

Abstract
Background
Fascin is crucial for cancer cell filopodium formation and tumor metastasis, and is functionally regulated by post‐translational modifications. However, whether and how Fascin is regulated by acetylation remains unclear. This study explored the regulation of Fascin acetylation and its corresponding roles in filopodium formation and tumor metastasis.
Methods
Immunoprecipitation and glutathione‐S‐transferase pull‐down assays were performed to examine the interaction between Fascin and acetyltransferase P300/CBP‐associated factor (PCAF), and immunofluorescence was used to investigate their colocalization. An in vitro acetylation assay was performed to identify Fascin acetylation sites by using mass spectrometry. A specific antibody against acetylated Fascin was generated and used to detect the PCAF‐mediated Fascin acetylation in esophageal squamous cell carcinoma (ESCC) cells using Western blotting by overexpressing and knocking down PCAF expression. An in vitro cell migration assay was performed, and a xenograft model was established to study in vivo tumor metastasis. Live‐cell imaging and fluorescence recovery after photobleaching were used to evaluate the function and dynamics of acetylated Fascin in filopodium formation. The clinical significance of acetylated Fascin and PCAF in ESCC was evaluated using immunohistochemistry.
Results
Fascin directly interacted and colocalized with PCAF in the cytoplasm and was acetylated at lysine 471 (K471) by PCAF. Using the specific anti‐AcK471‐Fascin antibody, Fascin was found to be acetylated in ESCC cells, and the acetylation level was consequently increased after PCAF overexpression and decreased after PCAF knockdown. Functionally, Fascin‐K471 acetylation markedly suppressed in vitro ESCC cell migration and in vivo tumor metastasis, whereas Fascin‐K471 deacetylation exhibited a potent oncogenic function. Moreover, Fascin‐K471 acetylation reduced filopodial length and density, and lifespan of ESCC cells, while its deacetylation produced the opposite effect. In the filipodium shaft, K471‐acetylated Fascin displayed rapid dynamic exchange, suggesting that it remained in its monomeric form owing to its weakened actin‐bundling activity. Clinically, high levels of AcK471‐Fascin in ESCC tissues were strongly associated with prolonged overall survival and disease‐free survival of ESCC patients.
Conclusions
Fascin interacts directly with PCAF and is acetylated at lysine 471 in ESCC cells. Fascin‐K471 acetylation suppressed ESCC cell migration and tumor metastasis by reducing filopodium formation through the impairment of its actin‐bundling activity.
Keywords: acetylation, actin‐bundling, esophageal cancer, fascin, filopodium formation, P300/CBP‐associated factor (PCAF), tumor metastasis
Abbreviations
- Ac‐K
acetylate‐lysine
- AcK471‐Fascin
Fascin‐K471 acetylation
- ATCC
American Type Culture Collection
- CBBCBB
CBBCoomassie brilliant blue
- CDS
coding sequence
- CK
cytokeratin
- CoA
coenzyme A
- DAPI
4’,6‐diamidino‐2‐phenylindole
- DFS
disease‐free survival
- DMEM
Dulbecco's modified Eagle's medium
- ESCC
esophageal squamous cell carcinoma
- FascinK471Q
a mimic of hyperacetylated Fascin
- FascinK471R
a mimic of acetylation‐defective Fascin
- FBS
fetal bovine serum
- FRAP
Fluorescence recovery after photobleaching
- G/F‐actin ratio
ratio of G‐actin to F‐actin
- GFP
Green fluorescent protein
- GST
glutathione‐S‐transferase
- HA
Hemagglutinin tag
- H&E
hematoxylin and eosin
- HAT
histone acetyltransferase
- IB
immunoblot
- IHC
immunohistochemistry
- IP
immunoprecipitation
- K471
lysine 471
- MS
mass spectrometry
- NC
negative control
- OS
Overall survival
- P
pellet
- PBST
PBS containing 0.1% Tween 20
- PCAF
P300/CBP‐associated factor
- Q
glutamine
- R
arginine
- RFP
Red Fluorescent Protein
- S
supernatant
- SDS‐PAGE
sodium dodecyl sulfate‐polyacrylamide gel electrophoresis
- SHEEC
Shantou Human Embryonic Esophageal Carcinoma
- siRNAs
Small interfering RNAs
- TMAs
Tissue microarrays
- WT
wild type
1. BACKGROUND
Filopodia are thin, spike‐like, cytoplasmic protrusions in migratory cells, composed of parallel and bundled actin filaments, and function as antenna during cell motility to probe their environment [1]. One of the most widely studied proteins critical for filopodium formation is the actin‐bundling protein Fascin [2, 3], which is a monomeric protein comprised of four structurally homologous tandem β‐trefoil folds. It is necessary for maximally cross‐linking the actin filaments into straight, compact, and rigid bundles to maintain the stiffness of filopodia [4, 5]. Fascin is thought to interact with actin through two actin‐binding sites, actin‐binding site 1 formed by the N‐ and C‐terminal of Fascin, including residues of β‐trefoil folds 1 and 4, and actin‐binding site 2, including residues of β‐trefoil folds 1 and 2 [6]. Fluorescence recovery after photobleaching (FRAP) analysis revealed that the dynamic life cycle of filopodia, which includes protrusion, retraction, and stationary stages, largely depends on the rapid association/disassociation cycles of Fascin from filopodial actin filaments [5, 7]. To date, Fascin has been found to be abnormally upregulated in all studied forms of human carcinoma, including colon, pancreatic, breast, lung, gastric, ovarian, cervical, tongue, brain, and esophageal cancers [8, 9], and associated with clinically aggressive phenotypes, poor prognosis, and short survival [10]. Thus, Fascin has emerged as a potential biomarker for cancer diagnosis and prognosis [10, 11, 12], as well as a promising molecular target for future cancer therapy [13].
As filopodia play an essential role in cell adhesion, migration, and invasion [14, 15], Fascin is strongly associated with cancer metastasis [16, 17]. Studies have shown that the rapid dynamic exchange of Fascin in filopodia is regulated by post‐translational modifications, such as phosphorylation and ubiquitination, which result in the inhibition of the actin‐bundling activity of Fascin and filopodium formation and a subsequent decrease in cell proliferation, migration, and invasion [5, 7, 18–23]. Lysine acetylation, which is a reversible post‐translational modification of histone and non‐histone proteins catalyzed by acetyltransferases and deacetylases, plays important roles in many biological processes [24], and its dysregulation has been implicated in various human diseases, such as metabolic disorders and cancer [25]. To date, several potential post‐translational modifications, including acetylation within Fascin, have been predicted by using mass spectrometry (MS) (http://www.phosphosite.org) in several cancer cell lines [24, 26]. The acetyltransferase KAT2 (lysine acetyltransferase 2) family, one of the three major families of acetyltransferases [27], consists of two acyltransferases, namely KAT2A and KAT2B/ PCAF (P300/CBP‐associated factor). It is considered to be involved in many cellular processes, including DNA replication and repair, cell cycle and death, and actin‐mediated cell contraction, according to a large‐scale MS‐based proteomics study exploring the acetylome in KAT2A/2B‐knockdown HeLa cells [28]. However, the mechanism by which Fascin is acetylated and its potential clinical significance in cancer progression remain unclear. In this study, we explored the acetyltransferase of Fascin, evaluated the acetylation mechanism by which Fascin is regulated, and determined the clinical significance of Fascin acetylation in esophageal squamous cell carcinoma (ESCC).
2. MATERIALS AND METHODS
2.1. Cell culture
The human ESCC cell lines KYSE140, KYSE150, KYSE180, KYSE450, and KYSE510 were established by Dr. Shimada Yutaka (Faculty of Medicine, Kyoto University, Kyoto, Japan) [29] and cultured in RPMI‐1640 medium (Thermo Scientific HyClone, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS; Life Technologies, Carlsbad, CA, USA). The human ESCC Shantou Human Embryonic Esophageal Carcinoma (SHEEC) cell line was previously established by our lab [30] and cultured in Dulbecco's modified Eagle's medium (DMEM; Thermo Fisher Scientific Gibco, Carlsbad, CA, USA)/F12 medium (Life Technologies) supplemented with 10% newborn bovine serum (Excell Biology Inc., Shanghai, China). HEK293T cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in DMEM supplemented with 10% FBS. All cell lines were tested for mycoplasma free and STR analysis (IGEbio, Guangzhou, China), and were cultured in a 5% CO2 atmosphere at 37°C.
2.2. Plasmids
Human full‐length Flag‐Fascin was generated by cloning the FSCN1‐coding sequence (CDS) to pcDNA3.1‐Flag vector for transient transfection. To construct the glutathione‐S‐transferase (GST)‐fused and His‐fused Fascin vectors for in vitro protein expression, the full‐length CDS sequence of FSCN1 (or the truncated FSCN1‐Δβ1, FSCN1‐Δβ1β2, FSCN1‐Δβ1β2β3, FSCN1‐Δβ4, FSCN1‐Δβ3β4, and FSCN1‐Δβ2β3β4) and PCAF were cloned into pGEX‐6P‐1 and pET‐32a vectors (MiaoLing Plasmid Sharing Platform, Wuhan, Hubei, China), respectively. The GST‐fused N‐terminal (1‐492), histone acetyltransferase (HAT) (493‐658), and C‐terminal BROMO (659‐832) domains of PCAF constructs were provided by Dr. Hongquan Zhang (Peking University Health Science Center, Beijing, China). The human full‐length HA‐PCAF, HA‐KAT2A, and HA‐KAT5 were generous gifts from Dr. Daming Gao (Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China).
Small interfering RNAs (siRNAs) against FSCN1 (siFSCN1‐#1: 5′‐GCGAGTCTGGCACCTCTTT‐3′, siFSCN1‐#2: 5′‐GAGCCTTATTTCTCTGGAA‐3′) and PCAF (siPCAF‐#1: 5′‐CCGCAUCAACUAUUGGCAUTT‐3′, siPCAF‐#2: 5′‐GCGACAACUCCUGGAACAATT‐3′) were used for Fascin and PCAF knockdown, respectively. All siRNAs were purchased from GenePharma (Shanghai, China) and transfected into cells using Lipofectamine® RNAiMAX Transfection Reagent (Life Technologies) following the manufacturer's instructions. The target short hairpin RNAs (shRNAs) of Fascin (shFSCN1‐#1: 5′‐CCCTTGCCTTTCAAACTGGAA‐3′ located in the 3′‐untranslated region, shFSCN1‐#2: 5′‐CGACTATAACAAGGTGGCCAT‐3′ located in the CDS region) (Hanbio, Shanghai, China) and the negative control (NC) sequences were cloned between the BamH I and EcoR I restriction sites in the pHBLV‐U6‐Luc‐T2A‐Puro vector (Hanbio). Since the shFSCN1‐#1 sequence is located in the 3′‐untranslated regions of FSCN1, its silencing effect on exogenous transfected FSCN1 gene (CDS sequence) could be avoided when re‐expressing Fascin in stable shFSCN1‐#1‐transfected cells. The coding region of Fascin was amplified and cloned into the Hind III and BamH I sites of pEGFP‐N1 to generate the Fascin‐GFP (green fluorescent protein) expression vector. A Fast Mutagenesis System kit (TransGen Biotech, Beijing, China) was used to generate site‐specific mutations at lysine 471 according to the manufacturer's instructions. The primers used for mutagenesis are as follows: 5′‐TACCTGAGGGGCGACCAC‐3′ and 5′‐CGTGGTCGCCCCTCAGGT‐3′ (K471R) and 5′‐TACCTGCAGGGCGACCAC‐3′ and 5′‐CGTGGTCGCCCTGCAGGT‐3′ (K471Q). LifeAct‐RFP (red fluorescent protein) construct was a gift from Brian Stramer (King's College London, England, UK). Wild‐type Fascin and FascinK471R sequences were subcloned into pBOBI‐3 × Flag‐N to construct stable expression cell lines for Flag immunoprecipitation (IP) assays by using anti‐Flag magnetic beads (36797; Thermo Fisher, Waltham, MA, USA) to detect anti‐AcK471‐Fascin antibody specificity.
2.3. Establishment of stable cell lines
KYSE150 cells and SHEEC cells were plated into 24‐well plates at 5 × 104 cells per well. After 24 h, the cells were infected with lentiviral particles containing shFSCN1‐#1 or NC sequences. After 48 h of transfection, the cells were selected in a growth medium containing 500 ng/mL puromycin (J593; Amresco, Houston, TX, USA). Stable puromycin‐resistant clones were obtained after 14‐16 days. The expanded cells were then used for further experiments. Knockdown of FSCN1 was evaluated by Western blotting. The stable FSCN1‐knockdown cells were transfected with the Fascin‐GFP, FascinK471R‐GFP, or FascinK471Q‐GFP constructs, using Lipofectamine 3000 reagent (Life Technologies) according to the manufacturer's instructions. After 24 h of transfection, the cells were selected in a culture medium containing 500 μg/mL G418 (Calbiochem, San Diego, CA, USA), and stable G418‐resistant clones were obtained after 14‐16 days. The expanded cells were used for xenograft assays in nude mice. The stable FSCN1‐knockdown cells were also transiently co‐transfected with Fascin‐GFP or its mutated constructs along with the LifeAct‐RFP construct using Lipofectamine 3000. After 24 h of transfection, the cells were plated on 10 μg/mL fibronectin‐coated chambers and visualized using Zeiss LSM880 confocal microscope (Carl Zeiss, Jena, Germany) for live‐cell imaging. To construct stable FSCN1‐Flag‐ and FSCN1K471R‐Flag‐expressing cells using pBOB‐FSCN1‐3 × Flag and pBOB‐FSCN1K471R‐3 × Flag, the infected cells were detected by Western blotting and immunofluorescence to evaluate the infection efficiency before using it for IP assays.
2.4. Immunoprecipitation and Western blotting
To determine the interaction of endogenous Fascin and PCAF, HEK293T and KYSE140 cells were lysed using IP buffer (50 mmol/L Tris pH 7.5, 150 mmol/L NaCl, and 0.5% NP‐40) supplemented with Protease and Phosphatase Inhibitor Cocktail (78445; Thermo Scientific). The protein concentration of the cell lysates was measured using the Pierce BCA protein assay kit (23225; Thermo Scientific). After pre‐clearing with isotype IgG and protein A/G magnetic beads, equal amounts of total protein (1 mg) of the cell lysates were immunoprecipitated by incubating with protein A/G magnetic beads (HY‐K0202; MCE, Shanghai, China) and PCAF (sc‐13124; Santa Cruz, Dallas, TX, USA) or Fascin antibodies (sc‐46675; Santa Cruz) or the control normal mouse IgG (sc‐2025; Santa Cruz) at 4°C overnight with gentle rocking. The immunoprecipitated proteins were washed five times with cold IP buffer and then denatured with 1 × SDS sample buffer. The denatured proteins were separated by sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) followed by Western blotting with anti‐Fascin rabbit polyclonal antibody (PA513696; Life Technologies). Flag‐tagged protein IP was performed using anti‐Flag magnetic beads (36797; Thermo Fisher). Primary antibodies, including monoclonal mouse anti‐human Fascin (M356701; Dako, Glostrup, Denmark), polyclonal rabbit anti‐Fascin (PA513696; Life Technologies), Flag (F3165; Sigma Aldrich, St. Louis, MO, USA), GFP (sc‐9996; Santa Cruz), GAPDH (sc‐32233; Santa Cruz), β‐Actin antibody (sc‐47778; Santa Cruz), and HA antibodies (3724; Cell Signaling Technology, Danvers, MA, USA) were used as indicated, followed by secondary antibodies, IRDye 680RD goat anti‐mouse IgG (926‐68070; LI‐COR Biosciences, Lincoln, NE, USA) and IRDye 800CW goat anti‐mouse IgG antibodies (926‐32211; LI‐COR Biosciences) for immunofluorescent blotting using Odyssey Sa Infrared Imaging System (LI‐COR Biosciences) or by secondary antibodies goat anti‐mouse IgG‐HRP (sc‐2005; Santa Cruz) and goat anti‐rabbit IgG‐HRP antibodies (sc‐2030; Santa Cruz) for chemiluminescence using ChemiDoc™ MP Imaging System (Bio‐Rad, Hercules, CA, USA).
2.5. Recombinant protein expression and purification
Recombinant GST‐tagged and His‐tagged Fascin, PCAF, and their truncated forms were transformed into Escherichia coli BL21 (DE3) strain, and protein expression was induced using 0.5 mmol/L isopropyl β‐D‐1‐thiogalactopyranoside (0487; Amresco) at 25°C for 6 h. Bacterial cultures were centrifuged at 5,000 × g for 10 min, and pellets were resuspended and lysed by sonification in GST lysis buffer (PBS containing 0.1% Triton X‐100) or His lysis buffer (50 mmol/L sodium phosphate, 300 mmol/L NaCl, and 10 mmol/L imidazole) followed by incubation with GST‐binding resin (70541‐3; Millipore, Billerica, MA, USA) or Ni‐NTA His‐bind resin (70666‐3; Millipore) at 4°C for 3 h. The GST‐resin and His‐resin were then washed four times using GST lysis and His wash buffer (50 mmol/L sodium phosphate, 300 mmol/L NaCl, and 20 mmol/L imidazole), respectively. To obtain non‐tagged Fascin, the GST‐Fascin binding resin was resuspended in 0.5 mL PreScission Buffer (P2303‐2; Beyotime Biotechnology, Shanghai, China). Non‐tagged Fascin was released from the beads by incubation with 20 units of PreScission Protease (P2303‐1; Beyotime Biotechnology) overnight at 4 °C to remove the GST tag. To obtain GST‐tag or His‐tag fused protein, the resin was eluted using GST (50 mmol/L Tris‐HCl pH 8.0, and 50 mmol/L reduced form of glutathione) or His elution buffer (50 mmol/L sodium phosphate, 300 mmol/L NaCl, and 200 mmol/L imidazole).
2.6. In vitro F‐Actin‐bundling Assay
The in vitro F‐actin bundling assays were performed as previously described [6]. Briefly, monomeric rabbit G‐actin (1 μmol/L) was polymerized in F‐actin buffer (20 mmol/L Tris‐HCl, pH 8.0, 1 mmol/L ATP, 1 mmol/L DTT, 2 mmol/L MgCl2, and 100 mmol/L KCl) at room temperature. Different forms (wild‐type or mutant) of purified fascin proteins (0.25 μmol/L) were incubated with F‐actin for 1 h at room temperature. Samples were centrifuged for 30 min at 10,000 × g. Both pellets and supernatants were dissolved in an equivalent volume of loading sample buffer, and equal volumes of pellet and supernatant were analyzed by using Coomassie bright blue staining.
2.7. n vitro acetylation assays
n vitro acetylation assays were performed as previously described [31] with modifications. Briefly, HA‐PCAF protein was immunoprecipitated from HEK293T cells using monoclonal anti‐HA‐agarose (A2095; Sigma Aldrich) after 48 h of transfection. The agarose was washed thrice in PBS containing 0.1% Tween 20 (PBST) buffer and once in acetylation buffer. Thereafter, the agarose was incubated with His‐Fascin in acetylation buffer with or without acetyl coenzyme A (CoA) at 30°C for 2 h. After the acetylation assays, the proteins were separated by SDS‐PAGE for Western blotting with acetylated lysine (anti‐Ac‐K, ICP0380; Immunechem, Columbia, Canada), HA (SC‐7392; Santa Cruz), and His antibodies (HT601‐01; Transgene, Beijing, China) using Odyssey Sa Infrared Imaging System (LI‐COR Biosciences). For acetylation site identification, the separated proteins were stained using Coomassie brilliant blue (24615; Thermo Fisher Scientific), and the protein bands were excised for MS analysis.
2.8. Identification of Fascin acetylation sites by mass spectrometry
Briefly, the bands of interest were excised from the gel and subjected to in‐gel tryptic digestion. The resulting peptides were separated by nano‐liquid chromatography on an easy‐nLC 1200 system (Thermo Fisher Scientific) and directly sprayed into a Q‐Exactive Plus mass spectrometer (Thermo Fisher Scientific). The MS analysis was carried out in data‐dependent mode with an automatic switch between a full MS and a tandem MS (MS/MS) scan in the Orbitrap. For full MS survey scan, the automatic gain control target was set to 3e6, and the scan range was from 330 to 1600 with a resolution of 70,000. The 10 most intense peaks with charge state ≥ 2 were selected for fragmentation by higher energy collision dissociation with normalized collision energy of 27%. The MS2 spectra were acquired with a resolution of 17,500, and the exclusion window was set at ± 2.2 Da. All MS/MS spectra were searched against the Uniport‐Human protein sequence database by using the PD search engine (v 2.1.0; Thermo Fisher Scientific) with an overall false discovery rate (FDR) for peptides of less than 1%. Peptide sequences were searched using trypsin specificity allowing a maximum of two missed cleavages. Carbamidomethylation on Cys was specified as a fixed modification. Acetylation on lysine and peptide N‐terminal and oxidation of methionine were specified as variable modifications. Mass tolerances for precursor ions were set at ± 10 ppm for precursor ions and ± 0.02 Da for MS/MS. The MS/MS spectra of interest were manually verified. The amino acid sequence alignment of Fascin was performed between humans and other species by DNAMAN software.
2.9. GST pull‐down assays
The control GST and purified GST‐tagged full‐length and truncated forms of Fascin and PCAF proteins were bound with GST resin in PBS binding buffer at 4°C for 3 h. After washing three times with PBST buffer, an equal amount of His‐tagged PCAF or Fascin proteins was added and rebound at 4°C overnight. The rebound GST resins were washed five times with PBST buffer and boiled in SDS loading buffer to release the bound proteins for SDS‐PAGE separation and Western blotting with anti‐His antibody.
2.10. Immunofluorescence staining
To determine the cellular localization of Fascin and PCAF, immunofluorescence assays were performed. Briefly, KYSE150 cells were stained with anti‐Fascin (PA513696; Life Technologies) and anti‐PCAF antibodies (sc‐13124; Santa Cruz) and with secondary antibodies Alexa Fluor 488 AffiniPure donkey anti‐rabbit IgG (711‐545‐152; Jackson ImmunoResearch, West Grove, PA, USA), and Alexa Fluor 647 AffiniPure donkey anti‐mouse IgG (715‐605‐150; Jackson ImmunoResearch), respectively, followed by counterstaining with 4´,6‐diamidino‐2‐phenylindole (D9564; Sigma Aldrich). Confocal imaging was carried out using a laser scanning confocal microscope (LSM800; Carl Zeiss).
2.11. Patients and specimens
A total of 265 tumor tissues from ESCC patients and 146 paired adjacent normal tissues were used for Fascin and AcK471‐Fascin immunohistochemistry (IHC), while 268 tumor tissues were used for PCAF. The median age of patients with ESCC was 59 years, with ages ranging from 39 to 88 years. The information on age, gender, and histopathological factors was collected from the medical records and is shown in Supplementary Table S1. All patients underwent curative resection, and none had distant metastasis. The tissue sections were histologically evaluated with hematoxylin and eosin (H&E) staining. All tumors were diagnosed with ESCC by pathologists in the Clinical Pathology Department of the Shantou Central Hospital between November 2007 and January 2010 and classified according to the 7th edition of TNM classification system of the International Union against Cancer. The follow‐up was terminated in September 2020, while patients with other malignant tumors and did not die of ESCC (such as postoperative complications) were excluded. Overall survival (OS) was defined as the interval between surgery and death from tumor at the last observation, while disease‐free survival (DFS) was defined as the interval between surgery and diagnosis of relapse. Ethical approval was obtained from the ethical committees of the Shantou Central Hospital and the Shantou University Medical College. Only resected samples from surgical patients who gave written informed consent were included for use in research.
2.12. Tissue microarrays and IHC
Tissue microarrays (TMAs) were constructed based on standard techniques, and IHC was performed using the PV‐9000 2‐step Polymer Detection System (ZSGB‐Bio, Beijing, China) and Liquid DAB Substrate Kit (Invitrogen, San Francisco, CA, USA) according to the manufacturer's instructions as previously described [32]. The primary antibodies, including anti‐human Fascin‐K471 acetylation (AcK471‐Fascin; rabbit polyclonal, 1:5000), anti‐Fascin (M3567; Dako), and anti‐PCAF antibodies (sc‐13124; Santa Cruz), were used in this study. We recorded protein expression using an automated quantitative pathology imaging system (Perkin Elmer, Waltham, MA, USA). Briefly, the automatically acquired color images were obtained using Vectra 2.0.8 software. Subsequently, the spectral libraries were constructed using Nuance 3.0 software, and the color images were evaluated by Inform 1.2 software as follows: 1) segmentation of the tumor region from the tissue compartments; 2) segmentation of one tumor region from the other; 3) calculation of the H score based on the optical density, which then produced a protein expression value in the range of 0 to 300.
2.13. Cell migration and invasion assays
Cell migration by wound‐healing assay and cell invasion in Matrigel invasion chambers (Corning, Bedford, MA, USA) were performed as described previously [33]. For real‐time monitoring of cell migration, experiments were carried out using the xCELLigence Real‐Time Cell Analysis DP instrument (Roche Applied Science, Mannheim, Germany), and 2 × 105 cells were washed once in serum‐free medium and inoculated into the upper chamber, which was placed in a humidified incubator at 37°C and 5% CO2. Cell impedance was monitored every 30 min for approximately 48 h. The slope indicated the increase in cell impedance per unit time (slope = impedance / time). Median values and standard deviations were calculated from individual triplicate wells. Experiments were repeated three times with similar results.
2.14. Establishment of footpad lymph node metastasis mouse model
Five‐week‐old male BALB/c nu/nu Mice were purchased from Weitonglihua Experimental Animal Technical Co., LTD (Beijing, China). The metastasis experiment in nude mice was performed as previously described [34, 35] with modifications. We previously reported that KYSE150 cells demonstrated good tumorigenicity in nude mice [36]. Therefore, KYSE150 cells were used to establish footpad lymph node metastasis mouse model. Specifically, KYSE150 cells transfected with pHBLV‐U6‐Luc‐T2A‐Puro vector (KYSE150‐shNC) or pHBLV‐U6‐Luc‐T2A‐Puro‐FSCN1 vector (KYSE150‐shFSCN1‐#1) and KYSE150‐shFSCN1‐#1 cells stably transfected with GFP‐Vector, Fascin‐GFP WT, FascinK471R‐GFP, or FascinK471Q‐GFP at 1 × 106 cells/inoculation were suspended in 100 μL PBS and injected into the left footpad of mice (8 mice per group of GFP‐Vector, Fascin‐GFP WT, FascinK471R‐GFP, and FascinK471Q‐GFP and 7 mice per group of KYSE150‐shNC and KYSE150‐shFSCN1‐#1). Mice were palpated every two days for tumors, and each mouse was imaged twice a week using an IVIS‐100 Imaging System (Caliper Life Sciences, Waltham, MA, USA) after intraperitoneal injection of Dc‐luciferin (150 mg/kg in PBS) under anesthesia for 5 min, 2 min, and 30 s exposure. The mice in KYSE150‐shNC and KYSE150‐shFSCN1‐#1 groups were euthanized 25 days after injection, and the other groups were euthanized 32 days after injection. The tumor from the footpad region was defined as the “primary tumor”, and tumor from the footpad to the ankle joint was defined as the “invaded tumor” (local tumor invasion). All inguinal lymph nodes and footpad tumors were resected and fixed in 10% formaldehyde/PBS for histological analysis. Lymph nodes and footpad tumors were subjected to H&E and IHC staining. IHC staining was performed as described previously [32] using mouse anti‐pan‐cytokeratin (CK) AE1/AE3 (ZM‐0069; ZSGB‐Bio) and mouse anti‐GFP antibodies (sc‐9996; Santa Cruz). All animal experiments were conducted after approval from the Institutional Animal Care and Use Committee of Shantou University.
2.15. Live‐cell imaging and FRAP
The behavior of GFP‐ and RFP‐tagged proteins in cell filopodia was visualized using a confocal microscope with an incubator (37°C and 5% CO2, LSM880; Carl Zeiss MicroImaging, Inc.) with a 40 ×, 1.43 numerical aperture (NA), Plan‐Apochromat oil objective (Carl Zeiss MicroImaging, Inc.). The number, length, and lifetime of filopodia were measured from three independent experiments using ZEN software (blue edition; Carl Zeiss MicroImaging, Inc.). FRAP experiments were performed as previously reported [5, 7] with modifications. GFP‐ and RFP‐tagged proteins at the base of protruding filopodia were bleached in a rectangular region with an area ranging from 1 to 3 μm2 for ∼0.5 s using the 488 nm and 543 nm laser lines at 100% laser power. Thereafter, fluorescence recovery within the bleached region was monitored every 4‐5 s over a period of 60‐100 s. The first frame post‐photobleaching corresponds to 0 s. FRAP data were analyzed in the FRAP module by ZEN software following the manufacturer's instructions. The average intensity over the time course in the bleached zone was measured and corrected for photobleaching by multiplying a correction factor. The recovery curve was fitted with a double‐exponential function as follows: I = IE ‐ I1 × exp (‐t/T1) – I2 × exp (‐t/T2), where I represents the average intensity of the bleached region at a given time t (s), IE is the final signal intensity in the analyzed bleach region following recovery, I1 is the amplitude of the fitted curve of a fast, I2 is the amplitude of a slowly recovering population of molecules, T1 is a constant that is associated with the rate of recovery of the fast fraction, and T2 is a constant associated with the recovery of a slow fraction. The half‐time of the recovery was calculated using the following formula: ‐thalf = (ln 0.5) × T1, where thalf is the halftime of recovery. The rate constant K (per second) for the exchange of molecules between the bleached region and the surrounding area was calculated using the formula: d[F1]/dt = −k1[F1] + k2[F2], where k1 is a disassociation constant, [F1] is the number of molecules in the cluster, k2 is an association constant, and [F2] is the number of molecules in the surrounding environment.
2.16. Analysis of G/F‐actin ratio
The ratio of G‐actin to F‐actin (G/F‐actin ratio) was determined using the G/F‐actin in vivo assay kit (Cytoskeleton, Inc., Denver, CO, USA) according to the manufacturer's instructions. Briefly, the KYSE150‐shNC, KYSE150‐shFSCN1‐#1 cells, and KYSE150‐shFSCN1‐#1 cells stably transfected with GFP‐vector, Fascin‐GFP (WT), FascinK471R‐GFP, or FascinK471Q‐GFP were homogenized in lysis and F‐actin stabilization buffers followed by centrifugation for 1 h at 100,000 × g at 37°C for F‐actin separation. The supernatants containing G‐actin were collected, and the pellets containing F‐actin were resuspended in F‐actin depolymerizing solution. Equal amounts of both the supernatant (G‐actin) and resuspended pellet (F‐actin) were subjected to Western blotting and probed with an anti‐actin antibody. The G/F‐actin ratio was determined by densitometry using the ImageJ software (National Institutes of Health, Bethesda, MD, USA). Experiments were repeated twice with similar results.
2.17. Generation of polyclonal rabbit anti‐human AcK471‐Fascin antibody
The polyclonal rabbit anti‐human AcK471‐Fascin antibody was produced by PTM Biolabs (Hangzhou, Zhejiang, China) and raised against two synthetic peptides with Fascin lysine 471 acetylated [CGGRYL‐(acetyl)K‐GDHAG and KVGGRYL‐(acetyl)K‐GDHAGC]. The peptides, including a non‐acetylated peptide (CGGRYLKGDHAG), were synthesized and examined by MS analysis. The acetylated peptides were conjugated to the carrier protein keyhole limpet hemocyanin and used to generate immune responses in pathogen‐free, barrier‐raised New Zealand White rabbits (PTM Biolab LLC). After immunization, sera were collected for enzyme‐linked immunosorbent assay and dot blotting assay. After sequential affinity chromatography using protein A and the corresponding acetyl‐peptide, the eluent was purified using corresponding non‐acetylated peptide (CGGRYLKGDHAG).
2.18. Statistical analysis
The SPSS v13.0 program (SPSS Inc., Chicago, IL, USA) was used for data analysis. Data are presented as the mean ± standard deviation or mean ± standard error of the mean as indicated in the figure legends. The Student's t‐test was used for unpaired, two‐tailed comparison. P < 0.05 was considered significant. Kaplan‐Meier survival analysis was used for patient OS and DFS, and the log‐rank test was used for survival comparison.
3. RESULTS
3.1. Fascin interacted with PCAF directly
To identify which acetyltransferase that interacted with Fascin, we co‐expressed Flag‐Fascin with several acetyltransferases, including PCAF, KAT2A, and KAT5, in HEK293T cells and tested their interactions via co‐IP. The results showed that PCAF and KAT2A, but not KAT5, were co‐immunoprecipitated by Flag‐Fascin (Supplementary Figure S1). As PCAF and KAT2A are homologs, we chose PCAF for subsequent analysis. To further verify the interaction between Fascin and PCAF, we performed IP assay in HEK293T and KYSE140 cells. The interaction of Fascin with PCAF was validated by the IP of endogenous Fascin and PCAF proteins (Figure 1A). Moreover, immunofluorescence staining showed that Fascin and PCAF colocalized in the cytoplasm (Figure 1B), indicating that they had the same distribution region in cells which is necessary for their direct cellular interaction. Furthermore, to examine whether Fascin and PCAF interact directly, we expressed GST‐ or His‐fused Fascin and PCAF in Escherichia coli and purified them for GST‐pull down assays. The results showed that His‐Fascin was pulled down by GST‐PCAF, and reciprocally, His‐PCAF was pulled down by GST‐Fascin (Figure 1C), indicating that Fascin interacted directly with PCAF. To further elucidate the interaction between Fascin and PCAF, the GST fusion proteins containing different PCAF domains, including the N‐terminal, HAT, and C‐terminal BROMO domains, were purified for GST‐pull down assays with His‐Fascin in vitro. Similar to the‐full length PCAF, the individual N‐ and C‐terminal of PCAF interacted with Fascin; however, when both the N‐ and C‐terminal domains were removed, only the HAT domain of PCAF was left, and the interaction between Fascin and PCAF was abrogated (Figure 1D). This result indicates that PCAF bounded directly to Fascin through its N‐ and C‐terminal domains but not the HAT domain. Reciprocally, different truncated Fascin forms (each β‐trefoil fold from the N‐ or C‐terminal individually removed) were fused to GST and purified for GST‐pull down assay. The results showed that removal of any β‐trefoil fold from either the N‐ or C‐terminal of Fascin markedly attenuated its interaction with PCAF (Figure 1E), indicating that all four β‐trefoil folds of Fascin were required for the interaction with PCAF. We deduced that an integrated structure of Fascin was required for its interaction with PCAF, and Fascin may be tightly packed with a unique protein configuration to form a substantial interdomain that interacts with PCAF. Removal of any of these domains will most likely disrupt protein configuration, leading to a loss of interaction between PCAF and these truncation mutants. Taken together, these findings demonstrate that full‐length Fascin interacts with PCAF directly via the N‐ and C‐terminal domains of PCAF.
FIGURE 1.
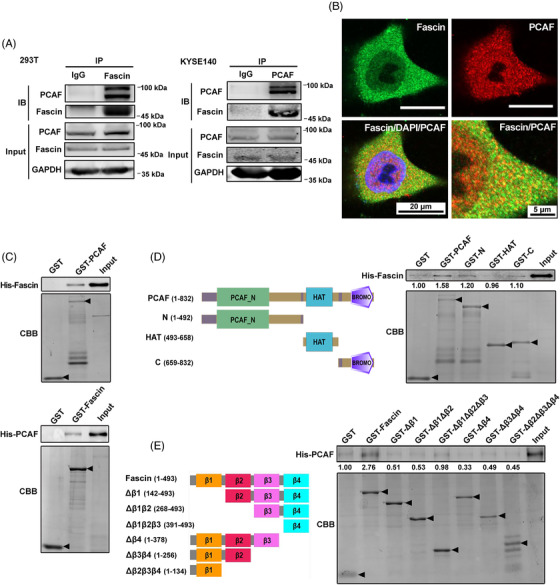
Fascin interacts with PCAF in ESCC cells. A, The interaction between Fascin and PCAF was investigated by immunoprecipitation assay using anti‐Fascin and anti‐PCAF antibodies in HEK293T and KYSE140 cells, respectively. B, Immunofluorescence analysis of the localization of endogenous Fascin and PCAF in ESCC KYSE150 cells. Endogenous Fascin (green) and PCAF (red) were stained with specific antibodies, and nuclei were stained with DAPI (blue). C, Fascin directly interacted with PCAF in in vitro GST‐pull down assays using GST‐PCAF and GST‐Fascin. D, The N‐terminal domain, acyltransferase domain (HAT), and C‐terminal BROMO domains of PCAF were fused to GST and purified for GST‐pull down assays with His‐fused Fascin (His‐Fascin). E, The GST‐fused truncated forms of Fascin were purified for GST‐pull down assays with His‐fused PCAF (His‐PCAF). The purified GST fusion proteins were examined with Coomassie brilliant blue staining, and the pulled‐down His proteins were examined by Western blotting. Arrows indicate proteins with correct molecular weights. Abbreviations: PCAF: P300/CBP‐associated factor; ESCC: esophageal squamous cell carcinoma; IP: immunoprecipitation; IB: immunoblot; GAPDH: glyceraldehyde‐3‐phosphate dehydrogenase; DAPI: 4’,6‐diamidino‐2‐phenylindole; CBB: Coomassie brilliant blue; GST: Glutathione S‐transferase; HAT: Histone acyltransferase; BROMO: bromodomain
3.2. Fascin was acetylated at lysine 471 by PCAF
We performed in vitro acetylation assays using immunoprecipitated HA‐PCAF from cells and in vitro purified His‐Fascin and found that Fascin was acetylated by PCAF (Supplementary Figure S2A). Using MS, we further identified that Fascin was acetylated at lysine 471 (K471) by PCAF (Figure 2A). The function of Fascin is conserved across organisms. Thus, to investigate the conservation of Fascin‐K471, we performed an amino acid sequence alignment of Fascin between humans and seven other species. Interestingly, we found that Fascin‐K471 was highly conserved in multiple species (Figure 2B), suggesting that it is an important acetylation site across organisms. To further characterize the K471 acetylation site, we generated an antibody (AcK471‐Fascin) that only recognizes acetylated Fascin. Antibody specificity was examined by dot blotting (Supplementary Figure S2B) and confirmed by Western blotting using proteins extracted from the KYSE140, KYSE150, KYSE180, KYSE450, and KYSE510 cells (Figure 2C), because the signal tested by AcK471‐Fascin antibody was significantly blocked by acetyl‐peptide but not by the unmodified‐Fascin peptide. Moreover, an acetylated K471 signal was detected in both endogenously (Figure 2D) and ectopically expressed Fascin (Supplementary Figure S2C) in ESCC cells; and when PCAF was overexpressed, the acetylated K471 signal was enhanced (Figure 2D) but was not detected in cells with FascinK471R mutant (K471R, a mimic of acetylation‐defective Fascin) (Supplementary Figure S2C). Furthermore, the siRNA‐mediated knockdown of endogenous PCAF decreased the K471 acetylation level of endogenous Fascin (Figure 2E). These results indicate that Fascin‐K471 is acetylated by PCAF.
FIGURE 2.
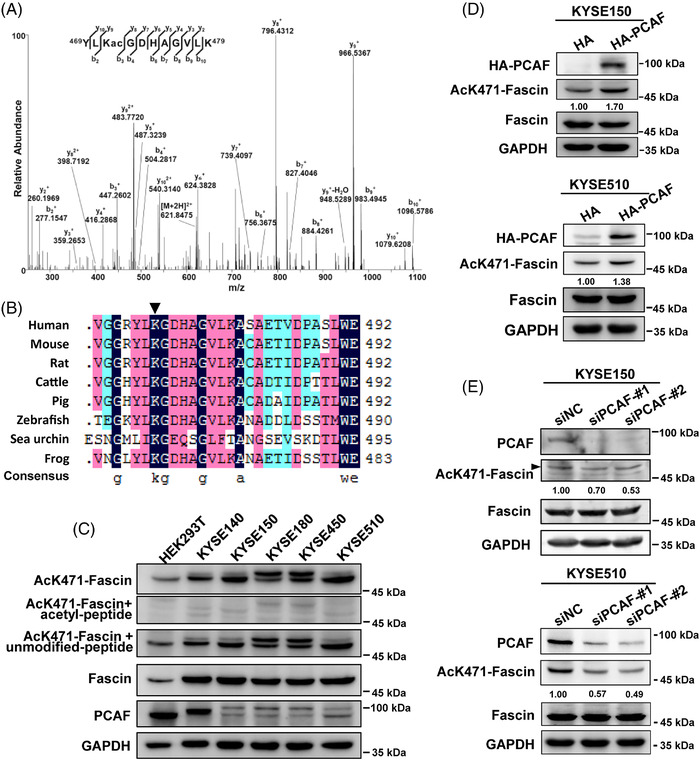
Fascin is acetylated at lysine 471 by PCAF. A, Mass spectrometry analysis showed that K471 was acetylated at the peptide 469‐YLKGDHAGVLK‐479. B, Sequence alignment of Fascin in human and other species for amino acid residues adjacent to human Fascin‐K471. C, The endogenous Fascin‐K471 acetylation was examined by specific AcK471‐Fascin antibody and AcK471‐Fascin antibody blocked by K471‐acetyl peptide or unmodified peptide. D, Endogenous Fascin‐K471 acetylation was examined in KYSE150 and KYSE510 cells overexpressing PCAF by Western blotting. E, The endogenous Fascin‐K471 acetylation was investigated under PCAF knockdown by siRNA in KYSE150 and KYSE510 cells. After 48 h of transfection, cells were collected and lysed, and lysates were used for Western blotting. GAPDH was used as a loading control. Abbreviations: PCAF: P300/CBP‐associated factor; GAPDH: glyceraldehyde‐3‐phosphate dehydrogenase. si: small interfering; NC: negative control; HA: Hemagglutinin
3.3. Acetylation of Fascin‐K471 by PCAF inhibited cancer cell migration and tumor metastasis
To further explore the physiological function of Fascin‐K471 acetylation in cancer cell migration, we first knocked down endogenous Fascin expression in KYSE150 and SHEEC cells via transient small RNA interference or stable short hairpin RNA silencing through lentivirus transfection. Knockdown of endogenous Fascin expression significantly inhibited the migration and invasion of ESCC cells (Supplementary Figure S3A‐C). We restored Fascin‐WT, FascinK471R, and FascinK471Q (K471Q, a mimic of hyperacetylated Fascin) in stable Fascin‐knockdown ESCC cells (Figure 3A). The restoration of Fascin‐WT recovered cell migration, restoration of K471R resulted in higher oncogenic abilities compared to Fascin‐WT, whereas restoration of FascinK471Q demonstrated the opposite, further suppressing cell migration in Fascin‐knockdown cells (Figures 3B and 3C). These results indicate that Fascin‐K471 acetylation plays a strong inhibitory role in cancer cell migration in vitro.
FIGURE 3.
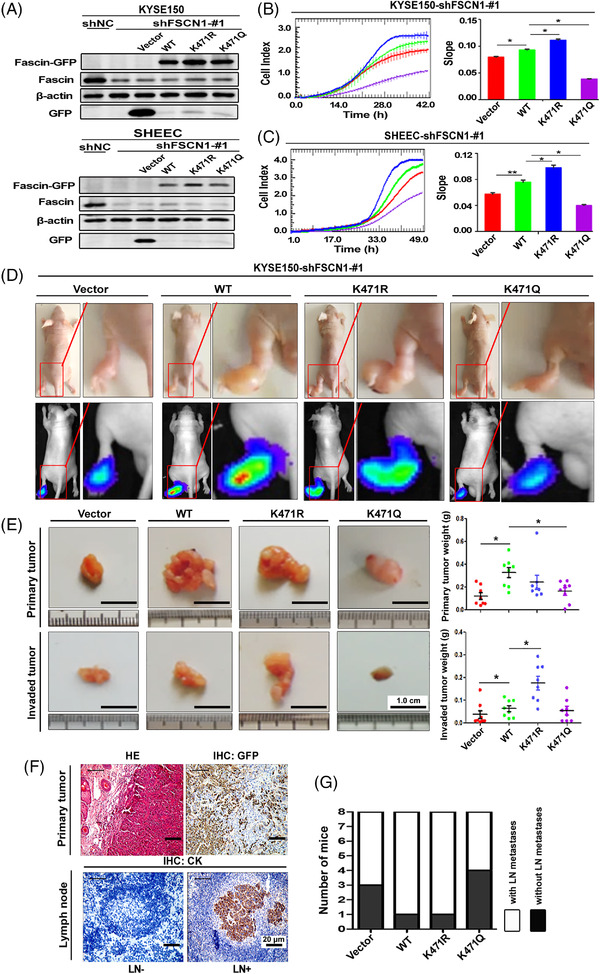
Acetylation of Fascin‐K471 by PCAF impairs cancer cell migration and tumor metastasis. A, Western blotting after restoration of GFP‐vector (Vector), Fascin‐GFP (WT), FascinK471R‐GFP (K471R), and FascinK471Q‐GFP (K471Q) expression in stable Fascin‐knockdown KYSE150 and SHEEC cells. Endogenous Fascin and Fascin‐GFP were detected using anti‐Fascin and GFP‐tag antibodies, respectively. β‐actin was used as a loading control. B and C, The migration of Fascin‐knockdown KYSE150 (B) and SHEEC (C) cells restored GFP‐vector (Vector), Fascin‐GFP (WT), FascinK471R‐GFP (K471R), and FascinK471Q‐GFP (K471Q) was investigated by using the xCELLigence Real‐Time Cell Analysis DP instrument. Left panel, real‐time detection of impedance response/cell index curves determined over 42 h (B) and 49 h (C). Right panel, the slope of the cell profiles. The experiments were performed independently in triplicate. Data are presented as the mean ± standard deviation (SD). The Student's t‐test was used for statistical analysis. D, Representative mice inoculated with cells in (B) (upper images), and bioluminescent images (lower images) at 32 days after injection in the left footpad. The invasion sites on the thigh are magnified. E, Representative excised primary tumors (from the footpad to the heel of the foot; upper images) and invaded tumors (from the heel of the foot to the inguinal lymph node; lower images) of mice in (D). Primary and invaded tumor weights were compared among the four groups. Data are presented as the mean ± SD of 8 mice in each group. The Student's t‐test was used for statistical analysis. F, Representative H&E and IHC staining for GFP was used to detect the primary tumor (upper images) and representative IHC staining of cytokeratin (CK) in the inguinal lymph nodes (lower images). G, Number of mice with and without lymph node metastases after 32 days of inoculation. *P < 0.05. Abbreviations: PCAF: P300/CBP‐associated factor; GFP: Green fluorescent protein; sh: short hairpin; NC: negative control; HE: hematoxylin‐eosin; IHC: immunohistochemistry; LN: lymph node; CK: cytokeratin; SD: standard deviation
Next, we utilized in vivo xenograft assays to examine the effect of Fascin‐K471 acetylation on tumor progression. The stable KYSE150 cells with endogenous Fascin knockdown, restoration of Fascin‐WT, FascinK471R, or FascinK471Q, or the control vector were transplanted into the left footpads of nude mice. Consistent with the results of a previous study [19], our results showed that depletion of endogenous Fascin strongly inhibited tumor growth and metastasis (Supplementary Figure S3D), with fewer mice (50%, 3/6) exhibiting lymph node metastasis (invasion to inguinal lymph nodes) compared to control mice (100%, 7/7) based on histopathological assessment (Supplementary Figure S3E‐F). Furthermore, the implantation assay showed that the recovery of Fascin‐WT significantly enhanced bioluminescence intensity, mean primary and invaded tumor weight, and lymph node metastasis rate compared to the vector control (Figure 3D‐G). In contrast to the restoration of Fascin‐WT, the invaded tumor weight was significantly higher after restoration of the acetyl‐defective form FascinK471R, although there was no significant difference in primary tumor weight and lymph node metastasis rate (Figure 3D‐G). Meanwhile, restoration of the acetylation‐mimicking form FascinK471Q significantly reduced the primary tumor weight and lymph node metastasis rate (Figure 3D‐G) compared to that of Fascin‐WT. These results indicate that acetylation of Fascin‐K471 inhibited tumor metastasis in vivo and the malignant phenotypes of ESCC cells both in vitro and in vivo.
3.4. Fascin‐K471 acetylation regulated filopodia dynamics by modulating its bundling to F‐actin
The biochemical function of Fascin‐K471 acetylation at the cellular level during filopodium formation was also investigated. Consistent with previous observations [5], live‐cell imaging assays revealed that the depletion of endogenous Fascin level significantly inhibited the density and length of filopodia in KYSE150 cells (Figure 4A and Supplementary Movie S1). Notably, the restoration of FascinK471Q and FascinK471R achieved opposite effects in terms of filopodium formation. Compared to the restoration of Fascin‐WT, that of FascinK471Q led to shorter and fewer filopodia extending from the cell edges, whereas that of FascinK471R promoted filopodium formation with longer and more filopodia protruding from the cell edges (Figure 4B and Supplementary Movie S2). Similar effects of restoration of Fascin‐WT, FascinK471R, and FascinK471Q in filopodium formation were also observed in SHEEC cells (Supplementary Figure S4A‐B and Supplementary Movie S3). These observations demonstrate that the acetylation of Fascin‐K471 suppresses filopodium formation. To further investigate the function of Fascin‐K471 acetylation in filopodia dynamics, we analyzed the filopodial lifetime during the extension‐retraction cycle. Live‐cell time‐lapse imaging showed that FascinK471Q significantly shortened both the lifetime and length of filopodia during the extension‐retraction cycle, although cells re‐expressing either Fascin‐WT or FascinK471R displayed similar filopodia lifetimes (Figures 4C and 4D, and Supplementary Movie S4). Taken together, these findings indicate that Fascin‐K471 acetylation regulates filopodial dynamics by reducing the length, density, and lifetime of filopodia.
FIGURE 4.
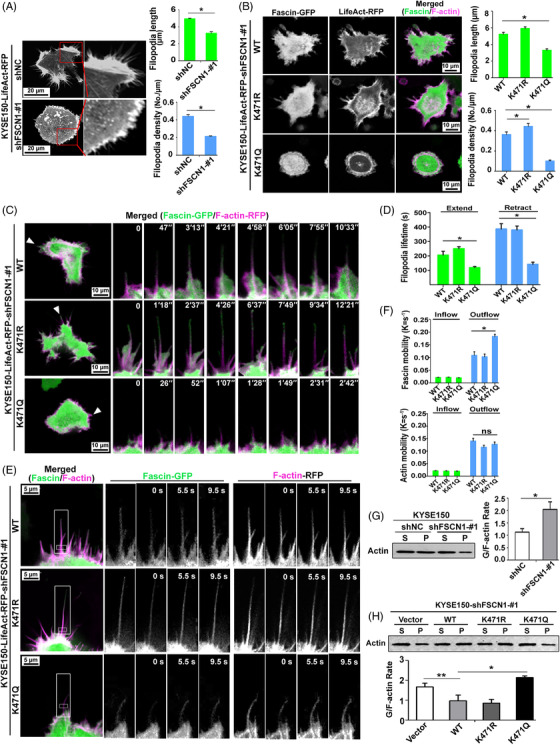
Fascin‐K471 acetylation regulates filopodia dynamics by modulating its bundling to F‐actin. A, The distribution of actin and the mean filopodial length and density (filopodium numbers per μm of cell perimeter; at least 50 cells were measured) were analyzed in KYSE150‐shNC and KYSE150‐shFSCN1 cells expressing LifeAct‐RFP. B, Fascin and F‐actin distribution and the mean filopodial length and density in KYSE150‐shFSCN1‐#1 cells that co‐expressed different types of Fascin‐GFP (WT, K471R, or K471Q) along with LifeAct‐RFP (at least 50 cells were measured). GFP and RFP fluorescence signals are shown in green and magenta, respectively. C and D, Representative confocal images of cell filopodia (arrowheads) during their lifetime (right; C). The lifetime of filopodia during one extension and retraction cycle was quantified (D; n ≥ 100 filopodia in at least 20 cells). E and F, FRAP assays were used to analyze the diffusion of different types of Fascin‐GFP (WT, K471R, or K471Q) and LifeAct‐RFP at the filopodia base of cells. GFP (green) and RFP (magenta) fluorescence signals of cell filopodia were obtained for an overview (left) with time‐lapse sequence images of the boxed region in one filopodium before and after photo‐bleaching (right; E). The recovery profile is curve‐fit by a double exponential function with recovery constant Kinflow and Koutflow for intra‐filopodia exchange and filopodia‐cytoplasmic exchange, respectively (F; n = 30). G and H, F‐actin bundling activity of Fascin and its mutant forms was analyzed by Western blotting. The pellet (P) and supernatant (S) represent F‐actin and G‐actin, respectively. Bar charts show the densitometric G‐actin/F‐actin ratio. Data were obtained from three independent experiments and are presented as the mean ± standard error of the mean (SEM). Student's t‐test was used for statistical analysis (*P < 0.05; **P < 0.01). Abbreviations: GFP: Green fluorescent protein; RFP: Red fluorescent protein; sh: short hairpin; NC: negative control; FRAP: fluorescence recovery after photobleaching; P: pellet; S: supernatant; SEM: standard error of the mean
Next, we sought to directly investigate whether the incorporation of Fascin in filopodia shafts was regulated by Fascin‐K471 acetylation. FRAP was performed with GFP‐tagged WT and mutant Fascin, along with RFP‐tagged actin, to measure the in vivo interaction between Fascin and F‐actin at the base of the filopodium (where filopodium connects to the cell). We used the rate constant K to quantify the exchange of molecules between the bleached region and the surrounding area (a per second unit) in order to assess protein dynamics and interactions with other cellular components [37]. In this study, the inflow and the outflow K values of Fascin‐GFP denote the association and dissociation rates of Fascin from F‐actin, respectively. By measuring the recovery of GFP fluorescence, we found no significant difference in either the inflow or the outflow K values between FascinK471R and WT Fascin (Figures 4E and 4F and Supplementary Figure S4C), suggesting that FascinK471R also underwent rapid association/dissociation cycles similar to Fascin. However, although the inflow K values of FascinK471Q did not show any changes, its outflow K values significantly increased compared to that of Fascin‐WT (Figures 4E and 4F, and Supplementary Figure S4C), implying that K471‐acetylated Fascin had even faster dynamic changes, as shown by its similar association rate, but had a faster dissociation rate than Fascin, which may be explained by the decreased binding affinity to F‐actin. Measuring the recovery of LifeAct‐RFP fluorescence demonstrated that there was no significant difference in the inflow and outflow K values of F‐actin between cells expressing WT Fascin and either mutant form (K471R or K471Q), suggesting that K471 acetylation had no effect on F‐actin dynamics (Figures 4E and 4F, and Supplementary Figure S4C).
Finally, to determine whether Fascin‐K471 acetylation affects actin‐bundling activity, we performed in‐cell actin‐bundling assays and found that the depletion of endogenous Fascin in KYSE150 and SHEEC cells significantly decreased F‐actin levels, leading to an increased G/F‐actin ratio (Figure 4G and Supplementary Figure S4D). Additionally, re‐expression of either WT or de‐acetylated FascinK471R restored F‐actin levels and reduced the G/F‐actin ratio, whereas the restoration of acetylated FascinK471Q significantly decreased F‐actin levels and resulted in a higher G/F‐actin ratio (Figure 4H and Supplementary Figure S4D). In addition, we performed a sedimentation (pelleting) assay at low‐speed centrifugation to determine the in vitro F‐actin bundling activity of Fascin and its mutants, as reported previously [6, 38]. We found that the K471Q mutation showed slightly reduced bundling activity, while the K471R mutation showed enhanced bundling activity (Supplementary Figure S4E). In general, these results provide both cellular and biochemical evidence that the K471 acetylation of Fascin inhibited its F‐actin‐bundling activity.
3.5. Acetylation of Fascin‐K471 by PCAF predicted favorable survival of ESCC patients
Fascin is highly expressed in several types of cancer, including esophageal cancer, and serves as a predictor of poor survival in patients [12, 35]. To evaluate the clinical significance of Fascin‐K471 acetylation, after testing the specificity of the AcK471‐Fascin antibody using IHC staining (Supplementary Figure S5), we examined the AcK471‐Fascin level in a cohort of 146 paired ESCC tissues and corresponding adjacent normal tissues using IHC staining. Interestingly, we found that AcK471‐Fascin was detected in both ESCC and adjacent normal tissues (Figure 5A). Quantification analysis showed that the protein level of AcK471‐Fascin in tumor tissues was significantly lower than that in the adjacent normal tissues (P = 0.006, Figure 5A). Remarkably, in contrast to our previous result that the high expression of Fascin was associated with poor outcome in patients with ESCC [12, 30], high levels of AcK471‐Fascin were strongly associated with increased OS (P = 0.004) and DFS (P = 0.007) (Figure 5B). As AcK471‐Fascin and total Fascin display prominent contrary prognostic values, we further performed combinatory analysis with AcK471‐Fascin and total Fascin in the same cohort which was analyzed previously [12]. As a result, low levels of total Fascin concomitant with high levels of AcK471‐Fascin in tumor tissues predicted favorable OS and DFS and was associated with the longest survival, whereas high levels of total Fascin together with low levels of AcK471‐Fascin were associated with the shortest survival (Figure 5C). In addition, we also evaluated the expression of PCAF in ESCC tissues by IHC to determine whether PCAF function is consistent with that of AcK471‐Fascin in predicting patient survival. Consistently, the clinical outcomes demonstrated that a high level of PCAF predicted favorable OS in patients with ESCC (Figure 5D). Collectively, the Fascin‐K471 acetylation levels in ESCC tissues were reduced and associated with patient survival, consistent with the association between PCAF expression and ESCC patient survival.
FIGURE 5.
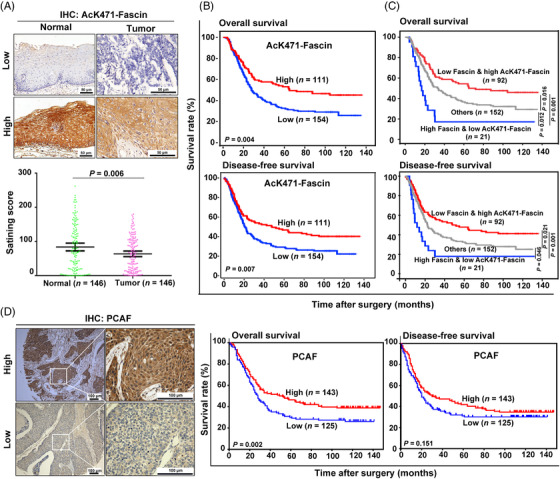
Fascin‐K471 acetylation is associated with ESCC patient survival. A, Representative IHC staining images of AcK471‐Fascin in paired ESCC tissues and adjacent normal tissues (n = 146). The staining scores are shown in the scatter diagram. Data are presented as mean ± SEM. The Student's t‐test was used for statistical analysis. B and C, Kaplan‐Meier survival analysis of ESCC patients with different expression levels of AcK471‐Fascin alone (B) or in combination with total Fascin (C). High expression of AcK471‐Fascin with concomitant low expression of total Fascin was significantly associated with the prolonged overall survival (OS) and disease‐free survival (DFS) of patients with ESCC. “Others” indicate patients concurrently expressing high or low levels of Fascin and AcK471‐Fascin. D, Representative IHC staining images of PCAF in ESCC tissues and Kaplan‐Meier analysis of OS and DFS of 268 patients with ESCC according to PCAF expression. Log‐rank test was used for survival analysis. Abbreviations: ESCC: esophageal squamous cell carcinoma; SEM: standard error of the mean; PCAF: P300/CBP‐associated factor; IHC: immunohistochemistry; OS: overall survival; DFS: disease‐free survival
4. DISCUSSION
In this study, we identified that Fascin K471 was acetylated by PCAF, which suppressed the actin‐bundling activity of Fascin, thereby inhibiting cancer cell filopodium formation and subsequent cancer migration and tumor metastasis. Therefore, we proposed a model in which Fascin may exist in two states through an acetylation‐deacetylation cycle mediated by the acetyltransferase PCAF and the unidentified deacetylases to regulate F‐actin bundling and further cell filopodium formation (Figure 6). In this context, PCAF functions as a tumor suppressor in ESCC, and when its levels are suppressed, Fascin remains in an unacetylated form with strong F‐actin‐bundling activity to promote filopodium formation, leading to cell migration and tumor metastasis. In contrast, high levels of PCAF may result in increased levels of Fascin‐K471 acetylation, which further weakens the bundling activity of Fascin to F‐actin, consequently inhibiting filopodium formation and tumor metastasis.
FIGURE 6.
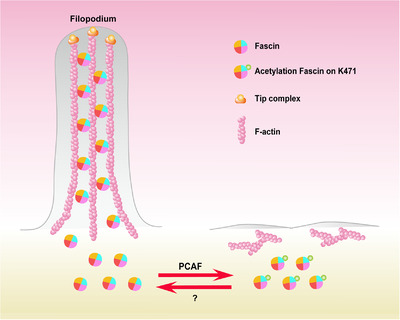
Working model of the PCAF‐mediated acetylation of Fascin at lysine 471 to inhibit tumor cell filopodium formation by impairing its bundling to F‐actin. PCAF functions as a tumor suppressor in ESCC. When the PCAF level is suppressed, Fascin remains in an unacetylated form with strong actin‐bundling activity to promote filopodium formation, leading to cell migration and tumor metastasis. In contrast, increased PCAF levels may result in increased levels of Fascin‐K471 acetylation, further weakening the bundling activity of Fascin to F‐actin and inhibiting filopodium formation and tumor metastasis. Abbreviations: ESCC: esophageal squamous cell carcinoma; PCAF: P300/CBP‐associated factor
Fascin interacted directly with PCAF, and the specific dependence on the N‐ and C‐terminal BROMO domains of PCAF was reconfirmed by the interaction of Fascin with PCAF homolog KAT2A, which has a similar structure to PCAF but not KAT5 (Supplementary Figure S1A). Both KAT2A and PCAF/KAT2B have been reported to be incorporated into large protein complexes [39], defined as ATAC and SAGA complexes, to carry out acetylation. Furthermore, all four subunits of the SAGA or ATAC HAT modules are needed for full and specific acetylation activity [40]. Therefore, in this study, we immunoprecipitated PCAF from cells and used it for in vitro acetylation assays to identify the acetylation residues of Fascin. We identified PCAF‐acetylated Fascin at the K471 residue, and this was verified using a specific antibody in ESCC cells by overexpressing and knocking down PCAF expression (Figures 2D and 2E).
Lysine 471 of Fascin is a conserved residue located at the C‐terminus of Fascin within β‐trefoil fold 4. Previous reports have demonstrated that a mutation at K471 (K471A) decreased the in vitro F‐actin‐bundling activity of Fascin [6] largely because K471 is located within a key actin‐binding site of Fascin, wherein the binding of Fascin to actin filaments interferes with the binding of a potential tumor metastasis inhibitor, macroketone [13]. The lysine acetylome analysis of epithelia‐originated A549 cells treated with the lysine deacetylase inhibitor MS275 [24] and HeLa cells treated with nicotinamide [26] by MS showed that many lysine residues of Fascin, including K471, were acetylated. In terms of the mechanism of protein lysine residue acetylation, the lysine ε‐amino group is activated for nucleophilic attack on the carbonyl group of acetyl CoA to form a tetrahedral intermediate, which then collapses with the loss of CoA to generate acetyl‐lysine. Lysine acetylation, catalyzed by acetyltransferases, neutralizes the positive charge on lysine residues. In this study, after identifying the acetylation of Fascin‐K471, we investigated how Fascin function is regulated by acetylation in ESCC. To this end, we used amino acid substitution of lysine to glutamine (Q), a polar uncharged amino acid, to mimic the acetylated lysine, and to arginine (R), another positively charged amino acid with a structure similar to lysine, to mimic the unacetylated lysine. The results of both in vivo and in vitro studies (Figure 3) indicated that Fascin‐K471 acetylation plays an important role in F‐actin bundling. Our FRAP experiments revealed that K471 acetylation inhibited Fascin binding to actin, leading to the fast dissociation of Fascin molecules from F‐actin in the filopodium shaft. These dissociated acetylated molecules mimicked by FascinK471Q mutants showed weaker interactions with F‐actin and moved more rapidly than deacetylated Fascin (K471R), leading to rapid and transient interactions with F‐actin. In addition, FRAP analysis showed that Fascin‐K471 acetylation had no effect on F‐actin kinetics, suggesting that at the base of the filopodium the mobile rate of monomers actin is not affected by Fascin‐K471 acetylation. According to the results that the G‐/F‐actin ratio was affected by the Fascin‐K471 acetylation, we speculated that acetylation of Fascin‐K471 results in a failure of the filopodium elongation, then reducing the content of F‐actin, while the filopodia initiation is independent of Fascin‐K471 acetylation, which is consistent with a previous report that Fascin did not participate in filopodia nucleation/initiation but only in elongation/retraction [41]. Interestingly, considering the similar effects of S39 phosphorylation and K471 acetylation in regulating Fascin bundling to F‐actin and their colocalization in the same actin‐binding site, further studies should be performed to elucidate whether crosstalk occurs between Fascin phosphorylation and acetylation and how they affect each other.
PCAF was found to acetylate Fascin‐K471, and although the level of Fascin‐K471 acetylation was decreased in ESCC cancer tissues compared to adjacent normal tissues, the high level of PCAF in ESCC tissues was associated with the favorable survival of patients with ESCC. There is currently a lack of consensus regarding the use of PCAF as a tumorigenic protein or suppressor in different cancers. However, based on our findings and previous studies, which demonstrated that PCAF was downregulated in ESCC tissues compared to adjacent normal tissues [42, 43], we deduced that PCAF plays an antitumorigenic role in ESCC. We proposed that the acetylation‐deacetylation cycle is critical for Fascin and, in this study, identified the acetylation writer PCAF. However, the deacetylases of Fascin and how they catalyze Fascin deacetylation need to be studied in the future. Interestingly, recent studies have revealed that PCAF is also a ubiquitination factor with intrinsic E3 ligase activity [44, 45, 46]. Considering that Fascin has also been found to be ubiquitinated at K471 under DNA damage conditions by MS analysis [47], it would be interesting to investigate whether PCAF ubiquitinates Fascin and how Fascin acetylation and ubiquitination coordinate in response to cancer progression and DNA damage in future studies.
5. CONCLUSIONS
Fascin was acetylated by PCAF at K471, and K471 acetylation inhibited the oncogenic function of Fascin in cancer progression. This study provides insights into the regulatory mechanism of acetylation on Fascin function and highlights the potential therapeutic value of mediating Fascin‐K471 acetylation against tumor metastasis.
DECLARATIONS
CONSENT FOR PUBLICATION
Informed consents were received from patients who participated in this study.
CONFLICT OF INTEREST
No potential conflicts of interest were disclosed
AUTHORS’ CONTRIBUTIONS
YWC, FMZ, LYX, and EML designed the research. YWC, FMZ, and DJL conducted the experiments and analyzed the data. SHW, JZH, ZCG, PJN, ZYW, WQS, and BW, XEX, LDL, JYW, and KZ acquired and analyzed the data. JZ and HQZ provided some vectors. YWC and FMZ wrote the manuscript. EML and LYX obtained the funding to support the project and supervised the development of the work.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
All animal experiments were conducted with the approval of the Institutional Animal Care and Use Committee of Shantou University. Ethical approval was obtained from the ethical committee of the Shantou Central Hospital and the ethical committee of the Shantou University Medical College, and only resected samples from surgical patients given written informed consent were included for use in research.
Supporting information
Supporting Information
MovieS1
MovieS2
MovieS3
MovieS4
ACKNOWLEDGMENTS
We are grateful to Dr. Daming Gao (Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences) for providing the acetyltransferase vectors, and Dr. Brian Stramer (King's College London, England, UK for providing the LifeAct‐RFP construct.
Cheng Y‐W, Zeng F‐M, Li D‐J, Wang S‐H, He J‐Z, Guo Z‐C, et al. P300/CBP‐associated factor (PCAF)‐mediated acetylation of Fascin at lysine 471 inhibits its actin‐bundling activity and tumor metastasis in esophageal cancer. Cancer Commun. 2021;41:1398–1416. 10.1002/cac2.12221
Contributor Information
Kai Zhang, Email: kzhang@tmu.edu.cn.
Li‐Yan Xu, Email: lyxu@stu.edu.cn.
En‐Min Li, Email: nmli@stu.edu.cn.
DATA AVAILABILITY STATEMENT
Data supporting the findings of this study are available within the article and its supplementary information files.
REFERENCES
- 1. Mattila PK, Lappalainen P. Filopodia: molecular architecture and cellular functions. Nat Rev Mol Cell Biol. 2008;9(6):446–54 [DOI] [PubMed] [Google Scholar]
- 2. Otto JJ, Kane RE, Bryan J. Formation of filopodia in coelomocytes: localization of fascin, a 58,000 dalton actin cross‐linking protein. Cell. 1979;17(2):285–93 [DOI] [PubMed] [Google Scholar]
- 3. Yamashiro‐Matsumura S, Matsumura F. Intracellular localization of the 55‐kD actin‐bundling protein in cultured cells: spatial relationships with actin, alpha‐actinin, tropomyosin, and fimbrin. J Cell Biol. 1986;103(2):631–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jaiswal R, Breitsprecher D, Collins A, Corrêa IR, Jr. , Xu MQ, Goode BL. The formin Daam1 and fascin directly collaborate to promote filopodia formation. Curr Biol. 2013;23(14):1373–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vignjevic D, Kojima S, Aratyn Y, Danciu O, Svitkina T, Borisy GG. Role of fascin in filopodial protrusion. J Cell Biol. 2006;174(6):863–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang S, Huang FK, Huang J, Chen S, Jakoncic J, Leo‐Macias A, et al. Molecular mechanism of fascin function in filopodial formation. J Biol Chem. 2013;288(1):274–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Aratyn YS, Schaus TE, Taylor EW, Borisy GG. Intrinsic dynamic behavior of fascin in filopodia. Mol Biol Cell. 2007;18(10):3928–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang H, Xu L, Xiao D, Xie J, Zeng H, Cai W, et al. Fascin is a potential biomarker for early‐stage oesophageal squamous cell carcinoma. J Clin Pathol. 2006;59(9):958–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cheng Y, Xie J, Zeng F, Nie P, Wu B, Du Z, et al. Fascin and esophageal squamous cell carcinoma. Precision Radiation Oncology. 2017;1(3):82‐7 [Google Scholar]
- 10. Ma Y, Machesky LM. Fascin1 in carcinomas: Its regulation and prognostic value. Int J Cancer. 2015;137(11):2534–44 [DOI] [PubMed] [Google Scholar]
- 11. Chen WX, Hong XB, Hong CQ, Liu M, Li L, Huang LS, et al. Tumor‐associated autoantibodies against Fascin as a novel diagnostic biomarker for esophageal squamous cell carcinoma. Clin Res Hepatol Gastroenterol. 2017;41(3):327–32 [DOI] [PubMed] [Google Scholar]
- 12. Tan H, Zhang H, Xie J, Chen B, Wen C, Guo X, et al. A novel staging model to classify oesophageal squamous cell carcinoma patients in China. Br J Cancer. 2014;110(8):2109–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen L, Yang S, Jakoncic J, Zhang JJ, Huang XY. Migrastatin analogues target fascin to block tumour metastasis. Nature. 2010;464(7291):1062–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jacquemet G, Hamidi H, Ivaska J. Filopodia in cell adhesion, 3D migration and cancer cell invasion. Curr Opin Cell Biol. 2015;36:23–31 [DOI] [PubMed] [Google Scholar]
- 15. Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, et al. Cell migration: integrating signals from front to back. Science. 2003;302(5651):1704–9 [DOI] [PubMed] [Google Scholar]
- 16. Adams JC. Roles of fascin in cell adhesion and motility. Curr Opin Cell Biol. 2004;16(5):590–6 [DOI] [PubMed] [Google Scholar]
- 17. Machesky LM, Li A. Fascin: Invasive filopodia promoting metastasis. Commun Integr Biol. 2010;3(3):263–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Anilkumar N, Parsons M, Monk R, Ng T, Adams JC. Interaction of fascin and protein kinase Cα: a novel intersection in cell adhesion and motility. EMBO J. 2003;22(20):5390–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hashimoto Y, Parsons M, Adams JC. Dual actin‐bundling and protein kinase C‐binding activities of fascin regulate carcinoma cell migration downstream of Rac and contribute to metastasis. Mol Biol Cell. 2007;18(11):4591–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li A, Dawson JC, Forero‐Vargas M, Spence HJ, Yu X, König I, et al. The actin‐bundling protein fascin stabilizes actin in invadopodia and potentiates protrusive invasion. Curr Biol. 2010;20(4):339–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ono S, Yamakita Y, Yamashiro S, Matsudaira PT, Gnarra JR, Obinata T, et al. Identification of an actin binding region and a protein kinase C phosphorylation site on human fascin. J Biol Chem. 1997;272(4):2527–33 [DOI] [PubMed] [Google Scholar]
- 22. Yamakita Y, Ono S, Matsumura F, Yamashiro S. Phosphorylation of human fascin inhibits its actin binding and bundling activities. J Biol Chem. 1996;271(21):12632–8 [DOI] [PubMed] [Google Scholar]
- 23. Zeng FM, Wang XN, Shi HS, Xie JJ, Du ZP, Liao LD, et al. Fascin phosphorylation sites combine to regulate esophageal squamous cancer cell behavior. Amino Acids. 2017;49(5):943–55 [DOI] [PubMed] [Google Scholar]
- 24. Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co‐regulates major cellular functions. Science. 2009;325(5942):834–40 [DOI] [PubMed] [Google Scholar]
- 25. Sheikh BN, Akhtar A. The many lives of KATs ‐ detectors, integrators and modulators of the cellular environment. Nat Rev Genet. 2019;20(1):7–23 [DOI] [PubMed] [Google Scholar]
- 26. Schölz C, Weinert BT, Wagner SA, Beli P, Miyake Y, Qi J, et al. Acetylation site specificities of lysine deacetylase inhibitors in human cells. Nat Biotechnol. 2015;33(4):415–23 [DOI] [PubMed] [Google Scholar]
- 27. Yang XJ. The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic Acids Res. 2004;32(3):959–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fournier M, Orpinell M, Grauffel C, Scheer E, Garnier JM, Ye T, et al. KAT2A/KAT2B‐targeted acetylome reveals a role for PLK4 acetylation in preventing centrosome amplification. Nat Commun. 2016;7:13227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shimada Y, Imamura M, Wagata T, Yamaguchi N, Tobe T. Characterization of 21 newly established esophageal cancer cell lines. Cancer. 1992;69(2):277–84 [DOI] [PubMed] [Google Scholar]
- 30. Shen Z, Shen J, Cai W, Chen J, Zeng Y. [Malignant transformation of the immortalized esophageal epithelial cells]. Zhonghua Zhong Liu Za Zhi. 2002;24(2):107–9 [PubMed] [Google Scholar]
- 31. Kuninger D, Lundblad J, Semirale A, Rotwein P. A non‐isotopic in vitro assay for histone acetylation. J Biotechnol. 2007;131(3):253–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhao Q, Shen JH, Shen ZY, Wu ZY, Xu XE, Xie JJ, et al. Phosphorylation of fascin decreases the risk of poor survival in patients with esophageal squamous cell carcinoma. J Histochem Cytochem. 2010;58(11):979–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhan XH, Jiao JW, Zhang HF, Xu XE, He JZ, Li RL, et al. LOXL2 Upregulates Phosphorylation of Ezrin to Promote Cytoskeletal Reorganization and Tumor Cell Invasion. Cancer Res. 2019;79(19):4951–64 [DOI] [PubMed] [Google Scholar]
- 34. Fang WK, Liao LD, Li LY, Xie YM, Xu XE, Zhao WJ, et al. Down‐regulated desmocollin‐2 promotes cell aggressiveness through redistributing adherens junctions and activating beta‐catenin signalling in oesophageal squamous cell carcinoma. J Pathol. 2013;231(2):257–70 [DOI] [PubMed] [Google Scholar]
- 35. Liu D, Li C, Trojanowicz B, Li X, Shi D, Zhan C, et al. CD97 promotion of gastric carcinoma lymphatic metastasis is exosome dependent. Gastric Cancer. 2016;19(3):754–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang XD, Huang GW, Xie YH, He JZ, Guo JC, Xu XE, et al. The interaction of lncRNA EZR‐AS1 with SMYD3 maintains overexpression of EZR in ESCC cells. Nucleic Acids Res. 2018;46(4):1793–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reits EA, Neefjes JJ. From fixed to FRAP: measuring protein mobility and activity in living cells. Nat Cell Biol. 2001;3(6):35078615 [DOI] [PubMed] [Google Scholar]
- 38. Jansen S, Collins A, Yang C, Rebowski G, Svitkina T, Dominguez R. Mechanism of actin filament bundling by fascin. J Biol Chem. 2011;286(34):30087–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bondy‐Chorney E, Denoncourt A, Sai Y, Downey M. Nonhistone targets of KAT2A and KAT2B implicated in cancer biology. Biochem Cell Biol. 2019;97(1):30–45 [DOI] [PubMed] [Google Scholar]
- 40. Helmlinger D, Tora L. Sharing the SAGA. Trends Biochem Sci. 2017;42(11):850–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Faix J, Rottner K. The making of filopodia. Curr Opin Cell Biol. 2006;18(1):18–25 [DOI] [PubMed] [Google Scholar]
- 42. Qin YR, Fu L, Sham PC, Kwong DL, Zhu CL, Chu KK, et al. Single‐nucleotide polymorphism‐mass array reveals commonly deleted regions at 3p22 and 3p14.2 associate with poor clinical outcome in esophageal squamous cell carcinoma. Int J Cancer. 2008;123(4):826–30 [DOI] [PubMed] [Google Scholar]
- 43. Zhu C, Qin YR, Xie D, Chua DT, Fung JM, Chen L, et al. Characterization of tumor suppressive function of P300/CBP‐associated factor at frequently deleted region 3p24 in esophageal squamous cell carcinoma. Oncogene. 2009;28(31):2821–8 [DOI] [PubMed] [Google Scholar]
- 44. Linares LK, Kiernan R, Triboulet R, Chable‐Bessia C, Latreille D, Cuvier O, et al. Intrinsic ubiquitination activity of PCAF controls the stability of the oncoprotein Hdm2. Nat Cell Biol. 2007;9(3):331–8 [DOI] [PubMed] [Google Scholar]
- 45. Mazza D, Infante P, Colicchia V, Greco A, Alfonsi R, Siler M, et al. PCAF ubiquitin ligase activity inhibits Hedgehog/Gli1 signaling in p53‐dependent response to genotoxic stress. Cell Death Differ. 2013;20(12):1688–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li Q, Liu Z, Xu M, Xue Y, Yao B, Dou C, et al. PCAF inhibits hepatocellular carcinoma metastasis by inhibition of epithelial‐mesenchymal transition by targeting Gli‐1. Cancer Lett. 2016;375(1):190–8 [DOI] [PubMed] [Google Scholar]
- 47. Povlsen LK, Beli P, Wagner SA, Poulsen SL, Sylvestersen KB, Poulsen JW, et al. Systems‐wide analysis of ubiquitylation dynamics reveals a key role for PAF15 ubiquitylation in DNA‐damage bypass. Nat Cell Biol. 2012;14(10):1089–98 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
MovieS1
MovieS2
MovieS3
MovieS4
Data Availability Statement
Data supporting the findings of this study are available within the article and its supplementary information files.