

Abstract
The enantioselective total synthesis of the rearranged spongian diterpenoid (–)-macfarlandin C is reported. This is the first synthesis of a rearranged spongian diterpenoid in which the bulky hydrocarbon fragment is joined via a quaternary carbon to the highly hindered concave face of the cis-2,8-dioxabicyclo[3.3.0]octan-3-one moiety. The strategy involves a late-stage fragment coupling between a tertiary carbon radical and an electrophilic butenolide resulting in the stereoselective formation of vicinal quaternary and tertiary stereocenters. A stereoselective Mukaiyama hydration that orients a pendant carboxymethyl side chain cis to the bulky octahydronapthalene substituent was pivotal in fashioning the challenging concave-substituted cis-dioxabicyclo[3.3.0]octanone fragment.
Keywords: C–C coupling, natural product synthesis, photoredox chemistry, terpene synthesis, radical chemistry
Graphical Abstract
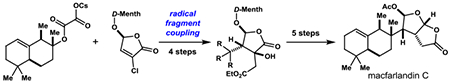
A diverse group of marine diterpenoids are believed to arise by fragmentation and rearrangement of the steroid-like spongian skeleton.[1] A distinctive set of these rearranged spongian diterpenoids harbor a cis-2,8-dioxabicyclo[3.3.0]octan-3-one fragment (1) attached at C-6 to a quaternary carbon of a hydrophobic fragment (Figure 1). These diterpenoids can be further subdivided into two families that differ by the orientation of the hydrocarbon fragment. In the largest collection, the hydrocarbon moiety resides on the more sterically hindered concave face of the cis-dioxabicyclo[3.3.0]octan-3-one fragment, exemplified by macfarlandin C (2),[2] whereas cheloviolene A (3)[3] is representative of members in which the hydrocarbon unit resides on the convex face.
Figure 1:
Rearranged spongian diterpenoids harboring the cis-2,6-dioxabicyclo[3.3.0]octan-3-one moiety, and a ball-and-stick representation of the X-ray model of macfarlandin C showing the unusually long C-8/C-14 σ-bond linking the two chiral fragments.
Our interest in these structures was initially piqued by Sütterlin’s observations of the unique Golgi-altering activity of macfarlandin E, a structurally related diterpenoid in which the cis-dioxabicyclooctanone fragment is replaced by a 2,7-dioxabicylo[3.2.1]-octan-3-one subunit.[4] Macfarlandin E, and some simplified congeners having either a cis-2,8-dioxabicyclo[3.3.0]octan-3-one or a 2,7-dioxabicylo[3.2.1]-octan-3-one subunit, uniquely induce irreversible fragmentation of the Golgi apparatus with retention of fragments in the pericentriolar region of the cell.[4,5] The fused and bridged dioxabicyclooctanone moieties degrade in the presence of primary amine functionalities to form pyrrole products via putative 1,4-dialdehyde intermediates. This mode of conjugation is suggested to be important for the Golgi phenotype of these natural products.[4–6]
The central challenge in the synthesis of the marine diterpenoids exemplified in Figure 1 is fashioning the σ-bond that links the two chiral fragments in a stereocontrolled fashion. This challenge is heightened significantly in members such as macfarlandin C (2) wherein the bulky hydrocarbon unit resides on the sterically demanding concave face of cis-2,8-dioxabicyclooctanone fragment. This steric congestion is apparent in the X-ray model of macfarlandin C (Figure 1),[2] and strikingly illustrated in the unusually long length (1.577 Å) of the C-8/C-14 σ-bond that joins the two fragments. In contrast, this bond in cheloviolene A (3) is quite standard (1.546 Å).[3a,7] In addition, this steric congestion results in significant distortion of the cis-2,8-dioxabicyclooctanone fragment of macfarlandin C (2) as compared to that of cheloviolene A (3).[7]
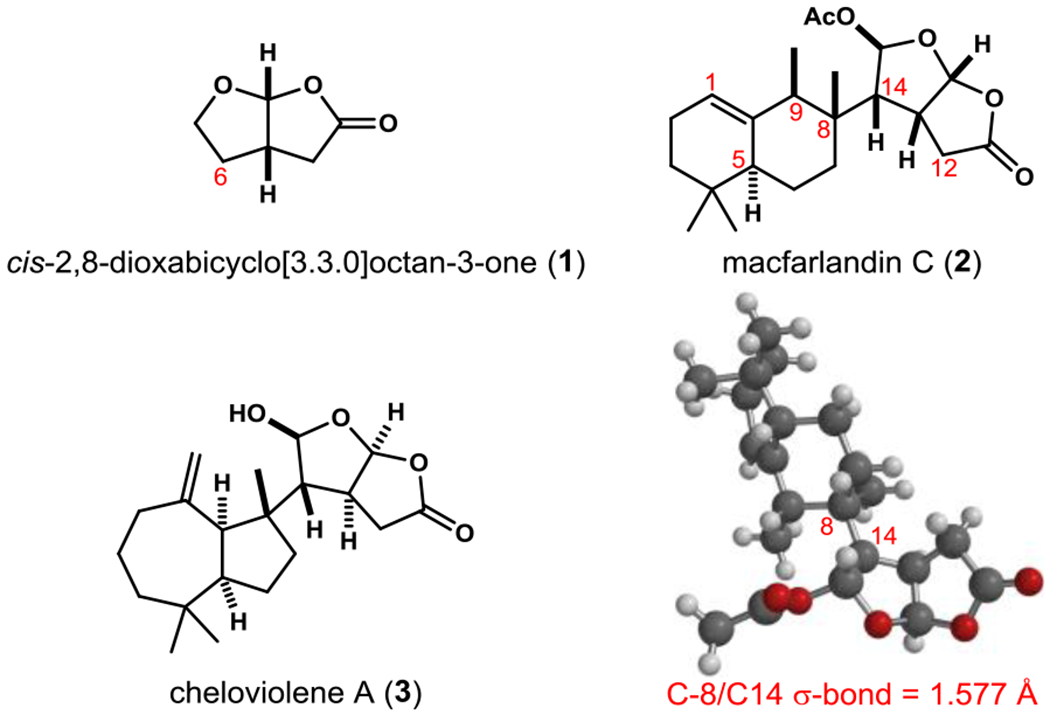
When we initiated studies to develop a chemical synthesis of macfarlandin C (2), only the related structural archetypes cheloviolene A (3) and cheloviolene B having the hydrocarbon fragment positioned on the convex face of the cis-2,8-dioxabicyclo[3.3.0]octan-3-one unit had been synthesized.[8] The approach employed in these syntheses to access the 6-substituted cis-2,8-dioxabicyclo[3.3.0]octan-3-one moiety relied on the coupling of a tertiary radical with a 5-alkoxy butenolide.[9] Although this approach allowed for facile access to diterpenoids bearing the C-6 hydrophobic fragment on the convex face of the cis-2,8-dioxabicyclo[3.3.0]octan-3-one unit, we were unable to tune this coupling to access the alternate stereoisomer.[8b] We report herein the development of a synthetic approach to cis-2,8-dioxabicyclo[3.3.0]octan-3-ones attached at C-6 to a quaternary carbon of a bulky hydrophobic fragment that allows for the divergent synthesis of either C-6 substituted stereoisomeric from the product of fragment coupling (Scheme 1). The utility of this strategy is exemplified by the enantioselective total synthesis of (–)-macfarlandin C (2).
Scheme 1:
General and divergent approach to 6-substituted cis-2,8-dioxabicyclo[3.3.0]octan-3-ones.
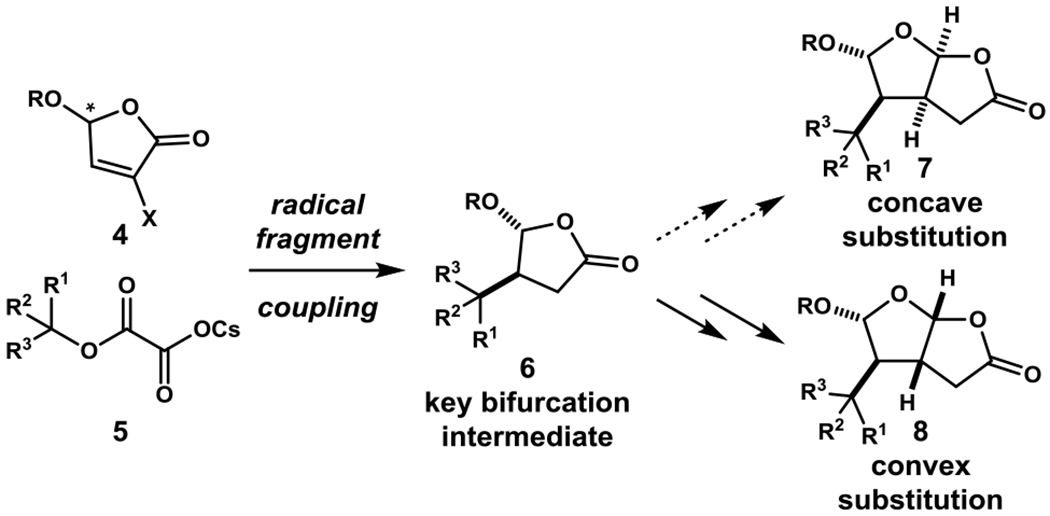
We initiated exploratory model studies with lactone 11, which is readily available from the coupling of cesium oxalate 9 and 5-methoxybutenolide (10).[8b] Our original aim was to explore the feasibility of directly installing a carboxymethyl substituent cis to the bulky 1-methylcyclohexyl substituent of 11. This goal has proven to be exceptionally challenging and has not yet been realized. One approach we examined was to introduce the side chain as an alkylidene fragment, with the hope that the double bond could be reduced selectively from the face anti to the adjacent hydrocarbon side chain. Aldol reaction of lactone 11 with ethyl glyoxylate yielded a mixture of aldol adducts, which was dehydrated to provide in high overall yield alkylidene product 12 as a mixture of E and Z stereoisomers. Unfortunately, under no conditions that we examined was the stereoisomeric hydrogenation product having the 1-methylcyclohexyl and carboxymethyl substituents cis formed selectively. Among the conditions examined were heterogeneous catalytic hydrogenation using Pd, Pt and Rh catalysts, homogeneous hydrogenation using Rh or Ir catalysts, Cu and Ni-promoted hydride reduction,[10] and several recent and older hydrogenation methods that likely proceed by initial hydrogen atom transfer.[11,12]
We turned to a strategy in which the ester side chain would be “locked” into a cis relationship with the bulky hydrophobic substituent by incorporation of a hydroxyl group at the α-position of a butyrolactone intermediate.[13] Mukaiyama hydration of alkylidene lactone 12 took place with complete regioselectivity from the lactone face opposite the 1-methylcyclohexyl substituent to give alcohol intermediate 13.[14–16] The transformation of alcohol 13 to concave-functionalized cis-2,8-dioxabicyclo[3.3.0]octan-3-one 17 was initially accomplished by way of three isolated intermediates. After initial silyl protection of the hydroxyl substituent, reaction with excess (iBu)2AlH provided a mixture of lactol epimers, which were directly oxidized to give cis-dioxabicyclooctanone 14 in high yield.[8b] Hydrolysis of 14 at room temperature in dilute HCl furnished diol 15, which was then allowed to react with excess acetic anhydride at room temperature. The intermediate diacetate, which could be observed in the crude product by NMR analysis, converted completely to elimination product 16 by simple treatment with silica gel. Conjugate-silane reduction of this unsaturated lactone by the method of Buchwald then provided cis-2,8-dioxabicyclo[3.3.0]octan-3-one 17 in good yield.[17]
Our application of this strategy to construct (–)-macfarlandin C (2) is summarized in Schemes 3 and 4. The route commences with the enantioselective synthesis of octahydronapthalene tertiary alcohol 28 in nine steps from 4,4-dimethylcyclohexen-1-one (18). Iodination of 18, followed by catalytic enantioselective reduction of α-iodocyclohexenone 19 by a variant of the Corey-Bakshi-Shibata reduction afforded (S)-cyclohexenol 20 in high yield and 98% ee.[18,19] After conversion to allylic phosphate 21, anti-SN2’ allylic displacement by reaction with an excess of the organocuprate intermediate generated in situ from CuCN and Grignard reagent 22 gave vinyl iodide 23 in high yield.[19] Enantioselective HPLC analysis showed that the displacement took place with complete transfer of chirality.
Scheme 3:
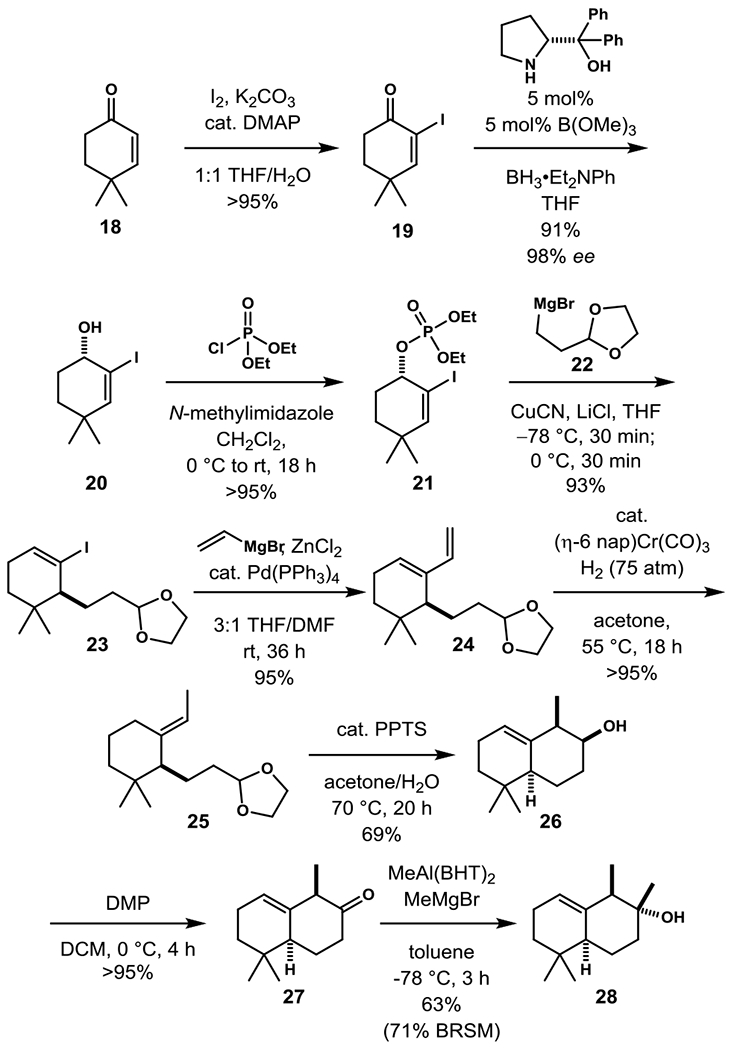
Enantioselective construction of octahydronapthalene oxalate coupling partner 28. (DMAP = N,N-dimethyl-4-aminopyridine, DMF = N,N-dimethylformamide, PPTS = pyridinium para-toluenesulfonate, DMP = Dess-Martin periodinane, BHT = 2,6-di-tert-butyl-4-methylphenol).
Scheme 4:
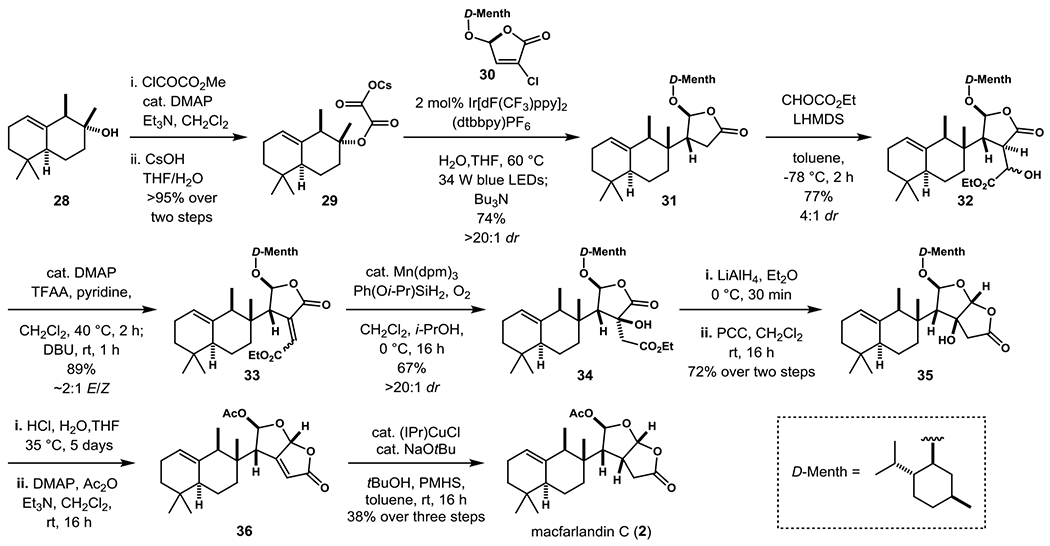
Photoredox-mediated fragment coupling for the generation of lactones 31 and elaboration to macfarlandin C (1). (DMAP = N,N-dimethyl-4-aminopyridine, dF(CF3)ppy = 2-(2,4-difluorophenyl)-5-trifluoromethylpyridine, dtbbpy = 4,4’-di-t-Bu-2,2’-bipyridine, LiHMDS = lithium hexamethyldisilazide, TFAA = trifluoroacetic anhydride, DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene, dpm = dipivaloylmethane, PCC = pyridinium chlorochromate, PMHS = polymethylhydrosiloxane).
As a prelude to forming the (E)-ethylidene side chain that is required for the pivotal intramolecular ene cyclization to fashion the octahydronapthalene fragment,[20] iodide 23 was advanced by Negishi vinylation to diene 24.[21] Exposure of 24 to 75 atm of hydrogen in the presence of catalytic (η6-napthalene)chromium tricarbonyl occasioned selective delivery of hydrogen to the termini of the diene to give exclusively the (E)-ethylidene product 25 in 95% yield from vinyliodide 23.[22] 70 °C promoted stereoselective intramolecular carbonyl-ene cyclization of the corresponding aldehyde to give alcohol 26 harboring the octahydronapthalene core of macfarlandin C in 69% yield.[20,23] The secondary alcohol of 26 was then oxidized using Dess-Martin periodinane to ketone 27,[24] which was transformed with high selectivity to equatorial tertiary alcohol 28 upon sequential treatment with an excess of Yamamoto’s MAD reagent (methylaluminum bis(2,6-di-tert-butyl-4-methylphenoxide) and methylmagnesium bromide.[25]
The pivotal fragment coupling step and advancement of the coupled product in eight steps to (–)-macfarlandin C are summarized in Scheme 4. Alcohol 28 was converted first to the oxalate radical precursor 29 by sequential reaction at room temperature with methyl chlorooxalate and cesium hydroxide. Irradiation of a solution of oxalate salt 29, D-menthol-derived chlorobutenolide 30,[8] and 2 mol% of the iridium photocatalyst with high-intensity blue LEDs for 20 h at 60 °C, followed by addition of excess tri-n-butylamine and irradiation for an additional 6 h gave coupled product 31 in 74% overall yield from alcohol 28.[26,27] This product was then advanced in high yield to vinylogous β-alkoxyacyl ester 33 by the aldol-dehydration sequence developed in our earlier model studies (Scheme 2). Mukaiyama hydration of 33 proceeded with high regio- and stereoselectivity to deliver alcohol intermediate 34, leaving the electron-rich trisubstituted double bond untouched. The highest yields in this conversion were realized using the more active catalytic system reported by Shenvi.[28] To our surprise, α-hydroxy lactone 34, and alcohol-protected variants thereof, proved remarkably resilient to reduction by a variety of hydride reagents. Fortunately, reaction with a large excess of lithium aluminum hydride at 0 °C gave rise to a mixture bicyclic lactols, which upon direct oxidation with excess PCC provided dioxabicyclooctanone 35 in 72% yield. Without purification of intermediates, the menthoxy group was removed under acidic conditions, the resulting diol product was peracetylated and then exposed to DMAP to provide butenolide intermediate 36. Silane reduction promoted by a N-heterocyclic-carbene copper complex[25] then delivered (–)-macfarlandin C (2) in 38% yield over three steps. Spectroscopic properties and optical rotation of synthetic (–)-macfarlandin C (2) are indistinguishable from those reported for the dorid nudibranch isolate.[2]
Scheme 2:
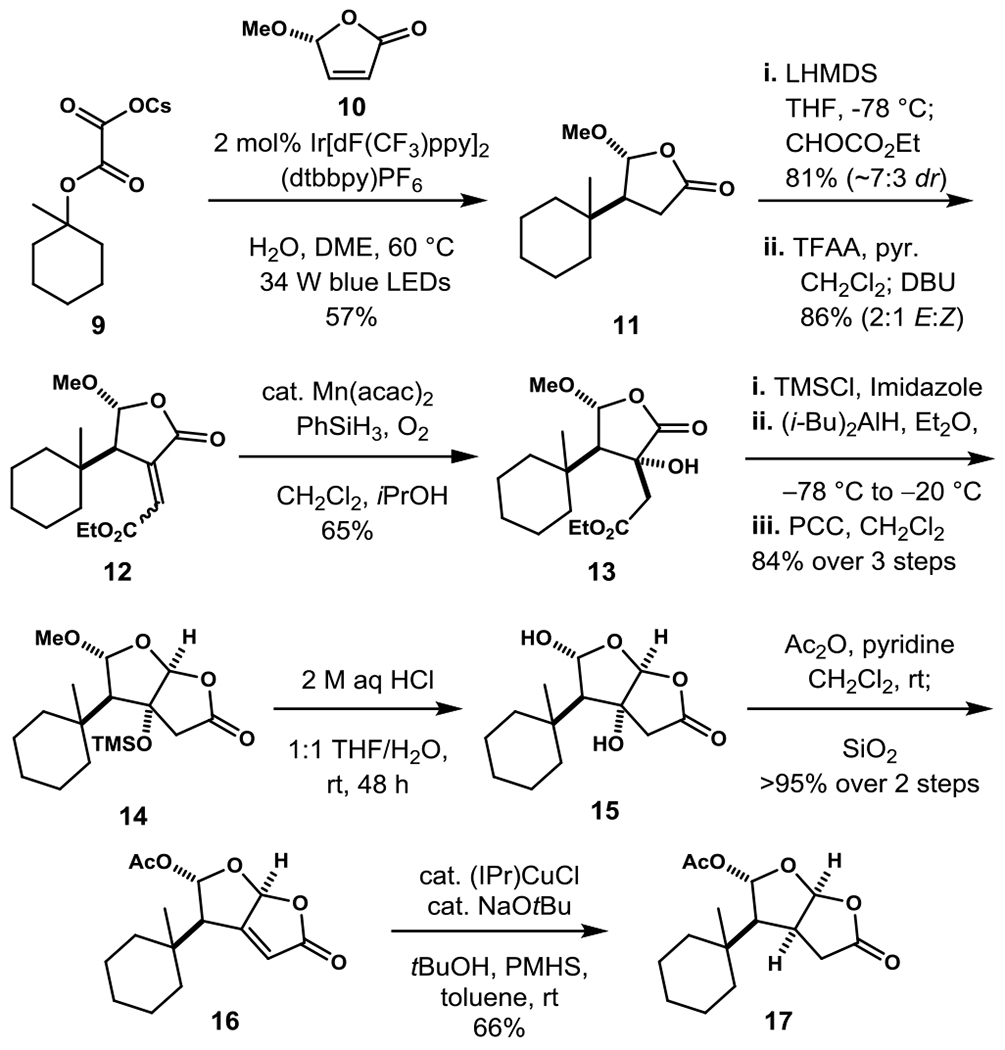
Model studies toward accessing concave 6-substituted cis-2,8-dioxabicyclo[3.3.0]octan-3-one 17. (dF(CF3)ppy = 2-(2,4-difluorophenyl)-5-trifluoromethylpyridine, dtbbpy = 4,4’-di-t-Bu-2,2’-bipyridine, DME = dimethoxyethane, THF = tetrahydrofuran, LHMDS = lithium hexamethyldisilazide, TFAA = trifluoroacetic anhydride, DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene, acac = acetylacetonate, TMSCl = chlorotrimethylsilane, PCC = pyridinium chlorochromate, Ipr = 1,3-bis(2,6-diisopropylphenyl)-1,3-dihydro-2H-imidazol-2-ylidene, PMHS = polymethylhydrosiloxane).
In summary, the first total synthesis of rearranged spongian diterpenoids having a bulky hydrocarbon positioned on the highly congested concave face of the cis-2,8-dioxabicyclo[3.3.0]octan-3-one fragment is reported. This sequence was exemplified in the first total synthesis of the structurally elaborate diterpenoid (–)-macfarlandin C (2), an enantioselective synthesis that rigorously establishes the absolute configuration of the natural product, which previously had been proposed only on the basis of biosynthetic conjecture. Three transformations are critical to the successful synthesis of 2: a) stereoselective carbonyl-ene cyclization to fashion the octahydronapthalene fragment, b) high-yielding fragment coupling between a tertiary alcohol-derived tertiary radical and an electron-deficient alkene resulting in the formation of a new quaternary and tertiary stereocenters, and c) a stereo-and diastereoselective Mukaiyama hydration that allows the concave-substituted cis-2,8-dioxabicyclo[3.3.0]octan-3-one unit to be elaborated from the product of fragment coupling.
Supplementary Material
Acknowledgements
Financial support was provided by the National Science Foundation (CHE-1265964 and CHE-1661612) and the National Institute of General Medical Sciences (R01-GM098601). We thank the ACS Organic Chemistry Division for partial support of G.L.L. by a Graduate Fellowship and the German Academic Exchange Service (DAAD) for postdoctoral fellowship support of A.P.D. NMR and mass spectra were determined at UC Irvine using instruments purchased with the assistance of NSF and NIH shared instrumentation grants. We are grateful to Eloisa Serrano and Yuriy Slutskyy for studies of alternate approaches to these targets.
References
- [1].For reviews, see:; a) Keyzers RA, Northcote PT; Davies-Coleman MT, Nat. Prod. Rep 2006, 23, 321–334. [DOI] [PubMed] [Google Scholar]; b) Gonzalez MA, Curr. Bioact. Compd 2007, 3, 1–36. [Google Scholar]
- [2].Molinski TF, Faulkner DJ, He C, Van Duyne GD, Clardy J, J. Org. Chem 1986, 51, 4564–4567. [Google Scholar]; B) CCDC ref code FAHYES for the X-ray structure of 2.
- [3].a) Buckleton JS, Cambie RC, Clark GR, Acta Crystallogr. Sect. C. Cryst. Struct. Commun 1991, 47, 1438–1440. [Google Scholar]; b) Bergquist PR, Bowden BF, Cambie RC, Craw PA, Karuso P, Poiner A, Taylor WC, Aust. J. Chem 1993, 46, 623–632. [Google Scholar]
- [4].Schnermann MJ, Beaudry CM, Egorova AV, Polishchuk RS, Sütterlin C, Overman LE, Proc. Natl. Acad. Sci 2010, 107, 6158–6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Schnermann MJ, Beaudry CM, Genung NE, Canham SM, Untiedt NL, Karanikolas BDW, Sütterlin C, Overman LE, J. Am. Chem. Soc 2011, 133, 17494–17503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].For a review of covalent modification of biological targets by Paal-Knorr pryrrole formation, see:; Kornienko A, La Clair JJ Nat. Prod. Rep 2017, 34, 1051–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a)Cheloviolene A CDCC ref. codes SIZZIK and SIZZIK01. b) The other diterpenoid in the convex-linked series whose structure has been determined by single-crystal X-ray analysis, cheloviolene B, also shows a typical value of 1.545 Å for the length of the linking bond (CDCC ref. code HEFVIA).[8b]
- [8].a) Slutskyy Y, Jamison CR, Zhao P, Lee J, Rhee Y-H, Overman LE, J. Am. Chem. Soc 2017, 139, 7192–7195. [DOI] [PubMed] [Google Scholar]; b) Garnsey MR, Slutskyy Y, Jamison CR, Zhao P, Lee J, Rhee Y-H, Overman LE, J. Org. Chem 2018, 83, 6958–6976. [DOI] [PubMed] [Google Scholar]
- [9].For a recent review of fragment coupling using carbon-based radicals in the synthesis of complex molecules, see:; Pitre SP, Weires NA, Overman LE, J. Am. Chem. Soc 2019, 141, 2800–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Many of these attempts are summarized in: a) the Supporting Information of reference 8b, and; Lackner GL, PhD thesis, University of California, Irvine (USA), 2016. [Google Scholar]
- [11].See the supporting information for details.
- [12].For an authoritative review, see:; Crossley SWM, Obradors C, Martinez RM, Shenvi RA, Chem. Rev 2016, 116, 8912–9000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].The partial isomerization of the desired (less stable) hydrogenation product formed from 12 under some of the reduction conditions we had examined, in part, led us to this approach.[10,11]
- [14].a) Isayama S, Mukaiyama T, Chem. Lett 1989, 18, 1071–1074. [Google Scholar]; b) Inoki S, Kato K, Isayama S, Mukaiyama T, Chem. Lett 1990, 19, 1869–1872. [Google Scholar]; c) Magnus P, Payne AH, Waring MJ, Scott DA, Lynch V, Tetrahedron Lett. 2000, 41, 9725–9730. [Google Scholar]; d) Tanaka M, Mukaiyama C, Mitsuhashi H, Maruno M, Wakamatsu T, J. Org. Chem 1995, 60, 4339–4352. [Google Scholar]
- [15].For selected examples where steric effects result in high stereoselection in Mukaiyama hydrations, see;; a) Renata H, Zhou Q, Dünstl G, Felding J, Merchant RR, Yeh C-H, Baran PS, J. Am. Chem. Soc 2015, 137, 1330–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen D, Evans PA, J. Am. Chem. Soc 2017, 139, 6046–6049. [DOI] [PubMed] [Google Scholar]; c) Ohtawa M, Krambis MJ, Cerne R, Schkeryantz JM, Witkin JM, Shenvi RA, J. Am. Chem. Soc 2017, 139, 9637–9644. [DOI] [PubMed] [Google Scholar]
- [16].a) Regio- and stereoselectivity was quite high, as no additional alcohol products were detected during chromatographic purification of 13. b) Single-crystal X-ray analysis confirmed the relative configuration of 13 (CCDC ID 1972398).
- [17].Jurkauskas V, Sadighi JP, Buchwald SL, Org. Lett 2003, 5, 2417–2420. [DOI] [PubMed] [Google Scholar]
- [18].a) Corey EJ, Bakshi RK, Shibata S, J. Am. Chem. Soc 1987, 109, 5551–5553. [Google Scholar]; b) Kamatani A, Overman LE, Org. Lett 2001, 3, 1229–1232. [DOI] [PubMed] [Google Scholar]
- [19].Soorukram D, Knochel P, Org. Lett 2004, 6, 2409–2411. [DOI] [PubMed] [Google Scholar]
- [20].For reviews, see:; a) Mikami K, Shimizu M, Chem. Rev 1992, 92, 1021–1050. [Google Scholar]; b) Oppolzer W, Snieckus V, Angew. Chem. Int. Ed 1978, 17, 476–486; [Google Scholar]; Angew. Chem 1978, 90, 506–516. [Google Scholar]; c) Clarke ML, France MB, Tetrahedron, 2008, 64, 9003–9031. [Google Scholar]
- [21].Baba S, Negishi E, J. Am. Chem. Soc 1976, 98, 6729–6731. [Google Scholar]
- [22].Sodeoka M, Shibasaki M, Synthesis, 1993, 643–658. [Google Scholar]
- [23].a) For an example of carbonyl ene cyclization of a related methylidene acetal, see;; Justicia J, Campaña AG, Bazdi B, Robles R, Cuerva JM, Oltra JE, Adv. Synth. Catal 2008, 350, 571–576. [Google Scholar]; For an early exploration of forming octahydronapthalene alcohols in this fashion, see:; Stoll M, Hinder M, Helv. Chim. Acta 1955, 38, 1593–1597. [Google Scholar]
- [24].Dess DB, Martin JC, J. Org. Chem 1983, 48, 4155–4156. [Google Scholar]
- [25].Maruoka K, Itoh T, Yamamoto H, J. Am. Chem. Soc 1985, 107, 4573–4576. [Google Scholar]
- [26].As we have observed earlier in related reactions,[8] the yield of the coupling reaction is enhanced when the butenolide contains an α-chloro substituent. Stereoselection in the fragment coupling was extremely high, as no stereoisomers were detected upon chromatographic purification of 31.
- [27].When the oxalate derived from the corresponding axial tertiary alcohol is subjected to the fragment coupling conditions, the major product arises from cyclization of the alkoxycarbonyl radical intermediate onto the pendent trisubstituted double bond to form a γ-butyrolactone.[10b]
- [28].Obradors C, Martinez RM, Shenvi RA, J. Am. Chem. Soc 2016, 138, 4962–4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.