

Abstract
Kratom, Mitragyna speciosa Korth., is being widely consumed in the United States for pain management and the reduction of opioid withdrawal symptoms. The central nervous system (CNS) active alkaloids of kratom, including mitragynine, 7-hydroxymitragynine, and numerous additional compounds, are believed to derive their effects through opioid receptor activity. There is no literature describing the systemic exposure of many of these alkaloids after the consumption of kratom. Therefore, we have developed and validated a bioanalytical method for the simultaneous quantitation of 11 kratom alkaloids (mitragynine, 7-hydroxymitragynine, corynantheidine, speciogynine, speciociliatine, paynantheine, corynoxine, corynoxine-B, mitraphylline, ajmalicine, and isospeciofoline) in rat plasma. The validated method was used to analyze oral pharmacokinetic study samples of lyophilized kratom tea (LKT) and a marketed product, OPMS liquid shot, in rats. Among the 11 alkaloids, only mitragynine, 7-hydroxymitragynine, speciociliatine, and corynantheidine showed systemic exposure 8 h postdose, and the dose-normalized systemic exposure of these four alkaloids was higher (1.6–2.4-fold) following the administration of the commercial OPMS liquid. Paynantheine and speciogynine levels were quantifiable up to 1 h postdose, whereas none of the other alkaloids were detected. In summary, the method was successfully applied to quantify the exposure of individual kratom alkaloids after an oral dose of traditional or commercial products. This information will contribute to understanding the role of each alkaloid in the overall pharmacology of kratom and elucidating the pharmacokinetic differences between traditional and commercial kratom products.
Graphical Abstract
Kratom (Mitragyna speciosa (Korth.) Havil., Rubiaceae) is a tropical tree native to Southeast Asia also known as biak-biak, kakuam, ketum, or krathom.1,2 The leaves of kratom are consumed as a self-treatment for the management of pain and opioid addiction in the United States.3,4 Natives of Southeast Asia use kratom for cough, diabetes, diarrhea, and hypertension, while laborers use it for physical endurance, to increase heat tolerance, and to gain energy.5,6 It has been reported that kratom results in stimulant activity at lower doses, while at higher doses it produces opioid-like effects.7 Traditionally, kratom leaves are consumed by making a decoction of fresh leaves, chewing fresh leaves, or by smoking dried leaves.1,8 In the United States (US) due to the unavailability of fresh leaf material, kratom is generally consumed as tablets, capsules of dried leaf powder, or concentrated extracts obtained from dried leaf material.4,9 There is little information available on the effects of long-term kratom use, though potential adverse effects reported among regular users include dark lips, dry skin, unhealthy complexions, and lean body mass. The main symptoms of acute overdose are vomiting and vertigo.1,8 Recently, the US Food and Drug Administration (FDA) issued a warning letter about kratom use due to the possible risk of addiction and dependence associated with regular kratom intake.10 Poison control centers in the US have reported that kratom overdoses present more similar to stimulant overdose symptoms, as opposed to opioid-like symptoms that might be expected on the basis of currently established pharmacology.11
Alkaloids are considered to be the main active constituents of kratom; the total alkaloidal content varies from 0.5% to 1.5%.1,9 Kratom has been reported to contain more than 40 different alkaloids.12 Of these, the indole-based alkaloids, mitragynine (MTG), and 7-hydroxymitragynine (7-HMG), constitute ~66% and ~2%, respectively, of the total alkaloidal content and are considered to play an important role in the pharmacological activity of kratom.1,6,13 Metabolism studies have revealed that 7-HMG is one of the major cytochrome P450 3A4 (CYP3A4) enzyme-mediated metabolites of MTG.14,15 Functionally, MTG (EC50 = 307 nM) and 7-HMG (EC50 = 7.6 nM) were found to be partial agonists at μ-opioid receptors.16 Additionally, MTG was found to have weak antagonistic effects (KB ranging from 1 to 3 μM) at α1-adrenergic receptor subtypes (α1A, -B, and -D) and 7-HMG had antagonistic effects at the κ-opioid receptor (KB =115 nM).16 There are numerous additional indole alkaloids in kratom, but sparse information is available for these compounds. Given the widespread use of kratom products for multiple indications, it is essential to understand the pharmacology of all kratom alkaloids. Among the other kratom alkaloids evaluated for pharmacological effects, corynantheidine (COR, ~1% of total alkaloidal content) has shown antagonism of μ-opioid receptors, demonstrated by reversing the morphine inhibited twitch contraction of guinea pig ileum.17 The affinity (Ki) of COR at μ-opioid receptors is demonstrated to be 118 nM, while at α1D-adrenergic receptors it is 41.7 nM, 131-fold higher affinity than that of MTG (Ki = 5480 nM).16 Speciociliatine (SPC, ~1% of total alkaloidal content),1 a diastereomer of MTG, more potently stimulates the μ-opioid receptor-mediated G protein-coupled signaling in vitro (EC50 = 39.2 nM) than MTG. The potency of SPC in vivo, as evidenced by antinociceptive effects in the rat hot plate test, is comparable to that of morphine.16 Furthermore, paynantheine (PAY) and speciogynine (SPG, another diastereomer of MTG), which, constitute ~9% and ~7% of the total alkaloidal content, respectively, were found to inhibit twitch contraction in a naloxone-insensitive (i.e., nonopioid receptor mediated) manner.1,18 Taken together, these results indicate, that although the relative natural abundance of these alkaloids is lower than that of MTG, they could have comparable or more potent pharmacological activity than MTG at opioidergic, adrenergic, and/or serotonergic receptors. Thus, it is essential to evaluate the systemic exposure of kratom alkaloids to predict their contribution to the overall therapeutic effects of kratom. Similar to the pharmacodynamic data, most of the reported pharmacokinetic information on kratom has focused on the purified, individual alkaloids. The vast majority of these studies only provide information on MTG and 7-HMG.19-23 Recently, King et al. in 2020 reported the pharmacokinetics of COR in male rats.24 It is noteworthy that the data obtained from the analysis of individual alkaloids will not always predict the pharmacology of the overall mixture of alkaloids contained in kratom products. This is due to the presence of alkaloids at different concentrations, which could influence the pharmacokinetics and pharmacodynamics of the other individual alkaloids contained in the overall matrix. In a previous report, MTG and COR have shown substantial inhibition of cytochrome P450 2D6 (CYP2D6) enzymes with Ki values of 1.1 and 2.8 μM, respectively.25 This suggests that the CYP2D6-mediated metabolism of some kratom alkaloids could be altered by the presence of others.25 Substantial differences in the pharmacokinetic profile of MTG were observed when MTG was administered as lyophilized kratom tea (LKT), an organic extract ofLKT, or MTG alone (purified MTG as an HCl salt) in rats, suggesting that the pharmacokinetics of other kratom alkaloids could also differ when administered alone or as a mixture.26
In the present study, we have developed and validated an ultraperformance liquid chromatography–tandem mass spectrometry (UPLC–MS/MS) bioanalytical method for the simultaneous quantification of 11 kratom alkaloids. These include MTG, 7-HMG, COR, SPG, SPC, PAY, corynoxine (COX), corynoxine-B (COX-B), mitraphylline (MTP), ajmalicine (AJM), and isospeciofoline (i-SPF) (Figure 1). The bioanalytical method was validated for quantification in rat plasma and applied for the quantitative analysis of plasma samples obtained from the pharmacokinetic study of different kratom formulations to understand the systemic exposure of the individual kratom alkaloids. The previously reported method for the simultaneous quantification of 10 kratom alkaloids was mainly developed as an analytical method for assaying kratom commercial and plant products. This published method was not optimized for the analysis of plasma samples and had a relatively long total run time of 22.5 min. Here, we sought to decrease the run time and increase the total number of kratom alkaloids included in the bioassay (i-SPF and AJM).
Figure 1.
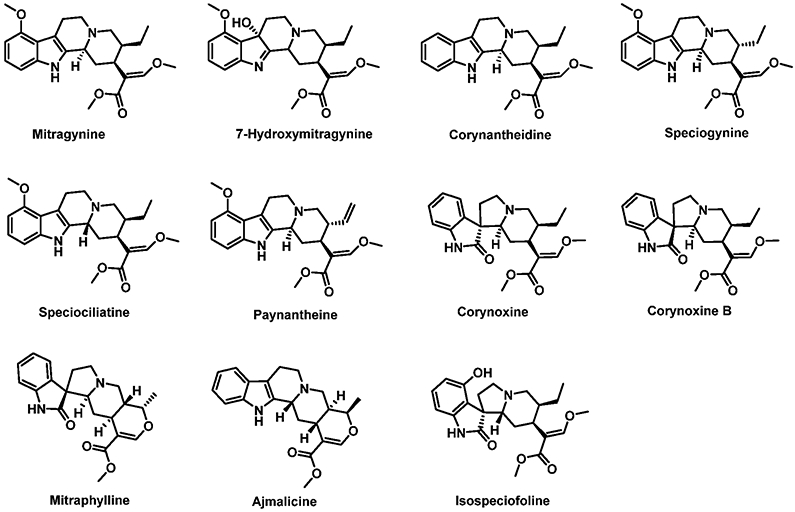
Chemical structures of kratom alkaloids.
RESULT AND DISCUSSION
UPLC–MS/MS Method Development and Validation.
A simple and sensitive UPLC–MS/MS bioanalytical method for the simultaneous quantification of 11 kratom alkaloids (MTG, 7-HMG, COR, SPG, SPC, PAY, COX, COX-B, MTP, AJM, and i-SPF) in rat plasma was successfully developed and validated consistent with the FDA guidelines.27 The representative chromatograms are shown in Figure 2. Initially, the chromatographic conditions for the simultaneous quantification of these alkaloids in rat plasma were adopted from the previously reported method.28 However, the chromatographic conditions were optimized to markedly reduce the total analysis time by one-half (11 min), to minimize the interference from the blank plasma, and to improve the retention time and resolution for the 7-HMG peak as the metabolism of MTG, SPG, and SPC in pharmacokinetic samples would result in multiple hydroxylated metabolite peaks sharing the same ion transition. Also, the simple, inexpensive, and high-throughput compatible protein precipitation method was employed for sample cleanup and the extraction of kratom alkaloids. The developed method was linear in the range 1–200 ng/mL for each alkaloid, and the observed coefficient of correlation was always >0.99 for all alkaloids.
Figure 2.

Representative chromatograms of 11 kratom alkaloids at LLOQ (1 ng/mL) and verapamil (internal standard, IS) in rat plasma.
Selectivity, Specificity, Carryover, and Sensitivity.
The selectivity and specificity were assessed in at least six individual sources of rat plasma for interference at the retention times of the analytes and the internal standard (IS, verapamil); no interference was observed for any of the alkaloids. There was no carryover noted for any of the alkaloids in the blank samples following high-concentration samples. The method was sensitive for all the alkaloids as the signal-to-noise ratio of the lower limit of quantitation (LLOQ, 1 ng/mL) for each alkaloid exceeded the recommended 10:1.
Recovery and Matrix Effect.
The recovery was determined by comparing the peak area of each analyte in prespike versus postspike samples at different concentrations. The mean percentage recovery of the individual alkaloids was consistent across different concentrations (n = 6, at each level) and was >90% in rat plasma (Table S1). The matrix effect was assessed by comparing the analyte peak area of each analyte in postspike versus samples prepared in a water–methanol mixture (1:1) (no matrix). The mean percentage recovery of each alkaloid was consistent across different concentrations (n = 6, at each level) and was >80% of the response in a water–methanol mixture (1:1) (Table S1), indicating minimal ion suppression due to the matrix.
Accuracy and Precision.
The inter- and intraday accuracy (% bias) and precision (% RSD) of the method for each alkaloid was determined by analyzing six sets of QCs [LLOQ, low-quality control (LQC), medium-quality control (MQC), and high-quality control (HQC)] on three different days in rat plasma. Both the accuracy and precision of the method for each alkaloid were within the acceptable limit of 15% of the nominal concentration (for LQC, MQC, and HQC) or 20% for the LLOQ (Table 1).
Table 1.
Accuracy and Precision of the Bioanalytical Method for the Quantification of Kratom Alkaloids
| accuracy (% bias) |
precision (% RSD) |
|||||
|---|---|---|---|---|---|---|
| alkaloid | quality control | nominal concentration (ng/mL) | intraday | interday | intraday | interday |
| MTG | LLQC | 1 | 10.0 | 8.6 | 7.3 | 8.5 |
| LQC | 2.5 | 0.9 | −2.8 | 4.9 | 4.0 | |
| MQC | 90 | 2.0 | 1.5 | 3.0 | 6.1 | |
| HQC | 160 | 1.8 | 2.8 | 2.5 | 5.5 | |
| 7-HMG | LLQC | 1 | 2.2 | 1.6 | 11.1 | 13.5 |
| LQC | 2.5 | −1.7 | −3.3 | 4.5 | 5.9 | |
| MQC | 90 | 4.4 | 2.8 | 3.3 | 5.9 | |
| HQC | 160 | 3.0 | 3.5 | 2.5 | 4.5 | |
| COR | LLQC | 1 | −0.4 | 0.0 | 4.8 | 4.8 |
| LQC | 2.5 | 0.2 | 0.6 | 3.4 | 2.8 | |
| MQC | 90 | 1.9 | 2.8 | 3.5 | 8.6 | |
| HQC | 160 | −0.1 | 2.2 | 2.3 | 8.4 | |
| SPG | LLQC | 1 | 0.0 | 0.0 | 3.0 | 4.0 |
| LQC | 2.5 | 0.2 | 0.6 | 2.6 | 4.8 | |
| MQC | 90 | 1.9 | 2.8 | 2.6 | 7.8 | |
| HQC | 160 | −0.1 | 2.2 | 2.4 | 7.4 | |
| SPC | LLQC | 1 | 0.0 | −2.5 | 5.7 | 7.4 |
| LQC | 2.5 | 0.3 | 0.6 | 2.1 | 3.9 | |
| MQC | 90 | 0.0 | 1.0 | 2.6 | 8.5 | |
| HQC | 160 | −1.7 | 0.9 | 2.4 | 7.9 | |
| PAY | LLQC | 1 | 5.2 | 3.7 | 5.6 | 8.1 |
| LQC | 2.5 | −1.1 | −1.9 | 2.2 | 2.5 | |
| MQC | 90 | 0.8 | 1.7 | 3.1 | 6.8 | |
| HQC | 160 | 0.8 | 2.2 | 2.2 | 5.8 | |
| COX | LLQC | 1 | −5.8 | −5.7 | 7.6 | 5.8 |
| LQC | 2.5 | −4.4 | −3.9 | 6.6 | 8.7 | |
| MQC | 90 | 0.6 | 0.7 | 3.1 | 8.4 | |
| HQC | 160 | −6.2 | −0.3 | 2.9 | 10.4 | |
| COX-B | LLQC | 1 | −0.6 | 0.7 | 3.8 | 8.5 |
| LQC | 2.5 | −1.7 | −2.4 | 2.6 | 2.6 | |
| MQC | 90 | 3.5 | 1.3 | 2.8 | 7.6 | |
| HQC | 160 | −2.6 | −1.4 | 2.6 | 7.0 | |
| AJM | LLQC | 1 | −0.6 | 0.7 | 7.4 | 7.0 |
| LQC | 2.5 | −2.4 | −0.8 | 3.4 | 3.7 | |
| MQC | 90 | 0.9 | 1.3 | 4.0 | 7.5 | |
| HQC | 160 | 3.2 | 3.6 | 2.8 | 7.4 | |
| MTP | LLQC | 1 | 2.0 | 2.8 | 2.8 | 4.7 |
| LQC | 2.5 | 0.4 | 0.1 | 3.8 | 3.8 | |
| MQC | 90 | 2.0 | 1.8 | 3.8 | 8.0 | |
| HQC | 160 | 0.0 | 0.5 | 3.3 | 9.0 | |
| i-SPF | LLQC | 1 | 0.0 | 2.1 | 4.5 | 7.2 |
| LQC | 2.5 | 3.2 | −0.1 | 5.7 | 7.3 | |
| MQC | 90 | 0.3 | −0.5 | 3.1 | 11.2 | |
| HQC | 160 | 0.2 | −2.1 | 2.7 | 9.6 | |
Stability.
The stability of the individual kratom alkaloids was assessed in the various conditions most likely to arise during the storage, sample processing, and analysis of samples. All the kratom alkaloids were stable in the autosampler up to 24 h (<15% deviation, for all alkaloids), on the benchtop up to 2 h (<15% deviation, for all alkaloids), for freeze–thaw up to three cycles (<15% deviation, for all alkaloids), and for up to 4 weeks (<10% deviation, for all alkaloids) at −80 °C at two different concentrations (LQC and HQC, n = 5). All data is shown as the mean percentage deviation from the nominal concentration (Table 2).
Table 2.
Stability of Kratom Alkaloids under Different Storage Conditions (n = 5, each concentration)
| % deviation |
|||||
|---|---|---|---|---|---|
| alkaloids | nominal concentration (ng/mL) |
benchtop (2 h) |
autosampler (24 h) |
freeze– thaw (3 cycles) |
long- term (4 weeks) |
| MTG | 2.5 | 4.0 | −0.8 | −8.0 | 4.0 |
| 160 | 9.8 | 14.8 | 9.0 | −0.3 | |
| 7-HMG | 2.5 | 6.0 | 1.6 | 6.4 | −9.6 |
| 160 | 13.3 | 6.6 | 9.1 | −6.8 | |
| COR | 2.5 | 3.3 | 6.4 | −0.8 | 5.6 |
| 160 | 11.4 | 14.2 | 9.5 | −4.6 | |
| SPG | 2.5 | 4.0 | 6.4 | −5.6 | 0.8 |
| 160 | 14.4 | 14.2 | 10.3 | −1.4 | |
| SPC | 2.5 | 1.3 | 3.2 | −2.4 | 0.0 |
| 160 | 14.7 | 13.6 | 11.5 | −1.5 | |
| PAY | 2.5 | 6.7 | 12.0 | −2.4 | 2.4 |
| 160 | 13.8 | 14.0 | 10.6 | −2.3 | |
| COX | 2.5 | 2.7 | 2.4 | 3.2 | 9.6 |
| 160 | 14.6 | 8.1 | 9.2 | −2.2 | |
| COX-B | 2.5 | 4.0 | 2.4 | 1.6 | 0.8 |
| 160 | 12.7 | 11.3 | 10.2 | −1.1 | |
| AJM | 2.5 | 2.0 | 1.6 | −4.8 | −5.6 |
| 160 | 13.9 | 12.2 | 12.2 | −3.2 | |
| MTP | 2.5 | 0.7 | 0.8 | 3.2 | 0.0 |
| 160 | 14.7 | 7.1 | 11.0 | −4.6 | |
| i-SPF | 2.5 | 0.7 | 12.0 | 12.8 | 1.6 |
| 160 | 14.6 | 10.1 | 13.2 | −4.2 | |
Pharmacokinetic Studies of Kratom Preparations.
The developed bioanalytical method was successfully applied to the simultaneous quantification of 11 kratom alkaloids in rat plasma samples obtained from the oral pharmacokinetic studies of LKT and a commercial kratom product (OPMS liquid kratom, Optimized Plant Mediated Solutions, Choice Organics, Los Angeles, CA). A comprehensive pharmacokinetic assessment of all detectable kratom alkaloids is needed because many possess an appreciable affinity at central nervous system (CNS) receptors (e.g., opioid receptors). Initially, LKT and OPMS liquid kratom preparations were analyzed to quantify the content of these 11 kratom alkaloids (Table S2). Mitragynine was the major alkaloid in both the preparation, while most of the alkaloids were detected in varying contents but the relative content normalized to mitragynine in each preparation was comparable. Further, COR was detected at a quantifiable level only in LKT preparation, while i-SPF was quantifiable only in OPMS liquid (Table S2). The MTG content in these preparations was used to calculate the human equivalent dose (HED). The HED normalized to MTG of LKT (5.7 mg/kg MTG corresponding to 366 mg/kg LKT) and OPMS liquid (9.6 mg/kg MTG corresponding to 0.8 mL/kg OPMS liquid) and the resultant doses of remaining individual alkaloids in these preparations, orally administered to male Sprague–Dawley (SD) rats as shown in Table 3.
Table 3.
Dose of Individual Alkaloids Administered to Rats (mg/kg)
| rat dose | MTG | 7-HMG | COR | SPG | SPC | PAY | COX | COX-B | MTP | AJM | i-SPF |
|---|---|---|---|---|---|---|---|---|---|---|---|
| OPMS_liquid (0.8 mL/kg) | 9.60 | <0.10 | 0.18 | 1.23 | 2.24 | 1.77 | 0.15 | 0.55 | <0.10 | <0.10 | 0.69 |
| LKT (366 mg/kg) | 5.73 | <0.10 | 0.13 | 0.79 | 2.03 | 1.10 | <0.10 | 0.14 | <0.10 | <0.10 | <0.10 |
The rat dose was calculated on the basis of the human dose of these kratom preparations using eq 1.
The plasma concentration–time profile and the pharmacokinetic parameters of kratom alkaloids following the oral administration of LKT and OPMS liquid are shown in Table 4 and Figure 3. As the oral doses of MTP, AJM, COX, COX-B, and i-SPF were below <0.1 mg/kg (Table 3), these alkaloids were not detected at any time point following the oral administration of LKT and OPMS liquid.
Table 4.
Pharmacokinetic Parameters of Kratom Alkaloids Following Oral Administration of LKT and OPMS Liquida
| MTG |
7-HMG |
COR |
SPC |
|||||
|---|---|---|---|---|---|---|---|---|
| LKT | OPMS liquid | LKT | OPMS liquid |
LKT | OPMS liquid | LKT | OPMS liquid | |
| Tmax (h) | 1.3 ± 0.3 | 3.1 ± 1.7 | 0.9 ± 0.2 | 3.1 ± 1.7 | 1.0 ± 0.2 | 3.1 ± 1.7 | 1.8 ± 0.7 | 3.2 ± 1.6 |
| Cmax (ng/mL) | 63.8 ± 6.2 | 111.9 ± 15.6 | 4.3 ± 0.8 | 4.0 ± 0.6 | 1.8 ± 0.3 | 3.1 ± 0.5 | 21.1 ± 3.3 | 23.8 ± 1.4 |
| AUC0–24h (h·ng/mL) | 479.6 ± 36.4 | 1306.8 ± 126.1 | 17.4 ± 4.8 | 41.0 ± 7.6 | 8.2 ± 2.3 | 30.4 ± 9.1 | 107.4 ± 25.4 | 222.7 ± 22.2 |
| Cmax/dose (kg·ng/mL/mg) | 11.1 ± 1.1 | 11.7 ± 1.6 | 14.0 ± 2.3 | 15.6 ± 2.3 | 10.4 ± 1.6 | 10.8 ± 0.6 | ||
| AUC0–24h/dose (h·kg·ng/mL/mg) | 83.7 ± 6.4 | 136.1 ± 13.1 | 63.1 ± 18 | 152.2 ± 45.5 | 52.9 ± 12.5 | 101.2 ± 10.1 | ||
| fold change in AUC0–24h/dose relative to LKT | 1.6 | 2.4 | 1.9 | |||||
All values represent mean ± SEM (n = 4). Abbreviations: AUC0–24h = area under the plasma concentration–time curve up to 24 h, Cmax = plasma peak concentration, Tmax = time to reach Cmax.
Figure 3.

Pharmacokinetic profiles of kratom alkaloids following an oral administration of 366 mg/kg (containing 5.7 mg/kg MTG, human dose equivalent) LKT (left) and 0.8 mL/kg (containing 9.6 mg/kg MTG, human dose equivalent) OPMS liquid (right) in male SD rats. The data represent the mean plasma concentration–time profiles, and the error bar represents the SEM (n = 4).
Even though the actual dose of 7-HMG was negligible (<0.1 mg/kg), there were measurable amounts of 7-HMG as observed in the plasma and maximum plasma concentrations (Cmax) of 4.3 ± 0.8 and 4.0 ± 0.6 ng/mL following the administration of LKT and OPMS liquid, respectively. These data are consistent with the metabolism of MTG into 7-HMG. The metabolic ratio of 7-HMG to MTG was calculated as % AUC0–24h, 7-HMG/AUC0–24h, MTG, MTG and was found to be 3.4 ± 0.9% and 3.1 ± 0.5% following the administration of LKT and OPMS liquid in rats, respectively. In female beagle dogs, as previously reported,29 the oral administration of mitragynine (5 mg/kg) alone resulted in a metabolic ratio of 12.6 ± 1.6%. SPG (administered at doses of 0.8 and 1.2 mg/kg in LKT and OPMS liquid, respectively, Table 3) and PAY (administered at doses of 1.1 and 1.8 mg/kg doses in LKT and OPMS liquid, respectively, Table 3) were detected in the systemic circulation at the earlier time points in some rats. The Cmax values for SPG were found to be 2.9 ± 0.8 (at a Tmax of 10.0 ± 0.0 min) and 2.9 ng/mL (at a Tmax of 5.0 min, quantifiable levels were observed in one rat only) following LKT and OPMS liquid administration, respectively. The Cmax values for PAY were observed at 5.3 ± 1.5 ng/mL (at a Tmax of 10.0 ± 0.0 min) and 2.0 ± 1.7 ng/mL (at a Tmax of 5.0 ± 0.0 min) following LKT and OPMS liquid dosing, respectively. The concentrations of both SPG and PAY dropped below the limit of quantitation (<1 ng/mL) within 1 h postdose. The plasma concentration–time profiles for MTG, 7-HMG, COR, and SPC were captured following LKT and OPMS liquid oral administration in rats. The dose-normalized Cmax values for MTG were 11.1 ± 1.1 ng/mL at a Tmax of 1.3 ± 0.3 h and 11.7 ± 1.6 ng/mL at a Tmax of 3.1 ± 1.7 h following LKT and OPMS liquid oral doses, respectively. Further, the dose-normalized area under the curve from 0 to 24 h (AUC0–24h/dose) for MTG was 83.7 ± 6.4 and 136.1 ± 13.1 h·kg·ng/mL/mg following LKT and OPMS liquid oral administration, respectively. These results indicate a slower rate of absorption and increased systemic exposure of MTG (1.6-fold) following OPMS liquid administration as compared to LKT. However, no change in the percentage ratio of AUC0–24h of 7-HMG to AUC0–24h of MTG (3.6% and 3.1% in LKT and OPMS liquid studies, respectively) was observed, suggesting that the extent of metabolism of MTG to 7-HMG with both the formulations was comparable. Further, SPC and COR also showed a similar trend where the Cmax/dose was comparable following LKT and OPMS liquid administration while the Tmax was delayed following OPMS liquid administration, as shown in Table 4. Also, the AUC0–24h/dose values for SPC (1.9-fold) and COR (2.4-fold) were higher after OPMS liquid dosing as compared to LKT, as shown in Table 4. Since the elimination phase was not achieved for MTG, 7-HMG, SPC, and COR and the percentage of extrapolated area under the curve zero to infinity (% extrapolated AUC0-inf) were greater than 20%, the plasma half-life and AUC0-inf were not calculated following the OPMS liquid dose. The pharmacokinetics of MTG and COR alone in rats has been reported previously.20-22,26,26 Avery et al. in 2019 compared the pharmacokinetics of MTG administered individually, as an ingredient in LKT or as an organic extract of LKT at an oral dose of 20 mg/kg equivalent of MTG to male rats, and showed an improved bioavailability of MTG when administered as LKT (1.5 fold) or organic extracts of LKT (1.8 fold) than pure MTG.26 The Cmax/dose and AUC/dose values for MTG following LKT (20 mg/kg equivalent MTG) oral dose were 46.5 ng/mL and 213.1 h·kg·ng/mL/mg, which is 4.2- and 2.5-fold higher than those of the present study (LKT 5.7 mg/kg equivalent MTG dose), suggesting that the dose and the alkaloidal composition of LKT influences the exposure of MTG. Further, the Cmax/dose value for COR following the pure COR oral administration to rats was reported to be 10.7 ng/mL, which is 0.76- and 0.62-fold lower following LKT and OPMS liquid oral administration, respectively. The AUC0–24h/dose following pure COR oral administration was 138.8 h·kg·ng/mL/mg, which is comparable to AUC0–24h/dose following OPMS liquid administration and it is 2.1-fold higher compared to LKT oral administration.24 None of the other kratom alkaloids pharmacokinetics in rats have been reported either alone or in combination.
In the present study, OPMS liquid showed an extended exposure of kratom alkaloids as compared to LKT. Among the tested alkaloids, only MTG, 7-HMG, COR, and SPC showed measurable systemic exposure following an oral dose. Having an understanding of the pharmacokinetics of individual kratom alkaloids following the oral administration of kratom products in preclinical species will facilitate the design of clinical trials evaluating kratom products. Additionally, the developed bioanalytical method can be implemented for the analysis of plasma samples obtained from a variety of animal species including humans using standardized kratom products. To the best of our knowledge, this is the first report that has simultaneously evaluated the systemic exposure of 11 kratom alkaloids following the administration of traditional and commercial kratom products. A simple and sensitive UPLC–MS/MS method was developed and validated as per FDA guidelines.27 This method required a small sample volume (25 μL) and simple sample preparation involving protein precipitation. The method has a dynamic range of 1–200 ng/mL and has a short run time (11 min) that allows for efficient analysis. The validated method was successfully applied for the simultaneous quantitation of kratom alkaloids in plasma samples obtained following the pharmacokinetic study of LKT and OPMS liquid in rats. Overall, the OPMS liquid has resulted in the delayed and greater systemic exposure of kratom alkaloids (MTG, 7-HMG, SPC, and COR) compared to LKT possibly due to the differences in the composition of other constituents. This method can be adopted for the analysis of kratom in human samples to include alkaloids beyond just MTG or 7-HMG.
EXPERIMENTAL SECTION
Chemicals and Reagents.
Individual kratom alkaloids MTG, COR, SPG, SPC, PAY, COX, COX-B, MTP, AJM, and i-SPF were isolated and purified from a kratom alkaloid rich extract as previously described. 28,30,31 The 7-HMG was semisynthetically obtained from MTG.28 All the alkaloids were >99% pure, with the structure and purity confirmed by proton (1H) nuclear magnetic resonance spectroscopy (NMR), carbon (13C) NMR, ultrahigh-performance liquid chromatography photodiode array detection (UHPLC-PDA), and liquid chromatography high-resolution quadrupole time-of-flight mass spectrometry (LC–Q-TOF) as previously described.28 LC–MS grade methanol, acetonitrile, isopropanol, formic acid, acetic acid, ammonium acetate, and sodium heparin were procured from Fisher Scientific (Fair Lawn, NJ). Verapamil (purity ≥98%, internal standard, IS) was purchased from Sigma-Aldrich (St Louis, MO). LC–MS grade water (J.T. Baker, Phillipsburg, NJ) and heparinized blank rat plasma were purchased from VWR International (Suwanee, GA). The sterile blood collection tubing and heparin-coated blood collection vials were obtained from BASi (West Lafayette, IN). Lyophilized kratom tea was prepared in-house from dried leaves of Mitragyna speciosa purchased from Pure Land Ethnobotanicals (described in the Supporting Information), and OPMS liquid vials (Optimized Plant Mediated Solutions, Choice Organics, Los Angeles, CA) were provided as gift samples.
Method Development and Validation.
The bioanalytical method was developed and validated according to the US FDA guidelines for selectivity, specificity, carryover, sensitivity, accuracy, precision, recovery, and stability.27
Instrumentation and Analytical Conditions.
A Waters Acquity I Class UPLC system coupled with a Waters Xevo TQ-S Micro triple quadrupole mass spectrometer (Milford, MA) was used for the simultaneous quantitative analysis of the kratom alkaloids. The chromatographic separation of kratom alkaloids sharing protonated monoisotopic masses (M + H+, m/z) was achieved on a Waters Acquity BEH C18 column (1.7 μm, 3.1 mm × 100 mm) with a shallow gradient using a mobile phase consisting of aqueous ammonium acetate buffer (2.5 mM, pH 3.5, (A)) and acetonitrile (B). The flow rate of the mobile phase was held at 0.35 mL/min, and a gradient was started with pumps A and B supplying a 90:10 composition of the mobile phase components up to 1 min. The composition of component B in the mobile phase was initially increased to 25% reaching 3.5 min and then to 40% reaching 10 min. Finally, the composition of component B in the mobile phase was sharply increased to 70% reaching 10.2 min and then decreased to 10% reaching 10.5 min and was maintained at 10% until 11 min. The sample injection volume was set to 2 μL. The strong needle wash consisted of acetonitrile, water, and isopropyl alcohol (7:1:2 proportion). The weak needle wash comprised acetonitrile, water, and methanol (1:1:1 proportion). The column oven and autosampler temperatures were maintained at 50 and 10 °C, respectively. The multiple reaction monitoring (MRM) method with electrospray ionization (ESI) in positive mode was used for the detection of kratom alkaloids. The compound parameters (MRM ion transitions, cone voltage, and collision energy) of the kratom alkaloids and the IS are shown in Table S3. The capillary voltage, source temperature, desolvation temperature, and desolvation gas and cone gas flow rates were held at 0.5 kV, 150 °C, 450 °C, 900 L/h, and 50 L/h, respectively. The dwell time was set to ensure that at least 12 data points per peak were recorded by the detector. Nitrogen and argon were used as the source and collision gases, respectively. The data acquisition of the UPLC–MS/MS system was controlled by MassLynx software version 4.1.
Preparation of Calibration and Quality Control Standards.
The primary stock solution of each kratom alkaloid (MTG, 7-HMG, COR, SPG, SPC, PAY, COX, COX-B, MTP, AJM, and i-SPF) was prepared by separately dissolving an accurately weighed quantity of each alkaloid standard in an appropriate volume of methanol to achieve a concentration of 1 mg/mL. The primary stock solutions of kratom alkaloids were mixed (20 μL, each) and diluted with methanol (up to 2 mL) to obtain a combined stock of 10 μg/mL of each alkaloid. The combined stock of kratom alkaloids was further diluted with methanol to prepare combined working stocks containing 12.5, 25, 125, 312.5, 625, 1250, 2250, and 2500 ng/mL of each analyte. The quality control (QC) standards of 12.5, 31.25, 1125, and 2000 ng/mL were prepared as described for calibration standards, except the primary stocks were prepared from separately weighed kratom alkaloids. The calibration standards (CS) were prepared by mixing 2 μL of each working stock with 23 μL of blank rat plasma to obtain a final concentration of 1, 2, 10, 25, 50, 10, 160, and 200 ng/mL. The QC standards were set at four concentrations: 1 (LLOQ), 2.5 (LQC), 90 (MQC), and 160 ng/mL (HQC) of each analyte. The QCs were prepared similarly as described for CS. All stock solutions were stored at −20 °C. The stock solutions were allowed to reach room temperature and vortex mixed for 5 min before use.
Sample Preparation.
The freshly prepared CS and QC plasma samples were vortex mixed using a BenchMixer (Benchmark, San Francisco, CA) for 5 min. To each of the 25 μL of standard samples, 100 μL of methanol containing 0.05% formic acid and 25 ng/mL IS (quenching solution) was added and again vortex mixed for 5 min. The quenched samples were then filtered through a multiscreen Solvinert (Millipore, St. Louis, MO) 96-well 0.45 μm filter plate under centrifugation at a speed of 2000g for 5 min maintained at 4 °C. The filtrate (2 μL) was subjected to UPLC–MS/MS analysis. The test plasma samples from the pharmacokinetic study stored at −80 °C were initially thawed at room temperature and vortex mixed for 5 min. An aliquot of 25 μL of each test sample was taken and then processed as described above. The CS, QC, and test samples were prepared on the same day and analyzed together.
Selectivity; Specificity; Carryover, and Sensitivity.
The selectivity and specificity were evaluated by analyzing blank plasma samples obtained from six different rats. The blank with the IS and the blank plasma samples were processed to assess for any interference peaks from endogenous substances that could have eluted at any of the same retention times for each kratom alkaloid or the IS. The interference for IS was evaluated by comparing the response in blank and IS spiked samples, and the average response should be within 5%. The carryover was evaluated by analyzing blank samples immediately following the highest concentration samples, and the response of any analyte peak detected in blank samples should be less than 20% of LLOQ. The sensitivity of the method was analyzed by evaluating the signal-to-noise ratio of at least 10:1 with the accuracy and precision of 20% of nominal concentration and standard deviation, respectively, for the LLOQ standard.
Recovery and Matrix Effect.
The recovery of each kratom alkaloid was assessed using four different QC standards (LLOQ, LQC, MQC, and HQC, n = 6) by comparing the analyte peak area in prespiked (before protein precipitation) and postspike (after protein precipitation) plasma samples. Similarly, the matrix effect for each kratom alkaloid was determined by comparing the analyte peak area of postspiked plasma QC standards with the QC standards prepared in water–methanol mixture (1:1) (aqueous QC standards). The recovery or matrix effect was calculated as the percentage ratio of the analyte peak area in prespike to postspike samples or the percentage ratio of the analyte peak area of the postspike plasma samples to the aqueous QC standards, respectively.
Accuracy and Precision.
The accuracy and precision of the method for each analyte were assessed by replicate analysis (n = 6) of four different QCs: LLOQ, LQC, MQC, and HQC against the calibration standards on three separate days. The accuracy (intra- and interday) was calculated as %bias, and the acceptance criteria were ≤20% at LLOQ and ≤15% for all other QC standards.
Similarly, the precision (intra- and interday) was calculated as percentage relative standard deviation (%RSD) for each QC standard, and the acceptance criteria were the same as described for accuracy.
Stability.
The stability of each alkaloid in plasma was assessed for the conditions most likely to arise during storage, sample preparation, and analysis. The LQC and HQC samples (n = 6) were analyzed for benchtop (2 h), autosampler (24 h), freeze–thaw (three cycles), and long-term (4 weeks) stability. For benchtop stability, the QC plasma samples were left at room temperature on the benchtop for 2 h. The autosampler stability was assessed by analyzing the extracted samples at 10 °C in the UPLC sample organizer left for 24 h and analyzed again. For freeze–thaw stability, samples were prepared and placed in a −80 °C freezer. Then, the following day, the samples were allowed to fully thaw and were stored back in the freezer. This cycle was repeated three times. For the long-term stability, the QC samples were stored at −80 °C for 4 weeks. All the QC samples for the stability studies were analyzed against a freshly prepared calibration curve.
Pharmacokinetic Studies of Kratom Preparations.
To evaluate the systemic exposure of individual kratom alkaloids following the administration of different kratom preparations, preclinical pharmacokinetic studies were performed. All the procedures were performed in accordance with a preapproved University of Florida Institutional Animal Care and Use Committee (IACUC) protocol. Right jugular vein cannulated healthy male Sprague–Dawley rats (225 ± 25 g) were obtained from Envigo (Indianapolis, IN). The rats were housed in single-occupancy ventilated cages with ad libitum access to food and water. For the pharmacokinetic study, the rats were housed in the BASi Culex (West Lafayette, IN) automated blood drawing metabolic cages.
The LKT (prepared in-house) or OPMS liquid, a commercial kratom preparation, was administered to rats (n = 4) orally by oral gavage at a human equivalent dose (HED) normalized for MTG content. The HED for rats was calculated using the FDA recommended body-weight-based formula:33
| (1) |
The human dose for LKT was previously reported,32 and the OPMS liquid human dose was assumed to be 8 mL (one 8 mL vial consumed per dose). The rats (n = 4) were fasted overnight (12–16 h) before the study and provided water ad libitum. A standard rodent diet was provided to rats at 2 h postdose. The formulation was prepared by mixing an appropriate amount of LKT powder or OPMS liquid with water to yield 4 mL/kg dosing strength, and the appropriate volume was administered orally to rats. The blood samples (100 μL) were collected at the following time points: predose and 0.083, 0.167, 0.25, 0.5, 0.75, 1, 1.5, 2, 4, 6, 8, 12, and 24 h postdose. The blood samples were centrifuged for 10 min at 2000g, and the separated plasma was collected and stored at −80 °C until analysis.
Data Analysis.
Following the acquisition of the CS, QCs, and test samples, the data was processed using TargetLynx (an application of MassLynx 4.1). The peak area ratio of individual alkaloids and IS from CS was plotted against the nominal concentration of alkaloids to obtain a calibration curve. The 1/x2 weighing was used to accommodate lower concentration samples for all the alkaloids. The equation of the line was used to quantify study samples. Following the quantification of the test samples, a plasma concentration of each quantified alkaloid vs time curve was constructed in Phoenix 6.4 (Certara, Princeton, NJ) and time (Tmax) to reach a maximum plasma concentration (Cmax) that was visualized from the concentration–time data. Further, the concentration versus time data was subjected to noncompartmental analysis Phoenix 6.4. The total systemic exposure of individual alkaloids was evaluated by measuring the area under the curve (AUC0–24h) using the linear-up log-down trapezoidal method.
Supplementary Material
Funding
This study was supported by R01 DA047855 and UG3 DA048353 grants from the National Institute on Drug Abuse and the University of Florida Clinical and Translational Science Institute, which is supported in part by the NIH National Center for Advancing Translational Sciences under award number UL1TR001427.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jnatprod.0c01163.
Discussion of preparation of lyophilized kratom tea and tables of recovery and matrix effect of each kratom alkaloids in rat plasma, individual and relative alkaloid content in OPMS liquid and lyophilized kratom tea, and source parameters for kratom alkaloids and internal standard (PDF)
The authors declare no competing financial interest.
Contributor Information
Shyam H. Kamble, Department of Pharmaceutics, College of Pharmacy and Translational Drug Development Core, Clinical and Translational Sciences Institute, University of Florida, Gainesville, Florida 32610, United States.
Erin C. Berthold, Department of Pharmaceutics, College of Pharmacy, University of Florida, Gainesville, Florida 32610, United States.
Tamara I. King, Department of Pharmaceutics, College of Pharmacy, University of Florida, Gainesville, Florida 32610, United States
Siva Rama Raju Kanumuri, Department of Pharmaceutics, College of Pharmacy and Translational Drug Development Core, Clinical and Translational Sciences Institute, University of Florida, Gainesville, Florida 32610, United States.
Raluca Popa, Department of Pharmaceutics, College of Pharmacy, University of Florida, Gainesville, Florida 32610, United States.
Julius R. Herting, Department of Pharmaceutics, College of Pharmacy, University of Florida, Gainesville, Florida 32610, United States
Francisco León, Department of Medicinal Chemistry, College of Pharmacy, University of Florida, Gainesville, Florida 32610, United States.
Abhisheak Sharma, Department of Pharmaceutics, College of Pharmacy and Translational Drug Development Core, Clinical and Translational Sciences Institute, University of Florida, Gainesville, Florida 32610, United States.
Lance R. McMahon, Department of Pharmacodynamics, College of Pharmacy, University of Florida, Gainesville, Florida 32610, United States
Bonnie A. Avery, Department of Pharmaceutics, College of Pharmacy and Translational Drug Development Core, Clinical and Translational Sciences Institute, University of Florida, Gainesville, Florida 32610, United States
Christopher R. McCurdy, Department of Pharmaceutics, College of Pharmacy, Translational Drug Development Core, Clinical and Translational Sciences Institute, and Department of Medicinal Chemistry, College of Pharmacy, University of Florida, Gainesville, Florida 32610, United States.
REFERENCES
- (1).Hassan Z; Muzaimi M; Navaratnam V; Yusoff NH; Suhaimi FW; Vadivelu R; Vicknasingam BK; Amato D; von Horsten S; Ismail NI; Jayabalan N; Hazim AI; Mansor SM; Muller CP Neurosci. Biobehav. Rev 2013, 37 (2), 138–51. [DOI] [PubMed] [Google Scholar]
- (2).Warner ML; Kaufman NC; Grundmann O Int. J. Legal Med 2016, 130 (1), 127–38. [DOI] [PubMed] [Google Scholar]
- (3).Boyer EW; Babu KM; Macalino GE; Compton W Am. J. Addict 2007, 16 (5), 352–356. [DOI] [PubMed] [Google Scholar]
- (4).Veltri C; Grundmann O Subst Abuse Rehabil 2019, 10, 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Assanangkornchai S; Muekthong A; Sam-Angsri N; Pattanasattayawong U Subst Use Misuse 2007, 42 (14), 2145–57. [DOI] [PubMed] [Google Scholar]
- (6).Cinosi E; Martinotti G; Simonato P; Singh D; Demetrovics Z; Roman-Urrestarazu A; Bersani FS; Vicknasingam B; Piazzon G; Li JH; Yu WJ; Kapitany-Foveny M; Farkas J; Di Giannantonio M; Corazza O BioMed Res. Int 2015, 2015, 968786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Bestha D Innov. Clin. Neurosci 2018, 15 (5–6), 11. [PMC free article] [PubMed] [Google Scholar]
- (8).Grewal KS J. Pharmcol. Exp. Ther 1932, 46 (3), 251–271. [Google Scholar]
- (9).Lydecker AG; Sharma A; McCurdy CR; Avery BA; Babu KM; Boyer EW J. Med. Toxicol 2016, 12 (4), 341–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).United States Food and Drug Administration. FDA and Kratom. https://www.fda.gov/news-events/public-health-focus/fda-and-kratom (accessed on 10-11-2020). [Google Scholar]
- (11).Post S; Spiller HA; Chounthirath T; Smith GA Clin. Toxicol 2019, 57 (10), 847–854. [DOI] [PubMed] [Google Scholar]
- (12).Adkins JE; Boyer EW; McCurdy CR Curr. Top. Med. Chem 2011, 11 (9), 1165–1175. [DOI] [PubMed] [Google Scholar]
- (13).Shellard EJ Bull. Narc 1974, 26 (2), 41–55. [PubMed] [Google Scholar]
- (14).Kamble SH; Sharma A; King TI; Leon F; McCurdy CR; Avery BA Xenobiotica 2019, 49 (11), 1279–1288. [DOI] [PubMed] [Google Scholar]
- (15).Kruegel AC; Uprety R; Grinnell SG; Langreck C; Pekarskaya EA; Le Rouzic V; Ansonoff M; Gassaway MM; Pintar JE; Pasternak GW; Javitch JA; Majumdar S; Sames D ACS Cent. Sci 2019, 5 (6), 992–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Obeng S; Kamble SH; Reeves ME; Restrepo LF; Patel A; Behnke M; Chear NJ; Ramanathan S; Sharma A; Leon F; Hiranita T; Avery BA; McMahon LR; McCurdy CR J. Med. Chem 2020, 63 (1), 433–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Takayama H; Ishikawa H; Kurihara M; Kitajima M; Aimi N; Ponglux D; Koyama F; Matsumoto K; Moriyama T; Yamamoto LT; Watanabe K; Murayama T; Horie SJ Med. Chem 2002, 45 (9), 1949–56. [DOI] [PubMed] [Google Scholar]
- (18).Takayama H Chem. Pharm. Bull 2004, 52 (8), 916–28. [DOI] [PubMed] [Google Scholar]
- (19).Vuppala PK; Jamalapuram S; Furr EB; McCurdy CR; Avery BA Biomed. Chromatogr 2013, 27 (12), 1726–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).de Moraes NV; Moretti RA; Furr EB 3rd; McCurdy CR; Lanchote VLJ Chromatogr. B: Anal. Technol Biomed. Life Sci 2009, 877 (24), 2593–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Trakulsrichai S; Sathirakul K; Auparakkitanon S; Krongvorakul J; Sueajai J; Noumjad N; Sukasem C; Wananukul W Drug Des., Dev. Ther 2015, 9, 2421–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Janchawee B; Keawpradub N; Chittrakarn S; Prasettho S; Wararatananurak P; Sawangjareon K Biomed. Chromatogr 2007, 21 (2), 176–83. [DOI] [PubMed] [Google Scholar]
- (23).Kong WM; Mohamed Z; Alshawsh MA; Chik ZJ Pharm. Biomed. Anal 2017, 143, 43–47. [DOI] [PubMed] [Google Scholar]
- (24).King TI; Sharma A; Kamble SH; Leon F; Berthold EC; Popa R; Cerlati O; Prentice BM; McMahon LR; McCurdy CR; Avery BA J. Pharm. Biomed. Anal 2020, 180, 113019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kamble SH; Sharma A; King TI; Berthold EC; Leon F; Meyer PKL; Kanumuri SRR; McMahon LR; McCurdy CR; Avery BA Toxicol. Lett 2020, 319, 148–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Avery BA; Boddu SP; Sharma A; Furr EB; Leon F; Cutler SJ; McCurdy CR Planta Med. 2019, 85 (4), 340–346. [DOI] [PubMed] [Google Scholar]
- (27).Guidance for Industry: Bioanalytical Method Validation; US Food and Drug Administration, 2018. [Google Scholar]
- (28).Sharma A; Kamble SH; Leon F; Chear NJ; King TI; Berthold EC; Ramanathan S; McCurdy CR; Avery BA Drug Test. Anal 2019, 11 (8), 1162–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Maxwell EA; King TI; Kamble SH; Raju KSR; Berthold EC; Leon F; Avery BA; McMahon LR; McCurdy CR; Sharma A Planta Med. 2020, 86 (17), 1278–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kitajima M; Misawa K; Kogure N; Said IM; Horie S; Hatori Y; Murayama T; Takayama H J. Nat. Med 2006, 60 (1), 28–35. [Google Scholar]
- (31).Ali Z; Demiray H; Khan IA Tetrahedron Lett. 2014, 55 (2), 369–372. [Google Scholar]
- (32).Singh D; Yeou Chear NJ; Narayanan S; Leon F; Sharma A; McCurdy CR; Avery BA; Balasingam VJ Ethnopharmacol. 2020, 249, 112462. [DOI] [PubMed] [Google Scholar]
- (33).Center for Drug Evaluation and Research (CDER). Guidance for industry: estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers; US Food and Drug Administration: Rockville, MD, 2005. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.