

ABSTRACT
The emergence of tet(X) genes has compromised the clinical use of the last-line antibiotic tigecycline. We identified 322 (1.21%) tet(X) positive samples from 12,829 human microbiome samples distributed in four continents (Asia, Europe, North America, and South America) using retrospective data from worldwide. These tet(X) genes were dominated by tet(X2)-like orthologs but we also identified 12 samples carrying novel tet(X) genes, designed tet(X45), tet(X46), and tet(X47), were resistant to tigecycline. The metagenomic analysis indicated these tet(X) genes distributed in anaerobes dominated by Bacteroidaceae (78.89%) of human-gut origin. Two mobile elements ISBf11 and IS4351 were most likely to promote the transmission of these tet(X2)-like orthologs between Bacteroidaceae and Riemerella anatipestifer. tet(X2)-like orthologs was also developed during transmission by mutation to high-level tigecycline resistant genes tet(X45), tet(X46), and tet(X47). Further tracing these tet(X) in single bacterial isolate from public repository indicated tet(X) genes were present as early as 1960s in R. anatipestifer that was the primary tet(X) carrier at early stage (before 2000). The tet(X2) and non-tet(X2) orthologs were primarily distributed in humans and food animals respectively, and non-tet(X2) were dominated by tet(X3) and tet(X4). Genomic comparison indicated these tet(X) genes were likely to be generated during tet(X) transmission between Flavobacteriaceae and E. coli/Acinetobacter spp., and ISCR2 played a key role in the transmission. These results suggest R. anatipestifer was the potential ancestral source of tet(X). In addition, Bacteroidaceae of human-gut origin was an important hidden reservoir and mutational incubator for the mobile tet(X) genes that enabled spread to facultative anaerobes and aerobes.
IMPORTANCE The emergence of the tigecycline resistance gene tet(X) has posed a severe threat to public health. However, reports of its origin and distribution in human remain rare. Here, we explore the origin and distribution of tet(X) from large-scale metagenomic data of human-gut origin and public repository. This study revealed the emergency of tet(X) gene in 1960s, which has refreshed a previous standpoint that the earliest presence of tet(X) was in 1980s. The metagenomic analysis from data mining covered the unculturable bacteria, which has overcome the traditional bacteria isolating and purificating technologies, and the analysis indicated that the Bacteroidaceae of human-gut origin was an important hidden reservoir for tet(X) that enabled spread to facultative anaerobes and aerobes. The continuous monitoring of mobile tigecycline resistance determinants from both culturable and unculturable microorganisms is imperative for understanding and tackling the dissemination of tet(X) genes in both the health care and agricultural sectors.
KEYWORDS: tigecycline resistance, tet(X), source tracking, human microbiome, Riemerella anatipestifer, Bacteroidaceae
INTRODUCTION
The first generation of tetracycline antibiotics consisted of tetracycline, chlortetracycline, and oxytetracycline and were put into clinical practice in 1952 (1), while the second generation derivatives doxycycline and minocycline were put into use in 1976 (2). These antibiotics have been incorporated into animal feed to improve growth and feed efficiency (3). However, bacterial resistance to the tetracyclines was observed from the very beginning of their usage. To date, more than 65 specific resistant determinants and nine MDR efflux pump genes of the root nodulation-division superfamily have been confirmed including AdeABC, AcrAB-TolC and MexAB-OprM (2). These determinants confer resistance to first and second generation tetracyclines and are widely distributed among 130 Gram-negative and Gram-positive bacteria (2).
A third-generation tetracycline (tigecycline) was approved in the United States in 2005 and its use in the European Union and China was authorized in 2006 and 2010, respectively (4, 5). Tigecycline has a robust treatment range and includes bacteria resistant to first- and second-generation tetracyclines (6) and is a last resort antibiotic used to treat severe infections caused by carbapenem- and colistin-resistant pathogens (5). Thus, this antibiotic was classified as a critically important antimicrobial by the World Health Organization and its usage is restricted (7). However as early as 1984, the transferable gene tet(X) displaying tigecycline insusceptibility was discovered on an R plasmid from a B. fragilis isolate of human origin. This was the earliest occurrence of an antibiotic resistance gene (ARG) that directly inactivated tetracyclines (8). The tet(X) gene was only functional under aerobic growth conditions because it is a flavin dependent monooxygenase that requires FAD, NADPH, Mg2+, and O2 to inactive almost all of the tetracycline class (9). In 2001, the existence of tet(X1) and tet(X2) were confirmed on a transposon from Bacteroides thetaiotaomicron of human origin and shared 61.7 and 99.5% amino acid identity with tet(X), respectively. To date, tet(X)/tet(X2) genes have already spread to 16 countries/regions covering five continents (Asia, Europe, North America, South America and Africa) (2). A small comfort was that these ARGs displayed low level tigecycline resistance (MIC ≤ 2 μg.ml−1) (2).
The emergence of plasmid-mediated high-level tigecycline resistance encoded by the tet(X3) and tet(X4) genes in 2019 posed a severe threat to public health (7, 10). In addition, 10 more tet(X) orthologs have been identified and include tet(X5)–tet(X14) (11–14). These orthologs were primarily found in food animals especially swine, including tet(X3), tet(X4), tet(X6), and tet(X14), first detected in Acinetobacter baumanii, Escherichia coli, Myroides phaeus, and Empedobacter stercoris, respectively. The tet(3.2) and tet(X5) gene were identified from an Empedobacter brevis isolate of shrimp origin and an Acinetobacter baumannii isolate of human origin, respectively. All the tet(X7)–tet(X13) orthologs were identified directly from gut-derived metagenomic libraries, but their host bacteria were unknown. Epidemiological studies (2, 15, 16) indicated the dissemination of these tet(X) orthologs were dominated by tet(X3) and tet(X4) that were primarily detected from Acinetobacter spp. and E. coli, respectively. Furthermore, tet(X3)/tet(X4) samples from humans, animals, and meat for consumption revealed a prevalence of 0.3–66.7% and the highest level of 66.7% was detected from pig cecum samples from abattoirs (7). Compared with the tet(X3)/tet(X4) in animal isolates (6.9%, 73/1,060) (7), lower prevalence from human (0.32%, 4/1,250) were observed in a retrospective screening of tet(X)-carrying clinical isolates (5).
The tet(X) genes have been primarily identified using traditional cultural methods and this imposed limitations on their identification including the loss of uncultured bacteria, low-throughput and long processing times. Although the tet(X7)–tet(X13) orthologs were found directly from gut-associated samples using metagenomic sequencing, the bacterial hosts, relative abundance and propagative characteristics were absent (14). Nonetheless, public repositories are a promising high-throughput resource for exploring antibiotic resistomes. For instance, a retrospective epidemiological study based on the available public bacterial gene data sets revealed that the food chain was a potential dissemination pathway for mcr-1 (17). Additionally, a metagenomic screening study based on public metagenome data sets revealed a high detection rate of tet(X3) (25.4%) in poultry samples (18). However, there are few studies that utilize data mining for tet(X) in public databases (18, 19). In addition, public data repositories including GenBank are a valuable resource for the exploration of novel bacterial species. For instance, a recent study utilized 9,428 metagenomes to reconstruct 154,723 microbial genome bins that generated 4,390 species-level genome bins including 77% of which were not present in public repositories (20). Identification of tet(X) genes from these species-level genome bins and tracing their distribution in assembly isolates from public repository may provide a new perspective for source tracking of the global spread of tet(X).
In the current study, we utilized these data mining techniques and discovered that tet(X) had emerged as early as 1960 and the Riemerella anatipestifer was its potential ancestral source. In addition, Bacteroidaceae of human gut origin were a hidden reservoir and mutational incubator for mobile tet(X) genes that enabled spread to facultative anaerobes and aerobes.
RESULTS
Identification of tet(X) orthologs.
A total of 202,265 metagenome-assembled genomes (MAGs) that were reconstructed from 12,829 metagenomic samples of human-microbiome origin in previous studies (20–22). We downloaded these MAGs and found a total of 322 (1.21%) encoded a 388 aa protein with 96.13–100% similarity with tet(X) orthologs reported in a previous study (11) (Text S1 and Table S2 in the supplemental material). All the assembled contigs in these MAGs have been constructed using a single-sample assembly strategy and passed strict quality control tailored at maximizing the quality. Of these, 96.27% (310/322) harbored 15 types of tet(X)-like orthologs that shared 98.71–100% identity with tet(X2) (Acc. No. AJ311171) (Fig. S1 and S2, Table S3). The remaining 3.73% (12/322) shared < 98.20% identical with the known tet(X). Since there was not a criterion for assignment of tet(X) orthologs in previous studies (11, 14), we temporarily designated these tet(X2)-like orthologs as tet(X2.2) to tet(X2.16). These orthologs represented a large proportion of samples (n = 12,829) from 19 countries, and they likely represent a relatively comprehensive assessment of tet(X2)-like orthologs. We therefore tentatively used the lowest cutoff of 98.20% between tet(X2)-like orthologs [tet(X2.15) vs tet(X2.16)] for assignment of novel tet(X) orthologs. In addition, when tet(X)s were annotated as one group of tet(X), such as tet(X2) (Fig. S1), they should be grouped into one clade using the neighbor joining typing method (23). According to data, 44 tet(X)s have been assigned (24). Therefore, we found three new tet(X) orthologs and their subtypes shared less than 98.20% amino acid identity with their closest neighbors in the phylogeny designed tet(X45), tet(X45.2), tet(X45.3), tet(X46), tet(X46.2), and tet(X47) (Fig. S1, Table S3). Most of these tet(X) orthologs found from metagenomic analysis were not present in the NCBI database with the exceptions of tet(X2) (Acc. No. AJ311171), tet(X2.4) (Acc. No. JQ990987) and tet(X46.2) (Acc. No. KU547718.1) (Table S3).
Resistance phenotypes of tet(X) orthologs.
All the tet(X45), tet(X46), and tet(X47) groups found from metagenomic analysis of human-gut origin (Fig. 1c) in the E. coli JM109 were resistant to tigecycline (MICs 8 to 16 ml/liter) and exhibited high MIC to the fourth-generation antibiotics omadacycline (MICs 16 to 32 ml/liter) and eravacycline (MICs 2 to 4 ml/liter) (Table S4 in the supplemental material). In addition, all the non-tet(X2) exhibited resistance to tetracycline (MICs 128 to 256 ml/liter), doxycycline (MICs 32 to 64 ml/liter), and minocycline (MICs 16 to 32 ml/liter) (Table S4). All the tet(X2) like orthologs were susceptible to tigecycline (MICs 0.25 to 1 ml/liter) but resistant to tetracycline (MICs 8 to 256 ml/liter). Among the 23 tet(X) variants, 22 of them were resistant to doxycycline (MICs 8 to 64 ml/liter) and minocycline (MICs 2 to 32 ml/liter), excluding tet(X2.10) that intermediate to doxycycline (MIC 1 ml/liter) and minocycline (MIC 0.5 ml/liter) (Table S4). All of these tet(X2) like orthologs exhibited MIC of omadacycline from 0.5 to 16 ml/liter, and eravacycline from 0.0125 to 4 ml/liter (Table S4). Three tet(X2) like orthologs tet(X2), tet(X2.4) and tet(X2.15) showed relatively higher MICs to omadacycline (8 to 16 ml/liter) and eravacycline (1 to 4 ml/liter).
FIG 1.
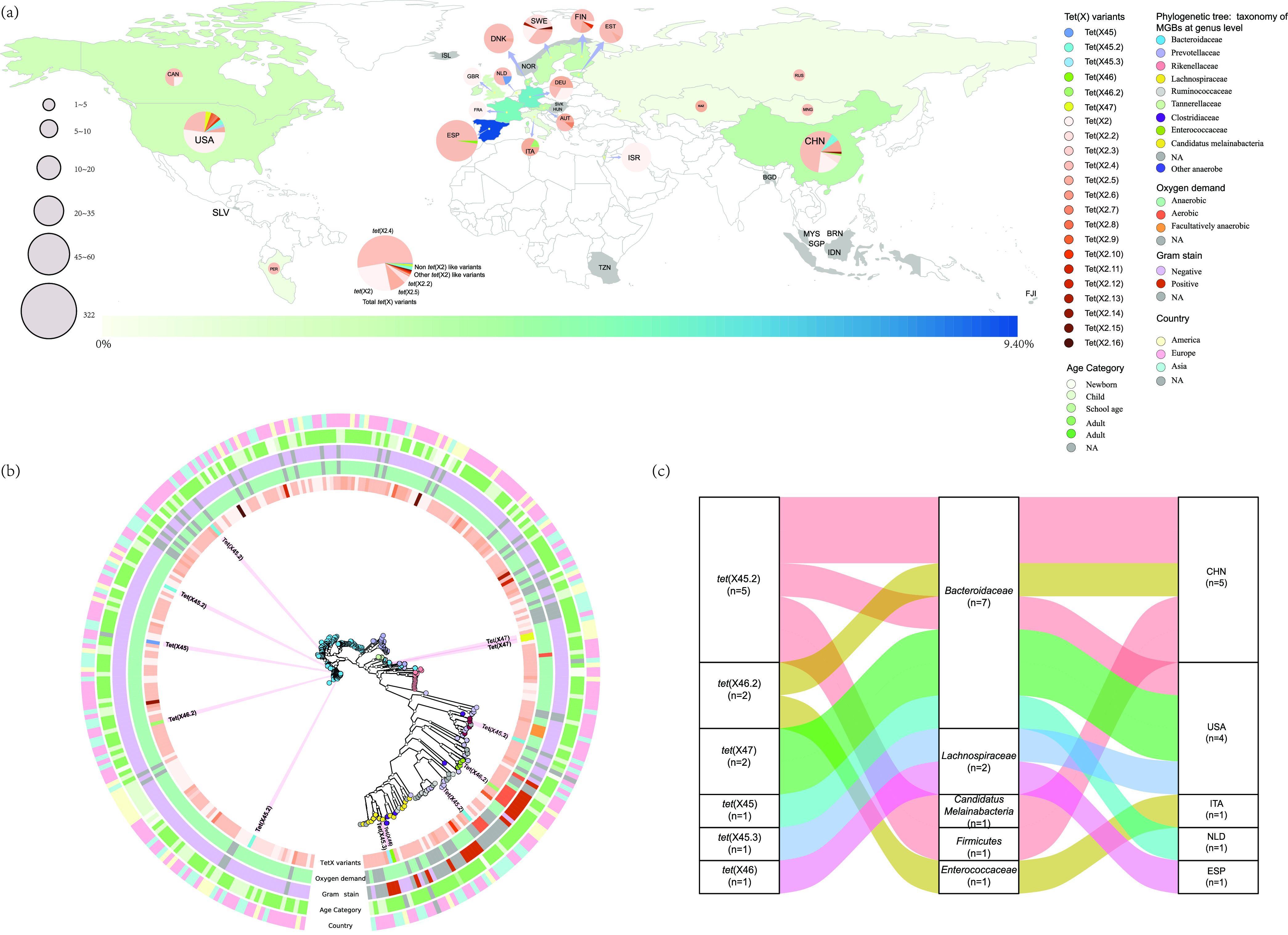
Global distribution of tet(X)s from human microbiome. (a) World map showed the positive rates of tet(X) gene in 19 countries and the colored countries represented the positive rates of tet(X) according to the hot map (>0–9.40%) at the bottom. The gray countries indicated they were negative for tet(X) gene. The size of the pie charts represented the numbers of tet(X)-positive MAGs and the colors in the pie charts indicated the composition of tet(X) variants. (b) PhyloPhlAn analysis of the tet(X) carrying MAGs. The taxonomic assignments of the tet(X)-carrying MAGs were depicted with colored circles in the phylogenetic tree. The tet(X) variants carried by the MAGs, as well as oxygen demand, Gram stain, age category and the countries of the tet(X)-carrying MAGs were showed in the five colored rings surrounding the phylogenetic tree. (c) Distribution of the 12 MAGs carried non-tet(X2) genes with tigecycline inactivate function.
Global distribution and taxonomic assignment of tet(X) carrying MAGs.
The 322 tet(X)s carrying MAGs were detected in 19 countries including Europe (n = 12), Asia (n = 4) and America (n = 3) but were absent in Oceanica and Africa (Fig. 1a, Table S5 in the supplemental material). The prevalence of tet(X) in European countries (0–9.40%) was more complex than Asian (0–3.17%) and American countries (0–2.44%) although the positive rates for tet(X) in these three continents were not significantly (P > 0.05) different (Fig. S3). The prevalence of tet(X) was highest in Spain (9.40%), followed by Germany (5.14%), France (3.82%), Denmark (3.34%) and China (3.17%) and the remaining countries were < 3% (Fig. 1a and Table S5).
The 322 tet(X)-carrying MAGs carried a range of 82–108,870 contigs (Table S8) and based on these contigs in each MAG the bacterial taxonomy assignment indicated that all the MAGs could be assigned to five phyla and was dominated by Bacteroidetes (78.89%, 254/322) followed by Firmicutes (16.78%, 47/322), Proteobacteria (1.24%, 4/322), Candidatus Melainabacteria (1.24%, 4/322), and Fusobacteria (0.62%, 2/322) (Fig. 1b). Of which, 96.68% (301/322) were classified to the family level and most of them were also belonging to Bacteroidaceae (70.10%, 211/301) (Fig. 1b and Table S6). Furthermore, 72.30% (154/211) of these tet(X) carrying Bacteroidaceae MAGs could be further divided into 14 Bacteroides species that were dominated by Bacteroides vulgatus (37.67%, 58/154), Bacteroides uniformis (20.78%, 32/154), Bacteroides dorei (14.94%, 23/154), Bacteroides ovatus (5.84%, 9/154), Bacteroides fragilis (3.90%, 6/154) and Bacteroides caccae (3.25%, 5/154) (Table S7).
The tet(X2)-like positive MAGs were distributed across these 19 tet(X) positive countries. tet(X2.4) (55.02%, 170/309), tet(X2) (26.86%, 83/309), tet(X2.5) (9.32%, 30/309), and tet(X2.2) (4.53%, 14/309) totaled 96.11% (297/309) and prevailed over other tet(X2)-like orthologs (≤0.6%, 2/309) (Fig. S4). Only 81.08% (240/296) of these four predominant tet(X) carrying MAGs could be assigned at genus level and most of them were also identified in Bacteroides (71.67%,172/240), followed by Prevotella (6.67%, 16/240) and Alistipes (5%, 12/240) (Table S8). In addition, the non-tet(X2) orthologs included 75% (9/12) that were distributed in China and United States. Of which, tet(X45.2) was the most prevalent ortholog (Fig. 1c). The remaining three non-tet(X2) carrying MAGs were distributed in Europe and the single tet(X46.2) ortholog was carried by an Enterococcaceae from Italy. Almost all tet(X)-carrying MAGs were collected from human stools excluding only one tet(X2.4)-carrying MAG identified as an Enterococcus spp. from an oral cavity sample (Table S8).
The 322 tet(X) carrying MAGs included 196 from the 4,390 MAGs and their average abundance in human microbiomes has been calculated in previous study (Table S8) (20). The average abundance of these 196 tet(X)-positive MAGs was 5.97 ± 3.89 and significantly higher than that of the total 4,390 MAGs (1.76 ± 3.74) (20).
Culturable isolates from public repository insight into the distribution and evolutionary timescale of tet(X)s.
To further trace the distribution of these tet(X)s in culturable isolates, we examined 774,435 whole genome sequences of bacterial isolates present in GenBank and only 0.12% (896/774,435) carried an ORF with > 70% amino acid identity with the known tet(X) genes including the novel ones found in the current study (Table S9 in the supplemental material). The 70% sequence identity cutoff was chosen to ensure that no other gene family would incorrectly annotate as new tet(X) members (24). The PhyloPhlAn analysis indicated that the facultative anaerobe clade was phylogenetically distinct between anaerobes and aerobes (Fig. 2a). These tet(X) genes were found in 17 bacterial families that were dominated by aerobes including Moraxellaceae (279/896, 31.17%), Enterobacteriaceae (208/896, 23.24%), and Weeksellaceae (16.20%, 145/896). The Bacteroidaceae (20.67%, 185/896) were also an important anaerobic carrier for tet(X2) like orthologs (Fig. 2a and Table S9). Three tet(X) orthologs were most prevalent and included tet(X2)-like (35.71%, 320/896), tet(X3) (27.34%, 245/896) and tet(X4) (21.99%, 199/896) (Table S9). Interestingly, different bacterial families from variant hosts were preference for carrying specific tet(X) ortholog (Fig. 2a). Almost all the Bacteroidaceae (98.91%, 182/184) and most Weeksellaceae (63.45%, 92/145) isolates carried tet(X2)-like orthologs and were primarily from human samples (38.64%, 114/295). In addition, all tet(X3) were detected from Acinetobacter spp. and almost all tet(X4) (88.89%, 176/197) were carried by E. coli and these were primarily from food animals (66.59%, 295/443) including pigs, chickens, ducks, cattle, and geese.
FIG 2.

Culturable isolates insight into tet(X) distribution patterns. (a) PhyloPhlAn analysis of the tet(X)-carrying isolates from the public repository. The species of the tet(X)-carrying isolates were depicted with colored circles in the phylogenetic tree. The information of the tet(X) carrying isolates including tet(X) variants, oxygen demand, host, collection date and country were showed in the six colored rings surrounding the phylogenetic tree. (b) Dates of lineage divergence of the earliest tet(X) orthologs as determined using Bayesian phylogenetic inference. The tet(X) variants, countries, host and species of these isolates were shown at the right region.
The tet(X2)-like orthologs were detected prior to 8.41 ± 6.17 years ago and earlier than tet(X3) (4.00 ± 1.12) and tet(X4) (2.38 ± 1.34). To be noted, the collection dates of three R. anatipestifer isolates from U.K. duck samples were prior to 1980. One isolate (BioSample: SAMN09912225) was collected in 1966 and was positive for tet(X12) and a tet(X2)-like gene that only differed in a single amino acid from tet(X2) was designed tet(X2.17). The two other isolates were collected in 1976. One (BioSample: SAMN09912224) carried a tet(X2.17) gene and another (BioSample: SAMN09912221) carried a tet(X) gene shared 99.45% similarity with tet(X47), designed tet(X47.2).
Date for lineage divergence of the earliest occurred tet(X) orthologs were produced by Bayesian phylogenetic inference (Fig. 2b). The analysis indicated a mean rate of 0.29 SNP per year for these tet(X) during 1966–2018. The most recent common ancestor (MRCA) of all tet(X) from this study was approximately from 1944 to1964, and the tracer analysis indicated that the presence of tet(X) most likely occurred in 1956 A.D. (95% highest posterior distributor). Two main lineages that originated from tet(X2.17) and tet(X12), respectively, were observed in this phylogeny and both tet(X) orthologs were collected from the Riemerella anatipestifer of duck origin from the United Kingdom in 1966 A.D.
Annotation and comparison of the tet(X) genomic environment.
A total of 1,218 tet(X)-carrying contigs ranging from 1,190 to 931,600 bp were retrieved from the metagenome and bacterial-isolate data. These contigs were grouped into 455 clusters that carried a range of 1–48 contigs (Table S10 in the supplemental material). In each cluster, longer contig shared more than 97% coverage and more than 97% similarity with shorter contig. The high coverage and similarity of these contigs indicated that these tet(X) could spread among each cluster (Table S10). This indicated tet(X) orthologs could spread among a great diversity of hosts including human, animal and environment (Fig. 3). Both humans and pigs were the primary tet(X) hosts. tet(X2)-like tet(X3), tet(X4), tet(X7), and tet(X47) have been found in humans as well as tet(X2)-like, tet(X3), tet(X4), and tet(X6) in pigs. However, only tet(X2)-like and tet(X3) orthologs could transfer between these two hosts (Fig. 3a). Interestingly, tet(X2)-like orthologs could hitch a great diversity of vehicles to spread between humans and pigs and these included Bacteroides spp., R. anatipestifer and Chryseobacterium spp. The remaining tet(X) genes were spread only via special species between different hosts. For instance, the tet(X3) gene could only be transited by Acinetobacter spp. and spread between pigs and other hosts including pigeons, cattle, geese, ducks and humans (Fig. 3a). In addition, the tet(X4) in the genomic array rdmC-tet(X4)-△ISCR2 could spread among wild birds, humans, pigs, chickens and the environment (Fig. 3b).
FIG 3.
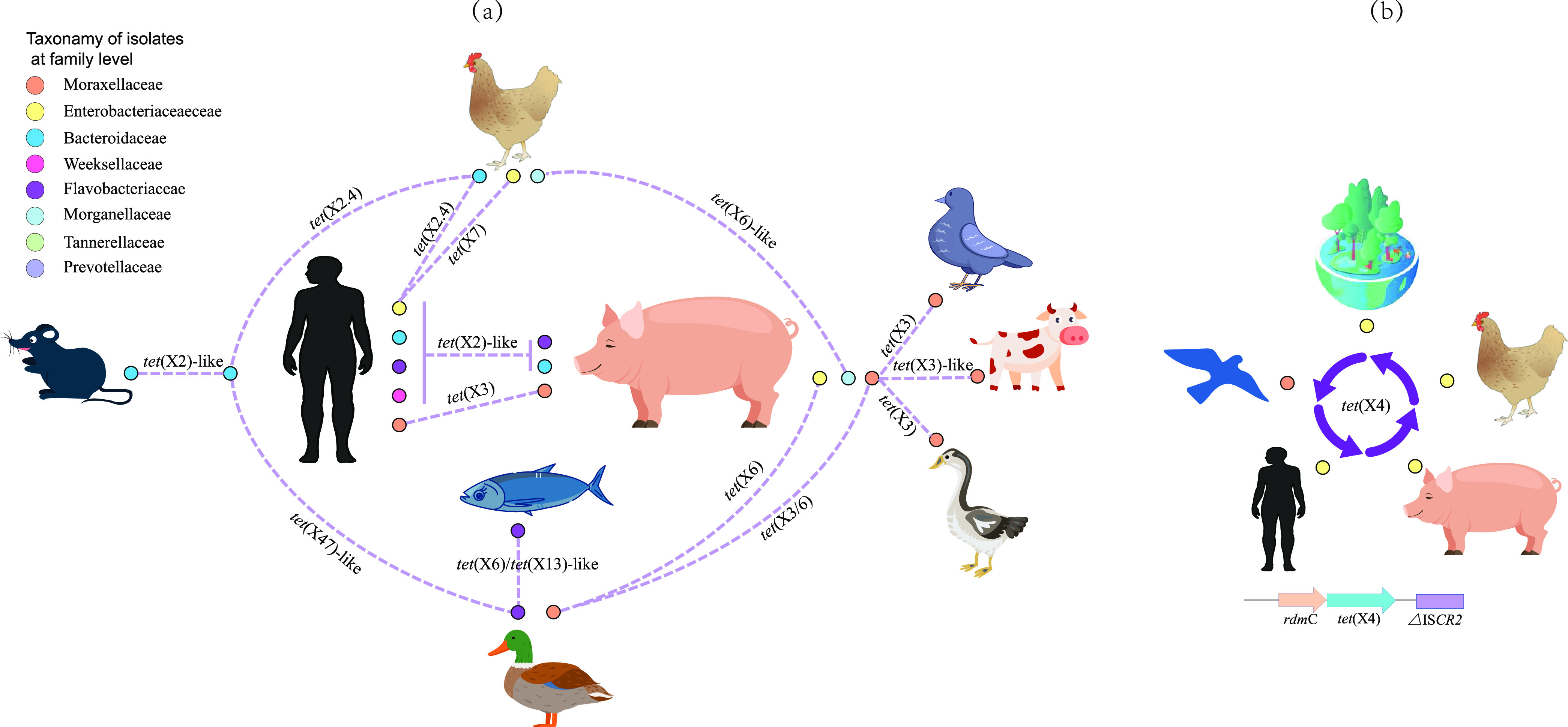
Possible transmission routes of the tet(X) genes. The colored circles surrounding the hosts represented the tet(X) carrying bacterial families. The dotted lines represented the possible transmission routes of the tet(X) genes between different hosts. (a) The transmission routes of the tet(X) gene between different hosts. (b) The transmission routes of the tet(X4) gene and their flanking genomic environment (rdmC-tet(X4)-△ISCR2) between different hosts.
Genomic annotation and comparisons indicated that other ARGs frequently flanked the tet(X) gene and these included ermF, aadK, tet(Q), and blaOXA-347 that conferred resistance to erythrocin, streptomycin, tetracycline, and ampicillin, respectively (Fig. 4a and b). Of these, the erm(F) gene was the most frequent to flank the tet(X) gene (n = 152), and their genomic environment were clustered into two types according to their relative position: ermF located upstream (15.79%, 24/152) and downstream (80.92%, 123/152) of tet(X) gene (Fig. 4a and b). The upstream ermF always formed a conserved structure tnpF-ermF-tet(X1)-tet(X2)/tet(X2.2)-aadK (n = 24) (Fig. 4a). This structure was also present in a conjugative transposon CTnDOT of Bacteroides origin (25), but the aadK (930 bp) was replaced by another aminoglycoside ARG aadS (903 bp) in CTnDOT and these two ARGs shared a 96.77% identity at the nucleotide level. Interestingly, the tet(X) orthologs and their genomic contexts were more diverse when ermF was located immediately downstream of tet(X). These tet(X) genes included 12 orthologs that were dominated by tet(X2.4) and tet(X2.5) (Fig. 4b), Text S1 in the supplemental material). In addition, these tet(X) genes were able to spread among anaerobes, aerobes and facultative anaerobes (Supplemental Text S1).
FIG 4.
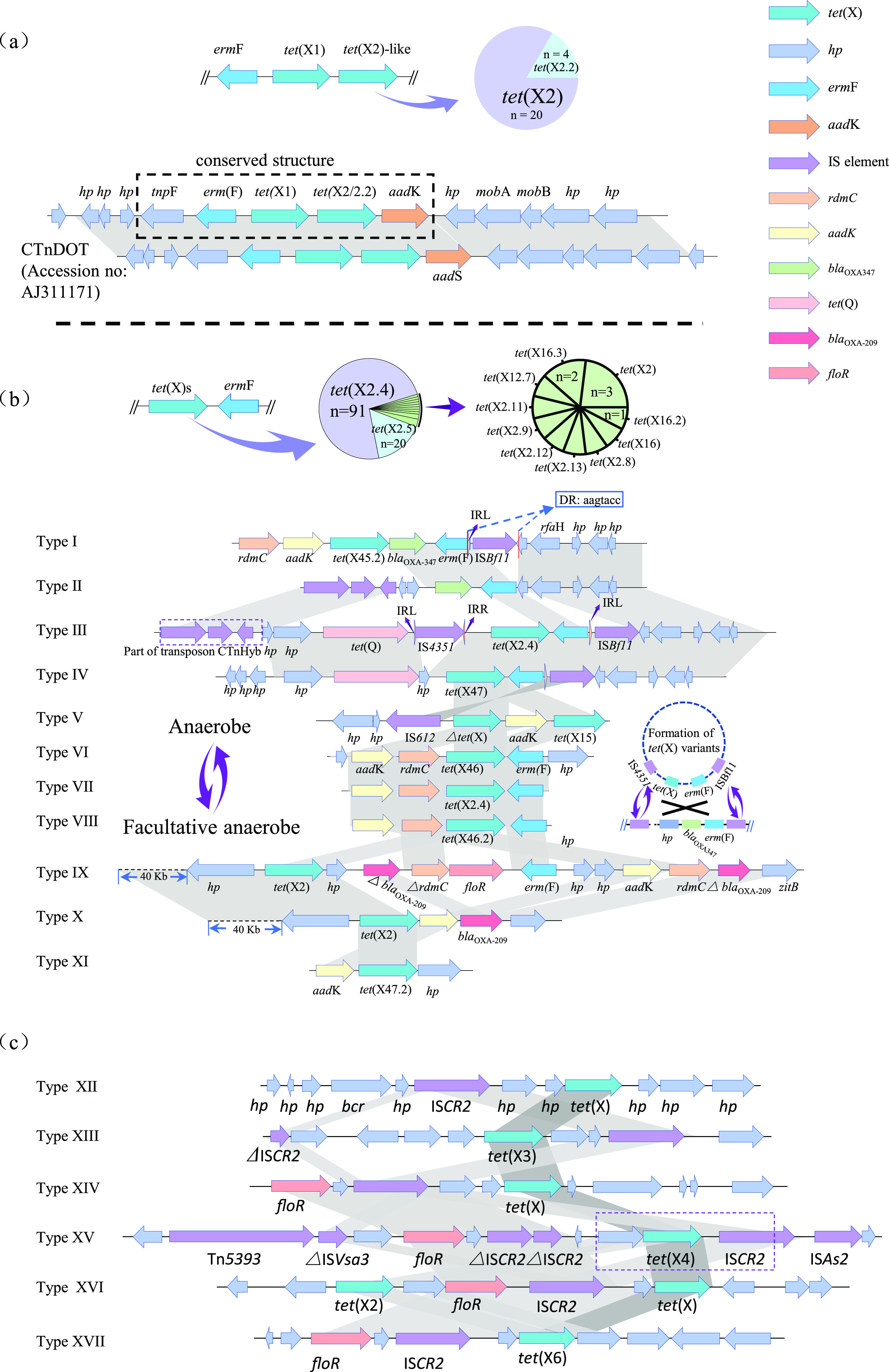
Comparison of tet(X) genomic environments. (a) The genomic comparison of erm(F) gene located upstream of tet(X2)-like genes. The proportions of the tet(X2) and tet(X2.2) located downstream tet(X1) were showed in the pie chart. (b) The genomic comparison of tet(X) genes located downstream erm(F). The proportions of the tet(X) variants located downstream of erm(F) were showed in the pie chart. The possible mechanisms of non-tet(X2) formations were showed in the two circles plotted with dotted line. (c) Genomic comparison of the regions flanking tet(X3) and tet(X4) among Flavobacteriaceae, Acinetobacter and E. coli. Arrows indicate the directions of transcription of the genes, and different genes are shown in different colors. Regions of ≥ 99.0% nucleotide sequence identity are shaded light gray. Regions of 77%–91% nucleotide sequence identity are shaded dark gray. The Δ symbol indicates a truncated gene. IS, insertion sequence. See Table S11 in the supplemental material for genomic Type I–XVII definitions.
DISCUSSION
Source tracking of the tet(X) orthologs.
One of the most significant findings in current study was that tet(X) emerged as early as 1960s in R. anatipestifer of duck origin, which was earlier than that had been previously reported in 1980s (8). A previous epidemiological study indicated the MRCA of these tet(X)s orthologs likely occurred 9900 years ago (7887 BC) (15). However, we also estimated the MRCA of these tet(X) genes but speculated it occurred only 65 years ago (1956 A.D.) that was 4 years following the introduction of tetracycline in clinics. This was consistent with the streptomycin, erythrocin and florfenicol, in which their resistance also emerged over the ensuing years following their introduction in clinic (4). In this study, we screened for the presence of tet(X) orthologs from public repositories on a large scale (774,435 isolates) and the earliest emergence of 28 tet(X) variants was selected to perform MRCA estimations (Fig. 2b). The time of the earliest occurrences of tet(X) variants was an important factor for the chronogram phylogeny construction using the Bayesian evolutionary analysis, which should be more reasonable than previous study (15). Meanwhile, this was likely to cause the discrepancy for the estimation of the tet(X) emergency between current and previous studies (15). However, this analysis was limiting in covering more tet(X) variants that spanned a longer time, and the coverages of some tet(X) variants carrying contigs were unknown (Table S3), which may affect the identification accuracy. Thus, these results could only provide an alternative model for the emergence of tet(X).
The Flavobacteriaceae have been recognized as a potential ancestral source of the tigecycline resistance gene tet(X) (5). We found that both the two earliest (1966) occurring tet(X) orthologs [tet(X2.17) and tet(X12)] harbored by R. anatipestifer that is also a bacterial species member belonging to Flavobacteriaceae family. Meanwhile, before 2000 only a total of seven isolates were confirmed as tet(X) gene carrier and five of them were also identified as R. anatipestifer (Table S9 in supplemental material). Among the 896 tet(X)-carrying isolates from public repository, 71 tet(X)-carrying were Flavobacteriaceae including R. anatipestifer that accounted for a large number (64.79%, 46/71) versus all other Flavobacteriaceae members (Fig. 2a). Additionally, R. anatipestifer harbored a great diversity of tet(X) variants and only 23.94% (17/71) carried the known tet(X)s including tet(X12), tet(X14), tet(X2.17) and tet(X47.2) (Fig. 2a). The remaining carried other tet(X) orthologs that shared 94.9–99.7% similarity with their most closely related tet(X) ortholog (Table S9). A recent study indicated the poultry pathogen R. anatipestifer appears to be a reservoir for tet(X) tigecycline resistance (26). These indicated that R. anatipestifer was most likely the ancestral source of the tigecycline resistance gene tet(X).
A great diversity of tet(X) and their flaking genomic contexts was observed when the ermF gene was present downstream of tet(X) (Fig. 4b and Text S2 in the supplemental material). A comparison of the tet(X) genomic contexts from MAGs and culturable isolates yielded 11 genomic backbones that were associated with the formation of non-tet(X2) orthologs found in the current study (Fig. 4b). The non-tet(X2) orthologs were likely to be generated from tet(X2)-like orthologs during their transmission between anaerobes or between anaerobes and facultative anaerobes (Fig. 4b and Text S2). ISBf11 and IS4351 played important roles in their transmission between anaerobes that was dominated by Bacteroides spp. where a mobile cyclic structure was speculated based on genomic Types I - IV (Fig. 4b and Text S2), and the tet(X45), tet(X46), and tet(X47) groups were likely to be generated during the transmission of these mobile structures. In addition, genomic comparisons indicated that these tet(X) genes were also able to spread between Flavobacteriaceae and E. coli as well as between Flavobacteriaceae and Acinetobacter sp. (Fig. 4c and Text S2). We have demonstrated that R. anatipestifer, a Flavobacteriaceae family member, was a potential ancestral source of tet(X) and the new tet(X) orthologs were likely to be produced during their transmission. The high similarity of the nucleotide sequences flanking tet(X3) and tet(X4) (Fig. 4c and Text S2) suggested that these two genes were also derived from Flavobacteriaceae and ISCR2 played a key role in this process (Fig. 4c and Text S2).
Global distribution of tet(X) orthologs.
The human microbiome plays an important role in public health. Here, we first determine the tet(X) prevalence in the human microbiome using a large-scale survey of 12,829 samples (20–22). A total of 16 tet(X2)-like and two new non-tet(X2) orthologs have been identified directly in the human stool samples. Since there was not standard for assignment of the newfound tet(X) orthologs, certain conflict for tet(X) numbering have been published in previous papers (13, 27), and it was necessary to distinguish the different tet(X) orthologs in current study. Thus, we temporarily set up a criterion for the assignment of tet(X) orthologs. This maybe not comprehensive as the assignment for mobile colistin resistance (mcr) genes (28) which have established a platform in NCBI (pd-help@ncbi.nlm.nih.gov) to confirm and allocate the allele numbers for new mcr gene. The assignment of new mcr alleles would not be confused in this platform and an allele numbers assignment platform for tet(X)s should be established urgently.
We found a prevalence for tet(X) at 1.21% (322/26,548) that was higher than for E. coli and K. pneumoniae from hospital isolates (0.32%, 4/1520) (5) indicating that traditional culture methods have underestimated the prevalence of tet(X). Our results were similar to an epidemiological study that detected the blaNDM and mcr-1 genes directly from samples that was higher than for the E. coli isolates (29).
The tet(X2)-like genes carrier in human microbiomes were dominated by the Bacteroidaceae in contrast to previous epidemiological studies where tet(X3) and tet(X4) were primarily carried by A. baumannii and E. coli, respectively (2, 15). The Bacteroides are predominant anaerobes estimated to account for 25–30% of human gut microflora (30) while the Enterobacteriaceae normally constitutes only 0.1–1% (31). We also found that the average abundance of tet(X)-carrying MAGs (5.97 ± 3.89) annotated as Bacteroidaceae prevailed over species-level genome bins (1.76 ± 3.74) in the human microbiome. This was likely the reason for the absence of tet(X3) and tet(X4) in our human microbiome analyses. Although tet(X) genes are inactive in anaerobes, the high abundance of tet(X2)-carrying MAGs and a variety of non-tet(X2)-like orthologs found in the current study indicated that the Bacteroidaceae were an important reservoir and mutational incubator for the mobile tet(X) orthologs in the human microbiome (Fig. 5). Furthermore, the Bacteroidaceae could generate new non-tet(X2) orthologs with tigecycline inactivation functions, and the comparison of the tet(X) genomic environment suggested that these non-tet(X2) enabled transfer to facultative anaerobes and aerobes (Fig. 5).
FIG 5.
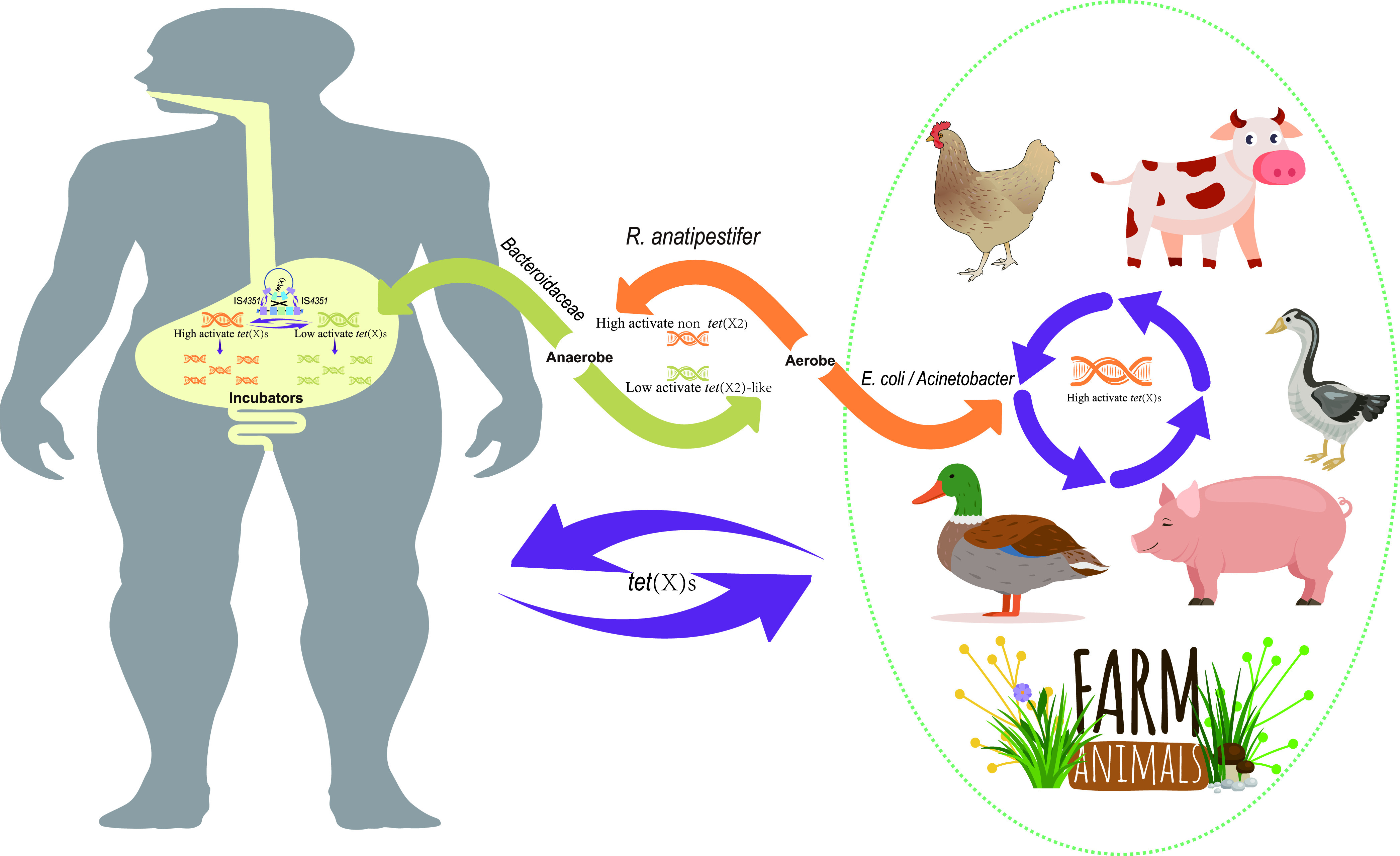
Potential origin and main transmission routes of the tet(X) genes.
We have demonstrated R. anatipestifer as potential ancestral source of tet(X) genes, and the earliest emergence of high-level tigecycline resistance genes tet(X3) and tet(X4) were likely to be 1975 and 1971, respectively, both of which were earlier than the clinical introduction of tigecycline in 2005. This was further evidence that the use of even older antibiotic tetracycline may contribute to the resistance to newer antibiotics (4). The tet(X) distributions from culturable isolates indicated that tet(X2)-like and non-tet(X2) orthologs were prevalent in anaerobes and aerobes respectively (Fig. 5). Since tet(X) is active only in an aerobic environment, the non-tet(X2)-like orthologs with tigecycline inactivate function tended to be captured by aerobes under tetracycline selective pressure (Fig. 5). This was likely to be the reason that non tet(X2) orthologs primarily distributed in aerobes but the high prevalence of tet(X2)-like orthologs in Bacteroidaceae from human microbiome need to be further explored.
Taxonomic assignments for tet(X)-carrying MAGs were estimated using bioinformatic methods that may not be as precise as cultural methods, but such an approach has proved feasible (20, 32). Another limitation of this approach was challenging to combine the chromosome with their respective plasmid sequences (33). Therefore, these tet(X)-carrying contexts from MAGs in the current study were likely to be chromosome-borne. This differed with tet(X3)/tet(X4) that are present in a variety of plasmids and ISCR2 was an essential element for their mobilization (2, 15). Transmission between Bacteroidaceae of these tet(X)s orthologs was primarily mediated by the CTnDOT-like conjugative transposon and ermF-related IS elements including ISBf11 and IS4351. CTnDOT has been reported to harbor ermF, tet(X1) and tet(X2) in Bacteroides (25) and conjugative transposons can also insert into co-resident plasmids in addition to the chromosome (34). Therefore, conjugative transposons have been found in numerous genera including Enterococcus, Streptococcus, Lactococcus, Butyrivibrio, Clostridium, Salmonella, Pseudomonas, Mezorhizobium, and Vibrio (25). The erythromycin resistance ermF gene is frequently reported in R. anatipestifer and Bacteroides spp. isolates (35, 36). The IS Bf11 and IS4351 flanking tet(X) in the Type III genomic contexts have also been previously identified (37) and reveals that this mobile structure has spread to China, the USA, France, Denmark, Sweden, and Belgium. In addition, the ISCR2 element belonging to the IS91 family has been described in the first report of tet(X3) and tet(X4), both downstream and upstream of tet(X3). This inserted sequence (IS) element could form a mobile amplicon and this was demonstrated using inverse PCR experiments (7) and in an Acinetobacter towneri isolate flanking region of tet(X6) (38). We also identified this IS element located upstream of tet(X6) and indicated that this IS element can play an important role in transmission of non-tet(X2) orthologs with tigecycline inactivation functions in the Flavobacteriaceae, E. coli and Acinetobacter.
In conclusion, we concluded an analysis on integrated human gut meta genome and global bacterial isolates to trace the origin and distribution of tet(X) gene. The tet(X) gene emerged as early as 1960 and the R. anatipestifer was an ancestral source of tet(X). The tet(X3)-carrying Acinetobacter spp. and tet(X4)-carrying E. coli were prevalent in food animals and these two tet(X)s were likely formed during the transmission of tet(X)s between Flavobacteriaceae and E. coli/Acinetobacter, and ISCR2 played a key role in the transmission. The tet(X2)-like orthologs enriched in the anaerobes that was dominated by Bacteroidaceae of human-gut origin and could transfer between these anaerobes. The mobile elements CTnDOT, ISBf11, and IS4351 played important roles in the transmission. The low-level tigecycline resistance tet(X2)-like gene could mutate to high-level tigecycline resistant determinants that could spread to facultative anaerobes and aerobes. Bacteroidaceae of human-gut origin was an important reservoir and mutational incubator for tet(X) that could spread to facultative anaerobes and aerobes.
MATERIALS AND METHODS
Collection of microbial genomic sequences from human microbiome in retrospective data.
A total of 26,728 metagenomic samples of human-microbiome origin deposited in public repositories were de novo-assembled and binned into metagenome-assembled genomes (MAGs) for the exploration of new bacterial species in previous studies (20–22). We removed the duplicative samples from these metagenomic samples according to their accession number and found a total of 12,829 non-duplicate metagenomic samples from 31 countries. These samples were reconstructed into 202,265 MAGs and online released in previous studies (20–22) (Table S1 and S2). We screened for the presence of all known tet(X) orthologs from these 202,265 MAGs using BLAST using an 80% identity and 70% hit length cutoff. The prevalence of tet(X) in 31 countries were plotted using R version 3.5.3. Phylogenetic analysis for amino acid sequences of all tet(X) gene products was constructed using neighbor joining with the default parameters in Mega X Version 10.0.5 (23) and alignments were constructed using ESPript 3 (39).
Functional identification of tet(X)s.
Tigecycline resistance for these gene products was assessed by synthesis of full-length nucleotide sequences of all detected tet(X) genes. EcoRI and a SalI sites were then added 5′ and 3′ respectively (Tsingke Biological Technology, Beijing, China). The synthesized tet(X) genes were cloned into plasmid vector pBAD24 and transformed into competent E. coli JM109 as described in our previous study (40). The transconjugants E. coli JM109+pBAD24-tet(X4) and E. coli JM109+pBAD24, were used as positive and negative controls, respectively, as previously described (40). The MIC for tetracycline, doxycycline, minocycline, tigecycline, eravacycline and omadacycline were determined by the broth microdilution method in accordance with Clinical and Laboratory Standards Institute (CLSI) guidelines. Tetracycline, doxycycline, and minocycline breakpoints were interpreted according to the European Committee on Antimicrobial Susceptibility Testing (EUCAST) guidelines (http://www.eucast.org/clinical_breakpoints). The United States FDA criteria was employed to interpret tigecycline breakpoints for E. coli and MIC ≥4 mg L−1 was considered non-susceptible while eravacycline and omadacycline were uninterpreted with no breakpoint. E. coli ATCC 25922 was used as the quality control strain.
Taxonomic assignment and phylogenetic analysis of tet(X)s-carrying MAGs.
We obtained 322 tet(X)-carrying MAGs from three previous studies (20–22). This group included taxonomic assignments for 196 that had been previously annotated (20), while the remaining 126 were annotated using metaWRAP-Annotate-bins module using the MetaWRAP pipeline and default parameters (41). Briefly, the assembly contigs from each tet(X)-carrying MAG was taxonomically profiled using Kraken2 (42) and then this entire metagenomic bin could conservatively and accurately estimate the taxonomic profiles (41).
The phylogenetic structure for the tet(X)-carrying MAGs were performed using an automatic PhyloPhlAn (3.0) pipeline (20, 43), through which the phylogeny in Fig. 1b was built using 400 universal PhyloPhlAn markers with parameter: “–diversity high –accurate –min_num_markers 80.” This pipeline integrates diamond (version 0.9.32), mafft (version 7.464) (44), trimal (version 1.4.rev15) (45) and RAxML (version 8.2.12) (46), and the parameters of these software were set as described previously (20). The phylogenetic tress in Fig. 1b were plotted using GraPhlan (version 1.1.3) (47).
Phylogenetic analyses of the tet(X) carrying isolates and evolutionary timescale for the tet(X)s from isolates.
To further trace the spread of all tet(X)s in culturable bacteria isolates, a total of 774,435 bacteria assembled whole genome sequences were downloaded from the NCBI database as of 7 November 2020. tet(X)-like open reading frames (ORFs) were determined using BLATX against all the tet(X)s variants mentioned in current study with a minimum similarity of 70 and 100% coverage. The collection date, origin, countries, and the bacterial host of the tet(X)-positive isolates were retrieved according to their Biosample Number. The phylogenetic structure for the tet(X)-carrying isolates were also performed using PhyloPhlAn (3.0) pipeline mentioned above.
To determine the evolutionary history of tet(X)s, the earliest emergency of tet(X) variants (with collection date) with a 388 amino acid (aa) length were applied to generate a chronogram using Bayesian evolutionary analysis version 1.10 (48). For all model combinations, three independent chains of 100 million generations each were run to ensure convergence with sampling every 1,000 iterations. Tracer v1.7.1 was used to assess convergence using all parameter effective sampling sizes of > 200 (49). LogCombiner v2.6.1 was used to combine tree files and a maximum clade credibility tree was created using TreeAnnotator v2.6.0 (49). Tree annotation was visualized using iTOL (50) and FigTree version 1.4.2.
Annotation and comparison of the genomic region flanking the tet(X) gene.
The tet(X)-carrying contig were extracted from the MAGs of metagenomic analysis and isolates from public repository. CD-HIT was employed to group tet(X)-carrying full length contigs using a cutoff with a minimum similarity of 97% over 97% of the query coverage (51). These tet(X)-carrying contigs were annotated using Prokka (52) and in conjunction with standalone BLAST analyses against the ResFinder (53) and ISfinder (54) databases to cross-validate ARGs and mobile genetic elements, respectively.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (31730097), the Program for Changjiang Scholars and Innovative Research Team in University of Ministry of Education of China IRT_17R39, the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2019BT02N054), and the 111 Project (D20008).
We are grateful to the previous studies that provide a large-scale of microbial genomic bins for the construction of the metagenomic analysis in current study.
Footnotes
Supplemental material is available online only.
Contributor Information
Xiao-Ping Liao, Email: xpliao@scau.edu.cn.
Ya-Hong Liu, Email: lyh@scau.edu.cn.
Wei-Hua Chen, Huazhong University of Science and Technology.
REFERENCES
- 1.Nelson ML, Levy SB. 2011. The history of the tetracyclines. Ann N Y Acad Sci 1241:17–32. doi: 10.1111/j.1749-6632.2011.06354.x. [DOI] [PubMed] [Google Scholar]
- 2.Fang LX, Chen C, Cui CY, Li XP, Zhang Y, Liao XP, Sun J, Liu YH. 2020. Emerging high-level tigecycline resistance: Novel tetracycline destructases spread via the mobile Tet(X). Bioessays 42:e2000014. doi: 10.1002/bies.202000014. [DOI] [PubMed] [Google Scholar]
- 3.Sarmah AK, Meyer MT, Boxall AB. 2006. A global perspective on the use, sales, exposure pathways, occurrence, fate and effects of veterinary antibiotics (VAs) in the environment. Chemosphere 65:725–759. doi: 10.1016/j.chemosphere. [DOI] [PubMed] [Google Scholar]
- 4.Aminov RI. 2013. Evolution in action: dissemination of tet(X) into pathogenic microbiota. Front Microbiol 4:192. doi: 10.3389/fmicb.2013.00192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang R, Dong N, Shen Z, Zeng Y, Lu J, Liu C, Zhou H, Hu Y, Sun Q, Cheng Q, Shu L, Cai J, Chan EW, Chen G, Chen S. 2020. Epidemiological and phylogenetic analysis reveals Flavobacteriaceae as potential ancestral source of tigecycline resistance gene tet(X). Nat Commun 11:4648. doi: 10.1038/s41467-020-18475-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bertrand X, Dowzicky MJ. 2012. Antimicrobial susceptibility among gram-negative isolates collected from intensive care units in North America, Europe, the Asia-Pacific Rim, Latin America, the Middle East, and Africa between 2004 and 2009 as part of the Tigecycline Evaluation and Surveillance Trial. Clin Ther 34:124–137. doi: 10.1016/j.clinthera.2011.11.023. [DOI] [PubMed] [Google Scholar]
- 7.He T, Wang R, Liu D, Walsh TR, Zhang R, Lv Y, Ke Y, Ji Q, Wei R, Liu Z, Shen Y, Wang G, Sun L, Lei L, Lv Z, Li Y, Pang M, Wang L, Sun Q, Fu Y, Song H, Hao Y, Shen Z, Wang S, Chen G, Wu C, Shen J, Wang Y. 2019. Emergence of plasmid-mediated high-level tigecycline resistance genes in animals and humans. Nat Microbiol 4:1450–1456. doi: 10.1038/s41564-019-0445-2. [DOI] [PubMed] [Google Scholar]
- 8.Guiney DG, Jr, Hasegawa P, Davis CE. 1984. Expression in Escherichia coli of cryptic tetracycline resistance genes from bacteroides R plasmids. Plasmid 11:248–252. doi: 10.1016/0147-619X(84)90031-3. [DOI] [PubMed] [Google Scholar]
- 9.Yang W, Moore IF, Koteva KP, Bareich DC, Hughes DW, Wright GD. 2004. TetX is a flavin-dependent monooxygenase conferring resistance to tetracycline antibiotics. J Biol Chem 279:52346–52352. doi: 10.1074/jbc.M409573200. [DOI] [PubMed] [Google Scholar]
- 10.Sun J, Yang RS, Zhang Q, Feng Y, Fang LX, Xia J, Li L, Lv XY, Duan JH, Liao XP, Liu YH. 2016. Co-transfer of blaNDM-5 and mcr-1 by an IncX3-X4 hybrid plasmid in Escherichia coli. Nat Microbiol 1:16176. doi: 10.1038/nmicrobiol.2016.176. [DOI] [PubMed] [Google Scholar]
- 11.Cheng Y, Chen Y, Liu Y, Guo Y, Zhou Y, Xiao T, Zhang S, Xu H, Chen Y, Shan T, Xiao Y, Zhou K. 2020. Identification of novel tetracycline resistance gene tet(X14) and its co-occurrence with tet(X2) in a tigecycline-resistant and colistin-resistant Empedobacter stercoris. Emerg Microbes Infect 9:1843–1852. doi: 10.1080/22221751.2020.1803769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang L, Liu D, Lv Y, Cui L, Li Y, Li T, Song H, Hao Y, Shen J, Wang Y, Walsh TR. 2019. Novel plasmid-mediated tet(X5) gene conferring resistance to tigecycline, eravacycline and omadacycline in clinical Acinetobacter baumannii. Antimicrob Agents Chemother 64:e01326-19. doi: 10.1128/AAC.01326-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu D, Zhai W, Song H, Fu Y, Schwarz S, He T, Bai L, Wang Y, Walsh TR, Shen J. 2020. Identification of the novel tigecycline resistance gene tet(X6) and its variants in Myroides, Acinetobacter and Proteus of food animal origin. J Antimicrob Chemother 75:1428–1431. doi: 10.1093/jac/dkaa037. [DOI] [PubMed] [Google Scholar]
- 14.Gasparrini AJ, Markley JL, Kumar H, Wang B, Fang L, Irum S, Symister CT, Wallace M, Burnham CD, Andleeb S, Tolia NH, Wencewicz TA, Dantas G. 2020. Tetracycline-inactivating enzymes from environmental, human commensal, and pathogenic bacteria cause broad-spectrum tetracycline resistance. Commun Biol 3:241. doi: 10.1038/s42003-020-0966-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen C, Cui C-Y, Yu J-J, He Q, Wu X-T, He Y-Z, Cui Z-H, Li C, Jia Q-L, Shen X-G, Sun R-Y, Wang X-R, Wang M-G, Tang T, Zhang Y, Liao X-P, Kreiswirth BN, Zhou S-D, Huang B, Du H, Sun J, Chen L, Liu Y-H. 2020. Genetic diversity and characteristics of high-level tigecycline resistance Tet(X) in Acinetobacter species. Genome Med 12:111. doi: 10.1186/s13073-020-00807-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li R, Lu X, Peng K, Liu Z, Li Y, Liu Y, Xiao X, Wang Z. 2020. Deciphering the structural diversity and classification of the mobile tigecycline resistance gene tet(X)-bearing plasmidome among bacteria mSystems 5:e00134-20. doi: 10.1128/mSystems.00134-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu Y, Liu F, Lin IY, Gao GF, Zhu B. 2016. Dissemination of the mcr-1 colistin resistance gene. Lancet Infect Dis 16:146–147. doi: 10.1016/S1473-3099(15)00533-2. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Liu F, Zhu B, Gao GF. 2020. Metagenomic data screening reveals the distribution of mobilized resistance genes tet(X), mcr and carbapenemase in animals and humans. J Infect 80:121–142. doi: 10.1016/j.jinf.2019.09.003. [DOI] [PubMed] [Google Scholar]
- 19.Zeng J, Pan Y, Yang J, Hou M, Zeng Z, Xiong WJEi. 2019. Metagenomic insights into the distribution of antibiotic resistome between the gut-associated environments and the pristine environments. Environ Int 126:346–354. doi: 10.1016/j.envint.2019.02.052. [DOI] [PubMed] [Google Scholar]
- 20.Pasolli E, Asnicar F, Manara S, Zolfo M, Karcher N, Armanini F, Beghini F, Manghi P, Tett A, Ghensi P, Collado MC, Rice BL, DuLong C, Morgan XC, Golden CD, Quince C, Huttenhower C, Segata N. 2019. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell 176:649–662.e20. doi: 10.1016/j.cell.2019.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Forster SC, Kumar N, Anonye BO, Almeida A, Viciani E, Stares MD, Dunn M, Mkandawire TT, Zhu A, Shao Y, Pike LJ, Louie T, Browne HP, Mitchell AL, Neville BA, Finn RD, Lawley TD. 2019. A human gut bacterial genome and culture collection for improved metagenomic analyses. Nat Biotechnol 37:186–192. doi: 10.1038/s41587-018-0009-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nayfach S, Shi ZJ, Seshadri R, Pollard KS, Kyrpides NC. 2019. New insights from uncultivated genomes of the global human gut microbiome. Nature 568:505–510. doi: 10.1038/s41586-019-1058-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berglund F, Böhm ME, Martinsson A, Ebmeyer S, Österlund T, Johnning A, Larsson DGJ, Kristiansson E. 2020. Comprehensive screening of genomic and metagenomic data reveals a large diversity of tetracycline resistance genes. Microb Genom 6:mgen000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whittle G, Hund BD, Shoemaker NB, Salyers AA. 2001. Characterization of the 13-KilobaseermF region of the Bacteroides conjugative transposon CTnDOT. Appl Environ Microbiol 67:3488–3495. doi: 10.1128/AEM.67.8.3488-3495.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Umar Z, Chen Q, Tang B, Xu Y, Wang J, Zhang H, Ji K, Jia X, Feng Y. 2021. The poultry pathogen Riemerella anatipestifer appears as a reservoir for Tet(X) tigecycline resistance. Environ Microbiol doi: 10.1111/1462-2920.15632. [DOI] [PubMed] [Google Scholar]
- 27.He D, Wang L, Zhao S, Liu L, Liu J, Hu G, Pan Y. 2020. A novel tigecycline resistance gene, tet(X6), on an SXT/R391 integrative and conjugative element in a Proteus genomospecies 6 isolate of retail meat origin. J Antimicrob Chemother 75:1159–1164. doi: 10.1093/jac/dkaa012. [DOI] [PubMed] [Google Scholar]
- 28.Partridge SR, Di Pilato V, Doi Y, Feldgarden M, Haft DH, Klimke W, Kumar-Singh S, Liu J-H, Malhotra-Kumar S, Prasad A, Rossolini GM, Schwarz S, Shen J, Walsh T, Wang Y, Xavier BB. 2018. Proposal for assignment of allele numbers for mobile colistin resistance (mcr) genes. J Antimicrob Chemother 73:2625–2630. doi: 10.1093/jac/dky262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y, Zhang R, Li J, Wu Z, Yin W, Schwarz S, Tyrrell JM, Zheng Y, Wang S, Shen Z, Liu Z, Liu J, Lei L, Li M, Zhang Q, Wu C, Zhang Q, Wu Y, Walsh TR, Shen J. 2017. Comprehensive resistome analysis reveals the prevalence of NDM and MCR-1 in Chinese poultry production. Nat Microbiol 2:16260. doi: 10.1038/nmicrobiol.2016.260. [DOI] [PubMed] [Google Scholar]
- 30.Moore WE, Cato EP, Holdeman LV. 1978. Some current concepts in intestinal bacteriology. Am J Clin Nutr 31:S33–S42. doi: 10.1093/ajcn/31.10.S33. [DOI] [PubMed] [Google Scholar]
- 31.Consortium HMP. 2012. Structure, function and diversity of the healthy human microbiome. Nature 486:207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang R-M, Liu X, Wang S-L, Fang L-X, Sun J, Liu Y-H, Liao X-P. 2021. Distribution patterns of antibiotic resistance genes and their bacterial hosts in pig farm wastewater treatment systems and soil fertilized with pig manure. Sci Total Environ 758:143654. doi: 10.1016/j.scitotenv.2020.143654. [DOI] [PubMed] [Google Scholar]
- 33.Sczyrba A, Hofmann P, Belmann P, Koslicki D, Janssen S, Dröge J, Gregor I, Majda S, Fiedler J, Dahms E, Bremges A, Fritz A, Garrido-Oter R, Jørgensen TS, Shapiro N, Blood PD, Gurevich A, Bai Y, Turaev D, DeMaere MZ, Chikhi R, Nagarajan N, Quince C, Meyer F, Balvočiūtė M, Hansen LH, Sørensen SJ, Chia BKH, Denis B, Froula JL, Wang Z, Egan R, Don Kang D, Cook JJ, Deltel C, Beckstette M, Lemaitre C, Peterlongo P, Rizk G, Lavenier D, Wu Y-W, Singer SW, Jain C, Strous M, Klingenberg H, Meinicke P, Barton MD, Lingner T, Lin H-H, Liao Y-C, et al. 2017. Critical assessment of metagenome interpretation-a benchmark of metagenomics software. Nat Methods 14:1063–1071. doi: 10.1038/nmeth.4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salyers AA, Shoemaker NB, Li LY. 1995. In the driver's seat: the Bacteroides conjugative transposons and the elements they mobilize. J Bacteriol 177:5727–5731. doi: 10.1128/jb.177.20.5727-5731.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xing L, Yu H, Qi J, Jiang P, Sun B, Cui J, Ou C, Chang W, Hu Q. 2015. ErmF and ereD are responsible for erythromycin resistance in Riemerella anatipestifer. PLoS One 10:e0131078. doi: 10.1371/journal.pone.0131078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnsen BO, Handal N, Meisal R, Bjørnholt JV, Gaustad P, Leegaard TM. 2017. erm gene distribution among Norwegian Bacteroides isolates and evaluation of phenotypic tests to detect inducible clindamycin resistance in Bacteroides species. Anaerobe 47:226–232. doi: 10.1016/j.anaerobe.2017.06.004. [DOI] [PubMed] [Google Scholar]
- 37.Pan Y, Awan F, Zhenbao M, Zhang X, Zeng J, Zeng Z, Xiong W. 2020. Preliminary view of the global distribution and spread of the tet(X) family of tigecycline resistance genes. J Antimicrob Chemother 75:2797–2803. doi: 10.1093/jac/dkaa284. [DOI] [PubMed] [Google Scholar]
- 38.Cheng Y, Chen Y, Liu Y, Song J, Chen Y, Shan T, Xiao Y, Zhou K. 2021. Detection of a new tet(X6)-encoding plasmid in Acinetobacter towneri. J Glob Antimicrob Resist 25:132–136. doi: 10.1016/j.jgar.2021.03.004. [DOI] [PubMed] [Google Scholar]
- 39.Robert X, Gouet P. 2014. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42:W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun J, Chen C, Cui CY, Zhang Y, Liu X, Cui ZH, Ma XY, Feng Y, Fang LX, Lian XL, Zhang RM, Tang YZ, Zhang KX, Liu HM, Zhuang ZH, Zhou SD, Lv JN, Du H, Huang B, Yu FY, Mathema B, Kreiswirth BN, Liao XP, Chen L, Liu YH. 2019. Plasmid-encoded tet(X) genes that confer high-level tigecycline resistance in Escherichia coli. Nat Microbiol 4:1457–1464. doi: 10.1038/s41564-019-0496-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Uritskiy GV, DiRuggiero J, Taylor J. 2018. MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6:158. doi: 10.1186/s40168-018-0541-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wood DE, Salzberg S. 2014. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol 15:R46. doi: 10.1186/gb-2014-15-3-r46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Asnicar F, Thomas AM, Beghini F, Mengoni C, Manara S, Manghi P, Zhu Q, Bolzan M, Cumbo F, May U, Sanders JG, Zolfo M, Kopylova E, Pasolli E, Knight R, Mirarab S, Huttenhower C, Segata N. 2020. Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat Commun 11:2500. doi: 10.1038/s41467-020-16366-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol Biol Evol 30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Asnicar F, Weingart G, Tickle TL, Huttenhower C, Segata N. 2015. Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ 3:e1029. doi: 10.7717/peerj.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. 2018. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst Biol 67:901–904. doi: 10.1093/sysbio/syy032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Letunic I, Bork P. 2019. Interactive Tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res 47:W256–W259. doi: 10.1093/nar/gkz239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fu L, Niu B, Zhu Z, Wu S, Li W. 2012. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28:3150–3152. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 53.Bortolaia V, Kaas RS, Ruppe E, Roberts MC, Schwarz S, Cattoir V, Philippon A, Allesoe RL, Rebelo AR, Florensa AF, Fagelhauer L, Chakraborty T, Neumann B, Werner G, Bender JK, Stingl K, Nguyen M, Coppens J, Xavier BB, Malhotra-Kumar S, Westh H, Pinholt M, Anjum MF, Duggett NA, Kempf I, Nykäsenoja S, Olkkola S, Wieczorek K, Amaro A, Clemente L, Mossong J, Losch S, Ragimbeau C, Lund O, Aarestrup FM. 2020. ResFinder 4.0 for predictions of phenotypes from genotypes. J Antimicrob Chemother 75:3491–3500. doi: 10.1093/jac/dkaa345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. 2006. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res 34:D32–D36. doi: 10.1093/nar/gkj014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download SPECTRUM01164-21_Supp_2_seq10.xlsx, XLSX file, 0.6 MB (611.8KB, xlsx)
Supplemental material. Download SPECTRUM01164-21_Supp_1_seq9.pdf, PDF file, 2.1 MB (2.1MB, pdf)
Supplemental material. Download SPECTRUM01164-21_Supp_3_seq11.xlsx, XLSX file, 1.8 MB (1.8MB, xlsx)