

Abstract
Cell death and the clearance of apoptotic cells are tightly regulated by various signaling molecules in order to maintain physiological tissue function and homeostasis. The phagocytic removal of apoptotic cells is known as the process of efferocytosis, and abnormal efferocytosis is linked to various health complications and diseases, such as cardiovascular disease, inflammatory diseases, and autoimmune diseases. During efferocytosis, phagocytic cells and/or apoptotic cells release signals, such as “find me” and “eat me” signals, to stimulate the phagocytic engulfment of apoptotic cells. Primary phagocytic cells are macrophages and dendritic cells; however, more recently, other neighboring cell types have also been shown to exhibit phagocytic character, including endothelial cells and fibroblasts, although they are comparatively slower in clearing dead cells. In this review, we focus on macrophage efferocytosis of vascular cells, such as endothelial cells, smooth muscle cells, fibroblasts, and pericytes, and its relation to the progression and development of cardiovascular disease. We also highlight the role of efferocytosis-related molecules and their contribution to the maintenance of vascular homeostasis.
Keywords: engulfment of apoptotic cells, phagocytosis, cardiovascular complications, endothelial cell, smooth muscle cell, macrophage
1. Introduction of efferocytosis
Billions of cells die daily in a healthy body. Therefore, proper clearance of apoptotic cells, a process called efferocytosis, is a critical homeostatic mechanism. Efferocytosis is defined as the engulfment and decomposition of apoptotic cells via phagocytes. Effective efferocytosis is orchestrated by a series of signaling cascades led primarily by “find me” signals, “eat me” signals, and “don’t eat me” signals (Fig. 1 and Table 1).
Fig. 1.
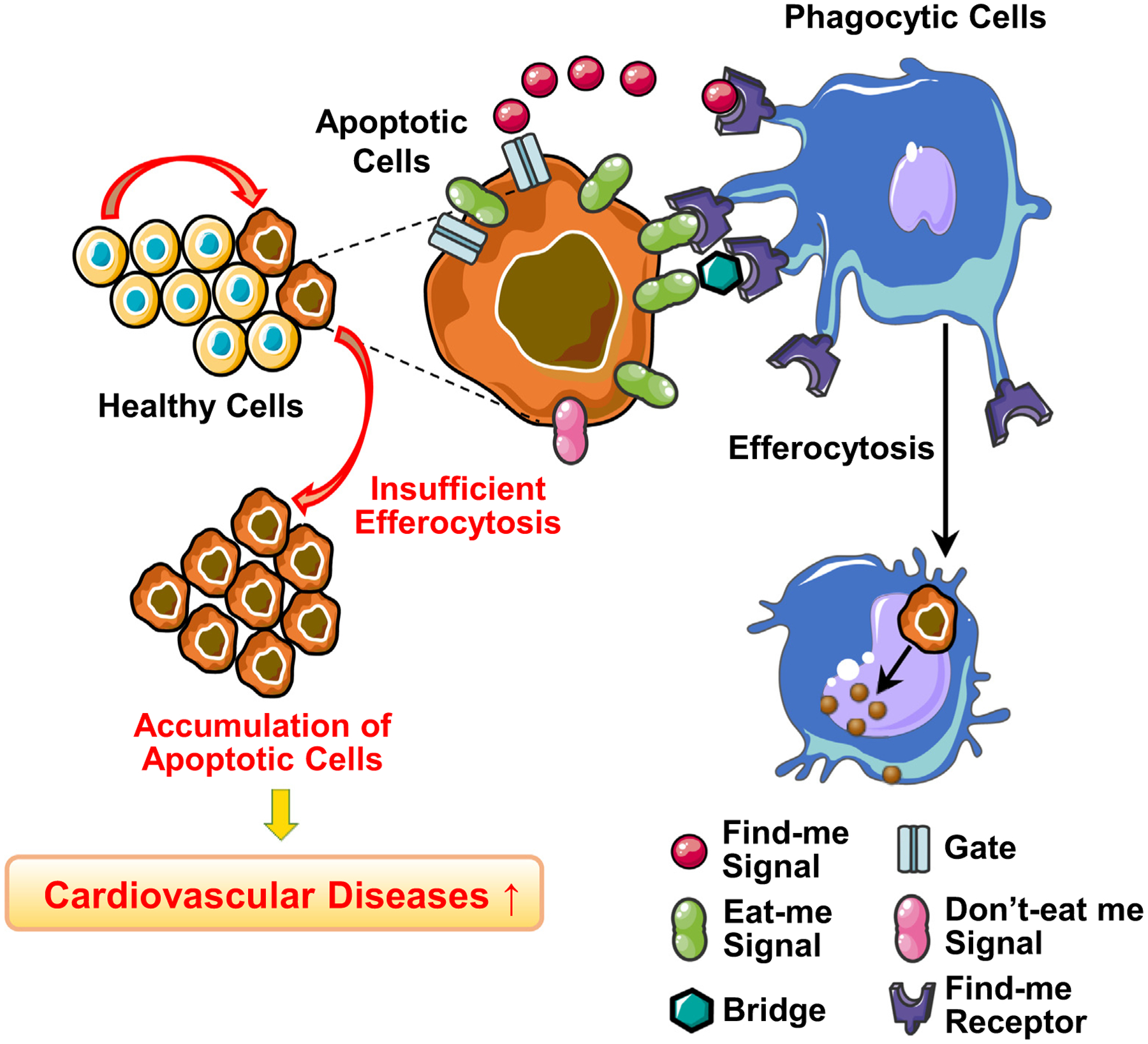
Efferocytosis and key molecules.
Table 1.
The list of molecules related to efferocytosis signaling.
| Role in Efferocytosis | Molecules | Other functions in vascular cells |
|---|---|---|
| Find me signal | C-X3-C motif ligand 1 (CX3CL1) (Truman et al., 2008) |
|
| Lysophosphatidylcholine (LPC) (Lauber et al., 2003) |
|
|
| Nucleotides (ATP,AMP,UTP) (Elliott et al., 2009) |
|
|
| Sphingosine-1-phosphate (S1P) (Gude et al., 2008) |
|
|
| Find me receptor | CX3C motif chemokine receptor 1 (CX3CR1) (Truman et al., 2008) |
|
| G2A receptor (L. V. Yang et al., 2005) |
|
|
| P2Y (Elliott et al., 2009) |
|
|
| S1P receptors (Gude et al., 2008) |
|
|
| Eat me signal | Annexin A1 (Arur et al., 2003) |
|
| Calreticulin (CRT) (Gardai et al., 2005) |
|
|
| Phosphatidylserine (PtdSer) (Katsumori Segawa & Nagata, 2015) |
|
|
| Thrombospondin 1 (TSP1) (Moodley et al.,2003) |
|
|
| Don’t eat me signal | CD24 (Barkal et al., 2019) |
|
| CD31 (Brown et al., 2002) |
|
|
| CD46 (Elward et al., 2005) |
|
|
| CD47 (Oldenborg et al., 2000) |
|
|
| Bridge molecules | C1q (Navratil et al., 2001) |
|
| Gas6 (Lemke & Burstyn-Cohen, 2010) |
|
|
| Milk fat globule-EGF-factor 8 (MFG-E8) (Hanayama et al., 2002) |
|
|
| Protein S (Lemke & Burstyn-Cohen, 2010) |
|
|
| Tubby, Tubby-like protein 1 (Tulp1) (Caberoy, Maiguel, Kim, & Li, 2010) |
|
|
| β-2-glycoprotein-1 (Maiti et al., 2008) |
|
|
| Gate for find me molecules | ATP-binding cassette transporter A1 (ABCA1) (C. Peter et al., 2012) |
|
| Pannexin-1 (Panx1) (Chekeni et al.,2010) |
|
“Find me” signal.
To initiate the clearance of dead cells, apoptotic cells must send signals, called “find me” signals, to the phagocytes in order to stimulate the migration of phagocytes to the site of cell death. The most notable “find me” signals are nucleotides (ATP/AMP/UTP) (Elliott et al., 2009), sphingosine-1-phosphate (S1P) (Gude et al., 2008), lipid lysophosphatidylcholine (LPC) (Lauber et al., 2003; Lee, Ko, Song, & Jung, 2018), and C-X3-C motif chemokine ligand 1 (CX3CL1, also known as fractalkine) (Truman et al., 2008). Nucleotides are released through pannexin-1 (Panx1) channels (Chekeni et al., 2010) by apoptotic cells and act as chemoattractants to the P2Y receptors (a family of purinergic G protein-coupled receptors) on monocytes and macrophages to induce phagocytosis (Elliott et al., 2009). The activity of Panx1 channels is regulated by the cleavage of its carboxyl-terminal; apoptotic cells increase caspases (i.e., caspase-3 and caspase-7), which remove the carboxyl-terminal and open Panx1 channels (Sandilos et al., 2012). Some apoptotic cells increase sphingosine kinase (SPK) 1 and 2 in a caspase-dependent manner that subsequently upregulates the production of S1P, another “find me” signal (Gude et al., 2008). There are several transporters for S1P (e.g., spinster homolog 2, ATP-binding cassette [ABC] transporters) (Nishi, Kobayashi, Hisano, Kawahara, & Yamaguchi, 2014); it is however not clear which transporter is responsible for apoptosis-induced S1P secretion. S1P in the extracellular space binds to S1P receptors on macrophages that drives the recruitment of phagocytes to the apoptotic cells. The release of S1P from apoptotic cells also helps promote macrophage survival and polarization (Weigert et al., 2006; Weigert et al., 2007). The activation of calcium-independent phospholipase A2 (iPLA2) is also controlled by caspase-3; the increase of caspase-3 in apoptotic cells leads to the production of LPC through iPLA2 activation (Lauber et al., 2003). LPC will then be released by ATP-binding cassette transporter A1 (ABCA1) (C. Peter et al., 2012). The binding of LPC to the G-protein-coupled receptor G2A on macrophages and monocytes initiates migration and phagocytosis of apoptotic cells (Christoph Peter et al., 2008; Yang, Radu, Wang, Riedinger, & Witte, 2005). CX3CL1 is the membrane protein expressed primarily in macrophages, dendritic cells, endothelial cells (ECs), and neurons (Lee, Lee, Song, Lee, & Chang, 2018). During cell apoptosis, CX3CL1 will be shed by a disintegrin and metalloprotease (Lee, Lee, et al., 2018). Secreted CX3CL1 then binds to the CX3C motif chemokine receptor 1 (CX3CR1) on macrophages and microglia, and this binding recruits phagocytes to the site of apoptotic cells (Truman et al., 2008).
“Eat me” signal.
In the meantime, apoptotic cells express “eat me” signals to let phagocytes engulf apoptotic cells. One of the most prominent “eat me” signals is the exposure of phosphatidylserine (PtdSer) on the cell surface that will be recognized by macrophages. Regulation of PtdSer exposure involves the work of two enzymes: flippase and scramblase. In healthy cells, ATP11 flippase works to keep PtdSer on the inner leaflet of the plasma membrane (K. Segawa et al., 2018). However, during the process of apoptosis, caspase-3 cleaves and inactivates ATP11 (K. Segawa et al., 2014). At the same time, scramblase XK-related protein 8 (Xkr8) promotes the exposure of PtdSer on the surface of apoptotic cells (Suzuki, Denning, Imanishi, Horvitz, & Nagata, 2013). Sustained PtdSer exposure on the outer leaflet leads to engulfment of apoptotic cells by phagocytes (Katsumori Segawa & Nagata, 2015). Macrophages express a wide array of PtdSer recognition receptors that come in a diverse range of molecular structures and properties (Park & Kim, 2017; Penberthy & Ravichandran, 2016). It is also known that PtdSer receptors vary between different phagocytic cells (Poon, Lucas, Rossi, & Ravichandran, 2014). Well-studied PtdSer receptors involved in efferocytosis are the members of CD300 family, Stabilin-1/2, the family of T-cell immunoglobulin mucin receptor (TIM), and brain-specific angiogenesis inhibitor 1 (BAI1) (Naeini, Bianconi, Pirro, & Sahebkar, 2020; Park & Kim, 2017). These PtdSer receptors help mediate efferocytosis in an “immunologically silent” manner, meaning that the clearance of apoptotic cells elicits the release of anti-inflammatory cytokines while suppressing proinflammatory responses (Kurosaka, Watanabe, & Kobayashi, 2002).
Although PtdSer is the traditional and most studied “eat me” signal, other ligands are also involved in mediating the efferocytic process. Gardai et al. showed that calreticulin (CRT) is upregulated on the surface of apoptotic cells and serves as a crucial “eat me” signal recognized by CD91 (also known as LRP1) on the phagocyte (Gardai et al., 2005). Panaretakis et al. identified that apoptotic cells undergo endoplasmic reticulum (ER) stress that results in the translocation of CRT from the ER to the outer membrane (Panaretakis et al., 2009). CRT is also known to bind to PtdSer as well as be expressed on the cell surface of macrophages; therefore, CRT also works as a bridge molecule and/or PtdSer receptor (Gardai et al., 2005). Another “eat me” signal is thrombospondin 1 (TSP1), which is significantly upregulated on apoptotic cells compared to healthy cells (Moodley et al., 2003). CD36 on macrophages recognizes TSP1, which leads to the clearance of the dead cells. Annexin A1 expressed on the plasma membrane has also been identified as an “eat me” signal (Arur et al., 2003). Arur et al. showed that during apoptosis Annexin A1 is recruited from the cytosol and exported to the outer plasma membrane leaflet in a calcium-and caspase-dependent manner. Annexin A1 then colocalizes with PtdSer and effectively stimulates engulfment of the apoptotic cells (Arur et al., 2003).
Bridge Molecules.
PtdSer enhances its efferocytic effectiveness not only by binding directly to its phagocytic receptors, but also by binding to a number of bridge molecules that help mediate the binding between PtdSer on the apoptotic cell and its receptor on the phagocyte. Gas6 and Protein S serve as a bridge molecule by binding to both PtdSer (exposed on the apoptotic cell) and Tyrosine-protein kinase Mer (MerTK) expressed on the macrophages. Both Gas6 and Protein S are vitamin K-dependent proteins that share similar homology and are produced by a range of different cell types. Protein S is produced by hepatocytes, ECs, and megakaryocytes (Maillard, Berruyer, Serre, Dechavanne, & Delmas, 1992); whereas Gas6 is expressed in monocytes, vascular smooth muscle cells (VSMCs), bone marrow, and ECs (Laurance, Lemarie, & Blostein, 2012). The binding of Gas6 or Protein S to PtdSer and MerTK activates apoptotic cell engulfment in a Ca2+ dependent manner (Lemke & Burstyn-Cohen, 2010). Caberoy and Zhou had identified other ligands for MerTK (i.e., Tubby and tubby-like protein 1 [Tulp1]). They are secreted mainly in retinal and neural tissues and act as novel bridge molecules that can facilitate efferocytosis through the binding to TAM receptors (Tyro3, Axl, and MerTK receptors) (Caberoy, Zhou, & Li, 2010). Milk fat globule-EGF-factor 8 (MFG-E8) is another known bridge molecule that recognizes PtdSer through its C-terminal and binds to integrin αvβ3 and αvβ5 receptors on the phagocyte. MFG-E8 is a glycoprotein produced by macrophages (Hanayama et al., 2002), ECs (Brissette et al., 2012), immature dendritic cells, and epithelial cells (Oshima, Yasueda, Nishio, & Matsuda, 2014). These cells generally produce and secret MFG-E8 under physiological conditions, and apoptotic stimuli increase the production of MFG-E8 to facilitate efferocytosis. Complement C1q is known as a recognition molecule of the classical complement pathway and plays a central role in immune clearance (Gaboriaud et al., 2004). However, there is increasing evidence showing the additional role of C1q as a bridge protein in efferocytosis (Navratil, Watkins, Wisnieski, & Ahearn, 2001; Taylor et al., 2000). C1q binds to both PtdSer on the apoptotic cell surface and CD91 expressed in the plasma membrane of macrophages, and this process promotes efferocytosis. C1q is generally produced and secreted by macrophages, monocytes, and immature dendritic cells (Thielens, Tedesco, Bohlson, Gaboriaud, & Tenner, 2017), but other cell types can also synthesize C1q, including fibroblasts and ECs (Bulla et al., 2016; Fleming, Reid, & McGee, 1983). It is, however, not clear whether these vascular cells can secrete a sufficient amount of C1q to regulate efferocytosis. β-2-glycoprotein-1 also serves as a bridge molecule during efferocytosis by binding to PtdSer on apoptotic cells and low-density lipoprotein receptor-related protein on phagocytes (Maiti, Balasubramanian, Ramoth, & Schroit, 2008). Together, these bridge molecules can assist the efficient clearance of dead cells.
“Don’t eat me” signal.
It is important to acknowledge that physiological efferocytosis is not only regulated by “eat me” signals but instead supported by a critical balance of “eat me” and “don’t eat me” signals. Healthy viable cells display high levels of “don’t eat me” signals such as CD31 (Brown et al., 2002), CD46 (Elward et al., 2005), CD47 (Oldenborg et al., 2000), and CD24 (Barkal et al., 2019). The clearance of dead and dying cells is more effectively signaled by expressing high levels of “eat me” signals, while suppressing “don’t eat me” signals to avoid miscommunication between the apoptotic cell and the phagocyte. It was found that homophilic ligation of CD31 promoted the detachment of viable cells from macrophages in order to prevent its engulfment. However, this CD31-mediated detachment was attenuated in apoptotic cells by decreasing the association between CD31 and protein tyrosine phosphatases SHP, thereby promoting the tethering to phagocytes (Brown et al., 2002). CD46 is uniformly distributed on the cell membrane in healthy cells. During apoptosis, CD46 will be rapidly accumulated into apoptotic blebs and secreted in microparticles. The decrease in CD46 ultimately increases the attraction of macrophages towards the apoptotic cell (Elward et al., 2005). CD47 is expressed on the membrane surface of healthy cells and is recognized by the inhibitory receptor, signal regulatory protein alpha (SIRPα), to inhibit engulfment by macrophages (Oldenborg et al., 2000). Recently, CD24 was identified to serve as a “don’t eat me” signal. CD24 is overexpressed in tumors, which allows cells to escape from the clearance by macrophages. CD24 expression can protect cells from phagocytosis through interaction with the inhibitory receptor Sialic Acid Binding Ig Like Lection 10 (Siglec-10), which is expressed by tumor-associated macrophages. The blockade of CD24-Siglec-10 interaction augments the phagocytosis of CD24 expressing cells, demonstrating a potential novel therapeutic target for cancer immunotherapy (Barkal et al., 2019).
Regulation of efferocytic peptides’ expression by apoptosis.
Prior to efferocytosis, cells must undergo apoptosis by producing apoptotic peptides, flipping the membrane inside-out, and changing their morphology and organelle functions, subsequently sending the messages of their need for rapid clearance to neighboring phagocytes (Saraste & Pulkki, 2000). There are many stimuli triggering apoptosis, and there are numerous peptides involved in apoptosis induction and its signaling. Therefore, we will not discuss the mechanisms of “apoptosis” in this review. The essential apoptotic peptides regulating the expression of efferocytosis-related molecules are caspases. It is known that cleavage and activation of caspase-3 and caspase-7 initiate irreversible changes of many enzyme activities, resulting in morphological and biochemical modifications for apoptosis. Those changes include the exposure of PtdSer as described in the “Eat me” signal paragraph (K. Segawa et al., 2014; Katsumori Segawa & Nagata, 2015; Suzuki et al., 2013). Caspase-3 also leads to membrane blebbing and the formation of apoptotic bodies (Coleman et al., 2001; Zhang, Chen, Gueydan, & Han, 2018). However, it has to be noted that the structural changes or membrane flipping could also be initiated without caspases (Abraham & Shaham, 2004; Borner & Monney, 1999; Broker, Kruyt, & Giaccone, 2005). The series of structural changes in apoptotic cells promotes the release of signaling molecules for their clearance by phagocytes. As indicated in the paragraph of “Find me” signal, activation of caspase-3 and -7 help release “find me” signals in order to enhance the localization of phagocytes to the apoptotic cells (Elliott & Ravichandran, 2016; Gude et al., 2008; Lauber et al., 2003; Sandilos et al., 2012). Altogether, caspase activation mediates this crosstalk between the apoptosis of cells and their eventual engulfment through efferocytosis. However, further studies might find other mechanisms involved in the release of efferocytic peptides.
Cytokines during efferocytosis.
The primary function of macrophages is to initiate an immune response by recognizing pathogens, releasing cytokines, and promoting T-cell recruitment. The major difference between phagocytosis of pathogens and apoptotic cells (efferocytosis) is that efferocytosis is commonly an immune-silent clearance. The question is whether cytokines are secreted from macrophages during efferocytosis. Macrophages do secret cytokines during efferocytosis, and different cytokines act differently depending on health status (Lin et al., 2020). The important cytokines released from macrophages during efferocytosis are anti-inflammatory cytokines, such as IL-10 and TGFβ. The increase of those cytokines helps accelerate the resolution of inflammation by increasing macrophage efferocytosis. However, inefficient clearance of apoptotic cells can lead to necrosis, resulting in the secretion of proinflammatory cytokines, such as TNF-α and IL-6, and leading to subsequent inflammation and tissue damage (Krysko, D’Herde, & Vandenabeele, 2006). Therefore, the number of apoptotic cells determines macrophages’ fate by releasing efferocytosis-promoting cytokines or inducing inflammation by secreting proinflammatory cytokines.
Efferocytosis is a multi-step mechanism that requires a critical balance of various signals, receptors, and phagocytes. Dysfunction in this homeostatic process leads to chronic inflammatory diseases, including but not limited to diabetes, lung disease, neurodegenerative disorders, and cancer. In this review, we aim to focus on the role of aberrant efferocytosis of vascular cells in the development of cardiovascular diseases (CVD). In addition, we discuss the effect of efferocytosis-related proteins on vascular function.
2. Efferocytosis of endothelial cells
ECs play crucial roles in regulating vascular tone, functioning as a mechanical barrier between the vascular walls and the bloodstream, and inducing angiogenesis. Therefore, EC dysfunction, such as decreased release of endothelium-derived relaxing factors, excess production of endothelium-derived contracting factors and proinflammatory molecules, unbalanced cell mobility, and apoptosis, can result in vascular damage and lead to the progression of CVD (e.g., coronary artery disease (Oikonomou et al., 2020), coronary microvascular diseases (Si et al., 2020; van den Oever, Raterman, Nurmohamed, & Simsek, 2010; Winn & Harlan, 2005), stroke (Cosentino et al., 2001), and peripheral artery disease (Vita & Hamburg, 2010). It has been shown that healthy ECs are constantly self-renewed; however, the speed of turnover varies from 47 days to over 23,000 days (Hobson & Denekamp, 1984). Some apoptotic or necrotic ECs can detach from the vascular wall, but the molecular mechanisms of endothelial detachment under healthy condition is yet unclear (Woywodt, Bahlmann, De Groot, Haller, & Haubitz, 2002). Most likely, apoptotic ECs would be cleared by neighboring phagocytes through efferocytosis (Lee et al., 2018). After removing apoptotic cells, the area where ECs were lost will be repaired by proliferation and migration of neighboring mature ECs or incorporation of circulating endothelial progenitor cells in the site of endothelial loss (Weihua, Tsan, Schroit, & Fidler, 2005). Under pathophysiological conditions, the number of apoptotic ECs is significantly increased, and insufficient efferocytosis leads to the accumulation of apoptotic ECs in the vascular lumen that subsequently causes endothelial dysfunction and exacerbates tissue damage (Tricot et al., 2000). Therefore, it is critical to understand the molecular mechanisms for effective clearance of apoptotic ECs and how defective efferocytosis of ECs can progress vascular disease. It has to be noted that we will not discuss programed capillary regression, which involves macrophage engulfment of ECs, since this process is required during tissue development and physiological phagocytosis.
2.1. Macrophage engulfment of apoptotic endothelial cells
Although apoptotic ECs are believed to be engulfed by macrophages, there are few reports showing efferocytosis of ECs in vivo, partly due to technical difficulty for visualizing macrophage engulfment of apoptotic ECs. To the best of our knowledge, Lee et al. is the only group that shows the statistical result of phagocytosis of apoptotic ECs by macrophages (Lee, Park, et al., 2018). They demonstrated that macrophages engulfed apoptotic ECs induced by LPS (lipopolysaccharide) treatment in vitro, and Stabilin-1, a phagocytic receptor, mediates macrophage phagocytosis of apoptotic ECs in a PtdSer dependent manner. They also showed that reduced phagocytosis of apoptotic ECs, via inhibition of Stabilin-1 in macrophages, led to disruption of vascular integrity through increased vascular permeability and enhanced transendothelial migration of inflammatory cells. It is of note that no image for efferocytosis of apoptotic ECs is shown, and the detailed method for the assessment of phagocytosis of apoptotic ECs is not available in the manuscript.
2.2. Endothelial engulfment of apoptotic cells
There are several manuscripts acknowledging that ECs are also capable of engulfing apoptotic cells. Dini et al. might be the first group to demonstrate endothelial engulfment of apoptotic cells using an electron microscope (Dini et al., 1995). Ultrastructural images clearly show that mouse liver sinusoidal ECs internalized apoptotic cells. EC phagocytic function was further observed by Oka et al., who demonstrated that ECs removed PtdSer-exposed aged/apoptotic cells (Oka et al., 1998). In this study, they found that bovine aortic ECs were capable of engulfing aged red blood cells (RBCs) and apoptotic Jurkat cells, which was mediated by Lectin-like oxidized low-density lipoprotein receptor 1 (LOX-1) on ECs. Lee et al. also showed evidence by imaging that sinusoidal ECs engulfed damaged RBCs in vivo during the depletion of macrophages. In the presence of macrophages, damaged RBCs were cleared mainly by macrophages instead of ECs (S. J. Lee, Park, Jung, Bae, & Kim, 2011). They also demonstrated that expression of Stabilin-1 and Stabilin-2 on ECs could further facilitate macrophages in sequestering damaged RBCs and augment engulfment in a PtdSer manner. Ramirez-Ortiz et al. evidenced endothelial engulfment of apoptotic mouse embryonic fibroblasts (MEFs) detected by the FACS analysis (Ramirez-Ortiz et al., 2013). Apoptotic MEFs were stained with pHrodo; therefore, MEFs uptaken by ECs changed their color from 488nm to 550nm due to acidosis inside the cells. They further revealed that the phagocytic receptor SCARF1 in ECs played a crucial role in the efferocytosis of MEFs. We were, however, unable to identify the source of ECs used in this study. Lastly, another study revealed that overexpression of αv-integrins in human umbilical vein ECs (HUVECs) enhanced endothelial phagocytosis of MFG-E8-opsonized apoptotic cells (Fens et al., 2008). They also found that MFG-E8 treatment significantly improved ECs ability to uptake apoptotic cells in vitro.
2.3. The effect of macrophage efferocytosis on endothelial function
A significant number of publications demonstrate the effect of macrophage efferocytosis on endothelial function without direct evidence of macrophage engulfment of apoptotic ECs. In other words, deletion and/or overexpression of efferocytosis-related molecules would modify the effectivity of macrophage engulfment of apoptotic cells (which might not be ECs) and result in alteration of endothelial function. Firstly, it was found that dysfunctional efferocytosis by macrophages is implicated in attenuated wound healing in diabetes (Khanna et al., 2010). Impaired efferocytosis was demonstrated by macrophages isolated from the wounds of diabetic mice, resulting in increased apoptotic cell burden at the wound site. Inefficient removal of apoptotic cells was also associated with increased levels of proinflammatory cytokines and decreased expression of anti-inflammatory cytokines. The same group further investigated the impaired wound closure in diabetic mice, by identifying the role of MFG-E8, a bridge molecule of efferocytosis, in resolving inflammation, angiogenesis, and accelerating diabetic wound closure (Das et al., 2016). Their results showed that MFG-E8 KO mice displayed attenuated macrophage efferocytosis, increased inflammatory response (evidenced by increased tumor necrosis factor-alpha (TNF-α), and decreased interleukin-10 (IL-10) in wound-edge tissues), and inadequate angiogenesis. MFG-E8 treatment exhibited a proangiogenic effect via augmented tube formation of ECs (Das et al., 2016). This work indicates that MFG-E8 not only augments macrophage efferocytosis of apoptotic cells and resolution of inflammation, but also promotes angiogenesis by bolstering signaling of vascular endothelial growth factor (VEGF). Another study showed that the application of Ac2-26 (an Annexin A1-derived short peptide) to wound sites facilitated macrophage engulfment of apoptotic neutrophils, improved angiogenesis, and healed wound skin tissue effectively in diabetic mice (Huang et al., 2020). In their study, Ac2-26 also decreased protein levels of TNF-α and IL-6 while increasing IL-10 and VEGFA expression in diabetic wound samples. Together, these results indicate that Ac2-26 treatment enhances macrophage efferocytosis of apoptotic neutrophils and augments angiogenesis, which subsequently ameliorates wound healing in diabetes.
2.4. The role of efferocytosis-related proteins in endothelial function
There is substantial evidence suggesting that a number of efferocytosis-related proteins play a pivotal role in endothelial function independent from efferocytosis. “Find me” signals. CX3CL1 has been implicated in the modulation of angiogenesis and vascular inflammation. Volin et al. identified the angiogenic properties of CX3CL1 in rheumatoid arthritis (Volin et al., 2001). Their results showed that CX3CL1 treatment induced migration of human dermal microvascular ECs, enhanced tube formation in vitro, and increased angiogenesis in vivo. They also found that CX3CL1/CX3CR1 interaction was required for CX3CL1-mediated angiogenesis. Similarly, another study also indicated that CX3CL1 increases proliferation, migration, and tube formation of HUVECs in vitro and stimulates angiogenesis in vivo by activating the G protein-coupled receptor (S. J. Lee et al., 2006). Zhang et al. showed a mechanistic interaction between CX3CL1 and endothelin B (ETB) receptor in pulmonary angiogenesis and monocyte migration in experimental hepatopulmonary syndrome (HPS) (Zhang, Yang, Hu, Wu, & Fallon, 2014). Their results demonstrate that Endothelin 1 (ET-1) treatment significantly increased tube formation in pulmonary microvascular ECs in vitro through CX3CL1 production. An administration of selective ETB receptor antagonist in rats with common bile duct ligation (an experimental animal model for HPS) significantly decreased lung angiogenesis, monocyte accumulation, and CX3CL1 levels. The beneficial effects of S1P on endothelial function have also been widely reported (Jozefczuk, Guzik, & Siedlinski, 2020). S1P is known to serve as a protective property for endothelial barrier function by maintaining endothelial integrity and preventing vascular leakage (Xiong & Hla, 2014). Also, S1P receptor stimulation in ECs leads to excessive angiogenesis and enhances adherent junction formation (Gaengel et al., 2012; M. J. Lee et al., 1999). Furthermore, it has been demonstrated that S1P promotes vasorelaxation through eNOS activation (Igarashi, Bernier, & Michel, 2001). The majority of efferocytosis-related proteins exhibit beneficial effects on endothelial function; however, it is important to include that there are efferocytic proteins that lead to endothelial dysfunction. The pretreatment of LPC reduces endothelium-dependent relaxation by inhibiting acetylcholine-induced Ca2+ uptake in aortic ECs (Miwa, Hirata, Kawashima, Akita, & Yokoyama, 1997). LPC also inhibits angiogenesis by attenuating EC migration and proliferation (Rikitake et al., 2000). In this study, the authors demonstrated that fibroblast growth factor (FGF-2)-mediated angiogenesis was reduced by LPC pretreatment in bovine aortic ECs via inhibiting Ras/ERK signaling cascade. “Find me” receptors. Efferocytosis-related receptors appear to impact endothelial function. For instance, an anti-inflammatory effect has been observed by G2A in the endothelium (Bolick et al., 2007). They found the deletion of G2A increased expression levels of ICAM-1, IL-6, and monocyte chemoattractant protein-1 in ECs. In addition, overexpression of G2A in G2A-deleted ECs significantly attenuated monocyte adhesion. Together, these results demonstrate that G2A deficiency can lead to EC activation and induce vascular inflammation. Downregulation of MerTK enhanced neutrophil transendothelial migration by altering the structure of adherent junction and decreased junction proteins in human pulmonary microvascular ECs (Y. Li et al., 2019). Although we introduced P2Y receptor as a “find me” receptor in the previous paragraph, this receptor is traditionally known as a receptor for nucleotides that provokes many signaling cascades in various cell types, including ECs (Strassheim et al., 2020). Endothelium-specific P2Y2 deletion decreased eNOS activation, followed by attenuated vasodilation and development of hypertension (Wang et al., 2015). Activation of P2Y exhibits anti-inflammatory effects as P2Y activation strengthens endothelial adherent junctions and enhances endothelial barrier function (Zemskov, Lucas, Verin, & Umapathy, 2011). Horckmans et al. demonstrate that P2Y deficient mice show decreased angiogenesis, vasculogenesis, and endothelial differentiation (Horckmans et al., 2012). These data suggest that P2Y plays a critical role in endothelial function. “Eat me” signals. TSP1 has diverse effects on endothelial function depending on the binding partner in ECs. For instance, TSP1 binding to CD36 on endothelial surface inhibits endothelial proliferation and motility (Dawson et al., 1997). However, TSP1 binding to α3β1 stimulates angiogenesis (Chandrasekaran et al., 2000). This study also shows that inhibition of TSP1 impairs EC proliferation and migration in vitro and attenuates angiogenesis in vivo. Annexin A1 has been identified in having anti-inflammatory effects on the endothelium by regulating leukocyte-endothelium interactions. Gavins et al. performed ischemia/reperfusion to promote leukocyte infiltration to the endothelium and found that mice treated with Ac2-26 (an Annexin A1 mimetic peptide) prior to ischemia/reperfusion exhibited attenuated leukocyte adhesion (Gavins, Yona, Kamal, Flower, & Perretti, 2003). These results indicate that Annexin A1 executes antiadhesive actions and can prevent inflammation in the vasculature. Bridge molecules. C1q has been demonstrated to play an essential role in endothelial function. Fan et al. discovered that overexpression of C1q increased VEGF and LAIR1 expression levels that subsequently enhanced angiogenesis, migration, and tube formation in rat and human brain microvascular ECs (Fan, Li, & Qian, 2019). Shah et al. investigated the effect of C1q deficiency on pulmonary endothelial function (Shah et al., 2015). C1q KO mice exhibited higher protein levels of vascular adhesion markers, such as ICAM-1 and VCAM-1, accompanied by enhanced proinflammatory cytokines and increased neutrophil recruitment to the vascular walls. They also found that C1q KO mice showed a decrease in endothelial barrier function. MFG-E8 is well known to act as a proangiogenic factor in ECs. Uchiyama et al. demonstrated that MFG-E8 knockdown by siRNA transfection in HUVEC inhibited formation of capillary-like structures (Uchiyama et al., 2014). They also showed that during vascular injury or wounding, MFG-E8 translocated and accumulated around damaged vessels and enhanced wound healing. Silvestre et al. found that MFG-E8 activates angiogenic pathways and promotes neovascularization via stimulation of αvβ3 or αvβ5-dependent AKT phosphorylation in cultured ECs (Silvestre et al., 2005). Gas6 is highly expressed and secreted by vascular ECs and plays a key role in granulocyte adhesion to ECs. Avanzi et al. showed that Gas6 treatment significantly inhibits polymorphonuclear (PMN) cell adhesion to ECs in vitro in a dose-dependent manner (Avanzi et al., 1998). Other studies examined the role of β-2-glycoprotein-1 on the endothelium and revealed that treatment of anti-β-2-glycoprotein-1 antibodies on HUVECs markedly increased the expression of adhesion molecules, such as E-selectin, ICAM-1, and VCAM-1 (Dunoyer-Geindre, de Moerloose, Galve-de Rochemonteix, Reber, & Kruithof, 2002). “Don’t eat me” signals. The proteins related to “Don’t eat me” signals also regulate endothelial function apart from efferocytosis. Wang et al. demonstrated that CD24 downregulation significantly attenuated HUVEC migration, tube formation, and proliferation (Wang et al., 2016). They revealed that CD24 promoted angiogenesis via activation of Heat shock protein 90, and STAT3-mediated transcription of VEGF. CD46 has been reported to protect the endothelium against complement-mediated cell lysis, by acting as an inhibitor for complement activation (Richards et al., 2007). Complement activation serves as a defensive immune response against pathogens; however, prolonged activation can subsequently damage the endothelium. Therefore inhibitory regulators, such as CD46, offer a protective role, and CD46 deficiency has been implicated in the pathogenesis of atypical hemolytic uremic syndrome, marked by EC injury with swelling, thickening, and detachment of ECs from the basement membrane. CD31 is a prominent “don’t eat me” signal and is commonly used as an EC marker. CD31 has displayed a multitude of beneficial effects on endothelial function, such as promoting cell migration and angiogenesis (Lertkiatmongkol, Liao, Mei, Hu, & Newman, 2016). Furthermore, CD31 protects ECs from apoptosis (Bird et al., 1999; Evans, Taylor, & Kilshaw, 2001). However, a major function of CD31 would be the maintenance of vascular integrity. CD31 is enriched in EC intercellular junctions where CD31 plays critical roles in modulating leukocyte trafficking and vascular permeability (Ferrero, Ferrero, Pardi, & Zocchi, 1995; Privratsky et al., 2011). Therefore, CD31 could be considered as a promising target for treating vascular disorders. These data suggest that efferocytosis-related proteins can moderate endothelial functions (e.g., angiogenesis, regulation of vascular tone, vascular integrity) independent of efferocytosis.
3. Efferocytosis of smooth muscle cells
VSMCs are the major constituents in the media of blood vessels, and they are responsible for regulating vascular tone, vascular elasticity, and vasculogenesis. During physiological vascular development, VSMCs exhibit a highly proliferative character and migrate along with ECs to form blood vessels (Owens, Kumar, & Wamhoff, 2004). After tissues are fully developed, VSMCs switch the phenotype to a quiescent state at a low proliferative rate, and they primarily function to regulate vascular tone through vasoconstriction and vasodilation (Steucke, Tracy, Hald, Hall, & Alford, 2015). However, in response to vascular stress and injury, VSMCs change their character back to a de-differentiated phenotype with increased proliferation, migration, and production of matrix components in the blood vessel wall (Owens, 1995). Importantly, these two states are not mutually exclusive and present a continuum of phenotypes that VSMCs can switch easily under different environmental cues such as growth factors, inflammatory mediators, and cell-cell and/or cell-matrix interactions (Owens et al., 2004). Proliferative VSMCs lead to the increase in VSMC plasticity, calcification, and senescence, and contribute to the development of vascular diseases such as coronary artery disease due to progressing atherosclerosis (Keul et al., 2019), restenosis (Clowes, Reidy, & Clowes, 1983), and hypertension (Touyz et al., 2018). Under pathophysiological conditions, VSMC apoptosis is also induced by proinflammatory cytokines, oxidized low-density lipoprotein (oxLDL), and high levels of nitric oxide (NO) (Grootaert et al., 2018). Apoptotic VSMCs are assumed to be cleared by neighboring phagocytes. However, the inefficient removal of apoptotic VSMCs will lead to secondary necrosis, excess production of inflammatory cytokines, and increased vascular injury (Grootaert et al., 2018; McCarthy & Bennett, 2000).
3.1. Macrophage engulfment of apoptotic smooth muscle cells
It should be noted that the majority of publications regarding efferocytosis of VSMCs are in relation to atherosclerosis. Briefly, atherosclerosis, marked by the deposition of lipid plaques, is an underlying cause of heart attack, stroke, and peripheral vascular disease, making it a leading cause of death in the United States (Mozaffarian, et al., 2015). In progressing atherosclerosis, lesional macrophages and smooth muscle cells (SMCs) undergo apoptosis. Chronic VSMC apoptosis accelerates both atherogenesis and inflammation by releasing proinflammatory cytokines such as IL-1 (Bennett, Sinha, & Owens, 2016). In normal physiological conditions, these apoptotic VSMCs could be rapidly cleared by neighboring phagocytes. However, delayed or defective efferocytosis under hyperlipidemia has been implicated as a causal factor of atherosclerosis; dysfunction of regulatory signals in efferocytosis could lead to expansion of the necrotic core in atherosclerotic plaques, stimulate plaque inflammation, increase foam cell accumulation and plaque vulnerability by making it susceptible to plaque rupture and atherothrombotic complications (Kojima, Weissman, & Leeper, 2017; Schrijvers, De Meyer, Kockx, Herman, & Martinet, 2005; Tajbakhsh, Rezaee, Kovanen, & Sahebkar, 2018). Accumulation of apoptotic VSMCs also changes atherosclerotic plaque vulnerability by thinning the fibrous cap, reducing collagen and matrix levels, and causing accumulation of cellular debris (Clarke et al., 2006). As dead VSMCs accumulate, more inflammatory cells are recruited, thereby triggering further apoptosis of neighboring cells and enhancing vascular damage (McCarthy & Bennett, 2000). It is thus vital to recognize the mechanistic details involving efferocytosis of VSMCs and how dysfunctional efferocytosis will lead to the development of vascular diseases.
Macrophage efferocytosis of VSMCs has been reported by a number of researchers. Kojima et al. detected engulfment of apoptotic VSMCs by macrophages in vitro using flow cytometry, and they identified the role of CDKN2B, an endogenous cell cycle inhibitor, in defective efferocytosis of VSMCs during atherosclerosis (Kojima et al., 2014). In this study, they found SNPs of CDKN2B in human atherosclerotic plaque and showed that this mutation significantly correlated with CRT expression (“eat me” signal). Furthermore, the deletion of CDKN2B decreased the level of CRT in mouse aorta, which subsequently reduced efferocytosis of VSMCs by macrophages and increased plaque formation in ApoE KO mice fed with a high-fat diet. The same group recently reported that VSMCs are de-differentiated in atherosclerotic plaques in a clonal fashion, and those de-differentiated VSMCs expressed CD47, a “don’t eat me” signal, to be resistant to efferocytosis by macrophages (Wang et al., 2020). They also showed that inhibition of CD47 reduced clonal VSMC expansion in atherosclerotic plaque in ApoE KO mice fed with a high-fat diet, and CD47 antibody increased VSMCs efferocytosis by macrophages in vitro. However, CD47 inhibition seemed to have no effect on the size of the atherosclerotic plaque in their study. In contrast, Kojima et al. demonstrated that CD47 blocking antibodies decreased the plaque size in the same atherosclerotic mouse model by increasing engulfment of apoptotic bodies (Kojima et al., 2016). Further study is required to identify the role of CD47 in the development of atherosclerotic plaque formation. Khan et al. investigated the effects of oxLDL on macrophage phagocytosis of apoptotic VSMCs in vitro using time-lapse microscopy (Khan et al., 2003). They found that in the presence of oxLDL, binding of the macrophages to apoptotic VSMC was not affected; however, engulfment was significantly delayed. This defective engulfment promoted inflammation through the release of IL-6. Taken together, this study provides evidence that oxLDL attenuates the uptake of apoptotic VSMCs by macrophages and thereby contributes to the persisting inflammation seen in the atherosclerotic artery.
3.2. Smooth muscle cell engulfment of apoptotic cells
The phagocytic capability of VSMCs in the engulfment of apoptotic cells has been widely reported. Fries et al. examined the uptake of apoptotic VSMCs by VSMCs using electron microscopy (Fries, Lightfoot, Koval, & Ischiropoulos, 2005). They demonstrated that apoptotic cells were tightly bound to the surface of VSMCs and were eventually internalized by VSMCs. It has also been indicated that neighboring VSMCs facilitate the clearance of apoptotic VSMCs by releasing cytokines (e.g., monocyte-chemoattractant protein-1, cytokine-induced neutrophil chemoattractant-1, and transforming growth factor beta 1 (TGFβ1)). These cytokines then recruit other phagocytic cells to the site of VSMC apoptosis, minimizing inflammation and vascular injury. Bennett et al. showed that the rapid phagocytosis of dead VSMCs by healthy VSMCs is mediated by the exposures of PtdSer on the membrane of the apoptotic VSMCs (Bennett, Gibson, Schwartz, & Tait, 1995). They also found that VSMC-mediated phagocytosis of apoptotic VSMCs could be inhibited by exogenous Annexin V application (but not by endogenous Annexin V). The efferocytic capability of VSMCs was further evidenced by the existence of PtdSer receptors on the VSMC surface using RT-PCR, western blot, and immunofluorescence (Kolb, Vranckx, Huisse, Michel, & Meilhac, 2007). Kolb et al. demonstrated that PtdSer receptors on VSMCs enabled the binding and engulfment of apoptotic RBCs using light and confocal microscopy (Kolb et al., 2007). Similarly, Delbosc et al. explored efferocytosis of senescent RBCs by VSMCs and investigated their role in initiating human atheroma (Delbosc et al., 2017). Interestingly, they claimed that engulfment of RBCs by VSMCs resulted in the accumulation of lipids and increased reactive oxygen species (ROS) production in VSMCs since RBCs carry cholesterol and phospholipids in their membranes. Therefore, phagocytosis of RBCs by VSMCs appears to serve as pathogenic mechanisms that trigger subsequent plaque formation.
3.3. The role of efferocytosis-related proteins in smooth muscle cell function
A significant number of publications demonstrates that molecules involved in efferocytosis have a profound impact on VSMC function independent from efferocytosis. “Find me” signals. CX3CL1 has been verified to have anti-apoptotic and proliferative effects on human coronary artery SMCs. White et al. showed that pretreatment with CX3CL1 on primary human coronary artery SMCs significantly blocked caspase 3 cleavage, whereas CX3CL1 induced SMC proliferation by increasing DNA synthesis through phosphorylation of ERK and Akt (White et al., 2010). They also showed that the synthesis of epiregulin, a ligand of epidermal growth factor receptor, is required for CX3CL1-mediated anti-apoptosis and proliferative effects on VSMCs. LPC is also known to stimulate DNA synthesis in VSMCs. This process is positively regulated by protein kinase C and fibroblast growth factor-2 (FGF-2) (Chai, Howe, DiCorleto, & Chisolm, 1996). Watanabe et al. showed that the synergistic interaction between LPC and ET-1 induces VSMC proliferation by activating both NADPH and MAPK pathways (Watanabe, Pakala, Katagiri, & Benedict, 2002). Another “find me” signal that has been extensively studied in VSMC function is S1P (Xing et al., 2015). S1P has profound effects on VSMC proliferation and differentiation; however, these effects are dependent on S1P receptor subtypes (Wamhoff, Lynch, Macdonald, & Owens, 2008). The stimulation of S1P receptors 1 and 3 (but not receptor 2) promotes S1P-induced VSMC proliferation. S1P binding to S1P receptor 2 increases the expression of VSMC differentiation marker genes, whereas S1P binding to S1P receptors 1 and 3 suppresses those gene expressions. Taken together, S1P receptor 1 and 3 have different effects from S1P receptor 2 on VSMC function. Bridge molecules. Yin et al. investigated the role of Gas6 in the proliferation of rabbit VSMCs in vitro and in vivo (Yin et al., 2000). Gas6 upregulation increased VSMC proliferation. Besides, Gas6 expression level was significantly increased in balloon-injured aorta compared to the normal aorta in rabbits. Gasic et al. reported Protein S as a mitogen in aortic SMCs (Gasic, Arenas, Gasic, & Gasic, 1992). This study showed that Protein S stimulates DNA synthesis in VSMCs, which may contribute to intimal VSMC proliferation after vascular injury. Together, these results suggested that Gas6 and Protein S play an important role in VSMC migration and proliferation, although the pathways involved remain elusive. Gates for “find me” molecules. ABCA1 has also been identified as a target protein that can regulate VSMC proliferation, migration, and apoptosis in pulmonary hypertension. Zhang et al. had reported that ABCA1 mRNA level was downregulated in hypoxia-exposed pulmonary arterial SMCs (Zhang et al., 2018), and ABCA1 overexpression decreased hypoxia-induced migration and proliferation, and increased apoptosis in SMCs. These results were supported by a similar study conducted by Zhou et al., who found attenuated ABCA1 expression in pulmonary arterial SMCs from mice with pulmonary hypertension and in human pulmonary arterial SMCs exposed to hypoxic conditions (Zhou, Fang, Zhang, Feng, & Wang, 2020). These studies suggest that ABCA1 is an essential molecule that negatively regulates proliferation, migration, and survival on pulmonary arterial SMCs. “Eat me” and “don’t eat me” signals. The binding of TSP1 to CD47 or CD36 has been shown to inhibit NO-dependent vascular relaxation (Isenberg, Frazier, & Roberts, 2008). TSP1 binding to CD47 or CD36 inhibits soluble guanylate cyclase (sGC) activity and reduces cGMP formation in VSMCs, thereby decreasing NO-dependent vascular relaxation. Another “eat me signal” that can modulate VSMC function is CRT. CRT had been reported to destabilize glucose transporter-1 (GLUT-1) mRNA in VSMCs under high glucose conditions (Totary-Jain et al., 2005). High glucose treatment increased CRT levels and downregulated GLUT-1 mRNA and protein levels in VSMCs. Further investigation revealed that CRT binds to the 3’ UTR of GLUT-1 mRNA and can destabilize the entire transcript, leading to reduced protein expression of GLUT-1 and, subsequently, to the downregulation of glucose transport. Altogether, these works demonstrate the importance of efferocytosis and efferocytosis-related proteins on VSMC function.
4. Efferocytosis of fibroblasts
Fibroblasts present another cell type critical for regulating vascular function. Fibroblasts exhibit heterogeneous phenotypes in various organs and anatomical sites throughout the body, giving fibroblasts the ability to perform an array of essential functions (Chang et al., 2002). Traditionally, fibroblasts primarily function as a structural support of the vasculature by producing extracellular matrix (ECM) proteins such as collagen and fibronectin. However, in pathophysiological conditions, such as myocardial infarction and hypertension, fibroblasts get activated and induce inflammation by the secretion of proinflammatory cytokines, growth factors, and proteases (Enzerink & Vaheri, 2011). In response to inflammatory signals, fibroblasts differentiate into myofibroblasts characterized by increased cell proliferation and migration, high expression of contractile protein α smooth muscle actin, and excess secretion of cytokines, vasoactive peptides, and growth factors. This response is initially adaptive for vascular restoration and remodeling after injury; however, if this response persists, it may lead to pathological remodeling and fibrosis (Porter & Turner, 2009). Abnormal fibroblasts also lead to dysregulation of ECM composition/turnover and apoptosis (Enzerink & Vaheri, 2011), delayed wound/ulcer healing (Wall et al., 2008), and progression of heart failure (Travers, Kamal, Robbins, Yutzey, & Blaxall, 2016). Under stress, such as prolonged hyperoxic conditions (Conconi et al., 2003) and enhanced ROS generation (S. Li et al., 2008), apoptosis of fibroblasts is induced, making efferocytosis of these cells essential for homeostatic maintenance in the vasculature. In this section, we will review the processes involved in efferocytosis of fibroblasts and how deficiencies in these processes can lead to adverse pathologies in the vasculature.
4.1. Macrophage engulfment of apoptotic fibroblasts
Induction of fibroblast apoptosis and the subsequent clearance by macrophages is essential for the regression of fibrosis. Moodely et al. were the first to provide evidence that apoptotic fibroblasts actively recruit macrophages for their uptake via releasing TSP1 and its receptor CD36 (Moodley et al., 2003). Flow cytometry data show that surface expression of both CD36 and TSP1 are upregulated on apoptotic fibroblasts compared to healthy cells. The data from immunoprecipitation and confocal microscopy studies confirmed that CD36 and TSP1 colocalized and physically associated on the surface on apoptotic fibroblasts. The blockade of these molecules inhibits phagocytosis, giving evidence that the TSP1/CD36 complex acts as a chemoattractant for macrophages. Further evidence revealed that this complex was recognized by αvβ3/CD36 complex on the macrophage surface. Interestingly, they found that blockade of PtdSer did not alter phagocytosis of apoptotic fibroblasts by macrophages. This study was the first to reveal TSP1/CD36/αvβ3 in teraction in the process of efferocytosis of apoptotic fibroblasts. Khan et al. found that in the presence of oxLDL in vitro, macrophage engulfment of apoptotic fibroblasts is impaired despite the binding of apoptotic fibroblast bodies to macrophages being unaffected. (Khan et al., 2003). Therefore, they suggested that the presence of oxLDL, as seen in the pathogenesis of atherosclerotic disease and rheumatoid arthritis, causes defective efferocytosis of fibroblasts and leads to chronic inflammation.
4.2. Fibroblast engulfment of apoptotic cells
A number of manuscripts have identified fibroblasts as the phagocyte in the uptake of apoptotic cells. In paticular, macrophage engulfment of apoptotic neutrophils have been well documented (Newman, Henson, & Henson, 1982; Savill et al., 1989). Hall et al. studied on the ability of fibroblasts in the clearance of apoptotic neutrophils and found that fibroblasts ingested apoptotic neutrophils through αvβ3 binding (Hall, Savill, Henson, & Haslett, 1994). Electron microscopy images confirmed that fibroblasts, but not epithelial cells or HUVECs, were able to uptake senescent neutrophils. αvβ3 inhibition reduced fibroblast phagocytosis of apoptotic neutrophils similarly as macrophages do. Banerjee et al. investigated the role of high mobility group box 1 (HMGB1, a chromatin protein) in the efferocytosis of apoptotic cells by fibroblasts (Banerjee et al., 2011). They demonstrated that HMGB1 inhibition in fibroblasts led to augmented uptake of apoptotic thymocytes and apoptotic neutrophils via Rac1-mediated FAK-Src activation. In another study, Hermetet et al. demonstrated that human fibroblasts clear apoptotic papillomavirus-positive cervical cancer cells by recognizing PtdSer with the PtdSer receptor BAI1 on fibroblasts (Hermetet et al., 2016). This study showed that fibroblasts identify the apoptotic stages of target cells, early and late apoptotic cells, in which fibroblasts mainly engulf late apoptotic cells. Furthermore, the uptake of apoptotic cells by fibroblasts occurs slower and less efficiently than macrophages. Gardai et al. revealed that apoptotic MEFs isolated from CRT-deficient mice were engulfed by neither macrophages nor fibroblasts; however, treatment of CRT to apoptotic MEFs stimulated the binding of apoptotic cells to CD91 on the phagocytes ex vivo (Gardai et al., 2005). They further examined the role of CRT in vivo and observed that clearance of MEFs by fibroblasts was significantly attenuated in CRT KO mice compared to WT control. Altogether, this study successfully showed the role of CRT/CD91 binding in fibroblast efferocytosis of apoptotic MEFs. Cardiac myofibroblasts had also been identified in regulating the clearance of apoptotic cells that is essential in the recovery from myocardial infarction (Nakaya et al., 2017). Apoptotic cells were engulfed by cardiac macrophages and cardiac myofibroblasts in vitro, although efferocytosis by myofibroblasts was slower than that in macrophages. Electron and confocal microscopy provided evidence of the engulfment of apoptotic cells by myofibroblasts. In addition, they found that MFG-E8 expression was increased in myofibroblasts isolated from the hearts with myocardial infarction in mice and humans. Myofibroblasts from MFG-E8 KO mice exhibited attenuated engulfment of apoptotic cells compared to the control, but it was recovered by MFG-E8 treatment in a dose-dependent manner. The results from this study reveal that myofibroblasts facilitate the removal of dead cells after myocardial infarction by secreting MFG-E8, which subsequently promotes anti-inflammatory responses and helps restore cardiac function.
4.3. The role of efferocytosis-related proteins in fibroblast function
Similar to VSMCs and ECs, efferocytosis-related proteins also impact fibroblast function independent of the efferocytosis mechanism. “Find me” signals and gates for “find me” molecules. Ishida et al. observed the role of fibroblast CX3CL1/CX3CR1 in skin wound healing (Ishida, Gao, & Murphy, 2008). They showed that mRNA levels of both CX3CL1 and CX3CR1 were significantly increased in wounded skin and increased CX3CR1 promoted myofibroblast accumulation and led to collagen deposition in wounded skin. Conversely, Vagnozzi et al. showed that in the ischemia/reperfusion-injured mouse hearts, the injection of mononuclear cells increased CX3CR1 expression, which subsequently attenuated cardiac fibroblast activity, reduced ECM formation in the border zone, and restored cardiac function (Vagnozzi et al., 2020). Also, co-culturing CX3CR1-upregulated macrophages with freshly isolated cardiac fibroblasts reduced the expression levels of α smooth muscle actin, collagen 1 alpha 2, and lysyl oxidase, but increased expression of connective tissue growth factor, in cardiac fibroblasts. These results imply that CX3CR1 from macrophages can modulate fibroblast activation and reduce adverse remodeling during cardiac damage. As discussed in the previous paragraphs, LPC affects the function of vascular cells such as ECs and VSMCs. Tseng et al. examined the role of LPC in modulating fibroblast phenotype. Their results indicated that LPC treatment induced excess production of mitochondrial ROS and cyclooxygenase 2 (COX-2), which subsequently increased extracellular collagen production by cardiac fibroblasts (Tseng, Lin, Hsiao, & Yang, 2019). S1P has been shown to have similar effects on human cardiac fibroblasts as S1P treatment on human cardiac fibroblasts also induces the expression of COX-2 by stimulating COX-2 promoter activity (Yang, Hsiao, Su, & Yang, 2020). Increased expression of COX-2 induced the secretion of proinflammatory Prostaglandin E2, suggesting that S1P could lead to enhanced inflammatory responses. Dolmatova et al. demonstrated that the induction of cardiac ischemia in canine upregulated Panx1 mRNA levels in cardiac myocytes in the border zone and that hypoxia exposure increased the open probability of Panx1 channels in cardiac myocytes, followed by increased release of ATP. ATP released from cardiac myocytes stimulated fibroblast differentiation into myofibroblast via the MAPK and p53 pathways (Dolmatova et al., 2012). “Eat me” signals. Annexin A1 has been implicated in diabetic wound healing by stimulating the migration of human skin fibroblasts in vitro (Bizzarro et al., 2012). This study showed that the expression and secretion of Annexin A1 were significantly decreased by high glucose treatment in normal human skin fibroblasts. Annexin A1 siRNA transfection in fibroblast cells significantly decreased fibroblast migration, while the treatment of fibroblast with Ac2-26 (an Annexin A1-derived peptide) restored attenuated migration in high glucose treated fibroblasts. Altogether, this study shows that Annexin A1 regulates the migration of fibroblasts, and downregulation of Annexin A1 by hyperglycemia attenuates wound contraction and restructuring of the ECM during wound healing (Bizzarro et al., 2012). Furthermore, CRT has been shown to regulate the expression, secretion, and production of ECM proteins by fibroblasts (Van Duyn Graham, Sweetwyne, Pallero, & Murphy-Ullrich, 2010). CRT deficient MEFs have reduced transcript levels of collagen I and III, which is followed by attenuated collagen deposition in the ECM, while CRT overexpression in fibroblasts leads to increased transcript and protein levels of collagen I. The same group further investigated the role of CRT in ECM regulation and found that ECM formation is mediated by TGFβ (Zimmerman, Graham, Pallero, & Murphy-Ullrich, 2013). In these studies, it was found that CRT is required for TGFβ to stimulate the production of collagen and fibronectin, in which failure of TGFβ to enhance the expression of collagen and fibronectin was seen in CRT deficient MEFs. These results suggest that CRT regulates the production of ECM proteins by fibroblasts. Along with other “eat me” signals, TSP1 has been reported to modulate fibroblast differentiation into myofibroblasts (Xia et al., 2011). Fibroblasts isolated from TSP1 KO mice exhibited impaired myofibroblast differentiation along with attenuated collagen synthesis. In the absence of TSP1, impaired TGFβ signaling in fibroblasts led to decreased collagen deposition, resulting in the formation of ECM that is inadequate in providing mechanical support to the ventricle. Bridge molecules. Rennard et al. revealed that C1q could bind to fibroblasts and stimulate fibroblast migration and growth ex vivo (Rennard, Chen, Robbins, Gadek, & Crystal, 1983). “Don’t eat me” signals. CD46 has been implicated in the protection of fibroblast from complement-mediated damage (Oglesby, Allen, Liszewski, White, & Atkinson, 1992). As previously mentioned, CD46 acts as a regulator for the complement activation pathways and protects host cells from inadvertent complement activation. Oglesby et al. had transfected mouse fibroblasts with human CD46 and found that CD46 overexpression offered a protective role against complement-mediated lysis. The protective role of CD46 from pathological complement-mediate damage is not only observed in fibroblasts, but could be extended to other cell types, such as ECs. In all, it is clearly evident that the regulation of fibroblast functions by efferocytic proteins is vital for the integrity of the vasculature.
5. Efferocytosis of pericytes
In the early 1870s, pericytes were first discovered by Rouget (Rouget, 1873). They were initially called Rouget cells and changed to pericytes by Zimmermann (Zimmermann, 1923). Pericytes are essential constituents of the basement membrane of capillary and postcapillary venules and wrap around endothelium. Despite a long history of pericyte investigations, its character has not been established until recent years, partly due to the difficulty to isolate pure pericyte because of the lack of pericyte-specific markers (Smyth et al., 2018). The primary functions of pericytes are blood vessel formation, capillary flow regulation, and maintaining capillary integrity (Attwell, Mishra, Hall, O’Farrell, & Dalkara, 2016; Birbrair, 2018). During pathophysiological conditions, an aberrant function of pericytes is observed. The dysfunction of pericytes contributes to the progression of diseases, such as ischemic heart disease (Avolio & Madeddu, 2016), diabetic retinopathy (Hammes, 2005), kidney disease (Shaw, Rider, Mullins, Hughes, & Peault, 2018), and Alzheimer’s disease (Jackson, ElAli, Virgintino, & Gilbert, 2017). This section will discuss the involvement of efferocytosis and its related peptides in the final vascular cell type to be covered, pericytes.
5.1. Pericyte engulfment of materials
To the best of our knowledge, there is no study demonstrating macrophage engulfment of pericytes or pericyte-mediated efferocytosis. This may be partly due to the difficulty in identifying and visualizing pericytes and their engulfment by macrophages in vivo. Due to its heterogeneous nature, the exact origin of pericytes remains to be under continuing research. Although pericytes were thought to be derived from mesenchymal or neural crest cells (Korn, Christ, & Kurz, 2002; Yamanishi, Takahashi, Saga, & Osumi, 2012), recent studies demonstrated that a subgroup of pericytes is actually derived from mature macrophages. Pericytes have also been reported to express multiple marker components of macrophages (Thomas, 1999). Yamamoto et al. had revealed that embryonic CD31+F4/80+ cells (= macrophage) could be differentiated into NG2/PDGFRβ/desmin-expressing pericytes (Yamamoto et al., 2017). CD31+F4/80+ cells exhibit the character of engulfment of apoptotic cells. Interestingly, the digestion of apoptotic cells and subsequent division drive the differentiation of CD31+F4/80+ cells into pericytes, confirmed by the expression of pericyte markers (PDGFRβ, desmin, and NG2). Other reports have also investigated the functional role of pericytes in the immune response, primarily due to their ability of having a macrophage-like phenotype. To study the immune potential of pericytes in the central nervous system (CNS), Balabanov et al. had found that pericytes expressed macrophage markers (e.g., ED-2 and CD11b) and showed the engulfment of fluorochrome-conjugated polystyrene beads and antibody-coated zymosan (Balabanov, Washington, Wagnerova, & Dore-Duffy, 1996). Cytokine stimulation has also been reported to induce pericyte-mediated immunological activity. Pieper et al. showed that the treatment with TNF-α and interferon γ increased pericytes’ phagocytic activity of fluorescent latex beads, while treatment with IL-1β and LPS did not alter phagocytic activity (Pieper, Marek, Unterberg, Schwerdtle, & Galla, 2014). TGFβ1 treatment on human brain pericytes attenuated the gene expression of CX3CL1, a “find me” signal, decreased pericyte-mediated phagocytosis and reduced pericyte proliferation rates (Rustenhoven et al., 2016). These studies indicate that pericytes contribute to the immune system by behaving like macrophages and exhibiting phagocytic activity. These findings imply the possibility of pericytes participating in efferocytosis; however, there is no direct evidence showing pericyte engulfment of apoptotic cells. Future studies are required to address the hypothesis.
5.2. The role of efferocytosis-related proteins in pericyte function
There are several manuscripts that discuss the role of efferocytosis-related peptides in the regulation of pericyte function, independent of the efferocytosis mechanism. “Find me” signals and “find me” receptors. Rahman et al. show the novel role of S1P in pericyte loss seen in sepsis (Abdel Rahman et al., 2021). This study found that S1P stimulates deposition of VE-cadherin in the sites of endothelial-pericyte junctions, which subsequently prevents pericyte loss and vascular hyper-permeability, leading to prolonged survival in sepsis-induced mice. The activation of P2Y receptor leads to contraction of capillary pericytes by increasing cytosolic calcium (Horlyck, Cai, Helms, Lauritzen, & Brodin, 2021). It is thus proposed to use P2Y receptor blockers as a potential therapeutic target for diseases related to microvessel constriction (e.g., peripheral artery disease, coronary microvascular disease). “Eat me” signals. TSP1 plays a vital role in regulating pericyte function. Canfield et al. demonstrated that TSP1 modulates osteogenesis of pericytes (Canfield, Sutton, Hoyland, & Schor, 1996). They revealed that inhibition of TSP1 in vitro promoted mineralized matrix production in vascular pericytes, while exogenous TSP1 treatment inhibited osteogenesis (Schor, Canfield, Sutton, Arciniegas, & Allen, 1995). Bridge molecules. Li et al. showed that Gas6 stimulated the migration of pericytes to endothelial tubes in a 3D ex vivo experiment, which promoted the maturation and stabilization of neovessels (X. Li et al., 2017). It was further demonstrated that Gas6 mediated stabilization of neovessels involved in Gas6/Axl and P13K/Akt/mTOR signaling pathways. Another bridge molecule that has been tested on pericyte-mediated angiogenesis is MFG-E8. Motegi et al. revealed that pericytes in tumors produce MFG-E8 and accelerate pathophysiological angiogenesis (Motegi et al., 2011). MFG-E8 KO mice exhibited attenuated pericyte coverage on neovessels and diminished tumor angiogenesis. Inhibition of MFG-E8 also reduced pericyte migration in vitro. The results from this study suggest that inhibition of MFG-E8 could be a potential target to treat tumor progression. The same group further investigated the role of MFG-E8 in PDGF (platelet-derived growth factor) signaling and found that MFG-E8 promotes angiogenesis, in part, by enhancing PDGF signaling in pericytes (Motegi, Garfield, Feng, Sardy, & Udey, 2011). The current literature on the regulatory roles of efferocytic proteins in pericyte function is primarily investigated in the central nervous system. Therefore, further research on pericyte function in other organs needs to be explored, and the finding will elucidate the potential role of pericytes in the progression of CVD.
6. Therapeutic aspects of efferocytosis
Efferocytosis is a critical mechanism in the maintenance of physiological function and homeostasis, and defective efferocytosis and dysregulation of efferocytic proteins can promote the onset of a variety of organ disorders marked by chronic inflammation such as immune diseases, metabolic disorders, and cancer. Therefore, the development of novel therapeutic approaches targeting efferocytosis is of considerable discussion.
Numerous signaling molecules play an essential role in the efferocytosis process; therefore, any of them could be a potential therapeutic target to resolve impaired efferocytosis. Atherosclerosis is a disease known to display decreased efferocytosis of apoptotic bodies (e.g., apoptotic macrophages, apoptotic SMCs). The molecular mechanisms of aberrant efferocytosis in atherosclerosis is discussed in many review articles; therefore, the details will not be discussed here (Kojima et al., 2017; Lin et al., 2020; Tajbakhsh et al., 2018; Wang, Li, Tang, & Yao, 2020). Basically, dysfunctional efferocytosis in the necrotic core is one of the causes of progressing atherosclerosis, and CD47, a “don’t eat me” peptide, is a promising therapeutic approach for atherosclerosis by enhancing phagocytosis of apoptotic bodies. However, it still requires more evidence to conclude it for the next step: pre-clinical trial. Sonodynamic therapy is on the clinical trial that focuses on efferocytosis in atherosclerosis (Clinical Trials.gov Identifier: NCT03382249). This novel methodology utilizes ultrasound to locally activate a sono-sensitizer which can induce cell death (Sun et al., 2019), and sonodynamic therapy has been shown to rapidly inhibit the progression of atherosclerotic plaques by inducing macrophage apoptosis, suppressing inflammation, and enhancing efferocytosis in the atherosclerotic plaque.
Another disease that is famous for being related to efferocytosis is lung disease. Defective efferocytosis and accumulated apoptotic cells in the lung are implicated in the development of lung diseases, including chronic obstructive pulmonary disease (COPD) (Lin et al., 2020; McCubbrey & Curtis, 2013; Zheng, Abou Taka, & Heit, 2021). Although it is not a “vascular cell”-initiated disease, we cite here for the example in which efferocytosis is used as a therapeutic target. COPD patients exhibit altered CD91 and CD31 expression in alveolar macrophages that contribute to attenuated efferocytosis and increased apoptotic epithelial cells in the lung (Hodge et al., 2007). There are several completed/ongoing clinical trials which aim to improve efferocytosis in COPD. Statins show a beneficial effect on outcomes in patients with many lung diseases (Lu et al., 2019). A clinical trial has been set to examine the impact of lovastatin on macrophage efferocytosis in COPD (ClinicalTrials.gov Identifier: NCT00700921). The mechanism of statins to increase efferocytosis is believed to mediate RhoA inhibition that leads to Rac1 activation and lamellipodia formation in macrophages and subsequently increases macrophage contact with apoptotic cells (Zheng et al., 2021). Glucocorticoid also enhances macrophage efferocytosis in the same way that statins do (Zheng et al., 2021). There is one ongoing clinical trial that investigates the effect of glucocorticoids on efferocytosis in alveolar macrophages isolated from COPD patients (ClinicalTrials.gov Identifier: NCT03034642). Hohlfeld et al. established the clinical trial to examine the impact of several compounds to increase efferocytosis of alveolar macrophages isolated from COPD patients (ClinicalTrials.gov Identifier: NCT04775394). However, detailed information on compounds is not available. There is not yet a result reported from these studies and the findings from these clinical trials would help understand further mechanisms of efferocytosis in lung diseases.
The clinical trials listed above are examples that aim to enhance efferocytosis as a treatment. However, it has to be noted that augmented efferocytosis is maladaptive in some diseases. For example, intact efferocytosis promotes tumor progression. Macrophages enriched in the microenvironment of tumors are called tumor-associated macrophages, and tumor-associated macrophages overexpress TAM receptors that accelerates efferocytosis. After efferocytosis, tumor-associated macrophages will produce and release more immunosuppressive cytokines, which results in accelerating efferocytosis, preventing immune response against cancer cells, and progressing tumor formation (Vaught, Stanford, & Cook, 2015). Therefore, inhibition of efferocytosis would exhibit a beneficial effect by attenuating progression of tumors.
The role of efferocytosis in cardiovascular disease is still underdeveloped. Further mechanistic understanding is required to develop a novel and effective therapeutic drug to alleviate inflammation in patients with aberrant efferocytosis.
7. Conclusion
In summary, this review highlights the importance of efferocytosis in the homeostatic maintenance of vasculature. We found that macrophages engulf a wide range of apoptotic cells (e.g., ECs, VSMCs, fibroblasts, RBCs, and neutrophils), and the process of efferocytosis, as well as regulation of the expression of efferocytosis-related proteins, exhibit beneficial effects on vascular functions, such as stimulating angiogenesis, protecting vessels from inflammation, and regulating vascular tone. Although the number of publications related to efferocytosis has greatly expanded during past decades, there are few reports that directly examine the macrophage engulfment of ECs, VSMCs, fibroblasts, or pericytes. This may be due to the difficulty in accurately visualizing and measuring the macrophage engulfment of vascular cells in vivo. Therefore, the development of a method could provide a more in-depth investigation of the mechanistic process of efferocytosis within the vasculature. It is apparent that a multitude of different signaling peptides leads to the proper engulfment of apoptotic cells. However, it still remains unclear what factors drive the expression of specific signaling molecules versus others. It is possible that the release of efferocytosis-related molecules depends on the apoptotic stage of the cells (early vs. late stage), how apoptosis was initially induced, or cell types. Therefore, it is too early to conclude that the peptides described above are all regulators in efferocytosis, or the signaling cascades listed in specific cell types are a unique pathway in those cell types. Further examinations are required to identify prominent and unique efferocytosis signaling cascades that occur in each vascular cell type. Our review well covered the current knowledge of efferocytosis in vascular cells and would offer the potential target in efferocytosis-related peptides for a novel treatment for cardiovascular disease.
Funding
This work was supported by grants from the National Heart, Lung, and Heart Institute of the National Institutes of Health (HL142214 and HL146764 to A. Makino).
Abbreviations:
- ABCA1
ATP-binding cassette transporter
- COX-2
cyclooxygenase 2
- CRT
calreticulin
- CVD
cardiovascular disease
- CX3CL1
C-X3-C motif chemokine ligand 1
- CX3CR1
CX3C motif chemokine receptor 1
- EC
endothelial cell
- ECM
extracellular matrix
- ER
endoplasmic reticulum
- GLUT-1
glucose transporter-1
- HUVEC
human umbilical vein endothelial cell
- IL
interleukin
- LPC
lysophosphatidylcholine
- MEF
mouse embryonic fibroblast
- MerTK
Mer Tyrosine Kinase
- MFG-E8
milk fat globule-EGF-factor 8
- NO
nitric oxide
- OxLDL
oxidized low density lipoprotein
- Panx1
pannexin-1
- PtdSer
phosphatidylserine
- RBC
red blood cell
- ROS
reactive oxygen species
- S1P
sphingosine-1-phosphate
- SMC
smooth muscle cell
- TGFβ
transforming growth factor beta
- TNF-α
tumor necrosis factor alpha
- TSP1
thrombospondin 1
- VEGF
vascular endothelial growth factor
- VSMC
vascular smooth muscle cell
Footnotes
Declaration of Competing Interest
The authors declare that there are no competing interests.
References
- Abdel Rahman F, d’Almeida S, Zhang T, Asadi M, Bozoglu T, Bongiovanni D, … Ziegler T (2021). Sphingosine-1-phosphate attenuates lipopolysaccharide-induced pericyte loss via activation of Rho-A and MRTF-A. Thrombosis and Haemostasis 121, 341–350. [DOI] [PubMed] [Google Scholar]
- Abraham MC, & Shaham S (2004). Death without caspases, caspases without death. Trends in Cell Biology 14, 184–193. [DOI] [PubMed] [Google Scholar]
- Arur S, Uche UE, Rezaul K, Fong M, Scranton V, Cowan AE, … Han DK (2003). Annexin I is an endogenous ligand that mediates apoptotic cell engulfment. Developmental Cell 4, 587–598. [DOI] [PubMed] [Google Scholar]
- Attwell D, Mishra A, Hall CN, O’Farrell FM, & Dalkara T (2016). What is a pericyte? Journal of Cerebral Blood Flow and Metabolism 36, 451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avanzi GC, Gallicchio M, Bottarel F, Gammaitoni L, Cavalloni G, Buonfiglio D, … Dianzani C (1998). Gas6 inhibits granulocyte adhesion to endothelial cells. Blood 91, 2334–2340. [PubMed] [Google Scholar]
- Avolio E, & Madeddu P (2016). Discovering cardiac pericyte biology: From physiopathological mechanisms to potential therapeutic applications in ischemic heart disease. Vascular Pharmacology 86, 53–63. [DOI] [PubMed] [Google Scholar]
- Balabanov R, Washington R, Wagnerova J, & Dore-Duffy P (1996). CNS microvascular pericytes express macrophage-like function, cell surface integrin alpha M, and macrophage marker ED-2. Microvascular Research 52, 127–142. [DOI] [PubMed] [Google Scholar]
- Banerjee S, de Freitas A, Friggeri A, Zmijewski JW, Liu G, & Abraham E (2011). Intracellular HMGB1 negatively regulates efferocytosis. Journal of Immunology 187, 4686–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, … Weissman IL (2019). CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 572, 392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MR, Gibson DF, Schwartz SM, & Tait JF (1995). Binding and phagocytosis of apoptotic vascular smooth muscle cells is mediated in part by exposure of phosphatidylserine. Circulation Research 77, 1136–1142. [DOI] [PubMed] [Google Scholar]
- Bennett MR, Sinha S, & Owens GK (2016). Vascular smooth muscle cells in atherosclerosis. Circulation Research 118, 692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birbrair A (2018). Pericyte biology: development, homeostasis, and disease. Advances in Experimental Medicine and Biology 1109, 1–3. [DOI] [PubMed] [Google Scholar]
- Bird IN, Taylor V, Newton JP, Spragg JH, Simmons DL, Salmon M, & Buckley CD (1999). Homophilic PECAM-1(CD31) interactions prevent endothelial cell apoptosis but do not support cell spreading or migration. Journal of Cell Science 112(Pt 12), 1989–1997. [DOI] [PubMed] [Google Scholar]
- Bizzarro V, Fontanella B, Carratu A, Belvedere R, Marfella R, Parente L, & Petrella A (2012). Annexin A1 N-terminal derived peptide Ac2-26 stimulates fibroblast migration in high glucose conditions. PLoS One 7, Article e45639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolick DT, Whetzel AM, Skaflen M, Deem TL, Lee J, & Hedrick CC (2007). Absence of the G protein-coupled receptor G2A in mice promotes monocyte/endothelial interactions in aorta. Circulation Research 100, 572–580. [DOI] [PubMed] [Google Scholar]
- Borner C, & Monney L (1999). Apoptosis without caspases: an inefficient molecular guillotine? Cell Death and Differentiation 6, 497–507. [DOI] [PubMed] [Google Scholar]
- Brissette MJ, Lepage S, Lamonde AS, Sirois I, Groleau J, Laurin LP, & Cailhier JF (2012). MFG-E8 released by apoptotic endothelial cells triggers anti-inflammatory macrophage reprogramming. PLoS One 7, Article e36368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broker LE, Kruyt FAE, & Giaccone G (2005). Cell death independent of caspases: a review. Clinical Cancer Research 11, 3155–3162. [DOI] [PubMed] [Google Scholar]
- Brown S, Heinisch I, Ross E, Shaw K, Buckley CD, & Savill J (2002). Apoptosis disables CD31-mediated cell detachment from phagocytes promoting binding and engulfment. Nature 418, 200–203. [DOI] [PubMed] [Google Scholar]
- Bulla R, Tripodo C, Rami D, Ling GS, Agostinis C, Guarnotta C, … Tedesco F (2016). C1q acts in the tumour microenvironment as a cancer-promoting factor independently of complement activation. Nature Communications 7, 10346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caberoy NB, Maiguel D, Kim Y, & Li W (2010). Identification of tubby and tubby-like protein 1 as eat-me signals by phage display. Experimental Cell Research 316, 245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caberoy NB, Zhou Y, & Li W (2010). Tubby and tubby-like protein 1 are new MerTK ligands for phagocytosis. The EMBO Journal 29, 3898–3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canfield AE, Sutton AB, Hoyland JA, & Schor AM (1996). Association of thrombospondin-1 with osteogenic differentiation of retinal pericytes in vitro. Journal of Cell Science 109(Pt 2), 343–353. [DOI] [PubMed] [Google Scholar]
- Chai YC, Howe PH, DiCorleto PE, & Chisolm GM (1996). Oxidized low density lipoprotein and lysophosphatidylcholine stimulate cell cycle entry in vascular smooth muscle cells. Evidence for release of fibroblast growth factor-2. The Journal of Biological Chemistry 271, 17791–17797. [DOI] [PubMed] [Google Scholar]
- Chandrasekaran L, He CZ, Al-Barazi H, Krutzsch HC, Iruela-Arispe ML, & Roberts DD (2000). Cell contact-dependent activation of alpha3beta1 integrin modulates endothelial cell responses to thrombospondin-1. Molecular Biology of the Cell 11, 2885–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HY, Chi JT, Dudoit S, Bondre C, van de Rijn M, Botstein D, & Brown PO (2002). Diversity, topographic differentiation, and positional memory in human fibroblasts. Proceedings of the National Academy of Sciences of the United States of America 99, 12877–12882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, … Ravichandran KS (2010). Pannexin 1 channels mediate “find-me” signal release and membrane permeability during apoptosis. Nature 467, 863–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke MC, Figg N, Maguire JJ, Davenport AP, Goddard M, Littlewood TD, & Bennett MR (2006). Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nature Medicine 12, 1075–1080. [DOI] [PubMed] [Google Scholar]
- Clowes AW, Reidy MA, & Clowes MM (1983). Mechanisms of stenosis after arterial injury. Laboratory Investigation 49, 208–215. [PubMed] [Google Scholar]
- Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, & Olson MF (2001). Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nature Cell Biology 3, 339–345. [DOI] [PubMed] [Google Scholar]
- Conconi MT, Baiguera S, Guidolin D, Furlan C, Menti AM, Vigolo S, … Nussdorfer GG (2003). Effects of hyperbaric oxygen on proliferative and apoptotic activities and reactive oxygen species generation in mouse fibroblast 3T3/J2 cell line. Journal of Investigative Medicine 51, 227–232. [DOI] [PubMed] [Google Scholar]
- Cosentino F, Rubattu S, Savoia C, Venturelli V, Pagannonne E, & Volpe M (2001). Endothelial dysfunction and stroke. Journal of Cardiovascular Pharmacology 38(Suppl. 2), S75–S78. [DOI] [PubMed] [Google Scholar]
- Das A, Ghatak S, Sinha M, Chaffee S, Ahmed NS, Parinandi NL, … Roy S (2016). Correction of MFG-E8 resolves inflammation and promotes cutaneous wound healing in diabetes. Journal of Immunology 196, 5089–5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson DW, Pearce SF, Zhong R, Silverstein RL, Frazier WA, & Bouck NP (1997). CD36 mediates the In vitro inhibitory effects of thrombospondin-1 on endothelial cells. The Journal of Cell Biology 138, 707–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delbosc S, Bayles RG, Laschet J, Ollivier V, Ho-Tin-Noe B, Touat Z, … Michel JB (2017). Erythrocyte efferocytosis by the arterial wall promotes oxidation in early-stage atheroma in humans. Frontiers in Cardiovascular Medicine 4, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dini L, Lentini A, Diez GD, Rocha M, Falasca L, Serafino L, & Vidal-Vanaclocha F (1995). Phagocytosis of apoptotic bodies by liver endothelial cells. Journal of Cell Science 108(Pt 3), 967–973. [DOI] [PubMed] [Google Scholar]
- Dolmatova E, Spagnol G, Boassa D, Baum JR, Keith K, Ambrosi C, … Duffy HS (2012). Cardiomyocyte ATP release through pannexin 1 aids in early fibroblast activation. American Journal of Physiology. Heart and Circulatory Physiology 303, H1208–H1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunoyer-Geindre S, de Moerloose P, Galve-de Rochemonteix B, Reber G, & Kruithof EK (2002). NFκB is an essential intermediate in the activation of endothelial cells by anti-beta2-glycoprotein 1 antibodies. Thrombosis and Haemostasis 88, 851–857. [PubMed] [Google Scholar]
- Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, … Ravichandran KS (2009). Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461, 282–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott MR, & Ravichandran KS (2016). The dynamics of apoptotic cell clearance. Developmental Cell 38, 147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elward K, Griffiths M, Mizuno M, Harris CL, Neal JW, Morgan BP, & Gasque P (2005). CD46 plays a key role in tailoring innate immune recognition of apoptotic and necrotic cells. The Journal of Biological Chemistry 280, 36342–36354. [DOI] [PubMed] [Google Scholar]
- Enzerink A, & Vaheri A (2011). Fibroblast activation in vascular inflammation. Journal of Thrombosis and Haemostasis 9, 619–626. [DOI] [PubMed] [Google Scholar]
- Evans PC, Taylor ER, & Kilshaw PJ (2001). Signaling through CD31 protects endothelial cells from apoptosis. Transplantation 71, 457–460. [DOI] [PubMed] [Google Scholar]
- Fan G, Li Q, & Qian J (2019). C1q contributes to post-stroke angiogenesis via LAIR1-HIF1alpha-VEGF pathway. Frontiers in Bioscience 24, 1050–1059. [DOI] [PubMed] [Google Scholar]
- Fens MH, Mastrobattista E, de Graaff AM, Flesch FM, Ultee A, Rasmussen JT, … Schiffelers RM (2008). Angiogenic endothelium shows lactadherin-dependent phagocytosis of aged erythrocytes and apoptotic cells. Blood 111, 4542–4550. [DOI] [PubMed] [Google Scholar]
- Ferrero E, Ferrero ME, Pardi R, & Zocchi MR (1995). The platelet endothelial cell adhesion molecule-1 (PECAM1) contributes to endothelial barrier function. FEBS Letters 374, 323–326. [DOI] [PubMed] [Google Scholar]
- Fleming KA, Reid KB, & McGee JO (1983). An assay for C1q biosynthesis in cultured human fibroblasts. Journal of Immunological Methods 56, 43–54. [DOI] [PubMed] [Google Scholar]
- Fries DM, Lightfoot R, Koval M, & Ischiropoulos H (2005). Autologous apoptotic cell engulfment stimulates chemokine secretion by vascular smooth muscle cells. The American Journal of Pathology 167, 345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaboriaud C, Thielens NM, Gregory LA, Rossi V, Fontecilla-Camps JC, & Arlaud GJ (2004). Structure and activation of the C1 complex of complement: unraveling the puzzle. Trends in Immunology 25, 368–373. [DOI] [PubMed] [Google Scholar]
- Gaengel K, Niaudet C, Hagikura K, Laviña B, Muhl L, Hofmann JJ, … Chen LL (2012). The sphingosine-1-phosphate receptor S1PR1 restricts sprouting angiogenesis by regulating the interplay between VE-cadherin and VEGFR2. Developmental Cell 23, 587–599. [DOI] [PubMed] [Google Scholar]
- Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, … Henson PM (2005). Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 123, 321–334. [DOI] [PubMed] [Google Scholar]
- Gasic GP, Arenas CP, Gasic TB, & Gasic GJ (1992). Coagulation factors X, Xa, and protein S as potent mitogens of cultured aortic smooth muscle cells. Proceedings of the National Academy of Sciences of the United States of America 89, 2317–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavins FN, Yona S, Kamal AM, Flower RJ, & Perretti M (2003). Leukocyte antiadhesive actions of annexin 1: ALXR- and FPR-related anti-inflammatory mechanisms. Blood 101, 4140–4147. [DOI] [PubMed] [Google Scholar]
- Grootaert MOJ, Moulis M, Roth L, Martinet W, Vindis C, Bennett MR, & De Meyer GRY (2018). Vascular smooth muscle cell death, autophagy and senescence in atherosclerosis. Cardiovascular Research 114, 622–634. [DOI] [PubMed] [Google Scholar]
- Gude DR, Alvarez SE, Paugh SW, Mitra P, Yu J, Griffiths R, … Spiegel S (2008). Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. The FASEB Journal 22, 2629–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall SE, Savill JS, Henson PM, & Haslett C (1994). Apoptotic neutrophils are phagocytosed by fibroblasts with participation of the fibroblast vitronectin receptor and involvement of a mannose/fucose-specific lectin. Journal of Immunology 153, 3218–3227. [PubMed] [Google Scholar]
- Hammes HP (2005). Pericytes and the pathogenesis of diabetic retinopathy. Hormone and Metabolic Research 37(Suppl. 1), 39–43. [DOI] [PubMed] [Google Scholar]
- Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, & Nagata S (2002). Identification of a factor that links apoptotic cells to phagocytes. Nature 417, 182–187. [DOI] [PubMed] [Google Scholar]
- Hermetet F, Jacquin E, Launay S, Gaiffe E, Couturier M, Hirchaud F, … Mougin C (2016). Efferocytosis of apoptotic human papillomavirus-positive cervical cancer cells by human primary fibroblasts. Biology of the Cell 108, 189–204. [DOI] [PubMed] [Google Scholar]
- Hobson B, & Denekamp J (1984). Endothelial proliferation in tumours and normal tissues: continuous labelling studies. British Journal of Cancer 49, 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge S, Hodge G, Ahern J, Jersmann H, Holmes M, & Reynolds PN (2007). Smoking alters alveolar macrophage recognition and phagocytic ability: implications in chronic obstructive pulmonary disease. American Journal of Respiratory Cell and Molecular Biology 37, 748–755. [DOI] [PubMed] [Google Scholar]
- Horckmans M, Robaye B, Leon-Gomicronmez E, Lantz N, Unger P, Dol-Gleizes F, … Communi D (2012). P2Y4 nucleotide receptor: a novel actor in post-natal cardiac development. Angiogenesis 15, 349–360. [DOI] [PubMed] [Google Scholar]
- Horlyck S, Cai C, Helms HCC, Lauritzen M, & Brodin B (2021). ATP induces contraction of cultured brain capillary pericytes via activation of P2Y-type purinergic receptors. American Journal of Physiology. Heart and Circulatory Physiology 320, H699–H712. [DOI] [PubMed] [Google Scholar]
- Howangyin KY, Zlatanova I, Pinto C, Ngkelo A, Cochain C, Rouanet M, … Silvestre JS (2016). Myeloid-Epithelial-Reproductive Receptor Tyrosine Kinase and Milk Fat Globule Epidermal Growth Factor 8 coordinately improve remodeling after myocardial infarction via local delivery of vascular endothelial growth factor. Circulation 133, 826–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JJ, Xia CJ, Wei Y, Yao Y, Dong MW, Lin KZ, … Fan YY (2020). Annexin A1-derived peptide Ac2-26 facilitates wound healing in diabetic mice. Wound Repair and Regeneration 28, 772–779. [DOI] [PubMed] [Google Scholar]
- Igarashi J, Bernier SG, & Michel T (2001). Sphingosine 1-phosphate and activation of endothelial nitric-oxide synthase. differential regulation of Akt and MAP kinase pathways by EDG and bradykinin receptors in vascular endothelial cells. The Journal of Biological Chemistry 276, 12420–12426. [DOI] [PubMed] [Google Scholar]
- Isenberg JS, Frazier WA, & Roberts DD (2008). Thrombospondin-1: a physiological regulator of nitric oxide signaling. Cellular and Molecular Life Sciences: CMLS 65, 728–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida Y, Gao JL, & Murphy PM (2008). Chemokine receptor CX3CR1 mediates skin wound healing by promoting macrophage and fibroblast accumulation and function. Journal of Immunology 180, 569–579. [DOI] [PubMed] [Google Scholar]
- Jackson S, ElAli A, Virgintino D, & Gilbert MR (2017). Blood-brain barrier pericyte importance in malignant gliomas: what we can learn from stroke and Alzheimer’s disease. Neuro-Oncology 19, 1173–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jozefczuk E, Guzik TJ, & Siedlinski M (2020). Significance of sphingosine-1-phosphate in cardiovascular physiology and pathology. Pharmacological Research 156, 104793. [DOI] [PubMed] [Google Scholar]
- Keul P, Polzin A, Kaiser K, Graler M, Dannenberg L, Daum G, … Levkau B (2019). Potent anti-inflammatory properties of HDL in vascular smooth muscle cells mediated by HDL-S1P and their impairment in coronary artery disease due to lower HDL-S1P: a new aspect of HDL dysfunction and its therapy. The FASEB Journal 33, 1482–1495. [DOI] [PubMed] [Google Scholar]
- Khan M, Pelengaris S, Cooper M, Smith C, Evan G, & Betteridge J (2003). Oxidised lipoproteins may promote inflammation through the selective delay of engulfment but not binding of apoptotic cells by macrophages. Atherosclerosis 171, 21–29. [DOI] [PubMed] [Google Scholar]
- Khanna S, Biswas S, Shang Y, Collard E, Azad A, Kauh C, … Roy S (2010). Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLoS One 5, Article e9539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima Y, Downing K, Kundu R, Miller C, Dewey F, Lancero H, … Leeper NJ (2014). Cyclin-dependent kinase inhibitor 2B regulates efferocytosis and atherosclerosis. The Journal of Clinical Investigation 124, 1083–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima Y, Volkmer JP, McKenna K, Civelek M, Lusis AJ, Miller CL, … Leeper NJ (2016). CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature 536, 86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima Y, Weissman IL, & Leeper NJ (2017). The role of efferocytosis in atherosclerosis. Circulation 135, 476–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb S, Vranckx R, Huisse MG, Michel JB, & Meilhac O (2007). The phosphatidylserine receptor mediates phagocytosis by vascular smooth muscle cells. The Journal of Pathology 212, 249–259. [DOI] [PubMed] [Google Scholar]
- Korn J, Christ B, & Kurz H (2002). Neuroectodermal origin of brain pericytes and vascular smooth muscle cells. The Journal of Comparative Neurology 442, 78–88. [DOI] [PubMed] [Google Scholar]
- Krysko DV, D’Herde K, & Vandenabeele P (2006). Clearance of apoptotic and necrotic cells and its immunological consequences. Apoptosis 11, 1709–1726. [DOI] [PubMed] [Google Scholar]
- Kurosaka K, Watanabe N, & Kobayashi Y (2002). Potentiation by human serum of anti-inflammatory cytokine production by human macrophages in response to apoptotic cells. Journal of Leukocyte Biology 71, 950–956. [PubMed] [Google Scholar]
- Lauber K, Bohn E, Krober SM, Xiao YJ, Blumenthal SG, Lindemann RK, … Wesselborg S (2003). Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell 113, 717–730. [DOI] [PubMed] [Google Scholar]
- Laurance S, Lemarie CA, & Blostein MD (2012). Growth arrest-specific gene 6 (gas6) and vascular hemostasis. Advances in Nutrition 3, 196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H-J, Ko H-J, Song D-K, & Jung Y-J (2018). Lysophosphatidylcholine promotes phagosome maturation and regulates inflammatory mediator production through the protein kinase a–phosphatidylinositol 3 kinase–p38 mitogen-activated protein kinase signaling pathway during mycobacterium tuberculosis infection in mouse macrophages. Frontiers in Immunology 9, 920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Lee Y, Song J, Lee J, & Chang S-Y (2018). Tissue-specific role of CX3CR1 expressing immune cells and their relationships with human disease. Immune Network 18, Article e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Thangada S, Claffey KP, Ancellin N, Liu CH, Kluk M, … Hla T (1999). Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell 99, 301–312. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Namkoong S, Kim YM, Kim CK, Lee H, Ha KS, … Kim YM (2006). Fractalkine stimulates angiogenesis by activating the Raf-1/MEK/ERK- and PI3K/Akt/eNOS-dependent signal pathways. American Journal of Physiology. Heart and Circulatory Physiology 291, H2836–H2846. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Park SY, Jung MY, Bae SM, & Kim IS (2011). Mechanism for phosphatidylserine-dependent erythrophagocytosis in mouse liver. Blood 117, 5215–5223. [DOI] [PubMed] [Google Scholar]
- Lee W, Park SY, Yoo Y, Kim SY, Kim JE, Kim SW, … Bae JS (2018). Macrophagic Stabilin-1 restored disruption of vascular integrity caused by sepsis. Thrombosis and Haemostasis 118, 1776–1789. [DOI] [PubMed] [Google Scholar]
- Lemke G, & Burstyn-Cohen T (2010). TAM receptors and the clearance of apoptotic cells. Annals of the New York Academy of Sciences 1209, 23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lertkiatmongkol P, Liao D, Mei H, Hu Y, & Newman PJ (2016). Endothelial functions of platelet/endothelial cell adhesion molecule-1 (CD31). Current Opinion in Hematology 23, 253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Tabar SS, Malec V, Eul BG, Klepetko W, Weissmann N, … Hanze J (2008). NOX4 regulates ROS levels under normoxic and hypoxic conditions, triggers proliferation, and inhibits apoptosis in pulmonary artery adventitial fibroblasts. Antioxidants &Redox Signaling 10, 1687–1698. [DOI] [PubMed] [Google Scholar]
- Li X, Chen M, Lei X, Huang M, Ye W, Zhang R, & Zhang D (2017). Luteolin inhibits angiogenesis by blocking Gas6/Axl signaling pathway. International Journal of Oncology 51, 677–685. [DOI] [PubMed] [Google Scholar]
- Li Y, Wittchen ES, Monaghan-Benson E, Hahn C, Earp HS, Doerschuk CM, & Burridge K (2019). The role of endothelial MERTK during the inflammatory response in lungs. PLoS One 14, Article e0225051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D, Kang X, Shen L, Tu S, Lenahan C, Chen Y, … Shao A (2020). Efferocytosis and its associated cytokines: a light on non-tumor and tumor diseases? Molecular Therapy Oncolytics 17, 394–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Chang R, Yao J, Xu X, Teng Y, & Cheng N (2019). Effectiveness of long-term using statins in COPD - a network meta-analysis. Respiratory Research 20, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maillard C, Berruyer M, Serre CM, Dechavanne M, & Delmas PD (1992). Protein-S, a vitamin K-dependent protein, is a bone matrix component synthesized and secreted by osteoblasts. Endocrinology 130, 1599–1604. [DOI] [PubMed] [Google Scholar]
- Maiti SN, Balasubramanian K, Ramoth JA, & Schroit AJ (2008). Beta-2-glycoprotein 1-dependent macrophage uptake of apoptotic cells. Binding to lipoprotein receptor-related protein receptor family members. The Journal of Biological Chemistry 283, 3761–3766. [DOI] [PubMed] [Google Scholar]
- McCarthy NJ, & Bennett MR (2000). The regulation of vascular smooth muscle cell apoptosis. Cardiovascular Research 45, 747–755. [DOI] [PubMed] [Google Scholar]
- McCubbrey AL, & Curtis JL (2013). Efferocytosis and lung disease. Chest 143, 1750–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwa Y, Hirata K, Kawashima S, Akita H, & Yokoyama M (1997). Lysophosphatidylcholine inhibits receptor-mediated Ca2+ mobilization in intact endothelial cells of rabbit aorta. Arteriosclerosis, Thrombosis, and Vascular Biology 17, 1561–1567. [DOI] [PubMed] [Google Scholar]
- Moodley Y, Rigby P, Bundell C, Bunt S, Hayashi H, Misso N, … Knight D (2003). Macrophage recognition and phagocytosis of apoptotic fibroblasts is critically dependent on fibroblast-derived thrombospondin 1 and CD36. The American Journal of Pathology 162, 771–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motegi S, Garfield S, Feng X, Sardy M, & Udey MC (2011). Potentiation of platelet-derived growth factor receptor-beta signaling mediated by integrin-associated MFG-E8. Arteriosclerosis, Thrombosis, and Vascular Biology 31, 2653–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motegi S, Leitner WW, Lu M, Tada Y, Sardy M, Wu C, … Udey MC (2011). Pericyte-derived MFG-E8 regulates pathologic angiogenesis. Arteriosclerosis, Thrombosis, and Vascular Biology 31, 2024–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, … Stroke Statistics S (2015). Heart disease and stroke statistics–2015 update: a report from the American Heart Association. Circulation 131 (e29–322). [DOI] [PubMed] [Google Scholar]
- Naeini MB, Bianconi V, Pirro M, & Sahebkar A (2020). The role of phosphatidylserine recognition receptors in multiple biological functions. Cellular & Molecular Biology Letters 25, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakaya M, Watari K, Tajima M, Nakaya T, Matsuda S, Ohara H, … Kurose H (2017). Cardiac myofibroblast engulfment of dead cells facilitates recovery after myocardial infarction. The Journal of Clinical Investigation 127, 383–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navratil JS, Watkins SC, Wisnieski JJ, & Ahearn JM (2001). The globular heads of C1q specifically recognize surface blebs of apoptotic vascular endothelial cells. Journal of Immunology 166, 3231–3239. [DOI] [PubMed] [Google Scholar]
- Newman SL, Henson JE, & Henson PM (1982). Phagocytosis of senescent neutrophils by human monocyte-derived macrophages and rabbit inflammatory macrophages. The Journal of Experimental Medicine 156, 430–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi T, Kobayashi N, Hisano Y, Kawahara A, & Yamaguchi A (2014). Molecular and physiological functions of sphingosine 1-phosphate transporters. Biochimica et Biophysica Acta 1841, 759–765. [DOI] [PubMed] [Google Scholar]
- van den Oever IA, Raterman HG, Nurmohamed MT, & Simsek S (2010). Endothelial dysfunction, inflammation, and apoptosis in diabetes mellitus. Mediators of Inflammation 2010, 792393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oglesby TJ, Allen CJ, Liszewski MK, White DJ, & Atkinson JP (1992). Membrane cofactor protein (CD46) protects cells from complement-mediated attack by an intrinsic mechanism. The Journal of Experimental Medicine 175, 1547–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikonomou E, Siasos G, Tsigkou V, Bletsa E, Panoilia ME, Oikonomou IN, … Tousoulis D (2020). Coronary artery disease and endothelial dysfunction: novel diagnostic and therapeutic approaches. Current Medicinal Chemistry 27, 1052–1080. [DOI] [PubMed] [Google Scholar]
- Oka K, Sawamura T, Kikuta K, Itokawa S, Kume N, Kita T, & Masaki T (1998). Lectin-like oxidized low-density lipoprotein receptor 1 mediates phagocytosis of aged/apoptotic cells in endothelial cells. Proceedings of the National Academy of Sciences of the United States of America 95, 9535–9540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, & Lindberg FP (2000). Role of CD47 as a marker of self on red blood cells. Science 288, 2051–2054. [DOI] [PubMed] [Google Scholar]
- Oshima K, Yasueda T, Nishio S, & Matsuda T (2014). MFG-E8: origin, structure, expression, functions and regulation. In Wang P (Ed.), MFG-E8 and Inflammation (pp. 1–31). Dordrecht: Springer. [Google Scholar]
- Owens GK (1995). Regulation of differentiation of vascular smooth muscle cells. Physiological Reviews 75, 487–517. [DOI] [PubMed] [Google Scholar]
- Owens GK, Kumar MS, & Wamhoff BR (2004). Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiological Reviews 84, 767–801. [DOI] [PubMed] [Google Scholar]
- Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund AC, Chapman DC, … Kroemer G (2009). Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. The EMBO Journal 28, 578–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SY, & Kim IS (2017). Engulfment signals and the phagocytic machinery for apoptotic cell clearance. Experimental & Molecular Medicine 49, Article e331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penberthy KK, & Ravichandran KS (2016). Apoptotic cell recognition receptors and scavenger receptors. Immunological Reviews 269, 44–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter C, Waibel M, Keppeler H, Lehmann R, Xu G, Halama A, … Lauber K (2012). Release of lysophospholipid ‘find-me’ signals during apoptosis requires the ATP-binding cassette transporter A1. Autoimmunity 45, 568–573. [DOI] [PubMed] [Google Scholar]
- Peter C, Waibel M, Radu CG, Yang LV, Witte ON, Schulze-Osthoff K, … Lauber K (2008). Migration to apoptotic “find-me” signals is mediated via the phagocyte receptor G2A. Journal of Biological Chemistry 283, 5296–5305. [DOI] [PubMed] [Google Scholar]
- Pieper C, Marek JJ, Unterberg M, Schwerdtle T, & Galla HJ (2014). Brain capillary pericytes contribute to the immune defense in response to cytokines or LPS in vitro. Brain Research 1550, 1–8. [DOI] [PubMed] [Google Scholar]
- Poon IK, Lucas CD, Rossi AG, & Ravichandran KS (2014). Apoptotic cell clearance: basic biology and therapeutic potential. Nature Reviews. Immunology 14, 166–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter KE, & Turner NA (2009). Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacology & Therapeutics 123, 255–278. [DOI] [PubMed] [Google Scholar]
- Privratsky JR, Paddock CM, Florey O, Newman DK, Muller WA, & Newman PJ (2011). Relative contribution of PECAM-1 adhesion and signaling to the maintenance of vascular integrity. Journal of Cell Science 124, 1477–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Ortiz ZG, Pendergraft WF 3rd, Prasad A, Byrne MH, Iram T, Blanchette CJ, … Means TK (2013). The scavenger receptor SCARF1 mediates the clearance of apoptotic cells and prevents autoimmunity. Nature Immunology 14, 917–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennard SI, Chen YF, Robbins RA, Gadek JE, & Crystal RG (1983). Fibronectin mediates cell attachment to C1q: a mechanism for the localization of fibrosis in inflammatory disease. Clinical and Experimental Immunology 54, 239–247. [PMC free article] [PubMed] [Google Scholar]
- Richards A, Kathryn Liszewski M, Kavanagh D, Fang CJ, Moulton E, Fremeaux-Bacchi V, … Atkinson JP (2007). Implications of the initial mutations in membrane cofactor protein (MCP; CD46) leading to atypical hemolytic uremic syndrome. Molecular Immunology 44, 111–122. [DOI] [PubMed] [Google Scholar]
- Rikitake Y, Kawashima S, Yamashita T, Ueyama T, Ishido S, Hotta H, & Hirata K. i., & Yokoyama M (2000). Lysophosphatidylcholine inhibits endothelial cell migration and proliferation via inhibition of the extracellular signal–regulated kinase pathway. Arteriosclerosis, Thrombosis, and Vascular Biology 20, 1006–1012. [DOI] [PubMed] [Google Scholar]
- Rouget C (1873). Mémoire sur le développment, la structure et les propertiés physiologiques des capillaries sanguins et lymphatiques. Arch Physiol Norm Pathol 5, 603–663. [Google Scholar]
- Rustenhoven J, Aalderink M, Scotter EL, Oldfield RL, Bergin PS, Mee EW, … Dragunow M (2016). TGF-beta1 regulates human brain pericyte inflammatory processes involved in neurovasculature function. Journal of Neuroinflammation 13, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandilos JK, Chiu YH, Chekeni FB, Armstrong AJ, Walk SF, Ravichandran KS, & Bayliss DA (2012). Pannexin 1, an ATP release channel, is activated by caspase cleavage of its pore-associated C-terminal autoinhibitory region. The Journal of Biological Chemistry 287, 11303–11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraste A, & Pulkki K (2000). Morphologic and biochemical hallmarks of apoptosis. Cardiovascular Research 45, 528–537. [DOI] [PubMed] [Google Scholar]
- Savill JS, Wyllie AH, Henson JE, Walport MJ, Henson PM, & Haslett C (1989). Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. The Journal of Clinical Investigation 83, 865–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schor AM, Canfield AE, Sutton AB, Arciniegas E, & Allen TD (1995). Pericyte differentiation. Clinical Orthopaedics and Related Research 313, 81–91. [PubMed] [Google Scholar]
- Schrijvers DM, De Meyer GR, Kockx MM, Herman AG, & Martinet W (2005). Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology 25, 1256–1261. [DOI] [PubMed] [Google Scholar]
- Segawa K, Kurata S, Yanagihashi Y, Brummelkamp TR, Matsuda F, & Nagata S (2014). Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 344, 1164–1168. [DOI] [PubMed] [Google Scholar]
- Segawa K, & Nagata S (2015). An apoptotic ‘eat me’signal: phosphatidylserine exposure. Trends in Cell Biology 25, 639–650. [DOI] [PubMed] [Google Scholar]
- Segawa K, Yanagihashi Y, Yamada K, Suzuki C, Uchiyama Y, & Nagata S (2018). Phospholipid flippases enable precursor B cells to flee engulfment by macrophages. Proceedings of the National Academy of Sciences of the United States of America 115, 12212–12217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah D, Romero F, Zhu Y, Duong M, Sun J, Walsh K, & Summer R (2015). C1q deficiency promotes pulmonary vascular inflammation and enhances the susceptibility of the lung endothelium to Injury. The Journal of Biological Chemistry 290, 29642–29651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw I, Rider S, Mullins J, Hughes J, & Peault B (2018). Pericytes in the renal vasculature: roles in health and disease. Nature Reviews. Nephrology 14, 521–534. [DOI] [PubMed] [Google Scholar]
- Si R, Zhang Q, Tsuji-Hosokawa A, Watanabe M, Willson C, Lai N, … Makino A (2020). Overexpression of p53 due to excess protein O-GlcNAcylation is associated with coronary microvascular disease in type 2 diabetes. Cardiovascular Research 116, 1186–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestre JS, Thery C, Hamard G, Boddaert J, Aguilar B, Delcayre A, … Mallat Z (2005). Lactadherin promotes VEGF-dependent neovascularization. Nature Medicine 11, 499–506. [DOI] [PubMed] [Google Scholar]
- Smyth LCD, Rustenhoven J, Scotter EL, Schweder P, Faull RLM, Park TIH, & Dragunow M (2018). Markers for human brain pericytes and smooth muscle cells. Journal of Chemical Neuroanatomy 92, 48–60. [DOI] [PubMed] [Google Scholar]
- Steucke KE, Tracy PV, Hald ES, Hall JL, & Alford PW (2015). Vascular smooth muscle cell functional contractility depends on extracellular mechanical properties. Journal of Biomechanics 48, 3044–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strassheim D, Verin A, Batori R, Nijmeh H, Burns N, Kovacs-Kasa A, … Gerasimovskaya E (2020). P2Y purinergic receptors, endothelial dysfunction, and cardiovascular diseases. International Journal of Molecular Sciences 21, 6855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Guo S, Yao J, Wang H, Peng C, Li B, … Tian Y (2019). Rapid inhibition of atherosclerotic plaque progression by sonodynamic therapy. Cardiovascular Research 115, 190–203. [DOI] [PubMed] [Google Scholar]
- Suzuki J, Denning DP, Imanishi E, Horvitz HR, & Nagata S (2013). Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science 341, 403–406. [DOI] [PubMed] [Google Scholar]
- Tajbakhsh A, Rezaee M, Kovanen PT, & Sahebkar A (2018). Efferocytosis in atherosclerotic lesions: Malfunctioning regulatory pathways and control mechanisms. Pharmacology & Therapeutics 188, 12–25. [DOI] [PubMed] [Google Scholar]
- Taylor PR, Carugati A, Fadok VA, Cook HT, Andrews M, Carroll MC, … Walport MJ (2000). A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. The Journal of Experimental Medicine 192, 359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thielens NM, Tedesco F, Bohlson SS, Gaboriaud C, & Tenner AJ (2017). C1q: A fresh look upon an old molecule. Molecular Immunology 89, 73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas WE (1999). Brain macrophages: on the role of pericytes and perivascular cells. Brain Research. Brain Research Reviews 31, 42–57. [DOI] [PubMed] [Google Scholar]
- Totary-Jain H, Naveh-Many T, Riahi Y, Kaiser N, Eckel J, & Sasson S (2005). Calreticulin destabilizes glucose transporter-1 mRNA in vascular endothelial and smooth muscle cells under high-glucose conditions. Circulation Research 97, 1001–1008. [DOI] [PubMed] [Google Scholar]
- Touyz RM, Alves-Lopes R, Rios FJ, Camargo LL, Anagnostopoulou A, Arner A, & Montezano AC (2018). Vascular smooth muscle contraction in hypertension. Cardiovascular Research 114, 529–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers JG, Kamal FA, Robbins J, Yutzey KE, & Blaxall BC (2016). Cardiac fibrosis: The fibroblast awakens. Circulation Research 118, 1021–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tricot O, Mallat Z, Heymes C, Belmin J, Leseche G, & Tedgui A (2000). Relation between endothelial cell apoptosis and blood flow direction in human atherosclerotic plaques. Circulation 101, 2450–2453. [DOI] [PubMed] [Google Scholar]
- Truman LA, Ford CA, Pasikowska M, Pound JD, Wilkinson SJ, Dumitriu IE, … Gregory CD (2008). CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood 112, 5026–5036. [DOI] [PubMed] [Google Scholar]
- Tseng HC, Lin CC, Hsiao LD, & Yang CM (2019). Lysophosphatidylcholine-induced mitochondrial fission contributes to collagen production in human cardiac fibroblasts. Journal of Lipid Research 60, 1573–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiyama A, Yamada K, Ogino S, Yokoyama Y, Takeuchi Y, Udey MC, … Motegi S (2014). MFG-E8 regulates angiogenesis in cutaneous wound healing. The American Journal of Pathology 184, 1981–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagnozzi RJ, Maillet M, Sargent MA, Khalil H, Johansen AKZ, Schwanekamp JA, … Molkentin JD (2020). An acute immune response underlies the benefit of cardiac stem cell therapy. Nature 577, 405–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Duyn Graham L, Sweetwyne MT, Pallero MA, & Murphy-Ullrich JE (2010). Intracellular calreticulin regulates multiple steps in fibrillar collagen expression, trafficking, and processing into the extracellular matrix. The Journal of Biological Chemistry 285, 7067–7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaught DB, Stanford JC, & Cook RS (2015). Efferocytosis creates a tumor microenvironment supportive of tumor survival and metastasis. Cancer Cell Microenviron 2, Article e666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vita JA, & Hamburg NM (2010). Does endothelial dysfunction contribute to the clinical status of patients with peripheral arterial disease? Canadian Journal of Cardiology 26(Suppl A), 45A–50A. [DOI] [PubMed] [Google Scholar]
- Volin MV, Woods JM, Amin MA, Connors MA, Harlow LA, & Koch AE (2001). Fractalkine: a novel angiogenic chemokine in rheumatoid arthritis. The American Journal of Pathology 159, 1521–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall IB, Moseley R, Baird DM, Kipling D, Giles P, Laffafian I, … Stephens P (2008). Fibroblast dysfunction is a key factor in the non-healing of chronic venous leg ulcers. The Journal of Investigative Dermatology 128, 2526–2540. [DOI] [PubMed] [Google Scholar]
- Wamhoff BR, Lynch KR, Macdonald TL, & Owens GK (2008). Sphingosine-1-phosphate receptor subtypes differentially regulate smooth muscle cell phenotype. Arteriosclerosis, Thrombosis, and Vascular Biology 28, 1454–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Li H, Tang Y, & Yao P (2020). Potential mechanisms and effects of efferocytosis in atherosclerosis. Front Endocrinol (Lausanne) 11, 585285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Iring A, Strilic B, Albarran Juarez J, Kaur H, Troidl K, … Offermanns S (2015). P2Y2 and Gq/G11 control blood pressure by mediating endothelial mechanotransduction. The Journal of Clinical Investigation 125, 3077–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhang Y, Zhao Y, Liang Y, Xiang C, Zhou H, … Peng L (2016). CD24 promoted cancer cell angiogenesis via Hsp90-mediated STAT3/VEGF signaling pathway in colorectal cancer. Oncotarget 7, 55663–55676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Nanda V, Direnzo D, Ye J, Xiao S, Kojima Y, … Leeper NJ (2020). Clonally expanding smooth muscle cells promote atherosclerosis by escaping efferocytosis and activating the complement cascade. Proceedings of the National Academy of Sciences of the United States of America 117, 15818–15826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Pakala R, Katagiri T, & Benedict CR (2002). Lysophosphatidylcholine is a major contributor to the synergistic effect of mildly oxidized low-density lipoprotein with endothelin-1 on vascular smooth muscle cell proliferation. Journal of Cardiovascular Pharmacology 39, 449–459. [DOI] [PubMed] [Google Scholar]
- Weigert A, Johann AM, von Knethen A, Schmidt H, Geisslinger G, & Brune B (2006). Apoptotic cells promote macrophage survival by releasing the antiapoptotic mediator sphingosine-1-phosphate. Blood 108, 1635–1642. [DOI] [PubMed] [Google Scholar]
- Weigert A, Tzieply N, von Knethen A, Johann AM, Schmidt H, Geisslinger G, & Brune B (2007). Tumor cell apoptosis polarizes macrophages role of sphingosine-1-phosphate. Molecular Biology of the Cell 18, 3810–3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihua Z, Tsan R, Schroit AJ, & Fidler IJ (2005). Apoptotic cells initiate endothelial cell sprouting via electrostatic signaling. Cancer Research 65, 11529–11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White GE, Tan TC, John AE, Whatling C, McPheat WL, & Greaves DR (2010). Fractalkine has anti-apoptotic and proliferative effects on human vascular smooth muscle cells via epidermal growth factor receptor signalling. Cardiovascular Research 85, 825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn RK, & Harlan JM (2005). The role of endothelial cell apoptosis in inflammatory and immune diseases. Journal of Thrombosis and Haemostasis 3, 1815–1824. [DOI] [PubMed] [Google Scholar]
- Woywodt A, Bahlmann FH, De Groot K, Haller H, & Haubitz M (2002). Circulating endothelial cells: life, death, detachment and repair of the endothelial cell layer. Nephrology, Dialysis, Transplantation 17, 1728–1730. [DOI] [PubMed] [Google Scholar]
- Xia Y, Dobaczewski M, Gonzalez-Quesada C, Chen W, Biernacka A, Li N, … Frangogiannis NG (2011). Endogenous thrombospondin 1 protects the pressure-overloaded myocardium by modulating fibroblast phenotype and matrix metabolism. Hypertension 58, 902–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing XQ, Li YL, Zhang YX, Xiao Y, Li ZD, Liu LQ, … Yang J (2015). Sphingosine kinase 1/sphingosine 1-phosphate signalling pathway as a potential therapeutic target of pulmonary hypertension. International Journal of Clinical and Experimental Medicine 8, 11930–11935. [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, & Hla T (2014). S1P control of endothelial integrity. In Olderstone MBA, & Rosen H (Eds.), Sphingosine-1-phosphate signaling in immunology and infectious diseases. Vol. 378. (pp. 85–105). Cham: Springer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Muramatsu M, Azuma E, Ikutani M, Nagai Y, Sagara H, … Sasahara M (2017). A subset of cerebrovascular pericytes originates from mature macrophages in the very early phase of vascular development in CNS. Scientific Reports 7, 3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanishi E, Takahashi M, Saga Y, & Osumi N (2012). Penetration and differentiation of cephalic neural crest-derived cells in the developing mouse telencephalon. Development, Growth & Differentiation 54, 785–800. [DOI] [PubMed] [Google Scholar]
- Yang CC, Hsiao LD, Su MH, & Yang CM (2020). Sphingosine 1-Phosphate induces Cyclooxygenase-2/Prostaglandin E2 expression via PKCα-dependent Mitogen-Activated Protein Kinases and NF-κB cascade in human cardiac fibroblasts. Frontiers in Pharmacology 11, 569802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang LV, Radu CG, Wang L, Riedinger M, & Witte ON (2005). Gi-independent macrophage chemotaxis to lysophosphatidylcholine via the immunoregulatory GPCR G2A. Blood 105, 1127–1134. [DOI] [PubMed] [Google Scholar]
- Yin J, McLachlan C, Chaufour X, McGuire MA, White G, Turner V, … Hambly BD (2000). Growth arrest-specific gene 6 expression in proliferating rabbit vascular smooth muscle cells in vitro and in vivo. Electrophoresis 21, 3851–3856. [DOI] [PubMed] [Google Scholar]
- Zemskov E, Lucas R, Verin AD, & Umapathy NS (2011). P2Y receptors as regulators of lung endothelial barrier integrity. Journal of Cardiovascular Disease Research 2, 14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yang W, Hu B, Wu W, & Fallon MB (2014). Endothelin-1 activation of the endothelin B receptor modulates pulmonary endothelial CX3CL1 and contributes to pulmonary angiogenesis in experimental hepatopulmonary syndrome. The American Journal of Pathology 184, 1706–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Shao R, Gao W, Sun G, Liu Y, & Fa X (2018). Inhibition of miR-361–5p suppressed pulmonary artery smooth muscle cell survival and migration by targeting ABCA1 and inhibiting the JAK2/STAT3 pathway. Experimental Cell Research 363, 255–261. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen X, Gueydan C, & Han J (2018). Plasma membrane changes during programmed cell deaths. Cell Research 28, 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng DJ, Abou Taka M, & Heit B (2021). Role of apoptotic cell clearance in pneumonia and inflammatory lung disease. Pathogens 10, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Fang XL, Zhang Y, Feng YN, & Wang SS (2020). miR-20a-5p promotes pulmonary artery smooth muscle cell proliferation and migration by targeting ABCA1. Journal of Biochemical and Molecular Toxicology 34, e22589. [DOI] [PubMed] [Google Scholar]
- Zimmerman KA, Graham LV, Pallero MA, & Murphy-Ullrich JE (2013). Calreticulin regulates transforming growth factor-beta-stimulated extracellular matrix production. The Journal of Biological Chemistry 288, 14584–14598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann KW (1923). Der feinere Bau der Blutkapillaren. Zeitschrift für Anatomie und Entwicklungsgeschichte 68, 29–109. [Google Scholar]