

Abstract
Lipoprotein(a) is a unique form of low-density lipoprotein. It is associated with a high incidence of premature atherosclerotic disease such as coronary artery disease, myocardial infarction, and stroke. Plasma levels of this lipoprotein and its activities are highly variable. This is because of a wide variability in the size of the apolipoprotein A moiety, which is determined by the number of repeats of cysteine-rich domains known as “kringles.” Although the exact mechanism of lipoprotein(a)-induced atherogenicity is unknown, the lipoprotein has been found in the arterial walls of atherosclerotic plaques. It has been implicated in the formation of foam cells and lipid deposition in these plaques. Pharmacologic management of elevated levels of lipoprotein(a) with statins, fibrates, or bile acid sequestrants is ineffective. The newer and emerging lipid-lowering agents, such as the second-generation antisense oligonucleotides, cholesteryl ester transfer protein inhibitors, and proprotein convertase subtilisin/kexin type 9 inhibitors offer the most effective pharmacologic therapy.
1. Introduction
Significant advances have been made in the treatment of dyslipidemia. However, many patients continue to experience clinical manifestations of atherosclerotic vascular diseases, such as myocardial infarction (MI), cerebrovascular diseases, and peripheral vascular disease [1]. Clinical dyslipidemias fall into four broad categories: high levels of low-density lipoprotein cholesterol (LDL-C), low levels of high-density lipoprotein cholesterol (HDL-C), elevated triglycerides, and elevated lipoprotein(a) [Lp(a)] [2]. Over time, more emphasis has been placed on pharmacologic and nonpharmacologic reduction of LDL-C as a method of reducing atherosclerotic vascular diseases. In addition, lifestyle modification and the treatment of modifiable diseases, such as hypertension and diabetes mellitus, have also been at the forefront of the management of dyslipidemia. Advances in our knowledge of non-LDL-C physiology and improved assay techniques have shed more light on the role of Lp(a) in the pathogenesis of atherosclerotic vascular diseases [3]. In this review, we focus on the emerging pharmacotherapeutic agents used to lower plasma levels of Lp(a).
Lp(a) was discovered in 1963 by the geneticist Kara Berg [4]. Initially described as a variant of LDL, Lp(a) is now widely recognized as a distinct plasma lipoprotein. Lp(a) is composed of two distinct parts: apolipoprotein-B (apoB) and apolipoprotein-A (apoA), a plasminogen-like glycoprotein (Fig. 1). ApoB is structurally and physicochemically similar to LDL-C, and apoA consists of carbohydrate-rich proteins [2, 5, 6]. Both molecules are covalently linked by a disulfide bond to form a single macromolecule. The origin of unbound plasma apoA is unknown, but the synthesis of this subunit takes place in the liver and appears to be independent of other lipoprotein synthesis. The actual assembly of Lp(a) is believed to take place within the hepatocytes [2, 7]. Although the size of the apoA moiety varies widely, it is mainly determined by the size and the number of repeats of cysteine-rich domains known as “kringles.” Evidence from DNA sequencing suggests that the kringle IV repeat shares a high degree of structural homology with the fibrinolytic enzyme precursor plasminogen [2, 7]. Plasminogen contains five kringles (KI–KV) and a protease domain. ApoA contains several subtypes of KIV repeat polymorphisms, so apoA protein size heterogeneity is extensive, resulting in different sizes of Lp(a) particles. Plasminogen is a protease zymogen; when activated, it cleaves fibrin to dissolve clots. Considering the striking molecular similarity between plasminogen and Lp(a), Lp(a)/apoA atherothrombotic properties are in part due to the competitive inhibition of tissue-type plasminogen activator-mediated binding, thus leading to a decrease in plasminogen activation, plasmin synthesis, and fibrinolysis [8].
Fig. 1.
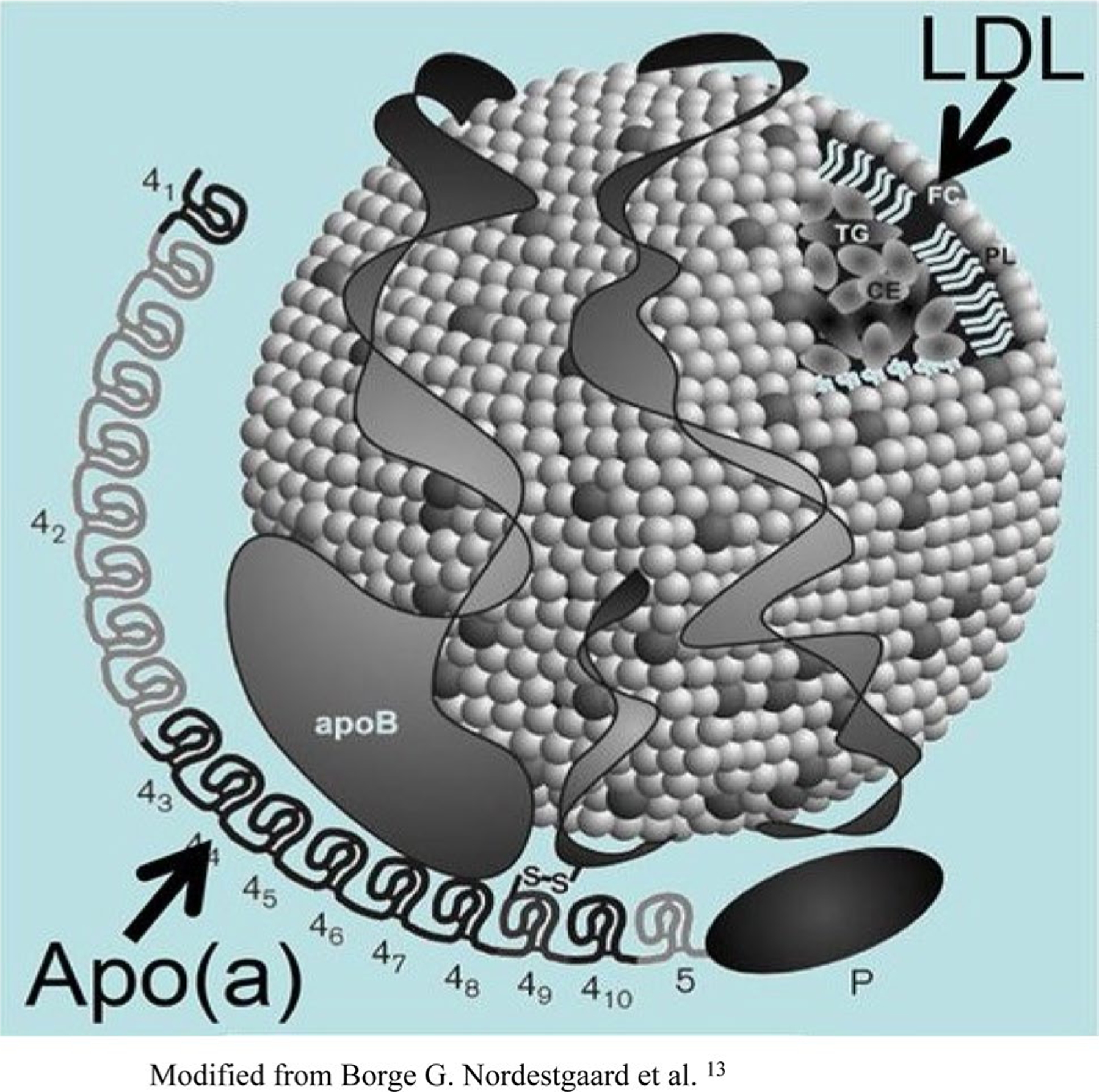
Structure of lipoprotein(a). Lipoprotein(a) consists of apolipoprotein(a) covalently bonded to the apolipoprotein(b)-100 component of a low-density lipoprotein (LDL)-like moiety by a single disulfide bond. Also depicted are the kringles. Modified from Nordestgaard et al. [8]
Although the exact mechanism of Lp(a) atherogenicity is unknown, Lp(a) is a known preferential carrier of oxidized phospholipids in humans and has been shown to bind pro-inflammatory-oxidized phospholipids [8].
1.1. Epidemiology
Population studies have revealed that plasma levels of Lp(a) vary amongst humans, ranging from 20 to > 2000 mg/dL between racial groups, with almost 20% of the population at the extreme levels [6]. These levels are not affected by age or sex. There are no differences in the serum levels of Lp(a) between Caucasian men and premenopausal women. Among the Caucasian, Asian, and Indian populations, Lp(a) distribution is highly skewed to the left, whereas the distribution is almost normal among African American and perhaps African populations. These variations in the distributions must be considered when interpreting studies involving Lp(a). Additionally, particle size varies widely, ranging from 180 to > 600 kDa. The number of kringle IV repeat genes in Lp(a) is thought to determine the size, which is inversely related to an increased risk of cardiovascular diseases (CVDs) [6].
1.2. Lipoprotein(a) [Lp(a)]: A Risk Factor for Atherosclerotic and Thrombogenic Events
An elevated serum level of Lp(a) is an independent risk factor for CVD [1, 6, 8, 9]. A residual risk of CVD remains in patients with low LDL-C goal, as demonstrated in a subgroup analysis of Caucasian participants in the JUPITER study [6]. In a meta-analysis of 29,069 patients with Lp(a) measurements, CVD risk was approximately linear with increased Lp(a) values [10]. Elevated Lp(a) of ≥ 30 mg/dL at baseline was associated with an increased hazard ratio of cardiovascular events independent of other cardiovascular risk factors [10]. Additionally, Willeit et al. [10] also reported that CVD risks were approximately linear with increased Lp(a) values in patients receiving statin treatment. Elevated Lp(a) of ≥ 50 mg/dL on treatment was associated with a linear increase of cardiovascular events, irrespective of statin therapy [10]. These data suggest that residual CVD risks remain in patients treated with maximally tolerated statin therapy and identifies elevated Lp(a) as one of the factors that may be modified to further reduce residual risk. Thus, statin-treated patients with elevated levels of Lp(a) represent a significant determinant of residual risks for CVD [6, 10]. These findings also highlight the importance of lowering plasma levels of Lp(a). Moreover, two prospective population studies—EPIC-Norfolk (The European Prospective Investigation of Cancer in Norfolk) and the Copenhagen City Heart Study—concluded that Lp(a) and LDL-C are independently associated with CVD risk [11]. Additionally, the Canadian Cardiovascular Society’s (CCS) guidelines for the management of dyslipidemia for the prevention of CVD in adults noted that Lp(a) is a marker of CVD risk [12].
Although the actionable clinical threshold value for Lp(a) is difficult to define, the European Atherosclerosis Society has proposed an optimal Lp(a) level of < 80th percentile, which approximates < 50 mg/dL, in Caucasian patients without any cardiovascular risks [8]. Furthermore, the CCS has recommended that particular attention be given to individuals with Lp(a) levels > 30 mg/dL, for whom CVD risk is increased approximately twofold [12].
Screening the general population for cardiovascular risk stratification is not recommended; however, Lp(a) screening should be considered in patients with premature coronary artery disease (CAD) and or in patients with dyslipidemia refractory to statins or bile acid sequestrants. Furthermore, other patient subgroups that may benefit from Lp(a) screening include those who may have particularly adverse clinical consequences secondary to elevated Lp(a) concentrations. These groups include patients with a history of coronary artery bypass grafting, percutaneous transluminal coronary angioplasty, heart transplantation, and familial dyslipidemia [13].
2. Pharmacological Treatment of Elevated Lp(a)
Clinical practice guidelines for the management of elevated Lp(a) are sparse. Available guidance, including the 2018 multisociety guideline on the management of blood cholesterol [14], focuses on the management of LDL-C and does not provide guidance on the management of elevated Lp(a) levels (Table 1). Pharmacotherapeutic agents used for the treatment of elevated Lp(a) differ significantly from the traditional management of other blood cholesterols in which HMG-CoA reductase inhibitors (statins) are the first line of therapy. In this review, we present the efficacy and safety of the newer antidyslipidemia agents in the treatment of elevated plasma Lp(a).
Table 1.
Summary of pharmacology for lowering elevated plasma lipoprotein(a)
| Agent/class | Target | Dose/administration | Major adverse effects | Contraindications | Extent of reduction |
|---|---|---|---|---|---|
| Statins [16, 17] | Inhibits HMG-CoA reductase | Varies, PO, daily | Arthralgia, diarrhea | Active liver disease, pregnancy | Ineffective |
| Fibric acid derivatives [18] | Induces PPAR-α; activates farnesoid X receptor | Varies, PO | Dyspepsia, GI pain, nausea, vomiting | Liver or severe kidney dysfunction | Limited reduction: WMD − 2.70 mg/dL; 95% CI − 4.56 to − 0.84; p = 0.004 |
| Omega-3 fatty acids (fish oil) [21] | Decreases apoA synthesis; increases Lp(a) catabolism | 8 gm PO, daily | AF, peripheral edema, atrial flutter, dysgeusia, constipation, gout, dyspepsia, hemorrhage | Hypersensitivity | Unpredictable effects |
| ER niacin [27] | Inhibits clearance of apoB, regulates Lp(a) turnover | ≤2 to ≥ 2 g/day, PO | Flushing, myopathy, transaminitis, hyperglycemia | Active liver disease, peptic ulcer disease | Significant decrease: ≤ 2 g/day (WMD − 21.85%; 95% CI − 30.61 to − 13.10, p < 0.001); ≥ 2 g/day (WMD − 23.21%; 95% CI − 28.41 to − 18.01; p < 0.001) |
| Tamoxifen/raloxifene [31, 32] | Possible modulation apoA expression | Varies, PO | Gynecologic malignancies, bone marrow suppression | Hypersensitivities | Significant reduction: ~ 30% |
| Mipomersen [33] | Binds to apoB-100 mRNA | 200 mg SC, weekly | Injection-site reactions, flu-like symptoms, elevated transaminases | Hypersensitivities, active liver disease (Child-Pugh class B or C) | Significant reduction: ~ 26% |
| Pelacarsen (AKCEA-APO(a)-LRx TQJ 230, ISIS 681257) [34] | Binds to apoB-100 mRNA | 20, 40, 60 mg SC Q4W; 20 mg SC Q2W; 20 mg SC weekly | Injection-site reactions, flu-like symptoms, elevated transaminases, myalgias | Unknown | Significant reduction: 35–80% |
| Anacetrapib [39]a | Inhibits CETP | 100 mg PO daily | Diarrhea, constipation, dyspepsia, myalgia | Unknown | Significant reduction: 25% |
| Obicetrapib (TA 8995, DEZ 001, AMG 899) [40] | Inhibits CETP | 1,2.5, 5, 10 mg PO daily | Headaches | Unknown | Significant reduction: 26.7–36.9% |
| Evolocumab [46] | Inhibits PCSK9 | 140 mg SC Q2W; 420 mg SC Q4W | Injection-site reactions, flu-like symptoms, sore throat, hyperglycemia | Hypersensitivity | Significant reduction: 21.7–24.7% |
| Alirocumab [43, 44] | Inhibits PCSK9 | 10, 20, 40 mg SC Q2W; 50, 100, 150 mg SC Q2W; 200 or 300 mg SC Q4W | Injection-site reaction, chest pain, difficulty breathing or swallowing, hives, myalgias, reddening of skin | Hypersensitivity | Absolute reduction: 27% |
| Tocilizumab [47] | IL-6 receptor antagonist | 8 mg/kg IV daily | Hypercholesterolemia, injection site reaction | Hypersensitivity | Modest reduction: 10–14.6 mg/dL at 1 and 3 months |
| Lomitapide [50] | Inhibits MTP | Median 40 mg PO daily | Fatigue, chest pain | Pregnancy, active liver disease (Child-Pugh class B or C) | Modest reduction: 15 and 19% at 26 and 56 weeks, respectively |
| Coenzyme Q10 [51] | Antioxidant | 100–300 mg PO daily | Nausea, fatigue, headaches, insomnia | Anticoagulants | Modest reduction: WMD − 3.54 mg/dL; 95% CI − 5.50 to − 1.58; p < 0.001 |
| Ezetimibe [52] | Anti-inflammatory effect, decreases LDL-C and apoB, through activation of LDL receptors | 10 mg PO daily | Fatigue, diarrhea, increased serum transaminases (with HMG-CoA reductase inhibitors; ≥ 3 × ULN), arthralgia, limb pain | Hypersensitivity, concomitant use with statin with active liver disease, unexplained persistent elevations in serum transaminases, pregnancy, breastfeeding | Modest reduction: 7.06% |
AF atrial fibrillation, apoA/B apolipoprotein A/B, CETP cholesteryl ester transfer protein, CI confidence interval, ER extended release, GI gastrointestinal, HMG-CoA hydroxymethylglutaryl coenzyme A (statin), IL interleukin, IV intravenous, LDL low-density lipoprotein, LDL-C LDL cholesterol, Lp(a) lipoprotein(a), mRNA messenger RNA, MTP microsomal triglyceride transport protein, PCSK9 proprotein convertase subtilisin/kexin type 9, PO oral administration, PPAR peroxisome proliferator activated receptor, Q2W every 2 weeks, Q4W every 4 weeks, SC subcutaneous, ULN upper limit of normal, WMD weighted mean difference
The manufacturer has halted further development of anacetrapib
2.1. Ineffective Pharmacotherapeutic Agents
2.1.1. HMG-CoA Reductase Inhibitors (Statins)
Statins are the most widely recommended and used therapy agent for patients with dyslipidemia. They are effective in lowering LDL-C and triglycerides, and they effectively increase HDL-C. Unlike in other cholesterol lipoproteins, statins are ineffective and/or may have a negative impact in the treatment of elevated serum Lp(a) levels [15]. Several studies have reported significant increases in Lp(a) levels and apoA production following statin therapy [16, 17]. A meta-analysis assessing the effect of statins on Lp(a) levels showed that statins significantly increased the plasma levels of Lp(a) compared with placebo (geometric mean 1.11; 95% confidence interval [CI] 1.07–1.14; P < 0.0001) [16].
2.1.2. Fibric Acid Derivatives
Fibric acid derivatives (fibrates) are another conventional class of pharmacotherapeutic agents that are often used for the treatment of dyslipidemia. They are most commonly used for the effective lowering of triglycerides and to increase HDL-C. Although effective in the aforementioned dyslipidemias, fibrates available in the USA, such as clofibrate, gemfibrozil, and fenofibrates, are not ideal treatment options for lowering Lp(a) levels. In fact, fibrates have been found to have an unpredictable or unfavorable effect on serum Lp(a). A randomized double-blind trial by Guyton et al. [18] evaluated the effects of extended-release niacin compared with those of gemfibrozil in the lowering of Lp(a) levels and reported an increase in serum levels of Lp(a) following therapy with gemfibrozil. A limited but significant reduction of Lp(a) levels using fibrates has been reported. A review of 16 head-to-head comparative trials with a total of 1388 subjects reported a significant reduction of plasma Lp(a) concentrations (weighted mean difference [WMD] − 2.70 mg/dL; 95% CI − 4.56 to − 0.84; p = 0.004) compared with statins. In the same analysis, combination therapy with fibrates and statins had a significantly greater effect compared with statin monotherapy (WMD −1.60 mg/dL; 95% CI − 2.93 to − 0.26; p = 0.019) but not fibrate monotherapy (WMD −1.76 mg/dL; 95% CI − 5.44 to 1.92; p = 0.349) in reducing plasma Lp(a) concentrations [19].
2.1.3. Omega-3 Fatty Acids
The successful use of omega-3 fatty acids therapy for the reduction of Lp(a) has been limited, and results have been conflicting [20, 21]. Herrmann et al. [20] investigated the effectiveness of omega-3 fatty acids in the lowering of Lp(a) levels in patients with CAD. Patients received omega-3 fatty acid concentrate with 76% omega-3 polyunsaturated fatty acid in doses of > 8 g/day and undertook aerobic exercise and a low-calorie diet. The omega-3 fatty acids treatment regimen had unpredictable effects on plasma Lp(a) levels. Only 23 of the 35 patients with elevated Lp(a) studied had an approximate 24% reduction in plasma Lp(a) [20]. Lp(a) levels in the remaining patients either increased or remained unchanged. However, in a randomized, double-blind, placebo-controlled trial involving 34 hemodialysis patients, serum concentrations of Lp(a) and total cholesterol did not significantly change in the omega-3 fatty acids group. However, significant increases were observed in levels of Lp(a) (p <0.01) and total cholesterol (p <0.05) in the placebo group [21].
Moreover, one study investigated the long-term effect of eicosapentaenoic acid (EPA) on serum levels of Lp(a) and lipids in patients with vascular disease [22]. In total, 24 patients with vascular disease received EPA 1800 mg/day for 24 months. Nine of the 24 patients had baseline elevated Lp(a) levels, and these patients experienced lowering of serum Lp(a) after administration for 12 and 18 months (p < 0.05) [22]. However, no significant change in Lp(a) levels in patients with normal baseline Lp(a) levels was observed [22].
The mechanism of omega-3 fatty acids in the reduction of Lp(a) is unclear, but it is believed to interfere with lipogenic enzymes and the secretion of apoB lipoproteins by decreasing the rate of apoA hepatic synthesis/secretion. Alternatively, omega-3 fatty acids may cause a reduction in Lp(a) level by increasing Lp(a) catabolism in patients who responded to omega-3 fatty acids therapy [20].
2.2. Effective Pharmacotherapeutic Agents
2.2.1. Nicotinic Acid
Extended-release (ER) niacin ≥ 3 gm/d has been shown to reduce plasma Lp(a) by as much as 24–38%, respectively [23–27]. In a meta-analysis comprising 14 randomized placebo-controlled trials and 9013 subjects, 5362 subjects in the niacin arm had a significant reduction of Lp(a) levels following ER niacin treatment (WMD −22.90%; 95% CI − 27.32 to − 18.48; p < 0.001). When the studies were categorized according to administered dose, there was a comparable effect between the subsets of studies with doses of < 2000 mg/day (WMD − 21.85%; 95% CI − 30.61 to − 13.10; p < 0.001) and ≥ 2000 mg/day (WMD − 23.21%; 95% CI − 28.41 to − 18.01; p < 0.001) [28]. In this analysis, several studies reported a mean niacin dose instead of a definite dosage for all treated subjects, in part because of the appearance of niacin-related side effects at different dosages. Common side effects of niacin include flushing, diarrhea, nausea, vomiting, myopathy, elevated transaminases, and hyperglycemia.
The molecular mechanism by which niacin reduces plasma levels of Lp(a) is unknown. Niacin may regulate lipoprotein turnover and inhibit clearance of apoB-containing lipoproteins, suggesting reduced production of very low-density lipoprotein as the principal mechanism of action [26]. Kamanna et al. [29] proposed that niacin’s action on Lp(a) is the result of its ability to directly and noncompetitively inhibit the activity of hepatocyte microsomal diacylglycerol acyltransferase-2. This enzyme catalyzes the final reaction in triglyceride synthesis [29].
2.2.2. Estrogen Therapy
Hormone-replacement therapy has been shown to reduce plasma levels of Lp(a) in postmenopausal women [30, 31]. At therapeutic doses, estrogen lowers plasma Lp(a) levels by up to 30% in postmenopausal women with plasma Lp(a) excess and may modulate menopause-associated dyslipidemia in women. The addition of progestin may diminish or even eliminate the effects of estrogen on HDL-C.
The synthetic estrogen receptor blocker tamoxifen, alone or in combination with estrogen, has been shown to reduce plasma levels of Lp(a) by as much as 24 and 34% below baseline in healthy postmenopausal women following 1 and 3 months of therapy [32].
Moreover, significant reductions in Lp(a) levels have been reported following treatment with raloxifene. In a meta-analysis of seven randomized placebo-controlled studies involving 1271 healthy postmenopausal and hypercholesterolemic women, therapy with the selective estrogen receptor modulator raloxifene (n = 634) resulted in a significant reduction of Lp(a) levels (standardized mean difference [SMD] − 0.42; 95% CI − 0.65 to − 0.19; p < 0.001). When the studies were categorized according to dose, a significant effect was seen in both subsets of studies with doses ≤ 60 mg/day (SMD − 0.43; 95% CI − 0.73 to − 0.13; p = 0.004) and > 60 mg/day (SMD −0.36; 95% CI − 0.68 to − 0.05; p = 0.025) [33].
2.2.3. Second-Generation Antisense Oligonucleotides
Second-generation antisense oligonucleotides (ASOs) such as mipomersen and AKCEA-APO(a)-LRx have been shown to significantly decrease plasma levels of Lp(a). ASO therapy exerts its effects specifically by binding to the apoB-100 messenger RNA (mRNA), thereby blocking translation of the apoB-100 protein, leading to reduced hepatic production of apoB-containing lipoproteins such as LDL-C and Lp(a) [34].
Four phase III trials have assessed the efficacy of mipomersen in the reduction of Lp(a). A total of 382 patients receiving maximum doses of lipid-lowering drugs were randomized to weekly subcutaneous injection of mipomersen 200 mg (n = 256) or placebo (n = 126) for 26 weeks. In the pooled analysis, the mean percent decrease in Lp(a) at 28 weeks was significantly larger with mipomersen than with placebo (median −26.4 [interquartile range {IQR} − 42.8 to − 5.4] vs. −0.0 [IQR − 10.7 to 15.3]; p < 0.001). In the combined groups, modest correlations were present between percent change in apoB-100 and Lp(a) (r = 0.43; p < 0.001) and LDL-C and Lp(a) (r = 0.36; p < 0.001) plasma levels [35].
A meta-analysis of six randomized controlled trials (RCTs) and a total of 444 patients examined the efficacy and safety of mipomersen in apoB-containing lipoproteins. Compared with the placebo group, patients who received mipomersen had a significant reduction in LDL-C (33.13%) and reductions in non-HDL-C (31.70%), apoB (33.27%), and Lp(a) (26.34%) [36].
Mipomersen therapy is associated with marked injection-site reactions, flu-like symptoms, and elevated transaminases.
AKCEA-APO(a)-LRx is a novel investigational second-generation N-acetyl-galactosamine-conjugated ASO agent. Its action is directed against apoA mRNA in the liver. A phase two multicenter randomized placebo-controlled dose-ranging study involving 286 subjects with preexisting CVD (CAD, MI, peripheral artery disease, and stroke) and baseline Lp(a) levels ≥ 60 mg/dL was divided into five dose-ranging groups [37]. Study participants were randomized in a 5:1 ratio to receive either the active drug or placebo in the following dosages and dosing intervals: 20 mg, 40 mg, 60 mg, or placebo every 4 weeks (Q4W), with another group receiving 20 mg active drug or placebo every 2 weeks (Q2W) or 20 mg active drug or placebo once weekly (QW). All subjects who received AKCEA-APO(a)-LRx had decreased levels of Lp(a) at 6 months: decrease by 35% for patients receiving 20 mg Q4W (n = 48); by 56% for patients receiving 40 mg Q4W (n = 48); by 72% for patients receiving 60 mg Q4W (n = 47); by 58% for patients receiving 20 mg Q2W (n = 48); and by 80% for those receiving 20 mg QW (n = 48). By comparison, Lp(a) decreased by 6% in the pooled placebo group (n = 47). The most frequent adverse event was injection-site reaction (26%). One patient discontinued treatment as a result of injection-site reaction; no safety concerns because of liver function, kidney toxic effects, or bleeding risk were reported [37]. A phase III study [Lp(a) HORIZONS; NCT04023552] is currently assessing the impact of Lp(a) lowering with pelacarsen (AKCEA-APO(a)-LRx TQJ 230, ISI 681257) on major cardiovascular events in patients with CVD [38].
2.2.4. Cholesteryl Ester Transfer Protein Inhibitors
Cholesteryl ester transfer protein (CETP) catalyzes the movements of cholesteryl esters and triglycerides between HDL-C and LDL-C particles in plasma. CETP inhibitors can substantially increase HDL levels and reduce non-HDL-C levels [39]. Anacetrapib is an orally active, selective CETP inhibitor with dose-dependent effects on LDL-C and HDL-C levels. It reduces LDL-C levels, including Lp(a), and increases HDL-C levels. In a phase III RCT evaluating the clinical efficacy, safety, and cardiovascular outcomes of anacetrapib in patients with preexisting atherosclerotic vascular disease, anacetrapib 100 mg daily reduced plasma Lp(a) by 25% [40]. However, the manufacturer has halted further clinical development of anacetrapib.
The investigational CETP inhibitor obicetrapib (TA 8995, DEZ 001, AMG 899) was evaluated in patients with mild dyslipidemia. Different doses of obicetrapib (1, 2.5, 5, or 10 mg as monotherapy or combined with a statin) resulted in reductions in Lp(a) ranging from 26.7 to 36.9% (p < 0.0064), whereas monotherapy with a statin resulted in Lp(a) reductions of 3.6 and 7.9% [41]. Although CEPTs have demonstrated Lp(a)-lowering benefits, none of the agents have been approved by the US FDA in this indication.
2.2.5. Proprotein Convertase Subtilisin/Kexin Type 9 Inhibitors
Proprotein convertase subtilisin/kexin type 9 (PCSK9) is a hepatic protease that attaches to and internalizes LDL receptors (LDLRs). PCSK9 regulates LDLRs, inhibiting LDLR recycling and thereby increasing LDL-C levels. PCSK9 inhibition is a target for pharmacologic management of Lp(a). Five monoclonal antibodies (mAbs) targeting PCSK9 inhibition are currently available or being developed and tested: alirocumab, evolocumab, RG 7652, LGT 209, and 1B20. Evolocumab and alirocumab have been approved by the FDA for use in patients with familial dyslipidemia and those receiving maximum antihyperlipidemic drugs who require additional therapy.
Evolocumab is a fully human mAb that binds PCSK9, inhibiting its interaction with the LDLR to preserve LDLR recycling and reduce LDL-C. Raal et al. [42] analyzed data from eight placebo-controlled, blinded phase II and III RCTs (MENDEL-1, MENDEL-2, LAPLACE-TIMI 57, LAPLACE-2, RUTHERFORD-1, RUTHERFORD-2, GAUSS-1, GAUSS-2) and two open-label extension trials (OSLER-1, OSLER-2). The data, from 3278 diverse patients with hypercholesterolemia who received evolocumab, showed a significant decrease in Lp(a) levels. Evolocumab 140 mg Q2W and 420 mg every month resulted in 24.7 and 21.7% reductions in Lp(a), respectively at 12 weeks. The analysis also showed that patients with low LDL-C (≤ 40 mg/dL) treated with evolocumab achieved greater reductions of Lp(a) than did patients with LDL-C ≥ 70 mg/dL, notwithstanding the presence or absence of background statin therapy [42]. Moreover, Lp(a) was measured in 25,096 patients in the FOURIER (Further Cardiovascular Outcomes Research with PCSK9 Inhibition in subjects with Elevated Risk) trial. Patients with established atherosclerotic CVD were randomized to receive evolocumab or placebo for a median follow-up period of 2.2 years. The median baseline Lp(a) concentration was 37 nmol/L (IQR 13–165). In the placebo arm, patients with a baseline Lp(a) in the highest quartile had a higher risk of coronary heart disease death, MI, or urgent revascularization independent of LDL-C: adjusted hazard ratio (HR) quartile 4:quartile 1 of 1.22; 95% CI 1.01–1.48. At 48 weeks of therapy, evolocumab significantly reduced Lp(a) by a median of 26.9% (IQR 6.2–46.7). Evolocumab reduced the risk of coronary heart disease death, MI, or urgent revascularization by 23% (HR 0.77; 95% CI 0.67–0.88) in patients with a baseline Lp(a) higher than the median and by 7% in patients with plasma Lp(a) levels lower than or equal to the median (HR 0.93; 95% CI 0.80–1.08; P interaction = 0.07) [43].
The efficacy of alirocumab on plasma levels of Lp(a) was evaluated in a phase II trial that assessed five different doses and two dosing regimens in patients with primary hypercholesterolemia while receiving stable doses of atorvastatin 10 mg, 20 mg, or 40 mg [44]. All eligible candidates received subcutaneous alirocumab 50 mg, 100 mg, or 150 mg Q2W or 200 or 300 mg Q4W for 12 weeks. Alirocumab exhibited a dose-dependent response pattern in reducing Lp(a) for both Q2W and Q4W administration. Lp(a) plasma levels decreased significantly from baseline by 13–29% at 12 weeks post-dosing across the Q2W regimens with alirocumab 50 mg (− 13.3%), 100 mg (− 26.1%), and 150 mg (− 28.6%), respectively [44]. The effects of alirocumab on plasma levels of Lp(a) were assessed from pooled data of three double-blind, placebo-controlled, phase II RCTs [45]. Patients with heterozygous familial hypercholesterolemia (HeFH) or nonfamilial hypercholesterolemia and LDL-C ≥ 100 mg/dL while receiving statin monotherapy or statin plus ezetimibe 10 mg (n = 352) were randomized to placebo (n = 77) or to alirocumab 50–300 mg Q2W or Q4W for 8 or 12 weeks (n = 275). This analysis focused on the dosing regimen of alirocumab 150 mg Q2W, which was common to all three phase II studies and was considered to provide consistent reduction in LDL-C. Treatment with alirocumab resulted in absolute and percentage median reductions from baseline in Lp(a) plasma levels of 27%. The median absolute reductions in Lp(a) from baseline were substantially greater in patients with a higher baseline Lp(a) level > 50 mg/dL than in patients with ≤ 50 mg/dL levels of Lp(a) (− 27 [IQR − 39 to − 16] vs. −3.5 [IQR − 12 to 1.5], respectively) [45]. Moreover, two double-blind, placebo-controlled identical phase III RCTs (ODYSSEY FH I and ODYSSEY FH II) evaluated the efficacy and safety of alirocumab over 78 weeks in patients with HeFH on a backdrop of statin therapy. In both trials, 735 patients were randomized: 486 in FH I (323 to alirocumab; 163 to placebo) and 249 in FH II (167 to alirocumab; 82 to placebo). Patients were randomized in a 2:1 fashion to receive either subcutaneous alirocumab 75 mg Q2W or placebo. The dose of alirocumab was increased in a blinded manner to 150 mg Q2W at week 12 if LDL-C level at week 8 was ≥ 70 mg/dL [46]. Following 24 weeks of treatment with alirocumab, plasma levels of Lp(a) were significantly reduced. In the FH I trial, plasma levels of Lp(a) decreased 25.2 vs. 7.5% with placebo. Similarly, the FH II trial reported a significant decrease of 30.3 vs. 10%.
The effects of PCSK9 mAbs on plasma levels of Lp(a) was assessed in a meta-analysis by Cao et al. [47]. They analyzed 27 RCTs with 11,864 participants. PCSK9 therapy demonstrated a significant efficacy in reducing Lp(a) (− 21.9%; 95% CI − 24.3 to − 19.5), irrespective of PCSK9 types, treatment duration, patient population, treatment methods, differences in control treatment, baseline Lp(a) levels, and Lp(a) assay methods. The greatest reduction was achieved with subcutaneous alirocumab 150 mg biweekly (− 24.6%; 95% CI − 28.0 to − 21.2) and subcutaneous evolocumab 140 mg monthly (− 26.8%; 95% CI − 31.6 to − 21.9). Meta-regression analyses found that lowered levels of LCL-C during PCSK9 treatment correlated with greater reductions in Lp(a) levels [47].
2.2.6. Interlukein-6 Receptor Antagonists
The interleukin (IL)-6 receptor antagonist tocilizumab has been shown to attenuate the proatherogenic Lp(a) in rheumatoid arthritis [48, 49]. However, tocilizumab did not affect Lp(a) plasma levels in patients with non-ST-elevation MI [50].
Tocilizumab 8 mg/kg daily for 4 weeks resulted in only a moderate reduction of Lp(a): 10.2 and 14.6 mg/dL at 1 and 3 months, respectively [48].
Inflammation is associated with an increase in plasma levels of Lp(a); thus, inhibition of inflammatory mediators such as IL-6 signaling decreases serum levels of Lp(a) [48].
2.2.7. Microsomal Triglyceride Transfer Protein Inhibitors
The microsomal triglyceride transfer protein inhibitor lomitapide was evaluated in homozygous familial hypercholesterolemia. Although Lp(a) levels were moderately reduced by 15 and 19% from baseline at week 26 and 56, respectively, following a median dose of 40 mg, no significant difference was observed at 78 weeks [51].
2.2.8. Supplementation with Coenzyme Q10
Studies of the efficacy of coenzyme Q10 (CoQ10) supplementation in patients with elevated levels of Lp(a) have generated conflicting results [52].
In a meta-analysis of six clinical trials of 409 subjects, CoQ10 supplementation elicited a modest but significant reduction of plasma Lp(a) levels (WMD − 3.54 mg/dL; 95% CI − 5.50 to − 1.58; p < 0.001). The extent of Lp(a) reduction was higher in subjects with higher baseline Lp(a) levels (slope − 0.44; 95% CI − 0.80 to − 0.08; p = 0.018). Reduction of plasma Lp(a) levels was consistent across different CoQ10 doses, with an inverse association between administered CoQ10 dose and Lp(a) lowering (slope 0.04; 95% CI 0.01–0.07; p = 0.004).
The exact mechanism by which C oQ10 reduces plasma levels of Lp(a) is unknown; however, inflammation has been linked to increased levels of plasma Lp(a). Given the anti-inflammatory effects of CoQ10, Sahebkar et al. [52] speculated that Lp(a) reduction following CoQ10 supplementation might be a result of its anti-inflammatory activity.
2.2.9. E zetimibe
A meta-analysis by Awad et al. [53] investigated the effects of ezetimibe 10 mg/day as monotherapy on Lp(a) [53]. The study included seven RCTs, with 2337 patients with primary hypercholesterolemia [53]. The patients randomly received ezetimibe 10 mg/day or placebo for 12 weeks [53]. The analysis suggested that ezetimibe 10 mg/day, compared with placebo, modestly reduced Lp(a) levels by 7.06% in patients with primary hypercholesterolemia (95% CI − 11.95 to − 2.18; p = 0.005) [53]. However, the leave-one-out sensitivity analysis found that the overall pooled analysis was sensitive to one study [54]; excluding this study resulted in the differences between the two groups becoming nonsignificant (p = 0.2) [53]. Moreover, it should be noted that none of the included studies investigated reductions of Lp(a) as a primary outcome and that the meta-analysis was unable to attribute the reduction of Lp(a) to cardiovascular risk reduction [53].
3. Conclusion
Lp(a) is a unique atherogenic and thrombogenic lipoprotein. Recent data indicate that it is an independent risk factor for premature atherosclerotic diseases, including CAD and stroke. Although the literature supports Lp(a) as a risk factor for CVD, no robust evidence is yet available to suggest that reducing Lp(a) levels reduces the occurrence of clinical events. Well-designed clinical studies are needed to discern Lp(a) clinical thresholds, particularly in non-Caucasians. In addition, studies addressing the role of reducing elevated plasma levels of Lp(a) as primary prevention of CVDs in patients with strong family history are warranted. Lp(a) plasma levels should be determined in patients at LDL-C goal who present with vascular events and those with strong family history of premature CAD. Many of the available agents traditionally used to treat dyslipidemia have been found to be ineffective or have modest effects on Lp(a) levels. However, several of the newer and emerging lipid-lowering agents, such as the second-generation ASOs, CETP inhibitors, and PCSK9 inhibitors, offer the most effective pharmacologic therapy and may be promising therapeutic agents in the reduction of elevated plasma levels of Lp(a). Additionally, conventional agents such as ER niacin and the selective estrogen receptor modulators tamoxifen and raloxifene appear efficacious and may be useful in select populations.
Key Points.
Lipoprotein(a) is associated with a high incidence of premature atherosclerotic disease such as coronary artery disease, myocardial infarction, and stroke.
Pharmacologic management of elevated levels of lipoprotein(a) with statins, fibrates, or bile acid sequestrants is ineffective.
The newer and emerging lipid-lowering agents, such as the second-generation antisense oligonucleotides, cholesteryl ester transfer protein inhibitors, and proprotein convertase subtilisin/kexin type 9 inhibitors, offer the most effective pharmacologic therapy.
Funding
No sources of funding were used to conduct this study or prepare this manuscript.
Glossary
- JUPITER
Rosuvastatin 20 mg versus placebo in prevention of cardiovascular (CV) EventsMENDEL-1: monoclonal antibody against PCSK9 to reduce elevated low-density lipoprotein cholesterol (LDL-C) in adults currently not receiving drug therapy for easing lipid levels
- MENDEL-2
Monoclonal antibody against PCSK9 to reduce elevated LDL-C in subjects currently not receiving drug therapy for easing lipid levels-2
- LAPLACE-TIMI 57
Low-density lipoprotein cholesterol (LDL-C) assessment with PCSK9 monoclonaL Antibody Inhibition Combined With Statin therapy (LAPLACE.
- LAPLACE-2
LDL-C assessment With PCSK9 monoclonal antibody inhibition combined with statin therapy-2
- RUTHERFORD-1
Reduction of low-density lipoprotein cholesterol (LDL-C) with PCSK9 inhibition in heterozygous familial hypercholesterolemia disorder study
- RUTHERFORD-2
Reduction of LDL-C with PCSK9 inhibition in heterozygous familial hypercholesterolemia disorder study-2
- GAUSS-1
Goal Achievement after utilizing an anti-PCSK9 antibody in statin intolerant subjects
- GAUSS-2
Goal achievement after utilizing an anti-PCSK9 antibody in statin intolerant subjects-2
- OSLER-1
Open label study of long term evaluation against LDL-C trial
- FOURIER
Further cardiovascular outcomes research with PCSK9 inhibition in subjects with elevated risk
- ODYSSEY
Evaluation of cardiovascular outcomes after an acute coronary syndrome during treatment with alirocumab
Footnotes
Conflict of interest Nathaniel Eraikhuemen, Dovena Lazaridis, and Matthew T. Dutton have no conflicts of interest that are directly relevant to the content of this article.
Availability of Data and Material Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
References
- 1.Tipping RW, Ford CE, Simpson LM, et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302(4):412–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsimikas S A test in context: lipoprotein(a): diagnosis, prognosis, controversies, and emerging therapies. J Am Coll Cardiol. 2017;69(6):692–711. 10.1016/j.jacc.2016.11.042. [DOI] [PubMed] [Google Scholar]
- 3.Marcovina SM, Albers JJ. Lipoprotein (a) measurements for clinical application. J Lipid Res. 2016;57(4):526–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lamon-Fava S, Marcovina SM, Albers JJ, Kennedy H, et al. Lipoprotein(a) levels, apo(a) isoform size, and coronary heart disease risk in the Framingham Offspring Study. J Lipid Res. 2011;52(6):1181–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Enkhmaa B, Anuurad E, Berglund L. Lipoprotein (a): impact by ethnicity and environmental and medical conditions. J Lipid Res. 2016;57(7):1111–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saeedi R, Frohlich J. Lipoprotein (a), an independent cardiovascular risk marker. Clin Diabetes Endocrinol. 2016;2(1):1–6. 10.1186/s40842-016-0024-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maranhão RC, Carvalho PO, Strunz CC, et al. Lipoprotein (a): structure, pathophysiology and clinical implications. Arq Bras Cardiol. 2014;103(1):76–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nordestgaard BG, Chapman MJ, Ray K, et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31(23):2844–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Virani SS, Brautbar A, Davis BC, et al. Associations between lipoprotein(a) levels and cardiovascular outcomes in black and white subjects: the Atherosclerosis Risk in Communities (ARIC) study. Circulation. 2012;125(2):241–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Willeit P, Ridker PM, Nestel PJ, et al. Baseline and on-statin treatment lipoprotein(a) levels for prediction of cardiovascular events: individual patient-data meta-analysis of statin outcome trials. Lancet. 2018;392(10155):1311–20. 10.1016/S0140-6736(18)31652-0. [DOI] [PubMed] [Google Scholar]
- 11.Verbeek R, Hoogeveen RM, Langsted A, et al. Cardiovascular disease risk associated with elevated lipoprotein(a) attenuates at low low-density lipoprotein cholesterol levels in a primary prevention setting. Eur Heart J. 2018;39(27):2589–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anderson TJ, Grégoire J, Pearson GJ, et al. 2016 Canadian Cardiovascular Society Guidelines for the Management of Dyslipidemia for the Prevention of Cardiovascular Disease in the Adult. Can J Cardiol. 2016;32(11):1263–82. [DOI] [PubMed] [Google Scholar]
- 13.Langlois MR, Chapman MJ, Cobbaert C, et al. Quantifying atherogenic lipoproteins: current and future challenges in the era of personalized medicine and very low concentrations of LDL cholesterol: A consensus statement from EAS and EFLM. Clin Chem. 2018;64(7):1006–33. [DOI] [PubMed] [Google Scholar]
- 14.Grundy SM, Stone NJ, Chair V, et al. Cholesterol clinical practice guidelines: executive summary. Circulation. 2018. 10.1161/CIR.0000000000000624. [DOI] [Google Scholar]
- 15.Miltiadous G, Saougos V, Cariolou M, et al. Plasma lipoprotein(a) levels and LDL-cholesterol lowering response to statin therapy in patients with heterozygous familial hypercholesterolemia. Ann Clin Lab Sci. 2006;36(3):353–5. [PubMed] [Google Scholar]
- 16.Tsimikas S, Gordts PLSM, Nora C, et al. Statin therapy increases lipoprotein(a) levels. Eur Heart J. 2019;1:1–10. [DOI] [PubMed] [Google Scholar]
- 17.Yahya R, Berk K, Verhoeven A, et al. Statin treatment increases lipoprotein(a) levels in subjects with low molecular weight apolipoprotein(a) phenotype. Atherosclerosis. 2019; p. 1–5. [DOI] [PubMed] [Google Scholar]
- 18.Guyton JR, Blazing MA, Hagar J, et al. Extended-release niacin vs gemfibrozil for the treatment of low levels of high-density lipoprotein cholesterol. Arch Intern Med. 2000;160(8):1177–84. [DOI] [PubMed] [Google Scholar]
- 19.Sahebkar A, Simental-Mendía LE, Watts GF, et al. Comparison of the effects of fibrates versus statins on plasma lipoprotein(a) concentrations: a systematic review and meta-analysis of head-to-head randomized controlled trials. BMC Med. 2017;15(1):1–14. 10.1186/s12916-017-0787-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herrmann W, Biermann J, Kostner GM. Comparison of effects of N-3 to N-6 fatty acids on serum level of lipoprotein(a) in patients with coronary artery disease. Am J Cardiol. 1995;76(459):462. [DOI] [PubMed] [Google Scholar]
- 21.Kooshki A, Taleban FA, Tabibi H, et al. Effects of omega-3 fatty acids on serum lipids, lipoprotein (a), and hematologic factors in hemodialysis patients. Ren Fail. 2011;33(9):892–8. [DOI] [PubMed] [Google Scholar]
- 22.Shinozaki K, Kambayashi J, Kawasaki T, et al. The long-term effect of eicosapentaenoic acid on serum levels of lipoprotein(a) and lipids in patients with vascular disease. J Atheroscler Thromb. 1996;2(2):107–9. [DOI] [PubMed] [Google Scholar]
- 23.Ballantyne CM, Davidson MH, McKenney JM, et al. Comparison of the efficacy and safety of a combination tablet of niacin extended-release and simvastatin with simvastatin 80 mg monotherapy: the SEACOAST II (high-dose) study. J Clin Lipidol. 2008;2(2):79–90. [DOI] [PubMed] [Google Scholar]
- 24.Ballantyne CM, Davidson MH, McKenney J, et al. Comparison of the Safety and Efficacy of a Combination Tablet of Niacin Extended Release and Simvastatin vs Simvastatin Monotherapy in Patients With Increased Non-HDL Cholesterol (from the SEACOAST I Study). Am J Cardiol. 2008;101(10):1428–36. [DOI] [PubMed] [Google Scholar]
- 25.Chen F, Maccubbin D, Yan L, et al. Lipid-altering efficacy and safety profile of co-administered extended release niacin/laropiprant and simvastatin versus atorvastatin in patients with mixed hyperlipidemia. Int J Cardiol. 2013;167(1):225–31. 10.1016/j.ijcard.2011.12.103. [DOI] [PubMed] [Google Scholar]
- 26.Insull W, Basile JN, Vo AN, et al. Efficacy and safety of combination therapy with niacin extended-release and simvastatin versus atorvastatin in patients with dyslipidemia: the SUPREME Study. J Clin Lipidol. 2009;3(2):109–18. 10.1016/j.jacl.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 27.Maccubbin D, Bays HE, Olsson AG, et al. Lipid-modifying efficacy and tolerability of extended-release niacin/laropiprant in patients with primary hypercholesterolaemia or mixed dyslipidaemia. Int J Clin Pract. 2008;62(12):1959–70. [DOI] [PubMed] [Google Scholar]
- 28.Sahebkar A, Reiner Ž, Simental-Mendía LE, et al. Effect of extended-release niacin on plasma lipoprotein(a) levels: a systematic review and meta-analysis of randomized placebo-controlled trials. Metabolism. 2016;65(11):1664–78. [DOI] [PubMed] [Google Scholar]
- 29.Kamanna VS, Kashyap ML. Mechanism of action of niacin. Am J Cardiol. 2008;101(8 SUPPL. 1). [DOI] [PubMed] [Google Scholar]
- 30.Suk Danik J, Rifai N, Buring JE, et al. Lipoprotein(a), hormone replacement therapy, and risk of future cardiovascular events. J Am Coll Cardiol. 2008;52(2):124–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shlipak MG, Simon JA, Vittinghoff E, et al. Estrogen and progestin, lipoprotein(a), and the risk of recurrent coronary heart disease events after menopause. JAMA. 2000;283(14):1845–52. [DOI] [PubMed] [Google Scholar]
- 32.Shewmon DA, Stock JL, Rosen CJ, et al. Tamoxifen and estrogen lower circulating lipoprotein(a) concentrations in healthy postmenopausal women. Arterioscler Thromb Vasc Biol. 1994;14(10):1586–93. [DOI] [PubMed] [Google Scholar]
- 33.Ferretti G, Bacchetti T, Simental-Mendía LE, et al. Raloxifene lowers plasma lipoprotein(a) concentrations: a systematic review and meta-analysis of randomized placebo-controlled trials. Cardiovasc Drugs Ther. 2017;31(2):197–208. [DOI] [PubMed] [Google Scholar]
- 34.Langsted A, Nordestgaard BG. Antisense oligonucleotides targeting lipoprotein(a). Curr Atheroscler Rep. 2019;21(8):1–7. [DOI] [PubMed] [Google Scholar]
- 35.Santos RD, Raal FJ, Catapano AL, et al. Mipomersen, an antisense oligonucleotide to apolipoprotein B-100, reduces lipoprotein(a) in various populations with hypercholesterolemia: results of 4 phase III Trials. Arterioscler Thromb Vasc Biol. 2015;35(3):689–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li N, Li Q, Tian XQ, et al. Mipomersen is a promising therapy in the management of hypercholesterolemia: a meta-analysis of randomized controlled trials. Am J Cardiovasc Drugs. 2014;14(5):367–76. [DOI] [PubMed] [Google Scholar]
- 37.Tsimikas S AKCEA-APO(a)-LRx Shows Promise of Reducing Lp(a) Levels in Patients With Preexisting CVD - American College of Cardiology. In: AHA Conference. Chicago, IL. https://www.acc.org/latest-in-cardiology/articles/2018/11/07/15/19/sat445pm-akcea-apo-lrx-to-lower-lipoprotein-aha-2018. Accessed 14 Oct 2019. [Google Scholar]
- 38.Assessing the Impact of Lipoprotein (a) Lowering With TQJ230 on Major Cardiovascular Events in Patients With CVD [NCT04023552]. Available online from: https://clinicaltrials.gov/ct2/show/NCT04023552. Accessed 2 Sep 2020
- 39.Gutstein DE, Krishna R, Johns D, et al. Anacetrapib, a novel CETP inhibitor: pursuing a new approach to cardiovascular risk reduction. Clin Pharmacol Ther. 2012;91(1):109–22. 10.1038/clpt.2011.271/nature06264. [DOI] [PubMed] [Google Scholar]
- 40.Bowman L, Hopewell JC, Chen F, et al. Effects of anacetrapib in patients with atherosclerotic vascular disease. N Engl J Med. 2017;377(13):1217–27. [DOI] [PubMed] [Google Scholar]
- 41.Hovingh GK, Kastelein JJP, Van Deventer SJH, et al. Cholesterol ester transfer protein inhibition by TA-8995 in patients with mild dyslipidaemia (TULIP): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet. 2015;386(9992):452–60. 10.1016/S0140-6736(15)60158-1. [DOI] [PubMed] [Google Scholar]
- 42.Raal FJ, Giugliano RP, Sabatine MS, et al. PCSK9 inhibitionmediated reduction in Lp(a) with evolocumab: an analysis of 10 clinical trials and the LDL receptor’s role. J Lipid Res. 2016;57(6):1086–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Donoghue ML, Fazio S, Giugliano RP, et al. Lipoprotein(a), PCSK9 inhibition, and cardiovascular risk. Circulation. 2019;139(12):1483–92. [DOI] [PubMed] [Google Scholar]
- 44.McKenney JM, Koren MJ, Kereiakes DJ, et al. Safety and efficacy of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease, SAR236553/REGN727, in patients with primary hypercholesterolemia receiving ongoing stable atorvastatin therapy. J Am Coll Cardiol. 2012;59(25):2344–53. 10.1016/j.jacc.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 45.Gaudet D, Kereiakes DJ, McKenney JM, et al. Effect of alirocumab, a monoclonal proprotein convertase subtilisin/kexin 9 antibody, on lipoprotein(a) concentrations (a pooled analysis of 150 mg every two weeks dosing from phase 2 trials). Am J Cardiol. 2014;114(5):711–5. 10.1016/j.amjcard.2014.05.060. [DOI] [PubMed] [Google Scholar]
- 46.Kastelein JJP, Ginsberg HN, Langslet G, et al. ODYSSEY FH i and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. Eur Heart J. 2015;36(43):2996–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cao YX, Liu HH, Li S, et al. A meta-analysis of the effect of PCSK9-monoclonal antibodies on circulating lipoprotein (a) levels. Am J Cardiovasc Drugs. 2019;19(1):87–97. 10.1007/s40256-018-0303-2. [DOI] [PubMed] [Google Scholar]
- 48.Schultz O, Oberhauser F, Saech J, et al. Effects of inhibition of interleukin-6 signalling on insulin sensitivity and lipoprotein (A) levels in human subjects with rheumatoid diseases. PLoS One. 2010;5(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.García-Gómez C, Martín-Martínez MA, Castañeda S, Sanchez-Alonso F, et al. Lipoprotein(a) concentrations in rheumatoid arthritis on biologic therapy: results from the CARdiovascular in rheuMAtology study project. J Clin Lipidol. 2017;11(3):749–756. e3. [DOI] [PubMed] [Google Scholar]
- 50.Ueland T, Kleveland O, Michelsen AE, et al. Serum lipoprotein(a) is not modified by interleukin-6 receptor antagonism or associated with inflammation in non-ST-elevation myocardial infarction. Int J Cardiol. 2019;274:348–50. 10.1016/j.ijcard.2018.06.093. [DOI] [PubMed] [Google Scholar]
- 51.Cuchel M, Meagher EA, du Toit TH, et al. Efficacy and safety of a microsomal triglyeride transfer protein inhibitor in homozygous familial hypercholesterolemia. Lancet. 2013;381(9860):40–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sahebkar A, Simental-Mendía LE, Stefanutti C, et al. Supplementation with coenzyme Q10 reduces plasma lipoprotein(a) concentrations but not other lipid indices: a systematic review and meta-analysis. Pharmacol Res. 2016;105:198–209. 10.1016/j.phrs.2016.01.030. [DOI] [PubMed] [Google Scholar]
- 53.Awad K, Mikhailidis DP, Katsiki N, et al. Effect of ezetimibe monotherapy on plasma lipoprotein(a) concentrations in patients with primary hypercholesterolemia: a systematic review and meta-analysis of randomized controlled Trials. Drugs. 2018;78(4):453–62. 10.1007/s40265-018-0870-1. [DOI] [PubMed] [Google Scholar]
- 54.Knopp RH, Gitter H, Truitt T, et al. Effects of ezetimibe, a new cholesterol absorption inhibitor, on plasma lipids in patients with primary hypercholesterolemia. Eur Heart J. 2003;24(8):729–41. [DOI] [PubMed] [Google Scholar]