

ABSTRACT
Fecal microbial community could not fully represent the intestinal microbial community. However, most studies analyzing diarrhea-dominant irritable bowel syndrome (IBS-D) were mainly based on fecal samples. We aimed to characterize the IBS-D microbial community patterns using samples at multiple intestinal sites. This study recruited 74 IBS-D patients and 20 healthy controls (HC). 22.34%, 8.51%, 14.89%, and 54.26% of them contributed to one, two, three, and four sites: duodenal mucosa (DM), duodenal lumen (DL), rectal mucosa (RM), and rectal lumen (RL) of intestinal samples, respectively. Then 16S rRNA gene analysis was performed on these 283 samples. The result showed that IBS-D microbial communities have specific patterns at each intestinal site differing from that of HC. Across hosts and sites, Bacillus, Burkholderia, and Faecalibacterium were the representative genera in duodenum of IBS-D, duodenum of HC, and rectum of HC, respectively. Samples from mucosa and lumen in rectum were highly distinguishable, regardless of IBS-D and HC. Additionally, IBS-D patients have lower microbial co-abundance network connectivity. Moreover, RM site-specific biomarker: Bacteroides used alone or together with Prevotella and Oscillospira in RM showed outstanding performance in IBS-D diagnosis. Furthermore, Bacteroides and Prevotella in RM were strongly related to the severity of abdominal pain, abdominal discomfort, and bloating in IBS-D patients. In summary, this study also confirmed fecal microbial community could not fully characterize intestinal microbial communities. Among these site-specific microbial communities, RM microbial community would be more applicable in the diagnosis of IBS-D.
IMPORTANCE Microbial community varied from one site to another along the gastrointestinal tract, but current studies about intestinal microbial community in IBS-D were mainly based on fecal samples. Based on 283 intestinal samples collected from DM, DL, RM, and RL of HC and IBS-D, we found different intestinal sites had their site-specific microbial patterns in IBS-D. Notably, RM site-specific microbes Bacteroides, Prevotella, and Oscillospira could be used to discriminate IBS-D from HC accurately. Our findings could help clinicians realize the great potential of the intestinal microbial community in RM for better diagnosis of IBS-D patients.
KEYWORDS: IBS-D, multiple intestinal sites, site-specific microbial patterns, microbial biomarkers
INTRODUCTION
Irritable bowel syndrome (IBS) is a functional gastrointestinal disorder (1), with recurrent abdominal pain or discomfort, stool irregularities, and bloating (2), which affected approximately 10% of the population worldwide (3). People with IBS suffer from low quality of life and considerable medication cost, imposing a great burden on society (3). Though the pathophysiology of IBS remains unclear, it has been found that intestinal microbial community dysbiosis contributes to the underlying mechanism of IBS (4).
In recent years, a growing number of researches supported that IBS patients showed altered gut microbial profiles (5), especially in diarrhea-predominant IBS (IBS-D).(6) The intestinal microbial community of these patients would result in the pathogenesis of IBS (7). To be specific, it has already been found that IBS patients exhibited an increased abundance of Enterobacteriaceae (8), Lactobacillaceae (9), and Bacteroides (10), and reduced abundance of Faecalibacterium (10) and Bifidobacterium (11) compared with healthy individuals.
Current studies on microbial communities of healthy individuals have revealed that the physiological function of different regions in the gastrointestinal (GI) tract would lead to distinct microbial profiles in different sites in GI tract (12–14). Vasapolli et al. identified distinct microbial communities among saliva, upper GI tract, lower GI tract, and fecal samples based on 21 healthy adults (13). Yuan et al. have revealed a location-specific microbial community along the intestinal tract based on mucosal samples collected from 10 adult purpose-bred female olive baboons (14). However, current knowledge about intestinal microbial community in IBS-D was principally based on analysis of fecal microbial community (15, 16). Few studies have described microbial communities combined with different intestinal sites, largely due to the difficulty of collecting samples from these sites, especially for healthy controls (HC).
Currently, IBS can be diagnosed using clinical symptoms according to Rome III (17) or the latest version (Rome IV) criteria (18), which was dependent on clinicians’ knowledge about IBS, so that this was a challenge for the clinicians lacking clinical experience with IBS. Moreover, exhaustive examinations need to be performed to exclude organic diseases before determining the diagnosis of IBS, which leads to substantial medical spending from patients. Additionally, the accurate diagnosis of IBS was challenging with numerous accompanying symptoms and manifestations (19). Furthermore, implementation of intestinal biomarkers in clinical practice is critical for accurate diagnosis of IBS. It was reported that intestinal microbial biomarkers have great potential in identifying inflammatory bowel disease (20), colorectal cancer (21), and autoimmune hepatitis (22). Combining with bioinformatic analysis, we could understand the role of intestinal microbes in IBS-D better, which was beneficial for the diagnosis of IBS-D.
To achieve this, in this study, we collected mucosa and lumen-associated microbial community samples in the duodenum and rectum of IBS-D patients and HC. We determined microbial biomarkers at these sites and produced an efficient classifier to identify IBS-D patients from HC.
RESULTS
Overview of sequencing data and clinical symptoms.
The study collected 283 samples from four representative intestinal sites (duodenal mucosa [DM], duodenal lumen [DL], rectal mucosa [RM], and rectal lumen [RL]) of 20 HC and 74 IBS-D patients, resulting in 3,943,124 reads in total and approximately 13,933 reads per sample on average. According to the rarefaction curves (Fig. S1), all curves reached saturation at around 10,000 sequences per sample, suggesting that the sequencing depth was sufficient enough for subsequent analysis. Based on the statistical analysis of clinical symptoms (Table S1), we found that age, gender, and BMI have no significant difference between HC and IBS-D patients. However, IBS-D patients were identified with increased stool consistency, frequency of defecation, and HAD anxiety and depression score compared with HC (P < 0.001, Table S1).
Microbial profile in multi-sites of the intestinal tract.
IBS-D microbial community was different from that of HC at multiple intestinal sites. We found that alpha diversity of IBS-D microbial community mainly varied across duodenum and rectum (P < 0.1), rather than across mucosa and lumen (Fig. 1A). On the contrary, no obvious difference was observed based on alpha diversity of microbial communities across these sites for HC (Fig. 1A). Moreover, IBS-D microbial community was more diverse among all collected intestinal sites compared with HC based on Bray Curtis distance, weighted and unweighted Unifrac distance (P < 0.001, Fig. 1B and C and Fig. S2A and B). In overall microbial composition, both IBS-D and HC microbial communities varied from duodenum to rectum, rather than between mucosa and lumen (Fig. 1 and 2, Fig. S3), indicating the disorder of duodenal and rectal microbial community in IBS-D patients. While both PCoA analysis and Procrustes analysis illustrated the mucosa and lumen-associated microbial communities in the duodenum were more distinguishable than those in rectum, regardless of IBS-D and HC (Fig. 2E to L), which again confirmed the fecal microbial community could not be representative of mucosal microbial community (13). These results also warranted the importance of recruiting samples from multiple intestinal sites for analyzing the IBS-D microbial community. Notably, IBS-D microbial community cannot be clearly differentiated from that of HC in PCoA analysis (Fig. 2M to P, Fig. S2C and D and Fig. S4), regardless of using all samples or using site-specific samples, emphasizing the difficulty of identifying IBS-D patients using simple clustering methods.
FIG 1.

Microbial diversity of duodenum and rectum in HC and IBS-D patients. (A) Comparison of alpha diversity (Shannon index) of microbial communities across intestinal sites in HC and IBS-D patients. (B) Comparison of beta diversity across intestinal sites in HC and IBS-D patients using Bray Curtis distance as the distance measurement. (C) Comparison of beta-diversity of microbial communities across intestinal sites in HC and IBS-D patients based on nonparametric multivariate analysis of variance (NPMANOVA) using Bray Curtis distance as the distance measurement. Kruskal-Wallis was used to detect the global difference, while the Wilcoxon test was used to detect variation across intestinal sites in HC and IBS-D patients by pairwise comparisons based on the microbial composition at genus level. DL_HC, duodenal luminal samples collected from HC; DM_HC, duodenal mucosal samples collected from HC; RL_HC, rectal luminal samples collected from HC; RM_HC, rectal mucosal samples collected from HC; DL_IBS-D, duodenal luminal samples collected from IBS-D patients; DM_IBS-D, duodenal mucosal samples collected from IBS-D patients; RL_IBS-D, rectal luminal samples collected from IBS-D patients; RM_IBS-D, rectal mucosal samples collected from IBS-D patients. *, P < 0.1; ***, P < 0.01; ****, P < 0.001.
FIG 2.

Comparison of microbial communities across intestinal sites in HC and IBS-D. Comparison of microbial communities between duodenum and rectum in all (A) HC and (B) IBS-D samples using PCoA analysis based on Jaccard coefficient as the distance measurement. Comparison of microbial communities between mucosa and lumen in all (C) HC and (D) IBS-D samples. Comparison of microbial communities between DM and DL in (E) HC and (F) IBS-D samples. Comparison of microbial communities between RM and RL in (G) HC samples and (H) IBS-D samples. Procrustes analysis for assessing the similarity of microbial community between DM and DL in (I) HC samples and (J) IBS-D samples, and between RM and RL in (K) HC samples and (L) IBS-D samples, respectively. PCoA analysis for comparison of microbial communities between HC and IBS-D samples within (M) duodenum, (N) rectum, (O) mucosa and (P) lumen. For Procrustes analysis, we utilized the Monte Carlo method with 999 permutations to measure the similarity of the mucosal and luminal microbial communities. The lower P value indicated more similarity between two compared microbial communities in Procrustes analysis. All results were based on microbial compositions at genus level. D, all duodenal samples; R, all rectal samples; M, all mucosal samples; L, all luminal samples; DM, duodenal mucosal samples; DL, duodenal luminal samples; RM, rectal mucosal sample; RL, rectal luminal samples; D_HC, duodenal samples collected from HC; D_IBS-D, duodenal samples collected from IBS-D patients; R_HC, rectal samples collected from HC; R_IBS-D, rectal samples collected from IBS-D patients.
To uncover the site-specific microbial community patterns, the microbial composition of IBS-D and HC samples collected from DM, DL, RM, and RL was illustrated at phylum level (Fig. S5) and genus level (Fig. S6). Microbial communities were dominated by the bacterial phyla Firmicutes (average relative abundance 39.64%), Proteobacteria (average relative abundance 24.13%), and Bacteroidetes (average relative abundance 21.95%), while their relative abundances in each site were different (Fig. 3A), especially between duodenum and rectum (Fig. S5 and Fig. S7).
FIG 3.
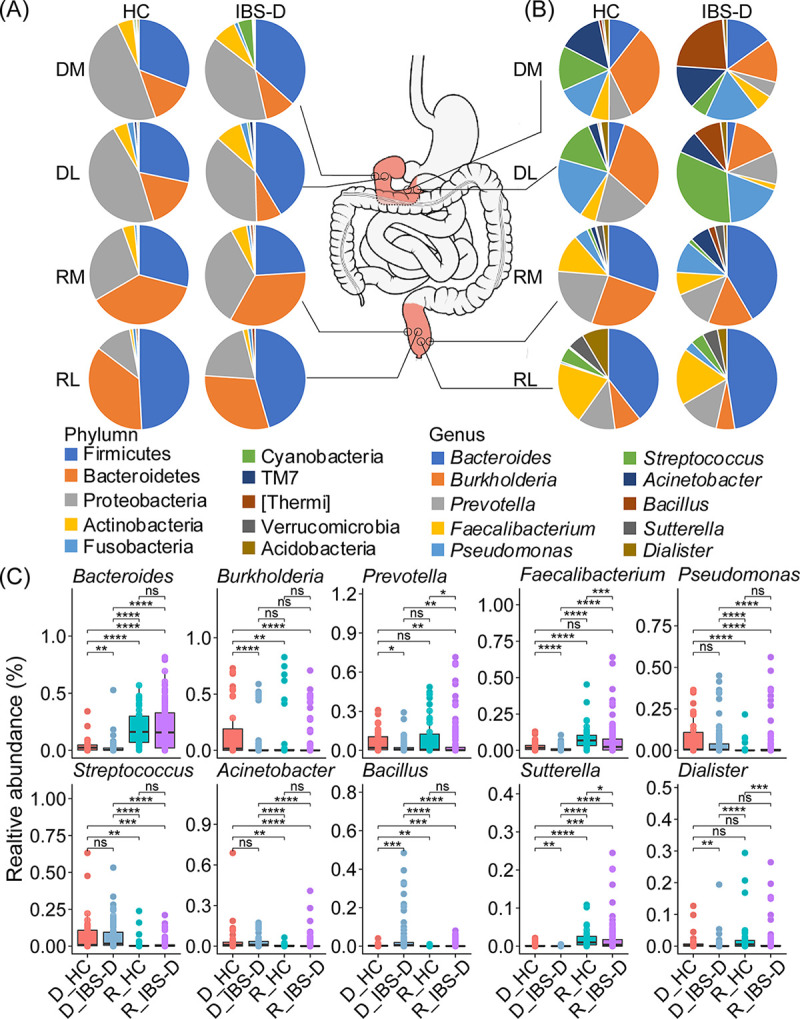
Comparison of microbial composition and enrichment patterns between HC and IBS-D patients in multiple intestinal sites. Composition of the top 10 microbes at (A) phylum level and (B) genus level across intestinal sites and hosts. (C) Comparison of relative abundance of the top 10 genera between duodenum and rectum of HC and IBS-D patients. Kruskal-Wallis was used to detect the global difference, while Wilcoxon test was used to detect the variation across intestinal sites in HC and IBS-D by pairwise comparisons. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant. HC, healthy controls; IBS-D, diarrhea-predominant irritable bowel syndrome. DL, duodenal lumen; DM, duodenal mucosa; RL, rectal lumen; RM, rectal mucosa. D_HC, duodenal samples collected from HC; D_IBS-D, duodenal samples collected from IBS-D patients; R_HC, rectal samples collected from HC; R_IBS-D, rectal samples collected from IBS-D patients.
At genus level, microbial compositions of duodenum were different from those of rectum in both HC and IBS-D samples (Fig. 3B). For duodenum, we detected Pseudomonas, Streptococcus, and Acinetobacter were significantly enriched in this site, while Burkholderia and Bacillus were significantly enriched in HC samples and IBS-D samples, respectively (P < 0.05, Fig. 3C and Table S2). For rectum, Faecalibacterium was significantly enriched in HC samples (P < 0.1, Fig. 3C). In IBS-D patients, Bacteroides, a genus belonging to phyla Bacteroidetes, was found to be significantly abundant in rectum (Fig. 3C and Table S2), while it was deficient in DL.
IBS-D patients have lower connectivity of microbial co-abundance network.
Decreased microbial co-abundance network connectivity was observed in IBS-D patients compared with HC at genus level (Fig. S8). Most correlations were positive 51.28% for HC microbial community (Fig. 4A) and 53.01% for IBS-D microbial community (Fig. 4B) within the cluster of duodenum-enriched genera and the cluster of rectum-enriched genera. However, several correlations were negative between the clusters dominated by duodenum-enriched genera and rectum-enriched genera. From these networks, we have also observed several genera whose relative abundances and connectivities were different among the four intestinal sites in IBS-D patients. For example, in IBS-D patients, Bacteroides, a genus enriched in rectum, has strong positive correlations (Spearman correlation coefficient > 0.4) with Parabacteroides and Phascolarctobacterium (Fig. 4B), which were potentially associated with intestinal inflammation (23–25). However, in HC samples, such profound correlations among these two genera and Bacteroides cannot be detected (Fig. 4A). Moreover, although most species of Bacteroides are commensals, several species of Bacteroides were considered to be pathobionts that can become pathogenic under specific environmental factors, such as antibiotic resistance usage (26, 27). In this network, Prevotella was negatively correlated with several genera in HC network, while it was positively correlated with other genera in the IBS-D patients (Fig. 4). Its colonization in the intestine could reduce the production of interleukin 18 (IL-18) (28), and thus probably exacerbate intestinal inflammation, and potential systemic autoimmunity (29).
FIG 4.

Genus-level co-abundance network of duodenum and rectum for HC and IBS-D microbial communities. Co-occurrence network of duodenum and rectum for (A) HC and (B) IBS-D microbial communities. Co-occurrence relationships between genera with Spearman correlations coefficient > 0.5 or <−0.2 and statistical significance (P < 0.05) were considered as strong correlations between genera and depicted in the Cytoscape. Nodes and edges represented microbial genera and their correlations, respectively. The size of nodes represents the relative abundance of genera. Red and blue edges represent positive and negative correlations, respectively. Each network was divided into two clusters: (left) cluster of duodenum-enriched genera and (right) cluster of rectum-enriched genera, according to the relative abundance of genera in duodenum and rectum. When DM and DL (or RM and RL) owned more proportion of a genus, this genus would be classified into the duodenal cluster (or rectal cluster).
IBS-D patients could be accurately predicted based on site-specific biomarkers.
The site-specific biomarkers, especially for biomarkers from RM and DL, could be used for IBS-D prediction with high fidelity. We identified 32 site-specific biomarkers by LEfSe method with LDA > 2.5 (list in Fig. 5A). Among them, 15 biomarkers were identified for DM (HC: two, IBS-D patients: 13), eight biomarkers for DL (HC: four, IBS-D patients: four), six biomarkers for RM (HC: three, IBS-D patients: three), and three biomarkers for RL (HC: two, IBS-D patients: one).
FIG 5.

Several combinations of site-specific biomarkers could be used for the prediction of host health status with high fidelity. (A) Site-specific biomarkers detected by LEfSe. (B) The performances of site-specific biomarkers using AUC measures. (C) List of site-specific biomarkers and comparison of their prediction accuracies using F1 score and AUC measures. HC, healthy controls; IBS-D, diarrhea-predominant irritable bowel syndrome. DL, duodenal lumen; DM, duodenal mucosa; RL, rectal lumen; RM, rectal mucosa. DL_HC, duodenal luminal samples collected from HC; DL_IBS-D, duodenal luminal samples collected from IBS-D; DM_HC, duodenal mucosal samples collected from HC; DM_IBS-D, duodenal mucosal samples collected from IBS-D; RL_HC, rectal luminal samples collected from HC; RL_IBS-D, rectal luminal samples collected from IBS-D; RM_HC, rectal mucosal samples collected from HC; RM_IBS-D, rectal mucosal samples collected from IBS-D. AUC, area under receiver operating curve.
To find out which site-specific biomarkers could best characterize the microbial community of IBS-D patients, a series of Random Forest (RF) models were built based on these site-specific biomarkers. For DM, the combination of Achromobacterr, Acinetobacter, Lactococcus, Ochrobactrum, and Bacillus could differentiate the HC and IBS-D samples with 80.44% area under receiver operating characteristic curve (AUC) (F1 = 0.67, Fig. 5B and C, red curve). For DL, the combination of Porphyromonas, Sphingomonas, Veillonella, Bulleidia, Leptotrichia, and Rothia showed outstanding performance with 95.11% AUC (F1 = 0.76, Fig. 5B and C, green curve). The combination of RM site-specific biomarkers including Bacteroides, Prevotella and Oscillospira, could be used to precisely distinguish whether the participant is an IBS-D patient (F1 = 0.90 and AUC = 97.36%, blue curve in Fig. 5B and C, and Fig. S9). For RL, according to the biomarkers’ performance, we can use Parabacteroides and Faecalibacterium to accurately differentiate IBS-D patients (F1 = 0.78 and AUC = 87.79%, Fig. 5B and C, yellow curve). Collectively, RM site-specific biomarkers showed the best performance compared with other intestinal sites, followed by DL, indicating the strong potential of RM biomarkers to serve as biomarkers for next-generation IBS-D diagnosis.
Again, RM’s site-specific IBS-D biomarker Bacteroides was included in the RF model, which also showed outstanding performance in differentiating IBS-D patients from HC (F1 = 0.84 and AUC = 91.01%, Fig. S9), confirming its key roles in IBS-D (30). Furthermore, among all sub-combinations of Bacteroides, Prevotella, and Oscillospira, the combination of Bacteroides and Prevotella could best differentiate the IBS-D patients from HC (F1 = 0.83 and AUC = 99.44%, Fig. S9), followed by the combination of Bacteroides and Oscillospira (F1 = 0.86 and AUC = 93.80%). These results suggested the distinct advantage of using RM biomarkers Bacteroides, Prevotella and Oscillospira to identify the IBS-D patients.
The site-specific biomarkers have profound associations with clinical symptoms in IBS-D patients.
Strong associations were observed among bacterial genera in four intestinal sites and clinical symptoms of IBS-D patients. Genera Bacillus and Sphingomonas in DM, DL, and RM were identified with positive correlations with abdominal symptoms (list in Fig. 6A) of IBS-D patients, such as the severity of abdominal pain, bloating, stool consistency (P < 0.05). On the contrary, Faecalibacterium, Dialister, and Sutterella in all intestinal sites were negatively correlated with clinical symptoms of IBS-D patients (P < 0.05), such as the severity of abdominal pain, bloating, and stool consistency (Fig. 6A).
FIG 6.

Microbial genera have profound associations with clinical symptoms in IBS-D patients. (A) Pearson correlations between clinical symptoms and microbial genera of DM, DL, RM and RL in IBS-D patients. Association between the severity of abdominal pain, bloating, abdominal discomfort and the relative abundance of (B) Bacteroides, (C) Prevotella, and (D) Oscillospira in RM. Only the adjusted P value < 0.05 was marked as “*” in Fig. 6A. Analysis of variance with least-significant-difference (ANOVA-LSD) was used to detect the differences between different groups in Fig. 6B. HC, healthy controls; IBS-D, diarrhea-predominant irritable bowel syndrome; DM, duodenal mucosa; DL, duodenal lumen; RL, rectal lumen; RM, rectal mucosa.
Considering the high prediction power of RM biomarkers including Bacteroides, Prevotella, and Oscillospira in identifying IBS-D patients, we also explored their association with clinical symptoms: the severity of abdominal pain, bloating, and abdominal discomfort. Notably, the increasing severity of abdominal pain and bloating was accompanied by a rising abundance of Bacteroides (Fig. 6B). The abundance of Prevotella was decreased when the severity of abdominal pain, bloating, and abdominal discomfort were aggravated (Fig. 6C). However, no significant association was detected between abdominal symptoms and Oscillospira in RM (Fig. 6D). Furthermore, based on RF model built by RM site-specific biomarkers, the misclassified IBS-D patients showed weaker clinical symptoms compared with patients that were classified correctly (Fig. S10). These results further confirmed the important role of RM site-specific biomarkers Bacteroides, Prevotella, and Oscillospira in IBS-D.
DISCUSSION
This study collected the largest number of samples from multiple intestinal sites of IBS-D patients and HC in China, aiming to generate a holistic understanding of mucosa and lumen-associated microbial profiles in duodenum and rectum of IBS-D patients and HC, as well as the potential role of RM microbes in IBS-D. The novel discoveries of this study could be understood in three contexts.
Firstly, microbial communities from different intestinal sites have different microbial compositions, largely due to the colonization of bacteria in different intestinal sites. This result again confirmed the fecal microbial community could not fully represent the intestinal microbial community (13). IBS-D patients and HC also have significantly different representative species even at the same intestinal sites. Bacillus, Burkholderia, Prevotella, and Faecalibacterium were the representative genera in duodenum of IBS-D, duodenum of HC, rectum of IBS-D, and rectum of HC, respectively. The reduction of Burkholderia in duodenum of IBS-D could affect the colonization of the probiotic strain (31), and thus facilitate intestinal inflammation (32). Faecalibacterium was reduced in IBS-D patients at RM site compared with HC (8, 10). It was reported that Faecalibacterium could produce butyrate (33) and anti-inflammatory protein (34), which could improve intestinal immunity and epithelial barrier (35). Hence, its reduction might contribute to intestinal inflammation, and thus promote the onset of IBS-D (36, 37). On the contrary, the relative abundance of Bacillus was increased in duodenum in IBS-D patients compared with HC. Bacillus could secret acetylcholine, which may potentially lead to abdominal pain and diarrhea in IBS-D patients (38, 39).
Secondly, we emphasize the importance of RM microbial community in IBS-D. RF modeling based on RM site-specific biomarkers Bacteroides, Prevotella, and Oscillospira could yield high prediction accuracy (F1 = 0.90 and AUC = 97.36%) for better diagnosis of IBS-D patients. However, fecal (RL) microbial biomarkers Parabacteroides and Faecalibacterium (F1 = 0.78 and AUC = 87.79%) could not perform as well as RM. Hence, rather than fecal samples (12, 13, 40). RM samples were able to reflect more predominant characteristics of intestinal microbial community of IBS-D patients. This phenomenon was consistent with a published study in analyzing intestinal microbial community of naive pediatric Crohn’s disease using ileal, rectal, and fecal samples (41). Moreover, the biomarkers detected by LEfSe in other intestinal sites were also important. For example, the biomarkers of DL (Porphyromonas, Sphingomonas, Veillonella, Bulleidia, Leptotrichia, and Rothia) showed an outstanding performance in identifying IBS-D patients (AUC = 95.11%, F1 = 0.76), and its predictive ability was second to RM. Notably, Porphyromonas, Veillonella, and Leptotrichia were reported to promote intestinal dysbiosis by influencing microbial communities (42). Additionally, the misclassified IBS-D patients by RF model using RM site-specific biomarkers, were also confirmed with weaker clinical symptoms compared with these correctly classified patients (Fig. S10), while some of these misclassified IBS-D patients could be correctly identified by the RF model using DL site-specific biomarkers (Table S3). All of these results emphasized the importance of RM microbial community in IBS-D. In summary, this is the first study to use the RM site-specific biomarkers Bacteroides, Prevotella, and Oscillospira to build a prediction model for the better diagnosis of IBS-D patients. These results also highlight the importance for exploring the potential link between IBS-D and RM microbial community.
Thirdly, RM site-specific biomarker Bacteroides was a genus that could serve well for fast and easy IBS-D diagnosis. The co-occurrence network has also shown its alteration in IBS-D patients compared with HC. In IBS-D patients, Bacteroides was positively correlated with Parabacteroides and Phascolarctobacterium, which were reported as intestinal inflammation contributors (23–25). This alteration might also indicate the potential role of Bacteroides in the pathophysiology of IBS-D. Using Bacteroides alone or together with Prevotella and Oscillospira from RM could result in high identification accuracy for IBS-D patients, among all RF models generated in this study, confirming its key role in IBS-D (30). As previously described (11, 43, 44), Bacteroides was a genus enriched in IBS-D patients, which was also found strongly associated with the severity of abdominal pain and bloating. Though this genus contains numerous commensal species that could secret immunomodulatory factors to regulate intestinal inflammation, such as Bacteroides vulgatus (45) and Bacteroides ovatus (46), several species of this genus could produce enterotoxins, such as Bacteroides fragilis (47) and Bacteroides melaninogenicus (48). Among them, Bacteroides fragilis could bind to IgA for colonization in intestinal mucosa, which might destroy the intestinal epithelial barrier and alter gut motility, leading to abdominal pain or bloating (49–51). This species was also resistant to antibiotics (52) and associated with diarrhea (50, 51). Taken together, monitoring Bacteroides from RM, though it might incur invasive sampling, would serve well in the next-generation diagnosis of IBS-D patients.
This study also has limitations. Firstly, the relatively small sample size prevented the extrapolation of our results to the general IBS-D population. However, considering the difficulty of endoscopy and biopsy for participants who offered their samples collected from multiple intestinal sites, this study has tried our best to collect the largest number of samples from multiple intestinal sites of IBS-D patients and HC in China. Secondly, our study revealed the site-specific microbial patterns and found the discriminative microbes for IBS-D diagnosis, although diet (such as high-fat diet) and lifestyle (such as smoking) play crucial roles in shaping intestinal microbial communities (53–55). Based on our analysis, further studies could explore the contribution of these factors to intestinal microbiota and the pathogenesis of IBS-D. Thirdly, due to the difference of intestinal microbial communities between Chinese and western people, most of the publicly available IBS-D data were from western individuals, the external validation test using the public data set was largely limited. With the increasing number of samples from multiple cohorts becoming available, universal patterns of intestinal microbial communities for IBS-D patients might be better understood.
Conclusion.
To conclude, our study collected the largest number of samples from multiple intestinal sites of IBS-D patients and HC in China. We found that IBS-D microbial community has specific patterns at each intestinal site differing from that of HC. Fecal microbial communities (that is RL samples) could not fully represent the intestinal microbial communities. Additionally, IBS-D microbial co-abundant network has lower interactions compared with HC. Moreover, using genera Bacteroides alone or together with Prevotella and Oscillospira of RM was found to have high prediction power to differentiate IBS-D from HC. Furthermore, Bacteroides and Prevotella in RM were closely related to the severity of abdominal pain, bloating, and abdominal discomfort in IBS-D patients.
Considering that most researches about the intestinal microbial community in IBS focused on fecal microbial community, this study is of particular importance in helping researchers understand the great potential of the microbial community in RM for better diagnosis of IBS-D. To better understand this potential, further researches are necessary to figure out the importance of the enrichment of Bacteroides, and decrease of Prevotella and Oscillospira in RM for IBS-D. All of these ongoing and future efforts could contribute to the realization of the next-generation IBS-D diagnosis.
MATERIALS AND METHODS
Ethics approval.
This study was approved by the Institutional Ethical Review Committee of Huazhong University of Science and Technology (2014-196) and Chinese Clinical Trials Registry (registration number: ChiCTR-OPC-15007624).
Study design and sample description.
Healthy individuals and IBS-D patients aged 35.89 on average (standard deviation:10.49), who expressed interest were invited to participate in this study, at the Division of Gastroenterology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology. For IBS-D patients, they were diagnosed according to Rome III criteria (17). Participants with the following situations were excluded from this study: (i) use antibiotics, prebiotics, probiotics, proton-pump inhibitors, colon-cleansing drugs, or any medications for IBS-D within a month before the start of this study; and (ii) acute gastroenteritis, coeliac disease, inflammatory bowel diseases, cancer, severe systemic diseases, abdominal surgery history, and/or psychiatric diseases. Healthy participants without personal history of immune-related diseases or gastrointestinal complaints were recruited as a control group. In total, this study recruited 74 IBS-D patients and 20 healthy participants.
Each participant signed informed consent and completed a questionnaire that included their clinical symptoms (Table S1). These clinical symptoms mainly included age, body mass index (BMI), stool consistency based on Bristol Stool Form (BSF) (56), frequency of defecation per day, onset frequency, course of IBS-D, the severity of abdominal pain, abdominal discomfort and bloating, and psychological state according to Hospital Anxiety and Depression (HAD) scale (57). The severity of abdominal pain, abdominal discomfort, and bloating were classified into not at all, mild, moderate and severe, which were represented by numbers 0, 1, 2, and 3, respectively. Onset frequency was divided into once or twice a month, three times a month, once a week, more than once a week and every day, which were represented by numbers 1, 2, 3, 4, and 5, respectively.
We investigated microbial profiles at multiple intestinal sites of IBS-D patients and HC, including DM, DL, RM, and RL samples. DM samples were collected by endoscopy from duodenum about 10 cm below the major duodenal papilla using biopsy forceps under the endoscope. DL samples were collected from the fluid in the same position with DM endoscopically using a sterile catheter. RM samples were collected from the rectum approximately 10 cm above the anus using biopsy forceps under the endoscope. RL samples (in this study, we collected fecal samples from rectum, and considered the fecal samples as RL samples) were obtained from participants after excretion as soon as possible. Then, these samples were carefully packed into sterile Eppendorf tubes and placed on ice. After that, these samples were immediately transported to the laboratory and stored at −80°C within 30 min. For mucosal biopsies, the HE staining has shown there was no inflammatory infiltration in all histological sections under the microscope (Fig. S11). We totally collected 283 samples from these four representative intestinal sites (Table S4). Among them, IBS-D samples contained 37 DM samples, 43 DL samples, 53 RM samples, and 74 RL samples. HC samples included 20 DM samples, 19 DL samples, 20 RM samples, and 17 RL samples (Table S4). The vast majority of participants contributed samples from at least two sites (77.66%), while 22.34%, 8.51%, 14.89%, and 54.26% of patients contributed one, two, three, and four sites of intestinal samples, respectively. All samples were preserved at −80°C until performing 16S rRNA gene sequencing.
DNA extraction and 16S rRNA gene sequencing.
DNA of all samples was extracted using a Fast DNA SPIN Kit (Tiangen Biotech). Then, the universal primers (forward: 5′-AGAGTTTGATCCTGGCTCAG-3′, reverse: 5′-TTACCGCGGCTGCTGGCAC-3′) and fusion primers (forward: 5′-454adapter-mid-AGAGTTTGATCCTGGCTCAG-3′, reverse: 5′-454adapter-TTACCGCGGCTGCTGGCAC-3′) were used to amplify the 16S rRNA V1-V3 regions. After that, all products were sent for 16S rRNA gene sequencing on a 454 Life Sciences Genome Sequencer FLX platform with titanium (Roche Life Sciences). Finally, all of these 283 samples were successfully sequenced for subsequent analysis.
Taxonomy annotation and microbial diversity analysis.
The low-quality reads which contained ambiguous base calls (N), or less than 300 bp were filtered using QIIME (version 1.9.1) (58). The primers were removed using the mothur (59) commands “chimera.uchime” and “remove.seqs” based on Silva database (Release 123) (60). These high-quality sequences were clustered into operational taxonomic units (OTUs) at 97% sequence similarity using QIIME (58) script pick_de_novo_otus.py. For OTUs, we filtered out the OTUs only detected in one sample with one read, and then these OTUs were assigned to the Greengenes database for taxonomy classification (61).
Before microbial diversity analysis, we subsampled all the samples to 187,000 reads using the QIIME script single_rarefaction.py (parameter: -d 187000), given the different sequences for each sample. Alpha (measured by Shannon index) and beta (measured by unweighted UniFrac and Bray Curtis distance) diversity metrics were used to analyze the microbial diversity using QIIME (58). Principal coordinates analysis (PCoA) based on Jaccard coefficient was used to determine the discrepancies between HC and IBS-D, or between different intestinal sites using R package “vegan.” Kruskal-Wallis was used to detect the global difference, while the Wilcoxon test was applied to detect the variation between HC and IBS-D microbial communities or the variation across intestinal sites by paired comparisons. The false discovery rate (FDR) adjusted P value was calculated using the Benjamini and Hochberg (BH) method. Procrustes analysis was utilized to assess the similarity of the two compared microbial communities based on R procrustes() function, and the results were tested by the Monte Carlo method with 999 permutations.
Co-abundant network analysis.
For co-abundance network analysis, we first selected the genera with relative abundance no less than 0.5% and coverage of at least 10% samples. Secondly, these genera were utilized for co-occurrence network analysis using Spearman correlation as correlation measurement. Thirdly, the co-occurrence relationships among these microbes with P value < 0.05 were considered as significant correlations (Spearman correlation coefficient: larger than 0.5 or smaller than −0.2), and depicted in the network diagram with genera as nodes and their correlations as edges (visualized by Cytoscape) (62). The FDR adjusted P value was calculated by BH method. Each network was divided into two clusters according to the abundance of the genera: When a genus was detected with a higher proportion of relative abundance in DM and DL, this genus would be classified into the duodenal cluster, or this genus was assigned to the rectal cluster.
Prediction model for distinguishing IBS-D patients and healthy participants.
To detect the species that could be used for better diagnosis of IBS-D patients, LEfSe (63) was utilized to identify the site-specific biomarker with LDA > 2.5. Then, a RF model (64) was developed to distinguish the IBS-D samples from HC based on these site-specific biomarkers. For the prediction model, important parameters ntree (number of decision trees in RF model) and mtry (variable sampling value for each iteration) were trained and estimated by the out-of-bag (OOB) values using R package “randomForest” (64). This process was iterated 15,000 times to construct the accurate model without overfitting. We also used the AUC and F1 score to evaluate the performance of the prediction model.
Association of intestinal bacteria and clinical symptoms in IBS-D patients.
Pearson correlation analysis was conducted to detect the association of intestinal bacteria and clinical symptoms (Table S1). The FDR adjusted P value was calculated using BH method. Only correlations identified with adjusted P < 0.05 were considered as significant correlations and visualized in the heatmap.
Data availability.
All the raw sequencing data in this study are available in the Genome Sequence Archive (GSA) database (GSA accession number: PRJCA004584), which can be accessed at https://bigd.big.ac.cn/bioproject/browse/PRJCA004584.
ACKNOWLEDGMENTS
This work was partially supported by National Natural Science Foundation of China (grant numbers: 32071465, 81573702, 81774008, 31871334, 31671374, 81330014, 81570486, 81800463, 81800480, 81500415), and National Key Research and Development Program of China (grant number: 2018YFC0910502).
X.H.H., K.N., and Z.D. designed the study. X.Z., P.S.Y., and G.C.H. performed analysis. X.Z., G.C.H., and P.S.Y. wrote the manuscript. Ying Li., L.Z., Y.J., G.P.L., M.Y., H.H.X., and W.Q. were involved in sample collection and preparation. K.N., X.Z., G.C.H., P.S.Y., M.Y.C., Y.X.L., L.J., C.Y.C., and C.F.Z. critically reviewed and proofed the manuscript.
Footnotes
Supplemental material is available online only.
Contributor Information
Zhen Ding, Email: docd720@126.com.
Kang Ning, Email: ningkang@hust.edu.cn.
Xiaohua Hou, Email: houxh@hust.edu.cn.
Kevin R. Theis, Wayne State University
REFERENCES
- 1.Enck P, Aziz Q, Barbara G, Farmer AD, Fukudo S, Mayer EA, Niesler B, Quigley EM, Rajilić-Stojanović M, Schemann M, Schwille-Kiuntke J, Simren M, Zipfel S, Spiller RC. 2016. Irritable bowel syndrome. Nat Rev Dis Primers 2:16014. doi: 10.1038/nrdp.2016.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sultan S, Malhotra A. 2017. Irritable bowel syndrome. Ann Intern Med 166:ITC81–ITC96. doi: 10.7326/AITC201706060. [DOI] [PubMed] [Google Scholar]
- 3.Black CJ, Ford AC. 2020. Global burden of irritable bowel syndrome: trends, predictions and risk factors. Nat Rev Gastroenterol Hepatol 17:473–486. doi: 10.1038/s41575-020-0286-8. [DOI] [PubMed] [Google Scholar]
- 4.Ford AC, Sperber AD, Corsetti M, Camilleri M. 2020. Irritable bowel syndrome. Lancet 396:1675–1688. doi: 10.1016/S0140-6736(20)31548-8. [DOI] [PubMed] [Google Scholar]
- 5.Pittayanon R, Lau JT, Yuan Y, Leontiadis GI, Tse F, Surette M, Moayyedi P. 2019. Gut microbiota in patients with irritable bowel syndrome-a systematic review. Gastroenterology 157:97–108. doi: 10.1053/j.gastro.2019.03.049. [DOI] [PubMed] [Google Scholar]
- 6.Liu T, Gu X, Li LX, Li M, Li B, Cui X, Zuo XL. 2020. Microbial and metabolomic profiles in correlation with depression and anxiety co-morbidities in diarrhoea-predominant IBS patients. BMC Microbiol 20:168. doi: 10.1186/s12866-020-01841-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Palma G, Lynch MD, Lu J, Dang VT, Deng Y, Jury J, Umeh G, Miranda PM, Pigrau Pastor M, Sidani S, Pinto-Sanchez MI, Philip V, McLean PG, Hagelsieb MG, Surette MG, Bergonzelli GE, Verdu EF, Britz-McKibbin P, Neufeld JD, Collins SM, Bercik P. 2017. Transplantation of fecal microbiota from patients with irritable bowel syndrome alters gut function and behavior in recipient mice. Sci Transl Med 9. doi: 10.1126/scitranslmed.aaf6397. [DOI] [PubMed] [Google Scholar]
- 8.Carroll IM, Ringel-Kulka T, Siddle JP, Ringel Y. 2012. Alterations in composition and diversity of the intestinal microbiota in patients with diarrhea-predominant irritable bowel syndrome. Neurogastroenterol Motil 24:521–530. doi: 10.1111/j.1365-2982.2012.01891.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ringel-Kulka T, Benson AK, Carroll IM, Kim J, Legge RM, Ringel Y. 2016. Molecular characterization of the intestinal microbiota in patients with and without abdominal bloating. Am J Physiol Gastrointest Liver Physiol 310:G417–26. doi: 10.1152/ajpgi.00044.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Y, Zhang L, Wang X, Wang Z, Zhang J, Jiang R, Wang X, Wang K, Liu Z, Xia Z, Xu Z, Nie Y, Lv X, Wu X, Zhu H, Duan L. 2016. Similar fecal microbiota signatures in patients with diarrhea-predominant irritable bowel syndrome and patients with depression. Clin Gastroenterol Hepatol 14:1602–1611.e5. doi: 10.1016/j.cgh.2016.05.033. [DOI] [PubMed] [Google Scholar]
- 11.Zhong W, Lu X, Shi H, Zhao G, Song Y, Wang Y, Zhang J, Jin Y, Wang S. 2019. Distinct microbial populations exist in the mucosa-associated microbiota of diarrhea predominant irritable bowel syndrome and ulcerative colitis. J Clin Gastroenterol 53:660–672. doi: 10.1097/MCG.0000000000000961. [DOI] [PubMed] [Google Scholar]
- 12.Vuik F, Dicksved J, Lam SY, Fuhler GM, van der Laan L, van de Winkel A, Konstantinov SR, Spaander M, Peppelenbosch MP, Engstrand L, Kuipers EJ. 2019. Composition of the mucosa-associated microbiota along the entire gastrointestinal tract of human individuals. United European Gastroenterol J 7:897–907. doi: 10.1177/2050640619852255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vasapolli R, Schutte K, Schulz C, Vital M, Schomburg D, Pieper DH, Vilchez-Vargas R, Malfertheiner P. 2019. Analysis of transcriptionally active bacteria throughout the gastrointestinal tract of healthy individuals. Gastroenterology 157:1081–1092. doi: 10.1053/j.gastro.2019.05.068. [DOI] [PubMed] [Google Scholar]
- 14.Yuan C, Graham M, Staley C, Subramanian S, Gibbons SM. 2020. Mucosal microbiota and metabolome along the intestinal tract reveal a location-specific relationship. mSystems 5:e00055-20. doi: 10.1128/mSystems.00055-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gu X, Song LJ, Li LX, Liu T, Zhang MM, Li Z, Wang P, Li M, Zuo XL. 2020. Fusobacterium nucleatum causes microbial dysbiosis and exacerbates visceral hypersensitivity in a colonization-independent manner. Front Microbiol 11:1281. doi: 10.3389/fmicb.2020.01281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chao G, Zhang S. 2020. The characteristics of intestinal flora of IBS-D with different syndromes. Immun Inflamm Dis 8:615–628. doi: 10.1002/iid3.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Longstreth GF, Thompson WG, Chey WD, Houghton LA, Mearin F, Spiller RC. 2006. Functional bowel disorders. Gastroenterology 130:1480–1491. doi: 10.1053/j.gastro.2005.11.061. [DOI] [PubMed] [Google Scholar]
- 18.Drossman DA. 2016. Functional gastrointestinal disorders: history, pathophysiology, clinical features, and Rome IV. Gastroenterology 150:1262–1279.e2. doi: 10.1053/j.gastro.2016.02.032. [DOI] [PubMed] [Google Scholar]
- 19.Palsson OS, Whitehead W, Tornblom H, Sperber AD, Simren M. 2020. Prevalence of Rome IV functional bowel disorders among adults in the United States, Canada, and the United Kingdom. Gastroenterology 158:1262–1273.e3. doi: 10.1053/j.gastro.2019.12.021. [DOI] [PubMed] [Google Scholar]
- 20.Pascal V, Pozuelo M, Borruel N, Casellas F, Campos D, Santiago A, Martinez X, Varela E, Sarrabayrouse G, Machiels K, Vermeire S, Sokol H, Guarner F, Manichanh C. 2017. A microbial signature for Crohn's disease. Gut 66:813–822. doi: 10.1136/gutjnl-2016-313235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu J, Feng Q, Wong SH, Zhang D, Liang QY, Qin Y, Tang L, Zhao H, Stenvang J, Li Y, Wang X, Xu X, Chen N, Wu WK, Al-Aama J, Nielsen HJ, Kiilerich P, Jensen BA, Yau TO, Lan Z, Jia H, Li J, Xiao L, Lam TY, Ng SC, Cheng AS, Wong VW, Chan FK, Xu X, Yang H, Madsen L, Datz C, Tilg H, Wang J, Brunner N, Kristiansen K, Arumugam M, Sung JJ, Wang J. 2017. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut 66:70–78. doi: 10.1136/gutjnl-2015-309800. [DOI] [PubMed] [Google Scholar]
- 22.Wei Y, Li Y, Yan L, Sun C, Miao Q, Wang Q, Xiao X, Lian M, Li B, Chen Y, Zhang J, Li Y, Huang B, Li Y, Cao Q, Fan Z, Chen X, Fang JY, Gershwin ME, Tang R, Ma X. 2020. Alterations of gut microbiome in autoimmune hepatitis. Gut 69:569–577. doi: 10.1136/gutjnl-2018-317836. [DOI] [PubMed] [Google Scholar]
- 23.Li R, Li L, Hong P, Lang W, Hui J, Yang Y, Zheng X. 2019. β-Carotene prevents weaning-induced intestinal inflammation by modulating gut microbiota in piglets. Asian-Australas J Anim Sci 34:1221–1234. doi: 10.5713/ajas.19.0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maillard F, Vazeille E, Sauvanet P, Sirvent P, Bonnet R, Combaret L, Chausse P, Chevarin C, Otero YF, Delcros G, Chavanelle V, Boisseau N, Barnich N. 2019. Preventive effect of spontaneous physical activity on the gut-adipose tissue in a mouse model that mimics Crohn’s disease susceptibility. Cells 8:33. doi: 10.3390/cells8010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bajer L, Kverka M, Kostovcik M, Macinga P, Dvorak J, Stehlikova Z, Brezina J, Wohl P, Spicak J, Drastich P. 2017. Distinct gut microbiota profiles in patients with primary sclerosing cholangitis and ulcerative colitis. World J Gastroenterol 23:4548–4558. doi: 10.3748/wjg.v23.i25.4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu Y. 2007. The study and application on bacteroides. Biotechnology Bull 1:66–69. [Google Scholar]
- 27.Sóki J, Wybo I, Hajdú E, Toprak NU, Jeverica S, Stingu CS, Tierney D, Perry JD, Matuz M, Urbán E, Nagy E, ESCMID Study Group on Anaerobic Infections . 2020. A Europe-wide assessment of antibiotic resistance rates in Bacteroides and Parabacteroides isolates from intestinal microbiota of healthy subjects. Anaerobe 62:102182. doi: 10.1016/j.anaerobe.2020.102182. [DOI] [PubMed] [Google Scholar]
- 28.Wawrocki S, Druszczynska M, Kowalewicz-Kulbat M, Rudnicka W. 2016. Interleukin 18 (IL-18) as a target for immune intervention. Acta Biochim Pol 63:59–63. doi: 10.18388/abp.2015_1153. [DOI] [PubMed] [Google Scholar]
- 29.Iljazovic A, Roy U, Gálvez EJC, Lesker TR, Zhao B, Gronow A, Amend L, Will SE, Hofmann JD, Pils MC, Schmidt-Hohagen K, Neumann-Schaal M, Strowig T. 2021. Perturbation of the gut microbiome by Prevotella spp. enhances host susceptibility to mucosal inflammation. Mucosal Immunol 14:113–124. doi: 10.1038/s41385-020-0296-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vaga S, Lee S, Ji B, Andreasson A, Talley NJ, Agréus L, Bidkhori G, Kovatcheva-Datchary P, Park J, Lee D, Proctor G, Ehrlich SD, Nielsen J, Engstrand L, Shoaie S. 2020. Compositional and functional differences of the mucosal microbiota along the intestine of healthy individuals. Sci Rep 10:14977. doi: 10.1038/s41598-020-71939-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Senan S, Prajapati JB, Joshi CG, Sreeja V, Gohel MK, Trivedi S, Patel RM, Pandya H, Singh US, Phatak A, Patel HA. 2015. Geriatric respondents and non-respondents to probiotic intervention can be differentiated by inherent gut microbiome composition. Front Microbiol 6. doi: 10.3389/fmicb.2015.00944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Didari T, Mozaffari S, Nikfar S, Abdollahi M. 2015. Effectiveness of probiotics in irritable bowel syndrome: Updated systematic review with meta-analysis. WJG 21:3072–3084. doi: 10.3748/wjg.v21.i10.3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang M, Zhou L, Wang Y, Dorfman RG, Tang D, Xu L, Pan Y, Zhou Q, Li Y, Yin Y, Zhao S, Wu J, Yu C. 2019. Faecalibacterium prausnitzii produces butyrate to decrease c-Myc-related metabolism and Th17 differentiation by inhibiting histone deacetylase 3. Int Immunol 31:499–514. doi: 10.1093/intimm/dxz022. [DOI] [PubMed] [Google Scholar]
- 34.Quevrain E, Maubert MA, Michon C, Chain F, Marquant R, Tailhades J, Miquel S, Carlier L, Bermudez-Humaran LG, Pigneur B, Lequin O, Kharrat P, Thomas G, Rainteau D, Aubry C, Breyner N, Afonso C, Lavielle S, Grill JP, Chassaing G, Chatel JM, Trugnan G, Xavier R, Langella P, Sokol H, Seksik P. 2016. Identification of an anti-inflammatory protein from Faecalibacterium prausnitzii, a commensal bacterium deficient in Crohn’s disease. Gut 65:415–425. doi: 10.1136/gutjnl-2014-307649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laval L, Martin R, Natividad JN, Chain F, Miquel S, Desclee de Maredsous C, Capronnier S, Sokol H, Verdu EF, van Hylckama Vlieg JE, Bermudez-Humaran LG, Smokvina T, Langella P. 2015. Lactobacillus rhamnosus CNCM I-3690 and the commensal bacterium Faecalibacterium prausnitzii A2-165 exhibit similar protective effects to induced barrier hyper-permeability in mice. Gut Microbes 6:1–9. doi: 10.4161/19490976.2014.990784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barbara G, Stanghellini V, De Giorgio R, Cremon C, Cottrell GS, Santini D, Pasquinelli G, Morselli-Labate AM, Grady EF, Bunnett NW, Collins SM, Corinaldesi R. 2004. Activated mast cells in proximity to colonic nerves correlate with abdominal pain in irritable bowel syndrome. Gastroenterology 126:693–702. doi: 10.1053/j.gastro.2003.11.055. [DOI] [PubMed] [Google Scholar]
- 37.Guilarte M, Santos J, de Torres I, Alonso C, Vicario M, Ramos L, Martinez C, Casellas F, Saperas E, Malagelada JR. 2007. Diarrhoea-predominant IBS patients show mast cell activation and hyperplasia in the jejunum. Gut 56:203–209. doi: 10.1136/gut.2006.100594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kawashima K, Misawa H, Moriwaki Y, Fujii YX, Fujii T, Horiuchi Y, Yamada T, Imanaka T, Kamekura M. 2007. Ubiquitous expression of acetylcholine and its biological functions in life forms without nervous systems. Life Sci 80:2206–2209. doi: 10.1016/j.lfs.2007.01.059. [DOI] [PubMed] [Google Scholar]
- 39.Dajas-Bailador F, Wonnacott S. 2004. Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol Sci 25:317–324. doi: 10.1016/j.tips.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 40.Yuan C, Graham M, Staley C, Subramanian S. 2020. Mucosal microbiota and metabolome along the intestinal tract reveal a location-specific relationship. mSystems 5:e00055. doi: 10.1128/mSystems.00055-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gevers D, Kugathasan S, Denson LA, Vázquez-Baeza Y, Van Treuren W, Ren B, Schwager E, Knights D, Song Se J, Yassour M, Morgan XC, Kostic AD, Luo C, González A, McDonald D, Haberman Y, Walters T, Baker S, Rosh J, Stephens M, Heyman M, Markowitz J, Baldassano R, Griffiths A, Sylvester F, Mack D, Kim S, Crandall W, Hyams J, Huttenhower C, Knight R, Xavier RJ. 2014. The treatment-naive microbiome in new-onset Crohn’s Disease. Cell Host Microbe 15:382–392. doi: 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koliarakis I, Messaritakis I, Nikolouzakis TK, Hamilos G, Souglakos J, Tsiaoussis J. 2019. Oral bacteria and intestinal dysbiosis in colorectal cancer. Int J Mol Sci 20:4146. doi: 10.3390/ijms20174146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tap J, Derrien M, Tornblom H, Brazeilles R, Cools-Portier S, Dore J, Storsrud S, Le Neve B, Ohman L, Simren M. 2017. Identification of an intestinal microbiota signature associated with severity of irritable bowel syndrome. Gastroenterology 152:111–123.e8. doi: 10.1053/j.gastro.2016.09.049. [DOI] [PubMed] [Google Scholar]
- 44.Shukla R, Ghoshal U, Dhole TN, Ghoshal UC. 2015. Fecal microbiota in patients with irritable bowel syndrome compared with healthy controls using real-time polymerase chain reaction: an evidence of dysbiosis. Dig Dis Sci 60:2953–2962. doi: 10.1007/s10620-015-3607-y. [DOI] [PubMed] [Google Scholar]
- 45.Hiippala K, Jouhten H, Ronkainen A, Hartikainen A, Kainulainen V, Jalanka J, Satokari R. 2018. The potential of gut commensals in reinforcing intestinal barrier function and alleviating inflammation. Nutrients 10:988. doi: 10.3390/nu10080988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ihekweazu FD, Engevik MA, Ruan W, Shi Z, Fultz R, Engevik KA, Chang-Graham AL, Freeborn J, Park ES, Venable S, Horvath TD, Haidacher SJ, Haag AM, Goodwin A, Schady DA, Hyser JM, Spinler JK, Liu Y, Versalovic J. 2021. Bacteroides ovatus promotes IL-22 production and reduces trinitrobenzene sulfonic acid-driven colonic inflammation. Am J Pathol 191:704–719. doi: 10.1016/j.ajpath.2021.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hwang S, Yi HC, Hwang S, Jo M, Rhee KJ. 2020. Dietary salt administration decreases enterotoxigenic bacteroides fragilis (ETBF)-promoted tumorigenesis via inhibition of colonic inflammation. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim EY, Baek YH, Jung DS, Woo KS. 2019. Concomitant liver and brain abscesses caused by Parvimonas Micra. Korean J Gastroenterol 73:230–234. doi: 10.4166/kjg.2019.73.4.230. [DOI] [PubMed] [Google Scholar]
- 49.Macfarlane S, Woodmansey EJ, Macfarlane GT. 2005. Colonization of mucin by human intestinal bacteria and establishment of biofilm communities in a two-stage continuous culture system. Appl Environ Microbiol 71:7483–7492. doi: 10.1128/AEM.71.11.7483-7492.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wick EC, Sears CL. 2010. Bacteroides spp. and diarrhea. Curr Opin Infect Dis 23:470–474. doi: 10.1097/QCO.0b013e32833da1eb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chambers FG, Koshy SS, Saidi RF, Clark DP, Moore RD, Sears CL. 1997. Bacteroides fragilis toxin exhibits polar activity on monolayers of human intestinal epithelial cells (T84 cells) in vitro. Infect Immun 65:3561–3570. doi: 10.1128/iai.65.9.3561-3570.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ghotaslou R, Yekani M, Memar MY. 2018. The role of efflux pumps in Bacteroides fragilis resistance to antibiotics. Microbiol Res 210:1–5. doi: 10.1016/j.micres.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 53.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, Biddinger SB, Dutton RJ, Turnbaugh PJ. 2014. Diet rapidly and reproducibly alters the human gut microbiome. Nature 505:559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tanaka S, Nemoto Y, Takei Y, Morikawa R, Oshima S, Nagaishi T, Okamoto R, Tsuchiya K, Nakamura T, Stutte S, Watanabe M. 2020. High-fat diet-derived free fatty acids impair the intestinal immune system and increase sensitivity to intestinal epithelial damage. Biochem Biophys Res Commun 522:971–977. doi: 10.1016/j.bbrc.2019.11.158. [DOI] [PubMed] [Google Scholar]
- 55.Frati F, Salvatori C, Incorvaia C, Bellucci A, Di Cara G, Marcucci F, Esposito S. 2018. The role of the microbiome in asthma: the gut-lung axis. Int J Mol Sci 20:123. doi: 10.3390/ijms20010123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heaton KW, O’Donnell LJD. 1994. An office guide to whole-gut transit time. Patients’ recollection of their stool form. J Clin Gastroenterol 19:28–30. doi: 10.1097/00004836-199407000-00008. [DOI] [PubMed] [Google Scholar]
- 57.Zigmond AS, Snaith RP. 1983. The Hospital Anxiety and Depression Scale. Acta Psychiatr Scand 67:361–370. doi: 10.1111/j.1600-0447.1983.tb09716.x. [DOI] [PubMed] [Google Scholar]
- 58.Lawley B, Tannock GW. 2017. Analysis of 16S rRNA gene amplicon sequences using the QIIME Software Package. Methods Mol Biol 1537:153–163. doi: 10.1007/978-1-4939-6685-1_9. [DOI] [PubMed] [Google Scholar]
- 59.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–6. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yilmaz P, Parfrey LW, Yarza P, Gerken J, Pruesse E, Quast C, Schweer T, Peplies J, Ludwig W, Glockner FO. 2014. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res 42:D643–8. doi: 10.1093/nar/gkt1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. 2011. Metagenomic biomarker discovery and explanation. Genome Biol 12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huang BF, Boutros PC. 2016. The parameter sensitivity of random forests. BMC Bioinformatics 17:331. doi: 10.1186/s12859-016-1228-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download SPECTRUM01255-21_Supp_1_seq8.pdf, PDF file, 1.9 MB (2MB, pdf)
Data Availability Statement
All the raw sequencing data in this study are available in the Genome Sequence Archive (GSA) database (GSA accession number: PRJCA004584), which can be accessed at https://bigd.big.ac.cn/bioproject/browse/PRJCA004584.