

Abstract
Colorectal cancer (CRC) represents the third most commonly diagnosed cancer and the second leading cause of cancer death worldwide. The modern concept of cancer biology indicates that cancer is formed of a small population of cells called cancer stem cells (CSCs), which present both pluripotency and self‐renewal properties. These cells are considered responsible for the progression of the disease, recurrence and tumor resistance. Interestingly, some cell signaling pathways participate in CRC survival, proliferation, and self‐renewal properties, and most of them are dysregulated in CSCs, including the Wingless (Wnt)/β‐catenin, Notch, Hedgehog, nuclear factor kappa B (NF‐κB), Janus kinase/signal transducer and activator of transcription (JAK/STAT), peroxisome proliferator‐activated receptor (PPAR), phosphatidyl‐inositol‐3‐kinase/Akt/mechanistic target of rapamycin (PI3K/Akt/mTOR), and transforming growth factor‐β (TGF‐β)/Smad pathways. In this review, we summarize the strategies for eradicating CRC stem cells by modulating these dysregulated pathways, which will contribute to the study of potential therapeutic schemes, combining conventional drugs with CSC‐targeting drugs, and allowing better cure rates in anti‐CRC therapy.
Keywords: colorectal, cancer stem cells, cell signaling, Wnt/β‐catenin pathway, Notch, Hedgehog, NF‐κB, JAK/STAT signaling, PI3K/Akt/mTOR signaling, targeted therapy
CRC represents the third most commonly diagnosed cancer and the second cause of cancer death;
Cell signaling pathways are dysregulated in CRC stem cells;
Cell signaling pathways are target to eradicate CRC stem cells.
Abbreviations
- 4‐AAQB
4‐acetyl‐antroquinonol B
- 5‐FU
5‐fluorouracil
- ABCG2
ATP binding cassette subfamily G member 2
- ABL
ABL proto‐oncogene 1, non‐receptor tyrosine kinase
- AC
adenylyl cyclase
- AF2
activation function 2
- ALDH1
aldehyde dehydrogenase 1
- AMPK
adenosine monophosphate‐activated protein kinase
- APC
adenomatous polyposis coli
- APCDD1
APC downregulated 1
- ATL‐1
atractylenolide I
- AXIN1
axin‐1
- Axin
axis inhibitor
- BCR
BCR activator of RhoGEF and GTPase
- BRAF
B‐Raf proto‐oncogene, serine/threonine kinase
- cAMP
cyclic adenosine monophosphate
- CAR‐T
chimeric antigen receptor T cell
- C‐B
cucurbitacin‐B
- CCND1
cyclin D1
- CD44s
CD44 standard isoform
- CD44v
CD44 variant isoform
- C‐I
cucurbitacin‐I
- CK1α
casein kinase 1α
- Co‐Smad
common partner Smad
- CRC
colorectal cancer
- CRC‐SCs
colorectal cancer stem cells
- CSCs
cancer stem cells
- CSL
CBF1–suppressor of hairless–LAG1
- CTLA4
cytotoxic T‐lymphocyte‐associated antigen 4
- CXXC4
CXXC finger protein 4
- DATS
diallyl trisulfide
- DCLK1
doublecortin‐like kinase‐1
- DKK3
dickkopf Wnt signaling pathway inhibitor 3
- DNMT1
DNA methyltransferase
- DVL
dishevelled
- EGCG
epigallocatechin‐3‐gallate
- EGFR
epidermal growth factor receptor
- EMT
epithelial‐mesenchymal transition
- EpCAM
epithelial cell adhesion molecule
- ERBB2
erb‐b2 receptor tyrosine kinase 2
- EZH2
enhancer of zeste 2 polycomb repressive complex 2 subunit
- FDMC
farnesyl dimethyl chromanol
- FOLFIRI
5‐FU, leucovorin plus irinotecan
- FOLFOX
5‐FU, folic acid plus oxaliplatin
- FZD
seven‐pass transmembrane receptor frizzled
- GLI
GLI family zinc finger
- GP130
glycoprotein 130
- GSK‐3β
glycogen synthase kinase‐3 beta
- HES1
split‐1
- HEY1
hes related with YRPW motif‐1
- HH
hedgehog
- HOXA5
homeobox A5
- IκB
inhibitor of NF‐κB
- IR
ionizing radiation
- I‐Smad
inhibitory Smad
- JAK
janus kinase
- KLF4
Krüppel‐like factor 4
- LEF
lymphoid enhancer factor
- LGR5
leucinerich repeat‐containing G‐protein‐coupled receptor 5
- LRP5/6
single‐pass low‐density lipoprotein receptor‐related protein 5 or 6
- LSD1
lysine‐specific demethylase 1
- MCM2
minichromosome maintenance complex component 2
- miRNA
microRNA
- MYC
MYC proto‐oncogene, bHLH transcription factor
- mTOR
mechanistic target of rapamycin
- mTORC1
mTOR complex 1
- NANOG
nanog homeobox
- NF‐κB
nuclear factor kappa B
- NIK
NF‐κB‐inducing kinase
- NKD1
NKD inhibitor of Wnt signaling pathway 1
- OCT4
octamer‐binding transcription factor 4
- PD‐1
programmed cell death protein 1
- PEITC
phenethyl isothiocyanate
- PI3K
phosphatidyl‐inositol‐3‐kinases
- PIK3C3
phosphatidylinositol 3‐kinase catalytic subunit type 3
- PIK3CA
phosphatidylinositol‐4,5‐bisphosphate 3‐kinase catalytic subunit alpha
- PIK3R2
phosphoinositide‐3‐kinase regulatory subunit 2
- PIP2
phosphatidylinositol 4,5‐bisphosphate
- PIP3
phosphatidylinositol (3,4,5)‐trisphosphate
- PKA
protein kinase A
- PLGA
poly(D,L‐lactic‐coglycolic acid
- PPAR
peroxisome proliferator‐activated receptor
- PTCH
patched
- PTEN
phosphatase and tensin homolog
- RAS
rat sarcoma virus
- R‐Smad
receptor‐regulated Smad
- RSPO
R‐spondin
- SHP‐1
Src homology 2 domain‐containing protein tyrosine phosphatase 1
- SMO
smoothened
- SOD2
superoxide dismutase 2
- SOX2
SRY‐box transcription factor 2
- SOX4
SRY‐box transcription factor 4
- SOX9
SRY‐box transcription factor 9
- STAT
signal transducer and activator of transcription
- TAp63α
transactivating p63 isoform α
- TCF
T‐cell factor
- TGFBR1
TGF‐β receptor type 1
- TGFBR2
TGF‐β receptor type 2
- TGF‐β
transforming growth factor‐β
- TNIK
TRAF2‐ and NCK‐interacting kinase
- TP53
tumor protein p53
- VEGF
vascular endothelial growth factor
- VHL
von Hippel‐Lindau tumor suppressor
- Wnt
wingless
1. BACKGROUND
Colorectal cancer (CRC) represents the third most commonly diagnosed cancer and the second leading cause of cancer death worldwide, representing 1,900,000 new cases and an estimated number of 935,000 deaths in 2020 [1]. Surgical resection is the preferred treatment for earlier‐stage CRC. Chemotherapy is indicated in patients at risk of cancer recurrence, as an adjuvant treatment, or when the patient has distant metastasis. A very important topic in the treatment of CRC is the response rate of anti‐neoplastic drugs in patients with metastases [2, 3, 4].
5‐Fluorouracil (5‐FU) is still the recommended first‐line drug treatment for advanced CRC and has achieved response rates of 10%‐15%, which are improved to approximately 40%‐50% when combined with irinotecan and oxaliplatin [3, 5]. Second‐line treatment for metastatic CRC includes some targeted therapies. These include inhibitors of vascular endothelial growth factor (VEGF), which reduce tumor angiogenesis. Other drugs target growth factor pathways such as epidermal growth factor receptor (EGFR) and abnormal B‐Raf proto‐oncogene serine/threonine kinase (BRAF) [2, 3]. The immune system also plays an important role in regulating the development of CRC. The immunological pathways that have been studied include cytotoxic T‐lymphocyte‐associated antigen 4 (CTLA4) and programmed cell death protein 1 (PD‐1) [6]. Drugs that promote the inhibition of CTLA4 produce a large increase in T helper cell‐dependent immune responses. Inhibition of the PD‐1 pathway can enhance the anti‐tumor immune response since PD‐1 is a cell surface receptor that limits T cell activity [7]. Despite advances in surgical and medical therapies, the mortality from CRC has changed little in recent decades, indicating the need for research on other pathways in advanced diseases [8].
The current concept of cancer biology indicates that cancer is a heterogeneous disease derived from a small subpopulation of undifferentiated cancer cells called cancer stem cells (CSCs) or tumor‐initiating cells. CSCs are defined as cells with three unique properties: the ability to renew themselves indefinitely; the ability to recreate the complete cancer cell repertoire of the original tumor; and the expression of a distinct set of surface biomarkers [9]. Some pluripotent transcription factors, such as octamer‐binding transcription factor 4 (OCT4) [10], SRY‐box transcription factor 2 (SOX2) [11], nanog homeobox (NANOG) [12], and MYC proto‐oncogene, bHLH transcription factor (MYC) [13], have been associated with the regulation of these CSC properties.
Self‐renewal involves the ability of CSCs to maintain their proportion through a combination of symmetric division (which produces two daughter cells with CSC properties) and asymmetric division (which produces a daughter cell with CSC properties and a daughter cell without CSC properties) [14]. In addition to the symmetric/asymmetric division theory, CSCs can differentiate into cells without CSC properties, and these cells can dedifferentiate back into CSCs through a bidirectional interconversion process [15]. This indicates that, for a good clinical outcome, cancer cells with or without properties of CSCs must be eradicated.
Although CSCs are self‐renewable, they are relatively quiescent. In fact, they have longer cell cycle times than cancer cells without properties of CSCs due to the permanence of CSCs in a phase of the cell cycle similar to G0 [16]. These unique properties of CSCs can explain the failure of many anti‐neoplastic drugs, which affect cells that divide rapidly, causing a reduction in the number of cancer cells, while CSCs divide slowly and are not sensitive to most cytotoxic drugs [16, 17].
An important feature of CSCs in culture is that they are able to form multicellular spheroid structures when grown in non‐adherent conditions without serum and supplemented with growth factors. These spheres are characterized by a well‐rounded morphology, ability to persist as floating cultures, and small size [18]. Thus, spheroid cultures are useful for enriching population cells that exhibit characteristics of CSCs. Likewise, the secondary and tertiary sphere formation method, in the absence of any agent, is useful to verify the self‐renewal capacity of cells in multiple passages [19, 20].
Several biomarkers have been identified in CRC stem cells (CRC‐SCs), including CD133 [21, 22], CD44 [22, 23], CD166 [23], CD29 [24], CD24 [24], aldehyde dehydrogenase 1 (ALDH1) [25], CD26 [26], and epithelial cell adhesion molecule (EpCAM) [27]. On the other hand, although these molecules are considered standard markers of CRC‐SCs, some of them are also found in stem cells from normal tissues and other types of cancer with different degrees of expression [28, 29]. CSCs can be identified and separated by combining specific biomarkers and separation techniques to increase the sensitivity and specificity of the assay. Normally, cells with positive/high expression of these biomarkers are considered to have stemness properties.
CD133 was the first biomarker identified, and although still contradictory, most clinical data support the hypothesis that CD133 expression is associated with a poor prognosis in CRC [30, 31, 32, 33]. Curiously, the CD133 protein is not lost in differentiating CRC‐SCs, but its AC133 epitope is [34]. Therefore, monoclonal antibodies against the AC133 epitope are most commonly used to appropriately identify CD133 in CRC‐SCs. Interestingly, CRC CD133+ cells, but not CD133– cells, have the ability to form solid tumors in immunodeficient NOD/SCID mice [21]. In particular, treatment with 5‐FU initially decreases the tumor mass; however, upon completion of treatment, enrichment of CD133+ cells in residual tumors is found, contributing to recurrence and decreasing progression‐free survival in patients with CRC [35, 36]. In addition, CD133 has also been used as a marker for CSCs in different types of cancer, such as brain [37], prostate [38], and liver cancers [39].
CD44 is also an important CRC‐SC marker that is a target of T‐cell factor (TCF)/β‐catenin‐mediated transcription [40]. In colorectal carcinogenesis in ApcMin/+ mice, CD44–/–ApcMin/+ mice had fewer intestinal adenomas than CD44+/+ApcMin/+ mice [41]. Likewise, CD44 expression is also associated with a poor prognosis in CRC [42]. Different CD44 isoforms have been identified that are generated by alternative splicing of 10 variant exons and are known as CD44 variant isoforms (CD44v) to differ from CD44 standard isoforms (CD44s) [43]. Interestingly, ApcMin/+ mice expressing only CD44v4‐10 could promote adenoma initiation, but not those expressing CD44s, indicating CD44v as a potential treatment target in CRC [44].
Despite advances in cancer therapy, to date, there are no clinically approved effective drugs targeting CRC‐SCs. Cell signaling pathways participate in CRC survival, proliferation, and self‐renewal properties, and most of them are dysregulated in CRC‐SCs. From this perspective, understanding the participation of the signaling pathways activated in CRC‐SCs is very promising, as their therapeutic targets can contribute to the improvement of anti‐CRC therapy. In this review, the roles of the Wingless (Wnt)/β‐catenin, Notch, Hedgehog, nuclear factor kappa B (NF‐κB), Janus kinase/signal transducer and activator of transcription (JAK/STAT), peroxisome proliferator‐activated receptor (PPAR), phosphatidyl‐inositol‐3‐kinase/Akt/mechanistic target of rapamycin (PI3K/Akt/mTOR), and transforming growth factor‐β (TGF‐β)/Smad pathways in CRC‐SCs were discussed as drug targets to eradicate this cell subpopulation.
2. CELL SIGNALING PATHWAYS
2.1. Wnt signaling pathway
The canonical and non‐canonical Wnt pathways have been associated with the development and progression of cancer. The canonical Wnt pathway acts by controlling the level of β‐catenin available to regulate gene expression (Figure 1). In the absence of Wnt signaling, β‐catenin does not accumulate in the cytoplasm. A multimeric protein complex composed of adenomatous polyposis coli (APC), axin‐1, glycogen synthase kinase‐3 beta (GSK‐3β) and casein kinase 1α (CK1α) is responsible for the degradation of β‐catenin by ubiquitination and digestion, thereby maintaining low levels of free β‐catenin in the cytoplasm and nucleus. On the other hand, in the presence of Wnt ligands, the destruction complex function is interrupted, since Wnt binds to the seven‐pass transmembrane receptor Frizzled (FZD) and single‐pass low‐density lipoprotein receptor‐related protein 5 or 6 (LRP5/6). The Wnt‐FZD‐LRP5/6 trimeric complex recruits dishevelled (DVL), resulting in dissociation of the β‐catenin phosphorylation complex, followed by events that stabilize and accumulate β‐catenin in the cytoplasm with subsequent translocation to the nucleus. This event allows direct binding to TCF/lymphoid enhancer factor (LEF) transcription factors for activation of Wnt‐responsive gene expression and subsequent transcription of several target genes of Wnt, such as AXIN2, SURVIVIN, ALDH, and cyclin D1 (CCND1) [45, 46, 47].
FIGURE 1.

The Wnt/β‐catenin signaling pathway. In the absence of Wnt signaling, β‐catenin is bound to a multimeric protein complex that contains APC, GSK‐3β, axin‐1 and CK1α, leading to proteasomal degradation of β‐catenin. In the presence of Wnt signaling, the destruction of the complex function is interrupted. Wnt binds to LRP5/6 and FZD and inhibits the activity of the multimeric protein complex, which makes β‐catenin enter the nucleus, with subsequent translocation to the nucleus, binds to TCF/LEF to form a complex, and then recruits cofactors to initiate downstream gene expression. Abbreviations: APC, adenomatous polyposis coli; CK1α, casein kinase 1α; DVL, dishevelled; EGCG, epigallocatechin gallate; FZD, seven‐pass transmembrane receptor Frizzled; GSK‐3β, glycogen synthase kinase‐3 beta; LEF; lymphoid enhancer factor; LGR5, leucine‐rich repeat‐containing G‐protein‐coupled receptor 5; LRP5/6, single‐pass low‐density lipoprotein receptor‐related protein 5 or 6; MYC, MYC proto‐oncogene, bHLH transcription factor; RSPO, R‐spondin; TCF, T‐cell factor; TNIK, TRAF2‐ and NCK‐interacting kinase; Wnt, wingless
The canonical Wnt pathway (the Wnt/β‐catenin signaling pathway) is essential for the normal homeostasis of intestinal stem cells, and aberrant activation or mutation is responsible for the establishment of approximately 90% of intestinal cancers through the loss of APC, leading to increased β‐catenin function. These events drive tumor initiation and maintenance of CSCs, regulating the capacity for self‐renewal, interacting with the microenvironment and immune system and promoting tumor invasion and metastasis [36, 48, 49]. Importantly, the CRC‐SC biomarkers leucine‐rich repeat‐containing G‐protein‐coupled receptor 5 (LGR5) and CD44 were identified as genes involved in Wnt signaling [50]. The Wnt pathway is also crucial to support the tumor initiation potential of CRC‐SC precursors that occur in the transition from ulcerative colitis to CRC [51].
Interestingly, Chen et al. [52] investigated the biological function of microRNAs (miRNAs), small non‐coding RNAs that regulate gene expression, and miR‐199a/b in cisplatin‐resistant CRC‐SCs. The expression of miR‐199a/b in CRC tissues was associated with short patient survival. Additionally, the overexpression of miR‐199a/b contributes to cisplatin resistance, upregulating ATP binding cassette subfamily G member 2 (ABCG2), an important multidrug resistance pump located downstream of the Wnt/β‐catenin pathway. Blocking the Wnt/β‐catenin pathway decreases ABCG2 levels in ALDH1+ CRC‐SCs. In addition, miR‐92a promotes the properties of CRC‐SCs, increases the expression levels of stem cell markers CD133, SOX2, and OCT4, and is upregulated by Wnt/β‐catenin signaling activity via downregulation of Krüppel‐like factor 4 (KLF4), GSK‐3β, and dickkopf Wnt signaling pathway inhibitor 3 (DKK3), negative regulators of Wnt/β‐catenin signaling [53].
The decreased expression of miR‐148a, a tumor suppressor, causes increased expression of CRC‐SC markers through the Wnt/β‐catenin signaling pathway, while its hyperexpression increases cisplatin chemosensitivity, reducing cell invasion and migration [54]. Additionally, miR‐30‐5p expression is normally reduced in CRC cells resistant to 5‐FU and in CD133+ CRC cells. In contrast, its overexpression significantly reduces cell viability and the expression of stem cell markers CD133 and SOX2, since miR‐30‐5p attenuates the expression of AXIN2 and MYC, target genes of Wnt/β‐catenin signaling [55]. Moreover, the 5‐FU‐resistant CRC HT‐29 cell line showed increased Wnt signaling and stemness properties [56].
Therefore, many aspects of how this pathway controls the growth and proliferation of normal cells and CSCs have been studied [51, 57, 58, 59]. Moreover, new studies focusing on the anti‐cancer properties of molecules that eradicate CRC‐SCs via the Wnt pathway have been carried out and are described below.
2.1.1. Natural products
2.1.1.1. Phenethyl isothiocyanate
Phenethyl isothiocyanate (PEITC) is a natural product present in many cruciferous vegetables with anti‐cancer and chemopreventive activities in clinical trials and has been associated with the induction of oxidative stress [60]. Interestingly, PEITC has also been reported to be able to eliminate CSCs of different types of cancer, including cervical [61, 62], lung [63], ovarian [64], and breast cancers [64, 65]. Importantly, PEITC inhibited the properties of CRC‐SCs through downregulation of the Wnt/β‐catenin pathway, inhibition of proliferation, and induction of apoptosis in CRC DLD‐1 and SW480 cell lines. PEITC decreased the percentage of CD133+ cells by approximately 3–16‐fold [66]. Activation of the Wnt/β‐catenin pathway with lithium chloride, an agonist of canonical Wnt signaling, elevated CD133 expression and suppressed the effects of PEITC on CRC‐SCs [66]. PEITC also suppressed the expression of pluripotency factors, including OCT4, SOX2, and NANOG, in NCCIT cells, a pluripotent stem cell line from mediastinal mixed germ cell tumors [67, 68]. In vitro self‐renewal capacity and clonogenicity and in vivo tumor growth and expression of pluripotency factors were also observed to be reduced after treatment with PEITC in an EpCAM‐expressing CSC model derived from the CRC HCT116 cell line [67, 68]. These results support that PEITC is a candidate to eradicate CRC‐SCs targeting Wnt signaling.
2.1.1.2. Sulforaphane
Sulforaphane is an isothiocyanate obtained from cruciferous vegetables with anti‐cancer properties. Some studies indicate that it can act on CSCs [69, 70, 71, 72]. Interestingly, sulforaphane reduced pancreatic CSCs by blocking the sonic hedgehog‐GLI family zinc finger (GLI) [70] and NF‐κB pathways [71] and enhanced imatinib against CSCs of chronic myelogenous leukemia by inhibiting Wnt/β‐catenin function [72]. In CRC, sulforaphane caused cell cycle arrest and apoptosis in CRC HT‐29 and Caco‐2 cell lines [73, 74]. Recently, Chen et al. [75] demonstrated that sulforaphane inhibited CRC‐SC properties by suppressing the transactivating p63 isoform α (TAp63α)/LGR5/β‐catenin axis in vitro and in vivo. In addition, activation of the Wnt/β‐catenin pathway leads to the expression of LGR5, which is also a promoter of the Wnt/β‐catenin pathway when binding to R‐spondin (RSPO), suggesting that LGR5 may be used not only as a marker but also as a target in CRC‐SCs. LGR5 acts through TAp63α to enhance Wnt signaling and the activation of β‐catenin targets, promoting the maintenance of stem cells. Sulforaphane treatment led to downregulation of CRC‐SC markers (CD133, CD44, NANOG, and OCT4), suppressing the formation of colonies in a spheroid cell model with CRC HCT116 and SW480 cell lines. Moreover, the upregulation of TAp63α blocked the inhibitory effect of sulforaphane. These data indicated that sulforaphane is a useful drug to eliminate CRC‐SCs by suppressing Wnt signaling.
2.1.1.3. Salinomycin
Salinomycin, an anti‐bacterial polyether isolated from Streptomyces albus, selectively eliminated CD133+ cells in CRC [76]. Salinomycin induced caspase activation, increased DNA damage and caused disruption of the Wnt/β‐catenin/TCF complex and apoptosis of human CRC‐SCs, and decreased tumor growth and expression of CSC‐related Wnt genes, including LGR5 [77, 78]. Treatment with salinomycin alone or in combination with the FOLFOX regimen (5‐FU, folic acid plus oxaliplatin) was also evaluated in primary CSCs of patients with hepatic metastases of CRC or primary CRC [79]. Salinomycin exhibited superior anti‐CSC activity and tumor growth inhibition when compared to the FOLFOX protocol, and salinomycin plus FOLFOX resulted in improvement of these effects when compared to salinomycin alone. Salinomycin increased the expression of LGR5 and mitigated the expression of ALDH1. Furthermore, salinomycin also caused apoptosis related to decreased cellular ATP production and the accumulation of dysfunctional mitochondria and reactive oxygen species [79]. Likewise, Singh et al. [80] demonstrated that salinomycin improved cell death receptors in CRC‐SCs by inhibiting enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2), a histone‐lysine N‐methyltransferase enzyme. Importantly, salinomycin has also been reported to target CSCs of various types of cancer, including melanoma [81], osteosarcoma [82], breast [83], liver [84], brain [85], ovarian [86], and oral cancers [87]. Salinomycin specifically eliminated epithelial CSCs [88].
Wang et al. [89] developed salinomycin nanocrystals to improve their oral bioavailability/solubility and reduce toxicity. Salinomycin nanocrystals exhibited greater cell uptake efficiency, cytotoxicity and Wnt‐inhibiting effects three times better than regular salinomycin, indicating that salinomycin nanocrystals have great potential for the clinical treatment of CRC. The combination of SN38, the active form of irinotecan, with salinomycin in nanoformulations also diminished the adverse effects and enhanced the treatment efficacy against CRC‐SCs [90]. These results suggest salinomycin as a drug capable of eliminating CRC‐SCs by targeting Wnt signaling and improving conventional chemotherapeutic effects, including the FOLFOX protocol.
2.1.1.4. Epigallocatechin gallate
Epigallocatechin gallate (EGCG), also known as epigallocatechin‐3‐gallate, is one of the catechins found in green tea. It has been widely studied as an anti‐CSC agent against different types of cancer, including lung [91], bladder [92], pancreas [93], and breast cancers [94]. In combination with cisplatin or oxaliplatin, EGCG increased the therapeutic effect on human CRC cells [95]. Furthermore, EGCG was also effective in suppressing CRC‐SCs by downregulating CD133, CD44, NANOG, OCT4, ALDH1, and Notch1 expression, inhibiting the ability to form spheres of CRC cells, and upregulating the expression of GSK‐3β, a negative regulator of Wnt/β‐catenin [96, 97]. In addition, EGCG induced the sensitivity of CRC‐SCs to 5‐FU [97].
In a pilot study with 125 patients, green tea extract reduced the recurrence of colorectal adenoma in approximately 50% of patients [98]. Moreover, in a randomized controlled clinical trial, 10 volunteers received 800 mg EGCG, and 11 volunteers received the same amount of EGCG plus Brazil nuts for 6 weeks. DNA methyltransferase (DNMT1) (DNA methylation is a molecular marker in CRC) and NF‐κB expression were decreased in rectal biopsies of both groups [99]. In a clinical trial with 223 healthy Japanese individuals, Escherichia coli containing polyketide synthase (tumorigenic bacteria for CRC) in the gut microbiota was downregulated by ingestion of green tea [100].
Taken together, these preclinical results support that EGCG reduces the population of CRC‐SCs and enhances the effect of cisplatin and oxaliplatin. Furthermore, clinical data suggest EGCG and/or green tea extract as a beneficial strategy in CRC therapy/prevention.
2.1.1.5. Farnesyl dimethyl chromanol
Farnesyl dimethyl chromanol (FDMC), a chemical structure that is part of the tocopherol and tocotrienol molecules (vitamin E), exhibits antioxidant properties [101]. Recently, Husain et al. [102] demonstrated that FDMC decreased the viability and self‐renewal of CRC‐SCs by downregulating Wnt/β‐catenin signaling in vitro and in vivo. FDMC reduced spheroid and organoid formation and downregulated the expression of the pluripotent transcription factors NANOG, OCT4 and SOX2 in CD24+/CD44+/LGR5+ CRC cells. Induction of DNA fragmentation, exposure to phosphatidylserine, cleaved caspase 3 and cleaved PARP and reduced migration, invasion, VEGF and NF‐κB and Wnt signaling were also observed in CRC‐SCs treated with FDMC in vitro. In an orthotopic xenograft metastasis model, FDMC inhibited tumor growth and liver metastasis and suppressed angiogenesis and NF‐κB and β‐catenin signaling. This indicates that FDMC is effective in eradicating CRC‐SCs.
2.1.1.6. 4‐Acetyl‐antroquinonol B
4‐Acetyl‐antroquinonol B (4‐AAQB), a mycelial from Antrodia camphorate, has been reported as a cytotoxic agent [103, 104]. In CRC, 4‐AAQB reduced migration, invasion and clonogenicity in CRC DLD1 and HCT116 cell lines, with less effect on the viability and proliferation of normal colon cells. Downregulation of N‐cadherin, vimentin, MYC and BcL‐xL and upregulation of E‐cadherin and BAX proteins were found in 4‐AAQB‐treated CRC cells. 4‐AAQB also reduced the superoxide dismutase 2 (SOD2)‐mediated CSC phenotype by hsa‐miR‐324. In vivo, 4‐AAQB reduced tumor growth and enhanced the FOLFOX protocol by inhibiting SOD2 and increasing hsa‐miR‐324‐5p expression [103]. In addition, 4‐AAQB also downregulated the LGR5/Wnt/β‐catenin, JAK/STAT, and non‐transmembrane receptor tyrosine kinase signaling pathways and stemness‐related factors in CRC cell lines. A reduction in tumor size was also observed after 4‐AAQB treatment in CRC animal models [104]. These results suggest that 4‐AAQB is useful to reduce the stemness properties of CRC cells and may improve the effect of conventional chemotherapeutics such as the FOLFOX protocol.
2.1.1.7. Diallyl trisulfide
Diallyl trisulfide (DATS) is a garlic‐derived organosulfur compound with anti‐tumor properties. Zhang et al. [105] reported that DATS reduced the size and number of spheroid forms of the CRC SW480 and DLD‐1 cell lines and decreased the expression of CD133, CD44, ALDH1, OCT‐4 and NANOG. The characteristics of cell death by apoptosis were found to be increased, including decreased Bcl‐2 levels and increased BAX, caspase‐3, ‐8 and ‐9. This effect was associated with the suppression of the activity of the Wnt/β‐catenin pathway, observed by the increased expression of GSK‐3β and decreased expression of β‐catenin, MYC and cyclin D1. Pretreatment of the culture with lithium chloride blocked the effects of DATS. These results indicate that DATS can inhibit proliferation and induce apoptosis of CRC‐SCs by suppressing Wnt signaling.
2.1.1.8. Zerumbone
Zerumbone, a sesquiterpene from Zingiber zerumbone, has anti‐tumor effects in various types of cancer. In the CRC HCT‐116 and SW‐48 cell lines, zerumbone suppressed epithelial‐mesenchymal transition (EMT), as observed by the increased expression of E‐cadherin and reduced expression of vimentin, ZEB1 and N‐cadherin [106]. A reduction in cell invasion, migration and spheroid formation was also found, as well as in the expression of CSC markers, including CD133, CD44, BMI1 and ALDH1. Zerumbon also reduced β‐catenin expression. Interestingly, silencing of miR‐200c inhibited these zerumbone effects, indicating that the inhibition of the β‐catenin pathway through miR‐200c may be a target of the anti‐CRC‐SC properties of zerumbone [106].
2.1.2. Small synthetic molecules
2.1.2.1. Retinoids
Homeobox A5 (HOXA5), a regulator of gene expression, morphogenesis, and differentiation, is downregulated in CRC. It inhibits Wnt signaling to reinforce differentiation through antagonists of APC downregulated 1 (APCDD1), CXXC finger protein 4 (CXXC4), and NKD inhibitor of Wnt signaling pathway 1 (NKD1) pathways [58]. All‐trans retinoic acid and 9‐cis retinoic acid upregulated HOXA5 levels, leading to tumor regression in the ALDH1+ HT‐29 cell line. In this context, tumor regression via HOXA5 induction by retinoids suppressed Wnt signaling, leading to the differentiation and elimination of CRC‐SCs, preventing tumor development, and inhibiting metastases [58]. All‐trans retinoic acid also eliminated CSCs of head and neck cancer by inhibiting the Wnt/β‐catenin pathway [107].
2.1.2.2. JIB‐04
JIB‐04 is an epigenetic modulator that acts as a pan‐selective inhibitor of histone demethylases [108]. JIB‐04 decreased the viability of HCT116 cells and reduced CD133 mRNA expression, CD133 protein expression levels, and clonogenic potential, indicating that JIB‐04 resulted in a reduction in the CRC‐SC population and self‐renewal to form colonies [109]. Beyond that, JIB‐04 mitigated invasion, migration, and growth/recurrence in an in vitro model. The expression level of E‐cadherin mRNA (an epithelial marker) was increased, while those of vimentin and N‐cadherin (mesenchymal markers) were decreased in cells treated with JIB‐04. It also reduced the tumorigenic activity of CRC‐SCs in vivo. These data were associated with increases in the expression of AXIN1 (axin‐1) and GSK3β, genes involved in the inhibition of β‐catenin signaling, and decreases in the expression of β‐catenin target genes, including ALDH1 and SRY‐box transcription factor 4 (SOX4) [109]. Importantly, JIB‐04 had less effect on the viability of normal cells [110]. These results suggest that JIB‐04 can be used to specifically target CRC‐SCs.
2.1.2.3. IC‐2
IC‐2, a derivative of ICG‐001 (a Wnt modulator), efficiently represses Wnt/β‐catenin signaling [111]. Interestingly, IC‐2 reduced the transcriptional activity of Wnt/β‐catenin in CRC cells more effectively than 5‐FU. IC‐2 reduced the expression levels of CSC marker proteins, such as CD44, CD133, OCT3/4, NANOG, and LGR5. Then, CD44high and CD44low DLD‐1 cells were isolated by cell sorting, and CD44high sphere numbers were selectively reduced by IC‐2, indicating that IC‐2 preferentially suppresses CRC‐SCs. Nevertheless, IC‐2 did not affect viability at concentrations below 10 μmol/L. IC‐2 also improved the cytotoxic effect of 5‐FU, suggesting that IC‐2 sensitizes CRC cells to 5‐FU through the suppression of CRC‐SCs [111]. Interestingly, IC‐2 has also been reported as an anti‐CSC agent against liver [112] and oral cancers [113].
2.1.2.4. NCB‐0846
NCB‐0846 is the first TRAF2‐ and NCK‐interacting kinase (TNIK) inhibitor available orally with high activity against Wnt signaling by binding to TNIK, an essential activator of Wnt target genes. The anti‐tumor and anti‐CRC‐SC activities of NCB‐0846 were demonstrated [57, 114]. Pharmaceutical companies have developed other TNIK inhibitors that are in clinical trials, including ON108600 (OnconovaTherapeutics, Newtown, PA, USA), which was efficient in acting on CSCs [115]. Mebendazole, an anthelmintic agent, has also been identified as a selective TNIK inhibitor [116]. The combination of mebendazole and sulindac, a non‐steroidal anti‐inflammatory agent, reduced tumor initiation in CRC in a preclinical model [117].
2.1.2.5. 36‐077
36‐077, an inhibitor of phosphatidylinositol 3‐kinase catalytic subunit type 3 (PIK3C3), was synthetized by Kumar et al. [118] as a potent autophagy inhibitor. In particular, 36‐077 improved the efficacy of 5‐FU in CRC Caco‐2 and HCT116 cell lines grown in monolayers. Treatment of Caco‐2 cells growing in spheroids or colon tumoroids derived from ApcMin/+ mice with 5‐FU plus 36‐077 also reduced spheroid growth more than 5‐FU or 36‐077 alone. This effect was associated with the inhibition of the CSC population through GSK‐3β/Wnt/β‐catenin signaling. These data indicate that 36‐077 can inhibit CRC‐SCs by suppressing Wnt/β‐catenin signaling and enhances 5‐FU treatment.
2.1.2.6. CBB1003
CBB1003, a lysine‐specific demethylase 1 (LSD1) inhibitor, has anti‐tumor properties. Treatment with CBB1003 in HCT116 cells inhibited proliferation and colony formation and decreased the levels of the CSC marker LGR5 [119]. Moreover, the overexpression of LGR5 reduced cell death caused by CBB1003. Inactivation of β‐catenin/TCF signaling was observed after treatment with CBB1003. The expression of LSD1, LGR5, β‐catenin and MYC was found to be lower in adjacent normal tissues than in CRC tissues as well as in CRC cell lines [119]. These data indicate LSD1 as a target to affect CRC‐SCs, inhibiting Wnt signaling. CBB1003 may be a useful anti‐CRC‐SC drug, acting by inhibiting LSD1.
2.1.2.7. Rimonabant
Rimonabant is an antagonist/inverse agonist of cannabinoid receptor 1 that can induce β‐catenin degradation in CRC cell lines. The target genes Wnt/β‐catenin, cyclin D1, MYC and cyclooxygenase‐2 were reduced after treatment with rimonabant [120]. Non‐canonical Wnt signaling was activated in CRC cells treated with rimonabant, as observed by the induction of Wnt5A and activation of calcium/calmodulin‐dependent protein kinase II. Rimonabant also inhibited tumor growth and β‐catenin transfer to cell nuclei in mice xenografted with HCT116 cells [120]. Fiore et al. [121] reported that rimonabant reduced primary CRC‐SCs in culture through downregulation of β‐catenin without affecting healthy colon epithelial cells. These results suggest rimonabant as an effective drug to eliminate CRC‐SCs.
2.1.2.8. FH535
FH535 is a dual inhibitor of β‐catenin/TCF/LEF and PPARs with anti‐tumor action [122]. It reduced the migration and invasion of SW480 cells and downregulated the expression of matrix metalloproteinases 7 and 9, vimentin, and snail. FH535 also inhibited the expression of the CSC markers CD24, CD44 and CD133 in HT29 cells by inhibiting the Wnt/β‐catenin pathway. FH535 also reduced tumor growth in mice xenografted with HT‐29 [123]. This suggests that FH535 is able to eradicate CRC‐SCs by targeting Wnt signaling.
2.1.3. Monoclonal antibody
2.1.3.1. HZ8CV2
Progastrin is overexpressed and detectable in the supernatant of CRC cell lines and patient‐derived CRC cells and stimulates the survival of CSCs [124]. Downregulation of progastrin decreases Wnt/β‐catenin activity and Wnt‐induced tumorigenesis [125]. Prieur et al. [126] created a humanized anti‐progastrin monoclonal antibody (HZ8CV2) that was effective against CRC with a KRAS mutation. Treatment of SW480 cells with HZ8CV2 for 96 h decreased the expression of survivin, a key component of the Wnt/β‐catenin signaling pathway, by 40.7%. Beyond that, combination chemotherapy with the HZ8CV2 antibody increased chemosensitivity and delayed recurrence. HZ8CV2 in combination with 5‐FU decreased 57.9% of the CSC population in KRAS‐mutated T84 cells. In the xenograft model, the combination of HZ8CV2 with irinotecan decreased the frequency of CSCs by 55.9% and 70.1% in T84 and SW620 tumors, respectively, inhibiting the migratory and invasive properties of these CRC cells. These data corroborate that progastrin is a target to eliminate CRC‐SCs and that HZ8CV2 is an effective anti‐progastrin monoclonal antibody that inhibits CRC‐SCs and improves 5‐FU and irinotecan treatments.
These data indicate that the Wnt/β‐catenin signaling pathway is a critical pathway to eradicate CRC‐SCs. On the other hand, crosstalk among pathways can lead to resistance to single‐pathway inhibitors and maintenance of the CSC phenotype [127, 128]. Therefore, multiple cellular pathways must be targeted to effectively eradicate CRC‐SCs.
2.2. Notch signaling pathway
Notch signaling is involved in the regulation of cell differentiation, proliferation, and tumorigenesis [129, 130]. Notch is a transmembrane receptor activated by series cleavage (Figure 2), and four different Notch receptors (Notch1, Notch2, Notch3, and Notch4) are known. While the non‐canonical pathway works independently of Notch receptors, the interaction between the ligands of the delta family (DLL1, DLL2, DLL3 and DLL4) or jagged‐4 family (JAG1/JAG2) and the receptors occurs in canonical pathway activation. Canonical Notch single‐strand precursors are cleaved by furin proteases in the Golgi complex to form a large fragment containing the extracellular domain and a small fragment containing transmembrane and intracellular regions, which combine to form a mature receptor. The results of this interaction expose the extracellular metalloprotease site that becomes susceptible to cleavage by a disintegrin and metallopeptidase/tumor necrosis factor‐alpha converting enzyme (ADAM/TACE) transmembrane family to release the extracellular fragment. The residual transmembrane Notch fragment acts as a substrate for proteolysis by the γ‐secretase complex, resulting in the release of the intracellular domain of the receptor that translocates to the nucleus and then binds to the CBF1 suppressor of hairless LAG1 (CSL) transcription factor and activates the expression of Notch target genes [129, 130, 131].
FIGURE 2.
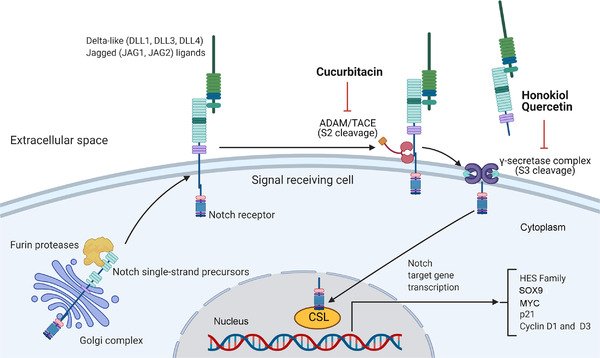
The Notch signaling pathway. The Notch single‐stranded pathways undergo proteolytic processing in the Golgi complex, which is furin protease‐mediated (S1 cleavage). The receptor is transported to the cell surface membrane. The extracellular domain of the Notch receptor in the signaling cell binds to the Notch ligands (Delta and Jagged) expressed by the adjacent cell. This induces the second proteolytic step by ADAM/TACE metalloproteases and γ‐secretase, which releases the intracellular Notch domain that translocates to the nucleus and then binds to the CSL transcription factor and activates the expression of Notch target genes. Abbreviations: ADAM, a disintegrin and metallopeptidase; CSL, CBF1 suppressor of hairless LAG1; MYC, MYC proto‐oncogene, bHLH transcription factor; SOX9, SRY‐box transcription factor 9; TACE, tumor necrosis factor‐alpha converting enzyme
The activation of Notch signaling was associated with short survival, CSC phenotype, and EMT, resulting in tumor progression in CRC [132]. Huang et al. [133] demonstrated that the CRC cell line HCT116 was resistant to 5‐FU or oxaliplatin and that parental HCT116 cells cultured in spheroids showed a greater number of CD133+ and CD44+ cells than parental HCT116 cells cultivated in monolayers. These effects were associated with enhanced Notch signaling. Moreover, inhibition of the Notch pathway reduced the growth of HCT116 cells resistant to 5‐FU or oxaliplatin and parental HCT116 cells cultured in spheroids in in vitro and in vivo models. In this context, several compounds that act on CRC‐SCs by inhibiting the Notch signaling pathway have been studied and point to promising strategies for the treatment of CRC.
2.2.1. Natural products
2.2.1.1. Honokiol
Honokiol, a naturally active biphenolic compound from Magnolia species used in traditional Chinese medicine, has anti‐tumor properties against various types of cancer [134]. Honokiol is a potent inhibitor of melanoma stem cells that suppresses Notch signaling [135]. It also inhibits tumor progression and stem cells in glioma [136], breast cancer [137], and oral carcinoma cells [138]. In CRC, the combination of honokiol with ionizing radiation (IR) inhibited the levels of expression of CSC proteins, such as doublecortin‐like kinase‐1 (DCLK1), SRY‐box transcription factor 9 (SOX9), CD133, CD44, Notch signaling‐related proteins, and proteins of the γ‐secretase complex in vitro, using HCT116 and SW480 cell lines, and in xenograft tissues using the HCT116 cell line [139]. Honokiol also slowed the growth of the tumor xenograft, inhibited proliferation, and promoted apoptosis. Furthermore, ectopic expression of the Notch intracellular domain partially rescues the honokiol effect, indicating that this compound is an anti‐CRC‐SC agent targeting Notch signaling [139].
Honokiol also suppressed the Hippo signaling pathway in CRC cells [140]. In this pathway, YAP1 is translocated to the nucleus to induce gene expression, which is inhibited by phosphorylation at Ser127. Honokiol inhibited YAP1 phosphorylation and caused apoptosis in HCT116 and SW480 cells. Inhibition of the expression of the CSC marker DCLK1 was also observed. In addition, honokiol prevented colitis‐associated cancer growth in a mouse model using azoxymethane and dextran sulfate sodium [140].
2.2.1.2. Cucurbitacins
Cucurbitacin‐B (C‐B) and cucurbitacin‐I (C‐I) are phytochemicals present in various fruits and vegetables, such as bitter melon [141]. C‐B and C‐I inhibited cell proliferation and induced cell cycle arrest and apoptosis in CRC HCT116 and SW480 cell lines by downregulating ADAM9, which participates in the cleavage of the Notch receptor at the extracellular domain [142]. In addition, C‐B and C‐I reduced the spheroid and colony formation of CRC cells and inhibited the expression of the CRC‐SC markers CD44, DCLK1, and LGR5. C‐B and C‐I also reduced tumor growth in mice xenografted with HCT116, as such reduced the expression of CSC markers and Notch signaling components in tumor tissues [142]. Together, these data suggest that cucurbitacins inhibit CRC, including CRC‐SCs, by downregulating the Notch signaling pathway. Additionally, C‐B was also able to eliminate liver [143] and gastric CSCs [144]. C‐I also eliminated CSCs of the lung [145], brain [146], and head and neck [147] by blocking the JAK2/STAT3 signaling pathway.
2.2.1.3. Evodiamine
Evodiamine, a derivative of the traditional herbal medicine Evodia rutaecarpa, has an antiproliferative effect in the CRC LoVo cell line, leading to the induction of caspase‐dependent apoptosis and S‐phase arrest [148, 149]. The surviving cells after treatment with evodiamine were plated using serial dilutions and under conditions for the enrichment of CSCs by spheroids or implanted subcutaneously in immunodeficient SCID mice [150]. The results showed that evodiamine decreased the self‐renewal of surviving cells in vitro and in vivo. Beyond that, the expression of the genes related to Wnt/β‐catenin and Notch signaling was suppressed by treatment with evodiamine, indicating the elimination of CRC‐SCs [150]. This indicates that evodiamine is an effective drug to eradicate CRC‐SCs, suppressing Wnt and Notch signaling.
2.2.1.4. Quercetin
Quercetin, a natural flavonoid, is a compound that enhances radiosensitivity [151]. Quercetin plus IR eliminated CRC‐SCs by inhibiting Notch1 signaling. Reductions in CSC markers (DCLK1, CD24, LGR5, CD29, CD44, and CD133) were observed in in vitro and in vivo studies. Quercetin plus IR suppressed the growth of both primary and secondary colonospheres and inhibited the Notch signaling pathway by downregulating γ‐secretase complex proteins, Jagged‐1, and Notch target genes hairy and enhancer of split‐1 (HES1) and Hes‐related with YRPW motif‐1 (HEY1)[151]. This indicates that quercetin can induce radiosensitivity and eliminate CRC‐SCs.
2.2.1.5. Portulaca oleracea extract
Portulaca oleracea is a grassy plant with succulent leaves and presents anti‐tumor activities [152]. Portulaca oleracea extract inhibited the proliferation of CSCs (CD133+ and CD44+) by inducing apoptosis and downregulating the expression of Notch1 and β‐catenin in CRC‐SCs [153]. This indicates that Portulaca oleracea extract is a source of anti‐CRC‐SC agents.
2.2.1.6. α‐Mangostine
α‐Mangostine is a natural xanthone from Garcinia mangostana. It has antioxidant, anti‐microbial, anti‐cancer and anti‐inflammatory properties [154]. α‐Mangostine encapsulated in poly(D,L‐lactic‐coglycolic acid) (PLGA) nanoparticles inhibited self‐renewal capacity, the expression of CSC markers, including CD133, CD44, Musashi, and LGR5, and pluripotency maintenance factors, such as OCT4, SOX2, KLF4, MYC, and NANOG. These effects were associated with inhibition of Notch signaling, suppression of the expression of Notch receptors (Notch1 and Notch2), their ligands (Jagged 1 and DLL4), protein of the γ‐secretase complex (Nicastrin), and downstream target (Hes‐1) in CRC HT‐29 and HCT116 cell lines [155]. These results indicate α‐mangostine as a drug with anti‐CRC‐SC properties.
2.2.1.7. Pien Tze Huang formula
Pien Tze Huang is a traditional Chinese herbal formula with anti‐tumor properties. It inhibited CRC‐CSs in the SW480 cell line [156]. In addition, Pien Tze Huang formula also decreased Notch1 and Hes1 expression in CRC‐SCs [156]. This indicates that Pien Tze Huang formula is able to eliminate CRC‐SCs by suppressing the Notch signaling pathway.
2.2.2. Monoclonal Antibodies
2.2.2.1. Anti‐DLL4 antibodies
Anti‐human and anti‐mouse DLL4 antibodies have been developed as anti‐tumor agents [157]. Moreover, anti‐DLL4 antibodies were effective against both KRAS wild‐type and mutant CRC cells. In combination with irinotecan, anti‐DLL4 antibodies also increased CRC growth inhibition in a patient‐derived xenograft model. They were also found to reduce the CRC‐SC population [158]. These data indicate that these antibodies against the Notch DDL4 ligand are effective in eradicating CRC‐SCs and in potentiating the effects of irinotecan. Notch signaling can interact and influence a large number of cancer‐relevant pathways, including Wnt. Furthermore, crosstalk between the Wnt and Hedgehog pathways can also determine the general effect of Notch signaling [159, 160]. In this context, all cross‐signaling networks in CSCs must be considered in a context‐dependent manner. Clinical trials with Notch pathway inhibitors are still limited in the treatment of CRC. As observed above, these targeted agents have already been shown to be effective in preclinical studies and should also be tested in combination with classical chemotherapy drugs or other inhibitors of the signaling pathway in clinical trials to better understand their role in CRC therapy.
2.3. Hedgehog signaling pathway
The Hedgehog (HH) signaling pathway is related to embryonic development, self‐renewal, migration, and stem cell population maintenance. Hyperactivation of this pathway is associated with CSCs, contributing to tumorigenesis and tumor progression. Conserved HH transmits the signal from the cell membrane to the nucleus [161, 162]. Abnormal dysfunction or activation of HH signaling has been implicated in the pathogenesis of several cancers, including CRC [163].
HH signaling can be classified as canonical or non‐canonical. In canonical HH signaling, HH ligands (sonic HH, Indian HH, or desert HH) bind to the patched (PTCH) transmembrane receptor, which leads to the release of smoothened (SMO) and consequent activation of a downstream cascade [161, 164]. This event culminates in the activation of the GLI family of transcription factors (GLI1, GLI2, and GLI3), which is responsible for the regulation of several target genes. In the absence of ligands, the PTCH receptor inhibits SMO, deactivating the pathway (Figure 3) [161, 164]. Non‐canonical HH signaling refers to all cellular and tissue responses to any of the HH isoforms that are independent of transcriptional changes mediated by GLI activation [165].
FIGURE 3.
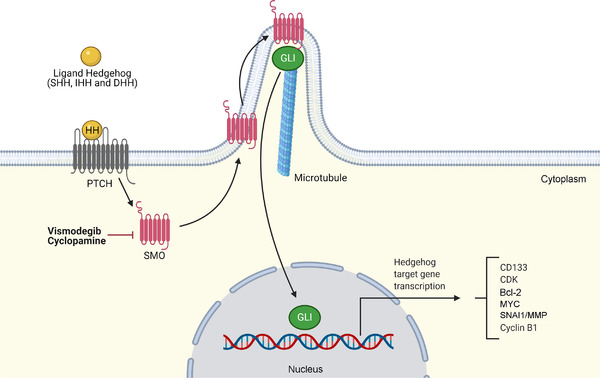
The Hedgehog signaling pathway. In canonical HH signaling, HH ligands (SHH, IHH or DHH) bind to the PTCH transmembrane receptor, which relieves the inhibition of the transmembrane protein SMO and induces the GLI family of transcription factors (GLI1, GLI2, and GLI3) to enter the nucleus to regulate downstream gene transcription. Abbreviations: CDK, cyclin‐dependent kinase; DHH, desert hedgehog; HH, hedgehog; IHH, Indian hedgehog; MMP, matrix metalloproteinase; MYC, MYC proto‐oncogene, bHLH transcription factor; PTCH, patched; SHH, sonic hedgehog; SMO, smoothened; SNAI1, snail family transcriptional repressor 1
The crosstalk between Wnt and HH signaling has been shown to be important in the development and progression of CRC. Some studies report that GLI‐dependent canonical HH signaling is a negative regulator of Wnt signaling in normal intestine and intestinal cancers [166, 167]. On the other hand, non‐canonical HH signaling in CSCs acts as a positive regulator of Wnt signaling to regulate the survival of CRC‐SCs [168]. HH signaling in CRC‐SCs is autocrine and sonic Hedgehog‐ and PTCH1‐dependent [168].
In primary cultures and CRC cell lines, HH signaling is critical for in vitro cell proliferation and survival, since CRC‐SCs require active HH signaling for self‐renewal and expansion in advanced cancers [169]. HH signaling is also necessary for the growth of CRC xenografts, recurrence, and induction of EMT necessary for metastases [169]. In contrast, arsenic trioxide, an anti‐leukemic drug known to inhibit the HH signaling pathway by targeting GLI1 [170], increased CRC‐SCs from patient samples [171].
2.3.1. Natural product
2.3.1.1. Cyclopamine
Cyclopamine, a naturally occurring alkaloid, is a classic inhibitor of the HH signaling pathway through the inhibition of SMO [172]. HCT‐116 cells grown in serum‐free non‐adherent spheroids were treated with cyclopamine and showed reduced mRNA levels of CSC markers and genes related to HH signaling, including PTCH1, SMO, and GLI1 [173].
Zhou et al. [174], using bioinformatics analysis, selected a series of non‐coding RNAs related to stemness in CRC‐SCs and identified long non‐coding RNAs (lncRNA‐cCSC1) that are highly expressed in CRC and CRC‐SCs and indicated a poor prognosis. After characterization, they observed that the depletion of lncRNA‐cCSC1 levels was proportional to the increased sensitivity of CRC cells to 5‐FU and to the inhibition of CRC‐SCs’ self‐renewal capacity via the HH pathway. The overexpression of lncRNA‐cCSC1 increased the protein levels of factors related to stemness, such as CD133, CD44, and NANOG, and effector proteins of the HH signaling pathway, SMO and GLI1. In contrast, the use of cyclopamine caused a reduction in the levels of biomarkers related to stemness. These data indicate the potential action of the anti‐SMO drug cyclopamine in eliminating CRC‐SCs.
2.3.2. Small synthetic molecule
2.3.2.1. Vismodegib
Vismodegib (also known as GDC‐0449) is a potent, orally bioavailable, specific SMO receptor antagonist. It is used to treat basal cell skin cancer in clinical practice approved by the United States‐Food and Drug Administration [175]. Although still controversial, vismodegib has been reported as a drug able to inhibit CRC‐SCs, since Wu et al. [176] showed that vismodegib inhibited cell proliferation and triggered apoptotic cell death with downregulation of Bcl‐2 in CRC cells. In addition, it reduced the expression of the CRC‐SC markers CD44 and ALDH by approximately 70%.
On the other hand, Chen et al. [177] demonstrated that vismodegib did not activate apoptosis in human CRC cells. Moreover, in a clinical trial of 199 patients with CRC treated with the FOLFOX or FOLFIRI (5‐FU, leucovorin plus irinotecan) protocol combined with vismodegib, no benefit associated with the combination was observed [178].
Although some negative results have been found with HH signaling inhibitors, new inhibitors must be evaluated in CRC‐SCs, especially compounds capable of inhibiting non‐canonical HH signaling. These new data can help understand the potential of HH signaling inhibitors in CRC therapy.
2.4. NF‐κB signaling pathway
NF‐κB is a transcription factor that plays a crucial regulatory role in the innate immune system and in the inflammatory response. NF‐κB signaling occurs canonically or non‐canonically. In the canonical pathway, an inhibitor of NF‐κB (IκB) prevents the translocation of the p65/p50 and c‐Rel/p50 dimers to the cell nucleus [179, 180]. The ubiquitination of IκB and its subsequent degradation release these proteins to be translocated to the cell nucleus, leading to the activation of the target genes. In the non‐canonical pathway, NF‐κB‐inducing kinase (NIK) induces the ubiquitination of p100 and its subsequent processing by proteasomes in p52. Then, RelB/p52 is translocated to the cell nucleus to activate the target genes (Figure 4) [179, 180].
FIGURE 4.
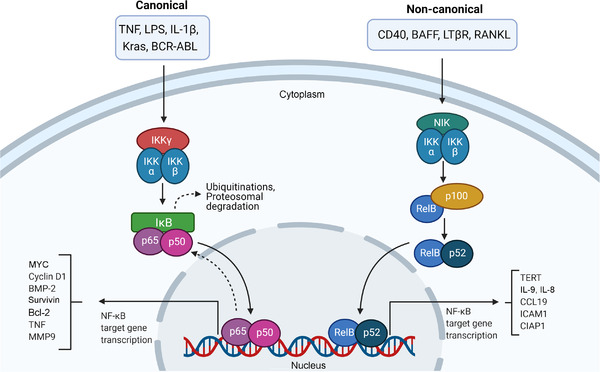
The NF‐κB signaling pathway. In the canonical pathway, the inhibitory protein IκB inhibits the translocation of the p65/p50 and c‐Rel/p50 dimers to the cell nucleus. The ubiquitination of IκB and its subsequent degradation release these proteins to be translocated to the cell nucleus, leading to the activation of the target genes. In the non‐canonical pathway, NIK induces the ubiquitination of p100 and its subsequent processing by proteasomes in p52. Then, RelB/p52 is translocated to the cell nucleus to activate the target genes. Abbreviations: ABL, ABL proto‐oncogene 1, non‐receptor tyrosine kinase; BAFF, B cell‐activating factor; BCR, BCR activator of RhoGEF and GTPase; BMP‐2, bone morphogenetic protein‐2; CCL19, C‐C motif chemokine ligand 19; CIAP1, cellular inhibitor of apoptosis protein 1; ICAM1, intercellular adhesion molecule 1; IKK, inhibitor of NF‐κB kinase; IκB, inhibitor of NF‐κB; Kras, Kirsten rat sarcoma viral oncogene homolog; LPS, lipopolysaccharide; LTβR, lymphotoxin beta receptor; MMP9, matrix metalloproteinase 9; MYC, MYC proto‐oncogene, bHLH transcription factor; NF‐κB, nuclear factor kappa B; NIK, NF‐κB‐inducing kinase; RANK, receptor activator of NF‐κB; TERT, telomerase reverse transcriptase; TNF, tumor necrosis factor
Several studies have shown that the NF‐κB pathway is largely associated with the biology of cancer and is upregulated in most hematological malignancies and solid cancers [181, 182]. The NF‐κB pathway can be activated in the loss of tumor suppressor genes, such as tumor protein p53 (TP53), von Hippel‐Lindau tumor suppressor (VHL), and phosphatase and tensin homolog (PTEN), and by the expression of some oncogenes, such as the rat sarcoma virus (RAS) family and BCR activator of RhoGEF and GTPase (BCR)/ABL proto‐oncogene 1, non‐receptor tyrosine kinase (ABL). Moreover, when NF‐κB is inhibited in cancer cells, apoptosis is increased, corroborating the pro‐survival and anti‐apoptotic role of NF‐κB in cancer cells [182, 183, 184, 185].
The activity of NF‐κB subunits arises from a prolonged chronic inflammatory microenvironment or from various oncogenic mutations [186]. In the tumor microenvironment, vascular disorganization causes tissue damage due to hypoxia, and inflammatory pathways are activated with increased NF‐κB activity [187]. Increased NF‐κB activity promotes the tumorigenic microenvironment of CRC [187].
The NF‐κB pathway participates in several processes in CRC‐SCs, such as stimulation of cell proliferation, prevention of apoptosis, EMT, angiogenesis, invasiveness, and metastasis [186]. The activation of NF‐κB can regulate the maintenance of CRC‐SCs by reducing the expression of miR‐195‐5p and miR‐497‐5p [188]. The elevation of miR‐195‐5p and miR‐497‐5p levels by a specific mimic inhibited the effects of NF‐κB on CRC‐SCs in vitro and in vivo. Minichromosome maintenance complex component 2 (MCM2) has been identified as the target gene for miR‐195‐5p and miR‐497‐5p in CRC‐SC culture. Overexpression of MCM2 restored the stem cell profile of CRC cells in the presence of miR‐195‐5p and miR‐497‐5p, suggesting that miR‐195‐5p and miR‐497‐5p may impair CRC‐SCs by inhibiting MCM2 transcription [188].
Even with only a few data on the role of NF‐κB in CRC‐SCs, these data suggest promising potential for NF‐κB inhibitors in CRC therapy.
2.5. JAK/STAT signaling pathway
JAK/STAT signaling is a fundamental signaling pathway for the development of various types of cancer and is directly involved in growth, progression, and metastasis and indirectly involved in immune surveillance modulation [189]. Activation of this cell signaling occurs when the JAK protein is recruited and activated by cytokine receptors. Subsequently, JAK catalyzes the tyrosine phosphorylation of the receptor, allowing the recruitment of STAT proteins. After phosphorylation of STAT, its dimers are translocated to the cell nucleus to bind to DNA, resulting in the transcription of target genes (Figure 5) [190, 191].
FIGURE 5.
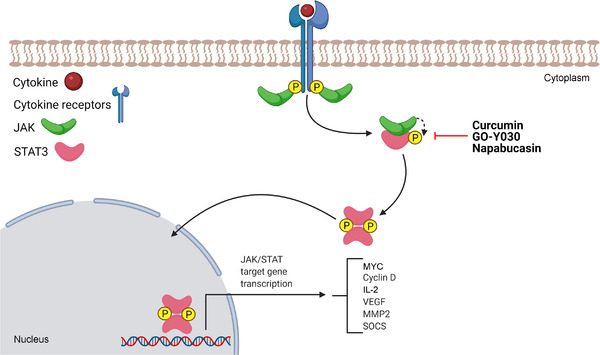
The JAK/STAT signaling pathway. JAK/STAT signaling starts with the interaction of cytokines or growth factors with their receptors, inducing the dimerization/oligomerization of these receptors and consequent activation. Activated JAKs autophosphorylate and phosphorylate their associated receptors. Therefore, cytoplasmic STATs bind to phosphorylated receptors and undergo homodimerization or heterodimerization after their phosphorylation, and they are able to translocate to the nucleus and activate the transcription of target genes. Abbreviations: JAK, Janus kinase; MMP2, matrix metalloproteinase 2; MYC, MYC proto‐oncogene, bHLH transcription factor; SOCS, suppressors of cytokine signaling; STAT, signal transducer and activator of transcription; VEGF, vascular endothelial growth factor
Activation of JAK/STAT signaling in cancer cells can occur by several mechanisms, the best known being mutations in STAT3 or glycoprotein 130 (GP130, also known as IL6‐beta), which promote activation independent of the STAT3 ligand in liver cancers, and the elevated expression of cytokines such as IL‐6 [192]. In CSCs, this pathway induces increased tumorigenic capacity, metastasis, and chemoresistance in cancer via increased EMT [193].
High STAT3 activity was found in CRC‐SCs and tumor‐infiltrating lymphocytes but not in non‐cancerous colon epithelia [194]. In addition, a relationship was observed between the JAK/STAT pathway and the tumor microenvironment. CRC‐derived cells lost STAT3 activity in culture; however, when these CRC cells were implanted in mice, STAT3 activity was restored. Furthermore, blocking STAT3 activation in CRC‐derived xenograft tumors reduced tumor development, corroborating the results that STAT3 participates directly in CRC growth [194].
2.5.1. Natural products
2.5.1.1. Curcumin and GO‐Y030
Curcumin is a polyphenol from Curcuma longa, and GO‐Y030 is a novel curcumin analog. ALDH+/CD133+ subpopulations from CRC express high levels of phosphorylated STAT3, which enhances the proliferation and survival of CRC‐SC lines [195]. Interestingly, curcumin and GO‐Y030 inhibited STAT3 phosphorylation and induced apoptosis, as observed by increased expression of cleaved PARP and cleaved caspase‐3 in CRC‐SCs. In addition, curcumin induced cell cycle arrest, inhibited cell viability, decreased the ability to form spheroids, and suppressed tumor growth in a xenograft model. The effects of GO‐Y030 were more potent than those of curcumin [195]. These data indicate curcumin and its analog GO‐Y030 as drug candidates to eliminate CRC‐SCs by suppressing STAT3 activity.
2.5.2. Small synthetic molecule
2.5.2.1. Napabucasin
Napabucasin (also known as BBI608) is an orally administered STAT3 inhibitor with anti‐cancer activity against various types of cancer. It killed CRC‐SC SW480 cells isolated by Hoechst‐33342 dye exclusion sorting [196]. In a phase I clinical trial with four patients with metastatic CRC, napabucasin (240 mg twice daily) plus FOLFIRI and bevacizumab showed a safety profile in Japanese patients with metastatic CRC [197]. All four patients had diarrhea, two patients reported decreased appetite, and decreased neutrophil counts were observed in three patients. Deaths or serious adverse effects were not reported. A phase III clinical trial with patients with advanced p‐STAT3+ CRC treated with napabucasin showed a significant survival benefit compared to placebo (median overall survival of 5.1 vs. 3 months, hazard ratio = 0.41, P = 0.0025) [198].
These preclinical and clinical data indicate that napabucasin is a promising anti‐CSC drug in CRC therapy. It is the most advanced drug in development that targets cell signaling pathways to eradicate CRC‐SCs and the only one with published results from phase III clinical trials. Napabucasin may be the first anti‐CRC drug approved for clinical use targeting CSCs.
2.5.2.2. SC‐43 and SC‐78
SC‐43 and SC‐78, STAT3 inhibitors that stimulate Src homology 2 domain‐containing protein tyrosine phosphatase 1 (SHP‐1) to inactivate STAT3, suppressed the activation of STAT3 in CRC HCT‐116 and HT‐29 cell lines [199]. They also reduced the ability to form spheroids in an assay with cells grown on non‐adherent spheroids without serum and supplemented with growth factors and to form colonies in a soft agar colony formation assay. A decrease in the CD133+/CD44+ subpopulation was also observed. Moreover, they synergize with oxaliplatin and/or irinotecan to inhibit sphere formation of these cells.
These data corroborate that STAT3 inhibitors may be useful to improve anti‐CRC therapy targeting CRC‐SCs.
2.6. PPAR signaling pathway
PPARs are nuclear receptor proteins that, when activated, act as transcription factors and play a critical role in the regulation of some diseases, including cancer (Figure 6). This pathway is activated by the interaction of a ligand with a G protein‐coupled receptor, which triggers a series of signal conductions, including adenylyl cyclase (AC), cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA) proteins, until culminating in the activation and translocation of PPAR for the nucleus to modulate the expression of the target genes [200, 201, 202]. There are three variants with different functions for these receptors: PPARα, PPARδ, and PPARγ. In tumor development, PPARs are involved in the regulation of cell proliferation, survival, apoptosis, and tumor growth and can inhibit or promote tumor development [200, 201, 202]. PPARs are also involved in the initiation and regulation of CSC functions and in the modulation of the EMT process [203]. They also regulate lipid droplets, which are organelles that store neutral lipids as an energy source in some CSCs [204].
FIGURE 6.
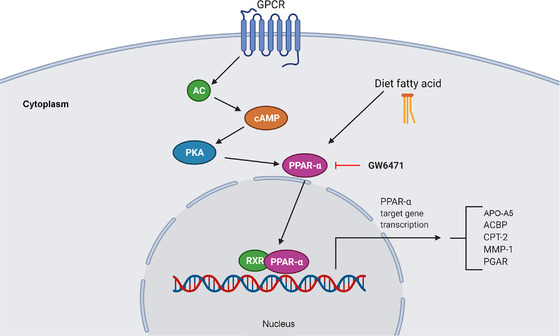
The PPAR signaling pathway. This signaling pathway begins through the interaction of a ligand with a GPCR, which triggers a series of signal conductions passed by AC, cAMP, and PKA until culminating in the activation and translocation of PPARα to the nucleus to modulate the expression of target genes. Abbreviations: AC, adenylyl cyclase; ACBP, acyl‐CoA‐binding protein; APO‐A5, apolipoprotein A5; cAMP, cyclic adenosine monophosphate; CPT‐2, carnitine palmitoyltransferase 2; GPCR, G protein‐coupled receptor; MMP1, matrix metalloproteinase 1; PGAR, PPAR gamma angiopoietin‐related gene; PKA protein kinase A; PPAR, peroxisome proliferator‐activated receptor; RXR, retinoid X receptor
2.6.1. Small synthetic molecule
2.6.1.1. GW6471
GW6471, a PPARα antagonist, interacts directly with PPARα activation function 2 (AF2) and prevents it from assuming an active conformation [204]. A CRC study using SW620, HT‐29, WiDr, and SW480 cell lines showed that the PPARα pathway is active in CRC‐SCs. Interestingly, GW6471 reduced the expression of CSC markers (SOX2, NANOG, and OCT4) and the ability of CRC cells to form spheroids [204]. These results suggest that GW6471 is a drug able to eliminate CRC‐SCs. The relationship between the PPAR pathway and CRC‐SCs is consistent, although more robust studies are needed to confirm the importance of this pathway in CRC‐SCs.
2.7. PI3K/Akt/mTOR signaling pathway
The PI3K/Akt/mTOR signaling pathway plays a key role in the cell cycle, metabolism, quiescence, and proliferation and is considered a master regulator of cancer [205, 206, 207]. The conversion of phosphatidylinositol (3,4,5)‐trisphosphate (PIP3) from phosphatidylinositol 4,5‐bisphosphate (PIP2) into the membrane begins after activation of the growth factor receptor protein tyrosine kinase, providing coupling sites to signal proteins, such as Akt. In the final activation of Akt, the mTORC2 complex phosphorylates Akt at Ser473. Akt activates mTOR complex 1 (mTORC1), improving the synthesis of target proteins (Figure 7).
FIGURE 7.
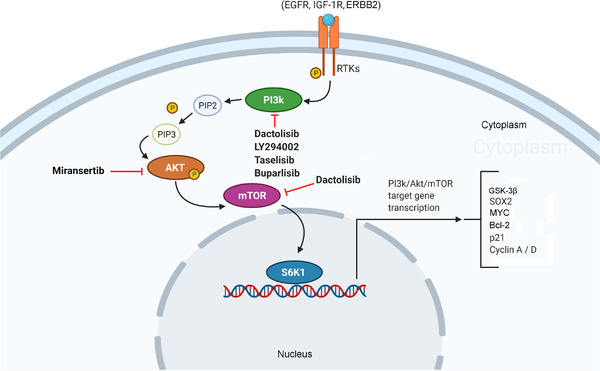
The PI3K/Akt/mTOR signaling pathway. In the pathway cascade, after growth factors bind to their RTKs, the PI3K signaling pathway is activated, converting PIP2 into PIP3 until Akt activation is reached. This process is downregulated by PTEN. Akt, in turn, stimulates mTOR to phosphorylate target proteins to modulate gene expression. Abbreviations: EGFR, epidermal growth factor receptor; ERBB2, erb‐b2 receptor tyrosine kinase 2; GSK‐3β, glycogen synthase kinase‐3 beta; IGF‐1R, insulin‐like growth factor 1 receptor; mTOR, mechanistic target of rapamycin; MYC, MYC proto‐oncogene, bHLH transcription factor; PI3K, phosphatidyl‐inositol‐3‐kinase; PIP2, phosphatidylinositol 4,5‐bisphosphate; PIP3, phosphatidylinositol (3,4,5)‐trisphosphate; PTEN, phosphatase and tensin homolog; RTKs, receptor tyrosine kinases; S6K1, ribosomal protein S6 kinase 1; SOX2, SRY‐box transcription factor 2
Some studies have shown the association between the PI3K/Akt/mTOR signaling pathway and the biology of CRC‐SCs [208]. Chen et al. [209] demonstrated that human CRC xenografts overexpressed various components of the PI3K/Akt/mTOR signaling pathway, including phosphoinositide‐3‐kinase regulatory subunit 2 (PIK3R2, a PI3K regulatory subunit), in CRC‐SCs. Through a collection of primary cell cultures growing on spheroids obtained from 60 CRC specimens, Mangiapane et al. [210] showed that the high expression of erb‐b2 receptor tyrosine kinase 2 (ERBB2) in CRC‐SCs is associated with the activation of the PI3K/Akt pathway, promoting acetylation in the regulatory elements of the ERBB2 gene. mTOR expression was also related to poor prognosis in patients with stage II CRC [211]. Moreover, a transcriptome of CRC‐SC primary cultures and their normal stem cell counterparts revealed enrichment of genes central to PI3K/Akt and Wnt signaling in CRC‐SCs [212].
2.7.1. Natural products
2.7.1.1. Piplartine/piperlongumine
Piplartine, also known as piperlongumine, is an alkaloid amide isolated from peppers (the Piperaceae family). It presents multiple pharmacological activities, including cytotoxicity and in vivo anti‐tumor effects against different types of cancers. The mechanism of action of piplartine has been associated with its capacity to induce oxidative stress by inhibiting the antioxidant system, including depletion of glutathione [213, 214, 215, 216, 217]. In particular, piplartine inhibited the Ras/PI3K/Akt/mTOR pathway and reduced tumor cell growth in a dimethylhydrazine/dextran sulfate sodium‐induced colon carcinogenesis animal model [218].
Auranofin is a clinically approved synthetic drug for the treatment of rheumatoid arthritis. Anticancer properties have been reported for this gold molecule, where its main mechanism of action is inhibition of cellular antioxidant enzymes, such as thioredoxin reductase, causing oxidative stress and cell death in cancer cells [219]. Auranofin demonstrated selective capacity to improve the inhibition of CT26 colon tumor growth in a mouse model of abdominal irradiation without enhancing radiation toxicity in the normal intestine [220]. Auranofin also sensitized HCT116 and SW620 cells to 5Z‐7‐oxozeaenol, an inhibitor of TGF‐β‐activated kinase 1 [221].
Moreover, Tanaka et al. [222] demonstrated that piplartine plus auranofin decreased the expression level of the CD44v9 surface marker and reduced CRC growth in a patient‐derived xenograft model. Piplartine also showed lower cytotoxicity to fibroblast cells than cancer cells growing in spheroids. This indicates that piplartine plus auranofin is able to eliminate CRC‐SCs. Interestingly, piplartine also inhibited stemness properties in leukemia [223] and oral cancer [224] and had less effect on the viability of normal cells [213, 214].
2.7.1.2. Atractylenolide I
Atractylenolide I (ATL‐1) is a plant‐based compound found in the rhizome of Atractylodes macrocephala used in traditional Chinese medicine. In a study using CRC COLO205 and HCT116 cell lines, ATL‐1 treatment induced apoptosis, inhibited invasion, downregulated the Akt/mTOR pathway by reducing the phosphorylation of pathway‐related proteins, and reduced the stem cell phenotype [225]. In an in vivo experiment, ATL‐1 reduced tumor weight and reduced CRC‐SCs. This suggests that ATL‐1 can inhibit CRC‐SCs in vitro and in vivo.
2.7.1.3. Rapamycin
Rapamycin, a macrolide from Streptomyces hygroscopicus, is clinically used due to its immunosuppressant functions. It is a known mTOR inhibitor. Interestingly, rapamycin treatment decreased the spheroid‐forming ability and ALDH1 activity in CRC Caco‐2 and SW480 cell lines [211]. These effects were also observed in co‐treatment with 5‐FU and oxaliplatin. Importantly, 5‐FU and oxaliplatin generally increased the CRC‐SC subpopulation, but co‐treatment with rapamycin reduced this cell subpopulation. These data indicate that rapamycin can inhibit CRC‐SCs and potentiate the effect of 5‐FU and oxaliplatin.
2.7.2. Small synthetic molecules
2.7.2.1. Dactolisib
Dactolisib (also known as BEZ235), a synthetic imidazoquinoline derivative, is a dual PI3K/mTOR inhibitor that selectively inhibits PI3K class I (p110α, β, δ, and γ), mTORC1, and mTORC2, inhibiting their catalytic activities and cell signaling [226]. Dactolisib suppressed the proliferation of CRC‐SCs and reduced the expression of CD133 and LGR5 in HCT116 cells. In particular, insulin, a positive regulator of the PI3K/Akt/mTOR pathway, reduced the effect of treatment with dactolisib. This result suggests that dactolisib is a drug with the ability to eliminate CRC‐SCs.
2.7.2.2. LY294002
LY294002, a synthetic compound developed based on the flavonoid quercetin, acts as a PI3K inhibitor. Treatment with LY294002 resulted in decreased proliferation, spheroid formation, and self‐renewal properties, together with reduced Akt phosphorylation and cyclin D1 expression in CRC‐SCs in vitro [209]. In an in vivo experiment, LY294002 reduced tumorigenicity, increased the detection of cleaved caspase 3, and increased the expression of the inflammatory chemokine IL‐8. These data indicate that LY294002 is capable of eliminating CRC‐SCs in preclinical models.
2.7.2.3. Taselisib, miransertib, and buparlisib
Taselisib, an inhibitor of PI3K class I, selectively inhibits phosphatidylinositol‐4,5‐bisphosphate 3‐kinase catalytic subunit alpha (PIK3CA) and its mutant forms in the PI3K/Akt/mTOR pathway. Treatment of CRC‐SCs from 60 samples from CRC patients with trastuzumab (an ERBB2 inhibitor), dabrafenib (a MEK inhibitor) or taselisib induced in vitro cell death and regression of tumor xenografts, including those with KRAS and PIK3CA mutations [210]. Furthermore, taselisib alone was also able to kill CRC‐SCs and inhibited the formation of liver metastases in immunocompromised mice after injection of CRC sphere cells. Buparlisib (also known as BKM120), another PI3K inhibitor, and miransertib, an Akt inhibitor, also reduced the viability of CD44v6+ cells in vitro [210].
In a phase Ib clinical trial in patients with KRAS wild‐type advanced CRC, buparlisib (60 mg given 5 out of 7 days per week) plus panitumumab (6 mg/kg injected intravenously every 2 weeks) was recommended for a phase II clinical trial [227].
2.7.2.4. Metformin
Metformin, an oral anti‐diabetic drug, has also been reported as a cytotoxic agent with the ability to reduce the CSC population in different types of cancer [228, 229, 230]. In both flow cytometric analysis (CD44+/CD133+) and tumor spheroid growth assays, CSCs decreased after treatment with metformin in CRC HT‐29 and DLD‐1 cell lines [231]. Augmentation of the phosphorylation of adenosine monophosphate‐activated protein kinase (AMPK) and reduction of mTOR (p‐S6) were also observed after treatment with metformin. In a xenograft mouse model using HT‐29 cells, tumor growth and CSC populations were decreased by metformin administration. Metformin effects were prevented by mevalonate treatment [231]. Interestingly, metformin also reduced CSCs in patients with colorectal and other gastrointestinal cancers in a pilot clinical trial [232].
These preclinical and clinical results indicate metformin as a promising anti‐CRC‐SC drug, but more clinical data are needed to use it in CRC therapy.
2.7.2.5. Torin 1, Torkinib and MK‐2206
Torin 1, a selective inhibitor of mTOR, has anti‐tumor properties. Torin 1 was able to induce cell death in CRC‐SCs in vitro and in vivo without affecting the proliferation of normal colon stem cells in vivo [233]. Torkinib (PP242) is an ATP‐competitive second‐generation inhibitor of mTOR. It reduced CRC‐SCs in in vitro models when it was used alone or in combination with 5‐FU and oxaliplatin [211]. MK‐2206 is an allosteric Akt inhibitor. It inhibited CRC‐SCs, as observed by the reduction of the capacity to form spheres in vitro and to initiate tumor formation in vivo using the CRC SW480 cell line [212]. MK‐2206 was also effective in reducing patient‐derived spheroid formation.
These data indicate the importance of the PI3K/Akt/mTOR pathway in the control of CRC‐SCs, showing that this pathway may be a target for therapeutic agents and can improve CRC therapy in the future.
2.8. TGF‐β/Smad signaling pathway
The TGF‐β/Smad signaling pathway plays an important role in tissue maintenance and is dysregulated in various cancers, including CRC [234, 235]. TGF‐β superfamily ligands include TGF‐β, activin A, and nodal. There are three distinct subtypes of Smads: receptor‐regulated Smads (R‐Smads, e.g., Smad2 and Smad3), common partner Smads (Co‐Smads, e.g., Smad4), and inhibitory Smads (I‐Smads, e.g., Smad7). Activation begins when TGF‐β ligands bind to TGF‐β receptor type 2 (TGFBR2) and then phosphorylate TGF‐β receptor type 1 (TGFBR1). This type I receptor phosphorylates R‐Smads that bind Co‐Smads. This complex is translocated to the cell nucleus and acts as a transcription factor to regulate target gene expression (Figure 8) [234, 235, 236].
FIGURE 8.
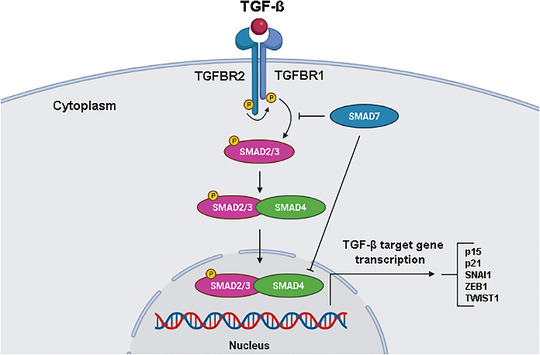
The TGF‐β/Smad signaling pathway. During activation, TGF‐β ligands bind to TGFBR2, which phosphorylates TGFBR1. TGFBR1 then phosphorylates receptor‐regulated Smads (Smad2/3) that bind to Smad4. This complex is translocated to the cell nucleus and acts as a transcription factor to regulate target gene expression. Abbreviations: SNAI1, snail family transcriptional repressor 1; TGFBR1, TGF‐β receptor type 1; TGFBR2, TGF‐β receptor type 2; TGF‐β, transforming growth factor‐β; TWIST1, twist family BHLH transcription factor 1; ZEB1, zinc finger e‐box binding homeobox 1
In CRC, mutations in TGFBR2, affected by DNA repair deficiency with microsatellite instability, increase the cell survival rate. Furthermore, when Smad4 undergoes mutations and loses its function, there is an increase in tumor progression [237, 238]. When Smad4 loss occurs together with mutations in the APC gene in intestinal epithelial cells, a malignant invasive phenotype arises in mouse models. The absence of Smad4 is found in approximately 20%‐40% of CRC patients [239]. Although the loss of heterozygosity on chromosome 18 may be the main cause of Smad4 loss in CRC, there are other post‐transcriptional and post‐translational mechanisms that can contribute to its loss or dysfunction, such as ubiquitylation, sumoylation, and microRNA interference [240]. In addition, TGF‐β signaling also interacts with the Wnt pathway, where the loss of Smad4 promotes β‐catenin expression [241]. Therefore, Wnt activation in the intestinal epithelium triggers the acquisition of stem cell properties and leads to dedifferentiation and rapid adenoma formation in the differentiated intestinal epithelium of a mouse model.
A study using miR‐4666‐3p and miR‐329 demonstrated that TGF‐β1 expression decreased with the weakening of stemness properties and that the activation of the TGF‐β1/Smad pathway could function as a tumor suppressor gene for inhibiting the stemness of CRC. The authors later tested both miR‐4666‐3p and miR‐329 in 73 tumor samples and paired normal tissues and revealed that these two miRNAs are related to poor prognosis and advanced tumor stage of CRC. This study confirms the importance of the TGF‐β/SMAD pathway for CRC‐SCs and provides a new epigenetic regulation mechanism in CRC‐SCs, which can be used in new therapeutic strategies for cancer treatment [242].
2.8.1. Natural product
2.8.1.1. Baicalin
Baicalin is a monomeric flavonoid from the root of Scutellaria baicalensis, a traditional Chinese herb. Baicalin has several pharmacological effects, including anti‐cancer, anti‐inflammatory, antioxidant and neuroprotective effects [243]. In CRC RKO and HCT116 cell lines, baicalin caused cell cycle arrest in the G1 phase, p53‐independent cell apoptosis, and EMT inhibition by inhibiting the TGF‐β/Smad pathway [244]. Baicalin also inhibited the CRC‐SC subpopulation in in vitro and in vivo models using the CRC HCT116 cell line.
Altogether, the TGF‐β/Smad signaling pathway also plays an important role in CRC‐SC maintenance, and novel selective inhibitors must be screened for CRC‐targeting stemness properties.
3. PERSPECTIVES
Tumors have high complexity and are composed of multiple morphologically and phenotypically distinct subpopulations of cancer cells. The CSC subpopulation plays a critical role in the recurrence, progression, and metastasis of CRC and consequent cell resistance to current treatments since this subpopulation appears to be more resistant to chemotherapy and radiotherapy. In this context, exploring new methods to inhibit CSCs seems essential for achieving therapeutic success in CRC. In this review, we highlight fundamental pathways for sustaining CRC‐SCs. Table 1 summarizes the reported drugs that eradicate CRC‐SCs by inhibiting these pathways. Among them, buparlisib, curcumin, epigallocatechin‐3‐gallate, metformin, MK‐2206, napabucasin, quercetin, rapamycin, taselisib, and vismodegib have been tested in clinical trials as anti‐CRC agents (Table 2). In particular, napabucasin, a STAT3 inhibitor, is the only agent that has been completed phase III clinical trials. It improved survival compared to placebo (median overall survival of 5.1 vs. 3 months) in advanced p‐STAT3+ CRC patients [198]. Clinical data with these CRC‐SC inhibitors are still limited. Currently, there is no clinically effective therapy to suppress CSCs.
TABLE 1.
Drugs that target cell signaling pathways in CRC‐SCs
| Drugs | Mechanism of action | Reference |
|---|---|---|
| 36‐077 | Suppresses GSK‐3β/Wnt/β‐catenin signaling | [118] |
| 4‐Acetyl‐antroquinonol B | Suppresses LGR5/Wnt/β‐catenin and JAK‐STAT signaling | [104] |
| Anti‐DLL4 antibodies | Suppresses Notch signaling | [158] |
| Atractylenolide I | Suppresses Akt/mTOR signaling | [225] |
| Baicalin | Suppresses TGF‐β/Smad signaling | [244] |
| Buparlisib (BKM120) | Suppresses PI3K/Akt/mTOR signaling by inhibition of PI3K | [210] |
| CBB1003 | Suppresses β‐catenin/TCF signaling | [119] |
| Cucurbitacins | Suppresses Notch signaling by downregulation of the expression of ADAM9 | [142] |
| Curcumin | Suppresses JAK/STAT signaling by inhibition of STAT3 | [195] |
| Cyclopamine | Suppresses HH signaling by inhibition of SMO | [171] |
| Dactolisib | Suppresses PI3K/Akt/mTOR signaling by inhibition of PI3K | [226] |
| Diallyl trisulfide | Suppresses Wnt/β‐catenin signaling | [105] |
| Epigallocatechin‐3‐gallate | Suppresses Wnt signaling by upregulation of GSK‐3β | [96] |
| Evodiamine | Suppresses Wnt and Notch signaling | [150] |
| Farnesyl dimethyl chromanol | Suppresses Wnt/β‐catenin signaling | [102] |
| FH535 | Suppresses Wnt/β‐catenin signaling | [123] |
| GO‐Y030 | Suppresses JAK/STAT signaling by inhibition of STAT3 | [195] |
| GW6471 | Suppresses PPAR signaling (PPARα antagonist) | [204] |
| Honokiol | Suppresses Notch signaling by inhibition of the levels of the γ‐secretase complex | [139] |
| HZ8CV2 | Suppresses Wnt signaling | [126] |
| IC‐2 | Suppresses Wnt signaling | [111] |
| JIB‐04 | Suppresses Wnt signaling by increasing the expression of AXIN1 and GSK3β | [109] |
| LY294002 | Suppresses PI3K/Akt/mTOR signaling by inhibition of PI3K | [209] |
| α‐Mangostine | Suppresses Notch signaling | [155] |
| Metformin | Suppresses PI3K/Akt/mTOR signaling by inhibition of mTOR | [231] |
| Miransertib | Suppresses PI3K/Akt/mTOR signaling by inhibition of Akt | [210] |
| MK‐2206 | Akt inhibitor | [212] |
| Napabucasin (BBI‐608) | Suppresses JAK/STAT signaling by inhibition of STAT3 | [196] |
| NCB‐0846 | Suppresses Wnt signaling by block TNIK | [57, 114] |
| Phenethyl isothiocyanate | Downregulates Wnt/β‐catenin | [66] |
| Pien Tze Huang formula | Suppresses Notch signaling | [156] |
| Piplartine/piperlongumine | Suppresses PI3K/Akt/mTOR signaling | [218, 222] |
| Portulaca oleracea extract | Downregulates the expression of Notch1 and β‐catenin | [153] |
| Quercetin | Suppresses Notch signaling by suppression of the γ‐secretase complex | [151] |
| Rapamycin | mTOR inhibitor | [211] |
| Retinoids | Suppresses Wnt signaling via differentiation therapy by HOXA5 induction | [58] |
| Rimonabant | Suppresses β‐catenin | [121] |
| Salinomycin | Disruption of the Wnt/β‐catenin/TCF complex | [78, 89] |
| SC‐43 and SC‐78 | Suppresses STAT3 signaling | [199] |
| Sulforaphane | Suppresses TAp63α/LGR5/β‐catenin axis | [75] |
| Taselisib | Suppresses PI3K/Akt/mTOR signaling by inhibition of PI3K | [210] |
| Torin 1 | mTOR inhibitor | [233] |
| Torkinib | mTOR inhibitor | [211] |
| Vismodegib | Suppresses HH signaling by inhibition of SMO | [176] |
| Zerumbone | Suppresses β‐catenin signaling | [106] |
Abbreviations: CRC‐SCs, colorectal cancer stem cells; AXIN1, axin‐1; GSK‐3β, glycogen synthase kinase‐3 beta; HH, hedgehog; HOXA5, homeobox A5; JAK/STAT, Janus kinase/signal transducer and activator of transcription; LGR5, leucinerich repeat‐containing G‐protein‐coupled receptor 5; NF‐κB, nuclear factor kappa B; PI3K/AKT/mTOR, phosphatidyl‐inositol‐3‐kinases/akt/mechanistic target of rapamycin; PPAR, peroxisome proliferator‐activated receptor; SMO, smoothened; TAp63α, transactivating p63 isoform α; TCF, T‐cell factor; TNIK, TRAF2‐ and NCK‐interacting kinase; Wnt, wingless.
TABLE 2.
Drugs that target cell pathways in CRC‐SCs and are tested in clinical trials as anti‐CRC agents*
| ID | Study Title | Conditions | Interventions | Phase | First Posted | Current Status |
|---|---|---|---|---|---|---|
| NCT01304602 | A trial of irinotecan and BKM120 in previously treated advanced colorectal cancer | Colorectal cancer |
Drug: Irinotecan Drug: BKM120 |
Phase 1 | February 25, 2011 | Completed |
| NCT01591421 | PI3Kinase inhibitor BKM120 in combination with panitumumab in metastatic/advanced RAS‐wild type colorectal cancer | Metastatic colorectal cancer |
Drug: BKM120 Drug: Panitumumab |
Phase 1 Phase 2 |
May 4, 2012 | Completed |
| NCT01571024 | BKM120 + mFOLFOX6 in advanced solid tumors with expansion cohort pancreatic cancer |
Advanced solid tumors Metastatic colorectal cancer Metastatic pancreatic cancer |
Drug: BKM120 Drug: mFOLFOX6 |
Phase 1 | April 4, 2012 | Completed |
| NCT01501604 | BKM120 in cancers with PIK3CA activating mutations |
Lung cancer Breast cancer Colorectal cancer Cholangiocarcinoma Solid tumors |
Drug: BKM120 | Phase 2 | December 29, 2011 | Withdrawn (the study has been closed due to lack of accrual) |
| NCT01576666 | Phase Ib, dose‐escalation study of oral LDE225 in combination with BKM120 in patients with advanced solid tumors |
Dose escalation Safety Preliminary efficacy Advanced solid tumors, including metastatic colorectal cancer |
Drug: LDE225 Drug: BKM120 |
Phase 1 | April 12, 2012 | Completed |
| NCT02439385 | Avastin/FOLFIRI in combination with curcumin in colorectal cancer patients with unresectable metastasis | Colorectal cancer |
Drug: Avastin/FOLFIRI Dietary supplement: Curcumin |
Phase 2 | May 8, 2015 | Completed |
| NCT00973869 | Curcumin in preventing colorectal cancer in patients undergoing colorectal endoscopy or colorectal surgery | Colorectal cancer | Dietary supplement: curcumin | Phase 1 | September 9, 2009 | Unknown |
| NCT01859858 | Effect of curcumin on dose‐limiting toxicity and pharmacokinetics of irinotecan in patients with solid tumors | Advanced colorectal cancer |
Dietary supplement: curcumin Drug: irinotecan |
Phase 1 | May 22, 2013 | Completed |
| NCT01333917 | Curcumin biomarkers | Colorectal cancer | Drug: Curcumin C3 tablet | Phase 1 | April 12, 2011 | Completed |
| NCT01490996 | Combining curcumin with FOLFOX chemotherapy in patients with inoperable colorectal cancer | Colonic cancer metastasis |
Drug: Oral complex C3 curcumin + chemotherapy Drug: Chemotherapy only |
Phase 1 Phase 2 |
December 13, 2011 | Completed |
| NCT00027495 | Curcumin for the prevention of colon cancer | Colorectal cancer | Dietary supplement: curcumin | Phase 1 | January 27, 2003 | Completed |
| NCT00003365 | Sulindac and plant compounds in preventing colon cancer | Colorectal cancer |
Dietary supplement: curcumin Dietary Supplement: rutin Drug: quercetin Drug: sulindac |
Not applicable | May 21, 2004 | Terminated (study completed) |
| NCT00118989 | Curcumin for the chemoprevention of colorectal cancer | Adenomatous polyps | Dietary Supplement: curcuminoids | Not applicable | July 12, 2005 | Terminated (could not be completed due to technical problems and cost constraints) |
| NCT00003365 | Sulindac and plant compounds in preventing colon cancer | Colorectal cancer |
Dietary supplement: curcumin Dietary supplement: rutin Drug: quercetin Drug: sulindac |
Not applicable | May 21, 2004 | Terminated (study completed) |
| NCT02891538 | Chemopreventive effects of epigallocatechin gallatein colorectal cancerpatients | Colon cancer | Drug: epigallocatechin gallate | Early phase 1 | September 7, 2016 | Recruiting |
| NCT02321969 | Green tea extracts for the prevention of colorectal adenomas and colorectal cancer | Neoplasms, colorectal | Dietary supplement: green tea extract | Not applicable | December 22, 2014 | Completed |
| NCT01941953 | Metformin and 5‐fluorouracil for refractory colorectal cancer | Metastatic colorectal cancer | Drug: Metformin and 5‐Fluorouracil | Phase 2 | September 13, 2013 | Completed |
| NCT02614339 | Effect of adjunctive metformin on recurrence of non‐DM colorectal cancer stage II high‐risk/III colorectal cancer |
Non‐DM stage II high‐risk colorectal cancer Non‐DM stage III colorectal cancer |
Drug: Metformin Drug: Control |
Phase 3 | November 25, 2015 | Recruiting |
| NCT01816659 | An open‐labeled pilot study of biomarker response following short‐term exposure to metformin | Colorectal carcinoma | Metformin extended release | Phase 1 | March 22, 2013 | Terminated (slow accrual) |
| NCT01312467 | Trial of metformin for colorectal cancer risk reduction for a history of colorectal adenomas and elevated BMI |
Adenomatous polyp Colorectal cancer Obesity |
Drug: Metformin hydrochloride | Phase 2 | June 25, 2015 | Completed |
| NCT01926769 | A phase II study to determine the safety and efficacy of second‐line treatment with metformin and chemotherapy (FOLFOX6 or FOFIRI) in the second line treatment of advanced colorectal cancer | Previously treated advanced colorectal cancer | Drug: Metformin | Phase 2 | August 21, 2013 | Terminated (slow accrual rate) |
| NCT01523639 | A randomized, placebo‐controlled, double‐blind phase II study evaluating if glucophage can avoid liver injury due to chemotherapy‐associated steatosis |
Colorectal cancer steatohepatitis |
Drug: Metformin/Placebo | Phase 2 | February 1, 2012 | Terminated (prematurely due to slow recruitment) |
| NCT03800602 | Nivolumab and metformin in patients with treatment‐refractory MSS colorectal cancer | Colorectal neoplasms |
Drug: Metformin Biological: Nivolumab |
Phase 2 | January 11, 2019 | Active, not recruiting |
| NCT01930864 | Metformin plus irinotecan for refractory colorectal cancer | Colorectal Neoplasms Adenocarcinoma |
Drug: Metformin Drug: Irinotecan |
Phase 2 | August 29, 2013 | Unknown |
| NCT03047837 | A randomized, 2×2 factorial design biomarker prevention trial of low‐dose aspirin and metformin in stage I‐III colorectal cancer patients | Tertiary prevention in colon cancer |
Drug: Aspirin + Metformin Drug: Aspirin Drug: Metformin Drug: Placebos |
Phase 2 | February 9, 2017 | Unknown |
| NCT01440127 | Impact of pretreatment with metformin on colorectal cancer stem cells and related pharmacodynamic markers | Colon cancer | Drug: Metformin | Phase 1 | September 26, 2011 | Terminated (unable to accrue all planned subjects in a timely fashion, but data collected will be analyzed) |
| NCT01340300 | Exercise and metformin in colorectal and breast cancer survivors |
Colorectal cancer Breast cancer |
Behavioral: Exercise training Drug: Metformin Other: Educational information |
Phase 2 | September 18, 2017 | Completed |
| NCT04033107 | High dose vitamin C combined with metformin in the treatment of malignant tumors |
Hepatocellular cancer Pancreatic cancer Gastric cancer Colorectal cancer |
Drug: Vitamin C Drug: Metformin |
Phase 2 | July 25, 2019 | Recruiting |
| NCT01632020 | Effect of metformin on biomarkers of colorectal tumor cell growth | Colorectal neoplasms |
Drug: Placebo Drug: Metformin |
Phase 2 | June 29, 2012 | Terminated (inadequate accrual) |
| NCT01333475 | MK‐2206 and AZD6244 in patients with advanced colorectal carcinoma | Colorectal neoplasms | Drug: MK‐2206 + AZD6244 | Phase 2 | April 12, 2011 | Completed |
| NCT01186705 | Clinical and translational study of MK‐2206 in patients with metastatic KRAS‐wild‐type, PIK3CA‐mutated, colorectal cancer |
Colon cancer Rectal cancer |
Drug: MK‐2206 | Phase 2 | August 23, 2010 | Terminated (lack of accrual) |
| NCT01802320 | Akt inhibitor MK‐2206 in treating patients with previously treated colon or rectal cancer that is metastatic or locally advanced and cannot be removed by surgery | Colorectal neoplasms |
Drug: Akt Inhibitor MK‐2206 Other: Laboratory Biomarker Analysis Other: Pharmacological Study |
Phase 2 | March 1, 2013 | Completed |
| NCT01243762 | A study of dalotuzumab + MK‐2206, dalotuzumab + MK‐0752, and dalotuzumab + ridaforolimus combination therapies in participants with advanced cancer (MK‐0646‐027) | Neoplasms malignant |
Drug: dalotuzumab Drug: MK‐0752 Drug: ridaforolimus Drug: MK‐2206 |
Phase 1 | November 19, 2010 | Terminated (the study was terminated for business reasons and not due to any safety or efficacy concerns related to dalotuzumab) |
| NCT03522649 | A Phase III clinical study of napabucasin (GB201) Plus FOLFIRI in adult patients with metastatic colorectal cancer | Previously treated metastatic colorectal cancer |
Drug: Napabucasin Drug: Fluorouracil Drug: Leucovorin Drug: Irinotecan |
Phase 3 | May 11, 2018 | Recruiting |
| NCT02753127 | A study of napabucasin (BBI‐608) in combination with FOLFIRI in adult patients with previously treated metastatic colorectal cancer (CanStem303C) | Colorectal cancer |
Drug: Napabucasin Drug: Fluorouracil Drug: Leucovorin Drug: Irinotecan Drug: Bevacizumab |
Phase 3 | April 27, 2016 | Completed |
| NCT01776307 | A study of BBI608 in adult patients with advanced colorectal cancer | Colorectal cancer |
Drug: BBI608 Drug: Panitumumab Drug: Capecitabine Drug: Cetuximab |
Phase 2 | January 28, 2013 | Completed |
| NCT01830621 | BBI608 and best supportive care vs placebo and best supportive care in pretreated advanced colorectal carcinoma | Colorectal carcinoma |
Drug: BBI608 Drug: Placebo Other: Best supportive care |
Phase 3 | April 12, 2013 | Completed |
| NCT02641873 | A study of BBI608 administered with FOLFIRI + bevacizumab in adult patients with metastatic colorectal cancer | Metastatic colorectal cancer |
Drug: BBI608 Drug: 5‐FU Drug: Irinotecan Drug: Levofolinate Drug: Bevacizumab |
Phase 1 | December 30, 2015 | Completed |
| NCT03647839 | Modulation of the tumor microenvironment using either vascular disrupting agents or STAT3 inhibition in order to synergize with PD1 inhibition in microsatellite stable, refractory colorectal cancer (MODULATE) | Colorectal cancer metastatic |
Drug: Nivolumab 10 MG/ML Drug: BNC 105 Drug: BBI608 |
Phase 2 | August 27, 2018 | Completed |
| NCT02851004 | A special combination of BBI608 and pembrolizumab | Metastatic colorectal cancer |
Drug: BBI608 (Napabucasin) Drug: Pembrolizumab |
Phase 1 Phase 2 |
August 1, 2016 | Terminated (on the request of the investigational drug provider) |
| NCT01606124 | Polyphenon E in treating patients with high‐risk of colorectal cancer | Advanced colorectal adenomas adenocarcinoma of the colon stage I colon cancer stage II colon cancer stage III colon cancer |
Drug: defined green tea catechin extract Other: placebo Other: questionnaire administration Other: laboratory biomarker analysis |
Phase 2 | May 25, 2012 | Terminated (pending expiration of the supply of study agent) |
| NCT03439462 | ABI‐009 (Nab‐rapamycin) in combination with FOLFOX and Bevacizumab as first‐line therapy in patients with advanced or metastatic colorectal cancer | Colorectal cancer metastatic | Drug: ABI‐009; nab‐rapamycin; albumin‐bound rapamycin |
Phase 1 Phase 2 |
February 20, 2018 | Recruiting |
| NCT00409994 | Safety study of rapamycin administered before and during radiotherapy to treat rectum cancer | Rectum cancer | Drug: Rapamycin |
Phase 1 Phase 2 |
December 12, 2006 | Completed |
| NCT03190174 | Nivolumab (Opdivo®) plus ABI‐009 (Nab‐rapamycin) for advanced sarcoma and certain cancers | Colorectal cancer and other cancers |
Drug: Nab‐Rapamycin Biological: Nivolumab |
Phase 1 Phase 2 |
June 16, 2017 | Recruiting |
| NCT01522820 | Vaccine therapy with or without sirolimus in treating patients with NY‐ESO‐1 expressing solid tumors |
Recurrent colorectal carcinoma Others cancers |
Biological: DEC‐205/NY‐ESO‐1 Fusion Protein CDX‐1401 Other: Laboratory Biomarker Analysis Other: Pharmacological Study Drug: Sirolimus |
Phase 1 | February 1, 2012 | Completed |
| NCT00375245 | Rapamycin with grapefruit juice for advanced malignancies |
Tumors Neoplasm metastasis |
Drug: rapamycin (sirolimus) Other: grapefruit juice |
Phase 1 | September 12, 2006 | Completed |
| NCT02465060 | Targeted therapy directed by genetic testing in treating patients with advanced refractory solid tumors, lymphomas, or multiple myeloma (the match screening trial) | Many tumors, including colorectal cancer and recurrent colorectal cancer | Many targeted drugs, including taselisib and vismodegib | Phase 2 | June 8, 2015 | Recruiting |
| NCT00636610 | A study of vismodegib (GDC‐0449, hedgehog pathway inhibitor) with concurrent chemotherapy and bevacizumab as first‐line therapy for metastatic colorectal cancer | Metastatic colorectal cancer |
Drug: Vismodegib 150 mg Drug: Placebo to vismodegib Drug: Bevacizumab Drug: Modified FOLFOX Drug: FOLFIRI |
Phase 2 | March 14, 2008 | Completed |
| NCT00959647 | A study of vismodegib (GDC‐0449) in patients treated with vismodegib in a previous Genentech‐sponsored phase I or II cancer study |
Ovarian cancer Basal cell carcinoma Metastatic colorectal cancer |
Drug: Vismodegib Drug: FOLFOX Drug: FOLFIRI Drug: Bevacizumab |
Phase 2 | August 14, 2009 | Completed |
*All studies were accessed at www.clinicaltrials.gov on August 28, 2021 by using the search term “CRC” and the drugs that are presented in Table 1.
Abbreviations: CRC‐SCs, colorectal cancer stem cells; BMI, body mass index; DM, diabetes mellitus; FOLFIRI, 5‐fluorouracil, leucovorin plus irinotecan; FOLFOX, 5‐fluorouracil, folic acid plus oxaliplatin; FOLFOX6, 5‐fluorouracil, leucovorin plus oxaliplatin; MSS, microsatellite stable; PI3Kinase, phosphatidyl‐inositol‐3‐kinase; PIK3CA, phosphatidylinositol‐4,5‐bisphosphate 3‐kinase catalytic subunit alpha.
The Wnt canonical pathway seems to be the most relevant for the growth of CRC‐SCs, also counting on the contribution of the Notch, HH, NF‐κB, JAK/STAT, PPAR, PI3K/Akt/mTOR, and TGF‐β/Smad pathways, which needs further study. It is important to note that these signaling pathways do not operate separately but function as a network. It is necessary to suppress more than one pathway to inhibit possible crosstalk with other pathways. Interestingly, these signaling pathways are aberrantly expressed in bulk CRC and CRC‐SCs, which allows the use of their protein expression levels as therapeutic biomarkers to guide the treatment of CRC patients with appropriate inhibitors.
On the other hand, many studies have reported that many of these pathways also regulate the maintenance of normal stem cell homeostasis [245, 246, 247, 248], indicating that inhibitors of these pathways may lead to toxicity to normal tissues and limit the use of anti‐CRC therapy. Although intestine and blood stem cells are in constant proliferation, most stem cells in the mammary glands, skin, and brain are usually quiescent [249, 250, 251, 252]. Thus, more data are needed on the toxicity of inhibitors of these signaling pathways.
Complementally, CAR‐T (chimeric antigen receptor T) cells, antibodies, and other molecules with different targets have also been developed to specifically target CSCs. Some of them are summarized in Table 3.
TABLE 3.
Other molecules/therapies that target CRC‐SCs
| Drug/Therapy | Pharmacological/Chemical class | Action | Reference |
|---|---|---|---|
| 5‐aminosalicylic acid | Anti‐inflammatory modulator drug | Suppress stemness features in many CRC cell lines | [256] |
| Antibody dual‐antigen‐binding specificity to CD133 and CD3 | Antibody | Reduces CD133+ CRC cells in vitro and in vivo | [257] |
| ASR352 | Chk1 inhibitor | Inhibits CRC‐SCs in preclinical models | [258] |
| CD133‐directed CAR T cells | CAR‐T cells | Eliminates CD133+ cells and reduced tumor growth in a phase I clinical trial with patients with advanced metastasis colorectal, liver and pancreatic cancers | [259] |
| CD133‐targeted oncolytic adenovirus | Oncolytic adenovirus | Eliminates CD133+ CRC in vitro and in vivo | [260] |
| Dabrafenib | BRAF inhibitor | Synergies with cetuximab (EGFR inhibitor) to decrease stem cell in BRAF(V600E)‐mutant CRC cells | [261] |
| Ellagic acid | Ellagitannin metabolites | Mixed with urolithins (gut microbiota‐derived) reduces CRC‐SCs of Caco‐2 cells and primary tumor cells from a patient with CRC | [262] |
| G2.2 | Sulfated nonsaccharide glycosaminoglycan mimetic | Reduces CRC‐SCs in cell lines via p38 MAPK activation | [263] |
| Gambogic acid | Xanthonoid from Garcinia hanburyi | Inhibits CRC‐SCs by upregulation of ZFP36 | [264, 265] |
| Ginsenoside | Major active component of ginseng | Reduced growth and stemness of CRC cells in vitro and in vivo | [266] |
| Heparan sulfate hexasaccharide | Non‐anticoagulant heparin derivative | Inhibits CRC‐SCs by activation of p38 MAPK | [267] |
| Mithramycin A | Polyketide antibiotic | Reduces CRC‐SCs in different cell lines | [268] |
| Nigericin | Antibiotic from Streptomyces hygroscopicus | Reduces CRC‐SCs in HT‐29 and SW116 cell lines | [269] |
| NSC30049 | Chk1 inhibitor | Inhibits CRC‐SCs in preclinical models | [270] |
| Parthenolide | Sesquiterpene lacton from Tanacetum parthenium | Eliminates CRC‐SCs in preclinical models | [271] |
| Polydatin | Glycoside of resveratrol found in Polygonum cuspidatum | Combination with radiation caused apoptosis of LGR5+ CRC cells | [272] |
| Silibinin | Flavonolignan from Silybum marianum | Inhibits the growth kinetics of CRC‐SCs in different cell lines | [273] |
| Thiostrepton | Thiazole antibiotic from Streptomycetes sp. | Induces cell death in CRC‐SCs in HCT‐15 and HT‐29 and synergizes with oxaliplatin | [274] |
| UCN‐01 | Staurosporin derivative | Inhibits CRC‐SCs growth and increases irinotecan action in vitro and in vivo | [275] |
Abbreviations: CRC‐SCs, colorectal cancer stem cells; BRAF, B‐Raf proto‐oncogene, serine/threonine kinase; CAR‐T, chimeric antigen receptor T cells; Chk1, checkpoint kinase 1; EGFR, epidermal growth factor receptor; LGR5, leucine‐rich repeat‐containing G‐protein‐coupled receptor 5; MAPK, mitogen activated protein kinases; ZFP36, ZFP36 ring finger protein.
Additionally, extracellular factors affecting the tumor microenvironment, such as vascular niches, hypoxia, tumor‐associated macrophages, cancer‐associated fibroblasts, and extracellular matrix, have emerged as an important topic in CSC regulation, and many strategies targeting the microenvironment of CSCs have been revealed [253, 254, 255]. Accordingly, effective treatment strategies for CRC‐SCs must emphasize not only intracellular signaling pathways but also reciprocal interactions between CRC‐SCs and extracellular factors.
4. CONCLUSIONS
This review highlights therapeutic targets for drugs that act on the signaling pathways activated in CRC‐SCs. Drugs under preclinical and clinical studies are summarized to direct future research. This knowledge will contribute to the study of potential therapeutic schemes, combining conventional drugs with CSC‐targeting drugs and allowing better cure rates in anti‐CRC therapy.
DECLARATIONS
ETHICS APPROVAL AND CONSENT TOPARTICIPATE
Not applicable.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
Not applicable.
CONFLICTS OF INTEREST
The authors have declared that there are no conflicts of interest.
AUTHORS’ CONTRIBUTIONS
VRS drafted the content of Wnt/β‐catenin, Notch and Hedgehog pathways; LSS drafted the content of NF‐κB, JAK/STAT, PPAR, PI3K/Akt/mTOR and TGF‐β/Smad pathways; CAQ drafted the content of clinical approaches for CRC; RBD and DPB planned this review and reviewed the whole text. All authors read and approved the final manuscript.
ACKNOWLEDGMENTS
VRS, LSS and RBD receive personal scholarship from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Brazil), and DPB receives personal scholarship from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brazil).
Silva VR, Santos LS, Dias RB, Quadros CA, Bezerra DP. Emerging agents that target signaling pathways to eradicate colorectal cancer stem cells. Cancer Commun. 2021;41:1275–1313. 10.1002/cac2.12235
REFERENCES
- 1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49. 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2. Wright M, Beaty JS, Ternent CA. Molecular markers for colorectal cancer. Surg Clin North Am. 2017;97(3):683–701. 10.1016/j.suc.2017.01.014. [DOI] [PubMed] [Google Scholar]
- 3. Xie YH, Chen YX, Fang JY. Comprehensive review of targeted therapy for colorectal cancer. Sig Transduct Target Ther. 2020;5(1):22. 10.1038/s41392-020-0116-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Biller LH, Schrag D. Diagnosis and treatment of metastatic colorectal cancer: a review. JAMA. 2021;325(7):669–85. 10.1001/jama.2021.0106. [DOI] [PubMed] [Google Scholar]
- 5. Giacchetti S, Perpoint B, Zidani R, Le Bail N, Faggiuolo R, Focan C, et al. Phase III multicenter randomized trial of oxaliplatin added to chronomodulated fluorouracil‐leucovorin as first‐line treatment of metastatic colorectal cancer. J Clin Oncol. 2000;18:136–47. 10.1200/JCO.2000.18.1.136. [DOI] [PubMed] [Google Scholar]
- 6. Lee LH, Cavalcanti MS, Segal NH, Hechtman JF, Weiser MR, Smith JJ, et al. Patterns and prognostic relevance of PD‐1 and PD‐L1 expression in colorectal carcinoma. Mod Pathol. 2016;29(11):1433–42. 10.1038/modpathol.2016.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–64. 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kuipers EJ, Grady WM, Lieberman D, Seufferlein T, Sung JJ, Boelens PG, et al. Colorectal cancer. Nat Rev Dis Primers. 2015;1:15065. 10.1038/nrdp.2015.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maenhaut C, Dumont JE, Roger PP, van Staveren WC. Cancer stem cells: a reality, a myth, a fuzzy concept or a misnomer? An analysis. Carcinogenesis. 2010;31:149–58. 10.1093/carcin/bgp259. [DOI] [PubMed] [Google Scholar]
- 10. Murakami S, Ninomiya W, Sakamoto E, Shibata T, Akiyama H, Tashiro F. SRY and OCT4 are required for the acquisition of cancer stem cell‐like properties and are potential differentiation therapy targets. Stem Cells. 2015;33:2652–63. 10.1002/stem.2059. [DOI] [PubMed] [Google Scholar]
- 11. Boumahdi S, Driessens G, Lapouge G, Rorive S, Nassar D, Le Mercier M, et al. SOX2 controls tumour initiation and cancer stem‐cell functions in squamous‐cell carcinoma. Nature. 2014;511:246–50. 10.1038/nature13305. [DOI] [PubMed] [Google Scholar]
- 12. Ibrahim EE, Babaei‐Jadidi R, Saadeddin A, Spencer‐Dene B, Hossaini S, Abuzinadah M, et al. Embryonic NANOG activity defines colorectal cancer stem cells and modulates through AP1‐ and TCF‐dependent mechanisms. Stem Cells. 2012;30:2076–87. 10.1002/stem.1182. [DOI] [PubMed] [Google Scholar]
- 13. Galardi S, Savino M, Scagnoli F, Pellegatta S, Pisati F, Zambelli F, et al. Resetting cancer stem cell regulatory nodes upon MYC inhibition. EMBO Rep. 2016;17:1872–89. 10.15252/embr.201541489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boman BM, Wicha MS. Cancer stem cells: a step toward the cure. J Clin Oncol. 2008;26:2795–9. 10.1200/JCO.2008.17.7436. [DOI] [PubMed] [Google Scholar]
- 15. Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem‐like state. Proc Natl Acad Sci USA. 2011;108:7950–5. 10.1073/pnas.1102454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boman BM, Huang E. Human colon cancer stem cells: a new paradigm in gastrointestinal oncology. J Clin Oncol. 2008;26:2828–38. 10.1200/JCO.2008.17.6941. [DOI] [PubMed] [Google Scholar]
- 17. Ricci‐Vitiani L, Fabrizi E, Palio E, De Maria R. Colon cancer stem cells. J Mol. Med. 2009;87:1097. 10.1007/s00109-009-0518-4. [DOI] [PubMed] [Google Scholar]
- 18. Lobo NA, Shimono Y, Qian D, Clarke MF. The biology of cancer stem cells. Annu Rev Cell Dev Biol. 2007;23:675–99. 10.1146/annurev.cellbio.22.010305.104154. [DOI] [PubMed] [Google Scholar]
- 19. Patel N, Baranwal S, Patel BB. A strategic approach to identification of selective inhibitors of cancer stem cells. Methods Mol Biol. 2015;1229:529–41. 10.1007/978-1-4939-1714-3_41. [DOI] [PubMed] [Google Scholar]
- 20. Gheytanchi E, Naseri M, Karimi‐Busheri F, Atyabi F, Mirsharif ES, Bozorgmehr M, et al. Morphological and molecular characteristics of spheroid formation in HT‐29 and Caco‐2 colorectal cancer cell lines. Cancer Cell Int. 2021;21(1):204. 10.1186/s12935-021-01898-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445(7123):106–10. 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 22. Haraguchi N, Ohkuma M, Sakashita H, Matsuzaki S, Tanaka F, Mimori K, et al. CD133+CD44+ population efficiently enriches colon cancer initiating cells. Ann Surg Oncol. 2008;15:2927–33. 10.1245/s10434-008-0074-0. [DOI] [PubMed] [Google Scholar]
- 23. Dalerba P, Dylla SJ, Park IK, Liu R, Wang X, Cho RW, et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci USA. 2007;104:10158–63. 10.1073/pnas.0703478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vermeulen L, Todaro M, de Sousa Mello F, Sprick MR, Kemper K, Perez Alea M, et al. Single‐cell cloning of colon cancer stem cells reveals a multi‐lineage differentiation capacity. Proc Natl Acad Sci USA. 2008;105:13427–32. 10.1073/pnas.0805706105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang EH, Hynes MJ, Zhang T, Ginestier C, Dontu G, Appelman H, et al. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res. 2009;69:3382–9. 10.1158/0008-5472.CAN-08-4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pang R, Law WL, Chu AC, Poon JT, Lam CS, Chow AK, et al. A subpopulation of CD26+ cancer stem cells with metastatic capacity in human colorectal cancer. Cell Stem Cell. 2010;6:603–15. 10.1016/j.stem.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 27. Liu D, Sun J, Zhu J, Zhou H, Zhang X, Zhang Y. Expression and clinical significance of colorectal cancer stem cell marker EpCAMhigh/CD44+ in colorectal cancer. Oncol Lett. 2014;7:1544–8. 10.3892/ol.2014.1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Munro MJ, Wickremesekera SK, Peng L, Tan ST, Itinteang T. Cancer stem cells in colorectal cancer: a review. J Clin Pathol. 2018;71:110–6. 10.1136/jclinpath-2017-204739. [DOI] [PubMed] [Google Scholar]
- 29. Yu X, Lin Y, Yan X, Tian Q, Li L, Lin EH. CD133, Stem Cells, and Cancer Stem Cells: Myth or Reality? Curr Colorectal Cancer Rep. 2011;7:253–9. 10.1007/s11888-011-0106-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li CY, Li BX, Liang Y, Peng RQ, Ding Y, Xu DZ, et al. Higher percentage of CD133+ cells is associated with poor prognosis in colon carcinoma patients with stage IIIB. J Transl Med. 2009;7:56. 10.1186/1479-5876-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Saigusa S, Tanaka K, Toiyama Y, Yokoe T, Okugawa Y, Koike Y, et al. Clinical significance of CD133 and hypoxia inducible factor‐1α gene expression in rectal cancer after preoperative chemoradiotherapy. Clin Oncol. 2011;23:323–32. 10.1016/j.clon.2010.09.012 [DOI] [PubMed] [Google Scholar]
- 32. Kemper K, Versloot M, Cameron K, Colak S, de Sousa e Melo F, et al. Mutations in the Ras‐Raf Axis underlie the prognostic value of CD133 in colorectal cancer. Clin Cancer Res. 2012;18:3132–41. 10.1158/1078-0432.CCR-11-3066. [DOI] [PubMed] [Google Scholar]
- 33. Pilati P, Mocellin S, Bertazza L, Galdi F, Briarava M, Mammano E, et al. Prognostic value of putative circulating cancer stem cells in patients undergoing hepatic resection for colorectal liver metastasis. Ann Surg Oncol. 2012;19:402–8. 10.1245/s10434-011-2132-2 [DOI] [PubMed] [Google Scholar]
- 34. Kemper K, Sprick MR, de Bree M, Scopelliti A, Vermeulen L, Hoek M, et al.The AC133 epitope, but not the CD133 protein, is lost upon cancer stem cell differentiation. Cancer Res. 2010;70:719–29. 10.1158/0008-5472.CAN-09-1820. [DOI] [PubMed] [Google Scholar]
- 35. Sprenger T, Conradi LC, Beissbarth T, Ermert H, Homayounfar K, Middel P, et al. Enrichment of CD133‐expressing cells in rectal cancers treated with preoperative radiochemotherapy is an independent marker for metastasis and survival. Cancer. 2013;119:26–35. 10.1002/cncr.27703 [DOI] [PubMed] [Google Scholar]
- 36. Cho YH, Ro EJ, Yoon JS, Mizutani T, Kang DW, Park JC, et al. 5‐FU promotes stemness of colorectal cancer via p53‐mediated WNT/β‐catenin pathway activation. Nat Commun. 2020;11:5321. 10.1038/s41467-020-19173-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 38. Maitland NJ, Collins AT. Prostate cancer stem cells: a new target for therapy. J Clin Oncol. 2008;26:2862–70. 10.1200/JCO.2007.15.1472. [DOI] [PubMed] [Google Scholar]
- 39. Shimada M, Sugimoto K, Iwahashi S, Utsunomiya T, Morine Y, Imura S, et al. CD133 expression is a potential prognostic indicator in intrahepatic cholangiocarcinoma. J Gastroenterol. 2010;45:896–902. 10.1007/s00535-010-0235-3. [DOI] [PubMed] [Google Scholar]
- 40. Wielenga VJ, Smits R, Korinek V, Smit L, Kielman M, Fodde R et al. Expression of CD44 in Apc and Tcf mutant mice implies regulation by the WNT pathway. Am J Pathol. 1999;154: 515–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zeilstra J, Joosten SP, Dokter M, Verwiel E, Spaargaren M, Pals ST. Deletion of the WNT target and cancer stem cell marker CD44 in Apc(Min/+) mice attenuates intestinal tumorigenesis. Cancer Res. 2008;68(10):3655–61. 10.1158/0008-5472.CAN-07-2940. [DOI] [PubMed] [Google Scholar]
- 42. Wang Z, Tang Y, Xie L, Huang A, Xue C, Gu Z, et al. The prognostic and clinical value of CD44 in colorectal cancer: a meta‐analysis. Front Oncol. 2019;9:309. 10.3389/fonc.2019.00309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mackay CR, Terpe HJ, Stauder R, Marston WL, Stark H, Günthert U. Expression and modulation of CD44 variant isoforms in humans. J Cell Biol. 1994;124(1‐2):71–82. 10.1083/jcb.124.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zeilstra J, Joosten SP, van Andel H, Tolg C, Berns A, Snoek M, et al. Stem cell CD44v isoforms promote intestinal cancer formation in Apc(min) mice downstream of Wnt signaling. Oncogene. 2014;33(5):665–70. 10.1038/onc.2012.611. [DOI] [PubMed] [Google Scholar]
- 45. Zhang X, Wang L, Qu Y. Targeting the β‐catenin signaling for cancer therapy. Pharmacol Res. 2020;104794. [DOI] [PubMed] [Google Scholar]
- 46. Zhang Y, Wang X. Targeting the Wnt/β‐catenin signaling pathway in cancer. J Hematol Oncol. 2020;13:165. 10.1186/s13045-020-00990-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rodrigues ACBDC, Costa RGA, Silva SLR, Dias IRSB, Dias RB, Bezerra DP. Cell signaling pathways as molecular targets to eliminate AML stem cells. Crit Rev Oncol Hematol. 2021;160:103277. 10.1016/j.critrevonc.2021.103277. [DOI] [PubMed] [Google Scholar]
- 48. Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36:1461–73. 10.1038/onc.2016.304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang M, Weng W, Zhang Q, Wu Y, Ni S, Tan C, et al. The lncRNA NEAT1 activates Wnt/β‐catenin signaling and promotes colorectal cancer progression via interacting with DDX5. J Hematol Oncol. 2018;11(1):113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. De Lau W, Peng WC, Gros P, Clevers H. The R‐spondin/Lgr5/Rnf43 module: regulator of Wnt signal strength. Genes Dev. 2014;28:305–16. 10.1101/gad.235473.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shenoy AK, Fisher RC, Butterworth EA, Pi L, Chang LJ, Appelman HD, et al. Transition from colitis to cancer: high Wnt activity sustains the tumor‐initiating potential of colon cancer stem cell precursors. Cancer Res. 2012;72:5091–5100. 10.1158/0008-5472.CAN-12-1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen B, Zhang D, Kuai J, Cheng M, Fang X, Li G. Upregulation of miR‐199a/b contributes to cisplatin resistance via Wnt/β‐catenin‐ABCG2 signaling pathway in ALDHA1+ colorectal cancer stem cells. Tumour Biol. 2017;39:1010428317715155. 10.1177/1010428317715155. [DOI] [PubMed] [Google Scholar]
- 53. Zhang GJ, Li LF, Yang GD, Xia SS, Wang R, Leng ZW, et al. MiR‐92a promotes stem cell‐like properties by activating Wnt/β‐catenin signaling in colorectal cancer. Oncotarget. 2017;8:101760–70. 10.18632/oncotarget.21667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shi L, Xi J, Xu X, Peng B, Zhang B. MiR‐148a suppressed cell invasion and migration via targeting WNT10b and modulating β‐catenin signaling in cisplatin‐resistant colorectal cancer cells. Biomed Pharmacother. 2019;109:902–9. 10.1016/j.biopha.2018.10.080. [DOI] [PubMed] [Google Scholar]
- 55. Jiang S, Miao D, Wang M, Lv J, Wang Y, Tong J. MiR‐30‐5p suppresses cell chemoresistance and stemness in colorectal cancer through USP22/Wnt/β‐catenin signaling axis. J Cell Mol Med. 2019;23:630–40. 10.1111/jcmm.13968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ayadi M, Bouygues A, Ouaret D, Ferrand N, Chouaib S, Thiery JP, et al. Chronic chemotherapeutic stress promotes evolution of stemness and WNT/beta‐catenin signaling in colorectal cancer cells: implications for clinical use of WNT‐signaling inhibitors. Oncotarget. 2015;6(21):18518–33. 10.18632/oncotarget.3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Masuda M, Uno Y, Ohbayashi N, Ohata H, Mimata A, Kukimoto‐Niino M, et al. TNIK inhibition abrogates colorectal cancer stemness. Nat Commun. 2016;7:12586. 10.1038/ncomms12586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ordóñez‐Morán P, Dafflon C, Imajo M, Nishida E, Huelsken J. HOXA5 counteracts stem cell traits by inhibiting Wnt signaling in colorectal cancer. Cancer Cell. 2015;28:815–29. 10.1016/j.ccell.2015.11.001 [DOI] [PubMed] [Google Scholar]
- 59. Krausova M, Korinek V. Wnt signaling in adult intestinal stem cells and cancer. Cell Signal. 2014;26:570–9. 10.1016/j.cellsig.2013.11.032. [DOI] [PubMed] [Google Scholar]
- 60. Gupta P, Wright SE, Kim SH, Srivastava SK. Phenethyl isothiocyanate: a comprehensive review of anti‐cancer mechanisms. Biochim Biophys Acta. 2014;1846:405–24. 10.1016/j.bbcan.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang D, Upadhyaya B, Liu Y, Knudsen D, Dey M. Phenethyl isothiocyanate upregulates death receptors 4 and 5 and inhibits proliferation in human cancer stem‐like cells. BMC Cancer. 2014;14:591. 10.1186/1471-2407-14-591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Upadhyaya B, Liu Y, Dey M. Phenethyl isothiocyanate exposure promotes oxidative stress and suppresses Sp1 transcription factor in cancer stem cells. Int J Mol Sci. 2019;20(5):1027. 10.3390/ijms20051027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang J, Luo B, Li X, Lu W, Yang J, Hu Y,et al. Inhibition of cancer growth in vitro and in vivo by a novel ROS‐modulating agent with ability to eliminate stem‐like cancer cells. Cell Death Dis. 2017;8(6):e2887. 10.1038/cddis.2017.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Koschorke A, Faraci S, Giani D, Chiodoni C, Iorio E, Canese R, et al. Phenethyl isothiocyanate hampers growth and progression of HER2‐positive breast and ovarian carcinoma by targeting their stem cell compartment. Cell Oncol. 2019;42(6):815–28. 10.1007/s13402-019-00464 [DOI] [PubMed] [Google Scholar]
- 65. Nguyen YT, Moon JY, Ediriweera MK, Cho SK. Phenethyl isothiocyanate suppresses stemness in the chemo‐ and radio‐resistant triple‐negative breast cancer cell line MDA‐MB‐231/IR via downregulation of metadherin. Cancers. 2020;12(2):268. 10.3390/cancers12020268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chen Y, Li Y, Wang XQ, Meng Y, Zhang Q, Zhu JY, et al. Phenethyl isothiocyanate inhibits colorectal cancer stem cells by suppressing Wnt/β‐catenin pathway. Phytother Res. 2018;32:2447–55. 10.1002/ptr.6183. [DOI] [PubMed] [Google Scholar]
- 67. Yun JH, Kim KA, Yoo G, Kim SY, Shin JM, Kim JH, et al. Phenethyl isothiocyanate suppresses cancer stem cell properties in vitro and in a xenograft model. Phytomedicine. 2017;30:42–9. 10.1016/j.phymed.2017.01.015. [DOI] [PubMed] [Google Scholar]
- 68. Shin JM, Lim E, Cho YS, Nho CW. Cancer‐preventive effect of phenethyl isothiocyanate through tumor microenvironment regulation in a colorectal cancer stem cell xenograft model. Phytomedicine. 2021;84:153493. 10.1016/j.phymed.2021.153493. [DOI] [PubMed] [Google Scholar]
- 69. Zhang Y, Talalay P, Cho CG, Posner GH. A major inducer of anticarcinogenic protective enzymes from broccoli: isolation and elucidation of structure. Proc Natl Acad Sci USA. 1992;89:2399–2403. 10.1073/pnas.89.6.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Li SH, Fu J, Watkins DN, Srivastava RK, Shankar S. Sulforaphane regulates self‐renewal of pancreatic cancer stem cells through the modulation of Sonic hedgehog‐GLI pathway. Mol Cell Biochem. 2013;373(1‐2):217–27. 10.1007/s11010-012-1493-6. [DOI] [PubMed] [Google Scholar]
- 71. Kallifatidis G, Rausch V, Baumann B, Apel A, Beckermann BM, Groth A, et al. Sulforaphane targets pancreatic tumour‐initiating cells by NF‐kappaB‐induced antiapoptotic signalling. Gut. 2009;58(7):949–63. 10.1136/gut.2008.149039. [DOI] [PubMed] [Google Scholar]
- 72. Lin LC, Yeh CT, Kuo CC, Lee CM, Yen GC, Wang LS, et al. Sulforaphane potentiates the efficacy of imatinib against chronic leukemia cancer stem cells through enhanced abrogation of Wnt/β‐catenin function. J Agric Food Chem. 2012;60(28):7031–9. 10.1021/jf301981n. [DOI] [PubMed] [Google Scholar]
- 73. Gamet‐Payrastre L, Li P, Lumeau S, Cassar G, Dupont MA, Chevolleau S, et al. Sulforaphane, a naturally occurring isothiocyanate, induces cell cycle arrest and apoptosis in HT29 human colon cancer cells. Cancer Res. 2000;60(5):1426–33. [PubMed] [Google Scholar]
- 74. Wang M, Chen S, Wang S, Sun D, Chen J, Li Y, et al. Effects of phytochemicals sulforaphane on uridine diphosphate‐glucuronosyltransferase expression as well as cell‐cycle arrest and apoptosis in human colon cancer Caco‐2 cells. Chin J Physiol. 2012;55(2):134–44. 10.4077/CJP.2012.BAA085. [DOI] [PubMed] [Google Scholar]
- 75. Chen Y, Wang MH, Zhu JY, Xie CF, Li XT, Wu JS, et al. TAp63α targeting of Lgr5 mediates colorectal cancer stem cell properties and sulforaphane inhibition. Oncogenesis. 2020;9:89. 10.1038/s41389-020-00273-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Dong TT, Zhou HM, Wang LL, Feng B, Lv B, Zheng MH. Salinomycin selectively targets ‘CD133+’ cell subpopulations and decreases malignant traits in colorectal cancer lines. Ann Surg Oncol. 2011;18:1797–1804. 10.1245/s10434-011-1561-2. [DOI] [PubMed] [Google Scholar]
- 77. Zhang C, Tian Y, Song F, Fu C, Han B, Wang Y. Salinomycin inhibits the growth of colorectal carcinoma by targeting tumor stem cells. Oncol Rep. 2015;34:2469–76. 10.3892/or.2015.4253 [DOI] [PubMed] [Google Scholar]
- 78. Wang Z, Zhou L, Xiong Y, Yu S, Li H, Fan J, et al. Salinomycin exerts anti‐colorectal cancer activity by targeting the β‐catenin/T‐cell factor complex. Br J Pharmacol. 2019;176:3390–3406. 10.1111/bph.14770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Klose J, Trefz S, Wagner T, Steffen L, A Preißendörfer Charrier, Radhakrishnan P, et al. Salinomycin: Anti‐tumor activity in a pre‐clinical colorectal cancer model. PLoS One. 2019;14:e0211916. 10.1371/journal.pone.0211916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Singh AK, Verma A, Singh A, Arya RK, Maheshwari S, Chaturvedi P, et al. Salinomycin inhibits epigenetic modulator EZH2 to enhance death receptors in colon cancer stem cells. Epigenetics. 2021;16(2):144–61. 10.1080/15592294.2020.1789270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhou J, Liu S, Wang Y, Dai W, Zou H, Wang S, et al. Salinomycin effectively eliminates cancer stem‐like cells and obviates hepatic metastasis in uveal melanoma. Mol Cancer. 2019;18(1):159. 10.1186/s12943-019-1068-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Mineo PG, Foti C, Vento F, Montesi M, Panseri S, Piperno A, et al. Salinomycin‐loaded PLA nanoparticles: drug quantification by GPC and wave voltammetry and biological studies on osteosarcoma cancer stem cells. Anal Bioanal Chem. 2020;412(19):4681–90. 10.1007/s00216-020-02721-6. [DOI] [PubMed] [Google Scholar]
- 83. Wang H, Zhang H, Zhu Y, Wu Z, Cui C, Cai F. Anticancer Mechanisms of Salinomycin in Breast Cancer and Its Clinical Applications. Front Oncol. 2021;11:654428. 10.3389/fonc.2021.654428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wang J, Zhuo J, Tao Y, Xu S, Chen Z, Yang F, et al. Salinomycin‐loaded small‐molecule nanoprodrugs enhance anticancer activity in hepatocellular carcinoma. Int J Nanomedicine. 2020;15:6839–54. 10.2147/IJN.S236928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Magrath JW, Raney WR, Kim Y. In vitro demonstration of salinomycin as a novel chemotherapeutic agent for the treatment of SOX2‐positive glioblastoma cancer stem cells. Oncol Rep. 2020;44(2):777–85. 10.3892/or.2020.7642. [DOI] [PubMed] [Google Scholar]
- 86. Harrington BS, Ozaki MK, Caminear MW, Hernandez LF, Jordan E, Kalinowski NJ, et al. Drugs targeting tumor‐initiating cells prolong survival in a post‐surgery, post‐chemotherapy ovarian cancer relapse model. Cancers. 2020;12(6):1645. 10.3390/cancers12061645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Patel S, Patel A, Nair A, Shah K, Shah K, Tanavde V, et al. Salinomycin mediated therapeutic targeting of circulating stem like cell population in oral cancer. J Biomol Struct Dyn. 2021;1–13. 10.1080/07391102.2021.1957018. [DOI] [PubMed] [Google Scholar]
- 88. Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, et al. Identification of selective inhibitors of cancer stem cells by high‐throughput screening. Cell. 2009;138(4):645–59. 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wang Z, Feng T, Zhou L, Jiang D, Zhang Y, He G, et al. Salinomycin nanocrystals for colorectal cancer treatment through inhibition of Wnt/β‐catenin signaling. Nanoscale. 2020;12:19931–8. 10.1039/d0nr04552g. [DOI] [PubMed] [Google Scholar]
- 90. Tsakiris N, Fauvet F, Ruby S, Puisieux A, Paquot A, Muccioli GG, et al. Combined nanomedicines targeting colorectal cancer stem cells and cancer cells. J Control Release. 2020;326:387–95. 10.1016/j.jconrel.2020.07.025. [DOI] [PubMed] [Google Scholar]
- 91. Jiang P, Xu C, Zhang P, Ren J, Mageed F, Wu X, et al. Epigallocatechin‐3‐gallate inhibits self‐renewal ability of lung cancer stem‐like cells through inhibition of CLOCK. Int J Mol Med. 2020;46(6):2216–24. 10.3892/ijmm.2020.4758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Sun X, Song J, Li E, Geng H, Li Y, Yu D, et al. (‐)‐Epigallocatechin‐3‐gallate inhibits bladder cancer stem cells via suppression of sonic hedgehog pathway. Oncol Rep. 2019;42(1):425–35. 10.3892/or.2019.7170. [DOI] [PubMed] [Google Scholar]
- 93. Kumar G, Farooqui M, Rao CV. Role of dietary cancer‐preventive phytochemicals in pancreatic cancer stem cells. Curr Pharmacol Rep. 2018;4(4):326–35. 10.1007/s40495-018-0145-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Gu HF, Mao XY, Du M. Prevention of breast cancer by dietary polyphenols‐role of cancer stem cells. Crit Rev Food Sci Nutr. 2020;60(5):810–25. 10.1080/10408398.2018.1551778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Hu F, Wei F, Wang Y, Wu B, Fang Y, Xiong B. EGCG synergizes the therapeutic effect of cisplatin and oxaliplatin through autophagic pathway in human colorectal cancer cells. J Pharmacol Sci. 2015;128:27–34. 10.1016/j.jphs.2015.04.003. [DOI] [PubMed] [Google Scholar]
- 96. Chen Y, Wang XQ, Zhang Q, Zhu JY, Li Y, Xie CF, et al. (‐)‐Epigallocatechin‐3‐gallate inhibits colorectal cancer stem cells by suppressing Wnt/β‐catenin pathway. Nutrients, 2017;9(6):572. 10.3390/nu9060572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Toden S, Tran HM, Tovar‐Camargo OA, Okugawa Y, Goel A. Epigallocatechin‐3‐gallate targets cancer stem‐like cells and enhances 5‐fluorouracil chemosensitivity in colorectal cancer. Oncotarget. 2016;7:16158–71. 10.18632/oncotarget.7567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Shimizu M, Fukutomi Y, Ninomiya M, Nagura K, Kato T, Araki H, et al. Green tea extracts for the prevention of metachronous colorectal adenomas: a pilot study. Cancer Epidemiol Biomarkers Prev. 2008;17:3020–5. 10.1158/1055-9965.EPI-08-0528. [DOI] [PubMed] [Google Scholar]
- 99. Hu Y, McIntosh GH, Le Leu RK, Somashekar R, Meng XQ, Gopalsamy G, et al. Supplementation with Brazil nuts and green tea extract regulates targeted biomarkers related to colorectal cancer risk in humans. Br J Nutr. 2016;116(11):1901–11. 10.1017/S0007114516003937. [DOI] [PubMed] [Google Scholar]
- 100. Watanabe D, Murakami H, Ohno H, Tanisawa K, Konishi K, Tsunematsu Y, et al. Association between dietary intake and the prevalence of tumourigenic bacteria in the gut microbiota of middle‐aged Japanese adults. Sci Rep. 2020;10(1):15221. 10.1038/s41598-020-72245-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Kannappan R, Gupta SC, Kim JH, Aggarwal BB. Tocotrienols fight cancer by targeting multiple cell signaling pathways. Genes Nutr. 2012;7:43–52. 10.1007/s12263-011-0220-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Husain K, Coppola D, Yang CS, Malafa MP. Farnesyl dimethyl chromanol targets colon cancer stem cells and prevents colorectal cancer metastasis. Sci Rep. 2021;11(1):2185. 10.1038/s41598-020-80911-z [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 103. Bamodu OA, Yang CK, Cheng WH, Tzeng DTW, Kuo KT, Huang CC, et al. 4‐Acetyl‐antroquinonol b suppresses SOD2‐enhanced cancer stem cell‐like phenotypes and chemoresistance of colorectal cancer cells by inducing hsa‐miR‐324 re‐expression. Cancers. 2018;10(8):269. 10.3390/cancers10080269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Chang TC, Yeh CT, Adebayo BO, Lin YC, Deng L, Rao YK, et al. 4‐Acetylantroquinonol B inhibits colorectal cancer tumorigenesis and suppresses cancer stem‐like phenotype. Toxicol Appl Pharmacol. 2015;288(2):258–68. 10.1016/j.taap.2015.07.025. [DOI] [PubMed] [Google Scholar]
- 105. Zhang Q, Li XT, Chen Y, Chen JQ, Zhu JY, Meng Y, et al. Wnt/β‐catenin signaling mediates the suppressive effects of diallyl trisulfide on colorectal cancer stem cells. Cancer Chemother Pharmacol. 2018;81(6):969–77. 10.1007/s00280-018-3565-0. [DOI] [PubMed] [Google Scholar]
- 106. Dermani FK, Amini R, Saidijam M, Pourjafar M, Saki S, Najafi R. Zerumbone inhibits epithelial‐mesenchymal transition and cancer stem cells properties by inhibiting the β‐catenin pathway through miR‐200c. J Cell Physiol. 2018;233(12):9538–47. 10.1002/jcp.26874. [DOI] [PubMed] [Google Scholar]
- 107. Lim YC, Kang HJ, Kim YS, Choi EC. All‐trans‐retinoic acid inhibits growth of head and neck cancer stem cells by suppression of Wnt/β‐catenin pathway. Eur J Cancer. 2012;48(17):3310–8. 10.1016/j.ejca.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 108. Wang L, Chang J, Varghese D, Dellinger M, Kumar S, Best AM, et al. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat Commun. 2013;4:2035. 10.1038/ncomms3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Kim MS, Cho HI, Yoon HJ, Ahn YH, Park EJ, Jin YH, et al. JIB‐04, a small molecule histone demethylase inhibitor, selectively targets colorectal cancer stem cells by inhibiting the Wnt/β‐catenin signaling pathway. Sci Rep. 2018;8:6611. 10.1038/s41598-018-24903-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Wang L, Chang J, Varghese D, Dellinger M, Kumar S, Best AM, et al. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat Commun. 2013;4:2035. 10.1038/ncomms3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Urushibara S, Tsubota T, Asai R, Azumi J, Ashida K, Fujiwara Y, et al. WNT/β‐Catenin signaling inhibitor IC‐2 suppresses sphere formation and sensitizes colorectal cancer cells to 5‐fluorouracil. Anticancer Res. 2017;37:4085–91. 10.21873/anticanres.11795. [DOI] [PubMed] [Google Scholar]
- 112. Seto K, Sakabe T, Itaba N, Azumi J, Oka H, Morimoto M, et al. A novel small‐molecule WNT inhibitor, IC‐2, has the potential to suppress liver cancer stem cells. Anticancer Res. 2017;37(7):3569–79. 10.21873/anticanres. [DOI] [PubMed] [Google Scholar]
- 113. Yokogi S, Tsubota T, Kanki K, Azumi J, Itaba N, Oka H, et al. Wnt/Beta‐Catenin signal inhibitor HC‐1 sensitizes oral squamous cell carcinoma cells to 5‐fluorouracil through reduction of CD44‐positive population. Yonago Acta Med. 2016;59(2):93–9. [PMC free article] [PubMed] [Google Scholar]
- 114. Mahmoudi T, Li VS, Ng SS, Taouatas N, Vries RG, Mohammed S, et al. The kinase TNIK is an essential activator of Wnt target genes. EMBO J. 2009;28:3329–40. 10.1038/emboj.2009.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Padgaonkar A, Cosenza S, Pallela V. The dual CK2/TNIK inhibitor, ON108600 targets cancer stem cells and induces apoptosis of paclitaxel resistant triple‐negative breast cancer cells. Cancer Res. 2015;75:4453. 10.1158/1538-7445.AM2015-4453. [DOI] [Google Scholar]
- 116. Tan Z, Chen L, Zhang S. Comprehensive modeling and discovery of mebendazole as a novel TRAF2‐ and NCK‐interacting kinase inhibitor. Sci Rep. 2016;6:33534. 10.1038/srep33534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Williamson T, Bai RY, Staedtke V, Huso D, Riggins GJ. Mebendazole and a non‐steroidal anti‐inflammatory combine to reduce tumor initiation in a colon cancer preclinical model. Oncotarget. 2016;7:68571–84. 10.18632/oncotarget.11851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Kumar B, Ahmad R, Sharma S, Gowrikumar S, Primeaux M, Rana S, et al. PIK3C3 inhibition promotes sensitivity to colon cancer therapy by inhibiting cancer stem cells. Cancers. 2021;13(9):2168. 10.3390/cancers13092168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Hsu HC, Liu YS, Tseng KC, Yang TS, Yeh CY, You JF, et al. CBB1003, a lysine‐specific demethylase 1 inhibitor, suppresses colorectal cancer cells growth through down‐regulation of leucine‐rich repeat‐containing G‐protein‐coupled receptor 5 expression. J Cancer Res Clin Oncol. 2015;141(1), 11–21. 10.1007/s00432-014-1782-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Proto MC, Fiore D, Piscopo C, Franceschelli S, Bizzarro V, Laezza C, et al. Inhibition of Wnt/β‐Catenin pathway and Histone acetyltransferase activity by Rimonabant: a therapeutic target for colon cancer. Sci Rep. 2017;7(1):11678. 10.1038/s41598-017-11688-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Fiore D, Ramesh P, Proto MC, Piscopo C, Franceschelli S, Anzelmo S, et al. Rimonabant kills colon cancer stem cells without inducing toxicity in normal colon organoids. Front Pharmacol. 2018;8:949. 10.3389/fphar.2017.00949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Handeli S, Simon JA. A small‐molecule inhibitor of Tcf/beta‐catenin signaling down‐regulates PPARgamma and PPARdelta activities. Mol Cancer Ther. 2008;7(3):521–9. 10.1158/1535-7163.MCT-07-2063. [DOI] [PubMed] [Google Scholar]
- 123. Chen Y, Rao X, Huang K, Jiang X, Wang H, Teng L. FH535 inhibits proliferation and motility of colon cancer cells by targeting Wnt/β‐catenin signaling pathway. J Cancer. 2017;8(16):3142–53. 10.7150/jca.19273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Giraud J, Failla LM, Pascussi JM, Lagerqvist EL, Ollier J, Finetti P, et al. Autocrine secretion of progastrin promotes the survival and self‐renewal of colon cancer stem‐like cells. Cancer Res. 2016;76:3618–28. 10.1158/0008-5472.CAN-15-1497. [DOI] [PubMed] [Google Scholar]
- 125. Pannequin J, Delaunay N, Buchert M, Surrel F, Bourgaux JF, Ryan J, et al. Beta‐catenin/Tcf‐4 inhibition after progastrin targeting reduces growth and drives differentiation of intestinal tumors. Gastroenterology. 2007;133:1554–68. 10.1053/j.gastro.2007.08.023 [DOI] [PubMed] [Google Scholar]
- 126. Prieur A, Cappellini M, Habif G, Lefranc MP, Mazard T, Morency E, et al. Targeting the Wnt pathway and cancer stem cells with anti‐progastrin humanized antibodies as a potential treatment for K‐RAS‐mutated colorectal cancer. Clin Cancer Res. 2017;23:5267–80. 10.1158/1078-0432.CCR-17-0533. [DOI] [PubMed] [Google Scholar]
- 127. Zhan T, Ambrosi G, Wandmacher AM, Rauscher B, Betge J, Rindtorff N, et al. MEK inhibitors activate Wnt signalling and induce stem cell plasticity in colorectal cancer. Nat Commun. 2019;10:2197. 10.1038/s41467-019-09898-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Espinosa‐Sánchez A, Suárez‐Martínez E, Sánchez‐Díaz L, Carnero A. Therapeutic Targeting of Signaling Pathways Related to Cancer Stemness. Front Oncol. 2020;10:1533. 10.3389/fonc.2020.01533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Fiúza UM, Arias AM. Cell and molecular biology of Notch. J Endocrinol. 2007;194:459–74. 10.1677/JOE-07-0242. [DOI] [PubMed] [Google Scholar]
- 130. Bray SJ. Notch signalling in context. Nat Rev Mol Cell Biol. 2016;17:722–35. 10.1038/nrm.2016.94. [DOI] [PubMed] [Google Scholar]
- 131. Li L, Tang P, Li S, Qin X, Yang H, Wu C, et al. Notch signaling pathway networks in cancer metastasis: a new target for cancer therapy. Med Oncol. 2017;34:180. 10.1007/s12032-017-1039-6. [DOI] [PubMed] [Google Scholar]
- 132. Yuan R, Ke J, Sun L, He Z, Zou Y, He X, et al. HES1 promotes metastasis and predicts poor survival in patients with colorectal cancer. Clin Exp Metastasis. 2015;32:169–79. 10.1007/s10585-015-9700-y [DOI] [PubMed] [Google Scholar]
- 133. Huang R, Wang G, Song Y, Tang Q, You Q, Liu Z, et al. Colorectal cancer stem cell and chemoresistant colorectal cancer cell phenotypes and increased sensitivity to Notch pathway inhibitor. Mol Med Rep. 2015;12:2417–24. 10.3892/mmr.2015.3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Ong CP, Lee WL, Tang YQ, Yap WH. Honokiol: a review of its anticancer potential and mechanisms. Cancers. 2019;12:48. 10.3390/cancers12010048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Kaushik G, Venugopal A, Ramamoorthy P, Standing D, Subramaniam D, Umar S, et al. Honokiol inhibits melanoma stem cells by targeting notch signaling. Mol Carcinog. 2015;54:1710–21. 10.1002/mc.22242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Fan Y, Xue W, Schachner M, Zhao W. Honokiol eliminates glioma/glioblastoma stem cell‐like cells via JAK‐STAT3 Signaling and inhibits tumor progression by targeting epidermal growth factor receptor. Cancers. 2018;11:22. 10.3390/cancers11010022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Sengupta S, Nagalingam A, Muniraj N, Bonner MY, Mistriotis P, Afthinos A, et al. Activation of tumor suppressor LKB1 by honokiol abrogates cancer stem‐like phenotype in breast cancer via inhibition of oncogenic Stat3. Oncogene. 2017;36:5709–21. 10.1038/onc.2017.164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Yao CJ, Lai GM, Yeh CT, Lai MT, Shih PH, Chao WJ, et al. Honokiol eliminates human oral cancer stem‐like cells accompanied with suppression of Wnt/β‐catenin signaling and apoptosis induction. Evid Based Complement Alternat Med. 2013;2013:10. 10.1155/2013/146136.146136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Ponnurangam S, Mammen JM, Ramalingam S, He Z, Zhang Y, Umar S, et al. Honokiol in combination with radiation targets notch signaling to inhibit colon cancer stem cells. Mol Cancer Ther. 2012;11:963–72. 10.1158/1535-7163.MCT-11-0999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Subramaniam D, Ponnurangam S, Ramalingam S, Kwatra D, Dandawate P, Weir SJ, et al. Honokiol affects stem cell viability by suppressing oncogenic YAP1 function to inhibit colon tumorigenesis. Cells. 2021;10(7):1607. 10.3390/cells10071607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Shang Y, Ma Y, Zhou Y, Zhang H, Duan L, Chen H, et al. Plant science. Biosynthesis, regulation, and domestication of bitterness in cucumber. Science. 2014;346:1084–8. 10.1126/science.1259215 [DOI] [PubMed] [Google Scholar]
- 142. Dandawate P, Subramaniam D, Panovich P, Standing D, Krishnamachary B, Kaushik G, et al. Cucurbitacin B and I inhibits colon cancer growth by targeting the Notch signaling pathway. Sci Rep. 2020;10:1290. 10.1038/s41598-020-57940-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Wang X, Bai Y, Yan X, Li J, Lin B, Dai L, et al. Cucurbitacin B exhibits antitumor effects on CD133+ HepG2 liver cancer stem cells by inhibiting JAK2/STAT3 signaling pathway. Anticancer Drugs. 2021;32(5):548–57. 10.1097/CAD.0000000000001062. [DOI] [PubMed] [Google Scholar]
- 144. Xu J, Chen Y, Yang R, Zhou T, Ke W, Si Y, et al. Cucurbitacin B inhibits gastric cancer progression by suppressing STAT3 activity. Arch Biochem Biophys. 2020;684:108314. 10.1016/j.abb.2020.108314. [DOI] [PubMed] [Google Scholar]
- 145. Hsu HS, Huang PI, Chang YL, Tzao C, Chen YW, Shih HC, et al. Cucurbitacin I inhibits tumorigenic ability and enhances radiochemosensitivity in nonsmall cell lung cancer‐derived CD133‐positive cells. Cancer. 2011;117(13):2970–85. 10.1002/cncr.25869. [DOI] [PubMed] [Google Scholar]
- 146. Stechishin OD, Luchman HA, Ruan Y, Blough MD, Nguyen SA, Kelly JJ, et al. On‐target JAK2/STAT3 inhibition slows disease progression in orthotopic xenografts of human glioblastoma brain tumor stem cells. Neuro Oncol. 2013;15(2):198–207. 10.1093/neuonc/nos302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Chen YW, Chen KH, Huang PI, Chen YC, Chiou GY, Lo WL, et al. Cucurbitacin I suppressed stem‐like property and enhanced radiation‐induced apoptosis in head and neck squamous carcinoma–derived CD44(+)ALDH1(+) cells. Mol Cancer Ther. 2010;9(11):2879–92. 10.1158/1535-7163.MCT-10-0504. [DOI] [PubMed] [Google Scholar]
- 148. Huang J, Chen ZH, Ren CM, Wang DX, Yuan SX, Wu QX, et al. Antiproliferation effect of evodiamine in human colon cancer cells is associated with IGF‐1/HIF‐1α downregulation. Oncol Rep. 2015;34:3203–11. 10.3892/or.2015.4309. [DOI] [PubMed] [Google Scholar]
- 149. Zhang C, Fan X, Xu X, Yang X, Wang X, Liang HP. Evodiamine induces caspase‐dependent apoptosis and S phase arrest in human colon lovo cells. Anticancer Drugs. 2010;21:766–76. 10.1097/CAD.0b013e32833d26a9. [DOI] [PubMed] [Google Scholar]
- 150. Kim H, Yu Y, Choi S, Lee H, Yu J, Lee JH, et al. Evodiamine eliminates colon cancer stem cells via suppressing notch and Wnt signaling. Molecules. 2019;24:4520. 10.3390/molecules24244520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Li Y, Wang Z, Jin J, Zhu SX, He GQ, Li SH, et al. Quercetin pretreatment enhances the radiosensitivity of colon cancer cells by targeting Notch‐1 pathway. Biochem Biophys Res Commun. 2020;523:947–53. 10.1016/j.bbrc.2020.01.048. [DOI] [PubMed] [Google Scholar]
- 152. Iranshahy M, Javadi B, Iranshahi M, Jahanbakhsh SP, Mahyari S, Hassani FV, et al. A review of traditional uses, phytochemistry and pharmacology of Portulaca oleracea L. J Ethnopharmacol. 2017;205:158–72. 10.1016/j.jep.2017.05.004. [DOI] [PubMed] [Google Scholar]
- 153. Jin H, Chen L, Wang S, Chao D. Portulaca oleracea extract can inhibit nodule formation of colon cancer stem cells by regulating gene expression of the Notch signal transduction pathway. Tumour Biol. 2017;39:1010428317708699. 10.1177/1010428317708699. [DOI] [PubMed] [Google Scholar]
- 154. Hafeez BB, Mustafa A, Fischer JW, Singh A, Zhong W, Shekhani MO, et al. alpha‐Mangostin: a dietary antioxidant derived from the pericarp of Garcinia mangostana L. inhibits pancreatic tumor growth in xenograft mouse model. Antioxid Redox Signal. 2014;21:682–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Chandra Boinpelly V, Verma RK, Srivastav S, Srivastava RK, Shankar S. α‐Mangostin‐encapsulated PLGA nanoparticles inhibit colorectal cancer growth by inhibiting Notch pathway. J Cell Mol Med. 2020;24(19), 11343–54. 10.1111/jcmm.15731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Qi F, Wei L, Shen A, Chen Y, Lin J, Chu J, et al. Pien Tze Huang inhibits the proliferation, and induces the apoptosis and differentiation of colorectal cancer stem cells via suppression of the Notch1 pathway. Oncol Rep. 2016;35(1):511–7. 10.3892/or.2015.4378. [DOI] [PubMed] [Google Scholar]
- 157. Hoey T, Yen WC, Axelrod F, Basi J, Donigian L, Dylla S, et al. DLL4 blockade inhibits tumor growth and reduces tumor‐initiating cell frequency. Cell Stem Cell. 2009;5(2):168–77. 10.1016/j.stem.2009.05.019. [DOI] [PubMed] [Google Scholar]
- 158. Fischer M, Yen WC, Kapoun AM, Wang M, O'Young G, Lewicki J, et al. Anti‐DLL4 inhibits growth and reduces tumor‐initiating cell frequency in colorectal tumors with oncogenic KRAS mutations. Cancer Res. 2011;71(5):1520–5. 10.1158/0008-5472.CAN-10-2817. [DOI] [PubMed] [Google Scholar]
- 159. Fre S, Pallavi SK, Huyghe M, Laé M, Janssen KP, Robine S, et al. Notch and Wnt signals cooperatively control cell proliferation and tumorigenesis in the intestine. Proc Natl Acad Sci USA. 2009;106:6309–14. 10.1073/pnas.0900427106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. 2015;12:445–64. 10.1038/nrclinonc.2015.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev. 2008;22:2454–72. 10.1101/gad.1693608. [DOI] [PubMed] [Google Scholar]
- 162. Sari IN, Phi LTH, Jun N, Wijaya YT, Lee S, Kwon HY. Hedgehog signaling in cancer: a prospective therapeutic target for eradicating cancer stem cells. Cells. 2018;7(11):208. 10.3390/cells7110208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Wu C, Zhu X, Liu W, Ruan T, Tao K. Hedgehog signaling pathway in colorectal cancer: function, mechanism, and therapy. Onco Targets Ther. 2017;10:3249–59. 10.2147/OTT.S139639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Kolterud A, Grosse AS, Zacharias WJ, Walton KD, Kretovich KE, Madison BB, et al. Paracrine Hedgehog signaling in stomach and intestine: new roles for hedgehog in gastrointestinal patterning. Gastroenterology. 2009;137:618–28. 10.1053/j.gastro.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Jenkins D. Hedgehog signalling: emerging evidence for non‐canonical pathways. Cell Signal. 2009;21:1023–34. 10.1016/j.cellsig.2009.01.033. [DOI] [PubMed] [Google Scholar]
- 166. Song L, Li ZY, Liu WP, Zhao MR. Crosstalk between Wnt/β‐catenin and Hedgehog/Gli signaling pathways in colon cancer and implications for therapy. Cancer Biol Ther. 2015;16:1–7. 10.4161/15384047.2014.972215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Van den Brink GR, Hardwick JC. Hedgehog Wnteraction in colorectal cancer. Gut. 2006;55:912–4. 10.1136/gut.2005.085902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Regan JL, Schumacher D, Staudte S, Steffen, A , Haybaeck J, Keilholz U, et al. Non‐Canonical Hedgehog signaling is a positive regulator of the WNT pathway and is required for the survival of colon cancer stem cells. Cell Rep. 2017;21:2813–28. 10.1016/j.celrep.2017.11.025. [DOI] [PubMed] [Google Scholar]
- 169. Varnat F, Duquet A, Malerba M, Zbinden M, Mas C, Gervaz P, et al. Human colon cancer epithelial cells harbour active HEDGEHOG‐GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol Med. 2009;1:338–51. 10.1002/emmm.200900039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Kim J, Aftab BT, Tang JY, Kim D, Lee AH, Rezaee M, et al. Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell. 2013;23(1):23–34. 10.1016/j.ccr.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Ning K, Ning W, Ning X, Wang X, Zhou F. Effect of As2O3 on colorectal CSCs stained with ALDH1 in primary cell culture in vitro. Oncol Lett. 2018;16(3):4008–12. 10.3892/ol.2018.9110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Chen JK, Taipale J, Cooper MK, Beachy PA. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002;16:2743–8. 10.1101/gad.1025302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Batsaikhan BE, Yoshikawa K, Kurita N, Iwata T, Takasu C, Kashihara H, et al. Cyclopamine decreased the expression of Sonic Hedgehog and its downstream genes in colon cancer stem cells. Anticancer Res. 2014;34:6339–44. [PubMed] [Google Scholar]
- 174. Zhou H, Xiong Y, Peng L, Wang R, Zhang H, Fu Z. LncRNA‐cCSC1 modulates cancer stem cell properties in colorectal cancer via activation of the Hedgehog signaling pathway. J Cell Biochem. 2020;121:2510–24. 10.1002/jcb.29473. [DOI] [PubMed] [Google Scholar]
- 175. Robarge KD, Brunton SA, Castanedo GM, Cui Y, Dina MS, Goldsmith R, et al. GDC‐0449‐a potent inhibitor of the hedgehog pathway. Bioorg Med Chem Lett. 2009;19:5576–81. 10.1016/j.bmcl.2009.08.049 [DOI] [PubMed] [Google Scholar]
- 176. Wu C, Hu S, Cheng J, Wang G, Tao K. Smoothened antagonist GDC‐0449 (Vismodegib) inhibits proliferation and triggers apoptosis in colon cancer cell lines. Exp Ther Med. 2017;13:2529–36. 10.3892/etm.2017.4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Chen Q, Zhang H, Wu M, Wang Q, Luo L, Ma H, et al. Discovery of a potent hedgehog pathway inhibitor capable of activating caspase8‐dependent apoptosis. J Pharmacol Sci. 2018;137:256–64. 10.1016/j.jphs.2018.07.001. [DOI] [PubMed] [Google Scholar]
- 178. Berlin J, Bendell JC, Hart LL, Firdaus I, Gore I, Hermann RC, et al. A randomized phase II trial of vismodegib versus placebo with FOLFOX or FOLFIRI and bevacizumab in patients with previously untreated metastatic colorectal cancer. Clin Cancer Res. 2013;19:258–67. 10.1158/1078-0432.CCR-12-1800. [DOI] [PubMed] [Google Scholar]
- 179. Gupta SC, Sundaram C, Reuter S, Aggarwal BB. Inhibiting NF‐κB activation by small molecules as a therapeutic strategy. Biochim Biophys Acta. 2010;1799:775–87. 10.1016/j.bbagrm.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Sun SC. The non‐canonical NF‐κB pathway in immunity and inflammation. Nat Rev Immunol. 2017;17:545–58. 10.1038/nri.2017.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Rinkenbaugh AL, Baldwin AS. The NF‐κB pathway and cancer stem cells. Cells. 2016;5:16. 10.3390/cells5020016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Kaltschmidt C, Banz‐Jansen C, Benhidjeb T, Beshay M, Förster C, Greiner J, et al. A role for NF‐κB in organ specific cancer and cancer stem cells. Cancers. 2019;11:655. 10.3390/cancers11050655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Mayo MW, Wang CY, Cogswell PC, Rogers‐Graham K S, Lowe SW, Der CJ, et al. Requirement of NF‐kappaB activation to suppress p53‐independent apoptosis induced by oncogenic Ras. Science. 1997;278:1812–5. 10.1126/science.278.5344.1812. [DOI] [PubMed] [Google Scholar]
- 184. Ying H, Elpek KG, Vinjamoori A, Zimmerman SM, Chu GC, Yan H, et al. PTEN is a major tumor suppressor in pancreatic ductal adenocarcinoma and regulates an NF‐κB‐cytokine network. Cancer Discov. 2011;1:158–69. 10.1158/2159-8290.CD-11-0031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Cooks T, Pateras IS, Tarcic O, Solomon H, Schetter AJ, Wilder S, et al. Mutant p53 prolongs NF‐κB activation and promotes chronic inflammation and inflammation‐associated colorectal cancer. Cancer Cell. 2013;23:634–46. 10.1016/j.ccr.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Taniguchi K, Karin M. NF‐κB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. 2018;18:309–24. 10.1038/nri.2017.142. [DOI] [PubMed] [Google Scholar]
- 187. Terzić J, Grivennikov S, Karin E, Karin M. Inflammation and colon cancer. Gastroenterology. 2010;138:2101–14.e5. 10.1053/j.gastro.2010.01.058. [DOI] [PubMed] [Google Scholar]
- 188. Wang L, Guo J, Zhou J, Wang D, Kang X, Zhou L. NF‐κB maintains the stemness of colon cancer cells by downregulating miR‐195‐5p/497‐5p and upregulating MCM2. J Exp Clin Cancer Res. 2020;39:225. 10.1186/s13046-020-01704-w. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 189. Brooks AJ, Putoczki T. JAK‐STAT signalling pathway in cancer. Cancers. 2020;12:1971. 10.3390/cancers12071971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Levy DE, Darnell JE Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–62. 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 191. Quintás‐Cardama A, Verstovsek S. Molecular pathways: Jak/STAT pathway: mutations, inhibitors, and resistance. Clin Cancer Res. 2013;19:1933–40. 10.1158/1078-0432.CCR-12-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Lokau J, Schoeder V, Haybaeck J, Garbers C. Jak‐Stat Signaling Induced by Interleukin‐6 Family Cytokines in Hepatocellular Carcinoma. Cancers. 2019;11:1704. 10.3390/cancers11111704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Jin W. Role of JAK/STAT3 signaling in the regulation of metastasis, the transition of cancer stem cells, and chemoresistance of cancer by epithelial‐mesenchymal transition. Cells. 2020;9:217. 10.3390/cells9010217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Corvinus FM, Orth C, Moriggl R, Tsareva SA, Wagner S, Pfitzner EB, et al. Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth. Neoplasia. 2005;7:545–55. 10.1593/neo.04571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Lin L, Liu Y, Li H, Li P K, Fuchs J, Shibata H, et al. Targeting colon cancer stem cells using a new curcumin analogue, GO‐Y030. Br J Cancer. 2011;105:212–20. 10.1038/bjc.2011.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Li Y, Rogoff HA, Keates S, Gao Y, Murikipudi S, Mikule K, et al. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc Natl Acad Sci USA. 2015;112:1839–44. 10.1073/pnas.1424171112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Taniguchi H, Masuishi T, Kawazoe A, Muro K, Kadowaki S, Bando H, et al. Phase I study of napabucasin in combination with FOLFIRI + bevacizumab in Japanese patients with metastatic colorectal cancer. Int J Clin Oncol. 2021; in press. 10.1007/s10147-021-01987-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Onker DJ, Nott L, Yoshino T, Gill S, Shapiro J, Ohtsu A, et al. Napabucasin versus placebo in refractory advanced colorectal cancer: a randomised phase 3 trial. Lancet Gastroenterol Hepatol. 2018;3:263–70. 10.1016/S2468-125330009-8. [DOI] [PubMed] [Google Scholar]
- 199. Chung SY, Chen YH, Lin PR, Chao TC, Su JC, Shiau CW, et al. Two novel SHP‐1 agonists, SC‐43 and SC‐78, are more potent than regorafenib in suppressing the in vitro stemness of human colorectal cancer cells. Cell Death Discov. 2018;4:25. 10.1038/s41420-018-0084-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. You M, Jin J, Liu Q, Xu Q, Shi J, Hou Y. PPARα promotes cancer cell glut1 transcription repression. J Cell Biochem. 2017;118:1556–62. 10.1002/jcb.25817. [DOI] [PubMed] [Google Scholar]
- 201. Hou Y, Moreau F, Chadee K. PPARγ is an E3 ligase that induces the degradation of NFκB/p65. Nat Commun. 2012;3:1300. 10.1038/ncomms2270. [DOI] [PubMed] [Google Scholar]
- 202. Zhang W, Xu Y, Xu Q, Shi H, Shi J, Hou Y. PPARδ promotes tumor progression via activation of Glut1 and SLC1‐A5 transcription. Carcinogenesis. 2017;38:748–55. 10.1093/carcin/bgx035. [DOI] [PubMed] [Google Scholar]
- 203. Zhang Y, Zhang X, Wang J, Shen Y, Tang X, Yu F, et al. Expression and function of PPARs in cancer stem cells. Curr Stem Cell Res Ther. 2016;11:226–34. 10.2174/1574888x10666150728122921 [DOI] [PubMed] [Google Scholar]
- 204. Kuramoto K, Yamamoto M, Suzuki S, Togashi K, Sanomachi T, Kitanaka C, et al. Inhibition of the lipid droplet‐peroxisome proliferator‐activated receptor α axis suppresses cancer stem cell properties. Genes. 2021;12:99. 10.3390/genes12010099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Morgensztern D, McLeod HL. PI3K/Akt/mTOR pathway as a target for cancer therapy. Anticancer Drugs. 2005;16:797–803. 10.1097/01.cad.0000173476.67239.3b. [DOI] [PubMed] [Google Scholar]
- 206. Polivka J Jr, Janku F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol Ther. 2014;142:164–75. 10.1016/j.pharmthera.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 207. Prossomariti A, Piazzi G, Alquati C, Ricciardiello L. Are Wnt/β‐Catenin and PI3K/AKT/mTORC1 Distinct Pathways in Colorectal Cancer? Cell Mol Gastroenterol Hepatol. 2020;10(3):491–506. 10.1016/j.jcmgh.2020.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Chen J. Potential value and limitation of dual inhibitors of PI3K and mTOR in the treatment of cancer. Curr Cancer Drug Targets. 2013;13:117–20. 10.2174/1568009611313020001. [DOI] [PubMed] [Google Scholar]
- 209. Chen S, Fisher RC, Signs S, Molina LA, Shenoy AK, Lopez MC, et al. Inhibition of PI3K/Akt/mTOR signaling in PI3KR2‐overexpressing colon cancer stem cells reduces tumor growth due to apoptosis. Oncotarget. 2016;8:50476–88. 10.18632/oncotarget.9919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Mangiapane LR, Nicotra A, Turdo A, Gaggianesi M, Bianca P, Di Franco S, et al. PI3K‐driven HER2 expression is a potential therapeutic target in colorectal cancer stem cells. Gut. 2021;gutjnl‐2020‐323553. 10.1136/gutjnl-2020-323553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Cai Z, Ke J, He X, Yuan R, Chen Y, Wu X, et al. Significance of mTOR signaling and its inhibitor against cancer stem‐like cells in colorectal cancer. Ann Surg Oncol. 2014;21(1):179–88. 10.1245/s10434-013-3146-8. [DOI] [PubMed] [Google Scholar]
- 212. Malkomes P, Lunger I, Luetticke A, Oppermann E, Haetscher N, Serve H, et al. Selective AKT inhibition by MK‐2206 represses colorectal cancer‐initiating stem cells. Ann Surg Oncol. 2016;23(9):2849–57. 10.1245/s10434-016-5218-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Adams DJ, Dai M, Pellegrino G, Wagner BK, Stern AM, Shamji AF, et al. Synthesis, cellular evaluation, and mechanism of action of piperlongumine analogs. Proc Natl Acad Sci USA. 2012;109(38):15115–20. 10.1073/pnas.1212802109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Bezerra DP, Pessoa C, de Moraes MO, Saker‐Neto N, Silveira ER, Costa‐Lotufo LV. Overview of the therapeutic potential of piplartine (piperlongumine). Eur J Pharm Sci. 2013;48(3):453–63. 10.1016/j.ejps.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 215. Costa COS, Araujo Neto JH, Baliza IRS, Dias RB, Valverde LF, Vidal MTA, et al. Novel piplartine‐containing ruthenium complexes: synthesis, cell growth inhibition, apoptosis induction and ROS production on HCT116 cells. Oncotarget. 2017;8(61):104367–92. 10.18632/oncotarget.22248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Oliveira MS, Barbosa MIF, de Souza TB, Moreira DRM, Martins FT, Villarreal W, et al. A novel platinum complex containing a piplartine derivative exhibits enhanced cytotoxicity, causes oxidative stress and triggers apoptotic cell death by ERK/p38 pathway in human acute promyelocytic leukemia HL‐60 cells. Redox Biol. 2019;20:182–94. 10.1016/j.redox.2018.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Baliza IRS, Silva SLR, Santos LS, Neto JHA, Dias RB, Sales CBS, et al. Ruthenium complexes with piplartine cause apoptosis through MAPK signaling by a p53‐dependent pathway in human colon carcinoma cells and inhibit tumor development in a xenograft model. Front Oncol. 2019;9:582. 10.3389/fonc.2019.00582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Kumar S, Agnihotri N. Piperlongumine, a piper alkaloid targets Ras/PI3K/Akt/mTOR signaling axis to inhibit tumor cell growth and proliferation in DMH/DSS induced experimental colon cancer. Biomed Pharmacother. 2019;109:1462–77. [DOI] [PubMed] [Google Scholar]
- 219. Marzano C, Gandin V, Folda A, Scutari G, Bindoli A, Rigobello MP. Inhibition of thioredoxin reductase by auranofin induces apoptosis in cisplatin‐resistant human ovarian cancer cells. Free Radic Biol Med. 2007;42(6):872–81. 10.1016/j.freeradbiomed.2006.12.021. [DOI] [PubMed] [Google Scholar]
- 220. Nag D, Bhanja P, Riha R, Sanchez‐Guerrero G, Kimler BF, Tsue TT, et al. Auranofin protects intestine against radiation injury by modulating p53/p21 pathway and radiosensitizes human colon tumor. Clin Cancer Res. 2019;25(15):4791–4807. 10.1158/1078-0432.CCR-18-2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Hrabe JE, O'Leary BR, Fath MA, Rodman SN, Button AM, Domann FE, et al. Disruption of thioredoxin metabolism enhances the toxicity of transforming growth factor β‐activated kinase 1 (TAK1) inhibition in KRAS‐mutated colon cancer cells. Redox Biol. 2015;5:319–27. 10.1016/j.redox.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Tanaka G, Inoue K, Shimizu T, Akimoto K, Kubota K. Dual pharmacological inhibition of glutathione and thioredoxin systems synergizes to kill colorectal carcinoma stem cells. Cancer Med. 2016;5(9):2544–57. 10.1002/cam4.844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Pei S, Minhajuddin M, Callahan KP, Balys M, Ashton JM, Neering SJ, et al. Targeting aberrant glutathione metabolism to eradicate human acute myelogenous leukemia cells. J Biol Chem. 2013;288(47):33542–58. 10.1074/jbc.M113.511170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Chen YJ, Kuo CC, Ting LL, Lu LS, Lu YC, Cheng AJ, et al. Piperlongumine inhibits cancer stem cell properties and regulates multiple malignant phenotypes in oral cancer. Oncol Lett. 2018;15(2):1789–98. 10.3892/ol.2017.7486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Wang K, Huang W, Sang X, Wu X, Shan Q, Tang D, et al. Atractylenolide I inhibits colorectal cancer cell proliferation by affecting metabolism and stemness via AKT/mTOR signaling. Phytomedicine. 2020;68:153191. 10.1016/j.phymed.2020.153191. [DOI] [PubMed] [Google Scholar]
- 226. Chen J, Shao R, Li F, Monteiro M, Liu JP, Xu ZP, et al. PI3K/Akt/mTOR pathway dual inhibitor BEZ235 suppresses the stemness of colon cancer stem cells. Clin Exp Pharmacol Physiol. 2015;42:1317–26. 10.1111/1440-1681.12493. [DOI] [PubMed] [Google Scholar]
- 227. Goodwin R, Jonker D, Chen E, Kennecke H, Cabanero M, Tsao MS, et al. A phase Ib study of a PI3Kinase inhibitor BKM120 in combination with panitumumab in patients with KRAS wild‐type advanced colorectal cancer. Invest New Drugs. 2020;38(4):1077–84. 10.1007/s10637-019-00814-3. [DOI] [PubMed] [Google Scholar]
- 228. Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009;69(19):7507–11. 10.1158/0008-5472.CAN-09-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Song CW, Lee H, Dings RP, Williams B, Powers J, Santos TD, et al. Metformin kills and radiosensitizes cancer cells and preferentially kills cancer stem cells. Sci Rep. 2012;2:362. 10.1038/srep00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Rattan R, Graham RP, Maguire JL, Giri S, Shridhar V. Metformin suppresses ovarian cancer growth and metastasis with enhancement of cisplatin cytotoxicity in vivo. Neoplasia. 2011;13(5):483–91. 10.1593/neo.11148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Seo Y, Kim J, Park SJ, Park JJ, Cheon JH, Kim WH,et al. Metformin suppresses cancer stem cells through AMPK activation and inhibition of protein prenylation of the mevalonate pathway in colorectal cancer. Cancers. 2020;12(9):2554. 10.3390/cancers12092554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Saif MW, Rajagopal S, Caplain J, Goodman MD, Popowich D, Orkin BA, et al. The first study evaluating the safety of pre‐surgery administration of metformin in patients with colorectal and other gastrointestinal cancers and effect on cancer stem cells. Cancer Med J. 2021;4(Suppl 4):1–10. [PMC free article] [PubMed] [Google Scholar]
- 233. Francipane MG, Lagasse E. Selective targeting of human colon cancer stem‐like cells by the mTOR inhibitor Torin‐1. Oncotarget. 2013;4(11):1948–62. 10.18632/oncotarget.1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Itatani Y, Kawada K, Sakai Y. Transforming growth factor‐β signaling pathway in colorectal cancer and its tumor microenvironment. Int J Mol Sci. 2019;20(23):5822. 10.3390/ijms20235822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Zonneville J, Safina A, Truskinovsky AM, Arteaga CL, Bakin AV. TGF‐β signaling promotes tumor vasculature by enhancing the pericyte‐endothelium association. BMC Cancer, 2018;18(1):670. 10.1186/s12885-018-4587-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Itatani Y, Kawada K, Yamamoto T, Sakai Y. Resistance to anti‐angiogenic therapy in cancer‐alterations to anti‐VEGF pathway. Int J Mol Sci. 2018;19(4):1232. 10.3390/ijms19041232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Takaku K, Oshima M, Miyoshi H, Matsui M, Seldin MF, Taketo MM. Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apc genes. Cell. 1998;92(5):645–56. 10.1016/s0092-8674(00)81132-0. [DOI] [PubMed] [Google Scholar]
- 238. Kitamura T, Kometani K, Hashida H, Matsunaga A, Miyoshi H, Hosogi H, et al. SMAD4‐deficient intestinal tumors recruit CCR1+ myeloid cells that promote invasion. Nat Genet. 2007;39(4):467–75. 10.1038/ng1997. [DOI] [PubMed] [Google Scholar]
- 239. Inamoto S, Itatani Y, Yamamoto T, Minamiguchi S, Hirai H, Iwamoto M, et al. Loss of SMAD4 promotes colorectal cancer progression by accumulation of myeloid‐derived suppressor cells through the CCL15‐CCR1 chemokine axis. Clin Cancer Res. 2016;22(2):492–501. 10.1158/1078-0432.CCR-15-0726. [DOI] [PubMed] [Google Scholar]
- 240. Cheng D, Zhao S, Tang H, Zhang D, Sun H, Yu F, et al. MicroRNA‐20a‐5p promotes colorectal cancer invasion and metastasis by downregulating Smad4. Oncotarget, 2016;7(29):45199–213. 10.18632/oncotarget.9900. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 241. Perekatt AO, Shah PP, Cheung S, Jariwala N, Wu A, Gandhi V, et al. SMAD4 Suppresses WNT‐driven dedifferentiation and oncogenesis in the differentiated gut epithelium. Cancer Res. 2018;78(17):4878–90. 10.1158/0008-5472.CAN-18-0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Ye J, Lei J, Fang Q, Shen Y, Xia W, Hu X, et al. miR‐4666‐3p and miR‐329 synergistically suppress the stemness of colorectal cancer cells via targeting TGF‐β/Smad pathway. Front Oncol. 2019;9:1251. 10.3389/fonc.2019.01251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Gao Z, Huang K, Xu H. Protective effects of flavonoids in the roots of Scutellaria baicalensis Georgi against hydrogen peroxide‐induced oxidative stress in HS‐SY5Y cells. Pharmacol Res. 2001;43(2):173–8. 10.1006/phrs.2000.0761. [DOI] [PubMed] [Google Scholar]
- 244. Yang B, Bai H, Sa Y, Zhu P, Liu P. Inhibiting EMT, stemness and cell cycle involved in baicalin‐induced growth inhibition and apoptosis in colorectal cancer cells. J Cancer. 2020;11(8):2303–17. 10.7150/jca.37242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Wu SM, Choo AB, Yap MG, Chan KK. Role of Sonic hedgehog signaling and the expression of its components in human embryonic stem cells. Stem Cell Res. 2010;4(1):38–49. 10.1016/j.scr.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 246. Mah AT, Yan KS, Kuo CJ. Wnt pathway regulation of intestinal stem cells. J Physiol. 2016;594(17):4837–47. 10.1113/JP271754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Siebel C, Lendahl U. Notch signaling in development, tissue homeostasis, and disease. Physiol Rev. 2017;97(4):1235–94. 10.1152/physrev.00005.2017. [DOI] [PubMed] [Google Scholar]
- 248. Clarke MF. Clinical and therapeutic implications of cancer stem cells. N Engl J Med. 2019;380(23):2237–45. 10.1056/NEJMra1804280. [DOI] [PubMed] [Google Scholar]
- 249. Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M, et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 2011;469:415–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Chapouton P, Skupien P, Hesl B, Coolen M, Moore JC, Madelaine R, et al. Notch activity levels control the balance between quiescence and recruitment of adult neural stem cells. J Neurosci 2010;30:7961–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Chen JY, Miyanishi M, Wang SK, Yamazaki S, Sinha R, Kao KS, et al. Hoxb5 marks long‐term haematopoietic stem cells and reveals a homogenous perivascular niche. Nature 2016;530:223–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Cai S, Kalisky T, Sahoo D, Dalerba P, Feng W, Lin Y, et al. A quiescent Bcl11b high stem cell population is required for maintenance of the mammary gland. Cell Stem Cell 2017;20(2): 247–60.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Hirata A, Hatano Y, Niwa M, Hara A, Tomita H. Heterogeneity of colon cancer stem cells. Adv Exp Med Biol. 2019;1139:115–26. 10.1007/978-3-030-14366-4_7 [DOI] [PubMed] [Google Scholar]
- 254. Yang L, Shi P, Zhao G, Xu J, Peng W, Zhang J,et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther. 2020 Feb 7; ;5(1):8. 10.1038/s41392-020-0110-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Jin MZ, Jin WL. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct Target Ther. 2020;5(1):166. 10.1038/s41392-020-00280-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Dixon SW, Collard TJ, Mortensson EMH, Legge DN, Chambers AC, Greenhough A, et al. 5‐Aminosalicylic acid inhibits stem cell function in human adenoma‐derived cells: implications for chemoprophylaxis in colorectal tumorigenesis. Br J Cancer. 2021;124(12):1959–69. 10.1038/s41416-021-01354-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Zhao L, Yang Y, Zhou P, Ma H, Zhao X, He X, et al. Targeting CD133high colorectal cancer cells in vitro and in vivo with an asymmetric bispecific antibody. J Immunother. 2015;38(6):217–28. 10.1097/CJI.0000000000000086. [DOI] [PubMed] [Google Scholar]
- 258. Narayan S, Ramisetti S, Jaiswal AS, Law BK, Singh‐Pillay A, Singh P, ASR352 Et al., A potent anticancer agent: Synthesis, preliminary SAR, and biological activities against colorectal cancer bulk, 5‐fluorouracil/oxaliplatin resistant and stem cells. Eur J Med Chem. 2019;161:456–67. 10.1016/j.ejmech.2018.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Wang Y, Chen M, Wu Z, Tong C, Dai H, Guo Y, et al. CD133‐directed CAR T cells for advanced metastasis malignancies: A phase I trial. Oncoimmunology. 2018;7:e1440169. 10.1080/2162402X.2018.1440169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Sato‐Dahlman M, Miura Y, Huang JL, Hajeri P, Jacobsen K, Davydova J, et al. CD133‐targeted oncolytic adenovirus demonstrates anti‐tumor effect in colorectal cancer. Oncotarget. 2017;8(44):76044–56. 10.18632/oncotarget.18340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Wu Z, Huang M, Gong Y, Lin C, Guo W. BRAF and EGFR inhibitors synergize to increase cytotoxic effects and decrease stem cell capacities in BRAF(V600E)‐mutant colorectal cancer cells. Acta Biochim Biophys Sin. 2018;50(4):355–61. 10.1093/abbs/gmy018. [DOI] [PubMed] [Google Scholar]
- 262. Núñez‐Sánchez MÁ, Karmokar A, González‐Sarrías A, García‐Villalba R, Tomás‐Barberán FA, García‐Conesa MT, et al. In vivo relevant mixed urolithins and ellagic acid inhibit phenotypic and molecular colon cancer stem cell features: A new potentiality for ellagitannin metabolites against cancer. Food Chem Toxicol. 2016;92:8–16. 10.1016/j.fct.2016.03.011. [DOI] [PubMed] [Google Scholar]
- 263. Patel NJ, Karuturi R, Al‐Horani RA, Baranwal S, Patel J, Desai UR, et al. Synthetic, non‐saccharide, glycosaminoglycan mimetics selectively target colon cancer stem cells. ACS Chem Biol. 2014;9:1826–33. 10.1021/cb500402f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Boothello RS, Patel NJ, Sharon C, Abdelfadiel EI, Morla S, Brophy DF, et al. A unique nonsaccharide mimetic of heparin hexasaccharide inhibits colon cancer stem cells via p38 MAP kinase activation. Mol Cancer Ther. 2019;18(1):51–61. 10.1158/1535-7163.MCT-18-0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Wei F, Zhang T, Yang Z, Wei JC, Shen HF, Xiao D, et al. Gambogic acid efficiently kills stem‐like colorectal cancer cells by upregulating ZFP36 expression. Cell Physiol Biochem. 2018;46(2):829–46. 10.1159/000488740. [DOI] [PubMed] [Google Scholar]
- 266. Tang YC, Zhang Y, Zhou J, Zhi Q, Wu MY, Gong FR, et al. Ginsenoside Rg3 targets cancer stem cells and tumor angiogenesis to inhibit colorectal cancer progression in vivo. Int J Oncol. 2018;52(1):127–38. 10.3892/ijo.2017.4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Patel NJ, Sharon C, Baranwal S, Boothello RS, Desai UR, Patel BB. Heparan sulfate hexasaccharide selectively inhibits cancer stem cells self‐renewal by activating p38 MAP kinase. Oncotarget. 2016;7:84608–22. 10.18632/oncotarget.12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Quarni W, Dutta R, Green R, Katiri S, Patel B, Mohapatra SS, et al. Mithramycin A inhibits colorectal cancer growth by targeting cancer stem cells. Sci Rep. 2019;9(1):15202. 10.1038/s41598-019-50917-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Zhou HM, Dong TT, Wang LL, Feng B, Zhao HC, Fan XK, et al. Suppression of colorectal cancer metastasis by nigericin through inhibition of epithelial‐mesenchymal transition. World J Gastroenterol. 2012;18(21):2640–8. 10.3748/wjg.v18.i21.2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Narayan S, Jaiswal AS, Sharma R, Nawab A, Duckworth LV, Law BK, et al. NSC30049 inhibits Chk1 pathway in 5‐FU‐resistant CRC bulk and stem cell populations. Oncotarget. 2017;8(34):57246–64. 10.18632/oncotarget.19778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Zhang S, Ju X, Yang Q, Zhu Y, Fan D, Su G, et al. USP47 maintains the stemness of colorectal cancer cells and is inhibited by parthenolide. Biochem Biophys Res Commun. 2021;562:21–8. 10.1016/j.bbrc.2021.05.017. [DOI] [PubMed] [Google Scholar]
- 272. Chen Q, Zeng YN, Zhang K, Zhao Y, Wu YY, Li G, et al. Polydatin increases radiosensitivity by inducing apoptosis of stem cells in colorectal cancer. Int J Biol Sci. 2019;15(2):430–40. 10.7150/ijbs.27050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Kumar S, Raina K, Agarwal C, Agarwal R. Silibinin strongly inhibits the growth kinetics of colon cancer stem cell‐enriched spheroids by modulating interleukin 4/6‐mediated survival signals. Oncotarget. 2014;5(13):4972–89. 10.18632/oncotarget.2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Ju SY, Huang CY, Huang WC, Su Y. Identification of thiostrepton as a novel therapeutic agent that targets human colon cancer stem cells. Cell Death Dis. 2015;6(7):e1801. 10.1038/cddis.2015.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Signore M, Buccarelli M, Pilozzi E, De Luca G, Cappellari M, Fanciulli M, et al. UCN‐01 enhances cytotoxicity of irinotecan in colorectal cancer stem‐like cells by impairing DNA damage response. Oncotarget. 2016;7(28):44113–28. 10.18632/oncotarget.9859. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.