycycline to prevent lethal disease (Figure 7J). MethoCult CFU assays revealed that IOE infection impaired hematopoietic function in WT mice (Figure 7K). While IL-18R-deficient mice also experienced reduced CFU activity during recovery, this was not significant, as seen in WT mice. However, MLKL deficiency completely protected against infection-induced loss in CFU activity. Thus, increased necroptotic cell death is an important driver of impaired hematopoietic function during the recovery of severe infection. We propose a model wherein IFN-αβ signals directly on HSCs to induce cell death (Smith et al., 2018), which proceeds via MLKL-dependent necroptosis, and IFN-αβ-dependent IL-18 signals on ST-HSCs to enforce quiescence (Figure 7L). Together, both quiescence and cell death contribute to BM aplasia and suppression of HSC/HSPCs during acute IOE.
Discussion
Shock-like disease caused by IOE infection induces similar pathology to what is seen in sepsis and trauma, including vascular leak, hepatic inflammation, and BM collapse (Ismail et al., 2010; Tominello et al., 2019), and defining mechanisms that contribute to hematopoietic dysfunction may identify targets for improving long-term outcomes of shock-like illnesses. IOE infection drives profound BM hypocellularity and hematopoietic suppression, and this is abrogated in the absence of type I IFN (Smith et al., 2018). We demonstrate specific roles for IL-18 and MLKL in hematopoietic suppression, which may prove beneficial to patient recovery as prolonged hematopoietic suppression is associated with poor outcomes in patients, likely due to the failure to mobilize new cells in response to injury and immune insults.
Quiescence is a hallmark of HSCs as it prevents stem cell exhaustion (Blank and Karlsson, 2015), differentiation to downstream progenitors (Pietras et al., 2015), and protects HSCs from mutations acquired during cell division that could lead to carcinogenic transformation (Walter et al., 2015). HSCs can respond directly to inflammatory insults, such as infection, however, and IL-1R-dependent signals can activate LT-HSCs and reduce quiescence (Chavez et al., 2021). Since type I IFNs can promote inflammasome activation (Kader et al., 2017), we examined the impact of IFNs and IOE on inflammasome-dependent cytokines, IL-1β and IL-18. IL-1β was upregulated in the absence of Ifngr−/− and did not correlate with changes in HSC quiescence. Our findings are consistent with previous work demonstrating the negative regulatory role for IFN-γ on IL-1β release (Masters et al., 2010), and suggest that prolonged innate inflammatory responses contribute to pathology in IOE infection. Elevated IL-18 during infection correlated with both reduced HSPCs and a maintenance of HSC quiescence during acute infection. IL-18 is an inflammatory cytokine that has been linked to ST-HSC quiescence during radiation-induced injury (Silberstein et al., 2016). In these studies, IL-18 was found be produced by osteolineage cells and thought to act directly on HSCs, limiting their proliferation. However, an indirect impact of IL-18 on other cell types in the marrow could not be ruled out. Therefore, we generated mixed BM chimeras, containing both WT and Il18r−/− cells, that repopulated the same marrow microenvironment. We determined that IL-18 directly enforces ST-HSC quiescence at steady state and during IOE infection, contributing to reduced HSPC function and myeloid output.
Enhanced HSC quiescence in the context of acute inflammation may limit hematopoietic function, but it may also provide protection to HSCs. In the context of mixed BM chimeras, Il18r−/− cells were less quiescent and exhibited robust CFU potential, relative to WT cells. However, IL-18R-deficient HSPCs exhibited reduced function upon IOE infection, suggesting that enforced quiescence was not the dominant driver of HSC/HSPC functional loss during infection. Whereas IL-18R-driven quiescence may be detrimental during acute infection by limiting blood cell production, enforced quiescence may be beneficial in long-term recovery by preventing cell death. IL-18 has been shown to induce apoptosis of various cell types under inflammatory conditions (Finotto et al., 2004; Inoue et al., 2020; Marino and Cardier, 2003). Leydig cells treated with recombinant IL-18 had increased Fas receptor, and cleaved caspases-3 and -8 (Inoue et al., 2020). Since we had previously found IFNAR signaling was required for IOE infection-induced HSC cell death (Smith et al., 2018), we predicted that IL-18 may also induce cell death. In contrast to this idea, however, WT:Il18r−/− mixed BM chimeras showed increased levels of cell death in Il18r−/− cells compared with WT. It is possible that, in this shared environment, WT cells had an advantage due to their quiescent state, rendering them less vulnerable to cell death stimuli.
IL-18 is a pro-inflammatory factor associated with pathology in various clinical contexts. IL-18 is increased in the blood of patients with sepsis (Grobmyer et al., 2000), COVID-19 (Tao et al., 2020), and hemophagocytic lymphohistiocytosis or macrophage activation syndrome (MAS) (Tsoukas et al., 2020). Elevated IL-18 levels in the urine of patients experiencing kidney failure and are a strong indicator of enhanced mortality following admittance to an intensive care unit (Parikh et al., 2005). Mechanisms of IL-18-dependent pathology in hyperinflammatory disorders are still being investigated, but in a context of MAS, excess IL-18 acts on T cells to drive CD8-dependent inflammation (Tsoukas et al., 2020). A role for IL-18 in gut dysbiosis has also been described where IL-18 equilibrium controls barrier function during colitis (Nowarski et al., 2015) and COVID-19 (Tao et al., 2020). IL-18 can induce liver injury in the context of IOE infection (Yang et al., 2015), but we extend our understanding of the broad actions of IL-18 by demonstrating a cell-autonomous role for IL-18R signaling in regulating ST-HSC quiescence during shock-like infection, which may be relevant in other inflammatory contexts.
Recently, the IL-18R signaling pathway has also been identified as a target for cancer therapy. IL-18R and IL-18BP are present in the tumor microenvironment, and were found to suppress anti-tumor immunity. Enhancing native IL-18R signaling via an engineered “decoy-resistant” IL-18, unable to bind IL-18BP, resulted in increased IL-18R-dependent T cell activation that was beneficial for the host to combat the tumor (Zhou et al., 2020). As recombinant IL-18 therapies become more clinically relevant, it is important to completely understand the impact of IL-18 in vivo, particularly its impact on stem cells.
A central finding of our study is that IFNs coordinate IL-18 signaling in HSPCs by regulating both IL-18 and its receptor. Ifnar−/− mice exhibited reduced BM protein levels of IL-18 during IOE infection, and the infection-induced increase of IL-18Rα on HSC/HSPCs required IFN-γ signaling. We also noted that Il18−/− mice showed reduced BM protein levels of IFN-β, suggesting the presence of a feedback loop where type I IFN enhances inflammasome activity and processing of IL-18, and IL-18 may act as a DAMP and upregulate type I IFNs (Martin, 2016). IL-18 is known as an inducer of IFN-γ (Dinarello et al., 2013), but, somewhat paradoxically, Ifnar−/− mice exhibited increased IFN-γ despite having reduced IL-18 (Smith et al., 2018; Zhang et al., 2014). While IFN-γ was necessary for pathogen control in Ifnar−/− mice, we found that it was dispensable for survival (Zhang et al., 2014). Treating Ifnar−/− mice with anti-IFN-γ improved their survival (Zhang et al., 2014), and here in Ifnar−/−;Ifngr−/− mice we observed nearly 100% survival, which was markedly improved compared with Ifnar−/− mice. These data support the notion that IFN-γ plays a critical role in controlling bacterial burden, but also contributes to hematopoietic suppression by regulating IL-18 receptor expression.
We previously showed that RIPK1 activity was necessary for type I IFN-dependent HSC/HSPC loss and BM suppression (Smith et al., 2018). Our data suggested a potential role for RIPK1-driven necroptotic cell death in HSPC loss as treatment with necrostatin-1s profoundly protected the HSPC compartment. RIPK1 can activate RIPK3, which phosphorylates MLKL, driving its conformational change and oligomerization of MLKL into pores in the cell membrane (Hildebrand et al., 2014). However, RIPK1 can also inhibit RIPK3/MLKL-dependent necroptosis and instead promote emergency hematopoiesis (Rickard et al., 2014). RIPK1 does not function solely in necroptosis (Wallach et al., 2016), as RIPK1 can also sensitize cells to undergo apoptosis or pyroptosis depending on cellular context and conditions (Malireddi et al., 2020; Mandal et al., 2014; Zhang et al., 2016b). Pyroptosis occurs via caspase-1 in systemic inflammatory disease, but it was ameliorated by IL-18 (Masters et al., 2012). We found that Mlkl−/− mice have a significantly protected hematopoietic compartment during infection. This revealed that in WT mice, MLKL contributed significantly to HSC/HSPC loss and loss of hematopoietic potential. It can be inferred that MLKL drives the loss of HSC/HSPCs via necroptosis, an inflammatory form of cell death involving the release of many different factors (Wallach et al., 2016). Our studies are in agreement with those observations in a model of sterile inflammation induced by TNF-α where transient HSC loss occurred via MLKL-dependent necroptosis (Yamashita and Passegue, 2019).
Tick-borne infections in humans are often overlooked due to vague, flu-like symptoms, and few sensitive diagnostic tests (Ismail et al., 2010; Tominello et al., 2019). Without early antibiotic treatment, severe infection can result in multi-organ failure (Ismail et al., 2010). Recovery from shock-like infection is often slow, accompanied by chronic illness and cognitive deficits (Hotchkiss et al., 2016).
Our study identifies a mechanism by which IFNs regulate hematopoiesis during shock-like infection via IL-18R-mediated quiescence of ST-HSCs. IL-18R deficiency directly increases CFU capacity at steady state and during IOE infection, and blocking IL-18 or its receptor may be a potential clinical treatment in conditions where myelopoiesis is impaired. Furthermore, type I IFNs and IL-18 appear to operate in a feedforward loop and blunting IL-18 signaling may result in reduced type I IFNs, which could have important implications in shock-like illness. Although enforced quiescence during IOE appeared to contribute to reduced HSC/HSPCs, increased proliferation alone was not sufficient to rescue CFU capacity of Il18r−/− BMCs. Type I IFNs also directly sensitized HSPCs to cell death (Smith et al., 2018) and this led to the discovery that MLKL is the key driver of HSC/HSPC loss during IOE infection. Our findings suggest that necroptotic cell death contributes to HSC/HSPC hypocellularity and results in long-term hematopoietic suppression. We previously showed that treatment with necrostatin-1s, an RIPK-1 inhibitor, preserved HSC/HSPCs during recovery from IOE infection (Smith et al., 2018) and this correlated with improved control of bacterial burden. Together, our studies suggest that therapeutic targeting of MLKL during acute shock-like disease may preserve the hematopoietic compartment and improve overall outcomes in patients recovering from severe infections.
Experimental procedures
Mice
C57BL/6 mice (B6NTac) and CD45.1 PepBoy (4007) were purchased from Taconic (Petersburgh, NY). Il18 knockout (stock no. 004130), Il18ra gene knockout mice (004131), UBC-GFP (004353), (Ifnar1)-receptor gene knockout (028288), Ifngr1-knockout (003288), gasdermin d-deficient (032410), and IFN-αβ receptor IFN-γ receptor double knockout (029098), were purchased from Jackson Laboratory (Bar Harbor, ME) and then bred in house. Mlkl-deficient mice were a kind gift of Dr. James Murphy and bred in house (Murphy et al., 2013). All experiments were approved by Albany Medical College's Animal Care and Use Committee.
Bacteria
IOE bacteria were obtained from splenocytes of infected mice, as described previously (Zhang et al., 2014). Mice, aged 6–8 weeks, both males and females, were inoculated via intraperitoneal (i.p.) injection with IOE (1 × 105 copies) in 500 μL sucrose-phosphate-glutamate (SPG) buffer. Mock-infected mice were i.p. injected with an equivalent volume of SPG buffer. Bacterial burden in tissues was determined by quantitative PCR for the IOE Dsb gene as described (Zhang et al., 2014).
Cytokine quantification
BM cells were flushed from pelvi and homogenized manually with a pestle in a buffer containing IGEPAL CA-630 and proteinase inhibitors. ELISA kits for IL-18 were purchased from Thermo Fisher Scientific, and type I IFN ELISAs for IFN-α and IFN-β were purchased from RayBiotech. To quantify many different cytokines simultaneously, a Bio-Plex Pro Mouse Cytokine 23-Plex Luminex assay from Bio-Rad was utilized. Results were normalized to total protein concentration per sample, as determined using the Pierce BCA kit.
BM processing and flow cytometry
BM was flushed from femora and tibiae and then filtered through a 70 μm filter. Following red blood cell lysis, single-cell suspensions were plated and stained. Data were collected on an LSR II or Symphony flow cytometer (BD Biosciences) and analyzed using FlowJo software (TreeStar). To determine levels of cell death, following surface staining cells were stained with Annexin V according to manufacturer’s guidelines (BioLegend). Immediately prior to analysis, 7AAD was added to samples for 5–10 min, and then data were acquired. Quiescence was determined by staining for Ki-67 and quantifying DNA. In brief, after surface staining cells were permeabilized (BD Cytofix/Cytoperm), followed by intracellular staining with Ki-67 (16A8 BioLegend), and 4′,6-diamidino-2-phenylindole (DAPI). DAPI was added to each sample 5–7 min before acquisition on the LSR II or Symphony (see Table S1).
Antibody neutralization
Mice were administered 75 μg/mouse of anti-IL-18Rα antibodies (clone AF856; R&D Systems) or IgG2 isotype control antibody via i.p. injection on days 4 and 6 post-infection.
Antibiotic treatment
Mice received 200 μg/μL doxycycline (i.p.) twice per day on days 6–9 post-infection, and once per day 10–13 days post-infection.
Generation of mixed BM chimeras
Mixed BM chimeric mice were generated by first lethally irradiating recipient mice (CD45.1 Pepbopy mice; 950 Rad, split-dose, 24 h apart), followed by adoptive transfer of a total of 10E6 whole BM cells at a 1:1 ratio. BM was resuspended in 1× PBS and injected via the intravenous route. Recipients were allowed to reconstitute their BM for 6 weeks, then screened to determine chimerism (CD19, Gr-1, CD11b, Ly6C, Ter119). Mice were infected with IOE as described above.
MethoCult assays
BM was harvested from hind limbs and single-cell suspensions were plated in triplicate in MethoCult medium (STEMCELL Technologies, GF M3434) at a concentration of 2E4 cells/dish. Plates were kept in an incubator at 37°C and 5% CO2 for 7–10 days until CFUs were visible. CFUs were counted using a confocal light microscope at 10–20× and using a spinning disc microscope sensitive for GFP when fluorescence detection was necessary.
Statistical analysis
Data were analyzed using GraphPad (version 7). Experiments with three or more groups were analyzed by one ANOVA with Tukey multiple comparison post-test, respectively. Experiments with two groups were analyzed by two-tailed Student's t test. Data are depicted as mean ± standard error of the mean. ∗p < 0.05, ∗∗p < 0.001, ∗∗∗p < 0.0001, ∗∗∗∗p < 0.00001.
Author contributions
J.E.H., J.N.P.S., G.F., and K.C.M. discussed and designed the research project and all experiments. J.E.H. and J.N.P.S. performed the experiments and analyzed the data. J.E.H. and K.C.M. wrote the manuscript.
Conflict of interests
The authors declare no competing interests.
Acknowledgments
The authors wish to thank Allison Seyfried for exceptional technical assistance. They would also like to acknowledge the Animal Resources Facility and Immunology Core staff for their support. This work was supported by the NIH NIGMS grant R35GM131842 to K.C.M. and HL141127 and HL153019 to G.F.
Published: November 18, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.stemcr.2021.10.011.
Supplemental information
References
- Baldridge M.T., King K.Y., Boles N.C., Weksberg D.C., Goodell M.A. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature. 2010;465:793–797. doi: 10.1038/nature09135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder D., Fehr J., Hengartner H., Zinkernagel R.M. Virus-induced transient bone marrow aplasia: major role of interferon-alpha/beta during acute infection with the noncytopathic lymphocytic choriomeningitis virus. J. Exp. Med. 1997;185:517–530. doi: 10.1084/jem.185.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank U., Karlsson S. TGF-beta signaling in the control of hematopoietic stem cells. Blood. 2015;125:3542–3550. doi: 10.1182/blood-2014-12-618090. [DOI] [PubMed] [Google Scholar]
- Chavez J.S., Rabe J.L., Loeffler D., Higa K.C., Hernandez G., Mills T.S., Ahmed N., Gessner R.L., Ke Z., Idler B.M., et al. PU.1 enforces quiescence and limits hematopoietic stem cell expansion during inflammatory stress. J. Exp. Med. 2021;218 doi: 10.1101/2020.05.18.102830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello C.A., Novick D., Kim S., Kaplinski G. Interleukin-18 and IL-18 binding protein. Front Immunol. 2013;4:289. doi: 10.3389/fimmu.2013.00289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essers M.A., Offner S., Blanco-Bose W.E., Waibler Z., Kalinke U., Duchosal M.A., Trumpp A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009;458:904–908. doi: 10.1038/nature07815. [DOI] [PubMed] [Google Scholar]
- Finotto S., Siebler J., Hausding M., Schipp M., Wirtz S., Klein S., Protschka M., Doganci A., Lehr H.A., Trautwein C., et al. Severe hepatic injury in interleukin 18 (IL-18) transgenic mice: a key role for IL-18 in regulating hepatocyte apoptosis in vivo. Gut. 2004;53:392–400. doi: 10.1136/gut.2003.018572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs A., Monlish D.A., Ghosh S., Chang S.W., Bochicchio G.V., Schuettpelz L.G., Turnbell I.R. Trauma induces emergency hematopoiesis through IL-1/MyD88-dependent production of G-CSF. J. Immunol. 2019;202:3020–3032. doi: 10.4049/jimmunol.1801456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grobmyer S.R., Lin E., Lowry S.F., Rivadeneira D.E., Potter S., Barie P.S., Nathan C.F. Elevation of IL-18 in human sepsis. J. Clin. Immunol. 2000;20:212–215. doi: 10.1023/a:1006641630904. [DOI] [PubMed] [Google Scholar]
- Gupta A., Fei Y.D., Kim T.Y., Xie A., Batai K., Greener I., Tang H., Ciftci-Yilmaz S., Juneman E., Indik J.H., et al. IL-18 mediates sickle cell cardiomyopathy and ventricular arrhythmias. Blood. 2021;137:1208–1218. doi: 10.1182/blood.2020005944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand J.M., Tanzer M.C., Lucet I.S., Young S.N., Spall S.K., Sharma P., Pierotti C., Garnier J.M., Dobson R.C., Webb A.I., et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc. Natl. Acad. Sci. U S A. 2014;111:15072–15077. doi: 10.1073/pnas.1408987111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotchkiss R.S., Moldawer L.L., Opal S.M., Reinhart K., Turnbull I.R., Vincent J.L. Sepsis and septic shock. Nat. Rev. Dis. Primers. 2016;2:16045. doi: 10.1038/nrdp.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T., Aoyama-Ishikawa M., Uemura M., Yamashita H., Koga Y., Terashima M., Usami M., Kotani J., Hirata J. Interleukin-18 levels and mouse Leydig cell apoptosis during lipopolysaccharide-induced acute inflammatory conditions. J. Reprod. Immunol. 2020;141:103167. doi: 10.1016/j.jri.2020.103167. [DOI] [PubMed] [Google Scholar]
- Ismail N., Bloch K.C., McBride J.W. Human ehrlichiosis and anaplasmosis. Clin. Lab Med. 2010;30:261–292. doi: 10.1016/j.cll.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kader M., Alaoui-El-Azher M., Vorhauer J., Kode B.B., Wells J.Z., Stolz D., Michalopoulos G., Wells A., Scott M., Ismail N. MyD88-dependent inflammasome activation and autophagy inhibition contributes to Ehrlichia-induced liver injury and toxic shock. PLoS Pathog. 2017;13:e1006644. doi: 10.1371/journal.ppat.1006644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacNamara K.C., Jones M., Martin O., Winslow G.M. Transient activation of hematopoietic stem and progenitor cells by IFNgamma during acute bacterial infection. PLoS One. 2011;6:e28669. doi: 10.1371/journal.pone.0028669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malireddi R.K.S., Gurung P., Kesavardhana S., Samir P., Burton A., Mummareddy H., Vogel P., Pelletier S., Burgula S., Kanneganti T.D. Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity-independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J. Exp. Med. 2020;217 doi: 10.1084/jem.20191644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal P., Berger S.B., Pillay S., Moriwaki K., Huang C., Guo H., Lich J.D., Finger J., Kasparcova V., Votta B., et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol. Cell. 2014;56:481–495. doi: 10.1016/j.molcel.2014.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino E., Cardier J.E. Differential effect of IL-18 on endothelial cell apoptosis mediated by TNF-alpha and Fas (CD95) Cytokine. 2003;22:142–148. doi: 10.1016/s1043-4666(03)00150-9. [DOI] [PubMed] [Google Scholar]
- Martin S.J. Cell death and inflammation: the case for IL-1 family cytokines as the canonical DAMPs of the immune system. FEBS J. 2016;283:2599–2615. doi: 10.1111/febs.13775. [DOI] [PubMed] [Google Scholar]
- Masters S.L., Gerlic M., Metcalf D., Preston S., Pellegrini M., O’Donnell J.A., McArthur K., Baldwin T.M., Chevrier S., Nowell C.J., et al. NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity. 2012;37:1009–1023. doi: 10.1016/j.immuni.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters S.L., Mielke L.A., Cornish A.L., Sutton C.E., O’Donnell J., Cengia L.H., Roberts A.W., Wicks I.P., Mills K.H., Croker B.A. Regulation of interleukin-1beta by interferon-gamma is species specific, limited by suppressor of cytokine signalling 1 and influences interleukin-17 production. EMBO Rep. 2010;11:640–646. doi: 10.1038/embor.2010.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy J.M., Czabotar P.E., Hildebrand J.M., Lucet I.S., Zhang J.G., Alvarez-Diaz S., Lewis R., Lalaoui N., Metcalf D., Webb A.I., et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39:443–453. doi: 10.1016/j.immuni.2013.06.018. [DOI] [PubMed] [Google Scholar]
- Nowarski R., Jackson R., Gagliani N., de Zoete M.R., Palm N.W., Bailis W., Low J.S., Harman C.C., Graham M., Elinav E., et al. Epithelial IL-18 equilibrium controls barrier function in colitis. Cell. 2015;163:1444–1456. doi: 10.1016/j.cell.2015.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh C.R., Abraham E., Ancukiewicz M., Edelstein C.L. Urine IL-18 is an early diagnostic marker for acute kidney injury and predicts mortality in the intensive care unit. J. Am. Soc. Nephrol. 2005;16:3046–3052. doi: 10.1681/ASN.2005030236. [DOI] [PubMed] [Google Scholar]
- Pietras E.M., Lakshminarasimhan R., Techner J.M., Fong S., Flach J., Binnewies M., Passegue E. Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J. Exp. Med. 2014;211:245–262. doi: 10.1084/jem.20131043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras E.M., Mirantes-Barbeito C., Fong S., Loeffler D., Kovtonyuk L.V., Zhang S., Lakshimarasimhan R., Chin C.P., Techner J.M., Will B., et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol. 2016;18:607–618. doi: 10.1038/ncb3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras E.M., Reynaud D., Kang Y.A., Carlin D., Calero-Nieto F.J., Leavitt A.D., Stuart J.M., Gottgens B., Passegue E. Functionally distinct subsets of lineage-biased multipotent progenitors control blood production in normal and regenerative conditions. Cell Stem Cell. 2015;17:35–46. doi: 10.1016/j.stem.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickard J.A., O’Donnell J.A., Evans J.M., Laloui N., Poh A.R., Rogers T., Vince J.E., Lawlor K.E., Ninnis R.L., Anderton H., et al. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell. 2014;157:1175–1188. doi: 10.1016/j.cell.2014.04.019. [DOI] [PubMed] [Google Scholar]
- Rodriguez S., Chora A., Goumnerov B., Mumaw C., Goebel W.S., Fernandez L., Baydoun H., HogenEsch H., Dombkowski D.M., Karlewicz C.A., et al. Dysfunctional expansion of hematopoietic stem cells and block of myeloid differentiation in lethal sepsis. Blood. 2009;114:4064–4076. doi: 10.1182/blood-2009-04-214916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scumpia P.O., Kelly-Scumpia K.M., Delano M.J., Weinstein J.S., Cuenca A.G., Al-Quran S., Bovio I., Akira S., Kumagai Y., Moldawer L.L. Cutting edge: bacterial infection induces hematopoietic stem and progenitor cell expansion in the absence of TLR signaling. J. Immunol. 2010;184:2247–2251. doi: 10.4049/jimmunol.0903652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberstein L., Goncalves K.A., Kharchenko P.V., Turcotte R., Kfoury Y., Mercier F., Baryawno N., Severe N., Bachand J., Spencer J.A., et al. Proximity-based differential single-cell analysis of the niche to identify stem/progenitor cell regulators. Cell Stem Cell. 2016;19:530–543. doi: 10.1016/j.stem.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirota J.C., Walcher A., Faubel S., Jani A., McFann K., Davarajan P., Davis C.L., Edelstein C.L. Urine IL-18, NGAL, IL-8 and serum IL-8 are biomarkers of acute kidney injury following liver transplantation. BMC Nephrol. 2013;14:17. doi: 10.1186/1471-2369-14-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J.N.P., Zhang Y., Li J.J., McCabe A., Jo H.J., Maloney J., MacNamara K.C. Type I IFNs drive hematopoietic stem and progenitor cell collapse via impaired proliferation and increased RIPK1-dependent cell death during shock-like ehrlichial infection. PLoS Pathog. 2018;14:e1007234. doi: 10.1371/journal.ppat.1007234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotomayor E.A., Popov V.L., Feng H.M., Walker D.H., Olano J.P. Animal model of fatal human monocytotropic ehrlichiosis. Am. J. Pathol. 2001;158:757–769. doi: 10.1016/S0002-9440(10)64018-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao W., Zhang G., Wang X., Guo M., Zeng W., Xu Z., Cao D., Pan A., Wang Y., Zhang K., et al. Analysis of the intestinal microbiota in COVID-19 patients and its correlation with the inflammatory factor IL-18. Med. Microecol. 2020;5:100023. doi: 10.1016/j.medmic.2020.100023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tominello T.R., Oliveira E.R.A., Hussain S.S., Elfert A., Wells J., Golden B., Ismail N. Emerging roles of autophagy and inflammasome in ehrlichiosis. Front Immunol. 2019;10:1011. doi: 10.3389/fimmu.2019.01011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoukas P., Rapp E., Van Der Kraak L., Weiss E.S., Dang V., Schneider C., Klein E., Picarsic J., Salcedo R., Stewart C.A., et al. Interleukin-18 and cytotoxic impairment are independent and synergistic causes of murine virus-induced hyperinflammation. Blood. 2020;136:2162–2174. doi: 10.1182/blood.2019003846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallach D., Kang T.B., Dillon C.P., Green D.R. Programmed necrosis in inflammation: toward identification of the effector molecules. Science. 2016;352:aaf2154. doi: 10.1126/science.aaf2154. [DOI] [PubMed] [Google Scholar]
- Walter D., Lier A., Geiselhart A., Thalheimer F.B., Huntscha S., Sobotta M.C., Moehrle B., Brocks D., Bayindir I., Kaschutnig P., et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature. 2015;520:549–552. doi: 10.1038/nature14131. [DOI] [PubMed] [Google Scholar]
- Yamashita M., Passegue E. TNF-alpha coordinates hematopoietic stem cell survival and myeloid regeneration. Cell Stem Cell. 2019;25:357–372 e357. doi: 10.1016/j.stem.2019.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q., Stevenson H.L., Scott M.J., Ismail N. Type I interferon contributes to noncanonical inflammasome activation, mediates immunopathology, and impairs protective immunity during fatal infection with lipopolysaccharide-negative ehrlichiae. Am. J. Pathol. 2015;185:446–461. doi: 10.1016/j.ajpath.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Rodriques S., Wang S., Serezani H., Kapur R., Cardoso A.A., Crlesso N. Sepsis induces hematopoietic stem cell exhaustion and myelosuppression through distinct contributions of TRIF and MYD88. Stem Cell Rep. 2016;6:940–956. doi: 10.1016/j.stemcr.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., He W., Sun L. Necrosome core machinery: MLKL. Cell. Mol. Life Sci. 2016;73:2153–2163. doi: 10.1007/s00018-016-2190-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zargarian S., Shlomovitz I., Erlich Z., Hourizadeh A., Ofir-Birin Y., Croker B.A., Regev-Rudzki N., Edry-Botzer L., Gerlic M. Phosphatidylserine externalization, “necroptotic bodies” release, and phagocytosis during necroptosis. PLoS Biol. 2017;15:e2002711. doi: 10.1371/journal.pbio.2002711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Thai V., McCabe A., Jones M., MacNamara K.C. Type I interferons promote severe disease in a mouse model of lethal ehrlichiosis. Infect Immun. 2014;82:1698–1709. doi: 10.1128/IAI.01564-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T., Damsky W., Weizman O.E., McGeary M.K., Hartmann K.P., Rosen C.E., Fischer S., Jackson R., Flavell R.A., Wang J., et al. IL-18BP is a secreted immune checkpoint and barrier to IL-18 immunotherapy. Nature. 2020;583:609–614. doi: 10.1038/s41586-020-2422-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.