

Abstract
Hexavalent chromium [Cr(VI)] is a global environmental pollutant that increases risk for several types of cancers and is increasingly being recognized as a neurotoxicant. Traditionally, the brain has been viewed as a largely post-mitotic organ due to its specialized composition of neurons, and consequently, clastogenic effects were not considered in neurotoxicology. Today, we understand the brain is composed of at least eight distinct cell types – most of which continue mitotic activity throughout lifespan. We have learned these dividing cells play essential roles in brain and body health. This review focuses on Cr(VI), a potent clastogen and known human carcinogen, as a potentially neurotoxic agent targeting mitotic cells of the brain. Despite its well-established role as a human carcinogen, Cr(VI) neurotoxicity studies have failed to find a significant link to brain cancers. In the few studies that did find a link, Cr(VI) was identified as a risk for gliomas. Instead, in the human brain, Cr(VI) appears to have more subtle deleterious effects that can impair childhood learning and attention development, olfactory function, social memory, and may contribute to motor neuron diseases. Studies of Cr(VI) neurotoxicity with animal and cell culture models have demonstrated elevated markers of oxidative damage and redox stress, with widespread neurodegeneration. One study showed mice exposed to Cr(VI)-laden tannery effluent exhibited longer periods of aggressive behavior toward an “intruder” mouse and took longer to recognize mice previously encountered, recapitulating the social memory deficits observed in humans. Here we conducted a critical review of the available literature on Cr(VI) neurotoxicity and synthesize the collective observations to thoroughly evaluate Cr(VI) neurotoxicity – much remains to be understood and recognized.
Keywords: hexavalent chromium, neurotoxicity, brain, metals, glia
Graphical Abstract
1. Introduction
Chromium (Cr) is the seventh most abundant element on earth and naturally occurs in two valence states – hexavalent [Cr(VI)] and trivalent [Cr(III)]. While Cr(III) is generally regarded as less toxic and argued to be potentially therapeutic, Cr(VI) is widely recognized as a human carcinogen and an environmental pollutant (Holmes et al., 2008; DesMarias and Costa, 2019; Moreira et al., 2019; Farrokhian et al., 2019; Peng and Yang, 2015; Speer and Wise, Sr., 2018). In the US alone, 900–970 tons of Cr(VI) are released into the air every year, largely due to industrial activities such as burning oil and coal, pigment oxidants, fertilizers, oil well drilling, metal electroplating, and leather tanning (ATSDR, 2012). Occupational exposure poses the most significant risk, and is often encountered in industries that use chromic acid, or are involved in chromite ore production, paint production, or steel welding. Inhalation of Cr(VI) increases incidence and risk of developing lung cancers, while ingestion via drinking water increases risk of liver cancers. As such, Cr(VI) is listed by the International Agency for Research on Cancer (IARC) as a group 1 human carcinogen and is listed 17th on the Agency for Toxic Substances and Disease Registry’s (ATSDR’s) Hazards Priority List. Drinking water contaminated with Cr(VI) presents the most wide-spread risk of exposure, though it is currently regulated as total Cr and thus reported drinking water levels for Cr(VI) are sparse. Average Cr(VI) drinking water levels range from 0.2–2 μg Cr(VI)/L in the United States and Canada (US EPA, 2017; Moffat et al., 2018). Sources for Cr(VI) in drinking water may come from erosion of naturally occurring rocks/sediments or anthropogenic industrial pollution (see reviews by Moffat et al., 2018 and McNeill et al., 2012 for further discussion).
Upon entry into the body, Cr(VI) particles dissociate and the Cr(VI) anion crosses cell membranes through ion channels or the entire particulate is phagocytosed. Due to Cr(VI) anion’s tetrahedral structure, it mimics sulfate and phosphate ions to passively cross the cell membrane through these ion channels. Interaction of Cr(VI) with arginine in these anion channels is critical for stabilizing the Cr(VI) anion through a network of hydrogen bonds (Saha et al., 2011). In the cytoplasm, Cr(VI) is rapidly reduced to Cr(III) with ephemeral intermediate valences states [Cr (V) and (IV)]. This reduction produces reactive oxygen species (ROS, such as superoxide, hydrogen peroxide, or hydroxyl radicals), which contribute to carcinogenesis. Cr(VI)’s clastogenic effects are well defined, but unknown in the brain (Newbold et al., 1979; Watanabe et al., 1985; Wise, S. et al., 2004). Further, Cr(VI) induces genomic instability, a hallmark of lung cancer, by disrupting DNA damage repair pathways (Wise, S. et al., 2018; Browning et al., 2016; Qin et al., 2014; Wise, S. et al., 2008; Zhitkovich, 2005; Martin et al., 2006; Reynolds et al., 2007).
The brain is a highly specialized organ that demands up to 20% of the body’s oxygen supply despite making up less than 2% of body mass. Most often people associate the brain with neurons, and neurotoxicity with how chemicals affect neuronal health. However, there are at least eight distinct cell types in addition to neurons conducting the complexity of the brain, including: astrocytes, microglia, oligodendrocytes, ependymal cells, pericytes, brain vascular endothelial cells, and smooth muscle cells. Collectively, these cell types make up the glia and cerebrovasculature that support the health and maintenance of neurons. Importantly, most of these cells remain mitotic throughout life and hence are potential targets of clastogenic toxicants.
Several brain cells (astrocytes, microglia, pericytes, endothelial cells, and some neurons) work together to form the neurovascular unit (NVU) (Muoio et al., 2014). The NVU detects the needs of the brain parenchyma, triggers appropriate chemotactic responses (e.g. vasodilation, vasoconstriction), and regulates the composition and tightness of the blood-brain barrier (BBB). In a healthy body and healthy brain, the vast majority of xenobiotics are kept out of the brain parenchyma by the BBB (Zheng et al., 2014; Haddad-Tóvolli et al., 2017; Gawdi and Emmady, 2020; Zheng and Ghersi-Egea, 2020). It is unknown if Cr(VI) can cross this barrier, or damage it. Alternatively, Cr(VI) could enter the brain through the olfactory bulb, which is unprotected by the BBB and is directly exposed to ambient air through its nerve fenestrations into the nasal septum (Hanson and Frey, 2008). Additionally, the hypothalamus and pituitary have a weaker BBB due to their roles in hormonal axes (e.g. hypothalamus-pituitary-gonad axis) and thus have regular interactions with blood components (Haddad-Tóvolli et al., 2017). Finally, Cr(VI) could enter the brain through cerebrospinal fluid (CSF). One study investigating metal-on-metal hip arthroplasty observed a small exchange of Co and Cr from blood plasma to CSF (Harrison-Brown et al., 2020). Yet, no data have been reported on Cr(VI) ability to cross the brain-CSF barrier either.
Hexavalent chromium is a widely recognized environmental pollutant with potential for carcinogenic, teratogenic and mutagenic effects. However, understanding the neurological health effects of Cr (VI) exposures are just beginning. Perhaps this is due to a lack of evidence for occupational Cr(VI) exposures linked to major neurodegenerative diseases (e.g. Alzheimer’s or Parkinson’s diseases). Regardless, we recognize the significant neurological health risks associated with exposure to non-essential heavy metals, and much is yet to be understood about most of these metals (Karri et al., 2016; dos Santos et al., 2016; Su et al., 2017). Our knowledge of brain health and function is growing exponentially with the development of newer imaging tools and more powerful computers to analyze them; meanwhile our understanding of how our environment affects brain health and function lags behind. Much is yet to be understood about neurotoxicology and how chemicals alter the health and function of the brain (especially non-neuronal cells), and our ability to treat or prevent many diseases will be limited until we do.
The aim of this review is to summarize and discuss the available literature on Cr(VI) neurotoxicity. Our discussion will begin summarizing reports with humans, considering: Cr levels in human brain regions, risk for brain cancer after occupational exposure, Cr metallosis from failed metal-on-metal hip replacements, Cr(VI) and autism spectrum disorder, other neurological disturbances linked to Cr exposures, and the lack of regulations for Cr(VI) neurotoxicity. Then we will discuss reports with animals, considering: dose- and time-dependent Cr deposition in the brain, impacts on animal behavior, oxidative damage, histopathology, and other neurotoxic endpoints. We will discuss comparisons across exposure routes, novel insights considering co-exposures with antioxidants or other metals, and limitations present in the literature for Cr(VI) neurotoxicity. We briefly discuss the observations from cell culture studies. We then discuss studies considering prevention or intervention of Cr(VI) toxicology, current limitations and new insights from Cr(VI) neurotoxicology. We conclude our discussion with future directions for Cr(VI) neurotoxicology research.
2. Methods
2.1. Search Strategy
International databases (PubMed and Science Direct) and online search engine (Google Scholar) were searched for relevant publications throughout the composition of this review. The first search was executed on 3/11/2020, and the final search was on 1/7/2021. Searches included: 1) ((chromium) and brain) and toxicology, 2) ((chromium) and brain) and disease, 3) (chromium) and brain levels, 4) ((chromium) and neurologic), 5) ((chromium) and neurotoxicity). Additionally, search terms “chromate”, “hexavalent chromium”, “Cr(VI)” were used in place of “chromium”, and “neuro” was used in place of “brain” to avoid missing papers. This search method was enhanced by following referenced literature under consideration in this review, and by considering articles listed under “Cited By” or “Related Articles” links under search results.
2.2. Exclusion Criteria
From all results obtained from searches we excluded: 1) articles not relevant to the review topic, 2) articles on pharmacology of trivalent chromium [Cr(III)], 3) articles that may have mentioned Cr as a measurement but did not report Cr data.
3. Assessment of Cr Levels in Human Brain
An understanding of the toxic effects of Cr(VI) on the brain starts with determining what normal, non-toxic levels of Cr are present. Here, we discuss brain Cr levels observed in individuals who are presumably not occupationally exposed (occupation was not reported for these studies). The earliest studies from 1975 and 1977 investigated the levels of trace elements including Cr in a few key regions of the brain – the cortex, basal ganglia, and pituitary (Kanabrocki et al., 1975; Gooddy et al., 1975; Höck et al., 1975; Persigehl et al., 1977). Gooddy et al. (1975) reported a mean brain Cr level of 0.03 μg/g based on 10 patients that died of accidents or cardiac infarction and resided in urban areas. Analysis of brain regions showed mean levels of 0.03 and 0.09 μg/g for the frontal lobe cortex and basal ganglia, respectively, from 2 patients. By analyzing similar brain regions in subjects aged 5 hours to 74 years, Höck et al. (1975) reported cortex and basal ganglia Cr levels ranging from 0.21–1.35 μg/g and 0.43–16.11 μg/g dry weight (0.65–4.09 μg/g and 1.32–28.82 μg/g wet weight), respectively. From their table, there were three individuals aged 64+ and three younger than 25; Cr levels in each of the older individuals was higher than each of the younger individuals, suggesting bioaccumulation across lifespan. Kanabrocki et al. (1975) measured levels of Cr in four of fourteen pituitaries sampled: 0.53, 0.67, 0.33, and 0.83 mg/g dry weight (160.6, 203.03, 100.0 and 251.15 μg/g wet weight) for individuals aged 54, 58, 63, and 72 years, respectively. Finally, Persigehl et al. (1977) measured trace elements in various human organs in three age groups: 0–1, 20–40, and 50–83 and reported the brain Cr levels as 0.8–4.2, <2, and 2.0–9.6 μg/g dry weight (0.24–1.27, <0.61, 0.61–2.91 μg/g wet weight), respectively. While these studies were not able to discriminate the Cr valence state, the >100-fold differences between pituitary and cortex or basal ganglia Cr levels is striking and may reflect the weaker BBB in pituitary or may reflect an unknown essential role for pituitary Cr. These results also may suggest some credence to studies investigating a pharmacological role for Cr(III) in modulating insulin signaling in diabetes and hyperglycemia (Moreira et al., 2019; Farrokhian et al., 2019; Peng and Yang, 2015).
Since these initial studies, several more reports on Cr levels in the brain have been published and are summarized in Table 1 and Figure 1. Markesbery et al. (1984) conducted a thorough examination of trace elements in multiple brain regions across ages from premature births to 85 years old. While the data in the paper was limited, we are able to make some key insights. Levels of Cr were gradually increased with age (except for the 60–79 age group where a decrease was observed), and when data were grouped as adults versus infants, a statistically significant increase in Cr levels was found in adults (0.13 ± 0.01 vs. 0.07 ± 0.02 μg/g in infants, respectively), suggesting lifespan bioaccumulation. When considering Cr levels regionally in the brain (regions ranged from <0.01 to 0.60 μg/g), considerably more variation across brain regions than other elements was noticed. The highest Cr levels were observed in regions of the cerebellum (0.27 and 0.26 μg/g in the cerebellar hemisphere and vermis, respectively) and lowest levels were observed in the caudate nucleus and putamen (0.05 and 0.08 μg/g, respectively). Unlike the rest of the brain, however, hippocampal Cr levels decreased with age. Rajan et al. (1997) reported a total brain Cr content of 0.19 ± 0.03 mg. Here, the highest Cr level observed was in the temporal cerebrum (0.18 μg/g), an area involved in auditory function and processing information such as speech and words; whereas the lowest level was observed in the hippocampus (0.10 μg/g). Calderón-Guardeñas et al. (2013) reported a unique study design, investigating metal levels in the frontal lobes of individuals living in a heavily-polluted city (Mexico City) vs. those living in low-pollution Mexican cities (Tlaxcala and Veracruz). All brain samples were collected from individuals that died suddenly in accidents and were otherwise healthy. Of the 11 metals measured, Cr was sixth highest reported at 910 ± 63 μg/g and 527 ± 85 μg/g dry weight (1210 ± 84 μg/g and 701 ± 113 μg/g wet weight) for Mexico city residents and low-pollution city residents, respectively.
Table 1.
Cr Levels Reported in Human Brain and CSF
| Brain Region | Age (n) | Cr Concentration (μg/g) | Quantification Method | Brain Health Status | Reference |
|---|---|---|---|---|---|
| Whole Brain | N.R. (10) | 0.01 | SSMS | Healthy | Gooddy et al., 1975 |
| Frontal Cortex | N.R. (2) | 0.03 | Healthy | ||
| Basal Ganglia | 0.09 | ||||
| Cerebral Cortex | 5 h – 74 y (8) | Range = 0.648–4.088* | INAA | Healthy | Höck et al., 1975 |
| Basal Ganglia | Range = 1.315–48.818* | ||||
| Pituitary | 42 – 83 (14) | Detected in 4/14, by age: 54 = 160.6* 58 = 203.03* 63 = 100.0* 72 = 251.15* |
INAA | Healthy | Kanabrocki et al., 1975 |
| Brain | 0 – 1 (7) 20 – 40 (6) 50 – 83 (6) |
Range = 0.242–1.273* <0.606* Range = 0.606–2.909* |
INAA | Healthy | Persigehl et al., 1977 |
| Brain | Adults (28) | 0.127 ± 0.013 Range = <0.006–0.602 |
INAA | Healthy | Markesbery et al., 1984 |
| Infants (7) | 0.067 ± 0.015 | ||||
| Cerebellar hemisphere | Adults (28) | 0.265 | |||
| Cerebellar vermis | 0.260 | ||||
| Putamen | 0.260 | ||||
| Caudate nucleus | 0.0520 | ||||
| Hippocampus | 0.0807 | ||||
| Cerebral cortex (L hemisphere) |
Age-matched (23) | 2.42 ± 1.61* | INAA | Healthy | Guisun et al., 1991 |
| Astrocytoma | 28 – 64 Mean = 45 (15) |
1.76 ± 1.67* | Cancer | ||
| Frontal cerebrum | N.R. (8) | 0.15 ± 0.05 | ICP-AES | Healthy | Rajan et al., 1997 |
| Temporal cerebrum | 0.18 ± 0.12 | ||||
| Parietal cerebrum | 0.16 ± 0.09 | ||||
| Somatosensory cortex | 0.13 ± 0.07 | ||||
| Occipital cerebrum | 0.14 ± 0.08 | ||||
| Cerebellum | 0.11 ± 0.06 | ||||
| Mid-brain | 0.12 ± 0.06 | ||||
| Pons | 0.13 ± 0.04 | ||||
| Hypothalamus | 0.11 ± 0.05 | ||||
| Thalamus | 0.14 ± 0.09 | ||||
| Hippocampus | 0.10 ± 0.08 | ||||
| Medulla oblongata | 0.12 ± 0.06 | ||||
| Frontal Cortex | 33.06 ± 4.8 (59) | 701 ± 113* | ICP-MS | Low-pollution cities | Calderón-Garcidueñas et al., 2013 |
| 1210 ± 84* | Mexico city (high pollution) | ||||
| Temporal cortex | 30 (1) | 1.36* | INAA | Healthy | Civit et al., 2000 |
| Oligodendroglioma | 38 (1) | 1.12* | Cancer | ||
| Meningioma | 64 (1) | <0.242* | Cancer | ||
| CSF | 63.8 ± 13.7 (13) | 1.39 ± 0.64 ng/mL | SF-ICP-MS | Control | Bocca et al., 2004 |
| 64.9 ± 10.8 (26) | 0.60 ± 0.47 ng/mL | Parkinson’s disease | |||
| CSF | 66.2 ± 14.7 (20) | 1.28 ± 0.59 ng/mL |
SF-ICP-MS | Control | Alimonti et al., 2007 |
| 64.5 ± 10.6 (42) | 0.65 ± 0.46 ng/mL | Parkinson’s disease | |||
| CSF | 63 (252) | 5–24 nmol/L (0.26–1.25 ng/mL) | ICP-MS | Control | Harrison-Brown et al., 2020 |
| 65 (141) | 6–36 nmol/L (0.31–1.87 ng/mL) | Well-function implant | |||
| 64 (68) | 7–32 nmol/L (0.36–1.66 ng/mL) | Hip Revision |
N.R. = Not Reported; SSMS = Spark-Source Mass Spectrometry; INAA = Instrumental Neutron Activation Assay; ICP-AES = Inductively Coupled Plasma-Atomic Emission Spectroscopy; SF-ICP-MS = Sector Field-Inductively Coupled Plasma-Mass Spectrometry
Cr values adjusted from dry weight to wet weight by adjusting for 77% brain water content, according to Keep et al., 2013
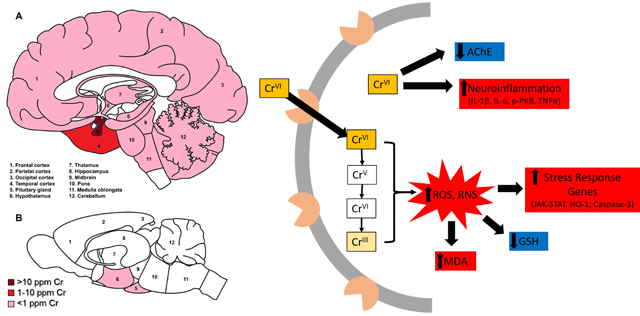
Figure 1. Relative distribution of reported Cr deposition in human (A) and rodent (B) brains.
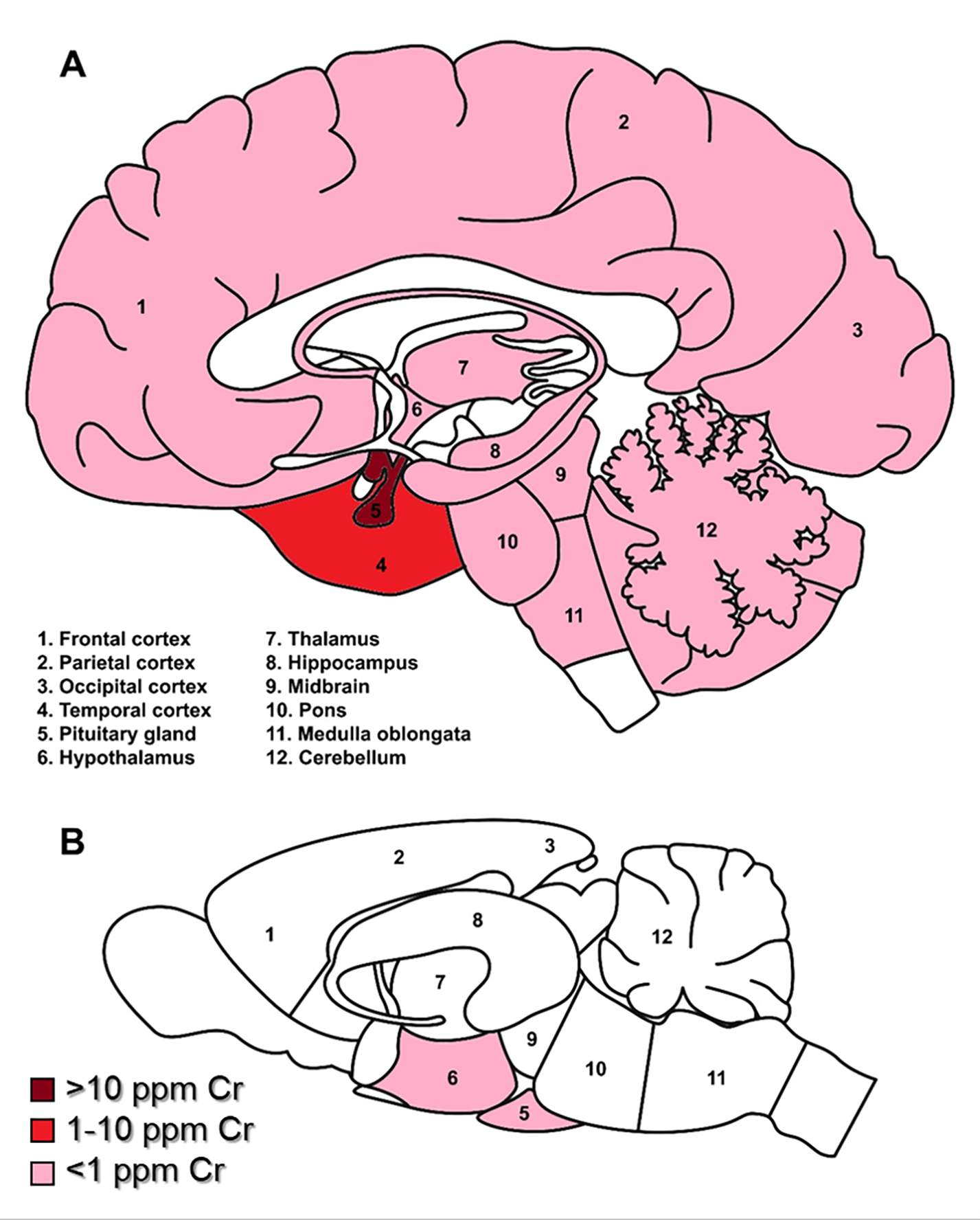
Dark red indicates Cr levels greater than 100 μg/g; red = 10–100 μg/g; pink indicates less than 1 μg/g; white areas indicate no data reported. NB: most rodent studies measured whole brain Cr, thus there is limited data for regional Cr deposition.
Only a few studies have reported Cr levels in abnormal brain conditions. Two early studies have reported Cr levels in a total of 17 brain tumors, but neither found a difference between Cr levels in control cortex tissue and either astrocytoma (n = 15), oligodendroglioma (n = 1), or meningioma (n = 1) tissue (Guisin et al., 1991; Civit et al., 2000). However, two recent studies compared levels of trace elements in cerebrospinal fluid (CSF) from Parkinson’s disease patients to age-matched controls (Alimonti et al., 2007; Bocca et al., 2004). Both studies observed lower Cr levels in CSF of Parkinson’s patients by approximately half that of controls.
In conclusion, Cr accumulates in the human brain and likely bioaccumulates over the course of a lifespan. Two studies observed high levels of Cr in the pituitary and hypothalamus, though it is not clear if this reflects a potential role for Cr in these regions, or if the elevated levels are due to a weaker BBB. Other brain regions observed with high Cr levels were cerebellum and temporal lobe. These areas are not known to be affected by major neurodegenerative diseases, but could have other detrimental impacts on an individual’s life if significantly damaged (e.g. impaired speech or locomotion). Further studies are needed to better understand Cr deposition and valence state in the human brain, what contributes to regional differences, and what potential symptoms or health effects might arise from Cr-induced damage.
4. Human Studies Involving Cr(VI) and Neurological or Neurocognitive Effects
The last few decades have seen a growing number of studies investigating Cr(VI) neurological endpoints in humans, pointing to the onset of paresthesia, depression, gait disturbance, essential tremor, short-term memory loss, difficulty concentrating, impaired olfactory function, and impaired child neuropsychological development (see Table 2 for summary).
Table 2.
Summary of Cr (VI) Neurological Studies in Humans
| Occupation/ Exposure | Geographic Region | Date(s) of Exposure | Number People Studied | Measured Cr(VI) Exposure | Health Effect(s) | Calculated Risk | Reference |
|---|---|---|---|---|---|---|---|
| Cancer Studies | |||||||
| Ferroalloy and Stainless Steel Production | France | 1 year, 1952–1982 |
2,269 | N.R. | Brain cancer | SMR = 3.36 (0.41–12.24 | Moulin et al., 1990 |
| Arc Welder | Germany | 6+ months, 1950–1970 |
2,416 | N.R. | Brain cancer | 2.7 (0.2–29.3) RR | Becker et al., 1991 |
| “Chromium and compounds” | Denver, CO | 1976–1983 | 252 cases 222 controls |
N.R. | Brain cancer | Father’s OR = 0.8 (0.3–2.8) | Feingold et al., 1991 |
| Boiler welder | Norway | 1942–1981 | 2,597 | N.R. | Brain cancer | SIR = 102 (49–188) | Danielson et al., 1996 |
| Chrome tannery worker | Italy | 6+ months, 1955–1988 | 1,244 | N.R. | Brain cancer | SMR = 168 (46–431) | Montanaro et al., 1997 |
| Concrete worker | Sweden | 1971–1986 | 33,503 | N.R. | Brain cancer | SIR = 107 (88–129) | Knuttson et al., 2000 |
| Turner, toolmaker | Finland | N.R. | 413,877 | 12 μg/g/m3 chromates | Brain cancer | SIR = 11.9 | Wesseling et al., 2002 |
| Occupational | All (meta-analysis) | N.R. | (Meta-analysis) | N.R. | Brain cancer | SMR = 88 (64–118) | Cole and Rodu, 2005 |
| Chromium salts | Canada | N.R. | 21,020 cases 5,039 controls |
N.R. | Brain cancer | Multivariable OR: Men = 1.40 (0.74–2.63) Women = 1.06 (0.22–5.12) Both = 1.35 (0.75–2.41) |
Pan et al., 2005 |
| Cr Plating | Japan | N.R. | 1,193 | N.R. | Brain cancer | SMR = 9.14 (1.81–22.09) | Hara et al., 2010 |
| Cement Production | Korea | 1988–2007 >1 day |
10,740 | N.R. | Brain cancer | 3 cases SMR = 3.58 (0.74–10.47) |
Koh et al., 2011 |
| Aircraft Manufacturing Worker | Burbank, CA | >1 year, 1960–1996 | 77,943 | 7,458 exposed workers (16.5%) | Brain/CNS cancer | Chromates SMR = 0.88 (0.54–1.36) Non-factory SMR = 1.09 (0.90–1.32) |
Lipworth et al., 2011 |
| Jet Fuel Manufacturing | Connecticut, USA | 1952–2004 | 210,784 | Mean = 0.2 μg/g/m3 | Glioblastoma | OR: >0–0.065 = 1.00 (0.55–1.68) 0.066–0.093 = 0.93 (0.56–1.50) 0.094+ = 0.81 (0.53–1.22) |
Marsh et al., 2013 |
| Jet Fuel Manufacturing | Connecticut, USA | 1952–2004 | 113,439 | Mean = 0.24 μg/g/m3 | Glioblastoma | RR: >0–0.065 = 1.17 (0.79–1.72) 0.066–0.093 = 0.75 (0.54–1.04) 0.094+ = 0.90 (0.67–1.23) |
|
| Ferrochromium and stainless steel production | Finland | 1967–2004 | 8,146 | 22–143 μg/g/m3 | Brain cancer | SIR: 0.93 (0.55–1.47) |
Huvinen and Pukkula, 2013 |
| Welding | 7 countries (mostly UK & Germany) | N.R. | 1,800 cases 5,160 controls |
N.R. | Glioma | No significant effect detected | Parent et al., 2017 |
| Occupational | All (meta-analysis) | N.R. | (Meta-analysis) | N.R. | Cancer | CNS cancer = 1.22 (0.67–2.23) Brain cancer = 1.04 (0.87–1.24) |
Deng et al., 2019 |
| Metal-on-Metal Hip Replacement Studies | |||||||
| Failed Metal-on-Metal Hip Replacement | Japan | 2 years | 1 | 221 μg/g/L | Polyneuropathy | N.R. | Ikeda et al., 2010 |
| Failed Metal-on-Metal Hip Replacement | United Kingdom | 8 ± 1.7 years | 25 | Blood = 1.42 (1.01–3.86) μg/g/L | Benign essential tremor; regional decreased gray matter | N.R. | Clark et al., 2014 |
| Failed Metal-on-Metal Hip Replacement | United Kingdom | 4.4 years | 10 | Blood = 338 nmol/L (144–164) | Depression, short term memory deficit, neurocognitive deficit | N.R. | Green et al., 2017 |
| Failed Non-Metal-on-Metal Hip Replacement | Netherlands | 6 months, fatal | 1 | Blood = 48.8 μg/L | Asymmetrical hearing loss, visual impairment, unintentional weight loss | N.R. | Peters et al., 2017 |
| Failed Metal-on-Metal Hip Replacement | France | <1 year | 1 | Blood = 65.9 μg/L | Rapid cognitive decline, behavioral disorders, lack of memory, brutal hearing and visual loss | N.R. | Leocoanet et al., 2019 |
| Other Health Endpoints | |||||||
| Chromate factory worker | Japan | 7+ years post-employment | 33 | N.R. | Impaired olfaction, perforated septum (unrelated to olfaction loss) | N.R. | Watanabe and Fukuchi, 1981 |
| Chromium Production Worker | Baltimore, MD | N.R. | 2,354 | N.R. | Cause of death: Mental & psych disorders; Nervous system disorder |
SMR: 1.42 (1.00–1.95) 0.53 (0.32–0.83) |
Gibb et al., 2015 |
| Cr Plating | Korea | 0.9–18.2 years (avg = 7.9) |
N.R. | Cr(III) = 0.059, 0.021, 0.0063, 0.0047 mg/m3 Cr(VI) = 0.013, 0.033, 0.0054, 0.0047 mg/m3 |
Impaired olfactory recognition, but detection unaffected | N.R. | Kitamura et al., 2003 |
| River Pollution | N Spain | Lifetime | 9,434 | N.R. | Motor Neuron Disease | SMR = 1.12 (1.08–1.17) 15.7% incr risk |
Sánchez-Díaz et al, 2018 |
| Environmental Pollution | S Spain | Lifetime (age 6–11, avg 7.8) |
393 | 0.44 (SD±1.32) μg/g/L urine 0.93 (SD±2.45) μg/g creat 0.33 (SD±1.13) μg/g hair |
Impaired neuropsychological development | N.R. | Caparros-Gonzalez et al., 2019 |
N.R. = Not reported; SMR = standard mortality ratio; SIR = standard incidence ratio; OR = odds ratio; RR = relative risk
4.1. Cr(VI) and Brain Cancer
Currently, eleven studies have considered links between occupational Cr(VI) exposure and brain cancers; of these, three reported a positive association. Becker et al. (1991) first reported two cases of German welders with at least a 6-month occupational exposure who died of brain tumors, with a risk ratio of 2.7 (95% CI = 0.2–29.3) for Cr(VI). Hara et al. (2010) conducted a cohort study of 1193 Japanese men employed at a Cr plating factory (mean age was 50) between 1970 and 1976, among which 626 workers (52%) had six months or longer working as a Cr plater, while 567 (48%) worked with other metals (i.e. nickel, zinc, precious metal, aluminum). This cohort showed an elevated mortality risk for brain tumors for Cr platers, with a standard mortality ratio (SMR) of 9.14 (95% CI = 1.81–22.09). In sum, three Cr platers had brain tumors compared to only one in the non-Cr plater population. A retrospective case study with Finnish women with brain/CNS cancers and employed during the 1970 census also showed women who were employed as turners or toolmakers had an estimated standard incidence ratio (SIR) of 11.9 after exposure to 12 μg/m3 chromates (Wesseling et al., 2002). Importantly, this was the second highest reported SIR in this study; second to aliphatic/alicyclic hydrocarbon solvents at 20 ppm (SIR = 28), but still 10-fold higher than the next risk (1.5 mg/m3 of oil mist, SIR 1.80).
Contrary to these reports, eight studies reported a lack of brain cancer risk with occupational Cr(VI) exposure, including two meta-analyses (Cole and Rodu, 2005; Deng et al., 2019). Marsh et al. (2013) evaluated risk of glioblastoma in jet engine manufacturers by a dual design to consider risk from a case-control study and an incidence cohort study from 8 plants in Connecticut, and considered 12 separate components of jet engine fuel, including Cr. They reported 304 cases of workers exposed to a mean of 0.2 μg/m3 Cr within their case-control study, and 113,439 exposed to 0.24 μg/m3 Cr within the incidence cohort study. However, their univariate exposure-risk analyses showed no significant risk for Cr in either study design. In a case-control study from the Canadian National Enhanced Cancer Surveillance System, Pan et al. (2005) evaluated the impact of occupational exposures on brain cancer risk. The analysis considered occupational exposure to Cr salts, but found no significance regardless of occupation duration or gender. In a population-based case-control study spanning seven countries (primarily Germany and United Kingdom), Parent et al. (2017) assessed occupational exposure to welding fumes and risk of developing glioma. They reported 178 (9.9%) cases and 359 (7.0%) controls were exposed to Cr, with an odds ratio of 0.9 (95% CI, 0.7–1.1), but no significant effect between any metal and glioma risk. Feingold et al. (1991) investigated the link between childhood cancers (age 0–14) and the parents’ job-exposure matrix during the year prior to the child’s birth. Their results showed no significant association between either parent’s exposure to Cr compounds and child brain cancer, but the authors noted a suggestive dose-response gradient for children with Cr-exposed fathers. Two other studies considered brain cancer risk in jobs known to have significant risk of Cr(VI) exposure. Moulin et al., 1990 considered a cohort of French workers producing ferroalloys and stainless steel, and reported a nonsignificant SMR of 3.36 for CNS tumors (3 CNS tumors observed in 2,269 workers). Danielsen et al. (1996) investigated cancer incidence among Norwegian boiler welders, observed 10 instances of brain cancers in 2,957 welders (SIR = 102; 49–188). Importantly, brain cancers are a common phenotype in metastatic lung cancers (Schuette, 2004). Two studies emphasize this issue, finding 20–30% of non-small cell lung cancer patients developed metastatic brain cancers and both the Kentucky Cancer Registry and the Alberta Cancer Registry reported the highest incidence of brain metastasis from lung cancers (Porta et al., 2011; Villano et al., 2015). Hence it may be the few studies that observed a significant link between Cr(VI) exposure and brain cancer incidence were actually observing metastatic lung cancers, especially considering the vast majority of Cr(VI)-induce lung cancers are squamous cell carcinomas occurring at lung bifurcation sites (Proctor et al., 2014; Speer and Wise, Sr., 2018).
4.2. Metal-on-Metal Hip Replacements and Co-Cr Metallosis
The systemic effects of Co-Cr metallosis after failed metal-on-metal (MoM) arthroplasty is thoroughly reviewed elsewhere, and discusses several neurological effects observed including: peripheral neuropathy, sensori-neural hearing loss, visual impairment, and cognitive decline (Bradberry et al., 2014). Importantly, ~12.5% of MoM replacements require revision surgery within ten years, mostly due to wearing down. With at least 500,000 people receiving MoM hip implants in the U.S. alone (as of 2012), at least 62,000 people will be faced with a MoM hip replacement failure (Rising et al., 2012). This presents a unique exposure route for Co and Cr, where the worn down metal particles can embed into neighboring tissue and slowly leach these metals into the blood. Further, most of these exposures will occur in elderly individuals, who may by differentially susceptible to metal toxicity due to natural aging or co-morbidities. When a patient presents with a variety of symptoms following MoM revision surgery, blood Co and Cr levels are routinely measured, providing a universal reference for comparison across studies. To discuss a few examples; several studies have reported neurological problems in patients exhibiting Cr and cobalt metallosis from 2, 4, or 8 years living with a failed MoM hip replacement (Ikeda et al., 2010; Clark et al., 2014; Green et al. 2017). One report was a case study of a 56-year-old woman exhibiting polyneuropathy two years after a failed MoM hip replacement (Ikeda et al., 2010). This patient exhibited a variety of neurological symptoms including progressive and painful dysesthesia in all extremities, impaired joint sensation, numbness and tingling in distal limbs, auditory disturbance, and was found to have moderate axonal degeneration without inflammation in a sural nerve biopsy. Blood Cr and cobalt levels were reported to be 221 and >400 μg/L, respectively. Two years after replacement surgery the blood metal levels were reduced and the neurological symptoms largely resolved. Green et al. (2017) reported 6 women and 4 men exhibiting neurological deficits following an average of 4 years with a MoM hip replacement failure. Mean blood Cr and cobalt levels were 338 and 669 nmol/L, respectively. Neurological symptoms included depressed mood, short-term memory deficit, location disorientation, difficulty concentrating, and neurocognitive deficit in 7 of the 10 patients. Finally, Clark et al. (2014) reported altered brain structure and function after 8 years of MoM hip replacement failure in 25 men (age 59 ±7) with 1.42 and 1.72 μg/L blood Cr and cobalt, respectively. This study reported benign essential tremor as well as decreased gray matter in the occipital cortex, putamen, and left head of the caudate nucleus. Importantly, Co toxicity can elicit many of the symptoms observed with MoM failed hip replacements, including: cardiomyopathy, hypothyroidism, polycythemia, cognitive dysfunction, neuropathy, and fatigue (Peters et al., 2017). Neurological symptoms observed in patients with failed MoM hip replacements also include essential tremor, peripheral neuropathy, auditory loss, and visual loss not typically observed with Co toxicity alone, and thus may be elicited by Cr neurotoxicity or the interaction of Co and Cr neurotoxicity.
4.3. Cr(VI) and Autism Spectrum Disorder
Heavy metal dyshomeostasis is a new perspective thought to contribute to autism spectrum disorders (ASD), as evidenced by multiple studies reporting differential metals levels in ASD children (Jory and McGinnis, 2008; Yasuda et al., 2005; Adams et al., 2006; Yorbik et al., 2010; Skalny et al., 2017a,b). Most of these studies assessed multiple metals in blood, hair, or serum, and frequently reported a significant association between Cr and ASD A Canadian study of 10 children diagnosed with autism and 15 age-matched control children found elevated Cr levels in red blood cells of autistic children (25.68 ±12.10 vs 21.15 ±13.31 nmol/L, respectively) (Jory and McGinnis, 2008). Six studies analyzed hair metals in association with ASD; five reported significantly lower Cr levels in ASD children, while one reported elevated levels (Yasuda et al., 2005; Adams et al., 2006; Al-Farsi et al., 2013; Skalny et al., 2017a; Tinkov et al., 2019; Al-Ayahdi, 2005). One study considered subgroups of autism with or without presence of pica, and reported Cr as the most statistically significant of 39 toxic metals assessed and concluded Cr was the most likely factor in etiology of autism with pica (Adams et al., 2006). Another considered ASD subgroups with or without catatonia; they observed elevated hair and serum Cr levels in ASD with catatonia (Tinkov et al., 2019). Their results observed the most significant associations between metal level and ASD with catatonia for Cu, Cr, and V. A Spanish study considered childhood Cr exposure (hair and urine Cr levels) and neuropsychological development for 393 children (171 males, 222 females, mean age 7.8) living in Cr-polluted areas of southern Spain (Caparros-Gonzalez et al., 2019). They concluded even low Cr levels can affect child attention development, and observed high Cr levels linked to poorer performance on neuropsychological tests. Similarly, an earlier study investigated urine levels of heavy metals including Cr, cadmium, and lead in children with autism (30 cases) and healthy children (20 controls). Urine cadmium and lead levels were significantly lower, but urine Cr levels were significantly higher in children with autism than healthy children, suggesting the potential risk of Cr exposure for the children to develop autism (Yorbik et al., 2010). In fact, a population-based case-control study from Pennsylvania identified an association between autism spectrum disorders and in utero exposure to elevated ambient Cr(VI) levels, as measured by the National Air Toxics Assessment (Talbott et al., 2015). In sum, there is provocative evidence that Cr plays a role in the etiology of ASD and further investigation is warranted.
4.4. Cr(VI) and Other Neurological Endpoints
The neurotoxicity of Cr(VI) to humans has been considered in a handful of other contexts. Two studies have shown chromate workers in Japan and Korea exhibited olfactory dysfunction correlated to employment duration (Watanabe and Fukuchi, 1981; Kitamura et al., 2003). Importantly, the two studies observed the olfactory impairment was unrelated to septal perforation. Sánchez-Díaz et al. (2018) conducted a geographical analysis of heavy metal pollution in rivers of Spain and 9,434 cases of motor neuron disease deaths. They reported increased risk for motor neuron disease with seven heavy metals; of these metals, Cr was reported to have a 15.7% increased risk and linked to 2,068 deaths (21.9% of total deaths). A recent meta-analysis demonstrated a link between higher blood Cr in patients with acute or newly diagnosed schizophrenia, but was not linked to patients with chronic or previously treated schizophrenia (Saghazadeh et al., 2019). Their results suggest increased Cr levels during early stages of schizophrenia development may cause or aggravate abnormalities of serotonin, contributing to disease burden. One study considered airborne metal emissions and adjudication of youth for felonies, reporting a statistically significant association for five metals considered, with the exception of Cr (Haynes et al., 2011).
4.5. Regulations Lacking Considerations for Cr(VI) Neurotoxicity
Neurotoxic effects are not considered in any safety regulations for Cr(VI) exposure. In fact, the “Toxicological Profile for Chromium” from the Agency for Toxic Substances and Disease Registry (ATSDR), which is considered the gold standard for toxicological reference, does not consider most of the literature discussed here and lacks much needed information on Cr(VI) neurotoxicology. Safety levels for inhalation exposures are determined with data pertaining to pulmonary health effects from epidemiology or animal studies. However, the results presented by Hegazy et al. (2021) suggest real-life exposures to Cr(VI) pose a significant neurotoxic risk. Here, the authors used intra-nasal instillation of potassium dichromate at concentrations that mimicked what an electroplater might be exposed to (2.4 mg Cr[VI]/kg/d), exposing rats to 0.125, 0.25 or 0.5 mg/kg/d for 5 consecutive days and evaluated neurotoxic endpoints after 2 weeks, 1 month, or 2 months of this exposure regiment. Their results demonstrated behavioral and neuropathological effects that are discussed later. EPA standards for drinking water defines a chronic oral reference dose of 0.003 mg Cr(VI)/kg/d, based on a study with rats exposed to 2.5 mg Cr(VI)/kg/d in drinking water for 1 year (ATSDR, 2012; MacKenzie et al., 1958). This study did not consider behavior or neurological effects, and there are currently no studies focused on Cr(VI) neurotoxicity at or below 2.5 mg/kg/d for comparison. Finally, two recent review articles reporting on Cr(VI) regulations and health effects failed to mention neurotoxicology as a potential health effect after Cr(VI) exposure (Vaiopoulou and Gikas., 2020; Alvarez et al., 2021). Hence, there is much needed consideration for neurotoxicology in our national and international discussion about Cr(VI) toxicology.
4.6. Summary of Cr(VI) Neurotoxicity Observed in Humans
In sum, these papers demonstrate the need for deeper understanding of Cr(VI) impacts on brain health, development, and neurobehavior (Table 2). Given Cr(VI)’s known roles in carcinogenesis, it is not surprising brain cancer has been considered most often as an endpoint in these human studies. While most of the evidence suggests a lack of association with cancers, some indeed raise the possibility that Cr(VI) may contribute to carcinogenesis in the brain by mechanisms yet to be explored. Two separate groups reported loss of olfaction in chromate workers. This may be an early warning sign for other neurological problems, as hyposmia is a prodromal symptom of Alzheimer’s disease and Parkinson’s disease. Results from the failed MoM hip replacements were unique in that the Cr(VI) exposure was internal and was in elderly individuals. We do not yet understand the interactions between aging and metals toxicology, but this collection of papers may provide us with a unique glimpse of what to expect for Cr(VI): social memory loss, spatial disorientation, depression, polyneuropathy, and loss of regional brain mass. While these neurological symptoms were recognizable in a clinical setting following surgery, such symptoms could be easily missed in someone environmentally or occupationally exposed to Cr(VI). The handful of other neurological conditions linked to Cr(VI) exposures further reflect our poor understanding of its neurotoxicity; autism spectrum disorder, schizophrenia and motor neuron disease are all distinct neurological conditions with different etiologies.
5. Experimental Studies and Potential Mechanisms for Cr(VI) Neurotoxicity
5.1. Animal Models Used for Studying Cr(VI) Neurotoxicity
The neurotoxicity of Cr(VI) has been tested in nine distinct species to date (rat, mouse, rabbit, guinea pig, chicken, zebrafish, rockfish, Japanese quail, and C. elegans), primarily with sodium chromate (Na2CrO4) or potassium dichromate (K2Cr2O7) administered intraperitoneally or in drinking water. Toxic endpoints are often inconsistent across studies, making comparisons between most studies difficult. The most common endpoints include brain Cr accumulation, oxidative damage (most often assessed by malondialdehyde), and effects on acetylcholinesterase. A few studies considered other endpoints including: behavioral changes, histopathology, neuroinflammation, and brain element homeostasis. The experimental details for animal models reported for Cr(VI) neurotoxicity are summarized in Table 3.
Table 3.
Animal Models in Cr (VI) Neurotoxicology Studies
| Species (Gender) | Cr Compound | Exposure Route | Dose(s) | Duration | Cr Level Reported (ug/g) | Method of Detection | Reference |
|---|---|---|---|---|---|---|---|
| Rabbit (M) | K2Cr2O7 | i.p. | 2 mg/kg | 3 and 6 weeks | 1.6 and 1.3 | Not reported | Mathur et al., 1977 |
| Wistar rat (M) | Na2CrO4 | d.w. | 700 mg/L | 28 d | N/A | Diaz-Mayans et al., 1986 | |
| 70 mg/L | 7 d | N/A | |||||
| i.p. | 2 mg/kg | 24 h | N/A | ||||
| Swiss mice (F) | K2Cr2O7 | d.w. | 25 mg/kg | 1–3 d | 0.5 | AAS | Travacio et al., 2000 |
| C57BL/6Ntac (F) | Na2CrO4 | Oral | 95 mg/kg | 24 h | N/A | Bagchi et al., 2001 | |
| C57BL/6TSG p53 (M) (p53 deficient) | |||||||
| Albino mice (M) (ddY strain) |
K2Cr2O7 | i.p. | 20 mg/kg | 15 min, 3 h, 24 h |
0.28 ± 0.13, 0.31 ± 0.27, 0.15 ± 0.03 |
AAS | Ueno et al., 2001 |
| C57BL/6Ntac (F) | Na2CrO4 | Oral | 1.9, 19, 95 mg/kg | 0, 12, 24, 48, 72, 96 h | N/A | Bagchi et al., 2002 | |
| Wistar rats (M) | K2Cr2O7 | d.w. | 500 ppm = 73.05 mg Cr/kg/d |
30 d | Pituitary = 0.64 Hypothalamus = 0.059 |
AAS | Quinteros et al., 2007 |
| Wistar rats (M) | K2Cr2O7 | d.w. | 100 ppm = 11.6 mg Cr/kg/d |
30 d | Pituitary = 0.274 Hypothalamus = 0.026 |
AAS | Nudler et al., 2009 |
| Swiss albino mice (M) | K2Cr2O7 | i.p. | 8.0 mg/kg | 12h | 1.40 | ICP-MS | Döker et al., 2010 |
| Wistar rats (F) | K2Cr2O7 | d.w. | 700 ppm | 21 d | N/A | Soudani et al., 2012 | |
| Wistar rats (M) | K2Cr2O7 | i.p. | 15 mg/kg | 24 h | N/A | García-Niño et al., 2015 | |
| Albino Wistar rats (M) | K2Cr2O7 | i.p. | 15 mg/kg | 24 h | 5.4 | AAS | Salama et al., 2016 |
| i.n. | 0.5, 1, 2 mg/kg | 24 h | 0.13, 0.41, 0.92 | ||||
| Rockfish, S. schlegelii | K2Cr2O7 | Diet | 30, 60, 120, 240 mg/kg | 2 or 4 weeks | N/A | Kim and Kang, 2016 | |
| Guinea pig (M) | K2Cr2O7 | i.p. | 60 μg/g/kg | 3 weeks | N/A | Fahmy, 2017 | |
| Wistar rat (M) | K2Cr2O7 | i.p. | 2 mg/kg | 30 d | N/A | Mahmoud and El-Twab, 2017 | |
| Wistar rat (M) | Cr2O3 NP | Oral w/ cannula | 0.5 or 2 mg/kg | 24 h, 7 d, 14 d | N/A | Fatima et al., 2017 | |
| Wistar rat (M) | Cr(OH)3 NP | Intratracheal instillation | 2 mg/kg welding fumes w/ Mn and Fe | once a day, 5x/week, for 4 weeks | N/A | Máté et al., 2017 | |
| Hyland brown chicken (M) | K2Cr2O7 | d.w. | (6% LD50) | 14, 28, 42 d | N/A | Hao et al., 2017 | |
| Swiss mice, nulliparous (F) | Tannery effluent | Environmental sludge | 500 mL (containing 859 mg/L Cr) |
2 h/d, 20 d | N/A |
Estrela et al., 2017
|
|
| Sprague-Dawley rats (M) | K2Cr2O7 | Oral | 25 mg/kg | 6 weeks | N/A | Abu Zeid et al., 2018 | |
| Zebrafish (Danio rerio) |
K2Cr2O7 | Water | 3, 271, 2700 μg/g/L | 24 h | N/A | Heffern et al., 2018 | |
| Drosophila melanogaster | K2Cr2O7 | Diet | 5, 10, or 20 μg/g/mL | 24 h or 48h | N/A | Singh and Chowdhuri, 2018 | |
| Wistar rats (M) | K2Cr2O7 | i.p. | 14 mg/kg (0.5 LD50) | 24 h | N/A | Iztleuov et al., 2018 | |
| Chicken | K2Cr2O7 | Oral | 22.14 mg/kg | 14, 28, 42 d |
0.1669 ± 0.0041, 0.2520 ± 0.0075, 0.2918 ± 0.0040 |
ICP-MS | Zhu et al., 2018 |
| Sprague-Dawley rats (F) | K2CrO4 | Oral gavage | 10 mg/kg | 2 weeks | N/A | Sarica et al., 2019 | |
| Zebrafish (Danio rerio) | K2CrO4 | Water | 2 mg Cr/L | 1, 7, 15, 30, or 60 d | N/A | Shaw et al., 2020 | |
| Japanese Quail (Coturnix japonica) | K2Cr2O7 | d.w. | 1.2, 2.4 μg/mL |
20 d | 10.58 ± 4.5 10.62 ± 6.94 |
USEPA Methods 3060A and 7196A: alkaline digestion and colorimetric spectrophotometry | Suljević et al., 2021 |
| Wistar rats (M) | K2Cr2O7 | i.n. | 0.125, 0.25, 0.5 mg/kg/d | 5 d per week, 2 weeks and 2 months | N/A | Hegazy et al., 2021 |
NP = nanoparticles; i.p. = intra peritoneal; d.w. = drinking water; i.p. = intra peritoneal; d.w. = drinking water; i.n. = intra-nasal AAS = atomic absorption spectrometry; ICP-MS = inductively coupled plasma-mass spectrometry; N/A = not assessed
5.1.1. Cr brain levels in animal models
Only one study has conducted a thorough assessment for regional distribution of normal brain Cr levels in an animal model; Wasim et al. (2014) reported Cr levels in cerebellum, cerebrum, medulla oblongata, meninges, midbrain, pons, and thalamus from five healthy goats. They reported a whole brain average Cr level of 0.54 μg/g and regional levels ranged from 0.10 to 1.20 μg/g, with the highest level observed in the meninges and the lowest in the pons. In addition, Zhu et al. (2018) reported brain Cr levels in chickens from a control group in their investigation of Se-Cr interactions. Groups of five chickens were euthanized after 14, 28, and 42 days in the study exhibited brain Cr levels of 0.0015 ± 0.0004, 0.0026 ± 0.0003, and 0.0036 ± 0.0004 μg/g, respectively. Intriguingly, their observations suggest Cr bioaccumulation in the chicken brain with age, similar to what was observed in humans.
Eight Cr(VI) neurotoxicity studies have considered brain Cr accumulation in their reports (Nota Bene: two studies used a trivalent nanoparticle form, Cr [NO3]3, but were included in this review because the focus was on neurotoxicity and not neuropharmacology). These studies included doses ranging from 2–73.05 mg/kg with Cr administered either via intra-peritoneal (i.p.) injection or drinking water and resulted in brain Cr levels ranging from 0.06 μg/g in the hypothalamus to 5.4 μg/g in whole brain. Generally, i.p. injection of Cr(VI) resulted in higher accumulation in the brain than exposure through drinking water. One study compared i.p. injection to intra-nasal administration, observing similar proportions of Cr deposition relative to administered dose (Salama et al., 2016). Two studies considered multiple exposure times in rabbits and mice and reported similar levels regardless of time, though much is yet to be learned about Cr deposition in the brain over time (Mathur et al., 1977; Ueno et al., 2001). Two studies from the same group considered regional differences of Cr accumulation in the brains of Wistar rats after administering 100 or 500 ppm Cr(VI) in drinking water (Quinteros et al., 2007; Nudler et al., 2009). These reported Cr levels in the pituitary and hypothalamus in comparison to liver levels; liver levels were about 100x higher than brain levels. Comparing the two studies, they observed a 2.3x higher Cr level in both pituitary and hypothalamus after 500 ppm vs 100 ppm Cr(VI) in drinking water. However, these reports did not include cortex or brain homogenate Cr levels, so comparison to other studies is currently limited. Altogether, these studies demonstrate Cr(VI) can readily enter the brain and accumulate with time and prolonged exposure, but there is a severe lack of regional analysis to determine where Cr deposits in the brains of laboratory animals.
5.1.2. Cr(VI) and neurobehavior
Behavior is an essential aspect of neurotoxicology analyses in animal studies, as it is the most informative way to link neurotoxicology health effects across species. Unfortunately, the Cr(VI) literature is brief regarding behavior analyses. Hegazy et al. (2021) used an intra-nasal model to expose rats to 0.125, 0.25, or 0.5 mg Cr/kg/d for five days each week, and assessed neurobehavior impacts after two weeks, one month, or two months of this exposure regiment. They reported dose- and time-dependent effects for: 1) significantly decreased locomotor activity in an activity cage, 2) significantly impaired learning and memory with an object recognition test, and 3) impaired motor coordination. Singh and Chowdhuri (2017) reported dose-dependent decreased climbing ability and jumping activity of fruit flies after 48 h Cr(VI) in their food, also supporting an effect of Cr(VI) on locomotor function and coordination. Heffern et al. (2018) observed no olfactory deficit in zebrafish after 24 hours exposure to potassium dichromate, even up to 2,700 μg/L. After a single intranasal doses of Cr(VI) of 0.5, 1.0 or 2.0 mg/kg, rats exhibited dose-dependent decreased motor activity by 42%, 59%, and 77%, respectively (Salama et al., 2016). One possible cause of this behavior the authors did not consider could be cerebellar toxicity, which was one of the highest Cr-laden regions in human brains; if the rats were uncoordinated they would likely move less. Estrela et al. (2017) observed short-term social memory deficits in female mice exposed to tannery effluent. Here, the authors used tannery effluent provided by an industry in Goiás State, Brazil and reported an effluent Cr level of 859 mg/L (at least 150x higher than other metals). This unique model exposed mice to 500 mL tannery waste for 2 hours per day for 20 consecutive days. In order to test social recognition, an “intruder” mouse was introduced to a resident mouse’s cage for 3 minutes, removed for 15 minutes, then re-introduced for 3 minutes again, with all encounters being recorded. A buried food test was also employed to test olfactory function. The tannery effluent-exposed mice exhibited no difference in the buried food test, but spent less time exploring the anogenital region and longer time displaying aggressive behavior toward an “intruder” mouse on the second encounter. Thus, the olfactory function was intact but their ability to recognize mice they recently encountered was impaired, and the authors concluded that direct contact with tannery effluent may cause short-term social memory deficit. While challenging to do, social memory would be an interesting parameter to test in humans employed at tanneries or other occupations with high Cr(VI) exposure (Takahashi et al., 2004). Intriguingly, these few behavior studies reflect some of the neurological effects observed in humans; e.g. social memory loss in patients with failed MoM hip replacements.
5.1.3. Cr(VI) brain oxidative stress, damage, and redox changes
Upon entering a cell, Cr(VI) is rapidly reduced to Cr(III) by ascorbate and biological thiols (e.g. glutathione). Along with ephemeral Cr(IV) and (V) species, the reduction also produces ROS such as superoxide anions, hydroxyl radicals and nitric oxide (Bagchi et al., 2001). The role of oxidative stress in neurodegenerative diseases has been thoroughly examined and is amply discussed in other reviews (see Niedzielska et al., 2015; Uttara et al., 2009; Chen et al., 2012). Most Cr(VI) neurotoxicity studies in rodents have measured redox changes, considering both enzymatic [superoxide dismutase (SOD), catalase (CAT)] and non-enzymatic [glutathione (GSH), ascorbate] antioxidants, and measuring malondialdehyde (MDA) levels for oxidative damage, providing a useful panel of biomarkers for comparing toxicity across studies. For a summary of redox data from Cr(VI) neurotoxicity in animal models literature (Table 4); to simplify comparisons we adjusted the data as percentage of control based on reported data.
Table 4.
Summary of Redox Data from Cr (VI) Neurotoxicity Animal Studies
| Species (Exposure Route) |
Dose, Duration | Lipid Peroxidation | Non-Enzymatic Antioxidant | Enzymatic Antioxidant Activity | Other Oxidative Stress or Damage Measure | Reference | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SOD | CAT | GPx/GR | |||||||||
| Mouse (oral) |
1.9, 19, 95 mg/kg, 12, 24, 48, 72, 96 h |
1MDA: 1.9 = unaffected; 19 = 1.6x incr after 72h; 95 = 3.2x incr after 48h |
N/A | N/A | N/A | N/A | N/A | Bagchi et al., 2002 | |||
| Mouse (d.w.) |
25 mg/kg, 3 d |
TBARS incr 29% | α-tocopherol decr 32% | Vit E and C unaffected | Incr 65% | Incr 74% | GPx unaffected | Travacio et al., 2000 | |||
| Mouse (oral) |
25 mg/kg, 6 wk |
MDA incr 1.92x | GSH decr 58% | N/A | N/A | GR decr 55% | Protein carbonyl incr 2x | Abu Zeid, 2018 | |||
| Rat (d.w.) |
100 ppm (11.6 mg Cr/kg/d), 30 d |
MDA incr: HT = 30% AP = 65% |
N/A | Incr 2x in AP | Unaffected | GR incr 2x in HT; GPx unaffected |
N/A | Nudler et al., 2009 | |||
| Rat (d.w.) |
700 ppm, 21 d |
MDA incr: CTX = 1.2x CB = 94% |
GSH decr: CTX = 52% CB = 15% |
NPSH decr: CTX = 37% CB = 46% |
Vit C decr: CTX = 56% CB = 53% |
N/A | N/A | N/A | Soudani et al., 2012 | ||
| Rat (oral) |
0.5 or 2 mg/kg, 24 h, 7 d, 14 d |
MDA incr: 24 h = unaffected 7 d (2 mg/kg) = 4x 14 d = 6.7x, 9.3x |
GSH decr: 24 h = 40%, 70% 7 d = 53%, 76% 14 d = 88%, 95% |
SOD: 24 h = Unaffected, 1.8x incr 7 d = unaffected, 33% decr 14 d = 50%, 75% decr |
N/A | N/A | N/A | Fatima et al., 2017 | |||
| Rat (i.p.) |
2 mg/kg, 30 d |
MDA incr 2.5x (CTX) | GSH decr 30% | Decr 50% | N/A | GPx decr 40% | NO incr 1.8x (CTX) | Mahmoud and El-Twab, 2017 | |||
| Rat (oral) |
10 mg/kg, 2 wk |
MDA incr 1.9x | GSH unaffected | Nonsig decr 23% | Nonsig decr 42% | N/A | N/A | Sarica et al., 2019 | |||
| Rat (i.p.) |
15 mg/kg, 24 h |
Plasma MDA incr 88% | N/A | Unaffected | Unaffected | Both unaffected; GPx non-significant 33% decr | N/A | García-Niño et al., 2015 | |||
| Rat (i.n.) |
0.5, 1.0, 2.0 mg/kg, 24 h |
MDA incr: 0.5 = 1.21x 1.0 = 1.61x 2.0 = 2.21x |
GSH decr: 1 = 88% 2 = 75% |
N/A | Decr: 1.0 = 72% 2.0 = 30% |
N/A | N/A | Salama et al., 2016 | |||
| Rat (i.p.) |
15 mg/kg, 24 h |
MDA +67% | GSH decr 42% | Non-protein thiols decr 36% | Unaffected | Decr 33% | GR decr 25% | Iztleuov et al., 2018 | |||
| Zebrafish (water) | 2 mg/L, 1, 7, 15, 30, 60d |
MDA incr 1.77x on day 15 | GSH incr 1.33x on day 60 | N/A | 15d = 1.58x 30d = 2.58x 60d = 2.35x |
N/A | N/A | Shaw et al., 2020 | |||
| Rat (i.n.) |
0.125, 0.25, 0.5 mg/kg/d, 2 weeks | MDA incr: 0.125 = 1.23x 0.250 = 1.77x 0.500 = 2.65x |
GSH decr: 0.125 = 18% 0.250 = 40% 0.500 = 42% |
N/A | N/A | N/A | N/A | Hegazy et al., 2021 | |||
| 0.125, 0.25, 0.5 mg/kg/d, 1 month | MDA incr: 0.125 = 4.34x 0.250 = 7.70x 0.500 = 7.98x |
MDA incr: 0.125 = 45% 0.250 = 52% 0.500 = 52% |
N/A | N/A | N/A | N/A | |||||
| 0.125, 0.25, 0.5 mg/kg/d, 2 months | MDA incr: 0.125 = 7.31x 0.250 = 8.00x 0.500 = 8.21x |
MDA incr: 0.125 = 54% 0.250 = 55% 0.500 = 59% |
N/A | N/A | N/A | N/A | |||||
Nota Bene: Data was adjusted to percent of control based on reported mean values.
N/A = not assessed; CTX = cortex/cerebrum; CB = cerebellum; HT = hypothalamus; AP = anterior pituitary; i.p. = intra peritoneal; d.w. = drinking water
Data reflect largest observed effect
Only a few papers have sought to quantify ROS associated with Cr(VI) neurotoxicity by various methods. Singh and Chowdhuri (2018) measured ROS using the fluorescent probe DHE and reported 1.7-fold increased fluorescence in brain ganglia of Drosophila after receiving dietary Cr(VI) for 48 h. Travacio et al. (2000) measured brain ROS with the DCFH-dAc reporter and observed 47% increased fluorescence in Swiss mice given 25 mg/kg Cr(VI) in drinking water for 3 days. Mahmoud and El-Twab (2017) reported 1.8-fold increased nitric oxide in rat cortex following 2 mg/kg Cr(VI) i.p. exposure for 30 days. The effects of ROS toxicity in the brain are widespread in neuropathologies, and thought to contribute to protein aggregations, mitochondrial dysfunction, autophagic dysfunction, and neurodegeneration (Tabner et al., 2005; Federico et al., 2012; Dasuri et al., 2013; Singh et al., 2019; Michalska and León, 2020).
Most studies demonstrate decreased non-enzymatic antioxidant levels after Cr(VI) intoxication, primarily considering GSH (Table 4). The relative decreased GSH levels observed across these studies were 15–95% decreased after Cr(VI) exposure. Yet, two papers observed GSH significantly increased following Cr(VI) exposure. One study observed increased GSH in rat cortex (52%) and cerebellum (15%) after Cr(VI) exposure in drinking water (Soudani et al., 2012). While the other instance of Cr(VI)-induced GSH increase (1.33x) occurred in a zebrafish model (Shaw et al., 2020). Another paper reported no effect on rat brain GSH levels after orally administered Cr(VI) (Sarica et al., 2019). Hegazy et al. (2021) observed a dose- and time-dependent decrease in GSH following intra-nasal instillation of potassium dichromate.
The reported effects of Cr(VI) on activity of enzymatic antioxidants [SOD, CAT, and glutathione peroxidase or reductase (GPx or GR)] are much more varied. Three papers reported no effect of Cr(VI) on brain SOD activity in rats and chickens (García-Niño et al., 2015; Iztleuov et al., 2018; Hao et al., 2017) and one paper reported ~50% decreased SOD activity in rats (Mahmoud and El-Twab, 2017). Fatima et al. (2017) conducted the most thorough analysis published to date for SOD activity after Cr(III) nanoparticle exposure in rats for 24 hours, 7 days, or 14 days. After 24 hours, they observed no effect in their low dose (0.5 mg/kg) and ~1.8-fold increased SOD activity in their high dose (2.0 mg/kg), but observed decreased levels at both doses in longer exposures (7–14 days). Two other rodent studies reported elevated SOD activity after Cr(VI) intoxication for 3 or 30 days (Travacio et al., 2000; Nudler et al., 2009). Seven studies considered Cr(VI) effects on CAT activity, with varying effects observed. Two studies in rats showed no effect after 30 days exposure in drinking water or i.p. 24 hours (Nudler et al, 2009; García-Niño et al., 2015). Salama et al. (2016) reported decreased CAT activity after 24 hours intranasal exposure to 1 or 2 mg/kg potassium dichromate, with a greater effect observed after 1 mg/kg (72% and 30% decreased, respectively). Similarly, rats given Cr(VI) for 24 hours via i.p. exhibited a 33% decreased CAT activity (Itzleuov et al., 2018). Conversely, two studies reported increased CAT activity after Cr(VI) exposure. Travacio et al. (2000) reported 74% increased CAT activity in mice given Cr(VI) in drinking water for 3 days. In a study with zebrafish exposed to Cr(VI) for up to 60 days there was a time-dependent increase of CAT activity (Shaw et al., 2020). Here, the authors observed an initial non-significant decrease of CAT activity after 7 days, then a significant increase after 15, 30 or 60 days (1.58x, 2.58x, 2.35x increase, respectively). Six studies considered Cr(VI) effects on the glutathione peroxidase (GPx) or glutathione reductase (GR) activity, predominantly reporting decreased activity for each. The largest effects observed were a 55% decrease in GR activity in mice exposed to 25 mg/kg orally for 6 weeks and a 40% decrease in GPx in rats given i.p. 2 mg/kg Cr(VI) for 30 days (Abu Zeid et al., 2018; Mahmoud and El-Twab, 2017). Only one study reported an increase in either; Nudler et al. (2009) reported 2-fold increased GR activity in hypothalamus of rats receiving 100 ppm Cr(VI) for 30 days.
Thus far, every study that has considered brain MDA levels after Cr(VI) exposure has reported elevated levels following exposure (Table 4). MDA is an end product of lipid peroxidation and often elevated in peripheral blood of patients with neurodegenerative diseases (Dib et al., 2002; Greilberger et al., 2008; Ozcankaya and Delibas, 2002). Importantly, MDA can have detrimental effects on neuronal mitochondrial respiration, targeting mitochondrial complexes I, II, and V and promote formation of ROS (Long et al., 2009). The highest reported Cr(VI)-induced MDA levels in a vertebrate model were from Hegazy et al. (2021). Using a rat model exposed to 0.5 mg Cr/kg/d, 5 d/week for two months, they observed a dose- and time-dependent increase in rat brain MDA levels that peaked at two months with 127.24 nmol MDA/g tissue (8.21x increased MDA). Concomitantly, 14 days also exhibited the greatest reduction in GSH levels, which were decreased by ~59%. Based on the reported data, MDA currently serves as the best biomarker to compare Cr(VI)-induced oxidative damage in rodent brain tissues and is consistently elevated upon exposure. Whereas antioxidant responses to Cr(VI) intoxication are variable across studies, possibly due to the sensitivity of redox balance or gender and age differences in brain ascorbic acid levels (Kume-Kick and Rice, 1998).
5.1.3. Cr(VI) Neuropathology
Nine papers reported histopathological assessments in their Cr(VI) neurotoxicity studies; only one reported a lack of histopathological effects in rats (García-Niño et al., 2015). Soudani et al. (2012) were the only to report tumor formation, an oligodendroglioma in a female rat. Widespread neurodegeneration was observed in multiple studies across multiple species, with notable toxicity to Purkinje cells of the cerebellum (Soudani et al., 2012; Fahmy, 2017; Hao et al., 2017). Hegazy et al. (2021) observed a dose- and time-dependent significant decrease in the number of viable neurons in cortical gray matter, with increased reactive astrogliosis. The authors noted GFAP+ astrocytes were significantly increased in number and characterized by cellular hypertrophy. Gliosis and satellitosis were also frequently observed. However, these effects were most likely responses to neurodegenerative effects and not due to direct Cr(VI)-induced activation, as these were typically observed at the highest doses and durations tested and were often associated with neurodegeneration. Other neurodegenerative changes including hemorrhage, neuronal vacuolation, edema, and neuronophagia were reported after Cr(VI) intoxication (Mathur et al., 1977; Soudani et al., 2012; Salama et al., 2016; Fahmy, 2017; Fatima et al., 2017; Hao et al., 2017; Abu Zeid et al., 2018; Shaw et al., 2020). Currently it appears Cr(VI) has a widespread neurotoxic effect with no clear evidence of regional or phenotypic specificity, but more detailed studies are needed to evaluate this further.
5.1.4. Other Endpoints Considered for Cr(VI) Neurotoxicity
A few other points are worth discussing from the available Cr(VI) animal neurotoxicity studies: effects on inflammation, acetylcholinesterase, and trace element homeostasis.
Three papers measured neuroinflammation and found elevated pro-inflammatory cytokines following Cr(VI) exposure in rats (Salama et al., 2016; Mahmoud and El-Twab, 2017; Hegazy et al., 2021). Salama et al. (2016) reported a dose-response increase in brain IL-1β and phospho-PKB after 0.5, 1.0, or 2.0 mg/kg Cr(VI) exposure, but they attributed the ~4x increased levels for both cytokines to widespread necrosis that was evident at the highest Cr(VI) concentration. Mahmoud and El-Twab (2017) reported ~3- and ~4.7-fold increased TNF-α and IL-6 following daily 2 mg/kg i.p. injection of Cr(VI) for 30 days. Hegazy et al. (2021) observed dose- and time-dependent increases in IL-1β, PI3K, and PKB following intra-nasal instillation of potassium dichromate.
Four studies considered Cr(VI) effects on brain acetylcholinesterase, a key enzyme for clearing the neurotransmitter acetylcholine from synapses. Importantly, alterations to the cholinergic system have been observed in Alzheimer’s disease and disrupted REM sleep, an early non-motor symptom of Parkinson’s disease (Francis et al., 1999; Platt and Riedel, 2011). In all cases, studies showed Cr(VI) decreased acetylcholinesterase activity by at least 40% (Soudani et al., 2012; Kim and Kang, 2016; Mahmoud and El-Twab, 2017; Abu Zeid et al., 2018).
Several studies considered changes in expression of stress response genes. Shaw et al. (2020) observed upregulation of stress response genes Nrf2, Nqo1, HO-1, and Ucp2 as well as apoptotic genes p53, Bax, Caspase-9, and Caspase-3 after 60 days Cr(VI) exposure in zebrafish. Nudler et al. (2009) reported elevated levels of metallothionein isoform 3 (MT3) in hypothalamus, of metallothionein isoform 1 (MT1) in anterior pituitary gland, and HO-1 in both regions of rats after 30 days exposure to 100 ppm Cr(VI) in drinking water. Importantly, they observed no effect on these genes in the liver, suggesting the brain is more vulnerable than the liver to Cr(VI) in drinking water. Mahmoud and El-Twab (2017) observed elevated levels of JAK-STAT genes JAK1 and STAT3, along with elevated SOCS3 in cerebrum of rats after 30 days daily i.p. 2 mg/kg Cr(VI). They suggested the SOCS3 elevation was in response to the JAK-STAT induction, and these effects were associated with the ongoing neuroinflammation. Abu-Zeid et al. (2018) also observed chronic Cr(VI)-induced elevated p51, Bax, and Caspase-3 in rats.
As a measure of Cr(VI)-induced DNA damage, Abu-Zeid et al. (2018) observed approximately 2-fold increased 8-hydroxy-2′-deoxyguanosine (8-Oxo-dG) in brain homogenates of rats exposed to Cr(VI) orally for 6 weeks.
Zhu et al. (2018) assessed trace element homeostasis in chickens following 14, 28, or 42 days of potassium dichromate exposure. Their results showed levels of calcium, copper, zinc, and magnesium increased following Cr(VI) exposure, while manganese and iron levels decreased with Cr(VI) exposure doses and time.
Singh and Chowdhuri (2017) utilized reporter strains of fruit flies to assess Cr(VI)-induced toxicity across neuronal subtypes (cholinergic or dopaminergic) and in glial cells of brain ganglia. They showed a dose- and time-dependent decrease in fluorescence intensity for flies with a pan-neuronal marker (ELAV-GAL4), but no change in fluorescence intensity for flies with a glial cell marker (Repo-GAL4). Fly strains with cholinergic neurons labeled (ChAT-GAL3) exhibited a dose- and time-dependent decrease in fluorescence intensity, while dopaminergic neurons (Ddc-GAL4) only exhibited a decrease in neuron count at the highest and longest Cr(VI) exposure studied. These observations were supported by their Annexin V staining, demonstrating significantly elevated apoptosis in their highest dose (20 μg/mL) and longest duration (48 h).
5.1.5. Summary of Neurotoxicity Observed in Animal Models
It is clear from these in vivo studies that Cr(VI) can accumulate in the brain and induce histological and biochemical damage. Impaired locomotor and memory functions were observed in multiple studies following Cr(VI) exposure. Oxidative damage, AChE levels, and neurodegeneration have been most often considered for Cr(VI) neurotoxic endpoints. Some studies have shown elevated pro-inflammatory cytokines in response to Cr(VI). Much work remains to determine what regions or cell types are most vulnerable, and what neurobehavioral deficits might arise.
5.2. Cell culture work
Three different groups have reported Cr(VI) neurotoxicity studies in cell culture, using concentrations ranging from 0.1–100 μM Cr(VI) for up to 72 h; the experimental details are summarized in Table 5. Quinteros et al. (2007, 2008) observed different sensitivities to Cr(VI) for different neuronal subtypes (lactotrophs and gonadotrophs) within the anterior pituitary. Using primary rat neuronal cultures, they demonstrated Cr(VI) induced ROS- and caspase-3-mediated apoptosis more severely in lactotrophs than gonadotrophs. Dashti et al. (2014) exposed mouse primary cerebellar granule cells (CGCs) at various stages of neuronal maturation, and identified immature CGCs were significantly more sensitive than cells undergoing maturation or fully matured. Subsequently, this same group observed elevated ROS production (DCFH-DA) and lipid peroxidation (MDA) in PC12 cells exposed to potassium dichromate for 24, 48, or 72 hours and reported IC50 values of 22.02, 1.88, and 1.85 μM, respectively. Altogether, these studies agree Cr(VI) induces ROS-mediated apoptosis in a concentration-dependent manner, but the impact of exposure duration was inconsistent. These studies assert the need for further in vivo investigation into Cr(VI) neurotoxicology to assess sensitivities to neuronal subtypes and toxicity across neurodevelopment. Already we can see a difference in Cr(VI) sensitivity between lactotrophs and gonadotrophs in the hypothalamus and different effects depending on neuron maturity (Quinteros et al. 2007, 2008).
Table 5.
Summary of Cr (VI) Cell Culture Neurotoxicology Studies
| Cell Line | Cell Type | Compound | Dose(s) | Duration | Observations | Reference |
|---|---|---|---|---|---|---|
| Primary anterior pituitary | Primary, rat | K2Cr2O7 | 0, 0.1, 1, 10 uM | 48 h, 72 h | Lactotrophs were more sensitive than gonadotrophs; 10 μM Cr induced ROS for first 2 h; caspase-3 activated after 10 μM Cr for 6, 12, 24 h | Quinteros et al., 2007 |
| Primary anterior pituitary | Primary, rat | K2Cr2O7 | 10 uM | Hours, up to 24 h | Increased PFTα suggests p53 is not involved in Cr(VI)-induce apoptosis; oxidative stress-mediated apoptosis | Quinteros et al., 2008 |
| Cerebellar granule cells | Primary, mouse | K2Cr2O7 | 0.1 – 100 uM | 24 h, 48 h | Mature neurons are more sensitive to Cr(VI) toxicity than immature neurons | Dashti et al., 2014 |
| PC12 | Rat pheochromocytoma | K2Cr2O7 | 1 – 100 uM | 24 h, 48 h, 72 h | ROS and lipid peroxidation significantly increased | Dashti et al., 2015 |
| SH-SY5Y | Human neuroblastoma | K2Cr2O7 | 1 – 10 uM | 24 h | Cr(VI) induces neuronal toxicity trough ROS-mediated mitochondrial-dependent apoptosis | Fu et al., 2020 |
5.3. Potential prevention and interventional studies
Several studies have considered antioxidant co-administration to alleviate Cr(VI) neurotoxicity. Selenium has a well-established role for quenching ROS in the brain (see review by Solovyev, 2015), and has been considered in three separate Cr(VI) neurotoxicity studies using rat, chicken, and guinea pig models (Soudani et al., 2012; Hao et al., 2017; Fahmy, 2017). In all three studies selenium co-treatment reduced neuronal loss, and in the rat study appeared to alleviate Cr(VI)-induced oxidative damage and pathology. The guinea pig study also observed an increase in the astrocyte number following Cr(VI), which was decreased by co-administration of selenium and interpreted as decreased astrocyte activation (Fahmy, 2017). These observations suggest selenium prevents ROS-mediated neurodegeneration, alleviating subsequent astrocyte activation. More recently, boron has received similar consideration as an antioxidant supplement (see review by Pizzorno, 2015). When co-treated with Cr(VI), two studies in rats showed boron attenuated Cr(VI)-induced oxidative damage and endogenous antioxidant depletion (Sarica et al., 2019; Iztleuov et al., 2018). Ascorbic acid is widely known for attenuating Cr(VI) toxicity and is well documented in the rodent brain, so it is surprising only one study has considered the effects of ascorbic acid on Cr(VI) neurotoxicity (Standeven et al., 1992; Wise et al., 1993; Xie et al., 2004; Kume-Kick and Rice, 1998). Abu Zeid et al. (2018) conducted a rat study involving groups given ascorbic acid for six weeks as either co-treatment (120 mg/kg ascorbate and 25 mg/kg potassium chromate co-administered) or pre- and co-treatment (ascorbate treatment started 2 weeks prior to chromate, and continued for all six weeks). The groups receiving ascorbic acid were found to have: (1) reduced oxidative damage, measured by levels of MDA, protein carbonylation, and 8-oxo-2’-deoxyguanosine, (2) reduced apoptotic markers p-53, Bax and caspase-3, (3) restored endogenous antioxidant enzymes and AChE levels, and (4) alleviated pathological effects (e.g. neuronal vacuolation). Other phytochemicals have been considered for alleviating Cr(VI) neurotoxicity, including caffeic acid phenethyl ester, rosmarinic acid, and curcumin (Mahmoud and El-Twab, 2017; Dashti et al., 2014; García-Niño et al., 2015). In each case, the antioxidant was shown to attenuate Cr(VI) toxicity, but whether this effect was from simply preventing Cr(VI) uptake was not determined in these studies. Singh and Chowdhuri (2018) considered a genetic approach to alleviate Cr(VI) neurotoxicity using a Drosophila model and modulating expression of the oxidative stress response gene sestrin. The authors found sestrin overexpression protected neurons from Cr(VI)-induced death by upregulating autophagy and reducing ROS levels, whereas sestrin knockdown increased neuronal vulnerability to Cr(VI). These studies demonstrate a possible pharmacological intervention for individuals exposed to Cr(VI), such as patients with failed MoM hip replacements; especially since most of these supplements are already commercially available.
6. Current Limitations and New Insights into Cr(VI) Neurotoxicology
Collectively, from human, animal, and cell culture studies it is clear that Cr(VI) is a sinister neurotoxicant that can affect behavior and health in subtle ways. These observations are generally summed up in Figure 2. While Cr(VI) is a known human carcinogen and poses a significant occupational health hazard, there is little evidence that Cr(VI) has a significant contribution to brain cancer, as two meta-analyses of the current literature failed to find an association (Cole and Rodu, 2005; Deng et al., 2019).
Figure 2. Summary of key findings from Cr(VI) neurotoxicity literature.
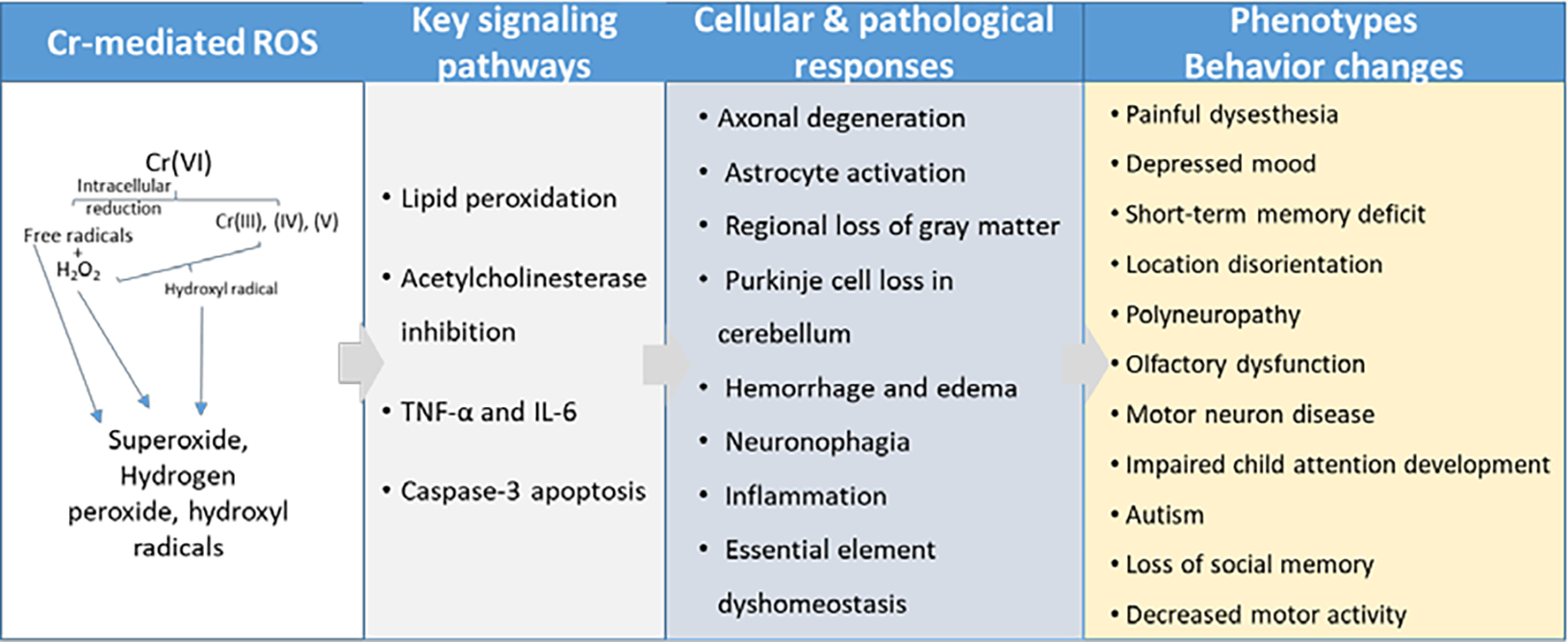
There is growing evidence suggesting Cr(VI) can have a variety of effects on neurobehavior. Various studies in human populations exposed to Cr(VI) exhibited social memory loss, depression, polyneuropathy, olfactory dysfunction, and neuropsychological impairments. These effects are particularly alarming for children and elderly individuals, who seem to be more susceptible to Cr(VI) behavioral effects. Importantly, it seems that neurons are more susceptible to Cr(VI) toxicity during neurodevelopment, as demonstrated in the child neurodevelopment study in Spain, the in utero exposure assessment for autism spectrum disorder, and the study using primary cerebellar granule cells (Caparros-Gonzalez et al., 2019; Dashti et al., 2014; Talbott et al., 2015). However, there is little to no neurotoxicology data investigating how age affects neurotoxic outcome of Cr(VI). Perhaps in children this vulnerability is due to the mitotic activity of a developing brain, as Cr(VI) is a well-defined clastogen. Whereas geriatric vulnerability may be due to clastogenic toxicity combined with life-long accumulation of neurodegenerative endpoints (e.g. protein aggregation, DNA damage accumulation, persistent neuroinflammation, weaker BBB). Data considering Cr(VI) exposure and neurologic morbidity in children and geriatrics would greatly help the Cr(VI) neurotoxicology field define the scope of Cr(VI) neurotoxic potential.
From the collection of animal studies available, it is clear that Cr(VI) is more neurotoxic via inhalation than i.p. injection or drinking water. Salama et al. (2016) demonstrated this with a comparison of i.p. versus intra-nasal administered Cr(VI) for 24 h, showing similar neuropathology with a much lower dose administered intra-nasally. However, i.p. injection does not adequately reflect a real life exposure, and lacking data for blood Cr in these models limits their translatability to occupational or environmental exposure settings. The impact of exposure route is also apparent when comparing across studies using similar concentrations and durations; for example, intra-nasal 0.5, 1.0 or 2.0 mg Cr/kg induced a greater fold increase in MDA than 15 mg Cr/kg i.p. as observed between Salama et al.’s study and others (Table 4). As is the case for most metals, Cr(VI) typically exhibited a dose-response effect for inducing ROS and oxidative damage in the brain. Interestingly, though, the duration of Cr(VI) exposure seemed to have little effect (Bagchi et al., 2002; Dashti et al., 2014). Perhaps the most limiting factors for understanding Cr(VI) neurotoxicity are: 1) the lack of recognizing what brain regions are most vulnerable to Cr(VI), and 2) the lack of recognizing which cell types are most vulnerable to Cr(VI).
Cr(VI) is most often encountered with other metals, but such metal mixtures have not been thoroughly considered. Two studies have considered the neurotoxicity of Cr in combination with other metals. In one study investigating neurotoxicity of welding fumes, rats were given an intratracheal dose of: (i) 2 mg/kg manganese nanoparticles in combination with (ii) 2 mg/kg chromium hydroxide nanoparticles, (iii) 2 mg/kg iron nanoparticles, or (iv) both, once a day, five times a week for four weeks (Máté et al., 2017); the design of this study was to use doses that replicate typical daily exposures for welders. Electrocorticogram readings shifted to higher frequencies and lengthened evoked potential latency following manganese nanoparticle exposure. These effects were strongly diminished by iron, but unaffected by Cr nanoparticles. In the other study, human neuroblastoma cells (SK-N-SH) were exposed to a combination of arsenic, Cr, and copper leached from chromated copper arsenate-treated wood (Hu et al., 2013). Here, the authors noted arsenic neurotoxicity was inhibited by Cr(III), suggesting a potential neuro-pharmacological role for the Cr(III).
7. Conclusions and Future Directions for Cr(VI) Neurotoxicity Studies
The neurotoxicity of Cr(VI) has been considered across a wide variety of species, including humans, rodents, chicken, fish, birds, and flies. The results largely describe to Cr(VI) induction of oxidative stress and oxidative damage, though the implications on long-term brain health and neurobehavior is unknown. Pathologic effects of Cr(VI) on the brain are likely to be regionally specific due to regional differences in GSH and ascorbic acid levels, reducers of Cr(VI) and oxidative damage, and differing vulnerability observed between gonadotrophs and lactotrophs in the anterior pituitary.
7.1. Cr(VI) Entry to the Brain
It’s clear that Cr(VI) can induce widespread oxidative damage, but the effects of that damage have yet to be fully understood, especially in the brain. This widespread effect may indicate Cr(VI) can cross the BBB or, alternatively, target the BBB.
7.2. Cr(VI) and Considerations for Age
We need a better understanding of how Cr(VI) affects neurodevelopment; we can see it is more toxic to immature neurons, exposed children may be neurodevelopmentally challenged, and exposed pregnant mothers have an increased risk of their child developing autism. On the flip side of the age spectrum, little is known how Cr(VI) might affect the brain in an aged individual, where Cr might pass the BBB more easily or might have bioaccumulated to a hazardous level in the brain. One study measured normal Cr levels, without any exposure, in an aging mouse model, observing a non-significant 19.9% decrease in Cr content (Massie et al., 1983). This outcome was a similar trend to Cr levels measured in CSF from Parkinson’s disease patients, begging the question if Cr has an essential role in some brain regions (Bocca et al., 2004; Alimonti et al., 2007). Studies have also shown Cr(VI) induces cellular senescence in bronchial fibroblasts, germ cells, and hepatocytes, suggesting a potential role as a gerontogen, i.e. a chemical that contributes to or accelerates the aging process (Zhang et al., 2016; Sivakumar et al., 2014; Val et al., 2015; Sorrentino et al., 2014). Indeed, a case-control study of welders exposed to Cr(VI) found blood Cr levels to be the main determinant for elevated levels of senescence biomarkers in blood samples, which replicated the observations from their earlier in vitro and in vivo studies (Alexopoulos et al., 2008; Katsiki et al., 2004). Hence, we need to understand more about how Cr(VI) might affect brain aging and an aged brain, which can inform us more about how exposures may contribute to age-related diseases.
7.3. Cr(VI) and Brain Cell Target
Thus far no studies have compared how Cr(VI) affects different types of brain cells, and most cell types in the brain have yet to be considered. There are at least 8 major cell types in the brain, including neurons, astrocytes, microglia, ependymal cells, pericytes, and brain endothelial cells. Most studies have not considered which cell types are responsible for oxidative damage observed in the brain following Cr(VI) exposure, and the implications for each cell type are strikingly different. Studies with homologous cell types in other tissues show the importance of this distinction. A study from Cao et al. (2020) reported Cr(VI) effects on human umbilical vein endothelial cells (HUVECs) and THP-1 cells (monocytes) increased expression of ICAM-1 and VCAM-1, and enhanced the adhesion of THP-1 cells to HUVECs. These results are useful to inform future Cr(VI) neurotoxicity studies investigating effects on brain vascular endothelial cells, and suggest Cr(VI) may affect endothelial cells to recruit peripheral monocytes/macrophages, inducing inflammatory effects. This study also reported Cr(VI)-induction of NF-κB, cleaved caspase-1, and IL-1β in THP-1 cells, which may be translatable to homologous microglial cells in the brain. Given the often observed “widespread neurodegeneration” in Cr(VI) animal studies, it would be informative to determine Cr(VI) effects on cells of the neurovascular unit and BBB. Results from such a study would further aid in identifying other potential neurological endpoints to consider in Cr(VI)-exposed human populations.
7.4. Cr(VI) Neurotoxic Mechanism
Thus far no studies have determined a mechanism for Cr(VI) neurotoxicity. Toxic effects have been pinpointed, such as lipid peroxidation, induction of apoptosis, altered acetylcholinesterase, and changes in neuroinflammatory markers – but no studies have pieced together a mechanism for Cr(VI)’s toxic effects in the brain or in any particular type of brain cells. Figure 3 summarizes the mechanistic data currently available for Cr(VI) neurotoxicology. In other organs and cell types, the mechanisms of Cr(VI) toxicity are much better developed and can provide useful insights for future Cr(VI) neurotoxicology studies (for reviews, see Holmes et al., 2008, Wise et al., 2008, Saha et al., 2011; DesMarias and Costa, 2019). Briefly, Cr(VI) is well characterized to induce ROS in a Fenton-like reaction (Shi and Dalal, 1990; Ding and Shi, 2002). Further details into Cr(VI)-mediated oxidative damage or damage from its resulting ROS may help inform neurodegenerative diseases affected by ROS-driven mechanisms. Cr(VI) is reduced in cells, ultimately to Cr(III) with Cr(V) and Cr(VI) produced during the reduction and these species rival ROS in mechanisms in other organs, but the role of these species is unknown in the brain. Epigenetics play an important role in neurodevelopment and brain organization. Several studies in other cell types illustrate Cr(VI) can disrupt methylation status of multiple genes in a variety of species and can increase expression of histone methyltransferases, key enzymes in modulating methylation status (Sun et al., 2009; Chen et al., 2016; Wang et al., 2018; Lv et al., 2018; Feng et al., 2018). Mitochondrial health is critical to neuronal health, given their high demand for ATP to maintain their polarized state, and many neurodegenerative diseases exhibit impaired mitochondrial dynamics. Recently there has been a resurgence of investigations into Cr(VI) and mitochondrial effects in other cell types, with many reporting altered expression of mitochondrial proteins, altered calcium signaling, and impaired mitochondrial respiration and ATP production (Xiao et al., 2012; Wise et al., 2018; Zhang et al., 2021; Yang et al., 2021). In sum of this review, it is clear Cr(VI) is neurotoxic, but there is much to be discovered.
Figure 3. Schematic of early events in Cr(VI) neurotoxicity.
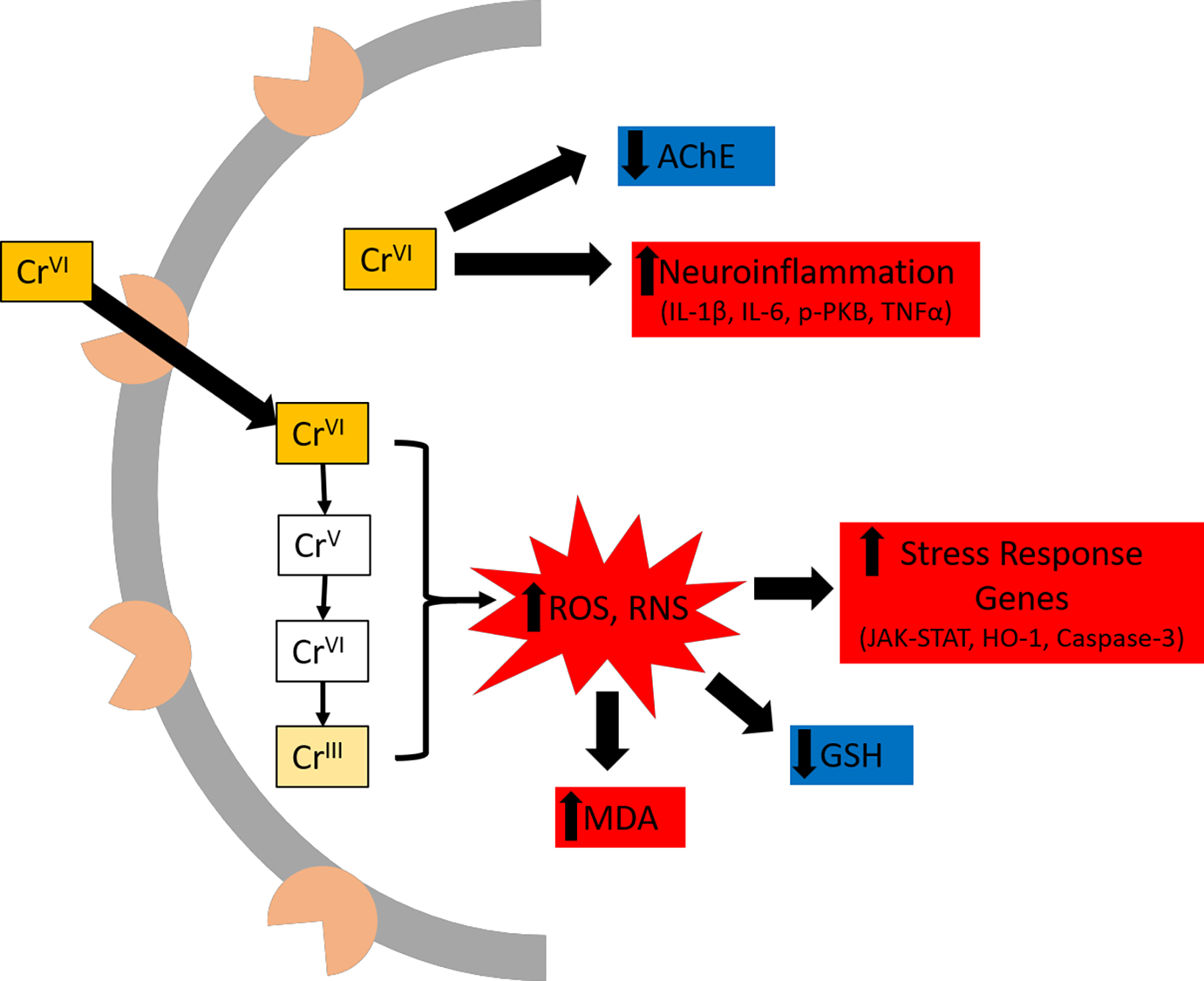
Cr(VI) enters a cell through phosphate or sulfate channels. In the cytoplasm, it is rapidly reduced to Cr(III), which also produces ROS. Oxidative stress and oxidative damage were most often reported as decreased GSH and increased MDA, while several studies have reported increased mRNA of stress response genes often induced by ROS. Cr(VI) increases pro-inflammatory cytokines (e.g. IL-1, IL-6, TNFα) and decreases AChE levels, though it is currently unclear by what mechanism(s). Gray semi-circle represents cell membrane, with pink shapes to represent sulphate/phosphate channels.
Acknowledgements
The authors of this review are supported in part by the National Institutes of Health grants T32-ES011564, R01-ES016893, R35-ES032876, and P30-ES030283, and American Diabetes Association grant 1–18-IBS-02. The authors thank Ami Cai for help producing Figure 1.
Footnotes
The authors have no competing financial interests to declare.
References
- Abu Zeid EH, Hussein MMA, Ali H (2018). Ascorbic acid protects male rat brain from oral potassium dichromate-induced oxidative DNA damage and apoptotic changes: the expression patterns of caspase-3, P 53, Bax, and Bcl-2 genes. Environmental Science and Pollution Research. 25: 13056–13066. [DOI] [PubMed] [Google Scholar]
- Adams JB, Holloway CE, George F, Quig D (2006). Analyses of Toxic Metals and Essential Minerals in the Hair of Arizona Children with Autism and Associated Condition, and Their Mothers. Biological Trace Element Research. 110: 193–209. [DOI] [PubMed] [Google Scholar]
- Al-Ayadhi LY (2005). Heavy metals and trace elements in hair samples of autistic children in Saudi Arabia. Neurosciences. 10(3): 213–218. [PubMed] [Google Scholar]
- Al-Farsi YM, Waly MI, Al-Sharbati MM, Al-Shafaee MA, Al-Farsi OA, Al-Khaduri MM, Gupta I, Ouhtit A, Al-Adawi S, Al-Said MF, Deth RC (2013). Levels of Heavy Metals and Essential Minerals in Hair Samples of Children with Autism in Oman: a Case-Control Study. Biol Trace Elem Res. 151: 181–186. [DOI] [PubMed] [Google Scholar]
- Alexopoulos EC, Cominos X, Trougakos IP, Lourda M, Gonos ES, Makropoulos V (2008). Biological Monitoring of Hexavalent Chromium and Serum Levels of the Senescence Biomarker Apolipoprotein J/Clusterin in Welders. Bioinorganic Chemistry and Applications. 2008: 420578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alimonti A, Bocca C, Pino A, Ruggieri F, Forte G, Sancesario G (2007). Elemental profile of cerebrospinal fluid in patients with Parkinson’s disease. Journal of Trace Elements in Medicine and Biology. 21: 234–241. [DOI] [PubMed] [Google Scholar]
- Alvarez CC, Gómez MEB, Zavala AH (2021). Hexavalent chromium: Regulation and health effects. Journal of Trace Elements in Medicine and Biology. 65: 126729. [DOI] [PubMed] [Google Scholar]
- Agency for Toxic Substances and Disease Registry (ATSDR). 2012. Toxicological profile for Chromium. Atlanta, GA: U.S. Department of Health and Human Services, Public Health Service. [PubMed] [Google Scholar]
- Bagchi D, Bagchi M, Stohs SJ (2001). Chromium (VI)-induced oxidative stress, apoptotic cell death and modulation of p53 tumor suppressor gene. Molecular and Cellular Biochemistry. 222: 149–158. [PubMed] [Google Scholar]
- Bagchi D, Balmoori J, Bagchi M, Ye X, Williams CB, Stohs SJ (2002). Comparative effects of TCDD, endrin, napthalene and chromium (VI) on oxidative stress and tissue damage in the liver and brain tissues of mice. Toxicology. 175: 73–82. [DOI] [PubMed] [Google Scholar]
- Becker N, Chang-Claude J, Frentzel-Beyme R (1991). Risk of cancer for arc welders in the Federal Republic of Germany: results of a second follow up (1983–8). British Journal of Industrial Medicine. 48: 675–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocca B, Amlimonti A, Petrucci F, Violante N, Sancesario G, Forte G, Senofonte O (2004). Quantification of trace elements by sector field inductively coupled plasma mass spectrometry in urine, serum, blood and cerebrospinal fluid of patients with Parkinson’s disease. Spectrochimica Acta Part B. 59: 559–566. [Google Scholar]
- Bradberry SM, Wilkinson JM, Ferner RE (2014). Systemic toxicity related to metal hip prostheses. Clinical Toxicology. 52(8): 837–847. [DOI] [PubMed] [Google Scholar]
- Browning CL, Qin Q, Kelly DF, Prakash R, Jasin M, Wise JP Sr. (2016) Prolonged particulate hexavalent chromium exposure suppresses homologous recombination repair in human lung cells. Toxicological Sciences. 153(1):70–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderón-Garcidueñas L, Serrano-Sierra A, Torres-Jardón R, Zhu H, Yuan Y, Smith D, Delgado-Chávez, Cross JV, Medina-Cortina H, Kavanaugh M, Guilarte TR (2013). The impact of environmental metals in young urbanites’ brains. Experimental and Toxicologic Pathology. 65: 503–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z, Bi R, Hao J, Wang S, Huo Y, Demoz RM, Banda R, Tian S, Xin C, Fu M, Pi J, Liu J (2020). A study on the protective effects of taxifolin on human umbilical vein endothelial cells and THP-1 cells damaged by hexavalent chromium: a probable mechanism for preventing cardiovascular disease induced by heavy metals. Food and Function. 11(5): 3851–3859. [DOI] [PubMed] [Google Scholar]
- Caparros-Gonzalez RA, Giménez-Asensio MJ, González-Alzaga B, Aguilar-Carduño C, Lorca-Marín JA, Algucil J, Gómez-Becerra I, Gómez-Ariza JL, García-Barrera T, Hernandez AF, et al. (2019). Childhood chromium exposure and neuropsychological development in children living in two polluted areas in southern Spain. Environmental Pollution. 252: 1550–1560. [DOI] [PubMed] [Google Scholar]
- Chen X, Guo C, Kong J (2012). Oxidative stress in neurodegenerative diseases. Neural Regeneration Research. 7(5): 376–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Kluz T, Fang L, Zhang X, Sun H, Jin C, Costa M (2016). Hexavalent Chromium (Cr(VI)) Down-Regulates Acetylation of Histone H4 at Lysine 16 through Induction of Stressor Protein Nupr1. PLoS One. 11(6): e0157317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civit T, Houdayer AJ, Kennedy G (2000). A Search for Trace Elements in Some Human Intracranial Tumors by Instrumental Neutron Activation Analysis. Biological Trace Element Research. 74: 203–210. [DOI] [PubMed] [Google Scholar]
- Clark MJ, Prentice JR, Hoggard N, Paley MN, Hadjivassiliou M, Wilkinson JM (2014). Brain Structure and Function in Patients after Metal-on-Metal Hip Resurfacing. American Journal of Neuroradiology. 35(9): 1753–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole P and Rodu B (2005). Epidemiologic studies of chrome and cancer mortality: A series of meta-analyses. Regulatory Toxicology and Pharmacology. 43: 225–231. [DOI] [PubMed] [Google Scholar]
- Costa DR, Pereira I, de Lima Rodrigues AS, Malafaia G (2017). Short-term social memory deficits in adult female mice exposed to tannery effluent and possible mechanism of action. Chemosphere. 184: 148–158. [DOI] [PubMed] [Google Scholar]
- Danielsen TE, Langård S, Andersen A (1996). Incidence of cancer among Norwegian boiler welders. Occupational and Environmental Medicine. 53: 231–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dashti A, Soodi M, Amani N (2014). Cr(VI) Induced Oxidative Stress and Toxicity in Cultured Cerebellar Granule Neurons at Different Stages of Development and Protective Effect of Rosmarinic Acid. Environmental Toxicology. 31(3): 269–277. [DOI] [PubMed] [Google Scholar]
- Dashti A, Soodi M, Amani N (2015). Evaluation of Cr(VI) induced Neurotoxicity and Oxidative Stress in PC12 Cells. Modares Journal of Medical Sciences: Pathobiology. 18(1): 55–65. [Google Scholar]
- Dasuri K, Zhang L, Keller JN (2013). Oxidative stress, neurodegeneration, and the balance of protein degradation and protein synthesis. Free Radical Biology and Medicine. 62: 170–185. [DOI] [PubMed] [Google Scholar]
- Deng Y, Wang M, Tian T, Lin S, Xu P, Zhou L, Dai C, Hao Q, Wu Y, Zhai Z, et al. (2019). The Effect of Hexavalent Chromium on the Incidence and Mortality of Human Cancers: A Meta-Analysis Based on Published Epidemiological Cohort Studies. Frontiers in Oncology. 9: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DesMarias TL and Costa M (2019). Mechanisms of chromium-induced toxicity. Current Opinion in Toxicology. 14: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Mayans J, Laborda R, Nuñez A (1986). Hexavalent Chromium Effects on Motor Activity and Some Metabolic Aspects of Wistar Albino Rats. Comparative Biochemistry and Physiology Part C: Comparative Pharmacology. 83C(1): 191–195. [DOI] [PubMed] [Google Scholar]
- Dib M, Garrel C, Favier A, Robin V, Desnuelle C (2002). Can malondialdehyde be used as a biological marker of progression in neurodegenerative disease? J. Neurol. 249: 367–374. [DOI] [PubMed] [Google Scholar]
- Ding M, and Shi X (2002). Molecular mechanisms of Cr(VI)-induced carcinogenesis. Molecular and Cellular Biochemistry. 234: 293–300. [PubMed] [Google Scholar]
- Döker S, Mounicou S, Doğan M, Lobinski R (2010). Probing the metal-homeostasis effects of the administration of chromium(VI) to mice by ICP MS and size-exclusion chromatography-ICP MS. Metallomics. 2: 549–555. [DOI] [PubMed] [Google Scholar]
- Dos Santos AA, Hort MA, Culbreth M, López-Granero C, Farina M, Rocha JBT, Aschner M (2016). Methylmercury and brain development: A review of recent literature. Journal of Trace Elements in Medicine and Biology. 38: 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrela FN, Rabelo LM, Vaz BG, Costa DRO, Pereira I, Rodrigues ASL, Malafaia G (2017). Short-term social memory deficits in adult female mice exposed to tannery effluent and possible mechanism of action. Chemosphere. 184: 148–158. [DOI] [PubMed] [Google Scholar]
- Fahmy A (2017). A histological study on the possible ameliorating effect of selenium on chromium (VI) induced neurotoxicity in the adult male Guinea pig cerebellar cortex. Journal of American Science. 13(5): 8–17. [Google Scholar]
- Farrokhian A, Mahmoodian M, Bahmani F, Amirani E, Shafabakhsh R, Asemi Z (2019). The Influences of Chromium Supplementation on Metabolic Status in Patients with Type 2 Diabetes Mellitus and Coronary Heart Disease. Biological Trace Element Research. 194(2): 313–320. [DOI] [PubMed] [Google Scholar]
- Fatima R, Akhtar K, Hossain MM, Ahmad R (2017). Chromium oxide nanoparticle-induced biochemical and histopathological alterations in the kidneys and brains of Wistar rats. Toxicology and Industrial Health. 33(12): 911–921. [DOI] [PubMed] [Google Scholar]
- Federico A, Cardaioli E, Da Pozzo P, Formichi P, Gallus GN, Radi E (2012). Mitochondria, oxidative stress and neurodegeneration. Journal of the Neurological Sciences. 322(1–2): 254–262. [DOI] [PubMed] [Google Scholar]
- Feingold L, Savitz DA, John EM (1991). Use of a job-exposure matrix to evaluate parental occupation and childhood cancer. Cancer Causes and Control. 3: 161–169. [DOI] [PubMed] [Google Scholar]
- Feng H, Liu J, Hu G, Jia G (2018). The Role of Epigenetics in the Toxic Effects Induced by Hexavalent Chromium. Reactive Oxygen Species. 5(14): 107–117. [Google Scholar]
- Francis PT, Palmer AM, Snape M, Wilcock GK (1999). The cholinergic hypothesis of Alzheimer’s disease: a review of progress. Journal of Neurology, Neurosurgery, and Psychiatry. 66(2): 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu SC, Liu JM, Lee KI, Tang FC, Fang KM, Yang CY, Su CC, Chen HH, Hsu RJ, Chen YW (2020). Cr(VI) induces ROS-mediated mitochondrial-dependent apoptosis in neuronal cells via the activation of Akt/ERK/AMPK signaling pathway. Toxicology in Vitro. 65: 104795. [DOI] [PubMed] [Google Scholar]
- García-Niño WR, Zatarain-Barrón ZL, Hernández-Pando R, Vega-García CC, Tapia E, Pedraza-Chaverri J (2015). Oxidative Stress Markers and Histological Analysis in Diverse Organs from Rats Treated with a Hepatotoxic Dose of Cr(VI): Effect of Curcumin. Biological Trace Element Research. 167: 130–145. [DOI] [PubMed] [Google Scholar]
- Gawdi R, Emmady PD (2020). Physiology, Blood Brain Barrier. [Updated 2020 Sep 27]. In: StatPearls[Internet]. Treasure Island (FL): StatPearls Publishing; 2020 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK557721/ [PubMed] [Google Scholar]
- Gibb HJ, Lees P.St.J., Wang J, O-Leary KG (2015). Extended Follow-up of a Cohort of Chromium Production Workers. American Journal of Industrial Medicine. 58: 905–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooddy W, Hamilton EI, Williams TR (1975). Spark-Source Mass Spectrometry in the Investigation of Neurological Disease. Brain. 98: 65–70. [DOI] [PubMed] [Google Scholar]
- Green B, Griffiths E, Almond S (2017). Neuropsychiatric symptoms following metal-on-metal implant failure with cobalt and chromium toxicity. BMC Psychiatry. 17: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greilberger J, Koidl C, Greilberger M, Lamprecht M, Schroecksnadel K, Leblhuber F, Fuchs D, Oettl K (2008). Malondialdehyde, carbonyl proteins and albumin-disulphide as useful oxidative markers in mild cognitive impairment and Alzheimer’s disease. Free Radical Research. 42(7): 633–638. [DOI] [PubMed] [Google Scholar]
- Guisun Z, Yinson W, Mingguang T, Min Z, Yongjie W, Fulin Z (1991). Preliminary study of trace elements in human brain tumor tissues by instrumental neutron activation analysis. Journal of Radioanalytical and Nuclear Chemistry. 151: 327–335. [Google Scholar]
- Haddad-Tóvolli, Dragano NRV, Ramalho AFS, Velloso LA (2017). Development and Function of the Blood-Brain Barrier in the Context of Metabolic Control. Frontiers in Neuroscience. 11: 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson LR, Frey WH II (2008). Intranasal delivery bypasses the blood-brain barrier to target therapeutic agents to the central nervous system and treat neurodegenerative disease. BMC Neuroscience. 9(Suppl. 3): S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao P, Zhu Y, Wang S, Wan H, Chen P, Wang Y, Cheng Z, Liu Y, Liu J (2017). Selenium Administration Alleviates Toxicity of Chromium(VI) in the Chicken Brain. Biological Trace Element Research. 178: 127–135. [DOI] [PubMed] [Google Scholar]
- Hara T, Hoshuyama T, Takahashi K, Delgermaa V, Sorahan T (2010). Cancer risk among Japanese chromium platers, 1976–2003. Scandinavian Journal of Work, Environment & Health. 36(3): 216–221. [DOI] [PubMed] [Google Scholar]
- Harrison-Brown M, Scholes C, Field C, McQuilty R, Farah SB, Nizam I, Kerr D, Kohan L (2020). Limited penetration of cobalt and chromium ions into the cerebrospinal fluid following metal on metal arthroplasty: a cross-sectional analysis. Clinical Toxicology. 58: 233–240. [DOI] [PubMed] [Google Scholar]
- Haynes EN, Chen A, Ryan P, Succop P, Wright J, Dietrich KN (2011). Exposure to airborne metals and particulate matter and risk for youth adjudicated for criminal activity. Environmental Research. 111(8): 10.1016/j.envres.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegazy R, Mansour D, Salama A, Hassan A, Saleh D (2021). Exposure to intranasal chromium triggers dose and time-dependent behavioral and neurotoxicological defects in rats. Ecotoxicology and Environmental Safety. 216: 112220. [DOI] [PubMed] [Google Scholar]
- Heffern K, Tierney K, Gallagher EP (2018). Comparative effects of cadmium, zinc, arsenic and chromium on olfactory-mediated neurobehavior and gene expression in larval zebrafish (Danio rerio). Aquat Toxicol. 201: 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höck A, Demmel U, Schicha H, Kasperek K, Feinendegen LE (1975). Trace Element Concentration in Human Brain: Activation Analysis of Cobalt, Iron, Rubidium, Selenium, Zinc, Chromium, Silver, Cesium, Antimony and Scandium. Brain. 98: 49–64. [DOI] [PubMed] [Google Scholar]
- Holmes AL, Wise SS, Wise JP Sr. (2008). Carcinogenicity of hexavalent chromium. Indian Journal of Medical Research. 128: 353–372. [PubMed] [Google Scholar]
- Hu L, Greer JB, Solo-Gabriele H, Fieber LA, Cai Y (2013). Arsenic toxicity in the human nerve cell line SK-N-SH in the presence of chromium and copper. Chemosphere. 91(8): 1082–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huvinen M and Pukkala E (2013). Cancer incidence among Finnish ferrochromium and stainless steel production workers in 1967–2011: a cohort study. BMJ Open. 3: e003819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda T, Takahashi K, Kabata T, Sakagoshi D, Tomita K, Yamada M (2010). Polyneuropathy caused by cobalt-chromium metallosis after total hip replacement. Muscle & Nerve. 42(1): 140–143. [DOI] [PubMed] [Google Scholar]
- Iztleuov Y, Abilov T, Zhanabayeva G, Ismailova I, Iztleuov M (2018). Protective Effect of Sodium Tetraborate on Chromium-induced Brain Damage in rats. Biomedical & Pharmacology Journal. 11(1): 227–236. [Google Scholar]
- Jory J and McGinnis WR (2008). Red-Cell Trace Minerals in Children with Autism. American Journal of Biochemistry and Biotechnology. 4(2): 101–104. [Google Scholar]
- Kanabrocki EL, Greco J, Graham LA, Kaplan E, Rubnitz ME, Oester YT (1975). Trace Elements in Human Pituitary. International Journal of Nuclear Medicine and Biology. 3: 73–76. [DOI] [PubMed] [Google Scholar]
- Karri V, Schuumacher M, Kumar V (2016). Heavy metals (Pb, Cd, As, MeHg) as risk factors for cognitive dysfunction: A general review of metal mixture mechanisms in brain. Environmental Toxicology and Pharmacology. 48: 203–213. [DOI] [PubMed] [Google Scholar]
- Katsiki M, Trougakos IP, Chondrogiann N, Alexopoulos EC, Makropoulos V (2004). Alterations of senescence biomarkers in human cells by exposure to Cr VI in vivo and in vitro. Experimental Gerontology. 39(7): 1079–1087. [DOI] [PubMed] [Google Scholar]
- Keep RF, Hua Y, Xi G (2013). Brain water content. A misunderstood measurement? Translational Stroke Research. 3(2): 263–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH and Kang JC (2016). Oxidative stress, neurotoxicity, and metallothionein (MT) gene expression in juvenile rock fish Sebastes schlegelii under the different levels of dietary chromium (Cr6+) exposure. Ecotoxicology and Environmental Safety. 125: 78–84. [DOI] [PubMed] [Google Scholar]
- Kitamura F, Yokoyama K, Araki S, Nishikitani M, Choi JW, Yum YT, Park HC, Park SH, Sato H (2003). Increase of Olfactory Threshold in Plating Factory Workers Exposed to Chromium in Korea. Industrial Health. 41: 279–285. [DOI] [PubMed] [Google Scholar]
- Knuttson A, Damber L, Järvholm B (2000). Cancers in concrete workers: results of a cohort study of 33 668 workers. Occupational and Environmental Medicine. 57: 264–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh DH, Kim TW, Jang SH, Ryu HW (2011). Cancer Mortality and Incidence in Cement Industry Workers in Korea. Safe Health Work. 2: 243–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kume-Kick J, Rice ME (1998). Estrogen-dependent modulation of rat brain ascorbate levels and ischemia-induced ascorbate loss. Brain Research. 803: 105–113. [DOI] [PubMed] [Google Scholar]
- Leocoanet P, Blangis M, Garcia M, Legallois Y, Fabre T (2019). Chromium-Cobalt Intoxication with Intense Systemic Complications following Total Hip Revision after Per-Operative Ceramic Fracture. Case Reports in Orthopedica. 2019: 4209796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipworth L, Sonderman JS, Mumma MT, Tarone RE, Marano DE, Boice JD Jr., McLaughlin JK (2011). Cancer Mortality Among Aircraft Manufacturing Workers: An Extended Follow-Up. Journal of Occupational and Environmental Medicine. 53(9): 992–1007. [DOI] [PubMed] [Google Scholar]
- Long J, Liu C, Sun L, Gao H, Liu J (2009). Neuronal Mitochondrial Toxicity and Malondialdehyde: Inhibitory Effects on Respiratory Function and Enzyme Activities in Rat Brain Mitochondria. Neurochemical Research. 34: 786–794. [DOI] [PubMed] [Google Scholar]
- Lv Y, Zhang P, Guo J, Zhu Z, Li X, Xu D, Zeng W (2018). Melatonin protects mouse spermatogonial stem cells against hexavalent chromium-induced apoptosis and epigenetic histone modification. Toxicology and Applied Pharmavology. 340: 30–38. [DOI] [PubMed] [Google Scholar]
- MacKenzie RD, Byerrum RU, Decker CF, Hoppert CA, Langham RF (1958). Chronic toxicity studies: II. Hexavalent and trivalent chromium administered in drinking water to rats. Arch Ind Health. 18: 232–234. [PubMed] [Google Scholar]
- Mahmoud AM and El-Twab SMA (2017). Caffeic acid phenethyl ester protects the brain against hexavalent chromium toxicity by enhancing endogenous antioxidants and modulating the JAK/STAT signaling pathway. Biomedicine & Pharmacology. 91: 303–311. [DOI] [PubMed] [Google Scholar]
- Markesbery WR, Ehmann WD, Alauddin M, Hossain TIM (1984). Brain Trace Element Concentrations in Aging. Neurobiology of Aging. 5: 19–28. [DOI] [PubMed] [Google Scholar]
- Marsh GM, Youk AO, Buchanich JM, Xu H, Downing S, Kennedy KJ, Esmen NA, Hancock RP, Lacey SE, Fleissner ML (2013). Long-Term Health Experience of Jet Engine Manufacturing Workers: VI: Incidence of Malignant Central Nervous System Neoplasms in Relation to Estimated Workplace Exposures. Journal of Occupational and Environmental Medicine. 55(6): 654–675. [DOI] [PubMed] [Google Scholar]
- Martin BD, Schoenhard JA, Hwang JM, Sugden KD (2006). Ascorbate Is a Pro-Oxidant in Chromium-Treated Human Lung Cells. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 610(1–2): 74–84. [DOI] [PubMed] [Google Scholar]
- Massie HR, Aiello VR, Williams TR (1983). Chromium Levels and Aging in Mice and Drosophila. Age. 6: 62–65. [Google Scholar]
- Máté Z, Horváth E, Papp A, Kovács K, Tombácz E, Nesztor D, Szabó T, Szabó A, Paulik E (2017). Neurotoxic effects of subchronic intratracheal Mn nanoparticle exposure alone and in combination with other welding fume metals in rats. Inhalation Toxicology. 29(5): 227–238. [DOI] [PubMed] [Google Scholar]
- Mathur AK, Chandra SV, Tandon SK (1977). Comparative Toxicity of Trivalent and Hexavalent Chromium to Rabbits. Toxicology. 8: 53–61. [DOI] [PubMed] [Google Scholar]
- McNeill LS, McLean JE, Parks JL, Edwards MA (2012). Hexavalent chromium review, part 2: Chemistry, occurrence, and treatment. Journal American Water Works Association. 104(7): E395–E405. [Google Scholar]
- Michalska P and León R (2020). When It Comes to an End: Oxidative Stress Crosstalk with Protein Aggregation and Neuroinflammation Induce Neurodegeneration. Antioxidants. 9(8): 740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat I, Martinova N, Seidel C, Thompson CM (2018). Hexavalent Chromium in Drinking Water. Journal American Water Works Association. 110(5): E22–E35. [Google Scholar]
- Montanaro F, Ceppi M, Demers PA, Puntoni R, Bonassi S (1997). Mortality in a cohort of tannery workers. Occupational and Environmental Medicine. 54: 588–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira L.d.P.D., Gomes JVP, Mattar JB, Chaves LO, Martino HSD (2019). Potential of trace elements as supplements for the metabolic control of Type 2 Diabetes Mellitus: A systematic review. Journal of Functional Foods. 57: 317–327. [Google Scholar]
- Moulin JJ, Portefaix P, Wild P, Mur JM, Smagghe G, Mantout B (1990). Mortality study among workers producing ferroalloys and stainless steel in France. British Journal of Industrial Medicine. 47: 537–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muoio V, Persson PB, Sendeski MM (2014). The neurovascular unit – concept review. Acta Physiol. 210: 790–798. [DOI] [PubMed] [Google Scholar]
- Newbold RF, Amos J, Connell JR (1979). The cytotoxic, mutagenic and clastogenic effects of chromium-containing compounds on mammalian cells in culture. Mutation Research/Genetic Toxicology. 67(1): 55–63. [DOI] [PubMed] [Google Scholar]
- Niedzielska E, Smaga I, Gawlik M, Moniczewski A, Stankowicz P, Pera J, Filip M (2015). Oxidative Stress in Neurodegenerative Diseases. Molecular Neurobiology. 53: 4094–4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nudler SI, Quinteros FA, Miler EA, Cabilla JP, Ronchetti SA, Duvilanski BH (2009). Chromium VI administration induces oxidative stress in hypothalamus and anterior pituitary gland from male rats. Toxicology Letters. 185: 187–192. [DOI] [PubMed] [Google Scholar]
- Ozcankaya R and Delibas N (2002). Malondialdehyde, Superoxide Dismutase, Melatonin, Iron, Copper, and Zinc Blood Concentrations in Patients with Alzheimer’s Disease: Cross-sectional Study. Croatian Medical Journal. 43(1): 28–32. [PubMed] [Google Scholar]
- Pan SY, Ugnat AM, Mao Y (2005). Occupational Risk Factors for Brain Cancer in Canada. Occupational Hazards and Brain Cancer. 47(7): 704–717. [DOI] [PubMed] [Google Scholar]
- Parent ME, Turner MC, Lavoué J, Richard H, Figuerola J, Kincl L, Richardson L, Benke G, Blettner M, Fleming S, et al. (2017). Lifetime occupational exposure to metals and welding fumes, and risk of glioma: a 7-country population-based case-control study. Environmental Health. 16: 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng M, Yang X (2015). Controlling diabetes by chromium complexes: The role of the ligands. Journal of Inorganic Biochemistry. 146: 97–103. [DOI] [PubMed] [Google Scholar]
- Persigehl M, Schicha H, Kasperek K, Klein HJ (1977). Trace Element Concentration in Human Organs in Dependence of Age. Beiträge zur Pathologie. 161: 209–220. [DOI] [PubMed] [Google Scholar]
- Peters RM, Willemse P, Rijk PC, Hoofendoorn M, Zijilstra WP (2017). Fatal Cobalt Toxicity after a Non-Metal-on-Metal Total Hip Arthroplasty. Case Reports in Orthopedics. 2017: 9123684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzorno L (2015). Northing Boring About Boron. Integrative Medicine. 14(4): 35–48. [PMC free article] [PubMed] [Google Scholar]
- Platt B and Riedel G (2011). The cholinergic system, EEG and sleep. Behavioural Brain Research. 221(2): 499–504. [DOI] [PubMed] [Google Scholar]
- Porta R, Sánchez-Torres JM, Paz-Ares L, Massutí B, Reguart N, Mayo C, Lianes P, Queralt C, Guillem V, Salinas P, et al. (2011). Brain metastases from lung cancer responding to erlotinib: the importance of EGFR mutation. European Respiratory Journal. 37: 624–631. [DOI] [PubMed] [Google Scholar]
- Prentice JR, Blackwell CS, Raoof N, Bacon P, Ray J, Kicman SJ, Wilkinson JM (2014). Auditory and Visual Health after Ten Years of Exposure to Metal-on-Metal Hip Prostheses: A Cross-Sectional Study Follow Up. PLoS One. 9(3): e90838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor DM, Suh M, Campleman SL, Thompson CM (2014). Assessment of the mode of action for hexavalent chromium-induced lung cancer following inhalation exposures. Toxicology. 325: 160–179. [DOI] [PubMed] [Google Scholar]
- Qin Q, Xie H, Wise SS, Browning CL, Thompson KN, Holmes AL, Wise JP Sr. (2014) Homologous Recombination Repair Signaling in Chemical Carcinogenesis: Prolonged Particulate Hexavalent Chromium Exposure Suppresses the Rad51 Response in Human Lung Cells. Toxicological Sciences, 142(1): 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinteros FA, Poliandri AHB, Machiavelli LI, Cabilla JP, Duvilanski BH (2007). In vivo and in vitro effects of chromium VI on anterior pituitary hormone release and cell viability. Toxicology and Applied Pharmacology. 218: 79–87. [DOI] [PubMed] [Google Scholar]
- Quinteros FA, Machiavelli LI, Miler EA, Cabilla JP, Duvilanksi BH (2008). Mechanisms of chromium (VI)-induced apoptosis in anterior pituitary cells. Toxicology. 249: 109–115. [DOI] [PubMed] [Google Scholar]
- Rajan MT, Rao KSJ, Mamatha BM, Rao RV, Shanmugavelu P, Menon RB, Pavithran MV (1997). Quantification of trace elements in normal human brain by inductively coupled plasma atomic emission spectrometry. Journal of the Neurological Sciences. 146: 153–166. [DOI] [PubMed] [Google Scholar]
- Reynolds M, Stoddard L, Bespalov I, Zhitkovich A (2007). Ascorbate Acts as a Highly Potent Inducer of Chromate Mutagenesis and Clastogenesis: Linkage to DNA Breaks in G2 Phase by Mismatch Repair. Nucleic Acids Research. 35(2): 465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rising JP, Reynolds IS, Sedrakyan A (2012). Delays and difficulties in assessing metal-on-metal hip implants. N Engl J Med. 367: e1. [DOI] [PubMed] [Google Scholar]
- Saghazadeh A, Mahmoudi M, Shahrokhi S, Mojarrad M, Dastmardi M, Mirbeyk M, Rezaei N (2019). Trace elements in schizophrenia: a systematic review and meta-analysis of 39 studies (N = 5151 participants). Nutrition Reviews. 78(4): 278–303. [DOI] [PubMed] [Google Scholar]
- Saha R, Nandi R, Saha B (2011). Sources and toxicity of hexavalent chromium. Journal of Coordination Chemistry. 64(10): 1782–1806. [Google Scholar]
- Salama A, Hegazy R, Hassan A (2016). Intranasal Chromium Induces Acute Brain and Lung Injuries in Rats: Assessment of Different Potential Hazardous Effects of Environmental and Occupational Exposure to Chromium and Introduction of a Novel Pharmacological and Toxicological Animal Model. PLoS One. 11(12): e0168688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Díaz G, Escobar F, Badland H, Arias-Merino G, de la Paz MP, Alonso-Ferreira V (2018). Geographic Analysis of Motor Neuron Disease Mortality and Heavy Metals Released to Rivers in Spain. International Journal of Environmental Research and Public Health. 15: 2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarica ZS, Eren M, Senturk M (2019). Effect of Boron on the Potassium Dichromate Induced Oxidative Damage in Brain Tissue of Sprague Dawley Rats. Pakistan Journal of Zoology. 51(5): 1905–1910. [Google Scholar]
- Schuette W (2004). Treatment of brain metastases from lung cancer: chemotherapy. Lung Cancer. 45(Suppl. 2): S253–S257. [DOI] [PubMed] [Google Scholar]
- Shaw P, Mondal P, Bandyopadhyay A, Chattopadhyay A (2020). Environmentally relevant concentration of chromium induces nuclear deformities in erythrocytes and alters the expression of stress-responsive and apoptotic genes in brain of adult zebrafish. Science of the Total Environment. 703: 135622. [DOI] [PubMed] [Google Scholar]
- Shi X and Dalal NS (1990). NADPH-dependent flavoenzymes catalyze one electron reduction of metal ions and molecular oxygen and generate hydroxyl radicals. FEBS Letters. 276(1–2): 189–191. [DOI] [PubMed] [Google Scholar]
- Singh P and Chowdhuri DK (2017). Environmental Presence of Hexavalent but Not Trivalent Chromium Causes Neurotoxicity in Exposed Drosophila melanogaster. Molecular Neurobiology. 54: 3368–3387. [DOI] [PubMed] [Google Scholar]
- Singh P and Chowdhuri DK (2018). Modulation of sestrin confers protection to Cr(VI) induced neuronal cell death in Drosophila melanogaster. Chemosphere. 191: 302–314. [DOI] [PubMed] [Google Scholar]
- Singh A, Kukreti R, Saso L, Kukreti S (2019). Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules. 24(8): 1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivakumar KK, Stanley JA, Arosh JA, Pepling ME, Burghardt RC, Banu S (2014). Prenatal exposure to chromium induces early reproductive senescence by increasing germ cell apoptosis and advancing germ cell cyst breakdown in the F1 offspring. Developmental Biology. 388(1): 22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalny AV, Simashkova NV, Klyushnik TP, Grabeklis AR, Bjorklund G, Skalnaya MG, Nikonorov AA, Tinkov AA (2017a). Hair toxic and essential trace elements in children with autism spectrum disorder. Metab Brain Dis. 32: 195–202. [DOI] [PubMed] [Google Scholar]
- Skalny AV, Simashkova NV, Klyushnik TP, Grabeklis AR, Radysh IV, Skalnaya MG, Nikonorov AA, Tinkov AA (2017b). Assessment of serum trace elements and electrolytes in children with childhood and atypical autism. Journal of Trace Elements in Medicine and Biology. 43: 9–14. [DOI] [PubMed] [Google Scholar]
- Solovyev ND (2015). Importance of selenium and selenoprotein for brain function: From antioxidant protection to neuronal signaling. Journal of Inorganic Biochemistry. 153: 1–12. [DOI] [PubMed] [Google Scholar]
- Sorrentino JA, Sanoff HK, Sharpless NE (2014). Defining the toxicology of aging. Trends in Molecular Medicine. 20(7): 375–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soudani N, Troudi A, Amara IB, Bouaziz H, Boudawara T, Zeghal N (2012). Ameliorating effect of selenium on chromium (VI)-induced oxidative damage in the brain of adult rats. Journal of Physiology and Biochemistry. 68: 397–409. [DOI] [PubMed] [Google Scholar]
- Speer RM, and Wise JP Sr. (2018). Current status on chromium research and its implications for health and risk assessment. Reference Module in Chemistry, Molecular Sciences and Chemical Engineering. 10.1016/B978-0-12-409547-2.14283-0 [DOI] [Google Scholar]
- Standeven AM, Wetterhahn KE, Kato R (1992). Ascorbate is the principal reductant of chromium(VI) in rat lung ultrafiltrates and cytosols, and mediates chromium-DNA binding in vitro. Carcinogenesis. 13(8): 1319–1324. [DOI] [PubMed] [Google Scholar]
- Su P, Aschner M, Chen J, Luo W (2017). Chapter 19 – Metals and Autophagy in Neurotoxicity. Biometals in Neurodegenerative Diseases: Mechanisms and Therapeutics P377–398. Academic Press. [Google Scholar]
- Suljević D, Sulejmanović J, Fočak M, Halilović E, Pupalović D, Hasić A, Alijagic A (2021). Assessing hexavalent chromium tissue-specific accumulation patterns and induced physiological responses to probe chromium toxicity in Coturnix japonica quail. Chemosphere. 266: 129005. [DOI] [PubMed] [Google Scholar]
- Sun H, Zhou X, Chen H, Li Q, Costa M (2009). Modulation of histone methylation and MLH1 gene splicing by hexavalent chromium. Toxicology and Applied Pharmacology. 237: 258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabner BJ, El-Agnaf OMA, German MJ, Fullwood NJ, Allsop D (2005). Protein aggregation, metals and oxidative stress in neurodegenerative diseases. Biochemical Society Transactions. 33(5): 1082–1086. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Ikeda K, Ishikawa M, Tsukasaki T, Nakama D, Tanida S, Kameda T (2004). Social stress-induced cortisol elevation acutely impairs social memory in humans. Neuroscience Letters. 363(2): 125–130. [DOI] [PubMed] [Google Scholar]
- Talbott EO, Marshall LP, Rager JR, Arena VC, Sharma RK, Stacy SL (2015). Air toxics and the risk of autism spectrum disorder: the results of a population based case-control study in southwestern Pennsylvania. Environmental Health. 14: 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinkov AA, Skalnaya MG, Simashkova NV, Klyushnik TP, Skalnaya AA, Bjørklund G, Notova SV, Kiyaeva EV, Skalny AV (2019). Association between catatonia and levels of hair and serum trace elements and minerals in autism spectrum disorder. Biomedicine & Pharmacology. 109: 174–180. [DOI] [PubMed] [Google Scholar]
- Travacio M, Polo JM, Llesuy S (2000). Chromium(VI) induces oxidative stress in the mouse brain. Toxicology. 150: 137–146. [DOI] [PubMed] [Google Scholar]
- Ueno S, Kashimoto T, Susa N, Furukawa Y, Ishii M, Yokoi K, Yasuno M, Sasaki YF, Ueda J, Nishimura Y, Sugiuama M (2001). Detection of Dichromate (VI)-Induced DNA Strand Breaks and Formation of Paramagnetic Chromium in Multiple Mouse Organs. Toxicology and Applied Pharmacology. 170: 56–62. [DOI] [PubMed] [Google Scholar]
- US EPA (US Environmental Protection Agency). (2017). Data Summary of the Third Unregulated Contaminant Monitoring Rule (UCMR 3). EPA 815-S-17-001. USEPA, Washington. [Google Scholar]
- Uttara B, Singh A, Zamboni P, Mahajan RT (2009). Oxidative Stress and Neurodegenerative Diseases: A Review of Upstream and Downstream Antioxidant Therapeutic Options. Current Neuropharmacology. 7(1): 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaiopoulou E and Gikas P (2020). Regulations for chromium emissions to the aquatic environment in Europe and elsewhere. Chemosphere. 254: 126876. [DOI] [PubMed] [Google Scholar]
- Val MM, Mendes LA, Alarcão A, Carvalho L, Carreira I, Rodrigues CFD, Alpoim MC (2015). Senescent bronchial fibroblasts induced to senescence by Cr(VI) promote epithelial-mesenchymal transition when co-cultured with bronchial epithelial cells in the presence of Cr(VI). Mutagenesis. 30: 277–286. [DOI] [PubMed] [Google Scholar]
- Villano JL, Durbin EB, Normandeau C, Thakkar JP, Moirangthem V, Davis FG (2015). Incidence of brain metastasis at initial presentation of lung cancer. Neuro-Oncology. 17(1): 122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Wu J, Humphries B, Kondo K, Jiang Y, Shi X, Yang C (2018). Upregulation of histone-lysine methyltransferases plays a causal role in hexavalent chromium-induced cancer stem cell-like property and cell transformation. Toxicology and Applied Pharmacology. 342: 22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasim M, Arif M, Iqbal MS (2014). Determination of elements in different parts of goat brain using k0 instrumental neutron activation analysis. Journal of Radioanalytical and Nuclear Chemistry. 300: 249–254. [Google Scholar]
- Watanabe S and Fukuchi Y (1981). Occupational Impairment of the Olfactory Sense of Chromate Producing Workers. Japanese Journal of Occupational Health. 23: 606–611. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Takayama Y, Koike M, Yamamoto M (1985). In vivo clastogenicity of lead chromate in mice. Tohoku J Exp Med. 146(3): 373–4. [DOI] [PubMed] [Google Scholar]
- Wesseling C, Pukkala E, Neuvonen K, Kauppinen T, Boffetta P, Partanen T (2002). Cancer of the Brain and Nervous System and Occupational Exposures in Finnish Women. Journal of Occupational and Environmental Medicine. 44(7): 663–667. [DOI] [PubMed] [Google Scholar]
- Wise JP, Orenstain JM, Patierno SR (1993). Inhibition of lead chromate clastogenesis by ascorbate: relationship to particle dissolution and uptake. Carcinogenesis. 14(3): 429–434. [DOI] [PubMed] [Google Scholar]
- Wise SS, Holmes AL, Ketterer ME, Hartsock WJ, Fomchenko E, Katsifis S, Thompson WD, Wise JP Sr. (2004). Chromium is the proximate clastogenic species for lead chromate-induced clastogenicity in human bronchial cells. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 560(1): 79–89. [DOI] [PubMed] [Google Scholar]
- Wise SS, Holmes AL, Wise JP Sr. (2008). Hexavalent chromium-induced DNA damage and repair mechanisms. Reviews on Environmental Health. 23(1): 39–57. [DOI] [PubMed] [Google Scholar]
- Wise SS, Aboueissa A, Martino J, and Wise JP Sr. (2018) Hexavalent chromium-induced chromosome instability drives permanent and heritable numerical and structural changes and a DNA repair-deficient phenotype. Cancer Research. 78: 4203–4214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise JTF, Wang L, Alstott MC, Ngalame NNO, Wang Y, Zhang Z, Shi X (2018). Investigating the Role of Mitochondrial Respiratory Dysfunction during Hexavalent Chromium-Induced Carcinogenesis. Journal of Environmental Pathology, Toxicology and Oncology. 37(4): 317–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao F, Li Y, Dai L, Deng Y, Zou Y, Li P, Yang Y, Zhong C (2012). Hexavalent chromium targets mitochondrial respiratory chain complex I to induce reactive species-dependent caspase-3 activation in L-02 hepatocytes. International Journal of Molecular Medicine. 30(3): 629–635. [DOI] [PubMed] [Google Scholar]
- Xie H, Holmes AL, Wise SS, Gordon N, Wise, Sr., J.P. (2004) Lead chromate-induced chromosome damage requires extracellular dissolution to liberate chromium ions but does not require particle internalization or intracellular dissolution. Chemical Research in Toxicology. 17(10): 1362–1367. [DOI] [PubMed] [Google Scholar]
- Yang D, Yang Q, Fu N, Li S, Han B, Liu Y, Tang Y, Guo X, Lv Z, Zhang Z (2021). Hexavalent chromium induced heart dysfunction via Sesn2-mediated impairment of mitochondrial function and energy supply. Chemosphere. 264(2): 128547. [DOI] [PubMed] [Google Scholar]
- Yasuda H, Yonashiro T, Yoshida K, Ishii T, Tsutsui T (2005). Mineral Imbalance in Children with Autistic Disorders. Biomed Res Trace Elements. 16(4): 285–292. [Google Scholar]
- Yorbik Ö, Kurt İ, Haşimi A, Öztürk Ö (2010). Chromium, Cadmium, and Lead Levels in Urine of Children with Autism and Typically Developing Controls. Biol Trace Elem Res. 135: 10–15. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zhang Y, Zhong C, Xiao F (2016). Cr(VI) induces premature senescence through ROS-mediated p52 pathway in L-02 hepatocytes. Scientific Reports. 6: 34578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wang Y, Chen M, Zeng M (2021). Hexavalent chromium-induced apoptosis in Hep3B cells is accompanied by calcium overload, mitochondrial damage, and AIF translocation. Ecotoxicology and Environmental Safety. 208: 111391. [DOI] [PubMed] [Google Scholar]
- Zheng W, Aschner M, Ghersi-Egea JF (2014). Brain barrier systems: a new frontier in metal neurotoxicological research. Toxicol Appl Pharmacol. 192(1): 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W and Ghersi-Egea JF (2020). ToxPoint: Brain Barrier Systems Play No Small Roles in Toxicant-induced Brain Disorders. Toxicol Sci. 175(2): 147–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhitkovich A (2005). Importance of chromium-DNA adducts in mutagenicity and toxicity of chromium(VI). Chemical Research in Toxicology. 18: 3–11. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Chen P, Wan H, Wang Y, Hao P, Liu Y, Liu J (2018). Selenium-Chromium(VI) Interaction Regulates the Contents and Correlations of Trace Elements in Chicken Brain and Serum. Biological Trace Element Research. 181: 154–163. [DOI] [PubMed] [Google Scholar]