

Abstract
Osteosarcoma (OS) is the most common primary malignancy of the bone that predominantly affects children and adolescents. Hippo pathway is a crucial regulator of organ size and tumorigenesis. However, how Hippo pathway regulates the occurrence of osteosarcoma is largely unknown. Here, we reported the regulator of G protein signaling protein 12 (RGS12) is a novel Hippo pathway regulator and tumor suppressor of osteosarcoma. Depletion of Rgs12 promotes osteosarcoma progression and lung metastasis in an orthotopic xenograft mouse model. Our data showed that knockdown of RGS12 upregulates Ezrin expression through promoting the GNA12/13-RhoA-YAP pathway. Moreover, RGS12 negatively regulates the transcriptional activity of YAP/TEAD1 complex through its PDZ domain function to inhibit the expression and function of the osteosarcoma marker Ezrin. PDZ domain peptides of RGS12 can inhibit the development of intratibial tumor and lung metastases. Collectively, this study identifies the RGS12 is a novel tumor suppressor in osteosarcoma through inhibiting YAP-TEAD1-Ezrin signaling pathway and provides a proof of principle that targeting RGS12 may be a therapeutic strategy for osteosarcoma.
Keywords: Osteosarcoma, Bone, RGS12, YAP, Ezrin
Introduction
Osteosarcoma (OS) is the most common malignant bone tumor in children and adolescents and is characterized by the production of osteoid, showing a high propensity for lung metastasis (90%) [1]. Up to now, traditional treatment for osteosarcoma has consisted of surgery and chemotherapy. Although the 5-year survival rate of non-metastatic osteosarcoma can reach ~70%, the metastatic osteosarcoma survival rate is only ~15% [1, 2]. Despite great advances in multi-agent chemotherapy and sophisticated surgery, the poor understanding of the molecular mechanisms of osteosarcoma pathogenesis and progression has restricted the improvement of patient survival rate over the past three decades. Therefore, elucidating the pathological mechanisms involved in osteosarcoma initiation, development and metastasis progression is crucial for its effective prevention, diagnosis and treatment.
Recent studies have reported that the Hippo pathway is crucial for cell fate determination, organ size control and tumorigenesis by regulating cell proliferation and apoptosis [3–5]. Yes-associated protein (YAP), a transcription cofactor, is an essential downstream effector of the Hippo pathway and can be inactivated through its cytoplasmic retention and phosphorylation by its upstream serine/threonine kinases such as the mammalian Ste20-like kinases 1/2 (Mst1/2) and large tumor suppressor kinases 1/2 (Lats1/2) [6]. When the Hippo pathway is inhibited, hypophosphorylated YAP will enter into the nucleus and function as a transcription coactivator by directly interacting with its major partner, the TEA domain DNA-binding family of transcription factors (TEADs), thereby promoting cell survival and proliferation [4, 6, 7]. In osteosarcoma, the previous study reported that TEAD1 is the major downstream transcriptional factor of YAP signaling [7]. Additionally, YAP has recently emerged as a critical oncogene that is overexpressed in many types of tumors and is also considered a novel prognostic marker and therapeutic target in osteosarcoma [3].
Regulators of G-protein signaling (RGS) constitute a large family of proteins that accelerate Gα GTP hydrolysis and inhibit the heterotrimeric G protein-coupled receptor (GPCR) signaling pathway [8, 9]. RGS12 is the largest protein in the RGS protein family. It expresses in bone, lung, brain and many other tissues and contains six domains, including PDZ domain, which interacts with GPCR chemokine receptors and the PDZ binding motif containing proteins [10–12]. The GPCR activator lysophosphatidic acid (LPA) has been shown to act through G12/13-coupled receptors to inhibit the kinases Lats1/2, thereby activating YAP, a transcription coactivator and oncogene [13]. RGS proteins have been reported to be relevant to tumor including regulating tumor cell proliferation, migration, and invasion. In prostate cancer, RGS12 has a lower expression in comparison with normal prostate tissues [14]. In many tumor cell lines, such as human breast cancer cell lines MCF7 and MDA-MB-231, cervical cancer cell line C-33A, adrenal cancer cell line SW-13, and osteosarcoma cell lines SaOS2 and U-2 OS, RGS12 can repress cell proliferation by inhibiting DNA synthesis to repress the cell cycle [15]. Our previous studies showed that RGS12 regulates osteoclast and osteoblast differentiation, function and bone homeostasis and development in vivo and in vitro [12, 16–18]. These observations suggested that RGS12 may play a role in bone cancer, such as osteosarcoma. However, the function and mechanism of RGS12 in osteosarcoma is completely unknown.
In this study, we explore for the first time about the role and molecular mechanisms by which RGS12 regulates osteosarcoma development and lung metastasis. Our findings present the first evidence that RGS12 is a novel tumor suppressor of osteosarcoma that inhibits YAP-TEAD1-Ezrin signaling, providing a proof of principle that targeting RGS12 may be a promising therapeutic strategy for osteosarcoma.
Results
RGS12 is downregulated in both human and mouse osteosarcoma tissues
Previous studies revealed that OSX-Cre;P53f/f/Rb1f/f animals could be used as a representative mouse model of osteosarcoma [19, 20]. Therefore, to study the potential role of RGS12 in osteosarcoma, we first evaluated the mRNA and protein expression profiles of Rgs12 in the OSX-Cre;P53f/f/Rb1f/f osteosarcoma mouse model. We found that osteosarcoma region of the bone derived from an OSX-Cre;P53f/f/Rb1f/f osteosarcoma mouse model expressed lower levels of Rgs12 mRNA and protein relative to mouse normal bone by qPCR analysis (Fig. 1a) and western blot (Fig. 1b). Immunohistochemistry staining analysis further confirmed that Rgs12 had a higher expression level in normal mouse bone compared to osteosarcoma bone (Fig. 1c). We also found that the expression of RGS12 decreased in the human osteosarcoma cell lines (SaOS2 and UMR106) compared to that in human normal dental pulp stem cells (DPSC) (Fig. S1). Next, we examined the expression level of RGS12 in human osteosarcoma specimens. Interestingly, we also found that the RGS12 expression level decreased in human osteosarcoma (Fig. 1d), which was parallel to the progression level of the osteosarcoma (Fig. 1e). Taken together, these results indicated that RGS12 is consistently downregulated in both human and mouse osteosarcoma compared with normal bone tissues.
Fig. 1. RGS12 is downregulated in both human and mouse osteosarcoma tissues.
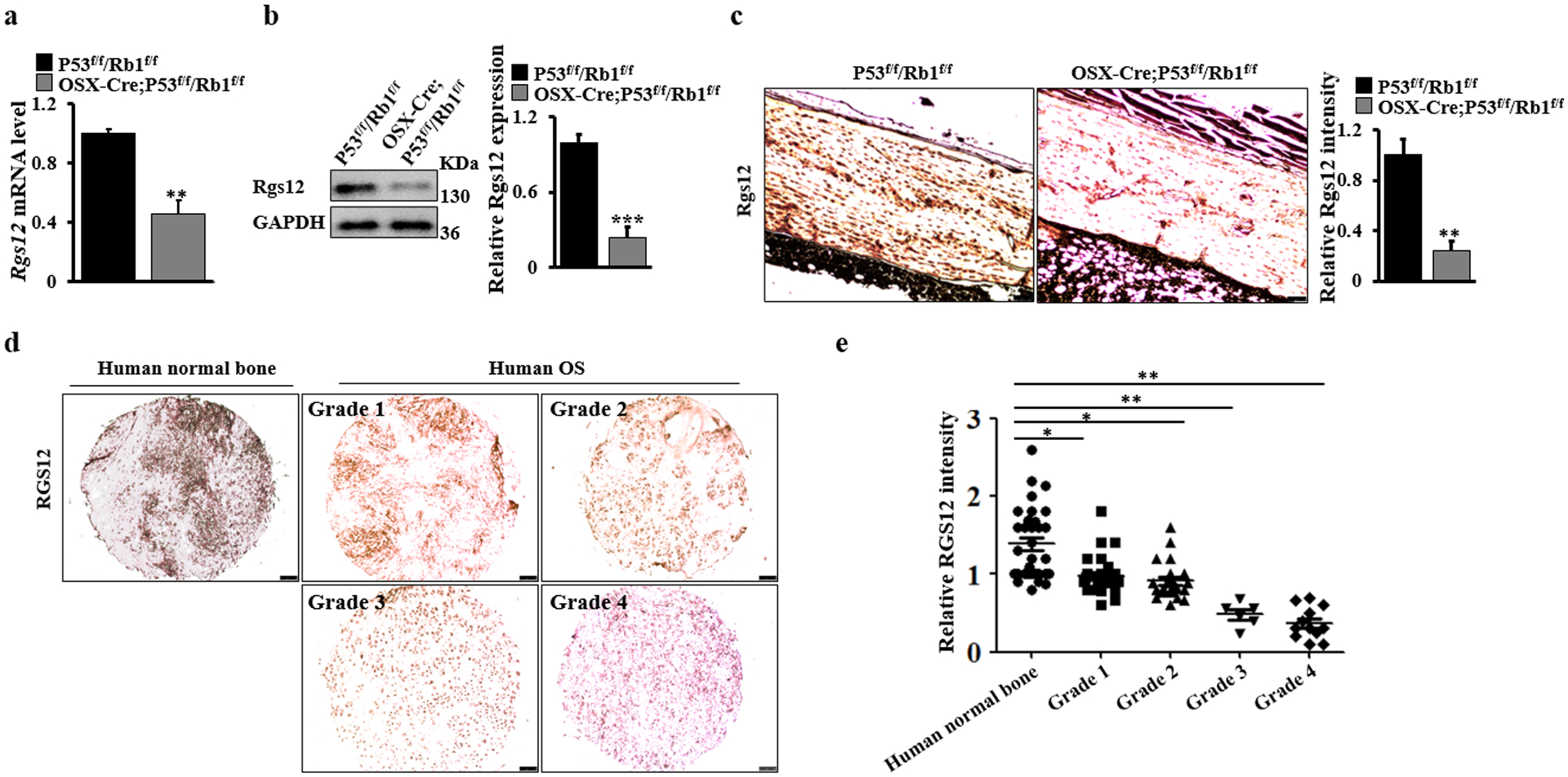
a Real-time PCR quantification of Rgs12 mRNA levels in the bones of OSX-Cre;P53f/f/Rb1f/f and P53f/f/Rb1f/f mice. b A representative image of western blot. Whole bone lysates from OSX-Cre;P53f/f/Rb1f/f and P53f/f/Rb1f/f mice were immunoblotted with antibodies against Rgs12 and GAPDH, respectively. N=3. The Rgs12 expression is quantified by ImageJ software in the corresponding column at right. c A representative image of immunohistochemical staining of Rgs12 on mouse osteosarcoma and normal bones. Scale bar, 75 μm. N=5. The Rgs12 expression is quantified by ImageJ software in the corresponding column at right. d A representative image of immunohistochemical staining of RGS12 on different grades of human osteosarcoma (tumor grades 1 to 4) and normal bones. Scale bar, 100 μm. OS, osteosarcoma. e, Quantification of RGS12 expression by ImageJ software based on IHC staining in human osteosarcoma (tumor grades 1 to 4; Grade 1, N=27; Grade 2, N=21; Grade 3, N=6; Grade 4, N=12) and normal bones (N=32). Error bars were the means ± standard error of the mean (SEM) of triplicates from a representative experiment. *P < 0.05, **P < 0.01 and ***P < 0.001.
RGS12 inhibits osteosarcoma cell migration, invasion and tumorsphere formation
To determine the role of RGS12 in osteosarcoma cell growth, we first established both RGS12 overexpression and knockdown stable cell lines in SaOS2 and UMR106, respectively (Fig. S2a, b). Our data showed that the ectopic expression of RGS12 significantly inhibited the proliferation of SaOS2 and UMR106; however, knockdown of RGS12 increased cell proliferation (Fig. 2a, b; Fig. S3). Additionally, knockdown of RGS12 promoted, while RGS12 overexpression inhibited anchorage-independent cell growth in soft agar (Fig. 2c, d). To further characterize the effect of RGS12 on the colony formation ability of osteosarcoma cells, colony-forming unit (CFU) assays were performed using bone marrow cells from Rgs12f/f (control) and CMV-Cre;Rgs12f/f mice. Of note, deletion of Rgs12 induced a severe increase in CFU compared to the control (Fig. S4a). These data demonstrated that RGS12 negatively regulates osteosarcoma cell proliferation and growth.
Fig. 2. RGS12 inhibits osteosarcoma cell migration, invasion and tumorsphere formation in vitro.
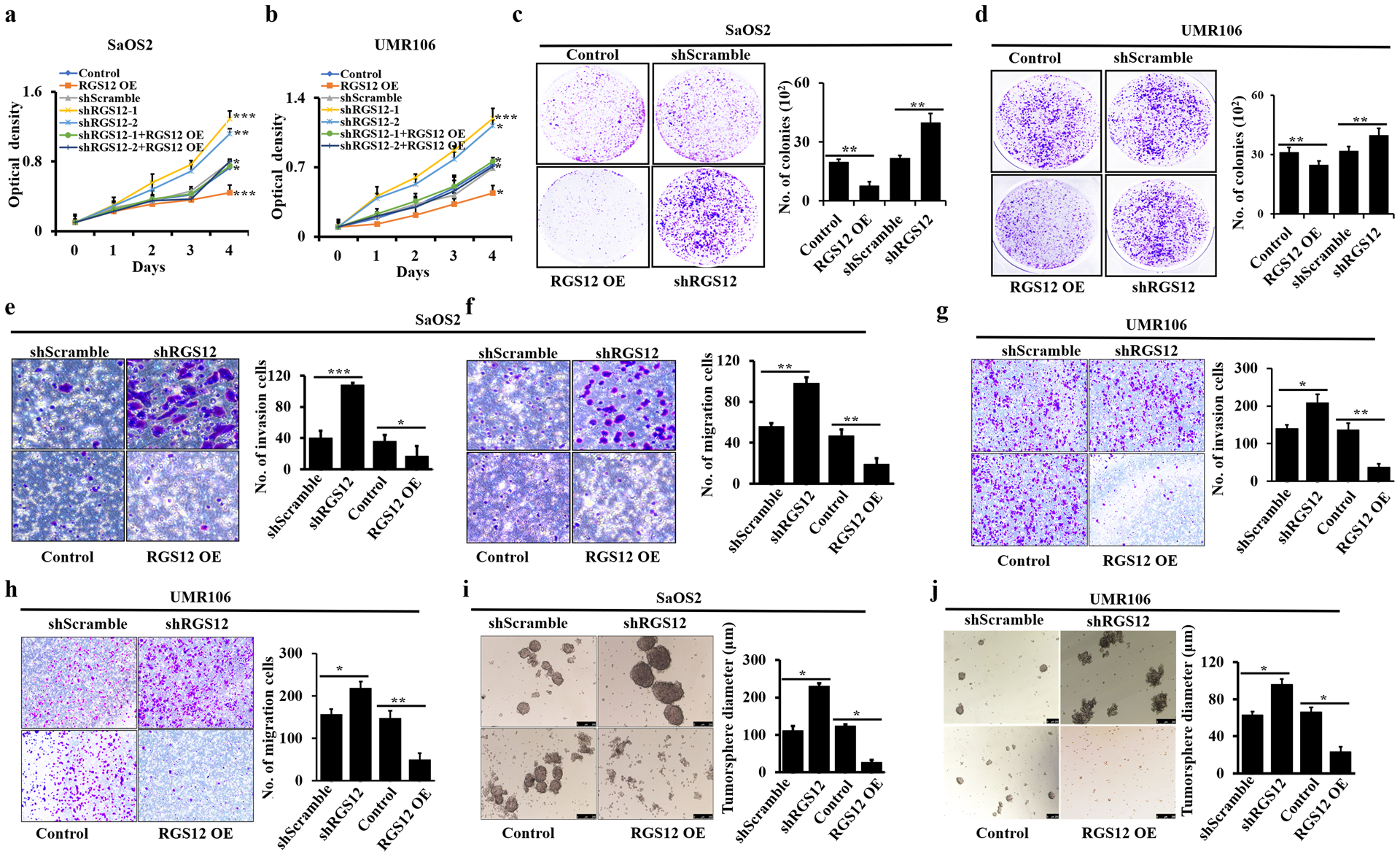
a-b The cell proliferation rates of indicated cells were detected by WST-1 assay. vs. shScramble or normal control cells. c, d The analyses of colony formation abilities in the indicated cells. Cell numbers are quantified in the corresponding column at right. e, g Cell invasion. Cell numbers are quantified in the corresponding column at right. f, h Cell migration. Cell numbers are quantified in the corresponding column at right. i-j Tumorspheres. Error bars were the means ± standard error of the mean (SEM) of triplicates from a representative experiment. *P < 0.05, **P < 0.01 and ***P < 0.001.
To explore the effect of RGS12 on cell migration and invasion in vitro, transwell assays with or without matrigel were performed in RGS12-overexpressed and -silenced SaOS2 and UMR106 cells, respectively. As shown in Fig. 2e–h, knockdown of RGS12 displayed a significant increase in cell migration and invasion, whereas RGS12 overexpression significantly inhibited cell migration and invasion compared with the controls in both cell lines. These results were further confirmed by the wound healing assay (Fig. S4b). The RGS12-silenced cells showed an advanced migration ability, while RGS12-overexpressing cells displayed a lower migration ability (Fig. S4b). To investigate the role of RGS12 on tumor function, we performed tumorsphere culture, which has been widely used as a tumor functional assay in vitro [21, 22]. Interestingly, we found that knockdown of RGS12 exhibited an increase in the formation and diameter of tumorspheres in both SaOS2 and UMR106, respectively; in contrast, RGS12 overexpression showed the opposite results (Fig. 2i, j; Fig. S4c). Given that the epithelial-mesenchymal transition (EMT) plays a critical role in cancer cell progression and metastasis [23, 24], we then detected the EMT-related factors in osteosarcoma cells. As expected, both immunofluorescence staining and western blot results showed that knockdown of RGS12 increased the expression of vimentin and N-cadherin but decreased the expression of E-cadherin in SaOS2 cells compared with those in the controls (Fig. S4d, e). Hence, these results indicated that RGS12 is a tumor suppressor of osteosarcoma.
Knockdown of RGS12 in SaOS2 promotes intratibial primary tumor growth and lung metastasis in SCID mice
To address whether RGS12 plays a tumor suppressive role in vivo, we employed an intratibial xenograft osteosarcoma model in SCID mice [25]. shScramble or shRGS12 cells were respectively injected orthotopically into the medulla of the left tibia of the mice (Fig. S5a). X-ray analysis of the tibia showed more extensive osteosarcoma with sclerosis and cortical destruction in the tibia of shRGS12 cell-injected group compared to that in the control (Fig. 3a). Consistently, leg swelling was apparent in the shRGS12-injected group but not in the control mice after tumor cells injection (Fig. S5b). Accordingly, Kaplan-Meier survival curves plotted for the mice indicated a significantly shorter mean survival rate in the shRGS12-injected mice compared with the control groups (Fig. 3b). Moreover, the results form X-ray and micro-CT analysis showed that in the groups with the injection of shRGS12 cells, more bone erosion were found compare to the group with the control cells (Fig. 3c, d). We also found that lung metastasis area in the shRGS12 cell-injected mice was much larger than that in the control mice (Fig. 3e, f). The immunofluorescence staining results showed a weaker RGS12+ signal in both human and mouse lung osteosarcoma metastasis regions compared to non-metastasis regions in the SCID mice (Fig. 3g, h). These findings demonstrated that knockdown of RGS12 promotes osteosarcoma growth and lung metastasis in SCID mice.
Fig. 3. Knockdown of RGS12 in SaOS2 promotes intratibial primary tumor growth and lung metastasis in SCID mice.
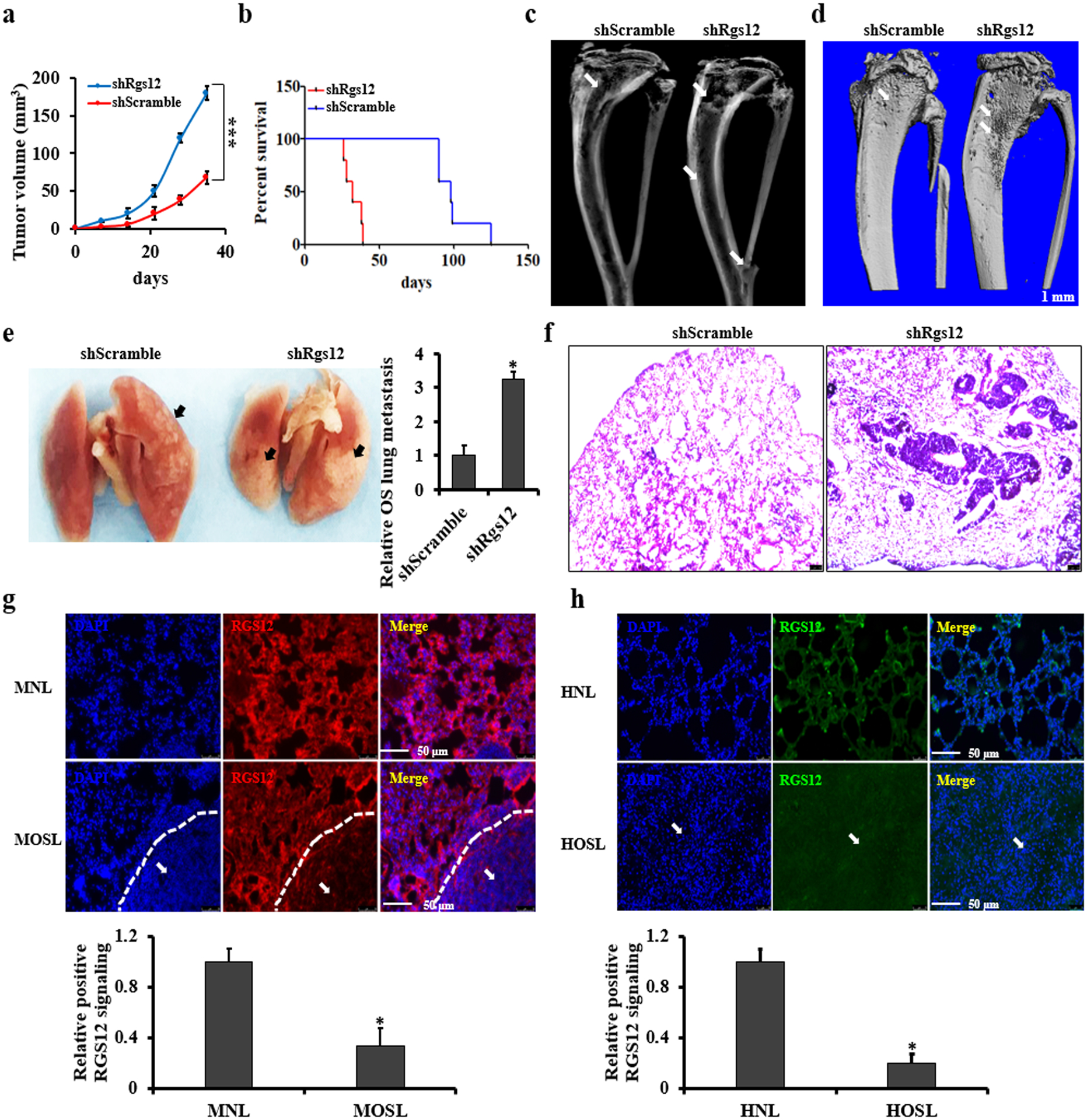
a Primary intratibial tumor growth over time in individual mice injected with shScramble or shRGS12 cells. b Kaplan-Meier survival analysis indicating the overall survival of mice injected with shScramble or shRGS12 cells. c, d Representative X-ray and micro-CT images of tumor-bearing legs after intratibial injection with shScramble or with shRGS12 cells at Day 28. The white arrow indicates the tumor in the bone. e Representative images of osteosarcoma lung metastasis of mice injected with shScramble or shRGS12 cells. Osteosarcoma lung metastasis is quantified in the corresponding column at right. The black arrow indicates the tumor in the lung. f Representative images of HE-stained lung sections of mice injected with shScramble or shRGS12 cells at Day 28. Scale bar, 100 μm. g Representative images of immunofluorescent staining of RGS12 in osteosarcoma lung metastasis sections of mice injected with shScramble or shRGS12 cells. The white arrow indicates the tumor in the lung. RGS12+ signaling is quantified in the corresponding column at lower panel. MNL, mouse normal lung tissues; MOSL, mouse osteosarcoma lung metastasis tissues. h Representative images of immunofluorescence staining for RGS12 in human osteosarcoma lung metastasis sections and controls. RGS12+ signaling is quantified in the corresponding column at lower panel. HNL, human normal lung tissues; HOSL, human osteosarcoma lung metastasis tissues. Error bars are the means ± standard error of the mean (SEM) of triplicates from a representative experiment. *P < 0.05 and ***P < 0.001.
RGS12 inhibits transcriptional YAP/TEAD1 activity through its PDZ domain function
Immunohistochemistry studies in a cohort containing human 32 normal and 66 osteosarcoma bones showed that 93.7% of osteosarcoma bones had higher nuclear expression of YAP, in contrast, only 58.2% of normal bones exhibited higher nuclear YAP expression (Fig. 4a). To further test the role of RGS12 in YAP activation, YAP phosphorylation was detected in shScramble and shRGS12 cells. As shown in Fig. 4b–d, knockdown of RGS12 inhibited YAP phosphorylation at Ser 127 through inhibiting phosphorylation of Mst1 and Lats1, YAP upstream kinases in osteosarcoma cell lines (SaOS2 and UMR106). Emerging evidence has shown that TEADs and YAP have higher expression levels in many human cancers, including osteosarcoma [24, 26, 27]. Moreover, TEAD1 is the major downstream transcriptional factor of Hippo-YAP signaling in osteosarcoma [7]. Given that knockdown of RGS12 could promote osteosarcoma progression and lung metastasis, we next asked whether RGS12 could regulate the expression and transcriptional activity of YAP and TEAD1. To address this, we first examined the expression of YAP and TEAD1 in three shRGS12 stable cell lines and found that knockdown of RGS12 significantly upregulated the expression of both genes (Fig. S6). By analyzing the protein structure of RGS12 with bioinformatics, we found that there was a PDZ binding motif in YAP and a PDZ domain in RGS12 protein (Fig. 4e), suggesting a potential interaction between RGS12 and YAP. To further test the interaction and importance of the PDZ domain, we carried out Co-IP and GST pull-down experiments. Here, we used RGS12-overexpressed 293T cells rather than normal osteosarcoma cell due to the poor antibody quality of RGS12 and lower level of RGS12 in osteosarcoma cells. As expected, we found that RGS12 was associated with YAP and that deletion of RGS12 PDZ domain abolished the interaction between RGS12 and YAP (Fig. 4f, g). Additionally, immunofluorescence staining result showed that YAP and TEAD1 translocated and colocalized into the nucleus after knockdown of RGS12 (Fig. 4j). Some studies have proved that YAP is a cofactor of the transcriptional factor TEAD1, and cannot directly regulate target genes expression due to lack of DNA binding motif [28, 29]. YAP usually needs to interact with TEAD1 to regulate the expression and function of downstream target genes [28, 30, 31]. Therefore, we further test whether knockdown of RGS12 affects YAP/TEAD1 transcriptional activity. By performing luciferase reporter assays as described [28, 30, 31], we found that knockdown of RGS12 increased YAP/TEAD1 transcriptional activity (Fig. 4h). In contrast, overexpression of RGS12 inhibited YAP/TEAD1 transcriptional activity (Fig. 4i). However, deletion of PDZ domain (RGS12ΔPDZ) impaired the RGS12 mediated inhibition of YAP/TEAD1 transcriptional activity (Fig. 4i). Furthermore, we also found that overexpression of YAP promotes TEAD1 transcriptional activity, which can be inhibited by overexpression of RGS12 rather than RGS12ΔPDZ, suggesting that RGS12 is associated with YAP through the PDZ domain to inhibit YAP and TEAD1 activation. To further determine the function of PDZ domain, we deleted the PDZ domain of RGS12 and test whether the RGS12-YAP association was required for tumor cell growth. Interestingly, as shown in Fig. 4k, deletion of PDZ domain significantly attenuated the tumor inhibitory function of RGS12. Thus, these findings suggested that RGS12 suppresses tumor cell growth through its PDZ domain binding with YAP to inhibit YAP activation.
Fig. 4. RGS12 inhibits transcriptional YAP/TEAD1 activity through its PDZ domain function.
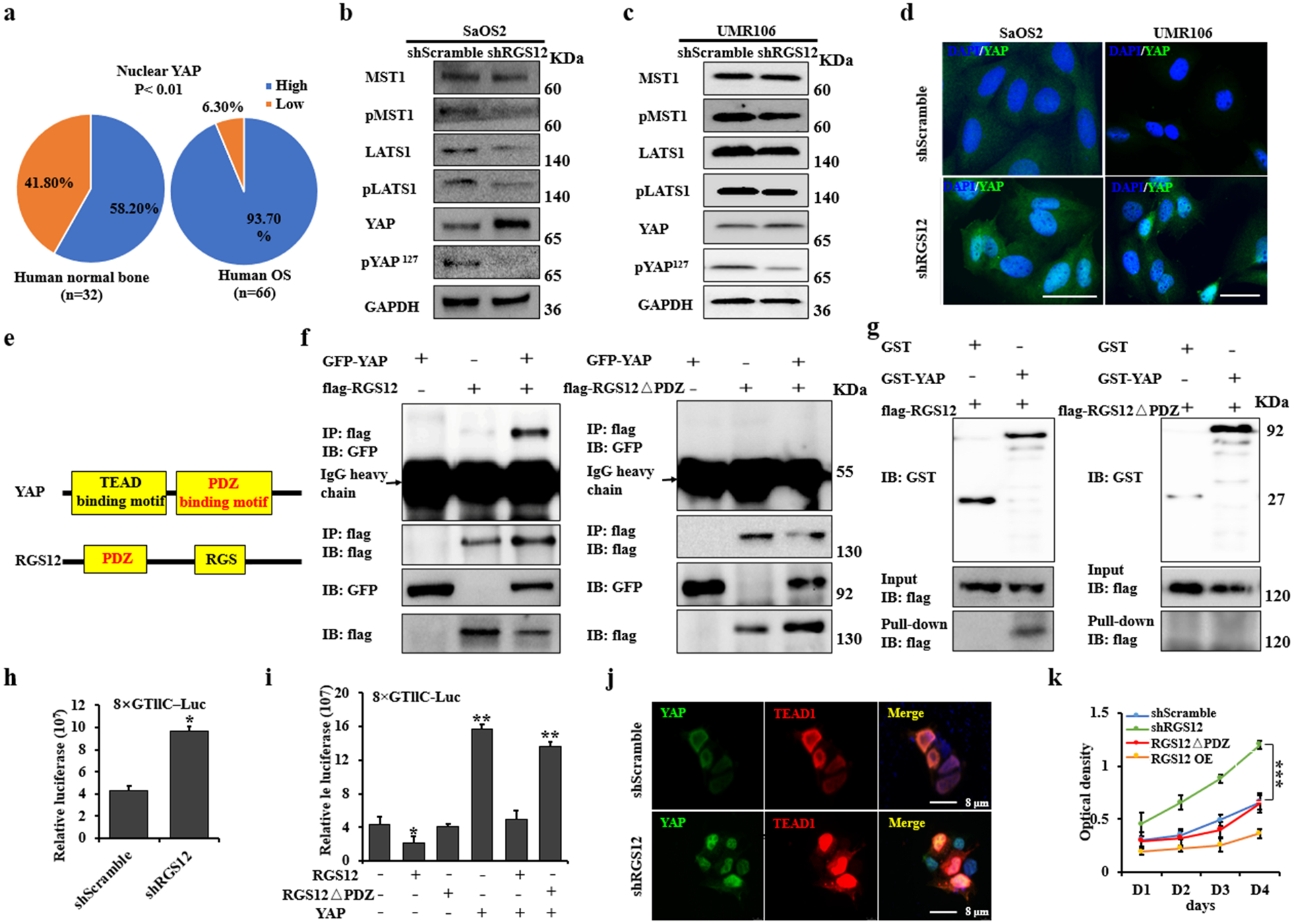
a Statistical analysis of nuclear YAP expression in human osteosarcoma and normal bone specimens. b, c Whole protein lysates of shScramble and shRGS12 cells were immunoblotted with the indicated antibodies. d Representative immunofluorescence-stained images of shScramble and shRGS12 cells for YAP expression, DAPI staining for nuclear. e Structures of YAP and RGS12. f Co-IP experiments of GFP-YAP, flag-RGS12 or flag-RGS12ΔPDZ in 293T cells. g 293T cells were transfected with flag-RGS12 or flag-RGS12ΔPDZ, respectively. Cells were lysed after 48 hr, and cell lysates were incubated with GST or GST-YAP protein on glutathione beads. The precipitated complexes were analyzed by western blot. h Scramble and shRGS12 cells were respectively seeded in 12-well plates. Luciferase reporter and pRL-TK vector (internal control) were co-transfected. Luciferase activities were measured after transfection of 48 hr. i Luciferase activity was measured in SaOS2 cells following co-transfection with flag-YAP, flag-RGS12 or flag-RGS12ΔPDZ, respectively. j Immunofluorescent staining of YAP and TEAD1 in shScramble and shRGS12 cells. k The proliferation of the indicated cells was detected by WST-1 assay. Error bars were the means ± standard error of the mean (SEM) of triplicates from a representative experiment. *P < 0.05, **P < 0.01, ***P < 0.001.
Knockdown of RGS12 promotes YAP/TEAD1-dependent Ezrin expression
Ezrin is a member of the ERM (ezrin, radixin and moesin) protein family and plays a positive role in maintaining cell polarity and shape, as well as in regulating tumor progression and metastasis in several cancers including osteosarcoma and rhabdomyosarcoma [32]. Current studies show that Ezrin is necessary for osteosarcoma lung metastasis [32]. To further test whether RGS12 affects osteosarcoma progression and metastasis through regulating YAP mediated Ezrin expression, we knocked down either YAP or RGS12 genes or both in SaOS2. As expected, silencing of RGS12 evoked Ezrin expression, on the contrary, silencing of YAP inhibited Ezrin expression. Moreover, when both YAP and RGS12 were silenced, increase of Ezrin expression caused by RGS12 silence was blocked by YAP silence (Fig. 5a). These data suggested that knockdown of RGS12 enhances Ezrin expression through YAP signaling (Fig. 4d; Fig. 5a). Because YAP is transcriptional coactivator that lack DNA-binding activity, it must interact with DNA-binding transcription factors to regulate target gene’s expression [5, 33]. In osteosarcoma, it’s reported that YAP/TEAD1 complex is a key upstream modulator [7]. To test whether YAP/TEAD1 binds with a specific region of Ezrin promoter to regulate Ezrin expression, we analyzed the YAP/TEAD1 binding motif in Ezrin promoter using Vector NTI software. Interestingly, we found 5 different YAP/TEAD1 DNA binding elements in Ezrin promoter (Fig. S7). By performing ChIP assay using YAP antibody and TEAD1 siRNA, we found that AATTCC, the DNA motif for TEAD1, is the most predominant hit (Fig. 5b–f; Fig. S8). Additionally, by subjecting immunoprecipitated chromosomal DNA to PCR using primers designed to amplify the Ezrin promoter region harboring the TEAD1 binding sites, we found a significant increase of AATTCC hit in Ezrin promoter region after knockdown of RGS12 (Fig. 5g), suggesting that knockdown of RGS12 enhances the binding of YAP/TEAD1 to the Ezrin promoter. Additionally, we also found that the human lung osteosarcoma metastasis tissues have higher expression level of Ezrin compared to normal lung, and YAP expression is maintained in metastatic tumor (Fig. S9). Overall, these data indicated that knockdown of RGS12 promotes YAP activation and YAP/TEAD1 mediated transcriptional regulation of Ezrin expression.
Fig. 5. Knockdown of RGS12 induces YAP-dependent Ezrin expression.
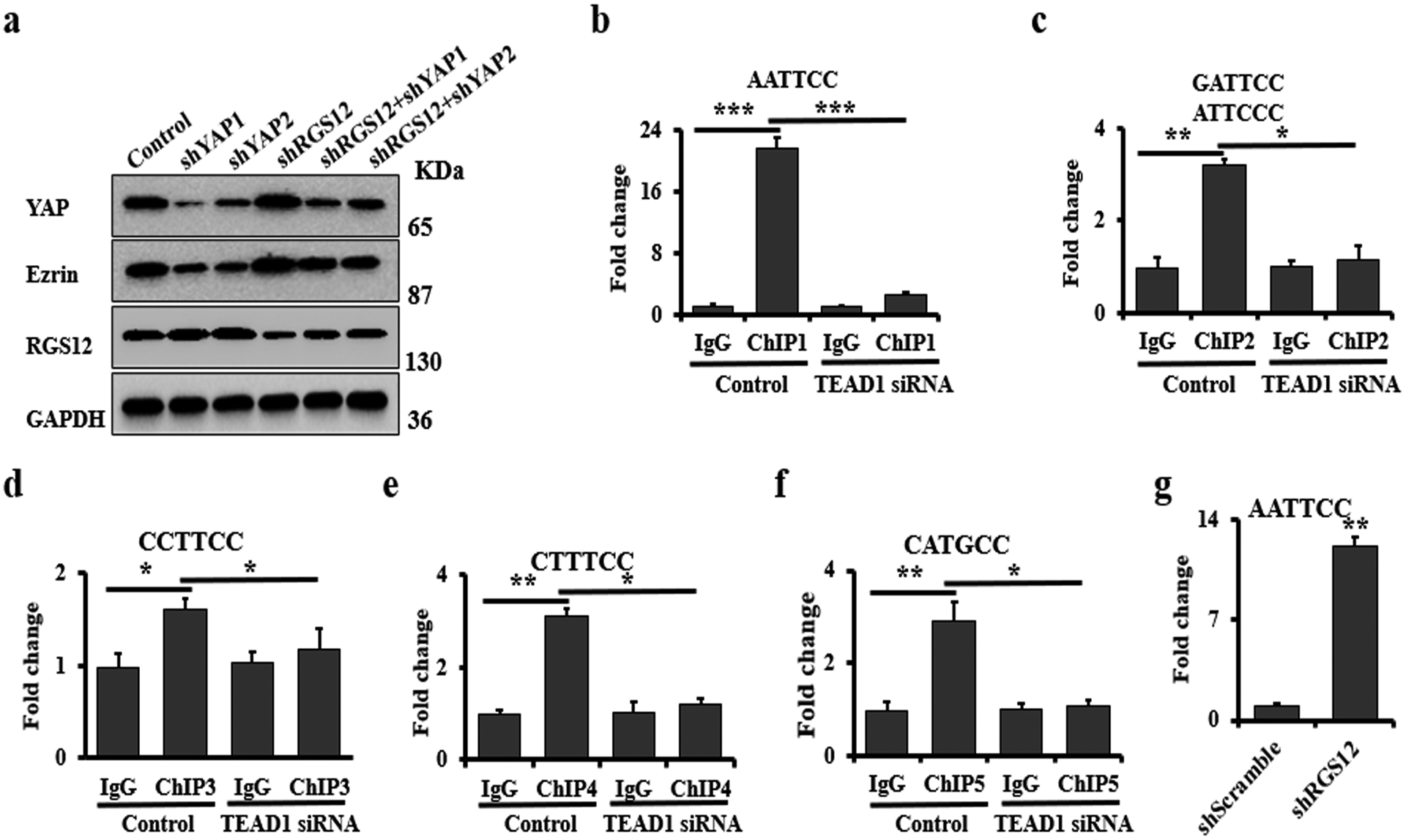
a Immunoblots of the whole protein lysates, which were isolated from the cells after co-transfection with shRGS12 or/both shYAP1/2 lentivirus for 48 hrs. b-f ChIP. Co-occupation of YAP/TEAD1 using YAP antibody in the Ezrin promoter. c ChIP. Co-occupation of YAP/TEAD1 using YAP antibody in the Ezrin promoter after silence of RGS12. Error bars were the means ± standard error of the mean (SEM). *P < 0.05, **P < 0.01, ***P < 0.001.
RGS12 negatively regulates Ezrin expression via GNA12/13-RhoA-YAP pathway
LPAs have been shown to act through membrane receptors, GNA12/13, and Rho GTPases to inhibit Lats1/2 activity and thereby promote YAP activation [13]; additionally, RGS12 couples to GNA12/13 [34] and activates RhoA to control tumor cell migration [35, 36]. Given that YAP activation promotes Ezrin expression, we test whether RGS12 modulates Ezrin expression through the GNA12/13-RhoA-YAP pathway in vivo and in vitro. Our data showed that loss of RGS12 upregulated the expression of GNA12/13, RhoA and Ezrin, and downregulated the phosphorylation of YAP in the bones of CMV-Cre;Rgs12f/f mice compared to those of Rgs12f/f mice (Fig. 6a). Consistent to the results in vivo, silencing RGS12 upregulated the expression of GNA12, GNA13, RhoA (Fig. 6b) and downstream Ezrin in SaOS2 cells (Fig. 6c), which was inhibited by C3 (Rho GTPases inhibitor), ki6425 (GPCR inhibitor) and Y27632 (Rock inhibitor) (Fig. 6c; Fig. S10). These results demonstrated that RGS12 negatively regulates Ezrin through the GPCR signaling pathway. To further investigate the role of RGS12 in the GNA12/13-RhoA-YAP pathway, we assessed the activity of RhoA. The results showed that knockdown of RGS12 significantly increased RhoA activity (Fig. 6d). Additionally, overexpression of RhoA promoted YAP and Ezrin expression in SaOS2 cells compared with the control group (Fig. 6e, f). Taken together, these results demonstrated that RGS12 negatively regulates Ezrin expression through inhibiting GNA12/13-RhoA-YAP pathway.
Fig. 6. RGS12 negatively regulates Ezrin expression via GNA12/13-RhoA-YAP pathway.
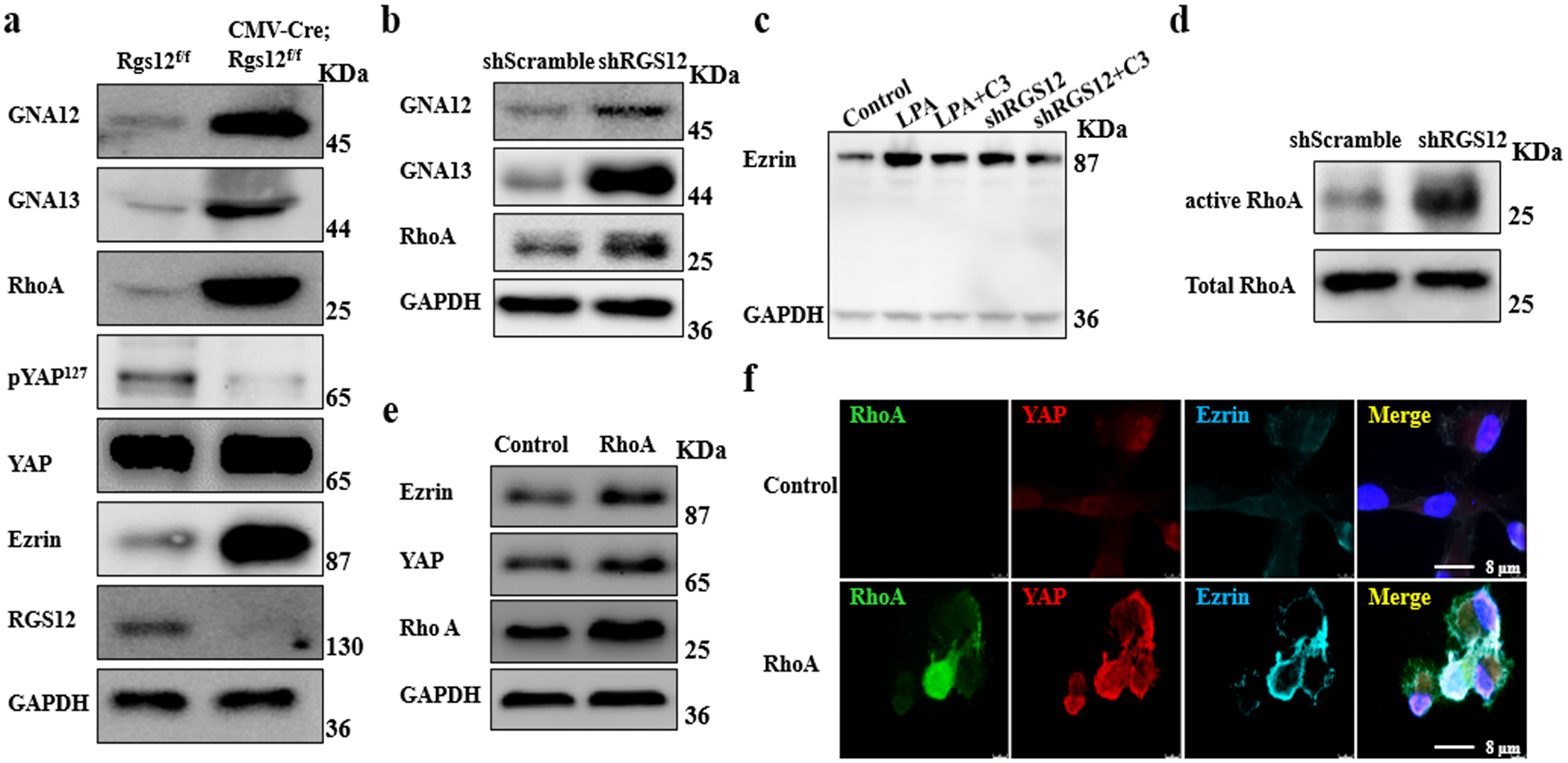
a Bone tissues from CMV-Cre;Rgs12f/f and control mice were analyzed by immunoblotting with the indicated antibodies. b Western blot for analyzing GNA12, GNA13 and RhoA expression in the lysates from shScramble and shRGS12 cells. c SaOS2 cells were starved for 12 hr and then treated with 2 mg/mL C3 (RhoA inhibitor) or 1 mM LPA (GPCR activator) for 48 hr. The whole protein lysates of the treated cells were analyzed by western blot. d RhoA activity. e Western blot analysis of the whole protein lysates isolated from the SaOS2 cells that were transfected with empty vector (Control) and pcDNA3.1-RhoA plasmid, respectively. f Immunofluorescent staining of RhoA, Ezrin and YAP in SaOS2 cells that were transfected with empty vector (Control) and pcDNA3.1-RhoA plasmid, respectively.
RGS12 PDZ domain peptides inhibit osteosarcoma formation and lung metastasis
PDZ domain is an abundant protein interaction module that often recognizes short amino acid motifs at the C-termini of target protein. Previous studies also showed that PDZ domain has a single binding site in a groove between the αB and βB structural elements and the amino acid residues “GYGF” of PDZ domain plays a key role in the ligand binding [37]. Based on our findings that the PDZ domain of RGS12 is important for inhibiting YAP activity, we synthesized the PDZ domain peptides of RGS12, termed “PDZ”. To test whether PDZ can bind with YAP to promote YAP phosphorylation, we used PDZ to treat SaOS2 cells. As expected, PDZ inhibited cell colony formation, as well as the tumorsphere and proliferation of SaOS2 cells (Fig. 7a, b), whereas PDZ peptides have no effect on osteosarcoma formation when the GYGF was mutated (Fig. 7a). Moreover, we also found that PDZ could repress YAP nuclear translocation (Fig. 7c), but RGS12 PDZ-GYGF-mutated control peptide didn’t inhibit its nuclear translocation (Fig. 7c). Further results showed that PDZ increased YAP phosphorylation, but did not affect the phosphorylation of Mob and Lats1 of the YAP upstream kinase (Fig. 7d). Next, to further test PDZ function in vivo, shRGS12 transfected cells with or without 100 nM of PDZ peptides were injected into the left tibia of 6-week-old SCID mice and then tumor formation ability was analyzed. As shown in Fig. 7e, PDZ peptides could significantly inhibit tumor formation. Accordingly, Kaplan-Meier survival curves plotted for the mice showed a significantly longer mean survival rate when having the PDZ peptide-injection compared with the control animals (Fig. 7f). Notably, the mice injected with shRGS12 cells mixed with the PDZ peptides exhibited less tumor formation and lung metastasis compared to the control mice (Fig. 7g, h; Fig. S11). Doxorubicin (DOX) and methotrexate (MTX) have been reported to be the most commonly used drugs for the treatment of osteosarcoma. Resistance to these drugs substantially decreases patient survival rates [3, 38]. Hence, we further investigated whether knockdown of RGS12 can affect osteosarcoma chemoresistance in SaOS2 cells. Our data showed that knockdown of RGS12 increased the chemoresistance of osteosarcoma cells (Fig. S11). Thus, these data suggested that RGS12 PDZ domain may be a promising therapeutic drug target for osteosarcoma.
Fig. 7. RGS12 PDZ peptides inhibit osteosarcoma formation and lung metastasis.
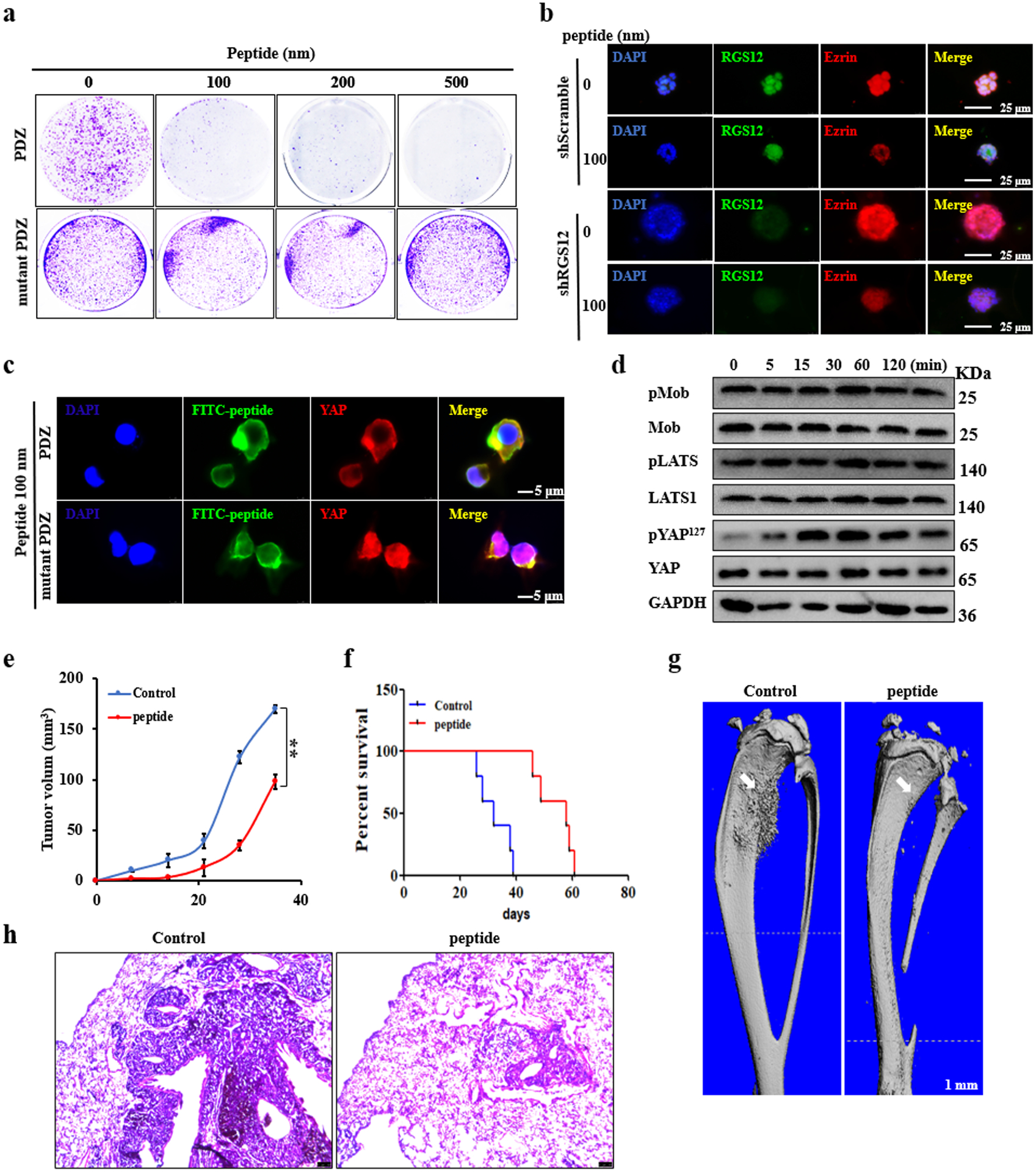
a Soft agar. Colony formation ability analysis after PDZ or PDZ mutated peptide treatment for 3 weeks in SaOS2 cells. b Immunofluorescent staining of RGS12 and Ezrin in shScramble and shRGS12 cells after co-culture of PDZ peptides and matrigel for 7 days. c, d PDZ peptides inhibited YAP nuclear translocation by increasing the phosphorylation level of YAP in SaOS2 cells. e Representative X-ray images of tumor-bearing legs after intratibial injection with shRGS12 cells mixed with 0 or 100 nm PDZ peptides. f Kaplan-Meier survival analysis indicating overall survival of mice injected with shRGS12 cells mixed with 0 or 100 nM of PDZ peptides. g Representative micro-CT images of tumor-bearing legs after intratibial injection with shRGS12 cells mixed with 0 or 100 nM of PDZ peptides at Day 35. The white arrow indicates the tumor in the bone. h Representative images of HE-stained lung sections of mice injected with shRGS12 cells mixed with 0 or 100 nM of PDZ peptides at Day 35. Scale bar, 100 μm. Error bars were the means ± standard error of the mean (SEM) of triplicates from a representative experiment. **P < 0.01.
Discussion
Abnormal expression of RGS proteins has been observed in different kinds of cancers [39]. For example, RGS1, RGS5, RGS6 and RGS19 are upregulated while RGS2 and RGS4 are downregulated in ovarian cancer [39]. The effect of RGS protein in promotion or inhibition of cancer progression are mainly depend on the type of cancer [39]. However, how RGS proteins regulate cancer formation, progression and metastasis are largely unknown.
RGS12 is the largest protein in RGS superfamily. Our previous studies showed that RGS12 regulates the differentiation and function of osteoclast and osteoblast, bone homeostasis and development in vivo and in vitro [12, 16, 17], and other studies reported that RGS12 plays key roles in neuronal function and behavior [40]; however, the role of RGS12 in cancer development, especially osteosarcoma, remains undefined. Our results showed that RGS12 expression markedly decreases in both human and mouse osteosarcoma specimens. Moreover, the expression of RGS12 is correlated with tumor severity. These findings are consistent with the recent finding that RGS12 is significantly downregulated in African American prostate cancer (AA PCa) accompanying with a higher propensity for tumorigenicity [14]. Moreover, RGS12 is also identified as one of the cell cycle inhibitors in some tumor cell lines (MCF7, SaOS2 and U-2 OS) due to the inhibition of DNA synthesis [15]. RGS14, a RGS12 homologous gene, has been also reported to be a p53 target gene that causes growth arrest in the G1 phase of the cell cycle [41]. Consistent to these findings, we found that knockdown of RGS12 increases osteosarcoma cell proliferation, migration, invasion and EMT in vitro and in vivo. We further found the RGS12 is a novel tumor suppressor in osteosarcoma through the regulation of RhoA/YAP/TEAD1/Ezrin pathway. Thus, our new findings demonstrate that targeting RGS12 may be an efficient therapeutic strategy for osteosarcoma.
Hippo pathway plays a critical role in human cancers, including osteosarcoma; and aberrant Hippo pathway eventually triggers the transcriptional activation of the YAP/TEAD1 complex and initiates the downstream target genes expression [5, 6, 42]. Consistent with the previous study [7], we also found YAP/TEAD1 complex plays a critical role in osteosarcoma. Recent studies also showed that YAP is highly expressed and predicts a poor prognosis in osteosarcoma [43]. Knockdown of YAP inhibits the proliferation, migration and invasion of osteosarcoma cells [3, 44]. Interestingly, we found that knockdown of RGS12 in osteosarcoma cells markedly upregulates YAP expression and activation. Additionally, our data showed that RGS12 associates with YAP through its PDZ domain to inhibit YAP nuclear translocation in osteosarcoma model. These findings are supported by the reports showing that PDZ domain can interact with GPCR chemokine receptors and the PDZ binding motif containing protein [11, 45] and that YAP activity can be modulated by the protein-protein interaction and cytosol retention [26]. Furthermore, we found that the PDZ domain of RGS12 inhibits osteosarcoma cell proliferation and colony formation; and increases the phosphorylation level of endogenous YAP at Ser 127 by binding with YAP. This is supported by the finding that YAP phosphorylation at Ser 127 shows higher level in the resting cells and normal tissues compared to tumor tissues by promoting 14-3-3 binding and subsequent cytoplasmic sequestration and inactivation [46]. Most importantly, in the in vivo animal model study, we found that the RGS12 PDZ domain peptides inhibit the development of intratibial tumors and lung metastasis, indicating RGS12 PDZ domain is critical for negatively regulating YAP activation, and RGS12 is a negative tumor suppressor for osteosarcoma.
The Rho GTPase family is a member of the Ras superfamily, which is critical for the invasion and metastasis of various cancers, including bone cancer. RhoA is one of the well-characterized Rho GTPases [47]. A recent study reported that RhoA is involved in the Hippo signaling pathway [48] and RhoA inactivation inhibits the invasion and migration of LM8 murine osteosarcoma cells [49]. Consistently, we found that RGS12 negatively regulates the transcriptional activity of YAP/TEAD1 complex in osteosarcoma through altering RhoA activity. This result is further supported by the findings that GPCRs transmit the extracellular signals by coupling to the heterotrimeric Gα12/13 proteins (encoded by GNA12 and GNA13, respectively) to activate RhoA, which results in YAP activation [50, 51]. Accumulating evidence indicates that a higher level of Ezrin expression is associated with poor clinical outcome and metastatic behavior of various solid tumors, including osteosarcoma lung metastasis [52]. Additionally, one study showed that Ezrin expression is regulated by YAP/TEAD complex to promote metastasis in pancreatic cancer cells [53]. Our results clearly demonstrated that knockdown of Rgs12 significantly enhances Ezrin expression via the activation of GNA12/13-RhoA-YAP pathway, which results in osteosarcoma growth and progression, as well as lung metastasis (Fig. 8).
Fig. 8. A proposed model to illustrate the regulatory mechanism of RGS12 on Ezrin expression in osteosarcoma.
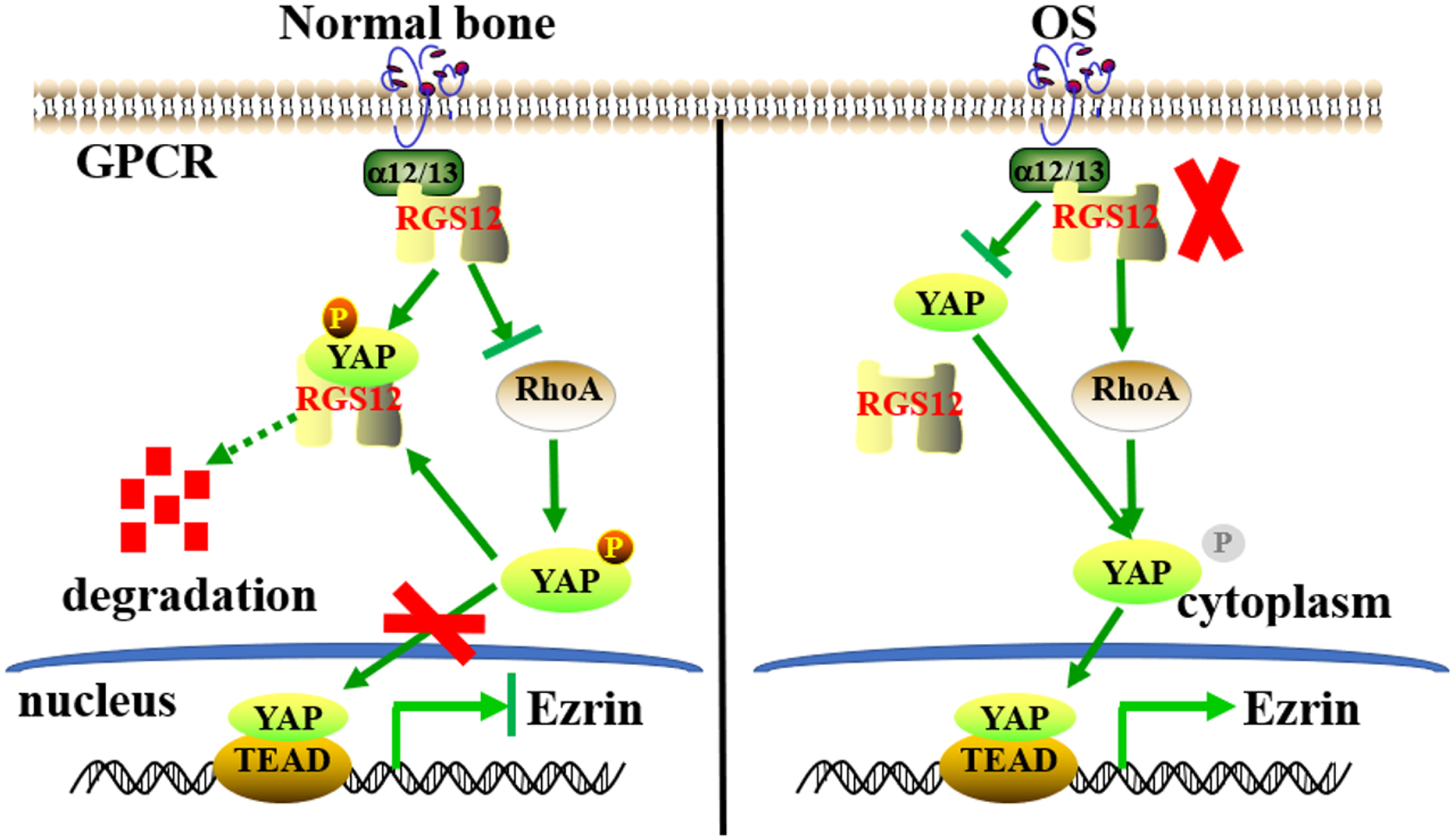
In normal bone, RGS12 promotes the growth arrest of osteosarcoma through inhibiting YAP nuclear translocation. In osteosarcoma bone, RGS12 has a lower expression and knockdown of RGS12 enhances RhoA activity and the transcriptional activity of YAP/TEAD1 to induce Ezrin expression.
In conclusion, this study reveals that RGS12 is a novel tumor suppressor in osteosarcoma via negatively regulating YAP/TEAD1-dependent Ezrin expression. This study highlights that RGS12 and its PDZ domain may be a promising drug target for osteosarcoma treatment.
Materials and Methods
Antibodies and reagents
Antibodies against p127YAP (D9W2I), YAP (D8H1X), MST1, pMST (E7UD1), LATS1 (C66B5), pLATS1, Lamin B1 (D9V6H) and GAPDH were from CST. Antibodies of Ezrin, GNA12, GNA13, RhoA, flag, TEAD1, GST and GFP were from Santa Cruz Biotechnology. RGS12 antibody (ab1) was from Sigma. The secondary fluorescent antibodies were from Abcam. PDZ domain peptides of RGS12 were purchased from GenScript. GPCR activator LPA, GPCR inhibitor Ki6425, Rho GTPases inhibitor C3 and Rock inhibitor Y2763 were obtained from Sigma. Doxorubicin hydrochloride (DOX), methotrexate 4-Amino-10-methylfolic acid hydrate (MTX) and EDTA-free cocktail inhibitor tablets were all obtained from Fisher Scientific™. TEAD1 siRNA and controls were from Santa Cruz Biotechnology. FuGENE® HD Transfection Reagent was purchased from Promega Corporation. The primers used for the quantification are listed in Supplementary Table 1.
The following plasmids: pRL-TK was generously provided by Dr. Zhen Zhang (University of Pennsylvania, Philadelphia, PA, USA); pcDNA3.1, shYAP1/2, pcDNA3.1-GFP-YAP, PET-GST-YAP and pcDNA3.1-RhoA were obtained from Addgene. Three human shRGS12 lentivectors (shRGS12–1, shRGS12–2 and shRGS12–3; Catalog # i019000) were ordered from ABM. pcDNA3.1-flag-RGS12 (flag-RGS12) and the pcDNA3.1-flag-RGS12 mutant with the deletion of the PDZ domain vectors (flag-RGS12ΔPDZ) were constructed in our lab.
Animals and human specimens
Rgs12f/f mice were created by our lab as previously reported [17]. P53f/f/Rb1f/f mice were kindly provided by Dr. David M. Feldser (University of Pennsylvania, USA). OSX-Cre, CMV-Cre and SCID mice were purchased from The Jackson Laboratory (Bar Harbor, USA). All the human osteosarcoma, normal bone, osteosarcoma lung metastasis and normal lung specimens were obtained from US Biomax (USA). All protocols were approved by the Institutional Animal Care and Use Committees at the University of Pennsylvania and complied with the National Research Council’s Guide for the Care and Use of Laboratory Animals.
Intratibial human osteosarcoma xenograft mouse model
We established the stable cell lines with different expression levels of RGS12 in SaOS2 and UMR106. They were named Control (transfected empty vector pcDNA3.1 in SaOS2 or UMR106), shScramble (transfected scramble shRNA lentivirus in SaOS2 or UMR106), RGS12 OE (pcDNA3.1-RGS12-overexpressing cells) and shRGS12 (RGS12-silenced cells), respectively. A total of 5×105 shScramble or shRGS12 cells mixed with 0 or 100 nM of PDZ peptides in 10 μL PBS were injected intramedullary with a Hamilton syringe into the left tibia of 6-week-old SCID mice, respectively. The healthy condition of the SCID mice was monitored 4 times per week, and the primary tumor growth was examined by X-ray and by calculating the osteosarcoma volume with the equation length × (width)2/2 of the osteosarcoma-bearing tibia minus the length × (width)2/2 of the control tibia after measurements of the respective widths and the lengths with a caliper every week [54, 55].
Cell culture, transfection and luciferase reporter assay
Human osteosarcoma cell lines SaOS2 or UMR106 were cultured in McCoy’s 5A Medium Modified (Gibco, USA) or DMEM (Gibco, USA) supplemented with Pen-Strep and 10% FBS (Gibco, USA), respectively. 293T and DPSC were cultured in DMEM supplemented with Pen-Strep and 10% FBS (Gibco, USA). The cells with shScramble or shRGS12 stable transfection were seeded in the 12-well plate and then co-transfected with luciferase reporter and the indicated plasmids. Luciferase activities were measured after 48 hrs of transfection using a Dual-Luciferase Assay Kit according to the manufacturer’s instructions.
Cellular functional assay
For the proliferation assay, logarithmically growing cells were trypsinized and were seeded in triplicates in 96-well plates (5×103 cells/well). The WST-1 assay was performed using the WST-1 Cell Proliferation Assay Kit (Cayman Chemical, USA) according to the manufacturer’s instructions after 24, 48, 72 and 96 hrs in culture.
For the adhesion assay, briefly, a 96-well plate was coated with fibronectin (Sigma, USA) overnight and was then blocked for 30 min with 0.5% BSA. The cells were suspended at a final concentration of 5×103 cells/mL in serum-free medium for seeding into the 96-well plate. The WST-1 assay (Cayman Chemical, USA) was used to determine the number of remaining cells (adherent cells).
The wound-healing assays were performed using a sterile 200 μL pipette tip to scratch the cells to form a wound when the cells were cultured to 100% confluence. Migration of wounded cells was evaluated at 0, 24 and 48 hr with a light microscope.
The migration assay was conducted using Transwell plates according to the manufacturer’s instructions. The top chambers of the transwells received 0.2 mL of cells (5×103 cells/mL) in serum-free medium, and the bottom chambers received 0.25 mL of McCoy’s 5A Medium Modified containing 10% FBS. The cells were incubated in the transwells at 37 °C in 5% CO2 for 24 hrs. Migrated cells were fixed and stained with 0.05% crystal violet. The migrated cells in each well were counted in three different fields per experiment under the microscope.
The cell invasion assay was performed using an EZCell™ Cell Invasion Assay Kit (BioVision, Inc, USA) according to the manufacturer’s instructions.
For the soft agar colony formation assay, cells were seeded with 0.2% agar and layered onto 1% agar beds in 6-well plates. The cells were fed with 1 mL of medium every five days. The colonies were stained with 0.05% crystal violet and counted after 3 weeks.
The 3D tumorsphere formation assay was performed using a MammoCult Human Medium Kit (Stemcell Technologies, USA) according to the manufacturer’s instructions [56]. Briefly, the Control, RGS12 OE, shScramble and shRGS12 cells were respectively seeded on low-attachment plates in a defined, serum-free culture medium at a density of 3 × 102 cells/well. Tumorspheres were cultured for 7 days. The tumorsphere diameter was measured by a Leica microanalysis system.
For the colony-forming assay, bone marrow cells from Rgs12f/f and CMV-Cre;Rgs12f/f were cultured at 37 °C in 5% CO2 for 2 weeks and then fixed in 4% PFA for 5 min and stained with 0.05% crystal violet.
Radiographic procedures and bone micro-computed tomography (CT) analysis
Radiographic procedures were performed in the Siemens X-ray equipment (Madison, WI, USA). The bone morphology and microarchitecture was carried out and analyzed using a micro-CT system as described previously [12] (School of Medicine, University of Pennsylvania, USA).
Immunofluorescence and immunohistochemistry
For immunofluorescence, briefly, cells cultured on coverslips were fixed with 4% PFA and permeabilized with TBST (0.3% Triton X-100 in TBS). Non-specific binding of antibodies to cells was blocked by 1% BSA and incubated with the corresponding primary antibody overnight at 4°C. Subsequently, the cells were washed 3 times with TBST and incubated with Alexa Fluor 488, 594 or 647 conjugated secondary antibodies for 1 hr at RT. Nuclei were counterstained with DAPI and washed 3 times with TBST. Then, the cells were mounted and visualized using a fluorescence microscope.
For immunohistochemistry, tissues previously fixed in 4% PFA were dehydrated through serial incubations in 75%, 95%, and 100% ethanol and xylene and then embedded in paraffin. The sections were mounted onto slides and deparaffinized. Endogenous peroxidase was inactivated by 3% H2O2 at RT for 10 min. The tissue sections were blocked at RT for 1 hr in goat serum. Subsequently, the primary antibody was added and incubated with sections at RT for 1 hr. After washing 3 times with TBST, the sections were incubated with corresponding secondary antibody at RT for 1 hr. After washing with TBST, the sections were incubated with the DAB (Dako, USA) for staining. Finally, the sections were counterstained with hematoxylin and visualized using a microscope.
Western blot, Co-IP and GST pull-down assay
Cells were lysed in modified RIPA lysis buffer (50 mM Tris–HCl, 150 mM NaCl, 5 mM EDTA, 0.5% NP-40 and 0.1% SDS) supplemented with a protease cocktail inhibitor (Fisher Scientific™, USA). The protein concentration was determined using the Pierce BCA Protein Assay Kit (Thermo Fisher, USA). 20 μg protein were subjected by SDS–PAGE, transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, USA), and immunoblotted with various antibodies. Following overnight incubation at 4°C, the membranes were washed 3 times by TBST (0.1% Tween-20 in TBS) and then incubated with corresponding secondary antibodies for 1 hr at RT. After washing 3 times by TBST, HRP-conjugated secondary antibodies were detected by chemiluminescence with ECL (Thermo Fisher, USA).
Regarding Co-IP, 293T cells were transfected with various expression vectors as indicated by FuGENE® HD transfection reagent (Promega, USA). After transfection of 48 hrs, the 293T cells were first lysed with IP buffer for 10 min at 4°C, and its supernatants were collected and incubated with anti-GFP antibody, anti-flag antibody and protein A/G agarose (Sigma, USA). After washing 3 times with TBST, the immune complexes were subjected to SDS-PAGE, and then analyzed by western blot.
For GST pull-down assay, 293T cells were transfected with the indicated plasmids using FuGENE® HD transfection reagent. After transfection of 48 hrs, the cells were lysed in GST lysis buffer including protein inhibitor. The cell lysis was pulled down by Glutathione Sepharose 4B (Thermo Fisher, USA) for 4 hrs at 4 °C and then analyzed by western blot.
Chromatin immunoprecipitation (ChIP)
The ChIP assay was conducted using the Imprint Chromatin Immunoprecipitation Kit (Sigma, USA). Briefly, shRGS12 cells or controls were fixed with 1% formaldehyde and nuclear extracts were isolated [57, 58]. The sonicated nuclear lysates were immunoprecipitated with anti-YAP antibody or rabbit IgG. Precipitated DNA fragments were amplified by qPCR using primers specific for the Ezrin promoter region. The primers’ sequences used for the quantification are listed in Supplementary Table 1.
RhoA activation assay
RhoA activation assays were respectively carried out using the Active Rho Pull-Down and Detection Kit (Thermo Fisher, USA) according to the manufacturer’s instructions. Briefly, shScramble or shRGS12 cells were seeded in 100 mm dishes; after culture of 48 hrs, the cells were lysed in Lysis/Binding/Wash Buffer on ice for 5 min and the cell supernatant was collected by centrifugation at 4 °C. Activated RhoA and total RhoA were purified using the kits mentioned above, and then analyzed by western blot.
Statistical analysis
All statistical analyses were carried out using the SPSS21 statistical software package, and data were analyzed by Student’s t-test. P < 0.05 was considered to be significant. Error bars represent the standard error of the mean.
Supplementary Material
Acknowledgements
This work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS, AR061052), the National Institute on Aging (NIA, AG048388) awarded to S. Yang.
Footnotes
Conflict of interest
The authors declare no competing interests.
References
- 1.Kansara M, Teng MW, Smyth MJ, Thomas DM. Translational biology of osteosarcoma. Nat Rev Cancer 2014; 14: 722–735. [DOI] [PubMed] [Google Scholar]
- 2.Tang QL, Lu JC, Zou CY, Shao Y, Chen Y, Narala S et al. CDH4 is a novel determinant of osteosarcoma tumorigenesis and metastasis. Oncogene 2018; 37: 3617–3630. [DOI] [PubMed] [Google Scholar]
- 3.Wang DY, Wu YN, Huang JQ, Wang W, Xu M, Jia JP et al. Hippo/YAP signaling pathway is involved in osteosarcoma chemoresistance. Chin J Cancer 2016; 35: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harvey KF, Zhang XM, Thomas DM. The Hippo pathway and human cancer. Nat Rev Cancer 2013; 13: 246–257. [DOI] [PubMed] [Google Scholar]
- 5.Yu FX, Guan KL. The Hippo pathway: regulators and regulations. Genes & development 2013; 27: 355–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao B, Tumaneng K, Guan KL. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nature cell biology 2011; 13: 877–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chai JW, Xu SJ, Guo FB. TEAD1 mediates the oncogenic activities of Hippo-YAP1 signaling in osteosarcoma. Biochem Bioph Res Co 2017; 488: 297–302. [DOI] [PubMed] [Google Scholar]
- 8.Abramow-Newerly M, Roy AA, Nunn C, Chidiac P. RGS proteins have a signalling complex: Interactions between RGS proteins and GPCRs, effectors, and auxiliary proteins. Cell Signal 2006; 18: 579–591. [DOI] [PubMed] [Google Scholar]
- 9.Stewart A, Fisher RA. Introduction: G Protein-coupled Receptors and RGS Proteins. Progress in molecular biology and translational science 2015; 133: 1–11. [DOI] [PubMed] [Google Scholar]
- 10.Yang SY, Li YP. RGS12 is essential for RANKL-evoked signaling for terminal differentiation of osteoclasts in vitro. Journal of Bone and Mineral Research 2007; 22: 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keinan D, Yang SY, Cohen RE, Yuan X, Liu TJ, Li YP. Role of regulator of G protein signaling proteins in bone. Front Biosci-Landmrk 2014; 19: 634–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Z, Liu T, Gilmore A, Gomez NM, Fu C, Lim J et al. Regulator of G Protein Signaling Protein 12 (Rgs12) Controls Mouse Osteoblast Differentiation via Calcium Channel/Oscillation and Galphai-ERK Signaling. J Bone Miner Res 2019; 34: 752–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH et al. Regulation of the Hippo-YAP Pathway by G-Protein-Coupled Receptor Signaling. Cell 2012; 150: 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang YQ, Wang JH, Zhang L, Karatas OF, Shao LJ, Zhang YQ et al. RGS12 Is a Novel Tumor-Suppressor Gene in African American Prostate Cancer That Represses AKT and MNX1 Expression. Cancer Res 2017; 77: 4247–4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chatterjee TK, Fisher RA. RGS12TS-S localizes at nuclear matrix-associated subnuclear structures and represses transcription: structural requirements for subnuclear targeting and transcriptional repression. Molecular and cellular biology 2002; 22: 4334–4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuan X, Cao J, Liu T, Li YP, Scannapieco F, He X et al. Regulators of G protein signaling 12 promotes osteoclastogenesis in bone remodeling and pathological bone loss. Cell Death Differ 2015; 22: 2046–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang S, Li YP, Liu T, He X, Yuan X, Li C et al. Mx1-cre mediated Rgs12 conditional knockout mice exhibit increased bone mass phenotype. Genesis 2013; 51: 201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ng AYH, Li Z, Jones MM, Yang S, Li C, Fu C et al. Regulator of G protein signaling 12 enhances osteoclastogenesis by suppressing Nrf2-dependent antioxidant proteins to promote the generation of reactive oxygen species. Elife 2019; 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walkley CR, Qudsi R, Sankaran VG, Perry JA, Gostissa M, Roth SI et al. Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes & development 2008; 22: 1662–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berman SD, Calo E, Landman AS, Danielian PS, Miller ES, West JC et al. Metastatic osteosarcoma induced by inactivation of Rb and p53 in the osteoblast lineage. P Natl Acad Sci USA 2008; 105: 11851–11856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ji Q, Hao X, Meng Y, Zhang M, Desano J, Fan D et al. Restoration of tumor suppressor miR-34 inhibits human p53-mutant gastric cancer tumorspheres. Bmc Cancer 2008; 8: 266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu JC, Deng T, Lehal RS, Kim J, Zacksenhaus E. Identification of tumorsphere- and tumor-initiating cells in HER2/Neu-Induced mammary tumors. Cancer Res 2007; 67: 8671–8681. [DOI] [PubMed] [Google Scholar]
- 23.Mittal V Epithelial Mesenchymal Transition in Tumor Metastasis. Annual review of pathology 2018; 13: 395–412. [DOI] [PubMed] [Google Scholar]
- 24.Lamar JM, Stern P, Liu H, Schindler JW, Jiang ZG, Hynes RO. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. P Natl Acad Sci USA 2012; 109: E2441–E2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fromigue O, Hamidouche Z, Vaudin P, Lecanda F, Patino A, Barbry P et al. CYR61 Downregulation Reduces Osteosarcoma Cell Invasion, Migration, and Metastasis. Journal of Bone and Mineral Research 2011; 26: 1533–1542. [DOI] [PubMed] [Google Scholar]
- 26.Zhang WJ, Gao YJ, Li PX, Shi ZB, Guo T, Li F et al. VGLL4 functions as a new tumor suppressor in lung cancer by negatively regulating the YAP-TEAD transcriptional complex. Cell research 2014; 24: 331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holden JK, Cunningham CN. Targeting the Hippo Pathway and Cancer through the TEAD Family of Transcription Factors. Cancers 2018; 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mohri Z, Del Rio Hernandez A, Krams R. The emerging role of YAP/TAZ in mechanotransduction. Journal of thoracic disease 2017; 9: E507–E509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang L, Yue T, Jiang J. Hippo signaling pathway and organ size control. Fly 2009; 3: 68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yuan X, Cao J, He XN, Serra R, Qu J, Cao X et al. Ciliary IFT80 balances canonical versus non-canonical hedgehog signalling for osteoblast differentiation. Nature communications 2016; 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang WQ, Dai YY, Hsu PC, Wang H, Cheng L, Yang YL et al. Targeting YAP in malignant pleural mesothelioma. Journal of cellular and molecular medicine 2017; 21: 2663–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ren L, Khanna C. Role of ezrin in osteosarcoma metastasis. Adv Exp Med Biol 2014; 804: 181–201. [DOI] [PubMed] [Google Scholar]
- 33.Meng Z, Moroishi T, Guan KL. Mechanisms of Hippo pathway regulation. Genes & development 2016; 30: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mao J, Yuan H, Xie W, Wu D. Guanine nucleotide exchange factor GEF115 specifically mediates activation of Rho and serum response factor by the G protein alpha subunit Galpha13. Proc Natl Acad Sci U S A 1998; 95: 12973–12976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jules J, Yang SY, Chen W, Li YP. Role of Regulators of G Protein Signaling Proteins in Bone Physiology and Pathophysiology. Prog Mol Biol Transl 2015; 133: 47–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cho H, Harrison K, Kehrl JH. Regulators of G protein signaling: potential drug targets for controlling cardiovascular and immune function. Current drug targets Immune, endocrine and metabolic disorders 2004; 4: 107–118. [DOI] [PubMed] [Google Scholar]
- 37.Lee HJ, Zheng JJ. PDZ domains and their binding partners: structure, specificity, and modification. Cell Communication and Signaling 2010; 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meyers PA, Schwartz CL, Krailo M, Kleinerman ES, Betcher D, Bernstein ML et al. Osteosarcoma: A randomized, prospective trial of the addition of ifosfamide and/or muramyl tripeptide to cisplatin, doxorubicin, and high-dose methotrexate. J Clin Oncol 2005; 23: 2004–2011. [DOI] [PubMed] [Google Scholar]
- 39.Hurst JH, Hooks SB. Regulator of G-protein signaling (RGS) proteins in cancer biology. Biochemical pharmacology 2009; 78: 1289–1297. [DOI] [PubMed] [Google Scholar]
- 40.Gross JD, Kaski SW, Schroer AB, Wix KA, Siderovski DP, Setola V. Regulator of G protein signaling-12 modulates the dopamine transporter in ventral striatum and locomotor responses to psychostimulants. Journal of psychopharmacology 2018; 32: 191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buckbinder L, Velasco-Miguel S, Chen Y, Xu N, Talbott R, Gelbert L et al. The p53 tumor suppressor targets a novel regulator of G protein signaling. Proc Natl Acad Sci U S A 1997; 94: 7868–7872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat Rev Cancer 2013; 13: 246–257. [DOI] [PubMed] [Google Scholar]
- 43.Bouvier C, Macagno N, Nguyen Q, Loundou A, Jiguet-Jiglaire C, Gentet JC et al. Prognostic value of the Hippo pathway transcriptional coactivators YAP/TAZ and beta1-integrin in conventional osteosarcoma. Oncotarget 2016; 7: 64702–64710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang Z, Zhang M, Xu K, Liu L, Hou WK, Cai YZ et al. Knockdown of YAP1 inhibits the proliferation of osteosarcoma cells in vitro and in vivo. Oncology reports 2014; 32: 1265–1272. [DOI] [PubMed] [Google Scholar]
- 45.Jules J, Yang S, Chen W, Li YP. Role of Regulators of G Protein Signaling Proteins in Bone Physiology and Pathophysiology. Progress in molecular biology and translational science 2015; 133: 47–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao B, Lei QY, Guan KL. The Hippo-YAP pathway: new connections between regulation of organ size and cancer. Curr Opin Cell Biol 2008; 20: 638–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang G, Beier F. Rac1/Cdc42 and RhoA GTPases antagonistically regulate chondrocyte proliferation, hypertrophy, and apoptosis. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 2005; 20: 1022–1031. [DOI] [PubMed] [Google Scholar]
- 48.Zhang YC, Xia HW, Ge XJ, Chen QJ, Yuan DD, Chen Q et al. CD44 acts through RhoA to regulate YAP signaling. Cell Signal 2014; 26: 2504–2513. [DOI] [PubMed] [Google Scholar]
- 49.Fukuda H, Nakamura S, Chisaki Y, Takada T, Toda Y, Murata H et al. Daphnetin inhibits invasion and migration of LM8 murine osteosarcoma cells by decreasing RhoA and Cdc42 expression. Biochem Bioph Res Co 2016; 471: 63–67. [DOI] [PubMed] [Google Scholar]
- 50.Vogt S, Grosse R, Schultz G, Offermanns S. Receptor-dependent RhoA activation in G(12)/G(13)-deficient cells - Genetic evidence for an involvement of G(q)/G(11). Journal of Biological Chemistry 2003; 278: 28743–28749. [DOI] [PubMed] [Google Scholar]
- 51.Bradley SJ, Wiegman CH, Iglesias MM, Kong KC, Butcher AJ, Plouffe B et al. Mapping physiological G protein-coupled receptor signaling pathways reveals a role for receptor phosphorylation in airway contraction. Proc Natl Acad Sci U S A 2016; 113: 4524–4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khanna C, Wan XL, Bose S, Cassaday R, Olomu O, Mendoza A et al. The membrane-cytoskeleton linker ezrin is necessary for osteosarcoma metastasis. Nature Medicine 2004; 10: 182–186. [DOI] [PubMed] [Google Scholar]
- 53.Rozengurt E, Sinnett-Smith J, Eibl G. Yes-associated protein (YAP) in pancreatic cancer: at the epicenter of a targetable signaling network associated with patient survival. Signal Transduct Tar 2018; 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sabile AA, Arlt MJ, Muff R, Bode B, Langsam B, Bertz J et al. Cyr61 expression in osteosarcoma indicates poor prognosis and promotes intratibial growth and lung metastasis in mice. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 2012; 27: 58–67. [DOI] [PubMed] [Google Scholar]
- 55.Igarashi K, Kawaguchi K, Murakami T, Miyake K, Kiyuna T, Miyake M et al. Patient-derived orthotopic xenograft models of sarcoma. Cancer Lett 2020; 469: 332–339. [DOI] [PubMed] [Google Scholar]
- 56.Brusgard JL, Choe M, Chumsri S, Renoud K, MacKerell AD Jr., Sudol M et al. RUNX2 and TAZ-dependent signaling pathways regulate soluble E-Cadherin levels and tumorsphere formation in breast cancer cells. Oncotarget 2015; 6: 28132–28150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li Y, Hu N, Yang D, Oxenkrug G, Yang Q. Regulating the balance between the kynurenine and serotonin pathways of tryptophan metabolism. Febs Journal 2017; 284: 948–966. [DOI] [PubMed] [Google Scholar]
- 58.Xu MZ, Chan SW, Liu AM, Wong KF, Fan ST, Chen J et al. AXL receptor kinase is a mediator of YAP-dependent oncogenic functions in hepatocellular carcinoma. Oncogene 2011; 30: 1229–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.