

Abstract
The presence of tight junction protein zonula occludens 2 (ZO-2) at the nucleus inhibits the transcription of genes regulated by TEAD transcription factor. Here, we analyzed whether the movement of ZO-2 into the nucleus modulates the nuclear concentration of TEAD. In sparse cultures of ZO-2 knockdown Madin–Darby canine kidney cells, nuclear TEAD was diminished, as in parental cells transfected with a ZO-2 construct without nuclear localization signals, indicating that ZO-2 facilitates the entry of TEAD into the nucleus. Inhibition of nPKCδ in parental cells triggers the interaction between ZO-2 and TEAD at the cytoplasm and facilitates TEAD/ZO-2 complex nuclear importation. Using proximity ligation, immunoprecipitation, and pull-down assays, TEAD/ZO-2 interaction was confirmed. Nuclear TEAD is phosphorylated, and its exit in parental cells is enhanced by activation of a ZO-2 nuclear exportation signal by nPKCε, while the nuclear accumulation of ZO-2 triggered by the mutation of ZO-2 nuclear export signals induces no change in TEAD nuclear concentration. In summary, our results indicate that the movements of ZO-2 in and out of the nucleus modulate the intracellular traffic of TEAD through a process regulated by nPKCδ and ε and provide a novel role of ZO-2 as a nuclear translocator of TEAD.
INTRODUCTION
Zonula occludens 2 (ZO-2) is a tight junction (TJ) protein member of the MAGUK (membrane-associated guanylate kinase homologue) protein family that translocates to the nucleus in response to low cell density (Islas et al., 2002) and stress induced by chemicals (CdCl2) and heat shock (Traweger et al., 2003). In sparse cultures, newly synthesized ZO-2 temporarily localizes to the nucleus before reaching the TJs at the cell borders (Chamorro et al., 2009). In contrast, in confluent monolayers, ZO-2 goes directly to the cell borders (Quiros et al., 2013). Movement of ZO-2 from the cytoplasm to the plasma membrane involves activation of the calcium-sensing receptor and the novel protein kinase Cε (nPKCε)/with no lysine kinase-4 (WNK4) downstream signaling cascade, which leads to ZO-2 serine phosphorylation, its release from 14-3-3 protein in the cytoplasm, and incorporation to TJs (Amaya et al., 2019). ZO-2 moves in and out of the nucleus with nuclear localization and exportation signals (NLS and NES) (Lopez-Bayghen et al., 2006). In canine ZO-2 (cZO-2), one monopartite and two bipartite (bp) NLS are present at the U2 segment located at the amino portion of the molecule between PDZ1 and PDZ2 domains (Quiros et al., 2013). In addition, 16 serine–arginine (SR) repeats are found at this segment, whose phosphorylation by the kinase SRPK induces protein entrance to the nucleus and its localization at nuclear speckles (Quiros et al., 2013). cZO-2 has four NES, two located at the PDZ2 domain and the other two at the guanylate kinase (GuK) module (Gonzalez-Mariscal et al., 2006).
ZO-2 has multiple protein-binding domains and motifs that allow it to function as a scaffold that concentrates a wide variety of proteins at both the TJ and the nucleus (for reviews, see Gonzalez-Mariscal et al., 2017, 2019). At the plasma membrane, ZO-2 and ZO-1 act as a platform for the polymerization of claudins into TJ strands (Umeda et al., 2006). In contrast, at the nucleus, even though ZO-2 has no DNA-binding sites, it inhibits the transcription of several genes regulated by AP-1 (activator protein 1) (Betanzos et al., 2004), TEAD (transcriptional enhanced associated domain) (Dominguez-Calderon et al., 2016) and TCF (T cell factor)/LEF (lymphoid enhancer family) (Tapia et al., 2009; Wetzel et al., 2017) transcription factors. Here we have studied if ZO-2 plays a role as a nuclear translocator of TEAD transcription factor.
Mammals express four TEAD genes, and the particular function of each one remains controversial. Thus, Tead1 null mice are embryonic lethal due to defective cardiac development (Chen et al., 1994); Tead2 knockout (KO) mice either show no phenotype (Sawada et al., 2008) or have defects in neural tube closure (Kaneko et al., 2007); Tead4 null mice are lethal due to embryo implantation failure (Yagi et al., 2007), and a KO of both Tead1 and Tead2 is embryonic lethal due to defects in neural tube closure (Sawada et al., 2008). In humans, an inactivation mutation of TEAD1 is associated with degeneration of the choroid and retina or Sveinsson’s chorioretinal atrophy (Fossdal et al., 2004).
Regulation of TEAD involves YAP (Yes-associated protein) and its paralogue TAZ (transcriptional activator with PDZ-binding domain). Thus, when YAP/TAZ are phosphorylated by the kinases of the Hippo pathway LATS1/2 (large tumor suppresor kinase 1/2), they remain in the cytoplasm by sequestration to 14-3-3 or are ubiquitinated and degraded. Instead, unphosphorylated YAP/TAZ move into the nucleus and bind to TEAD, driving the transcription of target genes (for review see Zhou et al., 2016).
Hippo-independent coactivators can also regulate TEAD. Such is the case of vestigial-like (VGLL) protein that competes with YAP/TAZ for TEAD binding (Pobbati et al., 2012; Jiao et al., 2014); of TCF4 (transcription factor 4), whose interaction with TEAD facilitates the expression of Wnt target genes (Jiao et al., 2017); of the p160 family of steroid receptor coactivators that potentiate TEAD transcriptional activity (Belandia and Parker, 2000); and of AP-1 that occupies the same chromatin sites as TEAD and is necessary for the activation of TEAD target genes (Zanconato et al., 2015; Liu et al., 2016).
Although TEAD function can also be regulated by posttranslational modifications, including palmitoylation (Chan et al., 2016) and phosphorylation by PKA (Gupta et al., 2000) and PKC (Jiang et al., 2001), it is noteworthy that TEAD subcellular localization regulates transcriptional programs that determine trophectoderm versus inner cell mass (ICM) lineage differentiation in embryos. Thus, in mouse embryonic stem cells (mESCs), TEAD4 localized exclusively in the cytoplasm, while in mouse trophoblast stem cells (mTSCs) TEAD4 was enriched in the nucleus, regulating a trophectoderm-specific transcriptional program (Home et al., 2012). Hyperosmotic stress also induces TEAD cytoplasmic localization through a mechanism where p38 binds to TEAD and subsequently translocates the transcription factor to the cytoplasm (Lin et al., 2017).
Previously we demonstrated that ZO-2 silencing in renal epithelial Madin-Darby canine kidney (MDCK) cells induced the nuclear accumulation of YAP and increased the activity of an artificial promoter driven by TEAD-binding sites. In contrast, the overexpression of ZO-2 abolished this activity (Dominguez-Calderon et al., 2016). Moreover, we showed that the promoter for the connective tissue growth factor (CTGF), regulated by TEAD-binding sites (Zhao et al., 2008), is more active in ZO-2 KD cells and that these changes correlated with increased CTGF mRNA (Dominguez-Calderon et al., 2016). These effects, in turn, were obliterated upon ZO-2 transfection (Dominguez-Calderon et al., 2016). These observations prompted us to explore whether the expression and subcellular localization of ZO-2 modulated the nuclear accumulation of TEAD.
Here, in epithelial cells we have found that the entry and departure of ZO-2 from the nucleus modulate TEAD intracellular traffic and that TEAD and ZO-2 form a complex that is negatively regulated by nPKCδ, which facilitates the nuclear importation of TEAD.
RESULTS
ZO-2 facilitates the entry of TEAD into the nucleus of epithelial cells
We first explored by immunofluorescence the subcellular localization of TEAD in parental and ZO-2 KD MDCK cells. For this purpose, we employed sparse and confluent cultures as we had previously shown that in the former, ZO-2 accumulates at the nucleus. In contrast, in confluent monolayers, the amount of nuclear ZO-2 is minimal (Islas et al., 2002). Figure 1A shows that the absence of ZO-2 induces a decreased expression of TEAD in the nuclei of sparse cultures. In contrast, in confluent cultures, an increased expression of nuclear TEAD is observed compared with parental cultures. A Western blot then revealed that in total cellular extracts, the amount of TEAD was diminished in sparse cultures in a ZO-2–dependent manner because the decrease in TEAD was reversed upon transfection with a human full-length ZO-2 construct with an altered short hairpin RNA (shRNA)-binding site (hZO-2). Instead, in confluent cultures, the cellular content of TEAD increased with the lack of ZO-2 and was diminished upon ZO-2 transfection (Figure 1B).
FIGURE 1:

ZO-2 facilitates the nuclear accumulation of TEAD. (A) In ZO-2 KD cells nuclear TEAD was diminished in sparse cultures, while cytoplasmic TEAD increased in confluent monolayers. Sparse and confluent cultures of parental and ZO-2 KD MDCK cells were processed for immunofluorescence with antibodies against TEAD. Left, representative images; right, quantitative analysis. The nuclei of 100 cells per condition derived from three independent experiments were quantitated. Statistical analysis done with Kruskal–Wallis test followed by Dunn’s multiple comparisons test. AU, arbitrary units. ***p < 0.001; ****p < 0.0001. (B) In ZO-2 KD cells, the amount of TEAD decreased in sparse cultures and increased in confluent cultures. Western blot of total cellular extracts derived from sparse and confluent cultures of parental and ZO-2 KD cells transfected or not with hZO-2. GADPH was employed as loading control. Top panel, representative image of three independent experiments; bottom panel, densitometric analysis. Statistics done with one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test, **p < 0.01; ***p < 0.001. (C) The lack of ZO-2 diminished the amount of TEAD at the nucleus of sparse cells and increased cytoplasmic TEAD in confluent monolayers. Western blot detection of TEAD in cytoplasmic (Cyt) and nuclear (Nuc) fractions derived from sparse and confluent cultures of parental and ZO-2 KD cells transfected or not with hZO-2. Lamin B1 and GAPDH were employed as markers of nuclear and cytosolic fractions, respectively. Top panels, representative images of three independent experiments; bottom panels, quantitative analysis of TEAD/lamin B1 in the nuclear fractions and of TEAD/GAPDH in the cytoplasmic fractions. Statistics done with two-way ANOVA followed by Tukey’s multiple comparisons test, *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. (D) Nuclear TEAD has a higher molecular weight than cytoplasmic TEAD due to phosphorylation. Left panel, Western blot detection of TEAD in cytoplasmic (Cyt) and nuclear (Nuc) fractions treated or not with alkaline phosphatase (AP). Antibodies against lamin B1 and GAPDH were employed as markers of nuclear and cytoplasmic fractions, respectively. Representative image of two independent experiments. Filled arrowhead, 49 kDa band; empty arrowhead, 51 kDa band. Right panel, nuclear (Nuc) fractions treated or not with AP were run on an SDS–PAGE with acrylamide-pendant Phos-tag, and blotted with antibodies against TEAD.
Next, we analyzed by Western blot the amount of TEAD present in nuclear and cytoplasmic fractions. Figure 1C reveals that nuclear TEAD was diminished in ZO-2 KD cells in sparse cultures and that this effect was reversed by hZO-2 transfection. However, the decrease of nuclear TEAD in ZO-2 KD cells was not accompanied by an increase in cytoplasmic TEAD. Instead, in confluent cultures, the lack of ZO-2 augments the cytoplasmic content of TEAD, and the transfection of hZO-2 in ZO-2 KD cells abolishes the presence of nuclear TEAD. These results suggest that ZO-2 regulates the entry of TEAD into the nucleus.
To further test this point, we next analyzed the expression of TEAD in the nucleus of sparse parental cells transfected with Flag-hZO-2 wild type (WT) or a construct lacking the NLS of the molecule (Flag-hZO-2 ΔNLS) present at the U2 segment located between the PDZ1 and PDZ2 domains. Figure 2 shows that the nuclear staining for TEAD is reduced in cells transfected with a ZO-2 that, as previously reported, cannot localize at the nucleus due to the lack of NLS (Jaramillo et al., 2004; Oka et al., 2010; Amaya et al., 2019). However, because the reduction in nuclear TEAD is small in cells transfected with Flag-hZO-2 ΔNLS, our results suggest that the nuclear importation of ZO-2 only favors the movement of TEAD into the nucleus but that other mechanism might also be involved, including signaling by the bpNLS present in TEAD (Magico and Bell, 2011).
FIGURE 2:

The abundance of nuclear TEAD is diminished in cells transfected with a ZO-2 construct lacking the NLS. (A) Representative image of parental MDCK cells 24 h after transfection with full-length Flag-hZO-2 (WT) or Flag-hZO-2 ΔNLS. Cells were fixed and processed for immunofluorescence with antibodies against Flag and TEAD. Nuclei were stained with DAPI. Arrows, nucleus of transfected cells. (B) Quantitative analysis of TEAD nuclear immunofluorescence in Flag-hZO-2 or Flag-hZO-2 ΔNLS transfected cells. The nuclei of 30 transfected cells per condition derived from three independent experiments were quantitated. AU, arbitrary units. Statistical analysis done with Student’s t test, *p < 0.05. Each bar shows the mean ± SEM.
Nuclear TEAD is phosphorylated
The Western blot in Figure 1C also revealed that the band of TEAD derived from the nuclear fraction had a slightly higher molecular weight (51 kDa) than the one derived from the cytosolic fraction (49 kDa). In silico analysis with GPS 3.0 revealed that TEAD has 58 putative phosphorylation sites. To determine whether the higher molecular weight of nuclear TEAD was due to phosphorylation, we next made a Western blot of nuclear extracts treated or not with alkaline phosphatase and observed that the molecular weight of the nuclear band of TEAD was diminished from 51 to 50 kDa (Figure 1D, left panel), suggesting that nuclear TEAD is more strongly phosphorylated than cytoplasmic TEAD. To confirm this observation, we next made a mobility shift detection assay of TEAD. To this end, we ran nuclear extracts of MDCK cells treated or not with alkaline phosphatase on phosphate-affinity SDS–PAGE with a dinuclear manganese complex of acrylamide-pendant Phos-tag, which binds to and retards the migration of phosphorylated proteins. The right panel of Figure 1D shows that the higher-molecular-weight band of TEAD is not present in the nuclear samples treated with alkaline phosphatase, thus confirming that this band corresponds to phosphorylated TEAD.
Inhibition of nPKCδ promotes the nuclear importation of TEAD and ZO-2
Next, we performed an in silico analysis with GPS 3.0, finding that TEAD has 23 and six putative PKC and PKA phosphorylation sites, respectively. Hence, we explored whether the inhibition of different PKC isozymes and PKA altered the nuclear/cytoplasmic distribution of TEAD in cultures of parental and ZO-2 KD cells. Figure 3A reveals that treatment with 27 nM Ro 31-8220, which inhibits cPKCα, γ, and β1 and nPKCε (Young et al., 2005), significantly increases the concentration of TEAD in the nuclei of ZO-2 KD cells but not of parental cells, thus suggesting that in the absence of ZO-2, cPKCα, γ, β1 or nPKCε induces TEAD nuclear exportation. Instead, treatment with 10 nM Gö-6983, which inhibits cPKCα, β, and γ and nPKCδ (Young et al., 2005), exerts no effect on the nuclear concentration of TEAD in ZO-2 KD cells. Thus, it is suggested that the inhibition of nPKCε blocks the nuclear exportation of TEAD in ZO-2 KD cells (Figure 3B).
FIGURE 3:

ZO-2 modulates the effect exerted by PKC δ on TEAD nuclear accumulation. (A) Confluent monolayers of parental and ZO-2 KD MDCK cells were treated with 10 μM H89 to inhibit PKA, 27 nM Ro 31-8220 to inhibit cPKC α, γ, and β1 and nPKCε, 10 nM Gö-6986 to inhibit cPKC α, β, and γ and nPKCδ, or 0.1% DMSO as vehicle. Cytoplasmic and nuclear fractions were isolated and run in a SDS–PAGE and blotted with antibodies against ZO-2 and TEAD. GAPDH and lamin B1 were employed as loading controls for cytoplasmic and nuclear fractions, respectively. Left, representative images of Western blots from three independent experiments; right, densitometric analysis. Statistics done with one-way ANOVA followed by Fisher’s least significant difference test, *p < 0.05; **p < 0.01; ***p < 0.001. Each bar shows the mean ± SEM. (B) Schematic representation showing that in ZO-2 KD MDCK cells Ro-31822 blocks the nuclear exportation of TEAD mediated by nPKCε. (C) Schematic representation depicting in parental cells ZO-2 entry to the nucleus triggered by the inhibition of PKA with 10 μM H89. (D) Parental MDCK cells were transfected with a nPKCδ construct, and 24 h later nuclear fractions were obtained and processed for Western blot employing antibodies against ZO-2 and TEAD. Lamin B1 was employed as loading control of nuclear fractions. Left, representative image of Western blots from three independent experiments; right, densitometric analysis. Statistics done with Students’ t test, **p < 0.01. Each bar shows the mean ± SEM. (E) MDCK ZO-2 KD cells were transfected with a nPKCδ construct, and 24 h later nuclear fractions were obtained and processed for Western blot employing antibodies against TEAD. Lamin B1 was employed as loading control of nuclear fractions. Left, representative image of Western blots from three independent experiments; right, densitometric analysis. Statistics done with Students’ t test, n.s., nonsignificant. Each bar shows the mean ± SEM. (F) Cartoon showing that nPKCδ blocks ZO-2-TEAD cytoplasmic interaction and nuclear importation.
To further understand the effect of nPKCε inhibition on TEAD function, we performed a luciferase reporter gene assay with the promoter of hCTGF regulated by three TEAD-binding sites (Zhao et al., 2008). Figure 4A confirms that, as we had previously reported (Dominguez-Calderon et al., 2016), hCTGF promoter activity is higher in ZO-2 KD than in parental cells. This has been explained previously in ZO-2 KD cells by the increased entry into the nucleus of YAP that associates to TEAD and functions as a transcriptional activator (Dominguez-Calderon et al., 2016). Figure 4B shows that treatment with permeable nPKCε inhibitor peptide εV1-2 increases hCTGF promoter activity in ZO-2 KD cells and not in parental cells. The increased amount of nuclear TEAD found in ZO-2 KD cells might explain this result as observed after treatment with 27 nM Ro 31-8220 but not with 10 nM Gö-6983 (Figure 3A).
FIGURE 4:

In ZO-2 KD cells inhibition of nPKCε increases the activity of a promoter regulated by TEAD-binding sites. Reporter gene assays done in parental and ZO-2 KD cells transiently transfected with hCTGF-Luc at a concentration of 100 ng/well (32 mm2). (A) hCTGF promoter activity is higher in ZO-2 KD than in parental cells. hCTGF promoter activity values were normalized to values observed in parental cells. Statistical analysis done with Student’s t test, *p < 0.05. (B) Inhibition of nPKCε increases the activity of hCTGF promoter in ZO-2 KD cells but not in parental cells. hCTGF promoter activity values were normalized to values observed in ZO-2 KD cells without the nPKCε inhibitor. Statistical analysis done with Student’s t test, *p < 0.05. In all cases data were normalized to protein content and transfection efficiency with pGL3-Control. Each bar shows the mean ± SEM from triplicates derived from two independent experiments.
Figure 3A also shows that the cytoplasmic content of TEAD in ZO-2 KD cells increases after treatment with 10 μM H89, which inhibits PKA, or 10 nM Gö-6983, but not with 27 nM Ro 31-8220, thus suggesting that inhibition of PKA and nPKCδ augments the content of TEAD but blocks its nuclear importation. In parental cells, PKA inhibition with H89 has no effect on TEAD in cytoplasmic and nuclear fractions but instead increases the amount of ZO-2 in the nuclei and decreases its content in the cytoplasm, thus suggesting that PKA inhibition induces ZO-2 nuclear accumulation (Figure 3C). To further confirm these results, we next employed two additional doses of H89, observing that in parental cells, the decrease in cytoplasmic ZO-2 was triggered when 1, 10, or 100 μM H89 was employed. The increase in nuclear ZO-2 was not observed when 1 μM H89 was used but was present upon treatment with 10 or 100 μM H89 (Supplemental Figure 1).
Figure 3A also shows that in parental MDCK cells, 10 nM Gö-6983 but not 27 nM Ro 31-8220 augments the nuclear content of TEAD, thus indicating that the inhibition of nPKCδ mediates the nuclear accumulation of this protein. However, this increase in the nuclear content of TEAD is accompanied by an increase in the cytoplasmic amount of TEAD, thus suggesting that inhibition of nPKCδ augments the synthesis or stability of this transcription factor and its nuclear importation. In parental cells, 10 nM Gö-6983 but not 27 nM Ro31-8220 also triggers an increase in nuclear ZO-2 accompanied by a decrease in the cytoplasmic fraction of ZO-2, thus suggesting that inhibition of nPKCδ induces the nuclear importation of ZO-2. To confirm these results, we next transfected parental MDCK cells with a construct for nPKCδ and analyzed by Western blot the content of ZO-2 and TEAD in the nuclear fractions. Figure 3D reveals a diminished amount of both ZO-2 and TEAD in the nuclei of parental cells transfected with nPKCδ. To further show the importance of ZO-2 in this process, we next transfected nPKCδ into ZO-2 KD cells and observed no change in the amount of nuclear TEAD compared with that in nontransfected cells (Figure 3E).
In summary, while in parental cells PKA inhibition induces the nuclear accumulation of ZO-2 but not TEAD, the inhibition of nPKCδ triggers the accumulation at the nucleus of both ZO-2 and TEAD. In cells that lack ZO-2, neither the inhibition nor the transfection of nPKCδ alters the nuclear content of TEAD, thus suggesting that phosphorylation mediated by nPKCδ blocks the interaction between ZO-2 and TEAD that facilitates the entry of TEAD and ZO-2 into the nucleus (Figure 3F).
ZO-2 and TEAD interaction is inhibited by nPKCδ
To test whether ZO-2 and TEAD interact and whether this process is inhibited by nPKCδ, we transfected MDCK cells with Flag-hZO-2, treated them or not with 3 μM röttlerin, which inhibits nPKCδ, and made a proximity ligation assay (PLA) with a mouse antibody against Flag and a rabbit antibody against TEAD (Figure 5A). At time 0 (6th hour after transfection), when as previously demonstrated 74–78% of transfected cells have nuclear ZO-2 (Chamorro et al., 2009; Quiros et al., 2013), positive red fluorescent spots are detected at the nucleus of Flag-hZO-2 transfected cells, identified by their green staining (Figure 5, B, a and a′, and C). The number of red spots was above the background present when the antibody against TEAD was omitted (Figure 5, B, e and e′, and C). The abundance of positive red spots at the nucleus of Flag-hZO-2 transfected cells increases when nPKCδ is inhibited with röttlerin (Figure 5, B, b and b′, and C). However, because röttlerin has also been found to inhibit Ca2+/calmodulin kinase II (CaM-kinase II), MAP kinase–activated protein 5 (PRAK/MK5), and MAP kinase–activated protein 2 (MAPKAP-K2) (Gschwendt et al., 1994; Davies et al., 2000), we next proceeded to test the interaction between TEAD and ZO-2 when instead of using röttlerin, nPKCδ was overexpressed by transfection. Figure 5 shows that the number of red spots decreases when nPKCδ was transfected together with Flag-hZO-2 (Figure 5, B, c and c′, and C) and achieves the highest value when transfected nPKCδ is inhibited with röttlerin. In this case, we also observe the appearance of abundant red spots in the cytoplasm (Figure 5, B, d and d′, and C), which further confirms that nPKCδ inhibition allows the interaction of TEAD and ZO-2 in the cytoplasm. Instead, 24 h posttransfection, when previous observations demonstrated that the percentage of cells with nuclear ZO-2 has diminished to 17% (Chamorro et al., 2009; Quiros et al., 2013), the red spots at the nuclei are barely detectable in all the experimental conditions (Figure 5, B, f, f′–j, j′, and C) and resemble the background signal obtained when no anti-TEAD antibody was added (Figure 5, B, j and j′, and C).
FIGURE 5:

Inhibition of nPKCδ with röttlerin augments ZO-2 and TEAD interaction at the nucleus. (A) Scheme of the experimental procedure followed. MDCK cells were transfected with Flag-hZO-2 or Flag-hZO-2 plus nPKCδ. Cells were fixed and processed for a PLA done with a mouse antibody against Flag and a rabbit antibody anti-TEAD at 0 and 24 h posttransfection (time 0 corresponds to the initial 6 h required after transfection for proteins to be expressed). Where indicated, 3 μM röttlerin was added 1 h before fixation. (B) Representative immunofluorescence images of PLA. Red fluorescent spots reveal the positive interaction between transfected Flag-hZO-2 and endogenous TEAD. Cells transfected with Flag-hZO-2 were identified with a mouse antibody against Flag, followed by a secondary goat anti-mouse IgG coupled to Alexa Fluor 488. Background corresponds to experiments done without the anti-TEAD antibody. Nuclei detected with DAPI. Transf, transfected. *, nuclei of transfected cells. Images obtained from two independent experiments. (C) Quantitative analysis of PLA done using BlobFinder. At least 25 cells were analyzed per experimental condition. Statistical analysis done with two-way ANOVA followed by Tukey’s multiple comparisons test, ****p < 0.0001. Each bar shows the mean ± SEM.
To further confirm that the interaction of TEAD and ZO-2 is mediated by nPKCδ, we analyzed the presence of TEAD in a ZO-2 immunoprecipitate, finding that inhibition of nPKCδ with röttlerin augments the amount of TEAD associated with ZO-2 in MDCK cells (Figure 6A). In addition, in intestinal epithelial cells IEC-18, we confirmed the presence of TEAD in a ZO-2 immunoprecipitate (Figure 6B). We also showed with a pull-down assay the interaction between TEAD and ZO-2 in the human kidney cell line HEK293T. For this purpose, the cells were transfected with the amino (coding PDZ domains 1, 2, and 3), or AP (coding the acidic and proline-rich regions) constructs of cZO-2, introduced in the pcDNA4/HisMax vector. Then, the corresponding proteins were purified from extracts of HEK293T cell using Ni affinity columns, run in SDS–PAGE, and stained with Coomassie blue. Figure 6C reveals the presence of bands of 62 and 45 kDa that respectively correspond to amino and AP segments of cZO-2. The identity of the amino pulled-down segment was confirmed in a blot with the anti–ZO-2 antibody that recognizes this section of the protein, as indicated by the manufacturer (Figure 6D, top panel). The blot with the antibody against TEAD revealed the presence of this transcription factor in the pull down of the amino and AP segments of ZO-2, thus suggesting that these segments of ZO-2 associate to TEAD (Figure 6D, bottom panel). We also transfected HEK293T cells with the 3PSG (coding PDZ-3, SH3, and GuK domains) segment of ZO-2. However, the identity of this ZO-2 segment could not be confirmed because it generated a very faint band upon staining with Coomassie blue (unpublished data) and cannot be recognized by any commercial anti–ZO-2 antibody.
FIGURE 6:

The interaction between TEAD and ZO-2 is inhibited by nPKCδ. (A) The amount of TEAD that coimmunoprecipitates with ZO-2 is augmented in cells treated for 1 h with 3 μM röttlerin. Blotting was done with antibodies against ZO-2 and TEAD. Top panel, representative image; bottom panel, densitometric analysis. Statistics done with Students’ t test, **p < 0.01. Each bar shows the mean ± SEM. Co-IP, coimmunoprecipitation; IP, immunoprecipitation. (B) In extracts derived from intestinal IEC-18 cells, TEAD coimmunoprecipitates with ZO-2. PIS, preimmune serum. (C) 6XHis-tagged amino (NH3) and AP segments of cZO-2 were purified with Ni affinity columns from extracts of HEK293T cells, run in SDS–PAGE, and stained with Coomassie blue. (D) Western blot analysis of pull downs of amino and AP segments of cZO-2. Top panel, anti–ZO-2 antibody recognizes only the 3PSG segment of ZO-2. Bottom panel, anti-TEAD antibody gives a positive signal in pull downs of amino and AP segments of cZO-2. (E) Cartoon showing that the inhibition of nPKCδ allows ZO-2-TEAD interaction, which facilitates the nuclear importation of these proteins. At the nucleus nPKCδ also ruptures ZO-2-TEAD association.
Altogether, these results indicate that ZO-2 and TEAD associate and that the inhibition of nPKCδ allows ZO-2 and TEAD to interact in the cytoplasm and enter together into the nucleus (Figure 6E).
Activation by nPKCε of NES in ZO-2 induces the nuclear exportation of ZO-2 and TEAD
Previously, we demonstrated the activation of ZO-2 NES-1 through the phosphorylation of Ser-369 by PKCε (Chamorro et al., 2009). Hence, we next analyzed in islets of cells what happened with TEAD after treatment with the nPKCε permeable activating peptide (εV1-7). In the islets of MDCK cells treated with peptide εV1-7, ZO-2 is no longer present at the nucleus (Figure 7A, b and e) in comparison to control cultures (Figure 7A, a and d), as had been previously reported (Quiros et al., 2013). Similarly, the expression of TEAD at the nucleus is barely detectable after treatment with the nPKCε permeable activating peptide εV1-7 (Figure 7A, h and k) in comparison to the control condition (Figure 7A, g and j). Treatment with εν1-2, a nPKCε permeable inhibitor peptide, concentrated ZO-2 at the nuclei (Figure 7A, c and f) and increased the amount of nuclear TEAD in comparison to control cells (Figure 7A, i and j). The same results were obtained when this experiment was done in intestinal epithelial cells IEC-18 (Figure 7B). Therefore, we conclude that nuclear exportation of ZO-2 triggers the translocation of nuclear TEAD to the cytoplasm, while the nuclear accumulation of ZO-2 facilitates the nuclear concentration of TEAD.
FIGURE 7:

Nuclear exportation of ZO-2 induced by PKCε triggers TEAD translocation from the nucleus to the cytoplasm. (A) Representative images obtained from three independent experiments of the immunofluorescence of ZO-2 (a–f) and TEAD (g–l) in control sparse cultures of parental MDCK cells (first column), treated for 24 h with 2 μM nPKCε permeable activating peptide (εV1-7) that activates the nuclear ZO-2 exportation (second column), or with 2 μM nPKCε permeable inhibitor peptide (εν1-2) that blocks the nuclear exportation of ZO-2 (third column). Nuclei were stained with DAPI. (B) Representative images obtained from three independent experiments of the immunofluorescence of intestinal epithelial cells IEC-18. ZO-2 (a–f) and TEAD (g–l) in control sparse cultures of IEC-18 cells (first column) treated for 24 h with 2 μM nPKCε permeable activating peptide (εV1-7) that activates the nuclear ZO-2 exportation (second column) or with 2 μM nPKCε permeable inhibitor peptide (εν1-2) that blocks the nuclear exportation of ZO-2 (third column). Nuclei were stained with DAPI. (C) Quantification of nuclear TEAD immunofluorescence per cell in arbitrary units (AU) of the experiment described in A. At least 50 nuclei were analyzed per experimental condition. Statistical analysis done with one-way ANOVA followed by Tukey’s multiple comparisons test, *p < 0.05; ****p < 0.0001. (D) Quantification of nuclear ZO-2 immunofluorescence per cell in arbitrary units (AU) of the experiment described in B. At least 50 nuclei were analyzed per experimental condition. Statistical analysis done with one-way ANOVA followed by Tukey’s multiple comparisons test, **p < 0.01; *** p < 0.001; ****p < 0.0001. (E) Quantification of nuclear TEAD immunofluorescence per cell in arbitrary units (AU), of the experiment described in B. At least 50 nuclei were analyzed per experimental condition. Statistical analysis done with one-way ANOVA followed by Tukey’s multiple comparisons test, *p < 0.05; ***p < 0.001.
We previously showed that newly synthesized ZO-2 translocates to the nucleus in sparse cultures. Then as the culture becomes confluent, it exits the nucleus and reaches the TJ region in the plasma membrane (Chamorro et al., 2009). Therefore, we next tested whether blocking the translocation of nuclear ZO-2 to the cytoplasm as the culture becomes confluent altered the nuclear content of TEAD. For this purpose, we transfected sparse parental cells with HA-cZO-2 or a construct where the four NES of cZO-2 are mutated (HA-cZO-2 MutNES) and analyzed 48 h later the expression of TEAD in the nucleus of the transfected cells. Figure 8 shows that the amount of TEAD at the nucleus is not altered when ZO-2 is kept at the nucleus due to the lack of functional NES.
FIGURE 8:

Nuclear accumulation of ZO-2 due to NES inactivation has no effect on nuclear concentration of TEAD. (A) Representative immunofluorescence images derived from three independent experiments of sparse parental MDCK cells 48 h after transfection with HA-cZO-2 or a construct where the four NES of cZO-2 were mutated (HA-cZO-2 MutNES 0,1,2,3). First column, HA staining; second column, TEAD staining. Observe the strong HA staining present in the nucleus of a cell transfected with HA-cZO-2 MutNES 0,1,2,3 in comparison to that of cells transfected with HA-ZO-2. Arrows, nuclei of transfected cells. (B) Quantification of nuclear TEAD immunofluorescence in cells transfected with HA-cZO-2 or HA-cZO-2 MutNES 0,1,2,3. AU, arbitrary units. Thirty-five nuclei of transfected cells were analyzed per experimental condition. Statistical analysis done with Students t test, ns, nonsignificant. Each bar shows the mean ± SEM.
Altogether, our results indicate that the movements of ZO-2 in and out of the nucleus modulate the intracellular traffic of TEAD in a process regulated by PKA and nPKC δ and ε (Figure 9).
FIGURE 9:
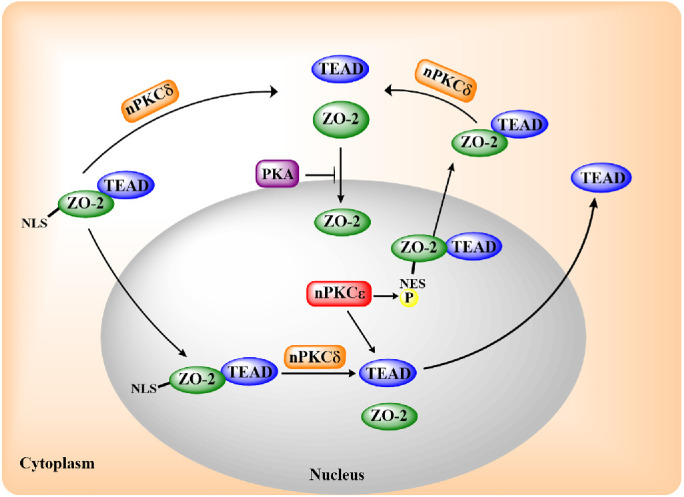
Schematic representation of TEAD and ZO-2 movement in and out of the nucleus, regulated by the NLS and NES of ZO-2 and by the activity of PKA and nPKC δ and ε. In epithelial MDCK cells, nPKCδ blocks ZO-2 and TEAD interaction, inhibiting nuclear importation and exportation of the ZO-2/TEAD complex. PKA blocks ZO-2 nuclear importation, while nPKCε activates a NES of ZO-2 and the exportation of TEAD either alone or associated to ZO-2.
DISCUSSION
In epithelial MDCK cells, the subcellular distribution of ZO-2 relies heavily on the degree of cell–cell contact. Thus, in sparse cultures, ZO-2 is found at the TJs present in the developing monolayer and abundant at the nuclei. In contrast, in confluent cultures with mature TJs, ZO-2 is mainly concentrated at the cell borders (Islas et al., 2002). Here, we have studied whether the nuclear concentration of the transcription factor TEAD is regulated by ZO-2 because the absence of ZO-2 increased the transcriptional activity of artificial and natural promoters driven by TEAD-binding sites, while the overexpression of ZO-2 abolished this activity (Dominguez-Calderon et al., 2016).
In sparse cultures of MDCK cells, we observe that both ZO-2 and TEAD accumulate at the nucleus and detect that the amount of nuclear TEAD is diminished in cells where the expression of ZO-2 has been silenced, as well as in sparse parental cells transfected with a ZO-2 construct lacking NLS, indicating that ZO-2 nuclear importation facilitates the entry of TEAD into the nucleus. This observation is reinforced with our finding that in parental cells, the inhibition of nPKCδ triggers the interaction between ZO-2 and TEAD at the cytoplasm, facilitating the entry and accumulation at the nucleus of both interacting proteins. The site phosphorylated by nPKCδ that blocks the interaction between ZO-2 and TEAD remains undefined and could be in either ZO-2 or TEAD, as both proteins display putative nPKCδ phosphorylation sites (ZO-2, 22 sites; TEAD, two sites, according to GPS 3.0), and experimental evidence reveals that TEAD is phosphorylated by several PKC isozymes (Jiang et al., 2001). Moreover, a previous report noted that cytoplasmic translocation of TEAD could be induced by hyperosmotic stress through a process mediated by p38 (Lin et al., 2017). However, TEAD is a poor substrate for p38 phosphorylation, and the mutation of the four putative p38 phosphorylation sites in TEAD did not alter its stress-induced cytoplasmic translocation, suggesting that other kinases different from p38 are involved (Lin et al., 2017).
Nevertheless, the blockade of the nuclear entry of TEAD and ZO-2 by nPKCδ could be explained by the fact that at the carboxyl basic region of the bpNLS-2 of ZO-2, three serine residues present there (S257, S259, and S261) are putative nPKCδ phosphorylation sites. Previously we demonstrated that their phosphomimetic mutation obstructs the nuclear accumulation of ZO-2, likely due to the neutralization of the positive charges present at the carboxyl-terminal segment of bpNLS-2 (Quiros et al., 2013).
Our results also indicate that in MDCK cells, the exportation of TEAD from the nucleus is triggered by the activation of nPKCε. This process can happen in a ZO-2–independent manner as observed in ZO-2 KD cells and in a ZO-2–dependent way in parental cells, which involves the activation of ZO-2 NES by nPKCε. The latter observation further confirms that the interaction with ZO-2 facilitates the movement of TEAD. Nevertheless, it should be mentioned that TEAD also contains a bipartite NLS and a NES, both conserved in numerous species whose functionality has been tested in Drosophila (Magico and Bell, 2011). Therefore, the interaction of ZO-2 with TEAD might only reinforce the movement of the transcription factor.
The role of ZO-2 as a nuclear translocator of proteins was previously observed. Thus, ZO-2 was found to enhance the nuclear localization of YAP-2 through a process that required the presence of the NLS of ZO-2 (Oka et al., 2010). YAP is a transcriptional activator of promoters regulated by the TEAD transcription factor (Zhao et al., 2008). It binds to the first PDZ domain of ZO-2 through its carboxyl-terminal PDZ-binding motif (Oka et al., 2010). Likewise, the movement of ARVCF into the nucleus requires the interaction of the PDZ-binding motif of this protein with the amino PDZ-containing region of ZO-2 and the presence of the NLS of ZO-2 (Kausalya et al., 2004). ARVCF protein at the nucleus binds to splicing factors and contributes to the regulation of alternating splicing (Rappe et al., 2014).
In summary, our observations indicate that ZO-2 acts as a platform that facilitates the nuclear importation and exportation of TEAD.
MATERIAL AND METHODS
Request a protocol through Bio-protocol.
Cell culture
Parental (control) and ZO-2 KD MDCK II cells were kindly provided by Alan Fanning (University of North Carolina, Chapel Hill, NC) and cultured as previously described (Van Itallie et al., 2009). These cells stably express a mixture of three different shRNAs against ZO-2 in the pSuper vector. Parental cells instead express only the empty vector. The stable clonal ZO-2 KD MDCK cell line here employed (IC5) was obtained based on zeocin resistance. Sparse cultures were plated at a density of 2.5 × 104 cells/cm2, whereas confluent cultures were seeded at a density of 5 × 105 cells/cm2.
IEC-18 epithelial cells derived from the rat small intestine and obtained from the American Type Culture Collection (Cat. CRL-1589; ATCC, Manassas, VA) were grown in DMEM F-12 (Cat. 12500-062; Thermo Fisher Scientific, Waltham, MA) supplemented with 5% fetal bovine serum (FBS) and penicillin–streptomycin 10,000 (U/μg/ml) (Cat. A-01; In Vitro, Mexico).
HEK293T epithelial cells derived from the human embryonic kidney (Cat. CRL-3216; ATCC, Manassas, VA) were grown in a high-glucose DMEM (Cat. 11965-118; Thermo Fisher Scientific, Waltham, MA) supplemented with 5% FBS and penicillin–streptomycin 10,000 (U/μg/ml) (Cat. A-01; In Vitro, Mexico).
Transfections
Transfections were done following the manufacturer’s instructions using Lipofectamine 2000 (Cat. 11668-019; Life Technologies, Carlsbad, CA). Because the Lipofectamine manufacturer indicates that 6 h is the optimal time required for the transfected construct to be expressed as protein, time 0 in Figure 5 corresponds to 6 h after transfection.
ZO-2 KD cells were transfected with a human full-length ZO-2 construct with altered shRNA-binding sites (pTRE-hZO-2), generously provided by Alan Fanning (University of North Carolina, Chapel Hill, NC).
To generate hZO-2 ΔU2 in a construct with CMV10 promoter and a tag of 3 × Flag, we liberated with KpnI and XbaI restriction enzymes the hZO-2 ΔNLS construct inserted into a vector containing CMV2 promoter and a tag of 2 × Flag (generously provided by Marius Sudol, National University of Singapore, Singapore). hZO-2 ΔNLS lacks base pairs 313–873 of hZO-2 that code for the U2 segment (amino acids 105–291). Full-length hZO-2 contained within a vector with CMV10 promoter and a 3 × Flag (Flag-hZO-2) (generously provided by Otmar Huber, Jena University, Germany) was cut with KpnI and XbaI restriction enzymes to eliminate full-length hZO-2 from the vector. The hZO-2 ΔNLS construct was then inserted into this empty vector containing CMV10 promoter and a 3 × Flag. Parental cells were transfected with either Flag-hZO-2 or hZO-2 ΔNLS within a vector with CMV10 promoter and three Flag tags (Flag-hZO-2).
In some experiments, parental and ZO-2 KD MDCK cells were transfected with mouse nPKCδ (plasmid 16386; Addgene, Watertown, MA) or Flag-hZO-2 plus nPKCδ.
HEK293T cells were transfected with amino (398–962 nucleotides [nt]), 3PSG (1595–3019 nt), or AP (3029–3923 nt) segments of cZO-2, in the pcDNA4/HisMax vector that had been previously reported (Betanzos et al., 2004).
Immunofluorescence
Immunofluorescence experiments on MDCK cells were done as previously described (Quiros et al., 2013) with the only modification that cells were fixed with 10% (vol/vol) formaldehyde in phosphate-buffered saline. As first antibodies, we employed a rabbit monoclonal against TEAD (Cat. 12292; dilution 1:200; Cell Signaling, Danvers, MA); mouse monoclonal antibodies against Flag (Cat. F3165; dilution 1:1500; Sigma Aldrich, Darmstadt, Germany); and rabbit polyclonal against ZO-2 (Cat. 71-1400; dilution 1:200; Invitrogen, Carlsbad, CA). As secondary antibodies, we employed donkey antibodies against rabbit immunoglobulin G (IgG) coupled to Alexa Fluor 488 (Cat. A21206; dilution 1:1000; Invitrogen, Carlsbad, CA) or Alexa Fluor 594 (Cat. A21207; dilution 1:1000; Invitrogen, Carlsbad, CA) and against mouse IgG coupled to Alexa Fluor 488 (Cat. A11001; Life Technologies, Eugene, OR).
Quantification of TEAD immunofluorescence intensity at the nucleus was done using ImageJ. Nuclei images stained with DAPI (4′,6-diamidino-2-phenylindole) were used to define the region to be subsequently quantitated for TEAD immunofluorescence.
Western blot
Western blot was done following a standard procedure previously described (Quiros et al., 2013), but using a different RIPA buffer (25 mM Tris-HCl, pH 7.6, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS) and a commercial sample buffer (Cat. NP0008; Invitrogen, Carlsbad, CA). The following primary antibodies were employed: a rabbit monoclonal against TEAD (Cat. 12292; dilution 1:2000; Cell Signaling, Danvers, MA); mouse monoclonals anti-GAPDH (Cat. Sc-32233; dilution 1:15,000; Santa Cruz Biotechnology, Santa Cruz, CA) and lamin B1 (Cat. 33-2000; dilution 1:2000; Invitrogen, Carlsbad, CA); and rabbit polyclonal against ZO-2 (Cat. 71-1400; dilution 1:2000; Invitrogen, Carlsbad, CA). As secondary antibodies, we employed goat polyclonals anti-rabbit IgG coupled to peroxidase (Cat. A9169; dilution 1:20,000; Sigma Aldrich, St. Louis, MO) or anti-mouse IgG coupled to peroxidase (Cat. 62-6420; dilution 1:10,000; Invitrogen, Carlsbad, CA). The procedure was followed by Immobilon chemiluminescence detection (Cat. WVKLS 0500; Merck KGaA, Darmstadt, Germany).
For the mobility shift detection assay of phosphorylated TEAD, phosphate affinity SDS–PAGE was done with the acrylamide-pendant Phos-tag ligand (Cat. AAL-107; Wako Pure Chemical Industries, Richmond, VA) following the manufacturer’s instructions. In Phos-tag SDS–PAGE, molecular weight estimations using molecular weight markers are not possible. Because the manufacturer recommends using a dephosphorylated sample of the target protein as a marker, in Figure 1D, right panel, we have placed the indication of 50 kDa in the lower band of TEAD that is still present after alkaline phosphatase treatment.
Cell fractionation assay
Cytoplasmic and nuclear fractions of parental and ZO-2 KD MDCK cells transfected or not with hZO-2 were obtained as previously reported by others (Shaiken and Opekun, 2014). Briefly, cells were lysed in isotonic buffer A (40 mM HEPES, pH 7.4, 120 mM KCl, 2 mM EGTA (ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid), 0.4% glycerol, 10 mM β-glycerophosphate, and 0.2% NP-40) while rotating for 30 min at 4°C. Nuclei were pelleted by centrifugation at 1000 × g for 5 min. The supernatant was centrifuged further at 10,000 × g for 10 min to obtain the cytosolic fraction. The pellet of nuclei was sequentially and gently washed with 0.1% NP-40 and non–detergent containing buffer A and centrifuged at 1000 × g for 5 min. The supernatant was discarded, and the nuclear pellet was resuspended in RIPA buffer.
Alkaline phosphatase treatment
Nuclear pellets obtained from confluent MDCK monolayers were resuspended in NEB buffer 3 (100 mM NaCl, 50 mM Tris-HCl, 10 mM MgCl2, and 1 mM dithiothreitol, pH 7.9; Cat. B7003S; New England Biolabs, Ipswich, MA). The nuclei were then sonicated and incubated for 60 min at 37°C with 20 U of calf intestine alkaline phosphatase (Cat. M0290S; New England Biolabs, Ipswich, MA). Samples were then processed for Western blot.
Proximity ligation assay
The Duolink in situ proximity ligation assay (PLA) (Cat. DUO92101; Sigma Aldrich, Darmstadt, Germany) was done according to the manufacturer’s instructions. Using a rabbit antibody against TEAD (Cat. 12292; dilution 1:200; Cell Signaling, Danvers, MA) and a mouse antibody anti-Flag (Cat. F3165; dilution 1:1500; Sigma Aldrich, Darmstadt, Germany). After the Duolink reaction was completed, the preparations were washed three times with Duolink B-buffer. The cells were stained with a donkey anti-mouse antibody coupled to Alexa Fluor 488 to detect cells transfected with Flag-hZO-2.
Quantitative analysis of PLAs was done using BlobFinder (Allalou and Wahlby, 2009) developed by the Centre for Image Analysis at Uppsala University.
Immunoprecipitation
Immunoprecipitation of ZO-2 was done using 1 μl of ZO-2 antibody (Cat. 71-1400; Invitrogen, Carlsbad, CA) per 300 μg of protein in the lysate of parental MDCK cells and following a protocol previously described (Raya-Sandino et al., 2017). The radioimmunoprecipitation assay buffer employed contained 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP-40 (vol/vol), and the protease inhibitor cocktail Complete (Cat. 11697498001; Roche Diagnostics, Mannheim, Germany). The blot for TEAD in the ZO-2 immunoprecipitate was done using the Tidy blot reagent (Cat. STAR209; BioRad, Hercules, CA) following the manufacturer’s instructions.
Protein purification
HEK293T cells were transfected with previously reported constructs containing the amino (398–962 nt), 3PSG (1595–3019 nt), or AP (3029–3923 nt) segments of cZO-2 in the pcDNA4/HisMax vector (Betanzos et al., 2004). After 24 h the cells were lysed and the extracts were subjected to affinity chromatography with Complete His-Tag Purification Columns (Cat. COHISC-RO; Sigma Aldrich, St. Louis, MO), following the manufacturer’s instructions. The purified fractions were run in a SDS–PAGE, stained with Coomassie blue, and blotted with antibodies against ZO-2 and TEAD.
Reporter gene assays
Parental and ZO-2 KD MDCK cells were plated in 96-well plates at a density of 2.5 × 104 cells/cm2. After 24 h, cells were transfected using Lipofectamine 3000 (Cat. L3000015; Thermo Fisher, Waltham, MA) with 100 ng of a construct of the human CTGF promoter region that contains three putative TEAD-binding sites cloned into the basic luciferase reporter vector pGL3-6xOSE-Luc (Zhao et al., 2008) generously provided by Kun-Liang Guan from the Moors Cancer Center, University of California at San Diego, or with 75 ng of the pGL3-Control (Cat. E1741; Promega, Madison, WI) plasmid with constitutive activity. After 6 h, the transfection medium was removed and replaced with a fresh culture medium or medium with 2 μM nPKCε permeable inhibitor peptide εv1-2. After 20 h of treatment, the cells were harvested and suspended in reporter lysis buffer (Cat. E3971; Promega, Madison, WI). Protein extraction was done by a heat shock lysis cycle of 5 min at 70°C followed by 1 min at 37°C and 3 min in agitation. Finally, luciferase activity was determined using the luciferase assay system (Cat. E1500; Promega, Madison, WI) and the Infinite M200 Pro series (Tecan, Männedorf, Switzerland) equipment. Luciferase activity values were normalized to protein content and expressed relative to those recorded in parental cells or in parental and ZO-2 KD cells without the nPKCε inhibitor. In all cases, transfection efficiency was normalized using pGL3-control as a reference.
Drugs
The nPKCε permeable inhibitor peptide εv1-2 and the nPKCε permeable activating peptide εv1-7 were kindly provided by Daria Mochly-Rosen (Stanford University, Stanford, CA) and dissolved in water as a stock at a concentration of 1 mM and used at a final concentration of 2 μM.
Röttlerin, an nPKCδ inhibitor (Cat. 557370; Calbiochem, Darmstadt, Germany), was prepared as a 12 μM stock in dimethyl sulfoxide (DMSO) and used at a final concentration of 3 μM.
Supplementary Material
Acknowledgments
This work was supported by grants to L.G.-M. from the Miguel Alemán Valdés Foundation 2018, SEP-Cinvestav FIDSC2018/33, and National Council of Science and Technology of Mexico (Conacyt) FORDECYT-PRONACES-140644/2020. H.G.-G. and L.G.-G. were recipients of doctoral fellowships from the Conacyt (282075 and 340209), and H.G.-G. was also the recipient of a scholarship from the Mexiquense Council of Science and Technology (2018AD0009-11).
Abbreviations used:
- AP
alkaline phosphatase
- AP-1
activator protein 1
- AU
arbitrary units
- bp
bipartite
- CAM-kinase II
Ca 2+/calmodulin kinase II
- Co-IP
coimmunoprecipitation
- CTGF
connective tissue growth factor
- cyt
cytoplasmic
- DAPI
4’,6-diamidino-2-phenylindole
- DMSO
dimethyl sulfoxide
- EGTA
ethylene glycol-bis(2-aminoethylether)- N, N, N’, N’-tetraacetic acid
- ICM
inner cell mass
- IP
immunoprecipitation
- KO
knockout
- LATS1/2
large tumor suppressor kinase 1/2
- LEF
lymphoid enhancer family
- MAGUK
membrane-associated guanylate kinase
- MAPKAP2
MAP kinase-activated protein 2
- MDCK
Madin-Darby canine kidney
- mESC
mouse embryonic stem cells
- mTSC
mouse trophoblast cells
- NES
nuclear exportation signal
- NLS
nuclear localization signal
- nPKC
novel protein kinase C
- ns
non significant
- nt
nucleotide
- nuc
nuclear
- PIS
preimmune serum
- PKA
protein kinase A
- PLA
proximity ligation assay
- PRAK/MK5
MAP kinase-activated 5
- shRNA
short hairpin RNA
- TAZ
transcriptional activator with PDZ-binding domain
- TCF
T cell factor
- TCF4
transcription factor 4
- TEAD
transcriptional enhanced associated domain
- TJ
tight junction
- Transf
transfected
- VGLL
vestigial like
- WNK4
with no lysine kinase 4
- WT
wild type
- YAP
Yes-associated protein
- ZO-2
zonula occludens.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E20-07-0470) on May 19, 2021.
REFERENCES
- Allalou A, Wahlby C (2009). BlobFinder, a tool for fluorescence microscopy image cytometry. Comput Methods Programs Biomed 94, 58–65. [DOI] [PubMed] [Google Scholar]
- Amaya E, Alarcon L, Martin-Tapia D, Cuellar-Perez F, Cano-Cortina M, Ortega-Olvera JM, Cisneros B, Rodriguez AJ, Gamba G, Gonzalez-Mariscal L (2019). Activation of the Ca(2+) sensing receptor and the PKC/WNK4 downstream signaling cascade induces incorporation of ZO-2 to tight junctions and its separation from 14-3-3. Mol Biol Cell 15, 2377–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belandia B, Parker MG (2000). Functional interaction between the p160 coactivator proteins and the transcriptional enhancer factor family of transcription factors. J Biol Chem 275, 30801–30805. [DOI] [PubMed] [Google Scholar]
- Betanzos A, Huerta M, Lopez-Bayghen E, Azuara E, Amerena J, Gonzalez-Mariscal L (2004). The tight junction protein ZO-2 associates with Jun, Fos and C/EBP transcription factors in epithelial cells. Exp Cell Res 292, 51–66. [DOI] [PubMed] [Google Scholar]
- Chamorro D, Alarcon L, Ponce A, Tapia R, Gonzalez-Aguilar H, Robles-Flores M, Mejia-Castillo T, Segovia J, Bandala Y, Juaristi E, et al. (2009). Phosphorylation of zona occludens-2 by protein kinase C epsilon regulates its nuclear exportation. Mol Biol Cell 20, 4120–4129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan P, Han X, Zheng B, DeRan M, Yu J, Jarugumilli GK, Deng H, Pan D, Luo X, Wu X (2016). Autopalmitoylation of TEAD proteins regulates transcriptional output of the Hippo pathway. Nat Chem Biol 12, 282–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Friedrich GA, Soriano P (1994). Transcriptional enhancer factor 1 disruption by a retroviral gene trap leads to heart defects and embryonic lethality in mice. Genes Dev 8, 2293–2301. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P (2000). Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 351, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Calderon A, Avila-Flores A, Ponce A, Lopez-Bayghen E, Calderon-Salinas JV, Luis Reyes J, Chavez-Munguia B, Segovia J, Angulo C, Ramirez L, et al. (2016). ZO-2 silencing induces renal hypertrophy through a cell cycle mechanism and the activation of YAP and the mTOR pathway. Mol Biol Cell 27, 1581–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fossdal R, Jonasson F, Kristjansdottir GT, Kong A, Stefansson H, Gosh S, Gulcher JR, Stefansson K (2004). A novel TEAD1 mutation is the causative allele in Sveinsson’s chorioretinal atrophy (helicoid peripapillary chorioretinal degeneration). Hum Mol Genet 13, 975–981. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Mariscal L, Gallego-Gutierrez H, Gonzalez-Gonzalez L, Hernandez-Guzman C (2019). ZO-2 is a master regulator of gene expression, cell proliferation, cytoarchitecture, cell size. Int J Mol Sci 20, 4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Mariscal L, Miranda J, Raya-Sandino A, Dominguez-Calderon A, Cuellar-Perez F (2017). ZO-2, a tight junction protein involved in gene expression, proliferation, apoptosis, and cell size regulation. Ann NY Acad Sci 1397, 35–53. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Mariscal L, Ponce A, Alarcon L, Jaramillo BE (2006). The tight junction protein ZO-2 has several functional nuclear export signals. Exp Cell Res 312, 3323–3335. [DOI] [PubMed] [Google Scholar]
- Gschwendt M, Muller HJ, Kielbassa K, Zang R, Kittstein W, Rincke G, Marks F (1994). Rottlerin, a novel protein kinase inhibitor. Biochem Biophys Res Commun 199, 93–98. [DOI] [PubMed] [Google Scholar]
- Gupta MP, Kogut P, Gupta M (2000). Protein kinase-A dependent phosphorylation of transcription enhancer factor-1 represses its DNA-binding activity but enhances its gene activation ability. Nucleic Acids Res 28, 3168–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Home P, Saha B, Ray S, Dutta D, Gunewardena S, Yoo B, Pal A, Vivian JL, Larson M, Petroff M, et al. (2012). Altered subcellular localization of transcription factor TEAD4 regulates first mammalian cell lineage commitment. Proc Natl Acad Sci USA 109, 7362–7367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islas S, Vega J, Ponce L, Gonzalez-Mariscal L (2002). Nuclear localization of the tight junction protein ZO-2 in epithelial cells. Exp Cell Res 274, 138–148. [DOI] [PubMed] [Google Scholar]
- Jaramillo BE, Ponce A, Moreno J, Betanzos A, Huerta M, Lopez-Bayghen E, Gonzalez-Mariscal L (2004). Characterization of the tight junction protein ZO-2 localized at the nucleus of epithelial cells. Exp Cell Res 297, 247–258. [DOI] [PubMed] [Google Scholar]
- Jiang SW, Dong M, Trujillo MA, Miller LJ, Eberhardt NL (2001). DNA binding of TEA/ATTS domain factors is regulated by protein kinase C phosphorylation in human choriocarcinoma cells. J Biol Chem 276, 23464–23470. [DOI] [PubMed] [Google Scholar]
- Jiao S, Li C, Hao Q, Miao H, Zhang L, Li L, Zhou Z (2017). VGLL4 targets a TCF4-TEAD4 complex to coregulate Wnt and Hippo signalling in colorectal cancer. Nat Commun 8, 14058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao S, Wang H, Shi Z, Dong A, Zhang W, Song X, He F, Wang Y, Zhang Z, Wang W, et al. (2014). A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell 25, 166–180. [DOI] [PubMed] [Google Scholar]
- Kaneko KJ, Kohn MJ, Liu C, DePamphilis ML (2007). Transcription factor TEAD2 is involved in neural tube closure. Genesis 45, 577–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kausalya PJ, Phua DC, Hunziker W (2004). Association of ARVCF with zonula occludens (ZO)-1 and ZO-2: binding to PDZ-domain proteins and cell-cell adhesion regulate plasma membrane and nuclear localization of ARVCF. Mol Biol Cell 15, 5503–5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KC, Moroishi T, Meng Z, Jeong HS, Plouffe SW, Sekido Y, Han J, Park HW, Guan KL (2017). Regulation of Hippo pathway transcription factor TEAD by p38 MAPK-induced cytoplasmic translocation. Nat Cell Biol 19, 996–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Li H, Rajurkar M, Li Q, Cotton JL, Ou J, Zhu LJ, Goel HL, Mercurio AM, Park JS, et al. (2016). Tead and AP1 coordinate transcription and motility. Cell Rep 14, 1169–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Bayghen E, Jaramillo BE, Huerta M, Betanzos A, Gonzalez-Marical L (2006). TJ proteins that make round trips to the nucleus. In: Tight Junctions, ed. Gonzalez-Mariscal L., Georgetown, TX: Landes Bioscience and Springer Science+Business Media, 76–100. [Google Scholar]
- Magico AC, Bell JB (2011). Identification of a classical bipartite nuclear localization signal in the Drosophila TEA/ATTS protein scalloped. PLoS One 6, e21431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka T, Remue E, Meerschaert K, Vanloo B, Boucherie C, Gfeller D, Bader GD, Sidhu SS, Vandekerckhove J, Gettemans J, et al. (2010). Functional complexes between YAP2 and ZO-2 are PDZ domain-dependent, and regulate YAP2 nuclear localization and signalling. Biochem J 432, 461–472. [DOI] [PubMed] [Google Scholar]
- Pobbati AV, Chan SW, Lee I, Song H, Hong W (2012). Structural and functional similarity between the Vgll1-TEAD and the YAP-TEAD complexes. Structure 20, 1135–1140. [DOI] [PubMed] [Google Scholar]
- Quiros M, Alarcon L, Ponce A, Giannakouros T, Gonzalez-Mariscal L (2013). The intracellular fate of zonula occludens 2 is regulated by the phosphorylation of SR repeats and the phosphorylation/O-GlcNAcylation of S257. Mol Biol Cell 24, 2528–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappe U, Schlechter T, Aschoff M, Hotz-Wagenblatt A, Hofmann I (2014). Nuclear ARVCF protein binds splicing factors and contributes to the regulation of alternative splicing. J Biol Chem 289, 12421–12434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raya-Sandino A, Castillo-Kauil A, Dominguez-Calderon A, Alarcon L, Flores-Benitez D, Cuellar-Perez F, Lopez-Bayghen B, Chavez-Munguia B, Vazquez-Prado J, Gonzalez-Mariscal L (2017). Zonula occludens-2 regulates Rho proteins activity and the development of epithelial cytoarchitecture and barrier function. Biochim Biophys Acta 1864, 1714–1733. [DOI] [PubMed] [Google Scholar]
- Sawada A, Kiyonari H, Ukita K, Nishioka N, Imuta Y, Sasaki H (2008). Redundant roles of Tead1 and Tead2 in notochord development and the regulation of cell proliferation and survival. Mol Cell Biol 28, 3177–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaiken TE, Opekun AR (2014). Dissecting the cell to nucleus, perinucleus and cytosol. Sci Rep 4, 4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia R, Huerta M, Islas S, Avila-Flores A, Lopez-Bayghen E, Weiske J, Huber O, Gonzalez-Mariscal L (2009). Zona occludens-2 inhibits cyclin D1 expression and cell proliferation and exhibits changes in localization along the cell cycle. Mol Biol Cell 20, 1102–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traweger A, Fuchs R, Krizbai IA, Weiger TM, Bauer HC, Bauer H (2003). The tight junction protein ZO-2 localizes to the nucleus and interacts with the heterogeneous nuclear ribonucleoprotein scaffold attachment factor-B. J Biol Chem 278, 2692–2700. [DOI] [PubMed] [Google Scholar]
- Umeda K, Ikenouchi J, Katahira-Tayama S, Furuse K, Sasaki H, Nakayama M, Matsui T, Tsukita S, Furuse M, Tsukita S (2006). ZO-1 and ZO-2 independently determine where claudins are polymerized in tight-junction strand formation. Cell 126, 741–754. [DOI] [PubMed] [Google Scholar]
- Van Itallie CM, Fanning AS, Bridges A, Anderson JM (2009). ZO-1 stabilizes the tight junction solute barrier through coupling to the perijunctional cytoskeleton. Mol Biol Cell 20, 3930–3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel F, Mittag S, Cano-Cortina M, Wagner T, Kramer OH, Niedenthal R, Gonzalez-Mariscal L, Huber O (2017). SUMOylation regulates the intracellular fate of ZO-2. Cell Mol Life Sci 74, 373–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi R, Kohn MJ, Karavanova I, Kaneko KJ, Vullhorst D, DePamphilis ML, Buonanno A (2007). Transcription factor TEAD4 specifies the trophectoderm lineage at the beginning of mammalian development. Development 134, 3827–3836. [DOI] [PubMed] [Google Scholar]
- Young LH, Balin BJ, Weis MT (2005). Go 6983: a fast acting protein kinase C inhibitor that attenuates myocardial ischemia/reperfusion injury. Cardiovasc Drug Rev 23, 255–272. [DOI] [PubMed] [Google Scholar]
- Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, Rosato A, Bicciato S, Cordenonsi M, Piccolo S (2015). Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol 17, 1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang CY, Chinnaiyan AM, et al. (2008). TEAD mediates YAP-dependent gene induction and growth control. Genes Dev 22, 1962–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Huang T, Cheng AS, Yu J, Kang W, To KF (2016). The TEAD family and its oncogenic role in promoting tumorigenesis. Int J Mol Sci 17, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.