

Abstract
Arsenic (As) is one of the most harmful and widespread groundwater contaminants globally. Besides the occurrence of geogenic As pollution, there is also a large number of sites that have been polluted by anthropogenic activities, with many of those requiring active remediation to reduce their environmental impact. Cost-effective remedial strategies are however still sorely needed. At the laboratory-scale in situ formation of magnetite through the joint addition of nitrate and Fe(II) has shown to be a promising new technique. However, its applicability under a wider range of environmental conditions still needs to be assessed. Here we use sediment and groundwater from a severely polluted coastal aquifer and explore the efficiency of nitrate-Fe(II) treatments in mitigating dissolved As concentrations. In selected experiments >99% of dissolved As was removed, compared to unamended controls, and maintained upon addition of lactate, a labile organic carbon source. Pre- and post experimental characterisation of iron (Fe) mineral phases suggested a >90% loss of amorphous Fe oxides in favour of increased crystalline, recalcitrant oxide and sulfide phases. Magnetite formation did not occur via the nitrate-dependent oxidation of the amended Fe(II) as originally expected. Instead, magnetite is thought to have formed by the Fe(II)-catalysed transformation of pre-existing amorphous and crystalline Fe oxides. The extent of amorphous and crystalline Fe oxide transformation was then limited by the exhaustion of dissolved Fe(II). Elevated phosphate concentrations lowered the treatment efficacy indicating joint removal of phosphate is necessary for maximum impact. The remedial efficiency was not impacted by varying salinities, thus rendering the tested approach a viable remediation method for coastal aquifers.
Keywords: arsenic remediation, in situ mineral precipitation, coastal aquifer, bioremediation
Graphical Abstract

1. Introduction
Arsenic (As) is one of the most serious groundwater contaminants that globally poses many challenges to the preservation and safe use of groundwater resources. Arsenate (As(V)) and arsenite (As(III)) are dominant valence states in groundwater, the latter in more reducing environments (Lei et al., 2018). As(III) is also more mobile and nearly 10 times more toxic for humans (Sharma and Sohn, 2009). As it can be taken up by plants and animals, it may affect the entire food chain (Morin and Calas, 2006). Potential health impacts include leathality, inhibition of growth, photosynthesis as well as reproduction and behavioural effects (Howe et al., 2001). Elevated As concentrations in groundwaters are mostly reported from geogenic sources (e.g., Fendorf et al., 2010; Podgorski and Berg, 2020), however, anthropogenic activities, particularly industrial, agricultural and mining are also common causes of As contamination (Qiu et al., 2017; Cantu et al., 2016; Edenborn,’ et al., 1986; Luther et al., 2012; Morin and Calas, 2006; Revesz et al., 2015). In recognition of its toxicity, widespread occurrence and diffuculty of remediation, As has been the highest ranked contaminant on the U.S. national substance priority list since 1997 (ATSDR, 2019). Growing awareness of its toxicity has resulted in the U.S. Environmental Protection Agency (USEPA) reducing the As drinking water standard from 50 μg/L to 10 μg/L, in line with the World Health Organisation (WHO) health-based recommendation (Missimer et al., 2018; Podgorski and Berg, 2020). Meanwhile, clean-up of As contaminated groundwaters remains extremely challenging. Many currently applied technologies have shown to be expensive and/or inefficient under conditions that commonly mobilize As (Akin et al., 2012; Liu et al., 2019; USEPA, 2002; Van Deuren et al., 2002). For example, pump-and-treat approaches require generally long periods of operation, often decades or more, with reducing effectiveness over time (Van Deuren et al., 2002).
Timely and cost-effective As remedial strategies that are broadly effective are sorely needed. One of the major alternatives to pump-and-treat schemes is in-situ immobilisation, which removes As from groundwater by stimulating its adsorption onto and co-precipitation within compatible minerals. Most in-situ immobilisation approaches target specifically the formation and transformation of iron (Fe) minerals, as they are known to be highly efficient As binding agents, specifically Fe oxides in oxic environments and Fe sulfides in anoxic environments (Beaulieu and Ramirez, 2013; Benner et al., 2002; Cañas Kurz et al., 2020; Dixit and Hering, 2003; Kneebone et al., 2002; O’Day et al., 2004; Pedersen et al., 2006; Saunders et al., 2008; Teclu et al., 2008). For example, Akin et al. (2012) reported a 99.2% reduction in dissolved As following the addition of synthesised magnetite nanoparticles. Also, Omoregie et al. (2013) found that nitrate (NO3−) amendments stimulated Fe(II) oxidation and formed fresh hydrous ferric oxide, in their case hematite, that remediated As contaminated groundwater to concentrations below the WHO guideline limits. Using batch and column experiments, Lien and Wilkin (2005) concluded Fe to be an effective in-situ As remedial reagent, with uptake occurring via adsorption and co-precipitation to green rust, a mixed valence Fe hydroxide. Kanel et al. (2005) reported the successful immobilisation of both As(III) and As(V) by co-precipitation on magnetite and maghemite. Sun et al. (2006) reported that Fe mineral co-precipitation is favoured for As(V), compared to As(III), while As(III) is adsorbed more rapidly than As(V) under anaerobic conditions. As(III) typically dominates in strongly reducing aquifers in which Fe(III) and sulfate reduction are taking place (Sasaki et al., 2009). Sulfate-reducing conditions were stimulated in an As contaminated aquifer treated with both Fe2+(aq) and sulfate and found to achieve long term (>15 months) removal of As below 5 μg/L in association with the inferred precipitation of both FeS and Fe (oxy)hydroxides (Beaulieu and Ramirez, 2013b). Beak and Wilkin (2009) reported As removal by both iron sulfides and iron (oxy)hydroxides whether conditions were sulfate-reducing or not. Deditius et al. (2014) observed As to not only be structurally incoporated into pyrite but to also occur as amorphous nanoparticle aggreates.
Overall, stimulated (trans)formation of iron minerals in affected groundwaters often effectively removes As from the dissolved phase (Akin et al., 2012; Cañas Kurz et al., 2020; Giles et al., 2011; Omoregie et al., 2013). Its reliability and durability, however, is contingent on the stability of the Fe minerals and the As bound to them. This is a central challenge for effective As remediation because many Fe oxides and sulfides are not stable over the typically broad ranging redox conditions that dominate at many As-polluted sites. Specifically, many less crystalline iron oxide minerals are unstable at the reducing environments typical of As-polluted groundwaters, e.g., at landfill sites, chemical plants (Beaulieu and Ramirez, 2013; Tufano et al., 2008) and other locations where the degradation of organic co-contaminants affects aquifer conditions. In fact, groundwater contamination often results from organics facilitating Fe(III) oxide substrate reduction (Bonte et al., 2013; Kocar et al., 2006; Rawson et al., 2016). The adsorbed or co-precipitated As formed from remedation efforts is thus also susceptible to re-mobilisation (Bonte et al., 2013; Revesz et al., 2016). Conversely, the oxidative dissolution of As-containing Fe sulfides, such as arsenopyrite and pyrite is another major pathway for As release (Fakhreddine et al., 2020; Prommer et al., 2018; Qiu et al., 2017; Rathi et al., 2017). In sulfate-reducing environments, sulfide production can also potentially lead to the formation of thioarsenic species and significantly enhance As mobility (e.g., Wang et al., 2020; Burton et al., 2013; Suess and Planer-Friedrich, 2012; Planer-Friedrich et al., 2007; Keimowitz et al., 2007).
The mixed valence iron oxide magnetite (Fe(II)Fe (III)2O4) is, by comparison, much more stable under a wider range of aquifer conditions, including both weakly oxic and iron(III)-reducing environments (Sun et al., 2016b; Yuan et al., 2009). The presence of magnetite facilitates multiple pathways for contaminant immobilisation, including adsorption, surface precipitation and structural incorporation (Coker et al., 2006; Lovley and Phillips, 1986; Schwertmann and Cornell, 2000). Not only does magnetite demonstrate high sorption capacity and specificity for As, but it can also absorb both As(III) and As(V) with similar affinity (Bujňáková et al., 2013; Giménez et al., 2007; Jönsson and Sherman, 2008; Root et al., 2009; Shipley et al., 2009; Yavuz et al., 2006). The in situ formation of magnetite has therefore the potential to be a more reliable treatment option than those creating Fe(III) oxides (Sun et al., 2016b; Yuan et al., 2009).
To date, the efficiency of magnetite as an As sequester has mostly been demonstrated in controlled laboratory experiments with simplified water and/or solid matrices (Farrell et al., 2014; Johnson et al., 2005; Kocar et al., 2006; Sun et al., 2018, 2016a). While these results established the feasibility of this approach, its efficiency and robustness during field-scale applications will depend on the ability to work well under a wide range of geochemical, in particular varying redox conditions. Among potential concerns is high phosphate, which can compete for sorption sites on neoformed Fe minerals (Su and Puls, 2001). Furthermore, high salinity, sulfate and chloride concentrations can also affect sorption and redox cycling (Jain and Loeppert, 2000; Mamindy-Pajany et al., 2011; Morelli et al., 2017).
Within this context, the main objective of the present study was to assess the remedial viability of stimulating Fe mineral formation and As uptake in amended microcosms containing natural sediments from highly As-polluted groundwaters. Secondary to this, was to assess the impacts of a high sulfate, phosphate and salinity on Fe and S mineralisation and the efficiency of As retention. The investigated microcosms contained sediments from a typical coastal aquifer with elevated salinity and phosphate contamination. A variety of microcosms of varying natural sediments were combined with groundwater collected on-site or synthesised to represent local groundwater to systematically explore the geochemical response to nitrate-Fe(II) amendment and elucidate the critical factors affecting the extent and duration of remediation. In all experiments, the stability of the sequestering Fe minerals was challenged through the addition of lactate as a labile organic substrate stimulating reducing conditions. We hypothesised that the nitrate-Fe(II) treatment would more effectively sequester As than the sulfate-lactate treatment. Further, increased salinity and elevated phosphate concentrations would both decrease the degree of As removal from groundwater.
2. Material and Methods
2.1. Site Sediment and Groundwater Sampling
The study site, located in coastal southern Italy, has hosted mutliple industrial activities since the 1970’s, resulting in severe accidental pollution of the underlying aquifer by petroleum and fertilizer products (Colombani et al., 2015; Sbarbati et al., 2020). The shallow coastal aquifer comprises Pleistocene–Holocene alluvial deposits of sandy and sandy-loam sediments, interbedded by silt and silty-clay lenses (Mastrocicco et al., 2012). A mixed contaminant plume of dissolved As, ammonia and phosphate extends across an area of over 4 ha (Sbarbati et al., 2020).The groundwater pH varies between pH 6 and 8. Redox conditions vary vertically, with an upper sub-oxic zone (at <5 m depth) and mildly reducing conditions developing with depth (especially at >15 m depth), as indicated by increases in dissolved Mn and Fe. Groundwater salinity varies spatially, ranging from fresh (< 600 mg L−1 TDS) to saline (>35,000 mg L−1 TDS) conditions due to (i) seawater infiltrating from a canal proximal to the contaminant plume and (ii) seawater intrusion that is locally enhanced by the ongoing remediation activities (Mastrocicco et al., 2012). An extensive pump-and-treat system has operated for almost two decades. Currently, groundwater is pumped from 70 wells across the site at a rate of 5000 m3 day−1.
Sediment and groundwater samples were collected during a 2018 sampling program (10 to 18 December) and retrieved from depths between 11 and 12 m below surface, i.e., targeting the depth of the highest As concentration. The samples were collected (i) adjacent to an identifed source zone (samples used in experiments E1.3 and E2, respectively, see Tables S1 and S2) and (ii) at two downstream locations where core sample positions corresponded with multilevel sampling (MLS) ports (samples used in experiments E1.1 and E1.2) located within the As plume. Groundwater salinities ranged between 1,500 and 4,900 mg L−1 while redox conditions were relatively stable with Eh values ranging between +300 mV and +350 mV. Full details of the site characteristics were previously reported in Sbarbati et al. (2020).
Sediments were collected by piston core sampler, immediately sealed in vacuum bags and stored at −18 °C for 2 months prior to use. Groundwater samples were collected from MLS ports using a low flow bladder pump after pH, DO and Eh measurements had stabilised. All field parameters were determined towards the end of purging using a multi-parameter probe WTW Multi 340i, which includes: a SentiTix 41 pH combined electrode with a built-in temperature sensor for pH measurement, a CellOx 325 galvanic oxygen sensor for DO measurement, a combined AgCl-Pt electrode for ORP measurement and a Tetracon 325 4-electrode conductivity cell for EC measurements. Groundwater samples were filtered to 0.45-μm, and those intended for major cation determination were acidified to pH 2.0 with ultrapure HCl. Groundwater samples were collected in air-tight containers (glass amber bottles) and kept in the dark at 4°C until analysed to determine groundwater composition or for use in the experiment.
2.2. Microcosm Experiments
Microcosms (Table S1) used sediments from three locations (E1.1, E1.2, E1.3) in combination with either corresponding groundwater samples from adjacent locations (GW) or artificial groundwater (AW) containing 7g NaCl, 1.2 g MgCl2 6H20, 0.5 g KCl and 0.1 g CaCl2 2H20, dissolved in 1 L deionised water (Table S3). Our microcosms were designed to test the feasibility of stimulated As sequestration in magnetite produced by oxidising Fe(II) (added as ferrous sulphate, 10 mM) with nitrate (10 mM NaNO3) (Sun et al., 2016b). Nitrate was selected instead of oxygen as oxidant because it is more soluble while also reacting more slowly with ferrous Fe (Sun et al., 2016b). This allows for a more dispersed distribution of reagents during in situ applications. After 12 days, 1mM of lactate, a labile reductant, was added to the treatment batches to stimulate the reduction of Fe(III) and thereby to test the stability of any existing As-Fe-oxide assemblages. A total of 6 treatments and their respective unamended control microcosms (lacking added Fe(II) and nitrate) were observed with three of each using groundwater and high salinity water (14.4 mS cm−1).
All microcosms were prepared, performed and sampled in a glove bag under a N2 atmosphere to limit oxygen exposure throughout the experiment. In each microcosm, 50 g sediments were mixed with 500 mL GW or AW in 500 mL HDPE bottles prepared without headspace. All microcoms were wrapped in foil to prevent light exposure and incubated at room temperature on an orbital shaker at 80 RPM within a N2-filled glove bag to maintain anoxic conditions for a period of 45 days. Solution sampling occurred once every three days within a N2-filled glove bag. Aliquots of sediments and solutions were removed to maintain a constant soil:water ratio. During subsampling, microcosms were monitored for physico-chemical parameters using pH indicator strips, a combined AgCl-Pt electrode for ORP and a Tetracon 325 4-electrode conductivity cell for electric conductivity (EC). Solutions were filtered to 0.2 μm and preserved as needed for water composition analysis. Pre- and post experiment sediment characterisation was performed via sequential chemical extraction.
An additional series of 9 microcosms was performed to compare the efficacy of oxic (NaNO3) versus anoxic (Na2SO4) treatments and to evaluate the effect of salinity and phosphate on As mobility, (Table S2). For these experiments, only source zone sediment (E2) was used, in combination with synthetic groundwater. Salinity effects were assessed using artificial ‘low’ and ‘high’ salinity water (Table S3). Phosphate competition was assessed in the presence and absence of 3 mM KH2PO4, typical of background levels at the site. The stability of any existing As-Fe-oxide assemblage was tested by the addition of 10 mM lactate on Day 12. Batches for the formation of reduced sulfides were treated with dissolved reactive organic carbon (serving as electron donor) at inception in the form of 10 mM sodium lactate and extra inorganic sulfate (SO42−) as 10 mM NaSO4. This method is a commonly used strategy to stimulate sulfate reduction (Omoregie et al., 2013; Sun et al., 2016c).
2.3. Analytical Procedure
2.3.1. Solution Analysis
Major cations and anions were determined by ion chromatography (IC) with a Dionex ICS 1100 and Dionex ICS 5000 (Thermo Fisher Scientific, Germany), respectively with detection limits at 10 ppb and an analytical precision of ±5%. Alkalinity was determined by acid (HCl) titration using Standard Methods 2320B (APHA, 1995). Sample solutions for dissolved metals were acidified to 1% HNO3 for analysis by inductively coupled plasma mass spectrometry (ICP-MS) with an X Series II instrument (Thermo Fisher Scientific, Germany), detection limits at 1 ppb (0.05 ppb for As). Rhodium was added as an internal response standard and used for monitoring instrument drift. For analysing As, the instrument was set at resolving power ~10,000, to resolve the As peak from argon-chloride interference. Quantification for each element was based on comparison to a six-point standard curve from a multi-element standard. Accuracy was within 100±5% based on certified NIST standards.
2.3.2. Sediment Characterisation
The elemental compositions of the sediments were assessed by acid digestion following the EPA Method 3050B (SW-846) procedure (USEPA, 1996). A homogenised sediment aliquot for each sample was weighed and transferred into a Teflon container that was previously washed with acid, placed in a digestion block, covered and digested by hot acid reflux in a mixture of HNO3, HCl, H2O2 and HF for several hours at 90–95°C. To remove any traces of undigested silicates, the obtained suspensions were filtered through a 0.45 μm nylon membrane. Dissolved metals in the solutions were analysed by ICPMS using standard methods (Sbarbati et al., 2020; Sun et al., 2016a).
Seven-step sequential extractions were conducted to quantify empirical Fe and As associations with solid phases. Full details of the employed 7 extraction steps are provided in Table S4 and in Sun et al. (2016a). Wet sediment samples were collected in vials. The extractions were all conducted at room temperature using a dry mass of 150 mg and an extractant volume of 10 mL. Each suspension was centrifuged for 25 min at 8500 rpm and the extractant then decanted and filtered using 0.45 μm nylon membrane filters. Dissolved Fe and As concentrations in the extractions were determined using ICP-MS, as previously described.
2.4. Data Handling Procedure
First series microcosm results were separated based on water type, either AW or GW, and averaged across the three sediment types. The nitrate-Fe(II) treated microcosms, referred to hereafter as treatments, are discussed in comparision with the corresponding unamended control microcosms (those without added FeSO4 or NaNO3), referred to hereafter as their corresponding controls. Statistically significant differences between data populations were assessed using single factor, (one-way) analysis of variance (ANOVA), or F-test. ANOVA tests determine whether there are differences in the means of two or more groups on the basis of normal population distributions. Population variance, or F-distriubtion, was assessed based on the independent variable of sediment type and dependant variables of pre and post experiment SE data, treatments and controls or AW and GW water types. A null hypothesis of all group means are equal, i.e., no difference, was rejected based on a significance level (p value) of less than 0.05.
3. Results
3.1. Microcosms Assessing the Feasibility of As Sequestration
3.1.1. Mineralogical Characterisation
3.1.1.1. Initial Sediment Mineralogy
The three sediment types used in these microcosm experiments were all medium to coarse-grained, weakly compacted sands ranging in colour from grey to orange-red, based on field observations. Bulk Fe contents, determined by total digestion, ranged between 174.6 and 242.1 mmol kg−1. Extractable Fe contents, determined by sequential chemical extraction, were <10% of the total Fe and ranged between 4.6 and 12.5 mmol kg−1 (Figure 1). Crystalline Fe oxides ranged from 2.8 to 8.6 mmol kg−1, the largest pool of extractable Fe while still representing only a small fraction of the total Fe. Small quantities of amorphous Fe oxides, 0.8 to 2.5 mmol kg−1 and Fe carbonates, 0.7 to 1.2 mmol kg−1 were also present. Recalcitrant Fe oxides, most likely magnetite, were <2%. Exchangeable and sulfidic Fe minerals were <0.5% and <5%, respectively.
Figure 1.
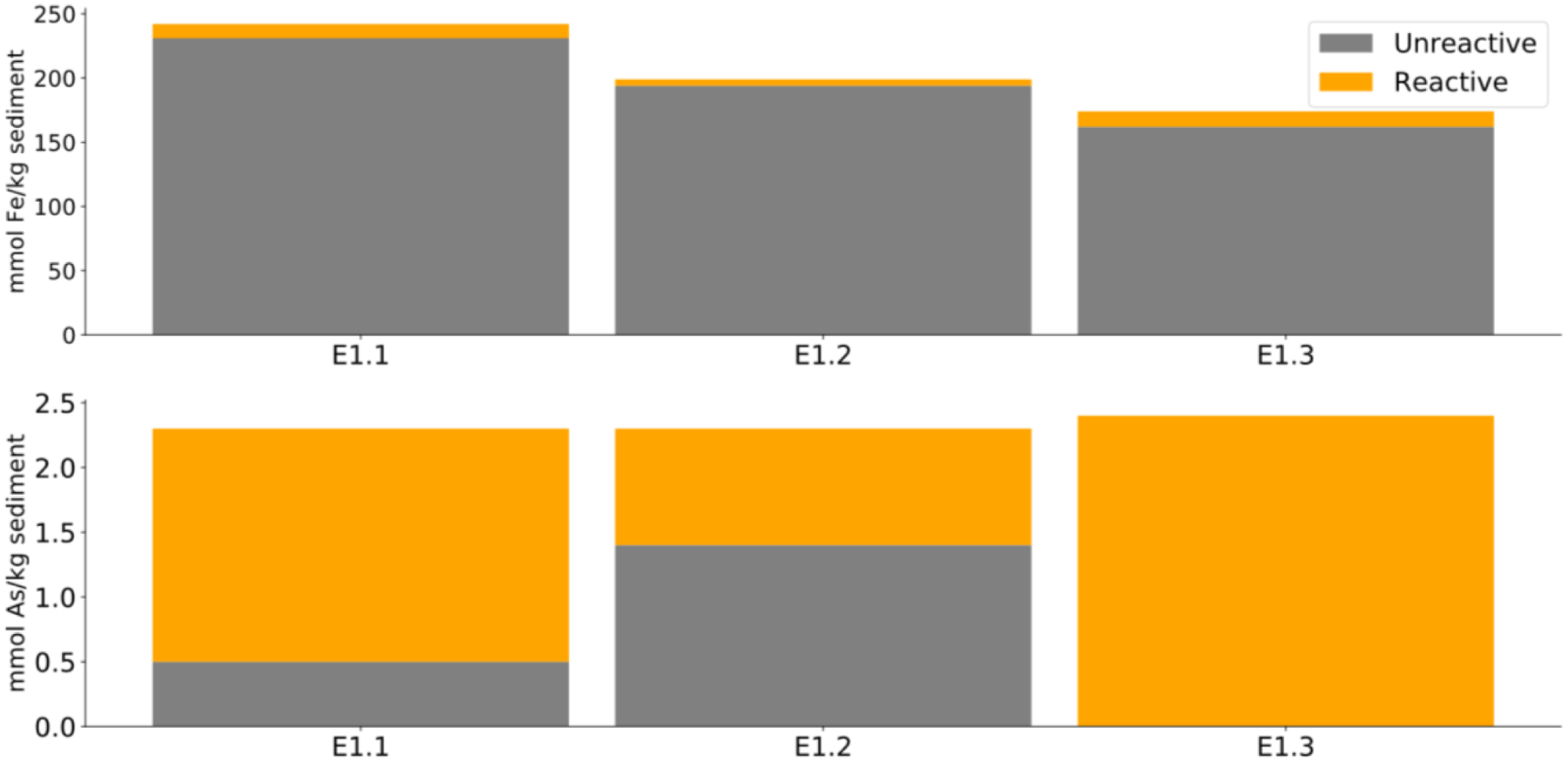
Total sediment Fe (top) and As (bottom) concentrations as determined by acid digestion for three site sediments, E1.1, E1.2 and E1.3 collected 11 to 12 m below surface. The reactive fraction is the sum of sequential extraction analysis results subtracted from acid digest totals (orange). The remainder is referred to as unreactive Fe and As (grey).
Bulk As contents ranged between 2.3 and 2.4 mmol kg−1, of which 0.9 to 2.4 mmol kg−1 was extractable (Figure 1). Extractable As was predominantly associated with amorphous Fe oxides with a concentration of 0.4 to 1.5 mmol kg−1, followed by crystalline Fe oxides, 0.2 to 0.5 mmol kg−1, and Fe carbonates, 0.1 to 0.2 mmol kg−1, respectively. Loosely bound As was <10%, As adsorbed on and co-precipitated with recalcitrant Fe oxides was <5% and As associated with sulfides was <1%.
3.1.1.2. Mineral Formation in Microcosms
Significant decreases (between 5.2–5.3 mmol kg−1, or about 45%) of the extractable Fe were observed in both the artificial water control (AWC) and groundwater control (GWC) (Figure 2). Extractable Fe was also lower than expected in the treatments with Fe amendment, ranging between 8.0 and 8.6 mmol kg−1. This implies that a significant quantity of the added Fe was not readily extractable after amendment. Changes in mineral assemblage composition (fractional representation) were found to be largely consistent across treatments and controls. Significant losses of amorphous and carbonate Fe minerals occurred while increases in recalcitrant Fe and Fe sulfide phases were observed. The proportion of crystalline Fe minerals increased from 68% to between 75 and 83% from pre- to post-treatment, while additionally recalcitrant and sulfide Fe minerals formed. A greater loss of amorphous Fe minerals was oberved in the controls compared to the treatments.
Figure 2.

Sequential extraction Fe mineralogy fractions (top) and As mineralogical associations (bottom) averaged from three site sediments (E1.1, E1.2 and E1.3). Initial fractions (far left) compared to final fractionations in microcosms (from left to right): artificial water treated with 10 mM FeSO4 and 10 mM NaNO3 (AWT), groundwater treated with 10 mM FeSO4 and 10 mM NaNO3 (GWT), the respective artificial water control (AWC) and the respective groundwater control (GWC).
No significant differences in absolute mineral concentrations were observed between the artificial water and groundwater types. Fe sulfides increased significantly from 0.1 mmol kg−1 in both the groundwater treatment (GWT) and artificial water treatment (AWT) to 0.9 and 1.5 mmol kg−1, respectively. Recalcitrant Fe oxides increased from 0.15 to 0.23 and 0.33 mmol kg−1 for GWT and AWT, respectively. Amorphous Fe oxides decreased from 1.75 mmol kg−1 to 0.05 and 0.07 mmol kg−1, a loss of between 92 and 99% in AWT and GWT, respectively.
Although extractable Fe concentrations decreased, no significant difference occurred between pre- and post-experiment total extractable As concentrations (Figure 2). However, the specific extraction pools did change. Prior to incubations, most As was associated with amorphous phases. Post experiment, As was mostly extracted with crystalline Fe oxides. As associated with amorphous Fe oxides decreased from 1 mmol kg−1 to 0.14 and 0.19 mmol kg−1 for AWT and GWT, respectively, with no significant difference between treatments and unamended controls. As associated with crystalline Fe oxides increased from 0.3 mmol kg−1 to 0.84 and 1.22 mmol kg−1 for AWT and GWT, respectively and to 1.06 and 1.34 mmol kg−1 for AWC and GWC, respectively.
As bearing sulfides also increased significantly in both treatments and unamended controls while being significantly higher in treatments (0.08–0.1 mmol kg−1) than both respective controls (~0.04 mmol kg−1). No significant difference was observed between treatment water types. As associated with recalcitrant Fe oxides increased in treatments, doubling from 0.06 mmol kg−1 to 0.13 mmol kg−1, but not in unamended controls, with no difference observed between treatment water types. As associated with Fe carbonates significantly reduced post experiment, across both FeSO4 and NaNO3 treatments (>90%) and their respective controls (~65%), consistent with the near total loss of Fe carbonates (Figure 2).
3.1.2. Hydrochemistry
3.1.2.1. Fe
Dissolved Fe concentrations in stored groundwater pre-amendment (Day 0) ranged from 0.02 to 0.07 μM, and were therefore up to 2 orders of magnitude lower than those measured at the time of field sampling (2 to 7 μM), indicating that some oxidation had likely occurred during sample handling and storage. Dissolved Fe concentrations in the treatments were depleted by Day 12, prior to lactate addition (Figure 3). GWT and AWT microcosms showed similar behaviour, despite highly variable salinities (2 to 15 mS cm−1), with a progressive colour change observed from clear to reddish-brown throughout the course of the experiment (Figure 4). No release of Fe occurred in the treatments for more than 30 days following the addition of lactate.
Figure 3.

Dissolved As (top) and Fe (bottom) concentrations in artificial water microcosms (left, circles) and site groundwater microcosms (right, triangles). Data presented is the average of the three E1 scenarios (Table S1). FeSO4 (10 mM) and NaNO3 (10 mM) treatment results are shown in solid blue and comparative controls (no FeSO4, NaNO3 or lactate) in hollow red. Concentrations are reported in mM. Grey shading indicates 1 mM lactate addition in treatments only.
Figure 4.

A: Day 3 FeSO4 (10 mM) and NaNO3 (10 mM) treatment (first, third and fifth pairs as duplicates) and respective control microcosms (second, fourth and sixth pairs as duplicates, no FeSO4 or NaNO3). B: Day 9 FeSO4 (10 mM) and NaNO3 (10 mM) treatments showing progressive colour change to red brown (first, third and fifth pairs as duplicates) and respective control microcosms (second, fourth and sixth pairs as duplicates, no FeSO4 or NaNO3) showing no colour change C: Day 9 (left), Day 12 (centre) and Day 34 (right) SO4-lactate (10 mM) microcosm duplicate pairs showing progressive colour change to black.
3.1.2.2. As
Initial dissolved As levels in GW ranged between 3 and 5 μM, comparable to concentrations at the time of sample collection in the field, where concentrations ranged between 2 and 6 μM (Sbarbati et al., 2020). These levels were significantly higher than concentrations in the AWT (0.01 μM), which had no As amended to the solution. An immediate reduction of ~99% of dissolved As concentration was observed within the GWT, with no re-release of As observed after the addition of 1mM lactate on day 12 (Figure 3). As concentrations in AWT also remained low following nitrate-Fe(II) amendment. In contrast, As concentrations in the unamended controls increased markedly within the first 3 days (e.g., from 0.085 μM to approximately 3.7μM for AWC), and remained elevated for the remainder of the experiment.
3.1.2.3. pH, Redox Potential and Alkalinity
All treatment microcosms showed a declining pH during the first 9 days, before returning to neutral conditions and finally becoming slightly alkaline after the addition of lactate at Day 12 (Figure 5). This trend was not evident in the controls, where generally stable pH levels persisted throughout the experiment. The treatments were slightly reducing with a redox potential (Eh) of −100 mV. Eh then increased sharply for both treatments and controls, from 0 mV on Day 9 to +200 mV by Day 12, consistent with Fe-controlled redox processes rather than denitrification. Initial alkalinity was 8.3 meq CaCO3 L−1 in GW, notably higher than the 0.2 meq CaCO3 L−1 measured in the AW microcosms. GWT alkalinity declined steeply by Day 3 to 1 meq CaCO3 L−1 before increasing to ~6.5 meq CaCO3 L−1 at Day 12 (Figure 5). The same increase occurred in AWT.
Figure 5.

Solution pH (top), alkalinity (middle) and calcium (bottom) concentrations in artificial water microcosms (left, circles) and site groundwater microcosms (right, triangles). Data presented is the average of the three E1 scenarios (Table S1). FeSO4 (10 mM) and NaNO3 (10mM) treatment results are shown in solid blue and comparative controls (no FeSO4, NaNO3 or lactate) in hollow red. Concentrations are reported in pH units, meq CaCO3 L−1 and mM, respectively. Grey shading indicates 1 mM lactate addition from Day 12 in the treatment microcosms only.
3.2. Additional Microcosms Assessing Ion Competition and Preferential Electron Donors and Acceptors
Nitrate-Fe(II) treatment on As mobility was evaluated for variable salinity and phosphate levels in AW. Additionally, nitrate-Fe(II) treatment effect was compared to a sulfate-lactate treatment to assess its performance.
The overall highest As release occurred in sulfate-lactate treatment microcosms. No significant difference in As release was observed between the lower and higher salinity microcosms of this treatment (Figure 6). Arsenic concentrations rose overall from 0.02 to 0.08 and 0.1 mM for AWT high salinity and AWT low salinity microcosms, respectively. Microcosms were observed to progressively change colour from clear to black (Figure 4). The measured redox conditions were mildly oxidising to reducing throughout the experiment (150 to −150 mV), and sulfate concentrations halved, decreasing from ~12 mM to ~7 mM in both AWT high and low salinity microcosms.
Figure 6.

Dissolved As (left) and Fe (right) concentrations in E.2 (Table S2) SO4-lactate (10 mM added from the start) treated microcosms of low salinity (5.3 mS cm−1) (solid stars) and high salinity (15.5 mS cm−1) (hollow triangles). Reported in mM.
Nitrate-Fe(II) treatments also showed no significant difference in dissolved Fe and As concentrations between lower (5.3 mS cm−1) and higher (15.5 mS cm−1) salinity AWTs (Figure 7). The 5 mM of amended Fe in these microcosms was almost completely removed from solution by Day 12. Dissolved As was <0.1 μM by Day 12 but remobilised upon 10 mM lactate addition, with concentrations increasing to 30 μM by Day 30 in both lower and higher salinity cases.
Figure 7.

Dissolved As (left) and Fe (right) concentrations from three E2 microcosms (Table S2) amended with FeSO4 (5 mM) and NaNO3 (10 mM). (a) E2.2 being low (1.5 mM) phosphate (1.5 mM) and low salinity (5.3 mS cm−1) (solid stars), (b) E2.3 being low phosphate (1.5 mM) and high salinity (15.5 mS cm−1) (hollow circles) and (c) E2.5 being high phosphate (4.5 mM) and low salinity (5.3 mS cm−1) (hollow triangles). Reported in mM. Grey shading indicates 10 mM lactate addition from Day 12.
The presence of phosphate decreased As retention, although this effect was moderated or reversed by the addition of Fe. Arsenic release was most pronounced (0.11 mM) in high phosphate (4.5 mM) microcosms lacking Fe amendment (Figure 7). In contrast, As release was halved (0.07 mM) with the Fe amendment, was more than 4 times lower (0.02 mM) with Fe amendment and lower phosphate (1.5 mM) and more than 4 orders of magnitude lower (0.03 μM) in microcosms with Fe amendment and no phosphate (Figure 3). No significant difference in dissolved Fe concentrations was observed across treatments. Post lactate addition, no re-release of As was observed in treatments without phosphate.
4. Discussion
4.1. As Immobilisation
Overall, the experimental results show that As was immobilised when (i) the treatment dose was sufficiently high (10mM Fe(II)) and (ii) the amount of reactive organic carbon was modest (1mM). Under these circumstances, As sequestration was achieved in both AW and GW treatments, with aqueous As concentrations decreasing by two to three orders of magnitude compared to the corresponding controls, to <0.1 μM. The enhanced As removal in the treatment microcosms is attributed to the transformation of Fe minerals into Fe sulfides and recalcitrant Fe mineral phases (such as magnetite), which are capable of incorporating As within their mineral structures (Coker et al., 2006; Huerta-Diaz and Morse, 1992; Langner et al., 2013; Sun et al., 2016a; Tufano and Fendorf, 2008) and via adsorption (Bostick and Fendorf, 2003; Farquhar et al., 2002; ThomasArrigo et al., 2016).
Importantly, the results also suggest that the addition of lactate did not destabilize any neo-formed As-Fe oxide assemblages within the treatments, as suggested by a lack of As and Fe release into solution, which would be indicative of Fe reduction. The neo-formed mineral phases consisted mostly of reduced Fe and crystalline phases, which are relatively resistant to destabilisation by lactate or other organic substrates. The absence of As release may point to the structural incorporation of As within crystalline minerals or incorporation into surface complexes rather than any type of more easily reversible sorption. While the general ability of the treatments to attenuate As was demonstrated by the substantial transformation of amorphous Fe oxides into more stable phases and the absence of As release, it should be noted that the 1 mM lactate amendment did not induce significant Fe(III) redution, likely due to an excess of other electron acceptors including nitrate and sulfate. Consumption of nitrate (decreased 0.8–2.5 mM) and sulfate (decreased 1–2 mM), see Figure S1, account, in principle, for 10 to 40% and 60 to 130% of the observed lactate oxidation, respectively, based on the stoichiometries for lactate oxidation coupled to nitrate reduction:
| (1) |
and lactate oxidation coupled to sulfate reduction:
| (2) |
Although nitrate reduction is expected, the comparatively large amount of sulfate reduction compared to Fe(III) reduction is unexpected as it does not adhere to the typical observed redox sequence.
Subsequent experiments tested the robustness of our treatment method further by (i) amending labile organic carbon in excess (i.e., 10 mM) and (ii) reducing the Fe(II) amendment from 10 mM to 5 mM. In these experiments the electron accepting capacity of nitrate was entirely consumed, oxidising approximately 20% of the lactate while sulfate reduction, by comparison, accounted for no more than 10% (~1 mM) of lactate oxidation, see Figure S2. We hypothesize that the higher nitrate to sulfate concentration ratio may have influenced the reduction processes. The surplus of lactate induced greater reducing conditions (Figure 8), which was accompanied by rising dissolved As concentrations (Figure 3) but did not increase dissolved Fe concentrations. Although sequential extractions were not conducted in these experiments, the lactate surplus was sufficient to reductively dissolve all amorphous phase Fe(III), which would be expected to cause spiking dissolved Fe and As (Chowdhury et al., 2011; Saalfield and Bostick, 2010). However, given that dissolved Fe concentrations did not increase, it appears, like in the 10 mM Fe treatments, that amorphous phases were no longer present. The release of As, therefore, may have resulted from As(V) desorption due topH values higher than 7.5 (Figure 5) (Dixit and Hering, 2003; Jain and Loeppert, 2000) or the reduction of As(V) to As(III), which desorbs more readily (Dixit and Hering, 2003; Giles et al., 2011; Herbel and Fendorf, 2006; Kocar et al., 2006; Meng et al., 2002).
Figure 8.

Solution Eh in artificial water microcosms (left, circles) and site groundwater microcosms (right, triangles). Data presented is the average of the three E1 scenarios (Table S1). FeSO4 (10 mM) and NaNO3 (10mM) treatment results are shown in solid blue and comparative controls (no FeSO4, NaNO3 or lactate) in hollow red. Concentrations are reported in mV. Grey shading indicates 1 mM lactate addition from Day 12.
4.2. Fe Mineral Host Phases for As
Prior to the treatments, As in the sediments was largely associated with amorphous Fe minerals, which are vulnerable to reductive dissolution under shifting redox conditions, and therefore are at risk of becoming a critical source of As (Muehe et al., 2013; Tufano and Fendorf, 2008). Yet, in our experiments, the overall oxidation state of Fe was relatively stable. Most mineralogical transformations appeared to involve the conversion of amorphous Fe(III) oxides to more crystalline Fe(III) phases. The observed loss of these amorphous phases would normally be expected to lead to a decrease in surface area, thereby causing a loss in sorption sites and thus an associated release of As into the aqueous phase (Dixit and Hering, 2003; Pedersen et al., 2006). However, As concentrations remained relatively low throughout the experiment, indicating that the new mineral phases contained sufficient sorption capacity to retain As. In fact, the fraction of As associated with crystalline Fe minerals increased considerably in both treatments (21%) and controls (75%), becoming the most important As hosting phase. The formation of the crystalline Fe minerals was likely driven by Fe(II)-catalysed recrystallisation of the amorphous Fe minerals (Handler et al., 2014; Hansel et al., 2005; Peiffer et al., 2021; Taylor et al., 2019; Yang et al., 2010). As the crystalline fraction increased in both the treatments and controls, the most likely source of Fe(II) was the dissolution of Fe(II) carbonates within the sediments, which were observed to significantly decrease in both treatment and control microcosms.
Further, As uptake occurred in association with recalcitrant phases including sulfides and magnetite. The quantity of As extracted in these fractions was twice as high in the treatment microcosms compared to the controls, consistent with the concomitant observed increase in the concentrations of Fe sulfide phases. Similarly, As associated with recalcitrant Fe phases increased in the treatment microcosms. Recalcitrant and sulfide phases, such as magnetite and arsenian or arsenopyrite, are preferred remedial hosts for As since it is incorporated within the crystal lattice and not prone to substitution by other anions (Huerta-Diaz and Morse, 1992; O’Day et al., 2004; Sun et al., 2016b). However, magnetite is the ideal targeted host given the greater susceptibility of Fe sulfides to changing redox conditions (Coker et al., 2006; Saalfield and Bostick, 2009; Sun et al., 2016b). Recalcitrant Fe minerals, under these conditions most likely prevailing as magnetite, form via topotactic or solid-state conversion of amorphous Fe minerals due to adsorption of Fe(II) (Hansel et al., 2015; Peiffer et al., 2021; Yang et al., 2010). The addition of Fe(II) within treatment microcosms has most likely enhanced its formation, along with pH rising to ~8.0, which would lead to a significantly faster rate of magnetite formation (Aeppli et al., 2019; Hansel et al., 2005). The increasing pH resulted from lactate mineralisation and concomitant calcite dissolution as evidenced by rising calcium concentrations and alkalinity (Figure 5). The formation of Fe sulfides within control microcosms suggests that sedimentary organic carbon or another, unidentified reductant was present within the sediments to facilitate sulfate reduction. Interestingly, neither sulfate nor nitrate concentrations decreased appreciably (Figure S1). The treatment experiments found 0.8 to 1.4 mmol Fe per kg of Fe sulfides to form in the sediments, which would require 0.08 to 0.14 mM of sulfate to be reduced from solution. This amount is negligible compared to the dissolved sulfate concentrations and consequently would not have been easily observable.
Iron oxide mineral formation in the nitrate-Fe(II) treatment experiments was less abundant than anticipated based on the amount of Fe(II) added. This may be due to a lack of available organic carbon substrates. Straub et al. (1996), among others, reported that the nitrate-dependent oxidation of Fe(II) to Fe(III) in brackish systems is not purely autotrophic and relies on the presence of organic substrates. Given that no exogenous OC was added before day 12 and DOC within the GW was only present at low concentrations while also being largely refractory (Sbarbati et al., 2020), the nitrate-Fe(II) reaction was likely inhibited. Given this limitation, it is likely that organic carbon should also be added to contaminated sediments low in carbon environments to enhance mineralisation.
4.3. As retention in different treatment environments
Aqueous As concentrations were highest in the sulfate treatments where they were actively released from sediments, peaking at concentrations that were four times above the Fe(II) treatment microcosms and well above water quality standards. Sulfate was rapidly consumed to below half of its initial concentration within these experiments and the microcosms turned black, clearly indicating Fe sulfide precipitation (Burton et al., 2011; Saalfield and Bostick, 2009). It is possible realgar also precipitated, albeit to a limited extent, given the measured Eh (150 to −150 mV) and pH (6.5 to 8.5) in these microcosms largely exceeded conditions for realgar stability (Langner et al., 2013; Smedley and Kinniburgh, 2002). The hydrogen sulfide that was generated during sulfate reduction may have caused the reductive dissolution of Fe-oxides and precipitation of mackinawite:
| (3) |
and/or pyrite:
| (4) |
Arsenic could, in principle, be mobilised in association with declining Fe-oxide concentrations, as the sorption affinity of As on ferrihydrite is 3 to 6 orders of magnitude greater than that of mackinawite (Burton et al., 2011; Smedley and Kinniburgh, 2002; Wolthers et al., 2005). However, the alkaline pH and sub-oxic conditions were also favourable for the mobilisation of As through the formation of thioarsenic species, which could have promoted enhanced As partitioning into the dissolved phase (Suess and Planer-Friedrich, 2012).
Sulfidic conditions have previously been shown to be conducive to the development of magnetite if there is abundant Fe(II) (Yang et al., 2010). In our case, no Fe(II) addition was made to the sulfate treatments, causing the sulfate-lactate microcosms to develop sulfidic conditions that mobilize As. Thus, in-situ immobilisation of As through sulfate amendments does not appear to be a viable strategy in this environment.
Salinity variations among the microcosms did not significantly affect As mobilisation. This indicates minimal competition of As with other anions for sorption sites as well as minimal effects on mineral transformation and As coprecipitation. This may have resulted from As(III)/As(V) forming strong inner-sphere complexes with Fe oxides and Fe sulfides (Huerta-Diaz and Morse, 1992; Tufano and Fendorf, 2008), which would not be easily displaced by weakly adsorbing anions. Our results are consistent with those of Meng et al. (2002), who also reported direct competition from chloride to be largely negligible.
In contrast to salinity variations, phosphate had a significant impact on As mobilisation. The effect of phosphate competition on As retention has been well documented (Dixit and Hering, 2003; Hug et al., 2008; Meng et al., 2002; Voegelin et al., 2019, 2010), with phosphate commonly outcompeting As for sorption sites. Increased As release was concurrent with rising bicarbonate concentrations, resulting from lactate mineralisation, which may have further enhanced the adverse influence of phosphate. Meng et al. (2002) reported such magnification of phosphate competition in combination with increased concentration of anions such as bicarbonate.
5. Conclusions
In our laboratory-based experiments, As was successfully removed from water as a result of Fe mineral (trans)formations that were largely stimulated by the addition of Fe(II). Our study was aimed to facilitate the direct formation of recalcitrant Fe phases, specifically magnetite. Magnetite, however, formed only to a limited extent, most likely due to the fast precipitation of Fe(II) sulfides, which too rapidly removed the supply of Fe(II) required for direct Fe-oxide transformation. Nitrate-dependent Fe-oxidation failed to occur, despite the abundance of nitrate as oxidant, possibly due to limited organic carbon availability which is usually necessary as a co-substrate. Instead, magnetite formed through the direct Fe(II)-catalysed transformation of pre-existing amorphous and crystalline minerals, whereby the amount that was transformed was limited by the exhaustion of dissolved Fe(II). Thus, in our study the joint addition of nitrate and Fe(II) appeared to be superfluous to the achieved remedial efforts and the quantity of amended Fe(II) proved to be the defining factor for the generation of recalcitrant Fe minerals and stable As immobilisation.
While As could be released in microcosms using insufficient Fe(II) (i.e., 5 mM amendments), most likely due to a residual association with remaining amorphous Fe minerals susceptible to reduction, the higher Fe(II) amendment (10 mM) induced a more complete transformation of the amorphous Fe phases and more stable retention in the solid phase during the reductive step. Importantly, the (trans)formation from amorphous to crystalline phases did not induce any observable release of As which is promising for field remediation application. The addition of labile organic carbon (lactate) as a reductant had no effect on As stability in the case of the higher Fe(II) amendments, most likely because a structural incorporation of As was achieved. Conversely, a decreased Fe(II) amendment resulted in less reliably sequestered As, most likely due to a less complete Fe mineral (trans)formation and lower levels of structural incorporation. Moreover, in contrast to previous studies that investigated chloride and sulfate effects on Fe mineral formation pathways (e.g., Hansel et al., 2005), our work did not show any considerable impact. This means that potentially there is no diminished remedial efficiency to be expected for in situ application in coastal and other saline aquifers.
Microcosm studies such as this represent the first step in developing appropriate remediation plans for contaminated sites. Understanding (and predicting) As concentrations and transport requires quantitative interpretation of the data. A crucial next step is the development of a process-based biogeochemical model that is capable of describing and quantifying the mineral (trans)formations and associated As behaviour that were observed in the present experimental study (Siade et al., 2021). This will allow the design of amendments that can maximise the generation of the target Fe phases under widely variable conditions that are common at contaminated sites. Models based on these data are thus vital to developing remediation approaches that are effective and sustainable.
Supplementary Material
Lab-scale experiments demonstrated feasibility of in situ As remediation
Fe(II) amendment stimulated formation of more recalcitrant Fe minerals
Nitrate-dependent Fe-oxidation presumably relied on organic carbon as co-substrate
Remedial efficiency was not deteriorated by increased salinity
Elevated phosphate concentrations lowered remediation efficacy
6. Acknowledgements
This paper refers to the research project titled “Proposal for lowering Arsenic concentration in groundwater: lab experiments, simulation models and field pilot test”, coordinated by the Earth Science Department of Sapienza University of Rome.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Appendix A Supplementary material
Microcosm conditions, artificial water constituents, the sequential extraction procedure and nitrate and sulfate graphics can be found in the Supporting Information.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
8 References
- Aeppli M, Vranic S, Kaegi R, Kretzschmar R, Brown AR, Voegelin A, Hofstetter TB, Sander M, 2019. Decreases in Iron Oxide Reducibility during Microbial Reductive Dissolution and Transformation of Ferrihydrite. Environ. Sci. Technol 53, 8736–8746. 10.1021/acs.est.9b01299 [DOI] [PubMed] [Google Scholar]
- Akin I, Arslan G, Tor A, Ersoz M, Cengeloglu Y, 2012. Arsenic(V) removal from underground water by magnetic nanoparticles synthesized from waste red mud. J. Hazard. Mater 235–236, 62–68. 10.1016/j.jhazmat.2012.06.024 [DOI] [PubMed] [Google Scholar]
- APHA, A.P.H.A., 1995. Standard Methods For the Examination of Water and Wastewater, 23nd edition, Standard Methods For the Examination of Water and Wastewater, 19th edition. New York. 10.2105/SMWW.2882.023 [DOI] [Google Scholar]
- ATSDR, A. for T.S. and D.R., 2019. Substance Priority List Resource Page | ATSDR. [Google Scholar]
- Beak DG, Wilkin RT, 2009. Performance of a zerovalent iron reactive barrier for the treatment of arsenic in groundwater: Part 2. Geochemical modeling and solid phase studies. J. Contam. Hydrol 106, 15–28. 10.1016/j.jconhyd.2008.12.003 [DOI] [PubMed] [Google Scholar]
- Beaulieu B, Ramirez RE, 2013. Arsenic Remediation Field Study Using a Sulfate Reduction and Zero-Valent Iron PRB. Groundw. Monit. Remediat 33, 85–94. 10.1111/gwmr.12007 [DOI] [Google Scholar]
- Benner SG, Hansel CM, Wielinga BW, Barber TM, Fendorf S, 2002. Reductive Dissolution and Biomineralization of Iron Hydroxide under Dynamic Flow Conditions. Environ. Sci. Technol 36, 1705–1711. 10.1021/es0156441 [DOI] [PubMed] [Google Scholar]
- Bonte M, van Breukelen BM, Stuyfzand PJ, 2013. Temperature-induced impacts on groundwater quality and arsenic mobility in anoxic aquifer sediments used for both drinking water and shallow geothermal energy production. Water Res. 47, 5088–5100. 10.1016/j.watres.2013.05.049 [DOI] [PubMed] [Google Scholar]
- Bostick BC, Fendorf S, 2003. Arsenite sorption on troilite (FeS) and pyrite (FeS2). Geochim. Cosmochim. Acta 67, 909–921. 10.1016/S0016-7037(02)01170-5 [DOI] [Google Scholar]
- Bujňáková Z, Baláž P, Zorkovská A, Sayagués MJ, Kováč J, Timko M, 2013. Arsenic sorption by nanocrystalline magnetite: An example of environmentally promising interface with geosphere. J. Hazard. Mater 262, 1204–1212. 10.1016/j.jhazmat.2013.03.007 [DOI] [PubMed] [Google Scholar]
- Burton ED, Johnston SG, Bush RT, 2011. Microbial sulfidogenesis in ferrihydrite-rich environments: Effects on iron mineralogy and arsenic mobility. Geochim. Cosmochim. Acta 75, 3072–3087. 10.1016/j.gca.2011.03.001 [DOI] [Google Scholar]
- Burton ED, Johnston SG, Planer-Friedrich B, 2013. Coupling of arsenic mobility to sulfur transformations during microbial sulfate reduction in the presence and absence of humic acid. Chem. Geol 343, 12–24. 10.1016/j.chemgeo.2013.02.005 [DOI] [Google Scholar]
- Cañas Kurz EE, Luong VT, Hellriegel U, Leidinger F, Luu TL, Bundschuh J, Hoinkis J, 2020. Iron-based subsurface arsenic removal (SAR): Results of a long-term pilot-scale test in Vietnam. Water Res. 181, 115929. 10.1016/j.watres.2020.115929 [DOI] [PubMed] [Google Scholar]
- Cantu J, Gonzalez LE, Goodship J, Contreras M, Joseph M, Garza C, Eubanks TM, Parsons JG, 2016. Removal of arsenic from water using synthetic Fe7S8 nanoparticles. Chem. Eng. J 290, 428–437. 10.1016/j.cej.2016.01.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury SR, Yanful EK, Pratt AR, 2011. Arsenic removal from aqueous solutions by mixed magnetite–maghemite nanoparticles. Environ. Earth Sci 64, 411–423. 10.1007/s12665-010-0865-z [DOI] [Google Scholar]
- Coker VS, Gault AG, Pearce CI, van der Laan G, Telling ND, Charnock JM, Polya DA, Lloyd JR, 2006. XAS and XMCD evidence for species-dependent partitioning of arsenic during microbial reduction of ferrihydrite to magnetite. Environ. Sci. Technol 40, 7745–50. 10.1021/es060990+ [DOI] [PubMed] [Google Scholar]
- Colombani N, Mastrocicco M, Prommer H, Sbarbati C, Petitta M, 2015. Fate of arsenic, phosphate and ammonium plumes in a coastal aquifer affected by saltwater intrusion. J. Contam. Hydrol 179, 116–131. 10.1016/j.jconhyd.2015.06.003 [DOI] [PubMed] [Google Scholar]
- Deditius AP, Reich M, Kesler SE, Utsunomiya S, Chryssoulis SL, Walshe J, Ewing RC, 2014. The coupled geochemistry of Au and As in pyrite from hydrothermal ore deposits. Geochim. Cosmochim. Acta 140, 644–670. 10.1016/j.gca.2014.05.045 [DOI] [Google Scholar]
- Dixit S, Hering JG, 2003. Comparison of Arsenic(V) and Arsenic(III) Sorption onto Iron Oxide Minerals: Implications for Arsenic Mobility. Environ. Sci. Technol 37, 4182–4189. 10.1021/es030309t [DOI] [PubMed] [Google Scholar]
- Edenborn HM, Belzile N, Mucci A, Lebel J, Silverberg N, 1986. Observations on the diagenetic behavior of arsenic in a deep coastal sediment. Biogeochemistry 2, 359–376. 10.1007/BF02180326 [DOI] [Google Scholar]
- Fakhreddine S, Prommer H, Gorelick SM, Dadakis J, Fendorf S, 2020. Controlling Arsenic Mobilization during Managed Aquifer Recharge: The Role of Sediment Heterogeneity. Environ. Sci. Technol 54, 8728–8738. 10.1021/acs.est.0c00794 [DOI] [PubMed] [Google Scholar]
- Farquhar ML, Charnock JM, Livens FR, Vaughan DJ, 2002. Mechanisms of Arsenic Uptake from Aqueous Solution by Interaction with Goethite, Lepidocrocite, Mackinawite, and Pyrite: An X-ray Absorption Spectroscopy Study. Environ. Sci. Technol 36, 1757–1762. 10.1021/es010216g [DOI] [PubMed] [Google Scholar]
- Farrell JW, Fortner J, Work S, Avendano C, Gonzalez-Pech NI, Zárate Araiza R, Li Q, Álvarez PJJ, Colvin V, Kan A, Tomson M, 2014. Arsenic Removal by Nanoscale Magnetite in Guanajuato, Mexico. Environ. Eng. Sci 31, 393–402. 10.1089/ees.2013.0425 [DOI] [Google Scholar]
- Fendorf S, Michael HA, van Geen A, 2010. Spatial and Temporal Variations of Groundwater Arsenic in South and Southeast Asia. Science (80-.). 328, 1123–1127. 10.1126/science.1172974 [DOI] [PubMed] [Google Scholar]
- Giles DE, Mohapatra M, Issa TB, Anand S, Singh P, 2011. Iron and aluminium based adsorption strategies for removing arsenic from water. J. Environ. Manage 92, 3011–3022. 10.1016/j.jenvman.2011.07.018 [DOI] [PubMed] [Google Scholar]
- Giménez J, Martínez M, de Pablo J, Rovira M, Duro L, 2007. Arsenic sorption onto natural hematite, magnetite, and goethite. J. Hazard. Mater 141, 575–80. 10.1016/j.jhazmat.2006.07.020 [DOI] [PubMed] [Google Scholar]
- Handler RM, Frierdich AJ, Johnson CM, Rosso KM, Beard BL, Wang C, Latta DE, Neumann A, Pasakarnis T, Premaratne WAPJ, Scherer MM, 2014. Fe(II)-Catalyzed Recrystallization of Goethite Revisited. Environ. Sci. Technol 48, 11302–11311. 10.1021/es503084u [DOI] [PubMed] [Google Scholar]
- Hansel CM, Benner SG, Fendorf S, 2005. Competing Fe(II)-Induced Mineralization Pathways of Ferrihydrite. Environ. Sci. Technol 39, 7147–7153. 10.1021/es050666z [DOI] [PubMed] [Google Scholar]
- Hansel CM, Lentini CJ, Tang Y, Johnston DT, Wankel SD, Jardine PM, 2015. Dominance of sulfur-fueled iron oxide reduction in low-sulfate freshwater sediments. ISME J. 9, 2400–2412. 10.1038/ismej.2015.50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbel M, Fendorf S, 2006. Biogeochemical processes controlling the speciation and transport of arsenic within iron coated sands. Chem. Geol 228, 16–32. 10.1016/j.chemgeo.2005.11.016 [DOI] [Google Scholar]
- Howe P, Hughes M, Kenyon E, Lewis DR, Moore M, Ng J, Aitio A, Becking G, 2001. Environmental Health Criteria 224 Arsenic and Asenic Compounds, Second edition, World Health Organization Geneva. [Google Scholar]
- Huerta-Diaz MA, Morse JW, 1992. Pyritization of trace metals in anoxic marine sediments. Geochim. Cosmochim. Acta 56, 2681–2702. 10.1016/0016-7037(92)90353-K [DOI] [Google Scholar]
- Hug SJ, Leupin OX, Berg M, 2008. Bangladesh and Vietnam: Different Groundwater Compositions Require Different Approaches to Arsenic Mitigation. Environ. Sci. Technol 42, 6318–6323. 10.1021/es7028284 [DOI] [PubMed] [Google Scholar]
- Jain A, Loeppert RH, 2000. Effect of Competing Anions on the Adsorption of Arsenate and Arsenite by Ferrihydrite. J. Environ. Qual 29, 1422–1430. 10.2134/jeq2000.00472425002900050008x [DOI] [Google Scholar]
- Johnson CM, Roden EE, Welch SA, Beard BL, 2005. Experimental constraints on Fe isotope fractionation during magnetite and Fe carbonate formation coupled to dissimilatory hydrous ferric oxide reduction. Elsevier. 10.1016/j.gca.2004.06.043 [DOI] [Google Scholar]
- Jönsson J, Sherman DM, 2008. Sorption of As(III) and As(V) to siderite, green rust (fougerite) and magnetite: Implications for arsenic release in anoxic groundwaters. Chem. Geol 255, 173–181. 10.1016/j.chemgeo.2008.06.036 [DOI] [Google Scholar]
- Kanel SR, Manning B, Charlet L, Choi H, 2005. Removal of Arsenic(III) from Groundwater by Nanoscale Zero-Valent Iron. Environ. Sci. Technol 39, 1291–1298. 10.1021/es048991u [DOI] [PubMed] [Google Scholar]
- Keimowitz AR, Mailloux BJ, Cole P, Stute M, Simpson HJ, Chillrud SN, 2007. Laboratory Investigations of Enhanced Sulfate Reduction as a Groundwater Arsenic Remediation Strategy. Environ. Sci. Technol 41, 6718–6724. 10.1021/es061957q [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneebone PE, O’Day PA, Jones N, Hering JG, 2002. Deposition and Fate of Arsenic in Iron- and Arsenic-Enriched Reservoir Sediments. Environ. Sci. Technol 36, 381–386. 10.1021/es010922h [DOI] [PubMed] [Google Scholar]
- Kocar BD, Herbel MJ, Tufano KJ, Fendorf S, 2006. Contrasting Effects of Dissimilatory Iron(III) and Arsenic(V) Reduction on Arsenic Retention and Transport. Environ. Sci. Technol 40, 6715–6721. 10.1021/es061540k [DOI] [PubMed] [Google Scholar]
- Langner P, Mikutta C, Suess E, Marcus MA, Kretzschmar R, 2013. Spatial Distribution and Speciation of Arsenic in Peat Studied with Microfocused X-ray Fluorescence Spectrometry and X-ray Absorption Spectroscopy. Environ. Sci. Technol 47, 9706–9714. 10.1021/es401315e [DOI] [PubMed] [Google Scholar]
- Lei L, Zhang G, Lin J, Wang X, Wang S, Jia Y, 2018. Co-adsorption of arsenite and arsenate on mixed-valence Fe(II,III) (hydr)oxides under reducing conditions. Appl. Geochemistry 98, 418–425. 10.1016/j.apgeochem.2018.10.015 [DOI] [Google Scholar]
- Lien H-L, Wilkin RT, 2005. High-level arsenite removal from groundwater by zero-valent iron. Chemosphere 59, 377–386. 10.1016/j.chemosphere.2004.10.055 [DOI] [PubMed] [Google Scholar]
- Liu T, Chen D, Luo X, Li X, Li F, 2019. Microbially mediated nitrate-reducing Fe(II) oxidation: Quantification of chemodenitrification and biological reactions. Geochim. Cosmochim. Acta 256, 97–115. 10.1016/j.gca.2018.06.040 [DOI] [Google Scholar]
- Lovley DR, Phillips EJP, 1986. Availability of Ferric Iron for Microbial Reduction in Bottom Sediments of the Freshwater Tidal Potomac River. Appl. Environ. Microbiol 52, 751–757. 10.1128/AEM.52.4.751-757.1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luther S, Borgfeld N, Kim J, Parsons J, 2012. Removal of arsenic from aqueous solution: A study of the effects of pH and interfering ions using iron oxide nanomaterials. Microchem. J 101, 30–36. 10.1016/j.microc.2011.10.001 [DOI] [Google Scholar]
- Mamindy-Pajany Y, Hurel C, Marmier N, Roméo M, 2011. Arsenic (V) adsorption from aqueous solution onto goethite, hematite, magnetite and zero-valent iron: Effects of pH, concentration and reversibility. Desalination 281, 93–99. 10.1016/j.desal.2011.07.046 [DOI] [Google Scholar]
- Mastrocicco M, Colombani N, Sbarbati C, Petitta M, 2012. Assessing the Effect of Saltwater Intrusion on Petroleum Hydrocarbons Plumes Via Numerical Modelling. Water, Air, Soil Pollut 223, 4417–4427. 10.1007/s11270-012-1205-6 [DOI] [Google Scholar]
- Meng X, Korfiatis GP, Bang S, Woong Bang K, Bang KW, 2002. Combined effects of anions on arsenic removal by iron hydroxides. Toxicol. Lett 133, 103–111. 10.1016/S0378-4274(02)00080-2 [DOI] [PubMed] [Google Scholar]
- Missimer T, Teaf C, Beeson W, Maliva R, Woolschlager J, Covert D, 2018. Natural Background and Anthropogenic Arsenic Enrichment in Florida Soils, Surface Water, and Groundwater: A Review with a Discussion on Public Health Risk. Int. J. Environ. Res. Public Health 15, 2278. 10.3390/ijerph15102278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli G, Rimondi V, Benvenuti M, Medas D, Costagliola P, Gasparon M, 2017. Experimental simulation of arsenic desorption from Quaternary aquifer sediments following sea water intrusion. Appl. Geochemistry 87, 176–187. 10.1016/j.apgeochem.2017.10.024 [DOI] [Google Scholar]
- Morin G, Calas G, 2006. Arsenic in Soils, Mine Tailings, and Former Industrial Sites. Elements 2, 97–101. 10.2113/gselements.2.2.97 [DOI] [Google Scholar]
- Muehe EM, Scheer L, Daus B, Kappler A, 2013. Fate of Arsenic during Microbial Reduction of Biogenic versus Abiogenic As–Fe(III)–Mineral Coprecipitates. Environ. Sci. Technol 47, 130711140829002. 10.1021/es400801z [DOI] [PubMed] [Google Scholar]
- O’Day PA, Vlassopoulos D, Root R, Rivera N, 2004. The influence of sulfur and iron on dissolved arsenic concentrations in the shallow subsurface under changing redox conditions. Proc. Natl. Acad. Sci 101, 13703–13708. 10.1073/pnas.0402775101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omoregie EO, Couture R-M, Van Cappellen P, Corkhill CL, Charnock JM, Polya DA, Vaughan D, Vanbroekhoven K, Lloyd JR, 2013. Arsenic Bioremediation by Biogenic Iron Oxides and Sulfides. Appl. Environ. Microbiol 79, 4325–4335. 10.1128/AEM.00683-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen HD, Postma D, Jakobsen R, 2006. Release of arsenic associated with the reduction and transformation of iron oxides. Geochim. Cosmochim. Acta 70, 4116–4129. 10.1016/j.gca.2006.06.1370 [DOI] [Google Scholar]
- Peiffer S, Kappler A, Haderlein SB, Schmidt C, Byrne JM, Kleindienst S, Vogt C, Richnow HH, Obst M, Angenent LT, Bryce C, McCammon C, Planer-Friedrich B, 2021. A biogeochemical–hydrological framework for the role of redox-active compounds in aquatic systems. Nat. Geosci 14, 264–272. 10.1038/s41561-021-00742-z [DOI] [Google Scholar]
- Planer-Friedrich B, London J, McCleskey RB, Nordstrom DK, Wallschläger D, 2007. Thioarsenates in Geothermal Waters of Yellowstone National Park: Determination, Preservation, and Geochemical Importance. Environ. Sci. Technol 41, 5245–5251. 10.1021/es070273v [DOI] [PubMed] [Google Scholar]
- Podgorski J, Berg M, 2020. Global threat of arsenic in groundwater. Science (80-.). 368, 845–850. 10.1126/science.aba1510 [DOI] [PubMed] [Google Scholar]
- Prommer H, Sun J, Helm L, Rathi B, Siade AJ, Morris R, 2018. Deoxygenation Prevents Arsenic Mobilization during Deepwell Injection into Sulfide-Bearing Aquifers. Environ. Sci. Technol 52, 13801–13810. 10.1021/acs.est.8b05015 [DOI] [PubMed] [Google Scholar]
- Qiu G, Gao T, Hong J, Tan W, Liu F, Zheng L, 2017. Mechanisms of arsenic-containing pyrite oxidation by aqueous arsenate under anoxic conditions. Geochim. Cosmochim. Acta 217, 306–319. 10.1016/j.gca.2017.08.030 [DOI] [Google Scholar]
- Rathi B, Neidhardt H, Berg M, Siade A, Prommer H, 2017. Processes governing arsenic retardation on Pleistocene sediments: Adsorption experiments and model-based analysis. Water Resour. Res 53, 4344–4360. 10.1002/2017WR020551 [DOI] [Google Scholar]
- Rawson J, Prommer H, Siade A, Carr J, Berg M, Davis JA, Fendorf S, 2016. Numerical Modeling of Arsenic Mobility during Reductive Iron-Mineral Transformations. Environ. Sci. Technol 50, 2459–2467. 10.1021/acs.est.5b05956 [DOI] [PubMed] [Google Scholar]
- Revesz E, Fortin D, Paktunc D, 2016. Reductive dissolution of arsenical ferrihydrite by bacteria. Appl. Geochemistry 66, 129–139. 10.1016/j.apgeochem.2015.12.007 [DOI] [Google Scholar]
- Root RA, Vlassopoulos D, Rivera NA, Rafferty MT, Andrews C, O’Day PA, 2009. Speciation and natural attenuation of arsenic and iron in a tidally influenced shallow aquifer. Geochim. Cosmochim. Acta 73, 5528–5553. 10.1016/j.gca.2009.06.025 [DOI] [Google Scholar]
- Saalfield SL, Bostick BC, 2010. Synergistic effect of calcium and bicarbonate in enhancing arsenate release from ferrihydrite. Geochim. Cosmochim. Acta 74, 5171–5186. 10.1016/j.gca.2010.05.022 [DOI] [Google Scholar]
- Saalfield SL, Bostick BC, 2009. Changes in Iron, Sulfur, and Arsenic Speciation Associated with Bacterial Sulfate Reduction in Ferrihydrite-Rich Systems. Environ. Sci. Technol 43, 8787–8793. 10.1021/es901651k [DOI] [PubMed] [Google Scholar]
- Sasaki K, Nakano H, Wilopo W, Miura Y, Hirajima T, 2009. Sorption and speciation of arsenic by zero-valent iron. Colloids Surfaces A Physicochem. Eng. Asp 347, 8–17. 10.1016/j.colsurfa.2008.10.033 [DOI] [Google Scholar]
- Saunders JA, Lee M-K, Shamsudduha M, Dhakal P, Uddin A, Chowdury MT, Ahmed KM, 2008. Geochemistry and mineralogy of arsenic in (natural) anaerobic groundwaters. Appl. Geochemistry 23, 3205–3214. 10.1016/j.apgeochem.2008.07.002 [DOI] [Google Scholar]
- Sbarbati C, Barbieri M, Barron A, Bostick B, Colombani N, Mastrocicco M, Prommer H, Passaretti S, Zheng Y, Petitta M, 2020. Redox Dependent Arsenic Occurrence and Partitioning in an Industrial Coastal Aquifer: Evidence from High Spatial Resolution Characterization of Groundwater and Sediments. Water 12, 2932. 10.3390/w12102932 [DOI] [Google Scholar]
- Schwertmann U, Cornell RM, 2000. Iron Oxides in the Laboratory. New York. [Google Scholar]
- Sharma VK, Sohn M, 2009. Aquatic arsenic: Toxicity, speciation, transformations, and remediation. Environ. Int 35, 743–759. 10.1016/j.envint.2009.01.005 [DOI] [PubMed] [Google Scholar]
- Shipley HJ, Yean S, Kan AT, Tomson MB, 2009. Adsorption of Arsenic to Magnetite Nanoparticles: Effect of Particle Concentration, pH, Ionic Strength, and Temperature. Environ. Toxicol. Chem 28, 509. 10.1897/08-155.1 [DOI] [PubMed] [Google Scholar]
- Siade AJ, Bostick BC, Cirpka OA, Prommer H, 2021. Unraveling biogeochemical complexity through better integration of experiments and modeling. Environ. Sci. Process. Impacts 10.1039/D1EM00303H [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedley P., Kinniburgh D., 2002. A review of the source, behaviour and distribution of arsenic in natural waters. Appl. Geochemistry 17, 517–568. 10.1016/S0883-2927(02)00018-5 [DOI] [Google Scholar]
- Straub KL, Benz M, Schink B, Widdel F, 1996. Anaerobic, nitrate-dependent microbial oxidation of ferrous iron. Appl. Environ. Microbiol 62, 1458–1460. 10.1128/aem.62.4.1458-1460.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su C, Puls RW, 2001. Arsenate and Arsenite Removal by Zerovalent Iron: Kinetics, Redox Transformation, and Implications for in Situ Groundwater Remediation. Environ. Sci. Technol 35, 1487–1492. 10.1021/es001607i [DOI] [PubMed] [Google Scholar]
- Suess E, Planer-Friedrich B, 2012. Thioarsenate formation upon dissolution of orpiment and arsenopyrite. Chemosphere 89, 1390–1398. 10.1016/j.chemosphere.2012.05.109 [DOI] [PubMed] [Google Scholar]
- SUN H, WANG L, ZHANG R, SUI J, XU G, 2006. Treatment of groundwater polluted by arsenic compounds by zero valent iron. J. Hazard. Mater 129, 297–303. 10.1016/j.jhazmat.2005.08.026 [DOI] [PubMed] [Google Scholar]
- Sun J, Chillrud SN, Mailloux BJ, Bostick BC, 2016a. In Situ Magnetite Formation and Long-Term Arsenic Immobilization under Advective Flow Conditions. Environ. Sci. Technol 50, 10162–10171. 10.1021/acs.est.6b02362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Chillrud SN, Mailloux BJ, Stute M, Singh R, Dong H, Lepre CJ, Bostick BC, 2016b. Enhanced and stabilized arsenic retention in microcosms through the microbial oxidation of ferrous iron by nitrate. Chemosphere 144, 1106–1115. 10.1016/j.chemosphere.2015.09.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Prommer H, Siade AJ, Chillrud SN, Mailloux BJ, Bostick BC, 2018. Model-Based Analysis of Arsenic Immobilization via Iron Mineral Transformation under Advective Flows. Environ. Sci. Technol 52, 9243–9253. 10.1021/acs.est.8b01762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Quicksall AN, Chillrud SN, Mailloux BJ, Bostick BC, 2016c. Arsenic mobilization from sediments in microcosms under sulfate reduction. Chemosphere 153, 254–261. 10.1016/j.chemosphere.2016.02.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SD, Liu J, Zhang X, Arey BW, Kovarik L, Schreiber DK, Perea DE, Rosso KM, 2019. Visualizing the iron atom exchange front in the Fe(II)-catalyzed recrystallization of goethite by atom probe tomography. Proc. Natl. Acad. Sci 116, 2866–2874. 10.1073/pnas.1816620116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teclu D, Tivchev G, Laing M, Wallis M, 2008. Bioremoval of arsenic species from contaminated waters by sulphate-reducing bacteria. Water Res. 42, 4885–4893. 10.1016/j.watres.2008.09.010 [DOI] [PubMed] [Google Scholar]
- ThomasArrigo LK, Mikutta C, Lohmayer R, Planer-Friedrich B, Kretzschmar R, 2016. Sulfidization of Organic Freshwater Flocs from a Minerotrophic Peatland: Speciation Changes of Iron, Sulfur, and Arsenic. Environ. Sci. Technol 50, 3607–3616. 10.1021/acs.est.5b05791 [DOI] [PubMed] [Google Scholar]
- Tufano KJ, Fendorf S, 2008. Confounding Impacts of Iron Reduction on Arsenic Retention. Environ. Sci. Technol 42, 4777–4783. 10.1021/es702625e [DOI] [PubMed] [Google Scholar]
- Tufano KJ, Reyes C, Saltikov CW, Fendorf S, 2008. Reductive Processes Controlling Arsenic Retention: Revealing the Relative Importance of Iron and Arsenic Reduction. Environ. Sci. Technol 42, 8283–8289. 10.1021/es801059s [DOI] [PubMed] [Google Scholar]
- USEPA, U.E.P.A., 1996. EPA Method 3050B: Acid Digestion of Sediments, Sludges, and Soils: Revision 2. Washington, DC. [Google Scholar]
- USEPA, U.S., 2002. Arsenic treatment technologies for solid, waste, and water. USEPA Rep. EPA-542-R-02–004. [Google Scholar]
- Van Deuren J, Lloyd T, Chhetry S, Liou R, Peck J, 2002. Remediation Technologies Screening Matrix and Reference Guide, 4th Edition. FRTR. [Google Scholar]
- Voegelin A, Kaegi R, Frommer J, Vantelon D, Hug SJ, 2010. Effect of phosphate, silicate, and Ca on Fe(III)-precipitates formed in aerated Fe(II)- and As(III)-containing water studied by X-ray absorption spectroscopy. Geochim. Cosmochim. Acta 74, 164–186. 10.1016/j.gca.2009.09.020 [DOI] [Google Scholar]
- Voegelin A, Senn A-C, Kaegi R, Hug SJ, 2019. Reductive dissolution of As(V)-bearing Fe(III)-precipitates formed by Fe(II) oxidation in aqueous solutions. Geochem. Trans 20, 2. 10.1186/s12932-019-0062-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Kerl CF, Hu P, Martin M, Mu T, Brüggenwirth L, Wu G, Said-Pullicino D, Romani M, Wu L, Planer-Friedrich B, 2020. Thiolated arsenic species observed in rice paddy pore waters. Nat. Geosci 13, 282–287. 10.1038/s41561-020-0533-1 [DOI] [Google Scholar]
- Wolthers M, Charlet L, van Der Weijden CH, van der Linde PR, Rickard D, 2005. Arsenic mobility in the ambient sulfidic environment: Sorption of arsenic(V) and arsenic(III) onto disordered mackinawite. Geochim. Cosmochim. Acta 69, 3483–3492. 10.1016/j.gca.2005.03.003 [DOI] [Google Scholar]
- Yang L, Steefel CI, Marcus MA, Bargar JR, 2010. Kinetics of Fe(II)-Catalyzed Transformation of 6-line Ferrihydrite under Anaerobic Flow Conditions. Environ. Sci. Technol 44, 5469–5475. 10.1021/es1007565 [DOI] [PubMed] [Google Scholar]
- Yavuz CT, Mayo JT, Yu WW, Prakash A, Falkner JC, Yean S, Cong L, Shipley HJ, Kan A, Tomson M, Natelson D, Colvin VL, 2006. Low-Field Magnetic Separation of Monodisperse Fe3O4 Nanocrystals. Science (80-.). 314, 964–967. 10.1126/science.1131475 [DOI] [PubMed] [Google Scholar]
- Yuan P, Fan M, Yang D, He H, Liu D, Yuan A, Zhu JX, Chen TH, 2009. Montmorillonite-supported magnetite nanoparticles for the removal of hexavalent chromium [Cr(VI)] from aqueous solutions. J. Hazard. Mater 166, 821–829. 10.1016/j.jhazmat.2008.11.083 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.