

Abstract
The synthesis of Cox1, the conserved catalytic-core subunit of Complex IV, a multisubunit machinery of the mitochondrial oxidative phosphorylation (OXPHOS) system under environmental stress, has not been sufficiently addressed. In this study, we show that the putative YihA superfamily GTPase, Mrx8, is a bona fide mitochondrial protein required for Cox1 translation initiation and elongation during suboptimal growth condition at 16°C. Mrx8 was found in a complex with mitochondrial ribosomes, consistent with a role in protein synthesis. Cells expressing mutant Mrx8 predicted to be defective in guanine nucleotide binding and hydrolysis were compromised for robust cellular respiration. We show that the requirement of Pet309 and Mss51 for cellular respiration is not bypassed by overexpression of Mrx8 and vice versa. Consistently the ribosomal association of Mss51 is independent of Mrx8. Significantly, we find that GTPBP8, the human orthologue, complements the loss of cellular respiration in Δmrx8 cells and GTPBP8 localizes to the mitochondria in mammalian cells. This strongly suggests a universal role of the MRX8 family of proteins in regulating mitochondrial function.
INTRODUCTION
Mitochondrial proteome is a composite of proteins encoded by its genome and the nuclear genome. Cells maintain at least two distinct translation systems to achieve this; one in the cytoplasm and one in the mitochondria. The cytosolic translation apparatus is responsible for the expression of the bulk of the mitochondrial proteome, while the mitochondrial translation apparatus is required for expression of only a small subset of open reading frames that are retained in the mitochondria (Ott et al., 2016). In Saccharomyces cerevisiae, mitochondrial DNA (mtDNA) encodes eight polypeptides, of which seven are involved in oxidative phosphorylation (OXPHOS) and ATP synthesis and one is the component of the small ribosome (Kurland and Andersson, 2000). Components that make up the mitochondrial translation system, including ribosomal proteins, are encoded by a set of nuclear genes that are separate from those encoding the cytosolic protein synthesis apparatus (Amunts et al., 2014; Desai et al., 2017). These are translated in the cytosol and imported into the mitochondria, where they are assembled into macromolecular complexes in a coordinated manner to incorporate mitochondrially expressed rRNAs (15S and 21S) at the correct stoichiometry. This allows for the tight regulation of the mitochondrial gene expression machinery by the nuclear genome (Couvillion et al., 2016).
Coordination between the synthesis of mitochondrial partners and import of nuclear-encoded proteins is the hallmark for a majority of multiprotein complexes required for electron transport chain and ATP synthesis. For example, Complex IV (COX) of the OXPHOS machinery that plays an essential role in energy production of aerobic cells has a dual genetic origin. It is composed of 11 subunits in yeast, three of which, COX1, COX2, and COX3, forming the catalytic core are encoded by mitochondrial genome while all others are nuclear encoded and imported from the cytosol. Cox1, which contains the two redox centers; one containing heme A and the other containing heme a3-CuB, is conserved in the mitochondrial genome of all aerobic organisms (Khalimonchuk and Rodel, 2005; Kim et al., 2012; Timon-Gomez et al., 2018). Cox1 translation and its assembly into complex IV are tightly coupled and are regulated by a large number of nuclear-encoded factors as incorrect assembly would generate free radicals that are detrimental to the cell (Fontanesi et al., 2008). Central to this process is Mss51, which along with Pet309 initiates Cox1 translation and remains associated with the newly synthesized Cox1 until it assembles with nuclear subunits of the preassembly complex (Decoster et al., 1990; Manthey and McEwen, 1995; Perez-Martinez et al., 2003; Barrientos et al., 2004; Tavares-Carreon et al., 2008). This allows Cox1 translation from the mitochondrial genome to be regulated with the rate of assembly of Complex IV that requires additional independent Cox2 and Cox3 assembly modules (McStay et al., 2013).
GTPases that belong to the family of P-loop NTPases form the largest class of accessory factors that regulate various aspects of ribosome biogenesis and protein synthesis (Sprang, 1997; Karbstein, 2007; Clementi and Polacek, 2010; Verstraeten et al., 2011; Wittinghofer and Vetter, 2011; Maracci and Rodnina, 2016; Maiti et al., 2021). Of the two families of NTPases, TRAFAC and SIMBI, the TRAFAC family comprises proteins that are thought to have evolved from an ancestral GTPase involved in translation (Leipe et al., 2002; Atkinson, 2015). The TRAFAC family of GTPases that function during ribosome assembly or protein synthesis generally have extensions at either their N- or C-terminus in addition to a GTPase domain. These proteins are hypothesized to function by a common mechanism whereby the energy released upon guanine nucleotide triphosphate hydrolysis powers protein conformational changes, allowing these additional domains to carry out a mechanical process (Karbstein, 2007; Strunk and Karbstein, 2009). A subset of the TRAFAC family are the translational GTPases (trGTPases) such as IF2, EF-Tu, EF-G, and RF3 with well-defined roles during protein synthesis (Maracci and Rodnina, 2016). All organisms in addition possess highly conserved GTPases classified as trGTPases, which are predicted to function during translation although their mechanism of action remains elusive. One such protein in S. cerevisiae is GUF1 (GTPase of unknown function 1), which belongs to LepA family of trGTPases, present in mitochondria. Absence of Guf1 alters the mitochondrial translation rates and assembly of cytochrome oxidase complex in the cell (Bauerschmitt et al., 2008).
Four members of the TRAFAC family of GTPases in addition to the trGTPases, namely, MTG1, MTG2, MTG3, and MSS1, regulate mitochondrial ribosome assembly and RNA modification in S. cerevisiae. Among them, MTG1 and MTG2 are involved in assembly of the large subunit (54S) of the mitochondrial ribosome (Barrientos et al., 2003; Datta et al., 2005). MTG3 is involved in the biogenesis of the small ribosomal subunit (37S) and regulates the processing and assembly of the 15S rRNA precursors (Paul et al., 2012). Mss1 in a heterodimer with Mto1 regulates modification of mitochondrial (mt)-tRNA (Decoster et al., 1993; Umeda et al., 2005). Previous studies of the mitochondrial proteome (Sickmann et al., 2003), genome-wide localization (Huh et al., 2003), phenotypic analysis of yeast knockout strains (Giaever et al., 2002), and genome-wide TAP purification (Gavin et al., 2002) have implicated a novel GTPase, MRX8 (MIOREX complex component 8) encoded by YDR336w, to have a significant role in mitochondrial function. Mrx8 belongs to the YihA family of GTPases, which in bacteria regulates large ribosomal subunit biogenesis/stability and is essential for cell growth (Schaefer et al., 2006; Cooper et al., 2009). Mrx8 is largely conserved with its bacterial family member YihA in the C-terminal GTPase domain with 58% similarity. Mrx8 contains an additional 132 amino acids at its N-terminus besides the conserved GTPase domain that contain a cryptic mitochondrial targeting sequence (Claros and Vincens, 1996). The human orthologue of MRX8 (GTPBP8-GTP binding protein 8) also contains a highly divergent N-terminal extension in addition to a conserved GTPase domain. Mrx8, which was found as a part of the MIOREX complex, is thus speculated to be involved in translation regulation (Kehrein et al., 2015).
In this study we show for the first time the role of this novel protein Mrx8 in regulating optimal Cox1 synthesis during cold stress. Deletion of MRX8 reduces the ability of cells to utilize carbon sources requiring robust cellular respiration when grown under suboptimal temperature. Consistent with a function during translation, Mrx8 peripherally localized to the inner mitochondrial membrane and associates with the mitochondrial ribosomes. Furthermore, ∆mrx8 cells were substantially defective for both translation initiation and elongation of Cox1, and mutations in mrx8 predicted to be deficient for guanine nucleotide binding were compromised for in vivo function. Finally, we show that the human orthologue of Mrx8 localizes to mitochondria in mammalian cells and partially rescues a glycerol growth defect under cold stress in Δmrx8 yeast cells, indicating functional conservation.
RESULTS
Mrx8 is localized to the mitochondrial matrix
To determine the cellular location of Mrx8, antibodies were raised against peptides within the N-terminus of Mrx8 as described in Materials and Methods. The specificity of the antibody was established by immunoblot analysis on isolated mitochondria from wild-type cells and compared with that of mitochondria from Δmrx8 cells. A novel band corresponding to the predicted size of Mrx8 (33.7 kDa) was observed in wild-type mitochondria and not in Δmrx8 mitochondria although the two samples expressed equivalent levels of a bona fide mitochondrial protein, Mtg2 (Figure 1A and Supplemental Figure S1 (Datta et al., 2005). Moreover, Mrx8 was specifically enriched in mitochondrial fractions as was Cox2, an integral membrane protein, and not in the cytosolic fraction (Figure 1B). To determine the submitochondrial location of Mrx8, a series of protease digestions were performed. To test its association with the outer membrane, intact mitochondria were treated with the indicated concentration of proteinase K. Mrx8 was resistant to externally added proteinase K, similar to inner membrane proteins F1β and Cox2, which reflects the presence of Mrx8 inside the mitochondria (Figure 1C). To determine the presence of Mrx8 in either the inner membrane space or the matrix, mitoplasts were generated and treated with the indicated concentration of proteinase K. Mrx8 as F1β, a known peripherally associated inner membrane protein facing the matrix, remained protease protected, whereas Cox2 having epitopes facing the inner membrane space was degraded (Figure 1D). Further, to determine whether Mrx8 is associated peripherally with the inner membrane or is an integral part of the membrane, mitochondria were treated with sodium chloride, sodium carbonate, and urea, which disrupt interactions between peripheral and integral membrane proteins using different mechanisms (Schook et al., 1979; Fujiki et al., 1982). Mrx8 remained in the membrane pellet as did Tim23, an integral membrane protein with four transmembrane helices (Bauer et al., 1996), upon treatment with NaCl and Na2CO3, while F1β was found in the supernatant fraction (Figure 1E). However, on treatment with urea Mrx8 fractionates in the supernatant as did F1β, while Tim23 remained in the pellet (Figure 1E), indicating Mrx8 to be a tightly bound peripheral membrane protein, consistent with the absence of transmembrane domain(s) as shown by in silico analysis (https://embnet.vital-it.ch/software/TMPRED_form.html). Taking the results together, we can conclude that Mrx8 is tightly attached to the inner mitochondrial membrane facing the matrix side.
FIGURE 1:
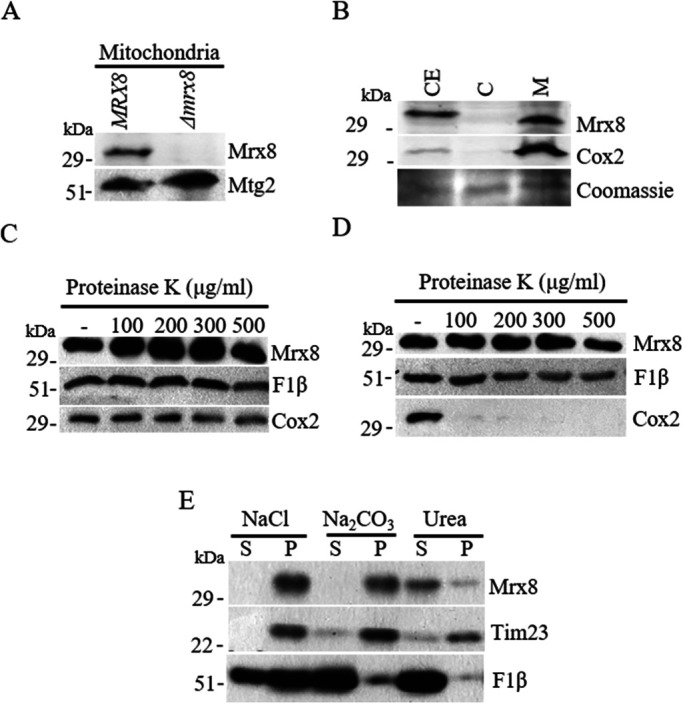
Mrx8 localizes to the mitochondrial inner membrane facing the matrix side. (A) Mitochondria were isolated from MRX8 and Δmrx8 cells. Equal amounts of mitochondrial protein were separated by SDS–PAGE and subjected to immunoblot analysis. (A larger area of the immunoblot is represented in Supplemental Figure S1.) (B) Yeast cell extract (CE) was fractionated into cytosol (C) and mitochondria (M). Protein fractions were separated by SDS–PAGE and subjected to immunoblot analysis. As control a Coomassie-stained gel is shown. (C) Intact mitochondria or (D) mitoplasts were treated with 0–500 μg/ml proteinase K as indicated. The reaction was terminated by the addition of TCA, and proteins were separated by SDS–PAGE and subjected to immunoblot analysis. (E) Mitochondria were treated with either 1 M NaCl, 0.1 M Na2CO3, or 6 M urea as indicated. Soluble (S) and membrane (P) protein fractions were separated on SDS–PAGE and subjected to immunoblot analysis. Samples were analyzed using antibodies to Mrx8, Mtg2, Cox2, Tim23, and F1β.
Mrx8 is required for growth on respiratory media during cold stress
To determine whether Mrx8 plays an essential role in optimal mitochondrial activity, the ability of Δmrx8 cells to utilize glycerol at different temperatures was examined, as cells require robust cellular respiration for its utilization. We observed that Δmrx8 cells were not able to grow in media containing glycerol as efficiently as MRX8 cells at 30°C (Figure 2, A and B). The severity of growth defect was significantly more pronounced when Δmrx8 cells were grown at a lower temperature (16°C), indicating that Mrx8 is required for growth on respiratory media during cold stress (Figure 2A and Supplemental Figure S2). During growth in fermentative media (glucose), the basal level of electron transport chain activity is reported (van Dijken et al., 1993). A significant up-regulation of electron transport chain components, especially those encoded by the mitochondrial genome, takes place when cells are shifted from fermentative to respiratory media (glycerol) (Couvillion et al., 2016; Morgenstern et al., 2017). This indicates that there are regulatory proteins that maintain mitochondrial translation at a basal level during growth in fermentative media and those that up-regulate mitochondrial translation during adaptation to respiratory media. We tested whether Mrx8 has a specific role during adaptation from fermentative to respiratory media or whether it was required for both adaptation and growth on respiratory media in a temperature-dependent manner. Wild-type and Δmrx8 cells were either cultured in glucose and shifted to glycerol or cultured in glycerol before inoculation in fresh glycerol medium.
FIGURE 2:

Mrx8 is required for efficient cellular respiration. (A) Shown are 10-fold serial dilutions of MRX8 and Δmrx8 cells on glucose and glycerol plates and incubated at the indicated temperatures. (B) MRX8 and Δmrx8 cells were initially cultured in either glucose (left) or glycerol (right) and then diluted into fresh glycerol media and incubated at 30°C. Optical densities were measured at 600 nm at the indicated time, and each data point is an average value of six independent colonies cultured in parallel. Tables indicate doubling time for each strain. (C) Mitochondria were isolated from wild-type cells cultured in glucose, galactose, and ethanol medium. Equal amounts of mitochondrial protein were separated by SDS–PAGE and subjected to immunoblot analysis. Samples were analyzed using antibodies to Mrx8 and Mtg2.
MRX8 and Δmrx8 cells show a similar growth lag when shifted from fermentative to respiratory media. However, Δmrx8 cells show slower utilization of glycerol as indicated by a difference of doubling time of approximately 3 h at 30°C (Figure 2B, left). Moreover, Δmrx8 cells adapted to respiratory media failed to utilize glycerol as efficiently as MRX8 cells (Figure 2B, right). Similar results were obtained when Δmrx8 cells were constantly cultured under cold stress at 16°C (Supplemental Figure S2). Taken together, the results indicate that Mrx8 is required for optimal utilization of respiratory media.
Given that the large-scale remodeling of the mitochondrial proteome takes place upon shifting cells from fermentative to respiratory media (Couvillion et al., 2016; Morgenstern et al., 2017), we examined whether Mrx8 is differentially regulated during such a shift. We observed no significant change in levels of Mrx8 in mitochondria from cells cultured under different carbon sources (Figure 2C). MRX8 transcript levels have been shown to remain constant in cells when shifted from fermentative to respiratory medium (Couvillion et al., 2016). Overall, our results reflect that Mrx8 promotes cellular adaptation to utilize a nonfermentative carbon source when glucose is exhausted.
De novo Cox1 synthesis is reduced in mrx8 null mutants
To examine whether Mrx8 is necessary for mitochondrial protein synthesis, wild-type and Δmrx8 cells were labeled with protein labeling mix (35S-l-methionine and 35S-l-cysteine) in the presence of cycloheximide to measure de novo mitochondrial translation as described in Materials and Methods. In Δmrx8 cells, no significant reduction in de novo synthesis of mitochondrially encoded proteins was observed in cells cultured at 30°C (Figure 3A and Supplemental Figure S3A). However, when newly synthesized mitochondrial proteins were labeled in cells cultured at 16°C, a severe defect in Cox1 synthesis was observed (Figure 3A and Supplemental Figure S3B). When Δmrx8 cells were grown at 30°C and shifted to 16°C before the addition of 35S protein–labeling mix, reduction in Cox1 synthesis was observed after 4 h of the shift to 16°C (Supplemental Figure S3C). To investigate whether a reduced Cox1 level is a consequence of a defect in mitochondrial protein synthesis at 16°C in Δmrx8 cells rather than protein turnover, wild-type and Δmrx8 cells were pulse labeled at 30°C and chased at 16°C for different time intervals. The level, and therefore the stability, of the labeled proteins were comparable in the wild-type and Δmrx8 cells at all time points during the chase at 16°C, indicating that preexisting translation products are equally stable (Figure 3B). Consistent with reduced de novo translation of Cox1 in Δmrx8 cells at 16°C, we observed a reduction in the steady state levels of Cox1 (Figure 3C). Interestingly, we also observed reduced levels of Cox2 and Cox3 at 16°C at the steady state (Figure 3C). Given that Cox1 synthesis is tightly linked to Complex IV assembly, the reduction in levels of Cox2 and Cox3 could be due to an indirect consequence of defective Complex IV biogenesis as has been previously reported (De Silva et al., 2017; Mays et al., 2019). However, the defect in Cox1 synthesis due to reduced COX1 mRNA could be ruled out as levels of COX1 mRNA were equivalent in mitochondria from MRX8 and Δmrx8 cells grown at 16°C, as were COX2, COX3, and COB mRNA levels (Figure 3D). Taken together, our results indicate that Mrx8 acts to promote Cox1 protein synthesis.
FIGURE 3:
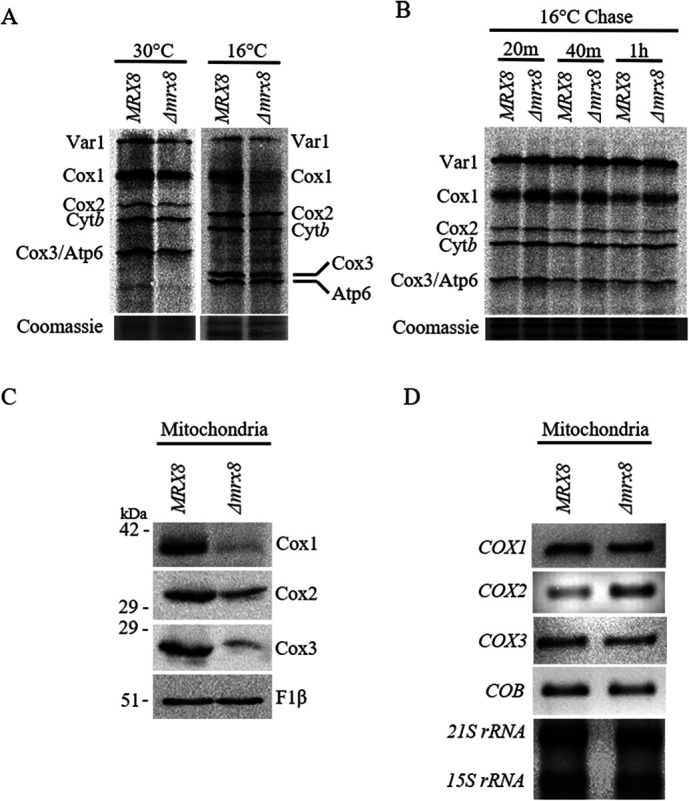
De novo Cox1 synthesis is reduced in Δmrx8 cells. (A) Newly synthesized mitochondrial protein products were labeled by incorporation of [35S]methionine and cysteine in the presence of cycloheximide to inhibit cytosolic protein synthesis in MRX8 and Δmrx8 cells at either 30°C or 16°C. (B) Newly synthesized mitochondrial protein products were labeled by incorporation of [35S]methionine and cysteine in the presence of cycloheximide to inhibit cytosolic protein synthesis in MRX8 and Δmrx8 cells at 30°C and chased at 16°C by the addition of casamino acid and sodium sulfate for the indicated time points. Mitochondria were isolated, and equal concentrations of mitochondrial proteins were separated on 17.5% SDS–PAGE, transferred onto a nitrocellulose membrane, exposed to BAS storage phosphor screen, and developed using phosphorimager. The positions of Var1, Cox1, Cox2, Cytb, and Cox3/Atp6 are indicated. As control a Coomassie-stained gel is shown. (C) Mitochondria were isolated from MRX8 and Δmrx8 cells cultured at 16°C. Equivalent amounts of mitochondrial proteins were separated via SDS–PAGE and subjected to immunoblot analysis. Samples were analyzed using antibodies to Cox1, Cox2, Cox3, and F1β. (D) Transcript levels of mitochondrially encoded genes were assayed in isolated mitochondria from MRX8 and Δmrx8 cells. RNA samples were subjected to RT-PCR using primers specific for COX1, COX2, COX3, and COB. 21s rRNA and 15s rRNA levels as detected by EtBr staining. Representative images of multiple trials are shown.
MRX8 is required for translation initiation and elongation of Cox1
The defect in de novo Cox1 synthesis in Δmrx8 cells could be due to either defective translation initiation or elongation or both. To examine this aspect, we used engineered strains XPM78a, and XPM171a carrying Δmrx8 (Supplemental Figure S4). Both strains are deleted for the nuclear gene ARG8, which encodes an essential arginine biosynthetic enzyme localized to the mitochondria. XPM78a is engineered to express 512 nucleotides of intronless COX1 fused to ARG8m under the control of the COX1 promoter (Perez-Martinez et al., 2003) (Supplemental Figure S4A). This allows translation to be scored as a function of growth on synthetic media lacking arginine and on YPG. In comparison to wild-type cells, Δmrx8 cells were defective for growth on synthetic media lacking arginine and also on YPG at 16°C but not at 30°C (Figure 4A). Aberrant de novo Cox1 synthesis and reduced steady state levels of Arg8m were observed in Δmrx8 cells in comparison to wild-type cells at 16°C (Figure 4B). However, we did not detect a Cox1-Arg8m fusion product. This is likely due to the labile nature of the Cox1-Arg8m fusion product containing an internal cleavage site such that Cox1 and Arg8 can function independently (Steele et al., 1996; Perez-Martinez et al., 2003). XPM171a has a functional reengineered ARG8m in place of the COX1 open reading frame in the mtDNA. This strain is further engineered such that COX1 and COX2 open reading frames are placed downstream of the COX2 promoter (Perez-Martinez et al., 2003) (Supplemental Figure S4B). The viability of this engineered strain on synthetic media lacking arginine allows us to score for translation initiation, while the viability of cells on YPG allows us to score for translation elongation. In comparison to cells harboring the wild-type allele of MRX8, Δmrx8 cells failed to grow either on synthetic media lacking arginine or on YPG at 16°C but not at 30°C, indicating the requirement of Mrx8 in Cox1 translation initiation and elongation during cold stress (Figure 4A). Consistently, both newly synthesized Cox1 and steady state levels of Arg8m were reduced in Δmrx8 cells at 16°C in comparison to wild-type cells (Figure 4B). Further, we examined the consequences of introducing Δmrx8 in RGV140, an engineered strain deleted for nuclear gene ARG8 carrying modified mtDNA, where the COX3 promoter controls Arg8m synthesis (Mays et al., 2019) (Supplemental Figure S5A). Growth on synthetic media lacking arginine or Arg8m accumulation at 16°C remained unperturbed in Δmrx8 cells, indicating that Mrx8 preferentially promotes Cox1 synthesis (Supplemental Figure S5, B and C). Thus, taken together, these results indicate that Mrx8 governs optimal Cox1 translation initiation and elongation during cold stress.
FIGURE 4:

MRX8 is essential for COX1 translation initiation and elongation. (A) Tenfold serial dilutions of Δmrx8 cells in either XPM78a or XPM171a expressing either wild-type MRX8 or vector were spotted on YPD, SD-Arg. and SGly. (B) Top: Newly synthesized mitochondrial protein products were measured in either XPM78a or XPM171a with the ∆mrx8 allele in their nuclear genome expressing either wild-type MRX8 or vector at 16°C by incorporation of [35S]methionine and cysteine in the presence of cycloheximide to inhibit cytosolic translation. Mitochondria from labeled cells were isolated, and proteins were separated on 17.5% SDS–PAGE. Radiolabeled proteins were transferred onto a nitrocellulose membrane and visualized by phosphoimaging. The positions of mtDNA-encoded proteins are indicated. As control a Coomassie-stained gel is shown. Representative images of multiple trials are shown. Bottom: Mitochondria was isolated from Δmrx8 cells in either XPM78a or XPM171a expressing either wild-type MRX8 allele or vector cultured at 16°C. Equivalent amounts of mitochondrial proteins were separated via SDS–PAGE and subjected to immunoblot analysis. Samples were analyzed using antibodies to Arg8 and F1β.
Mrx8 requires nucleotide binding for its in vivo function
MRX8 belongs to the YihA/YsxC family of GTPases, with highly conserved G-domains responsible for the binding and hydrolysis of guanine nucleotide. It is well documented among GTPases involved in ribosome function that nucleotide binding and GTPase activity often power an essential function (Verstraeten et al., 2011). Therefore, we investigated the consequences of mutations in the G-domain of MRX8 that are predicted to abrogate nucleotide binding and hydrolysis in vivo. The choice of the residue for mutation was based on the bacterial YihA protein as well as other TRAFAC family members, which are either biochemically characterized or for which detailed structural information is available (Lehoux et al., 2003; Ruzheinikov et al., 2004).The G1 box with the GX2NXGK(S/T) consensus is required for nucleotide binding, wherein the GKS motif is conserved across species and is responsible for binding with the α and β phosphates of GTP or GDP (Bourne et al., 1991; Sprang, 1997). Mutation of the GKS motif to AAA in MRX8 is predicted to result in a complete loss of nucleotide binding and thus inhibit in vivo function as shown for several Obg family proteins in S. cerevisiae and Caulobacter crescentus (Datta et al., 2004; Fuentes et al., 2007). Cells expressing the mrx8GKS145-147AAA mutant protein were defective for growth on glycerol at 16°C, similar to Δmrx8 cells, and de novo Cox1 synthesis is defective in cells expressing mrx8GKS145-147AAA at 16°C (Figure 5, A and C). A reduced accumulation of steady state protein as a cause for defects observed in cells expressing mrx8GKS145-147AAA could be ruled out, as similar levels of Mrx8 were observed in wild-type and mutant mitochondria (Figure 5B). Therefore, our results indicate that nucleotide binding and hydrolysis are required for the in vivo function of Mrx8.
FIGURE 5:
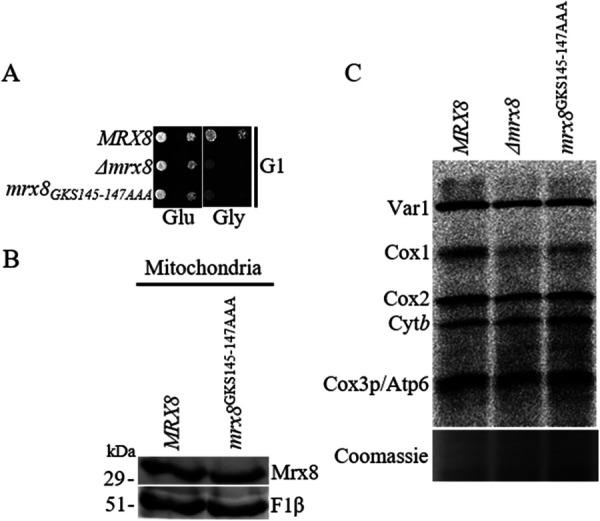
Putative nucleotide binding/hydrolysis is essential for in vivo Mrx8 function. (A) Shown are 10-fold serial dilutions of Δmrx8 cells expressing either wild-type MRX8, empty vector, or the mrx8GKS145-147AAA mutant allele on YPD and YPG at 16°C. (B) Mitochondria from Δmrx8 cells episomally expressing either wild-type MRX8 or the mrx8GKS145-147AAA mutant allele were isolated. Equivalent amounts of mitochondrial proteins were separated via SDS–PAGE and subjected to immunoblot analysis. Samples were analyzed using antibodies to Mrx8 and F1β. (C) Newly synthesized mitochondrial protein products were measured in Δmrx8 cells episomally expressing either wild-type MRX8, vector, or the mrx8GKS145-147AAA mutant allele at 16°C by incorporation of [35S]methionine and cysteine in the presence of cycloheximide to inhibit cytosolic translation. Mitochondria from labeled cells were isolated, and proteins were separated on 17.5% SDS–PAGE. Radiolabeled proteins were transferred onto a nitrocellulose membrane and visualized by phosphoimaging. The positions of mtDNA-encoded proteins are indicated. As control a Coomassie-stained gel is shown.
Mrx8 associates with mitochondrial ribosomes
Given that MRX8 plays a role in optimal mitochondrial translation regulation, we examined whether Mrx8 associates with mitochondrial ribosomes. Mitochondrial ribosomal subunits were separated on sucrose gradient as described in Materials and Methods. The identity of individual subunits was confirmed by immunoblot analysis using the antibody to the small subunit (37S) protein Mrp13 (Partaledis and Mason, 1988) and the large subunit (54S) protein Mrp7 (Fearon and Mason, 1988). In the presence of magnesium ions and low salt (10 mM MgOAc, 100 mM NH4Cl), Mrx8 cofractionated with both Mrp7 and Mrp13, indicating its association with the mitochondrial ribosomes (Figure 6A). Increasing the salt concentration (10 mM MgOAc, 500 mM NH4Cl) resulted in a minor pool fractionating with ribosomal proteins while the majority of the Mrx8 were in the low-molecular-weight protein fractions (Figure 6B). To further test whether Mrx8 requires intact RNA–protein complex for migration into the sucrose gradient, we treated mitochondrial lysates with RNase A (100 µg/ml) before separation on a sucrose gradient. This led to the disruption of mitochondrial ribosomes and prevented migration of Mrx8 into the sucrose gradient (Figure 6C). Furthermore, isolation of the Mrx8-containing complex by immobilized metal affinity chromatography (IMAC) from mitochondrial lysates expressing functional Mrx8-6xHis led to copurification of Mrp7 (Figure 6D). Taken together, the results indicate that Mrx8 associates with the 54S subunit either as a part of the 74S monosome or when the 54S subunit is free from the monosome.
FIGURE 6:
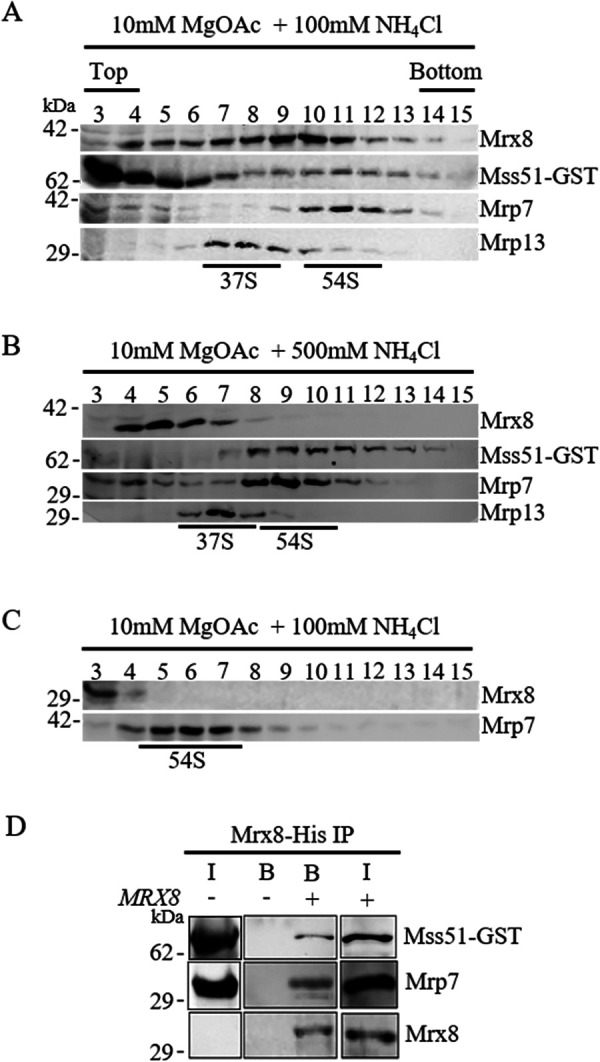
Mrx8 associates with mitochondrial ribosomes. Mitochondrial lysates from wild-type cells containing a functional MSS51-GST allele were separated by ultracentrifugation on a 5–30% sucrose gradient containing (A) 10 mM MgOAc, 100 mM NH4Cl, and (B) 10 mM MgOAc, 500 mM NH4Cl, at 135,000 × g for 4 h. (C) Mitochondrial lysates were incubated with RNase A before separation on a 5–30% sucrose gradient containing 10 mM MgOAc and 100 mM NH4Cl. Fractions were TCA precipitated, separated on SDS–PAGE, and subjected to immunoblot analysis. Antibodies used were against Mrx8, GST (to detect Mss51), Mrp7(bL27m), and Mrp13(mS44). The migration of the 37S and 54S peaks were labeled based on immunoblot analysis. (D) Mitochondria from cells expressing Mss51-GST and Mrx8-6xHis were solubilized and purified using metal ion chromatography. Fifteen percent of input (I), and bound protein (B) were separated by SDS–PAGE and subject to immunoblot analysis. Antibodies used were against Mrx8, GST (to detect Mss51), and Mrp7(bL27m).
Mrx8 does not govern Mss51 cofractionation with the ribosomes
Cox1 translation and its assembly into Complex IV are tightly coupled to a negative feedback loop involving Mss51. In addition to its role in Cox1 translation initiation, Mss51 also interacts with newly synthesized Cox1 and remains associated until Cox1 maturation and assembly into Complex IV is initiated (Perez-Martinez et al., 2003; Barrientos et al., 2004; Perez-Martinez et al., 2009). Defective assembly of newly synthesized Cox1 sequesters Mss51 in a higher-order Cox1 pre-assembly complex, which reduces its ability to initiate a fresh round of Cox1 translation (Perez-Martinez et al., 2003, 2009). To determine whether the absence of Mrx8 affects the Mss51-mediated feedback loop, we analyzed the mobility of a functional GST-tagged Mss51 on a sucrose density gradient in wild-type and Δmrx8 cells. When ribosomes from wild-type cells were fractionated on a sucrose gradient in the presence of magnesium ions and low salt, we observed that only a minor pool of Mss51 migrated with ribosomal marker proteins Mrp7 and Mrp13 while the majority were in the unbound fractions (Figure 6A). On increasing the salt concentration, the majority of Mss51 migrated with fractions containing ribosomal marker proteins, and minor pools migrated into deeper fractions than the large subunit (Figure 6B). These results were contrary to the mobility of Mrx8 in a sucrose gradient. Interestingly, isolation of Mrx8-containing complex led to copurification with Mrp7 and Mss51 (Figure 6D). Taken together, the results indicate that there exists a pool of Mrx8, Mss51, and ribosomes in the mitochondria. Our results indicate a condition that allows us to detect Mss51 cofractionating with ribosomes on a sucrose density gradient in a respiratory-competent cell that is not known currently. The high-salt condition might stabilize the hydrophobic interaction of Mss51 with the ribosome including the Cox1 preassembly complex. In Δmrx8 cells Mss51 migration with the ribosomes under low- and high-salt conditions remained unaltered at 16°C in comparison to wild-type cells (Figures 7A and 6, A and B). This indicates that the reduced Cox1 synthesis observed in Δmrx8 cells is not a result of sequestration of Mss51 into a higher-order complex.
FIGURE 7:
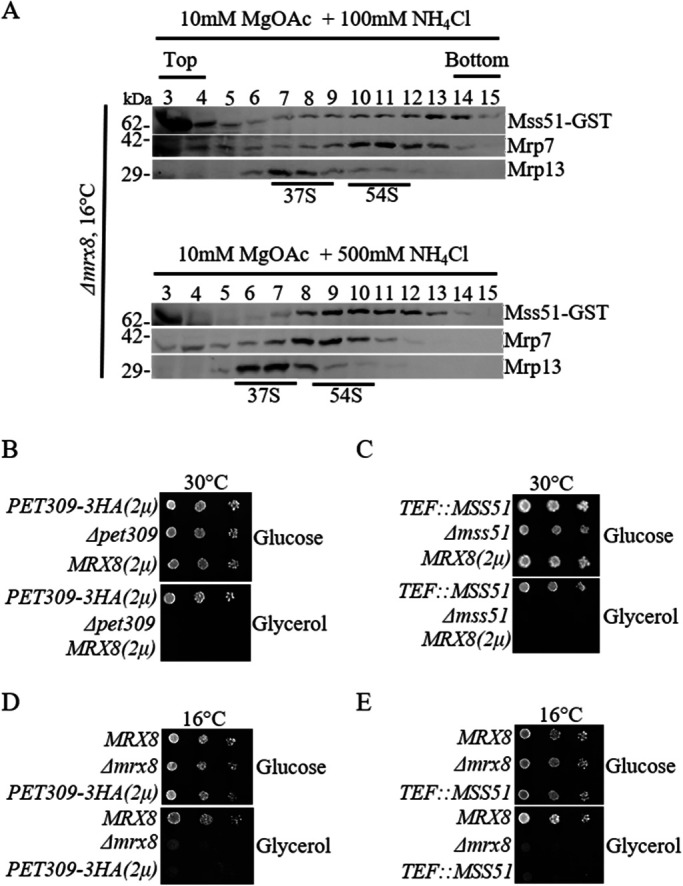
Mss51 migration on a sucrose density is independent of Mrx8. (A) Mitochondria from MSS51-GST cells carrying the Δmrx8 allele cultured at 16°C were lysed and separated by ultracentrifugation on a 5–30% sucrose gradient containing either (top) 10 mM MgOAc, 100 mM NH4Cl, or (bottom) 10 mM MgOAc, 500 mM NH4Cl. Fractions were TCA precipitated, separated by SDS–PAGE, and subjected to immunoblot analysis. Antibodies used were against GST (to detect Mss51), Mrp7(bL27m), and Mrp13(mS44). The migration of the 37S and 54S peaks were labeled based on immunoblot analysis. Shown are 10-fold serial dilutions of (B) Δpet309 cells expressing PET309-3HA(2µ), empty vector, or MRX8(2µ), (C) Δmss51 cells expressing MSS51-GST, empty vector, or MRX8(2µ), (D) Δmrx8 cells expressing MRX8, empty vector, or PET309-3HA(2µ), and (E) Δmrx8 cells expressing MRX8, empty vector, or MSS51-GST on glucose and glycerol media at either 30°C or 16°C.
Glycerol growth defects of Δmss51 and Δpet309 are not bypassed by multiple copies of Mrx8 and vice versa
Mss51 and Pet309 initiate Cox1 translation by binding to its 5′UTR in addition to Mss51’s role during Cox1 assembly (Manthey and McEwen, 1995; Zamudio-Ochoa et al., 2014). Mrx8 might function in conjugation with Pet309 and Mss51 or might serve in a redundant pathway to promote Cox1 synthesis. This was examined by testing the ability to restore glycerol growth defect in Δmss51 or Δpet309 cells upon introduction of multiple copies of MRX8 or vice versa. Under all circumstances, we did not observe restoration of cellular respiration in the deletion strains, arguing against functional redundancy (Figure 7, B–E).
GTPBP8, the human homologue of MRX8, complements Δmrx8 cells
The closest orthologue of MRX8 in humans is a GTPase of unknown function annotated as GTPBP8 with nearly 45% similarity over the entire protein length. We tested whether functional conservation exists between the two orthologous proteins. Expression of GTPBP8 in Δmrx8 cells failed to restore the growth defect on glycerol media at 16°C (unpublished data). This could be either because GTPBP8 is not functional in yeast or because the human mitochondrial targeting sequence on GTPBP8 is not recognized by the mitochondrial import machinery in yeast. To ensure mitochondrial localization, we fused the cleavable mitochondrial targeting sequence of MTG3 (amino acids 1–21) at the N-terminus of GTPBP8. Interestingly, there was partial restoration of growth on glycerol at 16°C by mitochondrially targeted GTPBP8 in Δmrx8 cells in comparison to cells expressing wild-type MRX8 (Figure 8A). This indicates that functional conservation exists between yeast and the human orthologue of Mrx8. The ability to restore mitochondrial function in Δmrx8 cells suggests that GTPBP8 is localized to the mitochondria and is involved in some aspect of cellular respiration. However, this does not rule out a differential localization of the endogenous GTPBP8. Using HEK293 cells transfected with enhanced green fluorescent protein (EGFP)-tagged GTPBP8, we demonstrated the localization of GTPBP8 in the mitochondria as it overlaps with MitoTracker red (Figure 8B). The specificity of GTPBP8-EGFP was confirmed by immunoblot analysis on mitochondria from HEK293 cells expressing EGFP-tagged GTPBP8 (63.2 kDa) or pEGFP-N1 (Figure 8C). Finally, when lysates from HEK293 cells expressing EGFP-tagged GTPBP8 were purified into cytosolic and mitochondrial fractions, we observed the enrichment of GTPBP8 in the mitochondrial fraction similar to COX IV, a bona fide mitochondrial protein, while GAPDH was enriched in the cytosolic fraction (Figure 8D). This shows that the endogenous GTPBP8 is localized to the mitochondria in mammalian cells and this function of mitochondria controlled by Mrx8 is conserved between yeast and humans. This is the first demonstration of conservation of this function of MRX8 to the best of our knowledge.
FIGURE 8:

Human MRX8 (GTPBP8) complements Δmrx8 cells and localizes to mitochondria in mammalian cell lines. (A) Shown are 10-fold serial dilutions of Δmrx8 cells expressing MRX8, empty vector, or MTS-tagged GTPBP8 on glucose and glycerol at 16°C. (B) Shown are representative confocal images of HEK293 cells transfected with either empty vector EGFP-N1 or EGFP-tagged GTPBP8. Scale bars: 10 µm (vector) and 5 µm (GTPBP8-EGFP). (C) Mitochondria were isolated from HEK293 cells transfected with EGFP-tagged GTPBP8 and empty vector EGFP-N1 as described in Materials and Methods. (D) HEK293 cell extracts (CE) were fractionated into cytosol (C) and mitochondria (M). Protein samples were separated by SDS–PAGE and subjected to immunoblot analysis. Antibodies used were against GFP (to detect GTPBP8), GAPDH, and COX IV.
DISCUSSION
Translation regulation in response to changing environmental signals such as temperature or nutrient availability is well documented in yeast as well as in bacteria. The process of mitochondrial translation is uniquely distinct from bacteria and cytosolic translation. Although mitochondrial translation is understood in some detail (Fox, 2012; Ott et al., 2016), the adaptation of the translational machinery to changing environmental conditions that might alter the spatiotemporal requirement for the expression of various genes is not clear.
Herein, we show that MRX8, a protein belonging to the YihA subfamily of GTPases, promotes Cox1 synthesis during cold stress. The only prior report showing temperature-dependent regulation of mitochondrially encoded Complex IV subunits and their subsequent assembly is in the case of Δguf1 cells (Bauerschmitt et al., 2008). We observed that Δmrx8 cells failed to utilize glycerol at the same rate as wild-type cells specifically at 16°C, indicating defective cellular respiration. The predicted mitochondrial localization (60% probability) (Claros and Vincens, 1996) and isolation of Mrx8 as a part of the MIOREX complex (Kehrein et al., 2015) strongly indicates that Mrx8 is likely to be a mitochondrial protein. Consistently we found Mrx8 to be peripherally associated with the mitochondrial inner membrane. It is well documented that the mitochondrial inner membrane serves as a platform for mitochondrial gene expression involving steps of DNA replication, repair, transcription, RNA processing/modification, RNA degradation, ribosome biogenesis, and mRNA turnover and translation (Ott et al., 2016; Singh et al., 2020). Thus, not surprisingly, in Δmrx8 cells at 16°C we observed that de novo Cox1 synthesis and steady state accumulation were significantly reduced, without a reduction in Cox1 transcript levels, indicating a requirement of Mrx8 for proper Cox1 translation (Figure 3). Mitochondrial transcripts are recognized by specific activator proteins that interact directly with the 5′ UTR and aid its association with mitochondrial ribosomes to initiate translation (Fox, 2012). Regulation of mitochondrial translation occurs by two different mechanisms. The first is by regulating the availability of nuclear-encoded translation activators in mitochondria, which in turn modulate rates of polypeptide formation from target mRNA (Fiori et al., 2005; Couvillion et al., 2016). Second, the regulation of mitochondrial translation is achieved by tightly coupling it to OXPHOS subunit assembly. If assembly fails, synthesis of key subunits expressed form mtDNA is also down-regulated (Fox, 2012). Thus, could the reduction observed in Cox1 synthesis in Δmrx8 cells be due to a direct role of Mrx8 in promoting Cox1 translation or a secondary consequence of sequestration of Mss51 in a higher-order complex, or a combination of both? Our results support a model whereby Mrx8 promotes Cox1 translation. First, we found Mrx8 to be associated with the ribosomes likely as a part of the 74S monosome. Second, using the Arg8m reporter system, we observed that Mrx8 is required for both translation initiation and elongation of Cox1 during growth under the suboptimal temperature of 16°C. Although this indicates that Mrx8 can act on UTRs and coding sequence (CDS) of Cox1 mRNA, further investigation is required to determine a direct binding of Cox1 mRNA with Mrx8. Moreover, Mss51 sequestration in a higher-order complex is not observed in Δmrx8 cells, arguing that reduction in Cox1 synthesis is not due to Mss51 limitation. Interestingly, we found Mrx8 to be in a complex containing Mss51 and the ribosome. However, overexpression of MRX8 did not rescue the defective cellular respiration in Δmss51 or Δpet309 or vice versa. Taken together, these results indicate that although there might be a complex within the mitochondria containing both Mrx8 and Mss51 in association with the ribosome, however they act independently to promote Cox1 synthesis.
Nuclear-encoded GTPases have been shown to regulate various aspects of ribosome assembly and protein translation in mitochondria presumably by utilization of energy released upon GTP hydrolysis to power a mechanistic step (Karbstein, 2007; Strunk and Karbstein, 2009; Maracci and Rodnina, 2016). Cells expressing mutant Mrx8 predicted to be compromised for nucleotide binding were defective in cellular respiration including Cox1 protein synthesis. One would wonder what the rationale would be for the cell to have a putatitve GTPase specifially promoting Cox1 synthesis especially during cold stress. An answer might lie in the way Mrx8 utilizes nucleotide binding/hydrolysis to carry out its in vivo function. This aspect is under investigation.
A mitochondrial gene-expression system, especially that of the mitochondrial ribosome, is the best example of diversity in the mitochondrial proteome (Desmond et al., 2011; Amunts et al., 2014). Mitochondrial ribosomal proteins, assembly factors, and regulatory proteins required for mitochondrial gene expression in S. cerevisiae can be classified in four categories: those that have orthologues 1) in prokaryotes and all eukaryotes, 2) only in eukaryotes, 3) in prokaryotes and in some eukaryotic lineages but not all, and 4) in a narrow, lineage-specific manner such as lower eukaryotes only and absent in higher eukaryotes sequenced (Kurland and Andersson, 2000; Szklarczyk and Huynen, 2010; Gray, 2015). MRX8 represents the third class, that is, it has orthologues in bacteria, in single cell eukaryotes such as S. cerevisiae, and in vertebrates including humans but in no other kingdom of life sequenced so far. Mss51 and Pet309, which are essential for Cox1 translational activation and assembly, are only yeast specific and no clear orthologues have been identified in metazoans (Perez-Martinez et al., 2003; Barrientos et al., 2004; Zamudio-Ochoa et al., 2014; Dennerlein and Rehling, 2015). Thus, in order to maintain the synthesis of COX1 in humans, a different mechanism must exist. Significantly, we found that targeting of GTPBP8 to the mitochondria of Δmrx8 cells is able to largely restore growth at a reduced temperature on media requiring cellular respiration. Consistent with functional conservation, we found GTPBP8 to localize to the mitochondria in mammalian cells. Thus, it appears that MRX8 represents a conserved pathway that has been retained during mitochondrial evolution in certain eukaryotic life forms. We are currently examining whether COX1 translation in humans is governed by GTPBP8 and whether it has a similar role in linking optimal translation with cold stress.
MATERIALS AND METHODS
Request a protocol through Bio-protocol.
Yeast strains and media
Yeast strains used in this study are listed in Table 1. Complete media used were YEP (1% yeast extract, 2% peptone) containing 2% glucose (YPD) or 3% glycerol (YPG) as the carbon source. Synthetic minimal media (0.67% yeast nitrogen base) containing 2% glucose (SD), 3% glycerol (SGly) 2% galactose (SGal), 2% glucose and 0.1% 5-fluoroorotic acid (5′FOAD), and 2% glucose and 0.006% canavanine sulfate (SCAN) were supplemented with appropriate amino acids when required, as described in Guthrie and Fink (1991). Semisynthetic lactate medium (0.3% yeast extract, 0.05% glucose, 0.05% CaCl2, 0.05% NaCl, 0.06% MgCl2, 0.1% KH2PO4, 0.1% NH4Cl, 2% ᴅl-lactic acid, 0.8% NaOH, pH 5.5) was prepared as described (Glick and Pon, 1995).
TABLE 1:
Genotypes and sources of yeast strains used in this study.
| Strain | Genotype | Source |
|---|---|---|
| BY4741 | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, ρ+ | American Type Culture Collection (ATCC) |
| BY4742 | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, ρ+ | ATCC |
| CRY1 | MATa, ura3-52, trp1Δ2, leu2-3112, his3-11, ade2-1, can1-100, ρ+ | Brickner and Fuller, 1997 |
| KD43 | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, mrx8::KanMX4, ρ+ | ATCC |
| KD337 | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, mrx8::KanMX, [CEN, MRX8,URA3], ρ+ | This study |
| KD749 | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, mrx8::KanMX4, [CEN, CYC1::MRX8, HIS3], ρ+ | This study |
| KD1069 | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, mrx8::KanMX4, [CEN, CYC1::mrx8GKS145-147AAA, HIS3], ρ+ | This study |
| KD757 | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, mrx8::KanMX4, [CEN, CYC1::GTPBP8, HIS3], ρ+ | This study |
| KD877 | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, mrx8::KanMX4, [CEN, CYC1::MTS-GTPBP8, HIS3], ρ+ | This study |
| KD878 | MATa, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, ρ+ | This study |
| KD884 | MATa, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, mrx8::KanMX4, ρ+ | This study |
| XPM171a | MATα, leu2-3,112, lys2, ura3-52, arg8::hisG, [ρ+,cox1::ARG8m, cox2::COX1 COX2] | Perez-Martinez et al., 2003 |
| KD1512 | MATα, leu2-3,112, lys2, ura3-52, arg8::hisG, mrx8::KanMX4 [ρ+,cox1::ARG8m, cox2::COX1 COX2] | This study |
| XPM78a | MATα, leu2-3,112, lys2, ura3-52, arg8::hisG,[ρ+,COX1(1-512)::ARG8m, Δ∑aI, Δ∑bI] | Perez-Martinez et al., 2003 |
| KD1562 | MATα, leu2-3,112, lys2, ura3-52, arg8::hisG, mrx8::KanMX4 [ρ+,COX1(1-512)::ARG8m, Δ∑aI, Δ∑bI] | This study |
| RGV140 | MATa, his3-1,15 leu2-3,112, trp1-1, ura3-1 Δarg8::KanMX 4[ρ+, cox3Δ::ARG8m] | Mays et al., 2019 |
| KD1516 | MATa, his3-1,15 leu2-3,112, trp1-1, ura3-1 Δarg8::KanMX4, mrx8::TRP1-1 [ρ+, cox3Δ::ARG8m] | This study |
| KD1532 | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, MSS51-GST, ρ+ | This study |
| KD1567 | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, MSS51-GST, mrx8::TRP1-1, ρ+ | This study |
| XPM76a | Mata, lys2, leu2-3,112, arg8::hisG, ura3-52, mss51∆::LEU2 [ρ+, COX1(1-512)::ARG8m, Δ∑aI, Δ∑bI] | Perez-Martinez et al., 2009 |
| KD1608 | Mata, leu2-3,112, ura3-52, mss51∆::LEU2, ρ+ | This study |
| KD1610 | Mata, leu2-3,112, ura3-52, mss51∆::LEU2, [TEF::MSS51-GST],ρ+ | This study |
| KD1611 | Mata, leu2-3,112, ura3-52, mss51∆::LEU2, [2µ, MRX8,URA3], ρ+ | This study |
| XPM232 | Mata, lys2, leu2-3,112, arg8::hisG, ura3-52, pet309∆::LEU2,[ρ+, Δ∑aI] | Tavares-Carreon et al., 2008 |
| KD1612 | Mata, lys2, leu2-3,112, arg8::hisG, ura3-52, pet309∆::LEU2, [pXP104: 2µ, PET309-3HA, URA3], [ρ+, Δ∑aI] | This study |
| KD1613 | Mata, lys2, leu2-3,112, arg8::hisG, ura3-52, pet309∆::LEU2, [2µ, MRX8,URA3], [ρ+, Δ∑aI] | This study |
| KD1614 | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, mrx8::KanMX4, [TEF::MSS51-GST]ρ+ | This study |
| KD1615 | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, mrx8::KanMX4, [pXP104: 2µ, PET309-3HA, URA3], ρ+ | This study |
Plasmid and strain construction
Isolation of yeast genomic DNA and yeast transformation were done as described previously (Guthrie and Fink, 1991). The MRX8 gene along with 1 kb upstream and 1 kb downstream sequences was amplified from genomic DNA of the CRY1 strain using primers 5′TCAAATGACTGAAGAAATTT3′ and 5′CCGCATCATTCAATCCTAGT3′. The resulting 2945-base-pair PCR product was cloned in pCR 2.1-TOPO to generate pDP2 (KD290) and confirmed by sequencing from both ends. This 2945-base-pair gene product was subcloned in pRS316 (CEN, URA3) and pRS426 (2µ, URA3) as an EcoRI fragment to generate pDP8 (KD337) and pYV31(KD1598), respectively.
To generate MRX8-6xHis, the MRX8 ORF without stop codon was PCR amplified using primers 5′CATGCCATGGTGGAACAACTGTGTAAG3′ and 5′CCGGAATTCGCTAAAATCAAACCACAGCT3′. The 945-base-pair PCR product was cloned into the pGEMT-Easy vector to generate pDP21 (KD466). MRX8 from pDP21 was shuttled into pET-28a as a NcoI-EcoRI fragment, resulting in pDP25 (KD546). To clone MRX8-6xHis under the CYC1 promoter, the 2242-base-pair gene product along with a downstream sequence from pET-28a was cloned into p413CYC1 (CEN, HIS3) as XbaI and SmaI fragments, resulting in pDP33 (KD686).
To express GTPBP8 (GTPBP8) in yeast, RNA isolated from a HeLa cell line was used to generate cDNA using the AMV First Strand cDNA Synthesis Kit (NEB,USA). The cDNA was then used to amplify the GTPBP8 gene fragment using primers 5′GATATCATGGCGGCGCCCGGGCTGCGG3′ and 5′CCCATCGATTTAGTCAAGACTTCCTGTTAC3′. The resulting 855-base-pair PCR product was cloned in pGEMT-Easy (Promega) to generate pDP55 (KD754). The 855-base-pair PCR product was also directly cloned in p413CYC1 as an EcoRV-ClaI fragment, resulting in pDP52 (KD751), which was confirmed by sequencing from both ends. MTS-GTPBP8 containing a cleavable mitochondrial targeting sequence from MTG3 upstream of GTPBP8 was generated by a two-step PCR with overlapping forward primers P1: 5′ATGTTGAATCTGTGTCATGCTCTTCGAGGCGTACGTCAGTTTTCCTGTTC3′, P2: 5′GTACGTCAGTTTTCCTGTTCTGTGATTGTGAAAATGGCGGCGCCCGGGCTGCG3′, and hYDR336wClaIdown: 5′CCCATCGATTTAGTCAAGACTTCCTGTTAC3′. Briefly, the first PCR amplification step used primers P2 and hYDR336wClaIdown to amplify the 855-base-pair GTPBP8 from pDP55. A second PCR, using the 855-base-pair PCR product obtained above as template was performed using primers P1 and hYDR336wClaIdown. The 906-base-pair PCR product obtained was cloned in p413CYC1 as a SpeI-ClaI fragment to generate pDP64. To create an GTPBP8 fused with GFP, the GTPBP8 gene product was amplified from pDP52 using primers 5′ACGCGTCGACATGGCGGCGCCCGGGCTGCG3′ and 5′CGGGGTACCCCGTCAAGACTTCCTGTTAC3′. The 855-base-pair PCR product was cloned in pEGFP N1 (Clontech) as a SalI-KpnI fragment to generate pDP77 (KD986).
Cell deleted for MRX8 were generated in a XPM171a, XPM78a, and RGV 140. Disruption of MRX8 was carried out by using a PCR-based gene replacement approach, where either KanMx or TRP1 (amplified from pFA6a-kanMX or pFA6a-TRP1) was placed between the upstream and downstream regions of MRX8 by PCR using primers MRX8F1: 5′ATATTCCCTAATATTTTAGCGAATAGGAACCATTGGCACGGATCCCCGGGTTAATTAA3′ and MRX8R1: 5′AGCTGAAATGCAATCAGCAATATATAGATACATATTGTGAGAATTCGAGCTCGTTTAAAC3′. The linear PCR product was transformed into XPM171a, XPM78a, and RGV 140 and selected for insertion of the marker allele in place of MRX8 to generate KD1512, KD1562, and KD1516, respectively.
Cells expressing MSS51-GST from its chromosomal loci was generated by transforming BY4742 with a linear PCR product obtained from pFA6a-GST-kanMX6 using primers MSS51F2: 5′CGGTTCAGAGGTAAAAGGTACCATACAATCAAGAGACAACGGATCCCCGGGTTAA TTAA3′ and MSS51R1: 5′TATATTATAAGATGAAGTTGGGCATGGCCTCCCGATAAGAATTCGAGCTCGTTTAAAC3′. Insertion of GST at the correct chromosomal position was verified by PCR using MSS51XbaIup: 5′GCTCTAGAATGACCGTGCTATATGCTCCT3′ and MSS51R1 to generate KD1532. KD1532 (MSS51-GST) was crossed with KD1516ρ0 (∆mrx8). Diploids were sporulated, and ∆mrx8 haploid spores carrying MSS51-GST were selected to generate KD1567.
To generate the Δmss51 strain, XPM76aρ0 was crossed with CRY1. Diploids were sporulated, and Δmss51ρ+ haploid spores were selected to generate KD1608. To episomally express MSS51-GST from a strong constitutive promoter, MSS51-GST was amplified from KD1532 using primers MSS51XbaIup: 5′GCTCTAGAATGACCGTGCTATATGCTCCT3′ and GSTClaIDown: 5′CCATCGATGGTCAACGCGGAACCAGATCCG3′. The 1968-base-pair PCR product was cloned in p416TEF (Mumberg et al., 1995) at the XbaI-ClaI sites to generate pYV29 (KD1596).
The mrx8GKS145-147AAA mutant allele was generated using QuikChange (Stratagene) according to the manufacturer’s instructions with variations described in Fuentes et al. (2007). The primers 5′TTTCTCGGAGGAACTAATGTGGCTGCAGCATCTATCTTGAACAACATAACC3′ and 5′GGTTATGTTGTTCAAGATAGATGCTGCAGCCACATTAGTTCCTCCGAGAAA3′ were used to incorporate specific mutation(s) in the GTPase domain of MRX8 to generate pDP67 (KD975).
Subcellular localization of MRX8
Mitochondria were isolated as described previously (Datta et al., 2005), except that cells were incubated with LongLife Zymolyase (2.5 mg/g of dry cell weight) in 1.2 M sorbitol, 20 mM potassium phosphate, pH 7.4, at 30°C for 2 h to produce spheroplasts. Mitochondria were subjected to extraction as described previously with sodium chloride or sodium carbonate (Fujiki et al., 1982) or urea (Schook et al., 1979). Soluble fractions containing peripherally associated membrane proteins were separated by centrifugation at 150,000 × g for 1 h in a Beckman 70Ti rotor from integral membrane proteins. Mitoplasts were generated by subjecting mitochondria to osmotic shock as described previously (Datta et al., 2005). Intact mitochondria or mitoplasts were treated with 0–500 µg/ml proteinase K and incubated on ice for 30 min. Proteinase K was inactivated by the addition of trichloroacetic acid (TCA) to 15%.
Separation of mitochondrial ribosome
Mitochondrial ribosomes were separated by sucrose density gradient as previously described (Fearon and Mason, 1992; Datta et al., 2005) with slight modification. Mitochondria were lysed by the addition of 10 mM Tris-Cl, pH 7.4, 10 mM MgOAc,100 mM NH4Cl, 7 mM β-mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride (PMSF), 2% NP-40. Lysates were clarified by centrifugation at 40,000 × g for 25 min at 4°C. Where indicated, lysates were incubated with 200 U/ml RNase A at room temperature for 30 min to disrupt RNA–protein complexes as described in Ott et al. (2006). Mitochondrial ribosomes were separated on a 5–30% sucrose density gradient containing 10 mM Tris-Cl, pH 7.4, 10 mM MgOAc, 7 mM β-mercaptoethanol, 0.1% NP-40, and 100 or 500 mM NH4Cl, as indicated, by ultracentrifugation at 135,000 × g for 4 h in a Beckman SW 41 Ti rotor (Datta et al., 2005). Equivalent fractions were collected, and proteins samples were precipitated by the addition of TCA to 15%.
Analysis of mitochondrial translation products
Mitochondrial translation products were labeled as previously described with slight modifications (Fox et al., 1991). Cells were grown in SGal lacking methionine at 30°C or 16°C to an OD600 of 1. Mitochondrial translation products were labeled by the addition of 0.1 mCi [35S]methionine and cysteine (Easy Tag Express35S protein labeling mix, Perkin Elmer NEG77200 [specific activity: 1175 Ci/mmol]) in the presence of 0.1 mg/ml cycloheximide to inhibit cytosolic protein synthesis at either 30°C or 16°C for 30 min (Fox et al., 1991). Labeled cells were chased by the addition of chase solution (1% casamino acids and 2 mg/ml sodium sulfate) for 10 min or as indicated, and mitochondria were isolated. The mitochondrial protein concentration was estimated by the Bradford method (Bradford, 1976). Equal amounts of mitochondrial proteins were separated on 17.5% SDS–PAGE and transferred onto a nitrocellulose membrane. The membrane was exposed to BAS storage phosphor screen (GE Healthcare Life Sciences) and developed using FUJIFILM FLA9000 phosphroimager (GE Healthcare Life Sciences).
Purification of Mrx8-6xHis using metal ion chromatography
Mitochondria (4 mg), isolated from cells expressing Mrx8-6xHis were lysed in buffer containing 50 mM Na2HPO4, 300 mM NaCl, 0.5 mM PMSF, 5 mM imidazole, pH 8.0, 1% Triton X-100, and 0.6% n-dodecyl-β-d-maltoside (DDM) for 30 min on ice. The lysate was clarified by centrifugation at 40,000 × g for 30 min. Clarified lysate was incubated with 75 µl of precalibrated Co-NTA resin (Cat #786932; G-Biosciences) for 2 h at 4°C. Subsequently, to remove unbound protein, beads were incubated with buffer containing 50 mM Na2HPO4, 300 mM NaCl, 0.5 mM PMSF, 10 mM imidazole, pH 8.0, for 5 min, and this step was repeated five times. Proteins bound to beads were eluted using buffer containing 50 mM Na2HPO4, 300 mM NaCl, 0.5 mM PMSF, 400 mM imidazole, pH 8.0. Eluted protein was concentrated by the addition of TCA to 15%, separated by SDS–PAGE, and subjected to immunoblot analysis.
Mitochondrial RNA isolation and RT-PCR analysis
Mitochondria were isolated from cells that were incubated with LongLife Zymolyase (2.5 mg/g of dry cell weight) in SB3 buffer (50 mM Tris-Cl, pH 8.0, 10 mM MgCl2, 3 mM dithiothreitol, and 1 M sorbitol) at 37°C for 2 h to produce spheroplasts. The cells were further lysed using 30–50 stokes of tight-fitting pestle in a Dounce homogenizer and centrifuged to remove any insoluble debris. Mitochondrial RNA was isolated from intact mitochondria using phenol chloroform as described previously (Turk and Caprara, 2010). Random hexamers as primers were used to generate cDNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific). For reverse transcript PCR (RT-PCR), sense and antisense primers specific to COX1, COX2, COX3, and COB are shown in Table 2. Equal amounts of total mitochondrial RNA were mixed with high-molecular-weight (HMW) RNA loading dye (95% formamide, 0.025% SDS, 0.1 µg/µl ethidium bromide, 0.5 mM EDTA, 1.25× MOPS buffer, 0.25% bromophenol blue, and 0.25% xylene cyanol) and separated on a 1.5% agarose formaldehyde denaturing gel. HMW RNA gel loading dye was prepared as described previously (Bhardwaj et al., 2012).
TABLE 2:
Primers used to quantify mRNA levels using RT-PCR.
| Gene | Sense primer | Antisense primer |
|---|---|---|
| COX1 | 5′GCTCTAATCCATGGTGGTTCAATTAGATTAGCACTACC3′ | 5′GAAAATGTCCCACCACGTAGTAAGT3′ |
| COX2 | 5′AAAGTTGATGCTACTCCTGGTAGA3′ | 5′TGCTTCGATCTTAATTGGCA3′ |
| COX3 | 5′TATGGTTCAGTATTCTATGC3′ | 5′TTAGACTCCTCATCAGTAGA3′ |
| COB | 5′TACTGATAGAAGTGTAGTAA3′ | 5′TTATTTATTAACTCTACCGA3′ |
Isolation of mitochondrial fractions from HEK293 cells
HEK293 cells were transfected with either pDP77 (GTPBP8-EGFP) or vector alone using Lipofectamine 2000 (Thermo Scientific) according to the manufacturer’s protocol. Five 100 mm culture dishes of transfected cells with approximately 100% confluency were used to isolate mitochondria. Cells were scraped and lysed by being passed through a narrow 25-guage needle 15 times. Cells were washed and lysed in mitochondrial lysis buffer containing 250 mM sucrose, 50 mM Tris-Cl, pH 7.4, 150 mM NaCl, 0.5 mM EDTA, 10 mM MgCl2, 1 mM PMSF, protease inhibitor cocktail, and phosphatase inhibitor. Mitochondrial enriched fractions were obtained using a differential centrifugation method described previously (Zhao et al., 2019).
Fluorescence microscopy
HEK293 cells were transfected using Lipofectamine 2000 (Thermo Scientific) with either pDP77 (GTPBP8-GFP) or vector alone. Cells were fixed and stained with MitoTracker Red CMXRos (Invitrogen) and 4′,6-diamidino-2-phenylindole (DAPI) (Roche) according to the manufacturer’s protocol. Images were captured using a Zeiss LSM 880 confocal laser-scanning microscope and analyzed using ZEN software.
Antibodies used for immunoblot analysis
Mrx8-specific polyclonal antibodies (Link Biotech) were raised in rabbits against epitopes ALKKKLNSRPKERLPNWLK and YEKPSNSDINKVNRFFNK, which correspond to amino acids 45–63 and 72–89, respectively, within the N-terminus, and used at a dilution of 1:500. The peptide sequence chosen were based on the likelihood of being after a cleavable mitochondrial targeting sequence. Antibodies used in this study, Mtg2 (1:2000) (Datta et al., 2005); Cox2 (1:50) (Pinkham et al., 1994); F1β (1:5000) (Emtage and Jensen, 1993); Tim23 (1:5000) (Emtage and Jensen, 1993); Cox1 (1:1000): mouse monoclonal (11D8B7), Cox3 (1:1000): mouse monoclonal (DA5BC4) and COX IV (1:5000): mouse monoclonal (20E8C12) from Abcam; GFP (1:2000): mouse monoclonal (sc-9996), GAPDH (1:4000): mouse monoclonal (sc-32233) from Santa Cruz Biotechnology; GST (1:5000): mouse monoclonal (GST.B6) from Epitope Biotech. Proteins were separated on either 10% or 12.5% SDS–PAGE and processed for immunoblot as described previously (Lin et al., 2004).
Supplementary Material
Acknowledgments
We are extremely grateful to Janine Maddock for strains, plasmids, and antibodies and Tom Fox for the Arg8 antibody. We also thank Vani Brahmachari and K. Natarajan for critical reading of the manuscript and Anagha Nair for technical assistance. Y. V. acknowledges University Grants Commission (UGC) for fellowship support. U. M. acknowledges Council Of Scientific And Industrial Research (CSIR), Science and Engineering Research Board (SERB), and Indian Council of Medical Research (ICMR) for fellowship support. D.K.P. acknowledges ICMR for fellowship support. We also acknowledge instrumentation facilities at Central Instrumentation Facility, University of Delhi South Campus (CIF-UDSC), and UGC:Special Assistance Programme/Department Of Science & Technology:Fund for Improvement of S&T Infrastructure (UGC-SAP/DST-FIST)–supported CIF, Genetics. This work was supported by grants from Department of Atomic Energy-Board of Research In Nuclear Sciences (DAE-BRNS) (Grant number: 37(1)/14/37/2016-brns/37272), and CSIR (Grant number: 38 (1297)/111EMR-II), SERB (CRG/2020/001932) and a Delhi University, Research and Development (DU-R&D) grant to K.D.
Abbreviations used:
- CDS
coding sequence
- EGFP
enhanced green fluorescent protein
- UTR
untranslated region
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E20-07-0457).
REFERENCES
- Amunts A, Brown A, Bai XC, Llacer JL, Hussain T, Emsley P, Long F, Murshudov G, Scheres SH, Ramakrishnan V (2014). Structure of the yeast mitochondrial large ribosomal subunit. Science 343, 1485–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson GC (2015). The evolutionary and functional diversity of classical and lesser-known cytoplasmic and organellar translational GTPases across the tree of life. BMC Genomics 16, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos A, Korr D, Barwell KJ, Sjulsen C, Gajewski CD, Manfredi G, Ackerman S, Tzagoloff A (2003). MTG1 codes for a conserved protein required for mitochondrial translation. Mol Biol Cell 14, 2292–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos A, Zambrano A, Tzagoloff A (2004). Mss51p and Cox14p jointly regulate mitochondrial Cox1p expression in Saccharomyces cerevisiae. EMBO J 23, 3472–3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer MF, Sirrenberg C, Neupert W, Brunner M (1996). Role of Tim23 as voltage sensor and presequence receptor in protein import into mitochondria. Cell 87, 33–41. [DOI] [PubMed] [Google Scholar]
- Bauerschmitt H, Funes S, Herrmann JM (2008). The membrane-bound GTPase Guf1 promotes mitochondrial protein synthesis under suboptimal conditions. J Biol Chem 283, 17139–17146. [DOI] [PubMed] [Google Scholar]
- Bhardwaj AR, Pandey R, Agarwal M, Katiyar-Agarwal S (2012). Northern blot analysis for expression profiling of mRNAs and small RNAs. Methods Mol Biol 883, 19–45. [DOI] [PubMed] [Google Scholar]
- Bourne HR, Sanders DA, McCormick F (1991). The GTPase superfamily: conserved structure and molecular mechanism. Nature 349, 117–127. [DOI] [PubMed] [Google Scholar]
- Bradford MM (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72, 248–254. [DOI] [PubMed] [Google Scholar]
- Brickner JH, Fuller RS (1997). SOI1 encodes a novel, conserved protein that promotes TGN-endosomal cycling of Kex2p and other membrane proteins by modulating the function of two TGN localization signals. J Cell Biol 139, 23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claros MG, Vincens P (1996). Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur J Biochem 241, 779–786. [DOI] [PubMed] [Google Scholar]
- Clementi N, Polacek N (2010). Ribosome-associated GTPases: the role of RNA for GTPase activation. RNA Biol 7, 521–527. [DOI] [PubMed] [Google Scholar]
- Cooper EL, Garcia-Lara J, Foster SJ (2009). YsxC, an essential protein in Staphylococcus aureus crucial for ribosome assembly/stability. BMC Microbiol 9, 266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couvillion MT, Soto IC, Shipkovenska G, Churchman LS (2016). Synchronized mitochondrial and cytosolic translation programs. Nature 533, 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta K, Fuentes JL, Maddock JR (2005). The yeast GTPase Mtg2p is required for mitochondrial translation and partially suppresses an rRNA methyltransferase mutant, mrm2. Mol Biol Cell 16, 954–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta K, Skidmore JM, Pu K, Maddock JR (2004). The Caulobacter crescentus GTPase CgtAC is required for progression through the cell cycle and for maintaining 50S ribosomal subunit levels. Mol Microbiol 54, 1379–1392. [DOI] [PubMed] [Google Scholar]
- Decoster E, Simon M, Hatat D, Faye G (1990). The MSS51 gene product is required for the translation of the COX1 mRNA in yeast mitochondria. Mol Gen Genet 224, 111–118. [DOI] [PubMed] [Google Scholar]
- Decoster E, Vassal A, Faye G (1993). MSS1, a nuclear-encoded mitochondrial GTPase involved in the expression of COX1 subunit of cytochrome c oxidase. J Mol Biol 232, 79–88. [DOI] [PubMed] [Google Scholar]
- Dennerlein S, Rehling P (2015). Human mitochondrial COX1 assembly into cytochrome c oxidase at a glance. J Cell Sci 128, 833–837. [DOI] [PubMed] [Google Scholar]
- Desai N, Brown A, Amunts A, Ramakrishnan V (2017). The structure of the yeast mitochondrial ribosome. Science 355, 528–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Silva D, Poliquin S, Zeng R, Zamudio-Ochoa A, Marrero N, Perez-Martinez X, Fontanesi F, Barrientos A (2017). The DEAD-box helicase Mss116 plays distinct roles in mitochondrial ribogenesis and mRNA-specific translation. Nucleic Acids Res 45, 6628–6643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmond E, Brochier-Armanet C, Forterre P, Gribaldo S (2011). On the last common ancestor and early evolution of eukaryotes: reconstructing the history of mitochondrial ribosomes. Res Microbiol 162, 53–70. [DOI] [PubMed] [Google Scholar]
- Emtage JL, Jensen RE (1993). MAS6 encodes an essential inner membrane component of the yeast mitochondrial protein import pathway. J Cell Biol 122, 1003–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon K, Mason TL (1988). Structure and regulation of a nuclear gene in Saccharomyces cerevisiae that specifies MRP7, a protein of the large subunit of the mitochondrial ribosome. Mol Cell Biol 8, 3636–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon K, Mason TL (1992). Structure and function of MRP20 and MRP49, the nuclear genes for two proteins of the 54 S subunit of the yeast mitochondrial ribosome. J Biol Chem 267, 5162–5170. [PubMed] [Google Scholar]
- Fiori A, Perez-Martinez X, Fox TD (2005). Overexpression of the COX2 translational activator, Pet111p, prevents translation of COX1 mRNA and cytochrome c oxidase assembly in mitochondria of Saccharomyces cerevisiae. Mol Microbiol 56, 1689–1704. [DOI] [PubMed] [Google Scholar]
- Fontanesi F, Soto IC, Barrientos A (2008). Cytochrome c oxidase biogenesis: new levels of regulation. IUBMB Life 60, 557–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox TD (2012). Mitochondrial protein synthesis, import, and assembly. Genetics 192, 1203–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox TD, Folley LS, Mulero JJ, McMullin TW, Thorsness PE, Hedin LO, Costanzo MC (1991). Analysis and manipulation of yeast mitochondrial genes. Methods Enzymol 194, 149–165. [DOI] [PubMed] [Google Scholar]
- Fuentes JL, Datta K, Sullivan SM, Walker A, Maddock JR (2007). In vivo functional characterization of the Saccharomyces cerevisiae 60S biogenesis GTPase Nog1. Mol Genet Genomics 278, 105–123. [DOI] [PubMed] [Google Scholar]
- Fujiki Y, Hubbard AL, Fowler S, Lazarow PB (1982). Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J Cell Biol 93, 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, et al. (2002). Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415, 141–147. [DOI] [PubMed] [Google Scholar]
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, Dow S, Lucau-Danila A, Anderson K, Andre B, et al. (2002). Functional profiling of the Saccharomyces cerevisiae genome. Nature 418, 387–391. [DOI] [PubMed] [Google Scholar]
- Glick BS, Pon LA (1995). Isolation of highly purified mitochondria from Saccharomyces cerevisiae. Methods Enzymol 260, 213–223. [DOI] [PubMed] [Google Scholar]
- Gray MW (2015). Mosaic nature of the mitochondrial proteome: implications for the origin and evolution of mitochondria. Proc Natl Acad Sci USA 112, 10133–10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie C, Fink GR (1991). Guide to yeast genetics and molecular biology. Methods Enzymol 194, 1–863. [PubMed] [Google Scholar]
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O’Shea EK (2003). Global analysis of protein localization in budding yeast. Nature 425, 686–691. [DOI] [PubMed] [Google Scholar]
- Karbstein K (2007). Role of GTPases in ribosome assembly. Biopolymers 87, 1–11. [DOI] [PubMed] [Google Scholar]
- Kehrein K, Schilling R, Moller-Hergt BV, Wurm CA, Jakobs S, Lamkemeyer T, Langer T, Ott M (2015). Organization of mitochondrial gene expression in two distinct ribosome-containing assemblies. Cell Rep 10, 843–853. [DOI] [PubMed] [Google Scholar]
- Khalimonchuk O, Rodel G (2005). Biogenesis of cytochrome c oxidase. Mitochondrion 5, 363–388. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Khalimonchuk O, Smith PM, Winge DR (2012). Structure, function, and assembly of heme centers in mitochondrial respiratory complexes. Biochim Biophys Acta 1823, 1604–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurland CG, Andersson SG (2000). Origin and evolution of the mitochondrial proteome. Microbiol Mol Biol Rev 64, 786–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehoux IE, Mazzulla MJ, Baker A, Petit CM (2003). Purification and characterization of YihA, an essential GTP-binding protein from Escherichia coli. Protein Expr Purif 30, 203–209. [DOI] [PubMed] [Google Scholar]
- Leipe DD, Wolf YI, Koonin EV, Aravind L (2002). Classification and evolution of P-loop GTPases and related ATPases. J Mol Biol 317, 41–72. [DOI] [PubMed] [Google Scholar]
- Lin B, Thayer DA, Maddock JR (2004). The Caulobacter crescentus CgtAC protein cosediments with the free 50S ribosomal subunit. J Bacteriol 186, 481–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti P, Lavdovskaia E, Barrientos A, Richter-Dennerlein R (2021). Role of GTPases in driving mitoribosome assembly. Trends Cell Biol 31, 284–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manthey GM, McEwen JE (1995). The product of the nuclear gene PET309 is required for translation of mature mRNA and stability or production of intron-containing RNAs derived from the mitochondrial COX1 locus of Saccharomyces cerevisiae. EMBO J 14, 4031–4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maracci C, Rodnina MV (2016). Review: translational GTPases. Biopolymers 105, 463–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mays JN, Camacho-Villasana Y, Garcia-Villegas R, Perez-Martinez X, Barrientos A, Fontanesi F (2019). The mitoribosome-specific protein mS38 is preferentially required for synthesis of cytochrome c oxidase subunits. Nucleic Acids Res 47, 5746–5760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McStay GP, Su CH, Tzagoloff A (2013). Modular assembly of yeast cytochrome oxidase. Mol Biol Cell 24, 440–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern M, Stiller SB, Lubbert P, Peikert CD, Dannenmaier S, Drepper F, Weill U, Hoss P, Feuerstein R, Gebert M, et al. (2017). Definition of a high-confidence mitochondrial proteome at quantitative scale. Cell Rep 19, 2836–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumberg D, Muller R, Funk M (1995). Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156, 119–122. [DOI] [PubMed] [Google Scholar]
- Ott M, Amunts A, Brown A (2016). Organization and regulation of mitochondrial protein synthesis. Annu Rev Biochem 85, 77–101. [DOI] [PubMed] [Google Scholar]
- Ott M, Prestele M, Bauerschmitt H, Funes S, Bonnefoy N, Herrmann JM (2006). Mba1, a membrane-associated ribosome receptor in mitochondria. EMBO J 25, 1603–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partaledis JA, Mason TL (1988). Structure and regulation of a nuclear gene in Saccharomyces cerevisiae that specifies MRP13, a protein of the small subunit of the mitochondrial ribosome. Mol Cell Biol 8, 3647–3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul MF, Alushin GM, Barros MH, Rak M, Tzagoloff A (2012). The putative GTPase encoded by MTG3 functions in a novel pathway for regulating assembly of the small subunit of yeast mitochondrial ribosomes. J Biol Chem 287, 24346–24355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Martinez X, Broadley SA, Fox TD (2003). Mss51p promotes mitochondrial Cox1p synthesis and interacts with newly synthesized Cox1p. EMBO J 22, 5951–5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Martinez X, Butler CA, Shingu-Vazquez M, Fox TD (2009). Dual functions of Mss51 couple synthesis of Cox1 to assembly of cytochrome c oxidase in Saccharomyces cerevisiae mitochondria. Mol Biol Cell 20, 4371–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkham JL, Dudley AM, Mason TL (1994). T7 RNA polymerase-dependent expression of COXII in yeast mitochondria. Mol Cell Biol 14, 4643–4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzheinikov SN, Das SK, Sedelnikova SE, Baker PJ, Artymiuk PJ, Garcia-Lara J, Foster SJ, Rice DW (2004). Analysis of the open and closed conformations of the GTP-binding protein YsxC from Bacillus subtilis. J Mol Biol 339, 265–278. [DOI] [PubMed] [Google Scholar]
- Schaefer L, Uicker WC, Wicker-Planquart C, Foucher AE, Jault JM, Britton RA (2006). Multiple GTPases participate in the assembly of the large ribosomal subunit in Bacillus subtilis. J Bacteriol 188, 8252–8258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schook W, Puszkin S, Bloom W, Ores C, Kochwa S (1979). Mechanochemical properties of brain clathrin: interactions with actin and alpha-actinin and polymerization into basketlike structures or filaments. Proc Natl Acad Sci USA 76, 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sickmann A, Reinders J, Wagner Y, Joppich C, Zahedi R, Meyer HE, Schonfisch B, Perschil I, Chacinska A, Guiard B, et al. (2003). The proteome of Saccharomyces cerevisiae mitochondria. Proc Natl Acad Sci USA 100, 13207–13212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AP, Salvatori R, Aftab W, Aufschnaiter A, Carlstrom A, Forne I, Imhof A, Ott M (2020). Molecular connectivity of mitochondrial gene expression and OXPHOS biogenesis. Mol Cell 79, 1051–1065.e1010. [DOI] [PubMed] [Google Scholar]
- Sprang SR (1997). G protein mechanisms: insights from structural analysis. Annu Rev Biochem 66, 639–678. [DOI] [PubMed] [Google Scholar]
- Steele DF, Butler CA, Fox TD (1996). Expression of a recoded nuclear gene inserted into yeast mitochondrial DNA is limited by mRNA-specific translational activation. Proc Natl Acad Sci USA 93, 5253–5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strunk BS, Karbstein K (2009). Powering through ribosome assembly. RNA 15, 2083–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk R, Huynen MA (2010). Mosaic origin of the mitochondrial proteome. Proteomics 10, 4012–4024. [DOI] [PubMed] [Google Scholar]
- Tavares-Carreon F, Camacho-Villasana Y, Zamudio-Ochoa A, Shingu-Vazquez M, Torres-Larios A, Perez-Martinez X (2008). The pentatricopeptide repeats present in Pet309 are necessary for translation but not for stability of the mitochondrial COX1 mRNA in yeast. J Biol Chem 283, 1472–1479. [DOI] [PubMed] [Google Scholar]
- Timon-Gomez A, Nyvltova E, Abriata LA, Vila AJ, Hosler J, Barrientos A (2018). Mitochondrial cytochrome c oxidase biogenesis: recent developments. Semin Cell Dev Biol 76, 163–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk EM, Caprara MG (2010). Splicing of yeast aI5beta group I intron requires SUV3 to recycle MRS1 via mitochondrial degradosome-promoted decay of excised intron ribonucleoprotein (RNP). J Biol Chem 285, 8585–8594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umeda N, Suzuki T, Yukawa M, Ohya Y, Shindo H, Watanabe K, Suzuki T (2005). Mitochondria-specific RNA-modifying enzymes responsible for the biosynthesis of the wobble base in mitochondrial tRNAs. Implications for the molecular pathogenesis of human mitochondrial diseases. J Biol Chem 280, 1613–1624. [DOI] [PubMed] [Google Scholar]
- van Dijken JP, Weusthuis RA, Pronk JT (1993). Kinetics of growth and sugar consumption in yeasts. Antonie Van Leeuwenhoek 63, 343–352. [DOI] [PubMed] [Google Scholar]
- Verstraeten N, Fauvart M, Versees W, Michiels J (2011). The universally conserved prokaryotic GTPases. Microbiol Mol Biol Rev 75, 507–542, second and third pages of table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittinghofer A, Vetter IR (2011). Structure-function relationships of the G domain, a canonical switch motif. Annu Rev Biochem 80, 943–971. [DOI] [PubMed] [Google Scholar]
- Zamudio-Ochoa A, Camacho-Villasana Y, Garcia-Guerrero AE, Perez-Martinez X (2014). The Pet309 pentatricopeptide repeat motifs mediate efficient binding to the mitochondrial COX1 transcript in yeast. RNA Biol 11, 953–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Sun X, Hu D, Prosdocimo DA, Hoppel C, Jain MK, Ramachandran R, Qi X (2019). ATAD3A oligomerization causes neurodegeneration by coupling mitochondrial fragmentation and bioenergetics defects. Nat Commun 10, 1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.