

SUMMARY
The endoplasmic reticulum (ER) depends on extensive association with the microtubule (MT) cytoskeleton for its structure and mitotic inheritance. However, mechanisms that underlie coupling of ER membranes to MTs are poorly understood. We have identified thousand and one amino acid kinase 2 (TAOK2) as a pleiotropic protein kinase that mediates tethering of ER to MTs. In human cells, TAOK2 localizes in distinct ER subdomains via transmembrane helices and an adjacent amphipathic region. Through its C-terminal tail, TAOK2 directly binds MTs, coupling ER membranes to the MT cytoskeleton. In TAOK2 knockout cells, although ER-membrane dynamics are increased, movement of ER along growing MT plus ends is disrupted. ER-MT tethering is tightly regulated by catalytic activity of TAOK2, perturbation of which leads to defects in ER morphology, association with MTs, and cell division. Our study identifies TAOK2 as an ER-MT tether and reveals a kinase-regulated mechanism for control of ER dynamics.
Graphical abstract
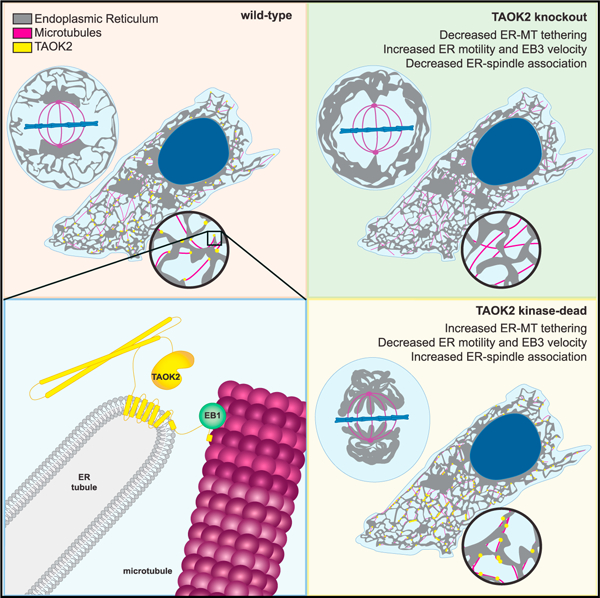
In brief
Nourbakhsh, Ferreccio, et al. reveal that the protein kinase TAOK2 is a molecular tether that couples the endoplasmic reticulum to the microtubule cytoskeleton. TAOK2-mediated tethering is autoregulated by its catalytic activity, which is essential for ER motility and ER-membrane dissociation from the mitotic spindle during cell division.
INTRODUCTION
Composed of a continuous interconnected web of membrane sheets and tubules, the endoplasmic reticulum (ER) has distinct domains; namely, the nuclear envelope, the rough ER sheets, and the smooth peripheral ER tubules (Voeltz et al., 2002). In addition, the ER makes specialized membrane contact sites with other organelles and the plasma membrane (Scorrano et al., 2019; Wu et al., 2018). Each of these structurally discrete regions of the ER serve specialized physiological functions. This vast network of ER membranes relies on the microtubule (MT) cytoskeleton for structurally supporting its intricate shape and functional domains, but also for its motility and remodeling. In animal cells, ER tubules align along MT (Terasaki et al., 1986). Disruption of MT by depolymerization agents collapses the reticulated ER network into primarily ER sheets around the nucleus (Terasaki and Reese, 1994). Further, ER membranes utilize the MT cytoskeleton as tracks for movement. Three distinct mechanisms of ER motility have been defined: (1) ‘‘sliding’’ movements are kinesin-based rapid movement, (2) ‘‘tip-attachment complex’’ (TAC) movements occur when the ER attaches to the MT plus ends and tracks along the growing MT (Friedman et al., 2010; Waterman-Storer and Salmon, 1998), and (3) ER ‘‘hitchhiking’’ where ER tubules co-migrate with an associated organelle such as endosome, peroxisome, or lipid droplets (Guo et al., 2018). Stabilization of MT by the drug Taxol prevents new MT growth, and also inhibits ER tubule extension (Terasaki and Reese, 1994). Motor-mediated ER ‘‘sliding’’ movement occurs on stable acetylated MT (Friedman et al., 2010). Motor-independent ER tubule extension along growing MT plus ends is also dependent on MTs and is carried out by the TAC composed of ER protein STIM1 and MT plus end protein EB1 (Waterman-Storer and Salmon, 1998; Grigoriev et al., 2008). Additionally, membrane contact sites between ER and organelles appear to be supported by MTs. For example, ER-mitochondria contact sites are preferentially aligned with acetylated MTs (Friedman et al., 2010). Endosome maturation occurs at junctions where ER-endosome contact sites and MTs converge (Wu and Voeltz, 2021). In geometrically complex cells such as neurons, the ER is dependent on MTs for its distribution throughout its processes including dendrites, dendritic spines, and axons. The presence of fine caliber ER tubules in axons is critical for neuronal polarity and is dependent on ER membrane interactions with MT (Farías et al., 2019). On the other hand, ER organization at dendritic branch points and dendritic spines is structurally complex and exhibits decreased MT association (Cui-Wang et al., 2012). Bidirectional regulation of ER-MT tethering, therefore, not only influences ER morphology but can also dictate cell shape and function.
Constantly in flux, ER membranes undergo a range of dynamic and structural changes including membrane extension, retraction, three-way junction formation, and tubule fusion (Pendin et al., 2011). Cell division brings about extensive ER remodeling, causing the ER membranes to coalesce around the spindle poles but remain largely absent from the mitotic MT spindle at metaphase (Jongsma et al., 2015; Smyth et al., 2015). Before insight into ER-MT dynamics and its physiological regulation can be gained, the mechanisms through which molecular tethers drive ER-MT association must be understood. Here, we investigate ER-MT association in interphase and mitotic human cells and identify thousand and one amino acid kinase 2 (TAOK2) as an ER-localized multifunctional protein kinase that serves as a molecular tether linking ER to MT.
TAO kinases are ubiquitously expressed serine/threonine protein kinases belonging to the Ste20 kinase family (Chen et al., 1999; Manning et al., 2002; Moore et al., 2000. While there is only one Tao kinase encoding gene in invertebrates, three distinct TAOK genes are expressed in humans (Chen et al., 2003; Manning et al., 2002). The encoded kinases TAOK1, TAOK2, and TAOK3 share a highly conserved N-terminal kinase domain, followed by distinct C-terminal domains (Chen et al., 2003; Manning et al., 2002). TAO kinases were originally identified as stress-sensitive kinases that activate the p38 kinase cascades through activation of MEK kinases (Chen et al., 1999). Among the TAO family of protein kinases, TAOK2 coordinates several aspects of neuronal development and function (Nourbakhsh and Yadav, 2021). TAOK2 is highly expressed during neuronal development and is important for basal dendrite formation as well as for axon elongation in cortical neurons (de Anda et al., 2012). TAOK2 is enriched in dendritic spines and is essential for their formation and stability through phosphorylation of the cytoskeletal protein septin 7 (Ultanir et al., 2014; Yadav et al., 2017). TAOK2 knockout (KO) mice exhibit cognitive and social-behavioral deficits, and show structural changes in brain size (Richter et al., 2019). Recently, mutations in TAOK2 have been associated with autism spectrum disorder, with several mutations present outside the kinase domain of the protein (Richter et al., 2019). Despite its clear relevance to human development and its disease association, little is known about the molecular and cellular functions of TAOK2 kinase.
In our study, we demonstrate that TAOK2 is a pleiotropic protein, with distinct catalytic kinase and ER-MT tethering functions. TAOK2 is enriched at junctions where the ER membrane makes contact with the MT cytoskeleton. We show that TAOK2 is embedded in the ER membrane through transmembrane helices and an amphipathic region that limits its localization to discrete subdomains of the ER. The cytoplasmic-facing C-terminal tail of TAOK2 directly binds MTs, tethering the ER membranes to MTs. TAOK2 KO cells display significantly reduced overlap of ER membrane with MTs and increased ER-membrane movement. Additionally, we found that STIM1-EB1-mediated extension of ER tubules on growing MT plus tips is significantly decreased in TAOK2 KO cells. We show that TAOK2 can associate with EB1 through the conserved SxIP motif in its C-terminal tail. Further, we show that the tethering function of TAOK2 is negatively regulated by its kinase activity. During mitosis, we find that TAOK2 is highly activated and inhibition of its catalytic function prevents ER disengagement from the mitotic spindle, causing profound mitotic defects. This study identifies TAOK2 as an ER protein kinase and elucidates a hitherto unknown autoregulated mechanism for ER-MT tethering important for ER dynamics and mitotic segregation.
RESULTS
TAOK2 is an ER-associated kinase
To investigate the unique role that nonkinase domains of TAO family members might impart on their biological function, we performed bioinformatic analysis of the secondary protein structure of TAO kinases. We found that TAOK2α (hereafter TAOK2) harbors unique hydrophobic regions in its C-terminal domain not present is its alternatively spliced isoform TAOK2β or paralogous genes TAOK1 and TAOK3. Sequence analysis of TAOK2 through the transmembrane helix prediction software TMHMM2.0 (Krogh et al., 2001) indicates that TAOK2 is a multipass membrane protein containing six distinct hydrophobic regions (Figures 1A and 1B). Additional predicted region of hydrophobicity following the predicted transmembrane domains was analyzed using AMPHIPASEEK (Combet et al., 2000) and HeliQuest (Gautier et al., 2008). This analysis revealed an amphipathic helical (AH) region with a sharply defined hydrophobic face (hydrophobicity <H> = 0.75, hydrophobic moment <mM> = 0.4) and a polar face rich in positively charged residues (net charge z = 4) (Figure 1A inset). To further gain insight into the structural domains of TAOK2, we used the machine-learning-driven protein-structure prediction software AlphaFold2.0 (Jumper et al., 2021). The prediction revealed a coiled-coil bundle that folded over the length of the kinase domain and transmembrane region, six transmembrane helices, followed by another helical region that was predicted to be amphipathic by AMPHIPASEEK. The C-terminal tail was predicted to be likely an intrinsically disordered region (Figure S1A).
Figure 1. TAOK2 is an ER-protein kinase.
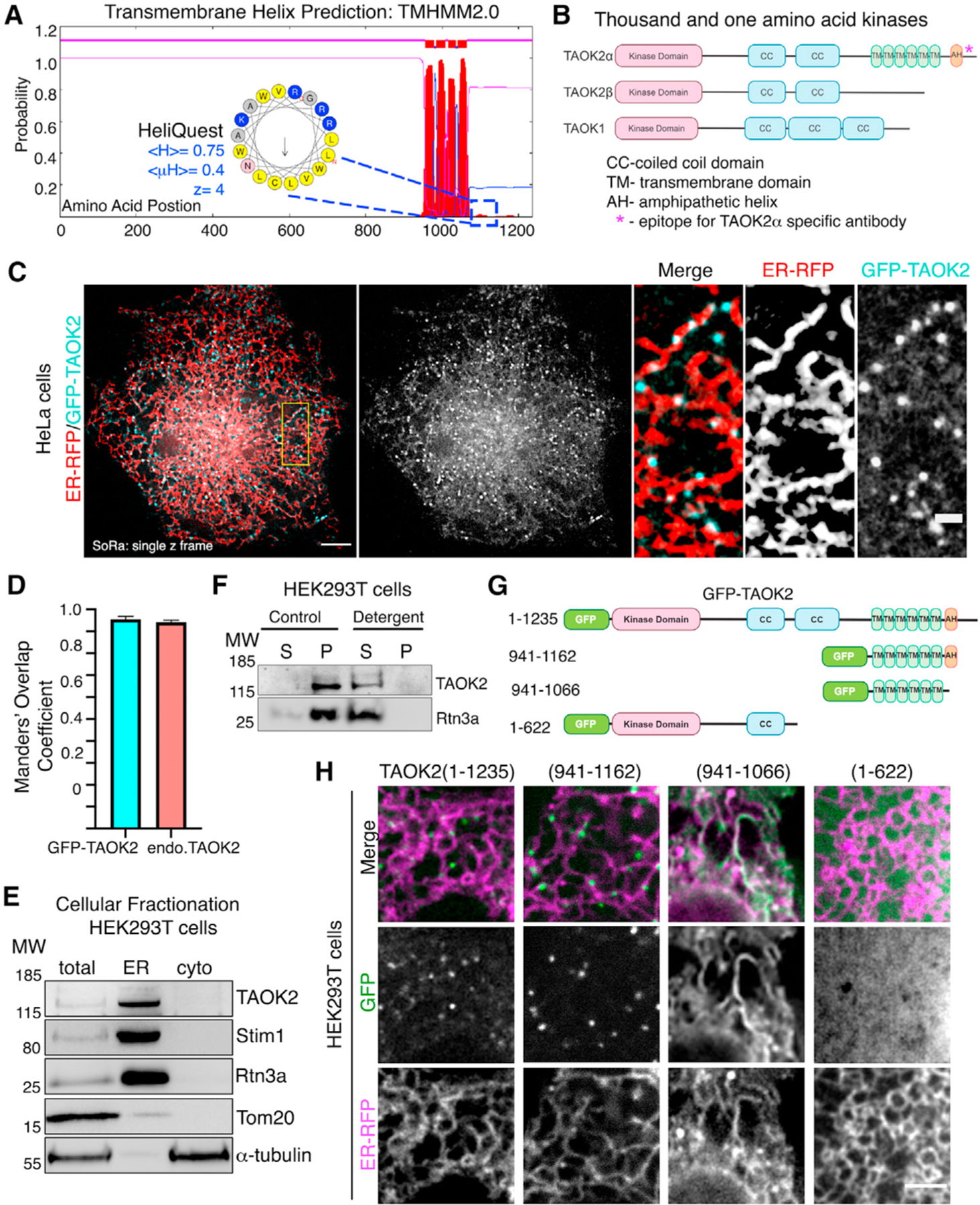
(A) Transmembrane Hidden Markov Model (TMHMM v2.0) prediction plot shows the posterior probabilities (y axis) of inside/outside(magenta)/TM helix (red) along the length of TAOK2 sequence (x axis). Hydrophobic region following the sixth predicted transmembrane domain (TMD) was analyzed through AMPHIPASEEK which predicted residues 1,146–1,162 to be amphipathic. Charged (blue) and hydrophobic (yellow) residues are indicated in the inset. HeliQuest was used to calculate hydrophobicity <H>, hydrophobic moment <μH> and net charge Z.
(B) Schematic representation of TAOK2 isoforms α, β and TAOK1. Coiled coils (CC), six transmembrane domains (TM), and amphipathic helix (AH) predicted in TAOK2 are depicted. The asterisk indicates the epitope used to generate the TAOK2α-specific antibody.
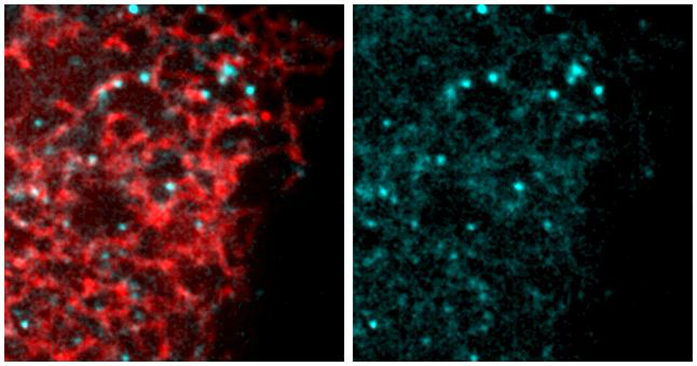
(C) SoRa (super resolution by optical reassignment) images of HeLa cells expressing GFPTAOK2 (cyan) and ER marker ER-mRFP (red). Scale bar: 5 μm. Grayscale images of TAOK2 and ER-RFP are shown separately. Scale bar: 1 μm.
(D) Manders’ overlap coefficient for co-localization of expressed GFP-TAOK2 and ER-mRFP and for endogenous TAOK2 and EGFP-Sec22b are plotted. Values indicate mean ± SEM; n = 10 for each condition.
(E) Western blot (n = 3) of cell homogenate fractionated into ER membrane and cytosol probed with antibodies against TAOK2, ER-membrane proteins Stim1 and Rtn3, mitochondrial protein Tom20, and tubulin.
(F) Western blot (n = 3) of cell homogenate fractionated into membrane pellet (P) and cytosolic supernatant (S) components, in the absence (control) or presence of detergent, probed with antibodies against TAOK2 and ER protein Rtn3a.
(G) GFP-tagged TAOK2 deletion constructs used in (H).
(H) Confocal images of cells transfected with distinct GFP-tagged TAOK2 deletion constructs (green) and ER-mRFP (magenta). Scale bar: 3 μm.
See also Figure S1.
To determine the cellular localization of the membrane-spanning TAOK2 kinase, we generated a rabbit polyclonal antibody against the unique C-terminal tail (residues 1,220–1,235) of TAOK2α (Figure 1B). Antibody staining in HEK293T and HeLa cells revealed that endogenous TAOK2 co-localized extensively with the expressed ER marker EGFP-Sec22b (Figure S1B). We noted that TAOK2 exhibited a striking localization on subdomains of the ER membrane in distinct punctate pattern. Therefore, we generated a GFP-tagged TAOK2 construct and performed super resolution microscopy (super resolution by optical reassignment using SoRa disk) to visualize the ER localization of TAOK2 at higher resolution (Figures 1C and 1D). GFP-TAOK2 localized on the ER membrane and was present in discrete membrane subdomains in both HeLa cells and HEK293T cells (Video S1). Manders’ overlap coefficient was used to quantify the overlap of the GFP-TAOK2 and endogenous TAOK2 with EGFP-Sec22b (Figure 1D). To test biochemically whether TAOK2 is an ER-membrane protein, we fractionated HEK293T cell homogenates into ER-membrane fractions using differential centrifugation (Hoyer et al., 2018). TAOK2 was enriched in the ER-membrane fraction along with other known ER-membrane proteins such as STIM1 (Grigoriev et al., 2008) and Rtn3a (Hu et al., 2009). 97.6% of the total TAOK2 in the postnuclear homogenate was enriched in the ER fraction, as compared with 6.17% of tubulin (Figure 1E). On differential centrifugation, TAOK2 partitioned in the membrane fraction, and treatment with detergent led to its release in the supernatant (Figure 1F). To identify the mechanism through which TAOK2 achieves its ER localization, we generated three GFP-tagged deletion constructs (Figure 1G), which were expressed in cells along with the ER-membrane marker ER-mRFP (Snapp et al., 2006). The truncated construct, lacking the transmembrane and amphipathic helices (1–622), was entirely cytosolic, while the six transmembrane helices alone (amino acids 941–1,066) were sufficient to target the kinase to the ER (Figure 1H). This construct containing just the TAOK2 transmembrane domain localized uniformly throughout the ER (Figure 1H) and was not restricted to ER subdomains like the full-length GFP-TAOK2 or endogenous TAOK2 (Figures 1C and S1B). We found that the predicted amphipathic helical region together with the transmembrane domains (amino acids 941–1,162) is required for localization of TAOK2 to discrete ER subdomains (Figure 1H) and captures the localization patterns of the endogenous protein. TAOK2 did not show significant overlap with other organelles that we tested (Figure S1C). These data show that TAOK2 is an ER-resident protein kinase that associates with the ER through its six transmembrane helices, while the amphipathic region confers its localization to distinct ER subdomains.
TAOK2 associates directly with assembled MTs via its C-terminal tail
Previously, TAOK2 has been shown to co-localize with MTs (Mitsopoulos et al., 2003); however, mechanism underlying this observation is unknown. We found that when GFP-TAOK2 or truncated C-terminal TAOK2 constructs were overexpressed, they led to bundling the of MT, and these MT bundles were extensively decorated with GFP-TAOK2 (Figures 2A and 2B). Co-localization of truncated C-terminal GFP-tagged TAOK2 constructs with tubulin immunostaining allowed us to map the MT-binding domain to 40 amino acids (1,196–1,235) in the extreme C-terminal tail of TAOK2 (Figures 2A and 2B). A truncation construct lacking this domain (1–1,195) did not associate with MTs (Figure 2A). Expression of truncation constructs containing the MT-binding domain led to increase in acetylated MTs, while constructs lacking MT-binding domain did not affect tubulin acetylation level (Figure S2A). We next tested whether TAOK2 can directly bind MTs using a biochemical binding assay with purified components. We bacterially purified GST-tagged TAOK2 C-terminal tail protein (residues 1,187–1,235) (Figure S2B). An in vitro MT-binding assay was used to test whether purified GST-TAOK2-C could bind MTs polymerized from purified tubulin protein. GST-TAOK2-C pelleted specifically with polymerized MTs, while the control GST protein remained in the supernatant (Figures 2C and 2D), suggesting that TAOK2 can directly associate with MTs via its C-terminal tail. The affinity of TAOK2-MT interaction was assessed by determining the fraction of MT bound TAOK2-C at increasing concentrations of tubulin. We found that the C-terminal tail of TAOK2 associates with MTs with a KD of 0.67 ± 0.19 μM (Figures 2E and 2F). These results show that TAOK2 can directly bind MTs through its cytoplasmic-facing C-terminal tail.
Figure 2. Direct binding of TAOK2 to MTs.
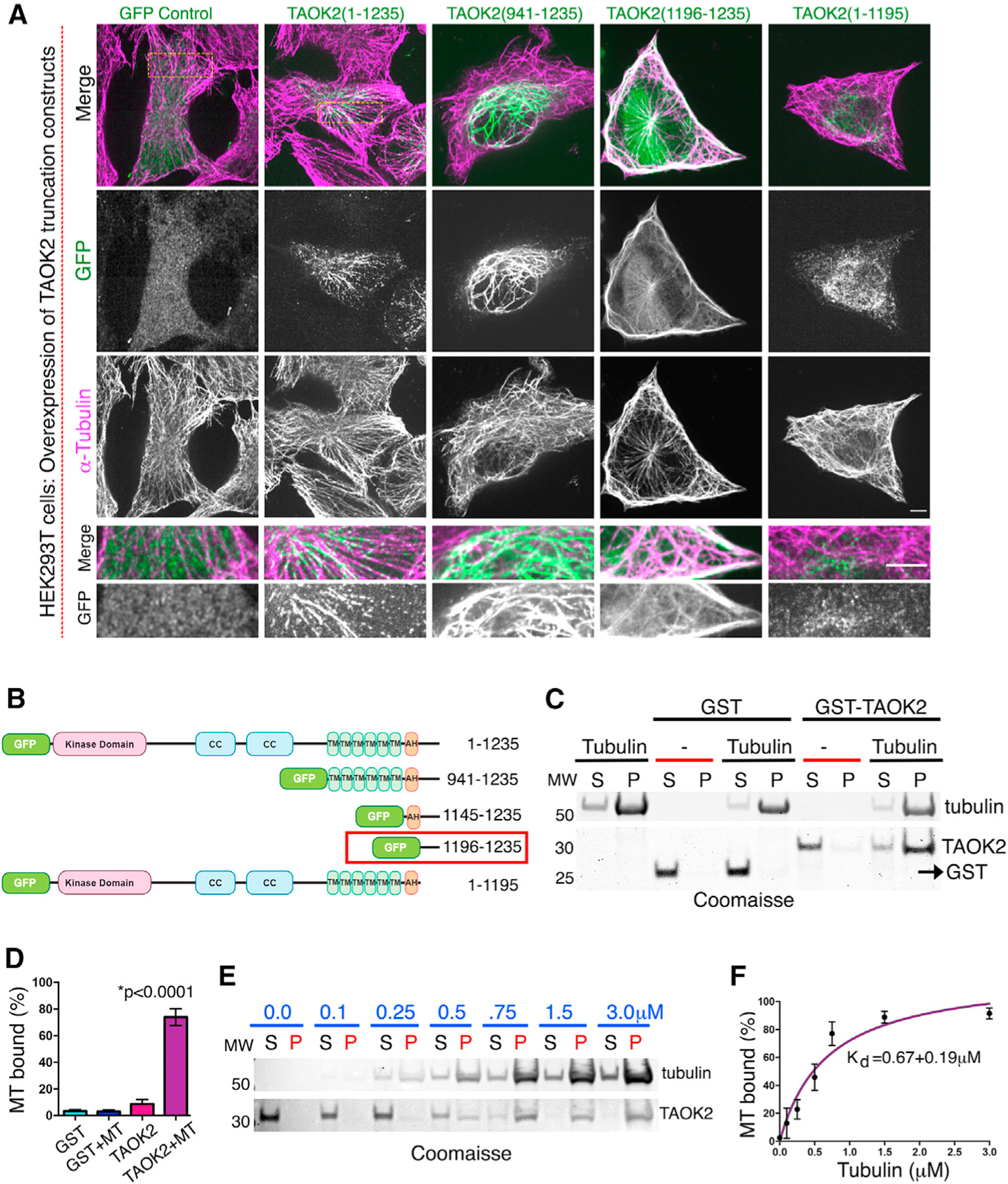
(A) Confocal images of cells transfected with indicated GFP-tagged TAOK2 constructs immunostained for α-tubulin. Scale bar: 3μm. Magnified images highlight overlap of GFP-tagged TAOK2 with MTs. Scale bar: 5 μm.
(B) GFP-tagged deletion constructs, coiled-coil (CC), transmembrane (TMD), amphipathic helix (AH) domains are marked. Domain necessary and sufficient for MT localization is indicated by red box.
(C) Coomassie-stained SDS-page gel shows co-sedimentation of indicated proteins with polymerized tubulin. MT binding assay (n = 3) performed with 5 μg of GST or GST-TAOK2-C (1,187–1,235) protein in presence of Taxol stabilized MT.
(D) Percentage of protein bound to MT plotted for indicated protein. Error bars: SEM; n = 3; p < 0.0001, one-way ANOVA.
(E) Coomassie-stained SDS-page gel shows amount of purified GST-TAOK2-(1,187–1,235) unbound (S) or co-sedimented with polymerized MT (P).
(F) Dissociation constant KD derived by the Michaelis-Menten equation. Error bars: SEM; n = 3 replicates. See also Figure S2.
ER-membrane tethering to the MT cytoskeleton is mediated by TAOK2
Our finding that TAOK2 is an ER kinase with the ability to directly bind to MTs led us to hypothesize that this kinase can act as a molecular tether linking the ER membrane to MTs (Figure 3A). To evaluate this possibility, we performed live-cell imaging with simultaneous visualization of the ER membrane and MT cytoskeleton. Cells transfected with GFP-TAOK2 and ER-mRFP were incubated with a cell-permeable MT-binding dye, and three-color time-lapse confocal microscopy was performed (Video S2). Additionally, in immunostained fixed cells, endogenous TAOK2 punctae were often localized at the end of ER tubules where the tubules made contact with MTs (Figures 3B and 3C). Quantification of live cell images revealed that 87% of GFP-TAOK2 punctae localized at the points of contact between the ER and the MT cytoskeleton (Figure 3D). These punctae tracked with the movement of the ER membrane on MTs (Figures S2C and S2D; Video S2). To determine the functional consequences of TAOK2 depletion on ER-MT tethering, we generated two independent TAOK2 knockout HEK293T cell lines using CRISPR/Cas9-mediated gene editing. Genetic KO and loss of protein was validated using genome sequencing and western blot, respectively (Figures 3E and S3A–S3C). Control and TAOK2 KO cells were transfected with MT-binding protein mEmerald-ensconsin and ER-mRFP to visualize MTs and ER membranes, respectively. The ER in control cells extended from the nuclear envelope to the cell periphery and made extensive contacts with the MTs (Figure 3F). In contrast in TAOK2 KO cells, while the nuclear envelope and the sheet like perinuclear ER appeared unaffected, the ER density and morphology was perturbed at the cell periphery (Figure 3F, inset). ER density at the cell periphery was significantly reduced in TAOK2 KO cells to 0.22 ± 0.03 compared to 0.43 ± 0.07 in control cells (Figure 3H). Further, area of the ER that overlapped with MTs was significantly decreased from 0.422 ± 0.06 in wild-type (WT) cells to 0.22 ± 0.05 in TAOK2 KO cells (n = 6, p = 0.009, two-tailed t test) (Figure 3G). Next, we ascertained which domains of TAOK2 were important for its tethering function. The expression of constructs that retained the ER-localization and MT-binding elements (amino acids 941–1,235) but lacked the kinase and coiled-coil domains (Figure 3I) led to aberrant overtethering of the ER membranes to MTs, perturbing ER morphology such that the ER appeared bundled alongside perinuclear MT cables. Expression of just the MT-binding domain (1,196–1,235) or of the constructs lacking the MT-binding domain (1–1,195) did not lead to ectopic tethering of the ER to MTs. These data together show that TAOK2 indeed is an ER-localized MT tether.
Figure 3. TAOK2 is an ER-MT tethering protein kinase.
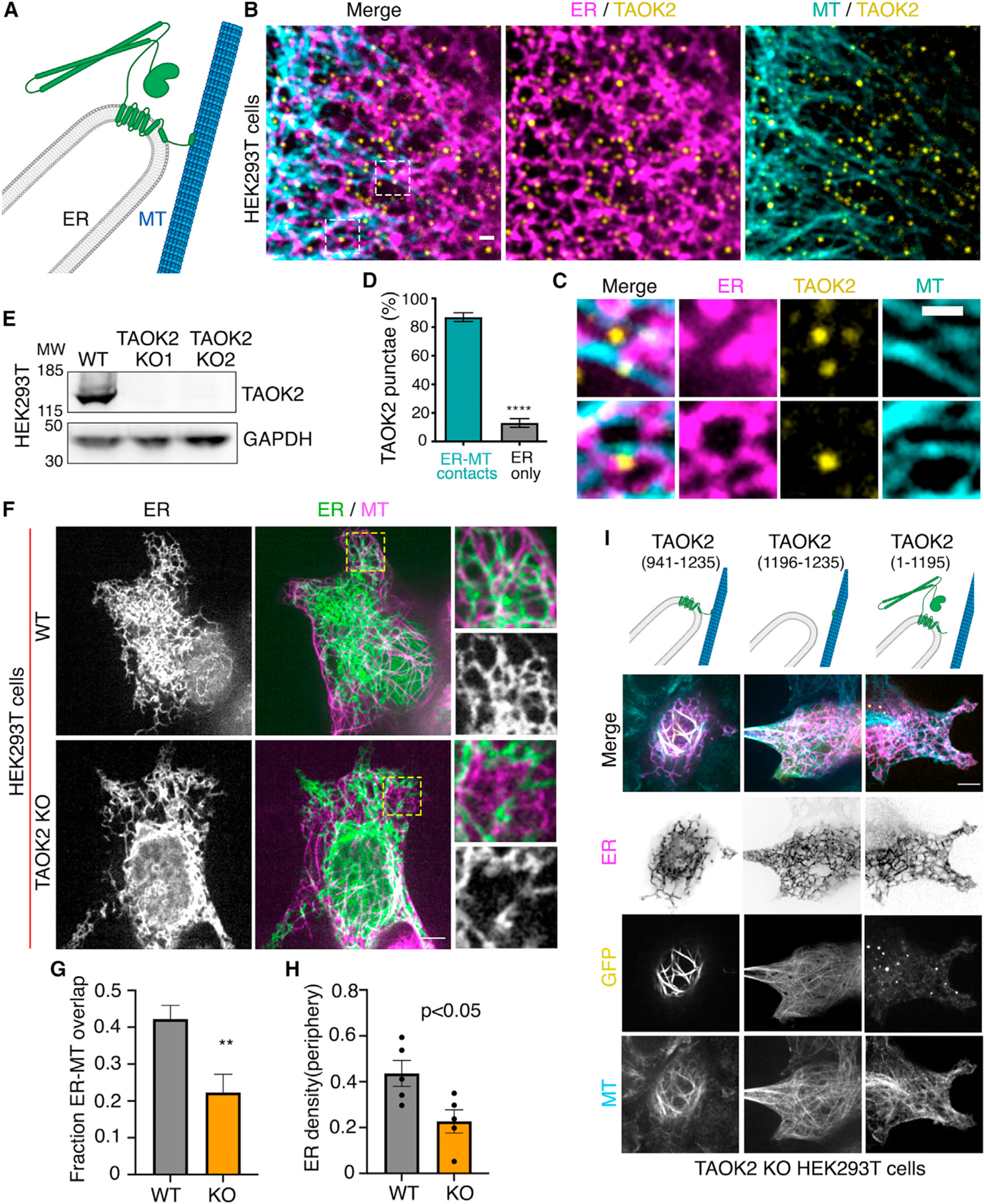
(A) Schematic representation of TAOK2 with cytoplasmic-facing kinase domain, membrane bound 6 transmembrane helices and amphipathic region, and C-terminal tail directly bound to MT.
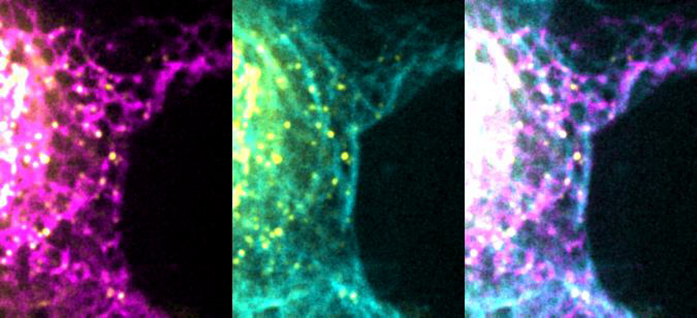
(B) Cells transfected with GFP-TAOK2 (yellow), ER-mRFP (magenta), and MT dye (cyan). Scale bar: 1 μm.
(C) Magnified images displays individual TAOK2 (yellow) punctae on ER tubules in contact with MT (cyan). Scale bar: 1mm.
(D) Percentage of TAOK2 punctae co-localized with ER and MTs in HEK293T cells. Values indicate mean ± SEM; n = 10 cells with at least 50 punctae per cell were analyzed. ****p < 0.0001
(E) Western blot of lysate from WT and TAOK2 KO cell lines generated using CRISPR/Cas9 gene editing.
(F) WT and TAOK2 KO cells transfected with ER-mRFP (green) and mEmerald-ensconsin (magenta) to visualize MTs. Peripheral ER is magnified and shown for both WT and TAOK2 KO cells. Scale bar: 5 μm.
(G) Ratio of peripheral ER area in contact with MT in 100-μm2 region for WT and TAOK2 KO cells. Values indicate mean SEM; n = 6 cells from 3 experiments, t test with Welch’s correction. **p < 0.01
(H) ER density measured in WT and TAOK2 knockout cells. Values indicate mean ± SEM, n = 6 cells from 3 experiments, t test with Welch’s correction.
(I) Topology of TAOK2 deletion constructs. KO cells expressing indicated TAOK2 constructs are shown where TAOK2 (yellow), ER-mRFP (magenta) and MTs (cyan) are shown in merged and grayscale. Scale bar: 5 μm. See also Figure S2.
TAOK2 is required for ER-MT plus end motility
To test whether loss of TAOK2-mediated tethering would impact overall ER motility, we measured ER-membrane movement over time. WT and TAOK2 KO cells were transfected with the ER marker EGFP-Sec22b and time-lapse confocal microscopy allowed us to visualize ER dynamics. ER motility in TAOK2 KO cells was significantly increased compared to WT cells (Figure 4A). Average normalized pixel differences over time allowed us to calculate the ER-motility index which increased from 0.287 ± 0.012 in WT cells to 0.351 ± 0.017 (n = 9, p = 0.0098) in TAOK2 KO cells (Figure 4B). To further investigate how the absence of TAOK2 might affect ER motility, we performed simultaneous confocal imaging of ER (EGFP-Sec22b) and MT-end-binding protein mCherry-EB3 (Figure 4C). In WT cells, both TAC movements on MT plus ends as well as rapid ER movements independent of EB3 comets were observed. However, in TAOK2 KO cells, TAC movements were severely disrupted and almost all observed movements were rapid and not associated with EB3 comets. Thus, the loss of TAOK2 specifically disrupts TAC movements of ER membrane on MT growing plus ends (Figures 4C and 4D). Further, we found that while the number of MT plus ends marked by EB3-mCherry were unaffected in TAOK2 KO cells (Figure 4E), MT growth assessed by measuring EB3 velocity was significantly increased in TAOK2 KO cells (Figure 4G). Additionally, in TAOK2 KO cells, EB3 comet tracks were less directed, exhibiting increased curvature and pausing more frequently compared to control (Figures 4F and 4H). These data show that TAOK2-mediated ER-MT tethering is essential for the dynamics of ER membranes as well as MTs.
Figure 4. TAOK2 knockout disrupts ER-MT dynamics.
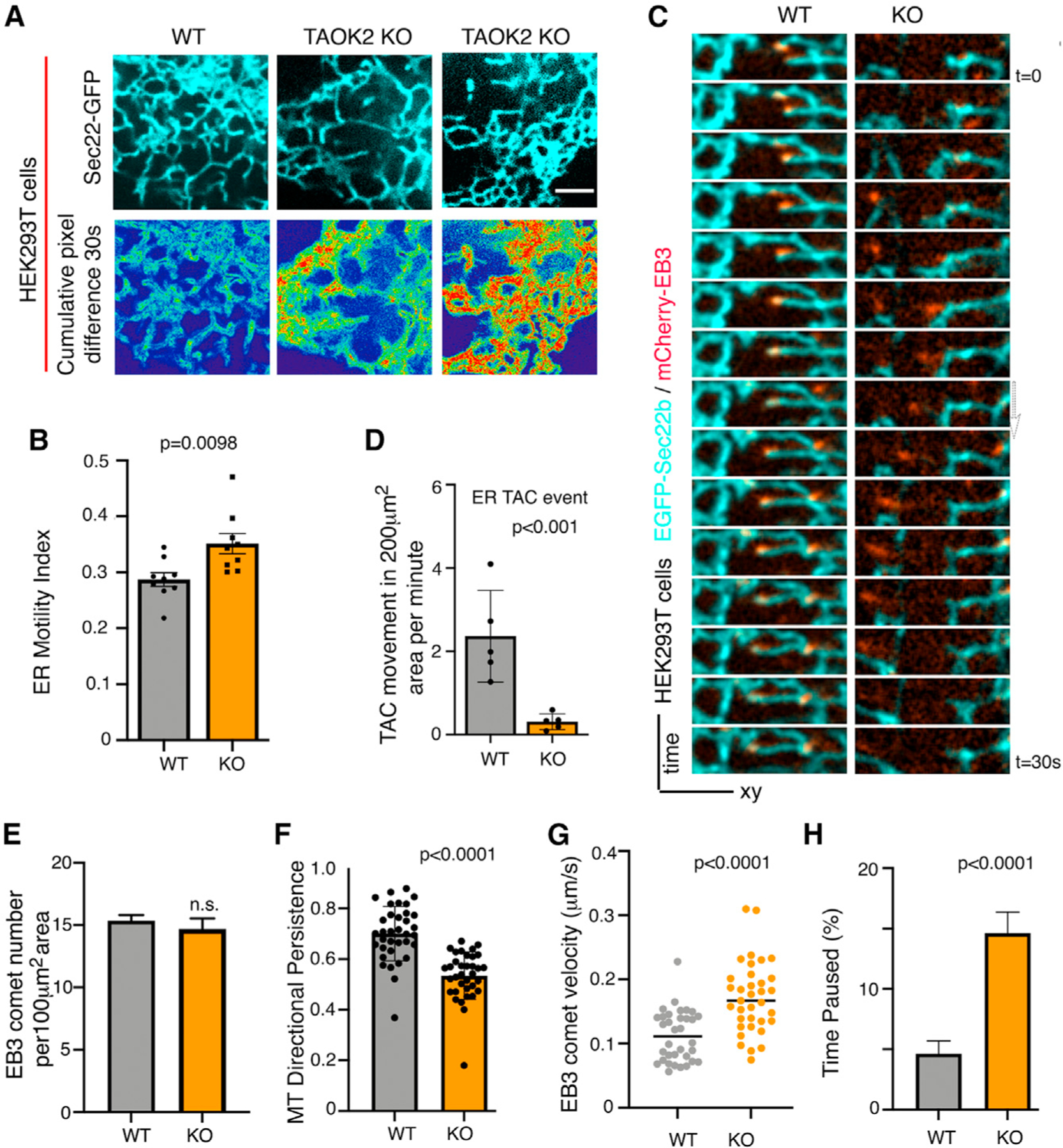
(A) Peripheral ER in WT and TAOK2 KO cells expressing EGFP-Sec22b at a single time point (left column). Sum slice projection of cumulative pixel difference in successive frames over a 30-s time period (right). Fluorescence intensity is pseudo-color coded; red represents increased ER motility. Scale bar: 3 μm.
(B) ER-motility index for WT and TAOK2 KO cells. Error bars: SEM; n = 9 cells from 3 experiments; t test.
(C) Montage of images acquired every 2 s, of WT and TAOK2 KO cells expressing EGFP-Sec22b and mCherry-EB3 shows movement of ER (cyan) associated with MT plus tips (red) in WT but not KO cells.
(D) ER-membrane movements classified as MT plus tip-associated TAC (co-localized with EB3 comet) in 200-μm2 area of the cell over 1-min time period. Values indicate mean ± SEM; n = 5 cells for each condition from 3 experiments; two-tailed t test with Welch’s correction.
(E) EB3 comet number in a 100-μm2 area in WT and knockout cells. Values indicate mean ± SEM; n = 10 cells; t test with Welch’s correction.
(F) MT directional persistence is plotted for WT and TAOK2 knockout cells. Values indicate mean ± SEM; n = 9 cells from 3 experiments with at least 5 comet paths measured per cell; two-tailed t test.
(G) Percentage of time spent by EB3 comet pausing (no growth) is plotted for WT and TAOK2 KO cells. Values indicate mean ± SEM; n = 9 cells from 3 experiments with at least 5 comet paths measured per cell; two-tailed t test.
(H) EB3 comet velocity for WT and TAOK2 KO cells, error bars: SEM; n = 9 cells from 3 experiments with 5 comets per cell; two-tailed t test. See also Figure S3.
TAOK2 regulates STIM1-mediated ER movement on MT growing ends
We further probed how TAOK2 kinase might contribute to the ER tip-associated movement or TAC. MT plus-end-associated movement of ER has been previously shown to be mediated by the interaction of ER transmembrane protein STIM1 with end-binding proteins EB1 and EB3, which bind the growing plus ends of MTs (Grigoriev et al., 2008). As TAOK2 KO cells exhibit defects in movement of ER on MT plus ends, we tested whether TAOK2 contributes to the STIM1-mediated TAC movements, or whether it constituted a distinct mechanism for TAC movement. We found no changes in protein levels of STIM1 and EB1 in TAOK2 KO cells (Figure S4A). Further, immunostaining for endogenous EB1 in control and TAOK2 KO cells revealed no differences in the density of EB1 comets (Figure S4B). If TAOK2 regulated STIM1 mediated TAC movements, we then hypothesized that STIM1-EB movements would be disrupted in absence of TAOK2. We transfected control and TAOK2 KO cells with STIM1-YFP, the ER marker Sec61-BFP, and MT plus tip marker EB3-mCherry. In control cells, we found that STIM1-YFP was ER localized and overlapped with Sec61-BFP as expected but importantly was also concentrated at the contact points between ER tubules and MT plus ends (Figure 5A). However, in TAOK2 KO cells, while STIM1-YFP still co-distributed with Sec61-BFP, its concentration at MT plus ends was dramatically reduced (Figure 5A). Time-lapse imaging revealed numerous TAC movements where STIM1-YFP coincided with EB3-mCherry comets that moved together over time in WT cells. However, both accumulation of STIM1 at MT plus ends and ER TAC movements were absent in TAOK2 KO cells (Figures 5B and 5C). While EB3/STIM1 comets mediating TAC movement declined drastically from 68.9% in WT to 18.5% in TAOK2 KO cells, the number of EB3 comets making no contact with STIM1 significantly increased from 21.3% in WT to 62.9% in TAOK2 KO HEK293T cells (n = 9 cells) (Figure 5C). The association of STIM1 with EB proteins is direct through interaction of the Ser-x-Ile-Pro (SxIP) motif in the C-terminal tail of Stim1, with a well-defined hydrophobic cavity in the C-terminal end-binding homology domain of EB1 and EB3 (Honnappa et al., 2009). We tested whether EB-binding motifs would be present in TAOK2. Indeed, we found that the C-terminal tail of TAOK2 harbored a highly conserved SxIP motif (TRIP amino acids 1,192–1,195) which is found in many other EB-binding proteins including CLASP1 and STIM1 (Figure 5D). The SxIP motif is typically present in a region rich in basic amino acids, which is also the case for TAOK2 (Figure 5D, highlighted in red). Therefore, we tested whether TAOK2 localized with EB1 on MT plus ends. Immunostaining for endogenous TAOK2 and EB1 proteins in HeLa cells transfected with EGFP-Sec22b revealed that TAOK2 punctae co-localized with 34.7% of EB1 comets (Figures 5E and 5H). Further, to test whether TAOK2 and EB1 interact biochemically, we immunoprecipitated EB1 from HeLa cell lysate and then probed for TAOK2 and STIM1. TAOK2 and STIM1 both co-immunoprecipitated with EB1 (n = 3 experiments, Figure 5F). To test whether the SxIP motif is important for binding of TAOK2 to EB1 on MT plus ends, we expressed the C-terminal tail of TAOK2 containing the SxIP motif and MT-binding domain (1,162–1,235) along with EB3-mCherry in TAOK2 KO HEK293T cells. This construct localized to the MT and accumulated at the MTs plus ends decorated by EB3. Further, the TAOK2 C-terminal tail construct, which lacks the SxIP motif 1192TRIP1195, can still bind MT but not MT plus tips. Therefore, deletion of SxIP motif was sufficient to disrupt localization of TAOK2 to the plus tips but did not affect its MT-binding properties (Figure 5G).
Figure 5. TAOK2 regulates ER TAC movement and interacts with EB1 via SxIP motif.
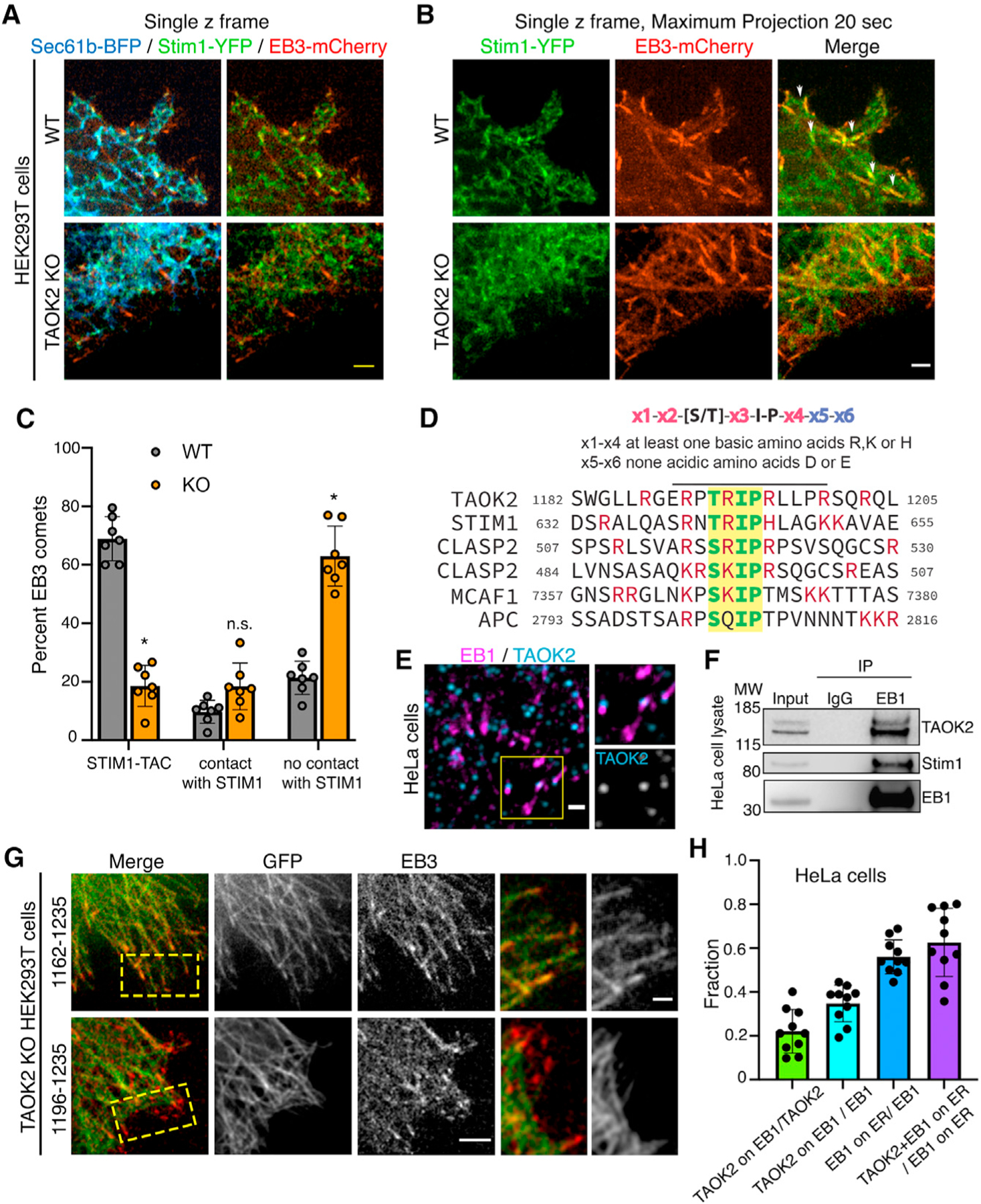
(A) Single z-frame confocal images of WT and TAOK2 KO cells expressing ER marker Sec61-BFP (blue), STIM1-YFP (green) and EB3-mcherry (red). Scale bar: 3 μm.
(B) Maximum projection of single z frames shown in (A) over a period of 20 s to visualize movement and paths of EB3 and STIM1 over time in WT and TAOK2 KO cells. STIM1-YFP (green) and EB3-mcherry (red). Scale bar: 3 μm.
(C) EB3 comets were classified as (1) comets interacting with STIM1/ER and performing TAC, (2) comets interacting with STIM1 transiently but not performing TAC, and (3) comets not contacting STIM1/ER. Values indicate mean ± SEM; n = 7 cells from 3 experiments; two-way ANOVA. *p < 0.05.
(D) Schematic depicts the SxIP motif of MT plus tip-interacting proteins that associate with EB proteins.
(E) HeLa cells immunostained for EB1 (magenta) and TAOK2 (blue). Scale bar: 1 μm.
(F) Western blot (n = 3) shows coimmunoprecipitation of TAOK2 and STIM1 with EB1 using anti-EB1 antibodies; 1% input is shown for comparison.
(G) TAOK2 KO cells transfected with EB3-mcherry (red) along with GFP (1,196–1,235) or GFP (1,162–1,235) containing the TRIP (SxIP) motif. Scale bar: 3 μm.
(H) Bar graph represents fractions of EB1-positive TAOK2 punctae to total TAOK2 punctae, number of TAOK2-positive EB1 punctae compared to total EB1 punctae, number of EB1 puncta on ER compared with total EB1 punctae, and number of TAOK2-positive EB1 punctae on ER compared with the total number of EB1s on the ER. Values indicate mean ± SEM; n = 10 cells from 3 experiments. See also Figure S4.
To determine the spatial distribution of endogenous TAOK2 and EB1 on MT plus ends and ER, respectively, we analyzed the co-distribution of these proteins in fixed HeLa cells expressing EGFP-Sec22b and immunostained for endogenous TAOK2 and EB1. Every TAOK2 puncta was on the ER membrane as expected, of which 22% were co-localized on EB1 comets (Figure 5H). Next, we tested the co-distribution of endogenous TAOK2 and STIM1 in HeLa cells transfected with EGFP-Sec22b. Both TAOK2 and STIM1 punctae were localized to the ER membrane (Figure S4D). About 18% of all TAOK2 punctae co-localized with STIM1 punctae, and 19% of total STIM1 punctae co-localized with TAOK2 (Figure S4C). Interestingly, 56% of EB1 comets appeared to be in contact with the ER membrane. Of these ER contacting MT plus tips, 62.5% were positive for TAOK2 punctae (Figure 5H). These data show that TAOK2 is important for TAC-mediated ER motility and that TAOK2 binds growing MT ends through interaction with EB1 mediated by the SxIP motif.
Aberrant TAOK2 tethering disrupts ER restructuring during mitosis
The ER undergoes dynamic restructuring during cell division (Carlton et al., 2020). During metaphase, as the mitotic spindle aligns the chromosomes at the metaphase plate, the ER is anchored at each end to the spindle poles but largely absent from chromosomes and the area between the spindle poles. Defects in ER clearance from the chromosomes and spindle elicit mitotic defects (Schlaitz et al., 2013). We investigated the role of TAOK2 in ER restructuring during mitosis. We performed superresolution confocal microscopy to visualize GFP-TAOK2 localization in mitotic cells. We found that GFP-TAOK2 was present in close apposition to the spindle poles at sites where the ER membranes converged (Figure 6A). TAOK2 localized in discrete punctae throughout the curvilinear peripheral ER surrounding the mitotic cell, as well as at the points of contact between ER and mitotic spindle (Figure 6A). To test whether TAOK2 was important for ER structural remodeling during cell division, we imaged WT and TAOK2-KO mitotic cells expressing EGFP-Sec22b and mCherry-EB3 to visualize ER membranes and mitotic spindles. WT cells exhibited the characteristic ER morphology in which ER membranes accumulated at spindle poles and fenestrated curvilinear peripheral ER surrounded the mitotic spindle. In contrast, TAOK2-KO cells had abnormal morphology with increased peripheral curvilinear ER membranes and a striking decrease in ER association with spindle poles (Figures 6B and 6C). To determine if this aberrant ER morphology affected cell division, we immunostained WT and TAOK2 KO cells with tubulin and DAPI. While 95.6% of WT cells showed normal bipolar spindles, only 44.8% KO cells had normal bipolar spindles. KO mitotic cells displayed a chromosomal misalignment defect, where 39.3% KO cells had bipolar spindles with misaligned chromosomes, and 15.8% KO cells had multipolar spindles (Figure 6D). We next queried whether these defects could be rescued by the expression of TAOK2. We quantified three distinct parameters, namely, the number of mitotic cells with (1) normal bipolar spindles, (2) ER associated with spindle poles, and (3) ER dissociated from mitotic spindles. Expression of the full-length TAOK2 in KO cells abrogated the mitotic spindle defects and the defects in ER association with the spindle pole (Figures 6E, top row, and 6F). Expression of the truncated TAOK2, lacking the N-terminal kinase and coiled-coil domains in TAOK2 KO cells, induced a dramatic collapse of the ER membranes on the mitotic spindle. TAOK2 binding to MTs created an extremely short and stable MT-ER bridge between the spindle poles. The chromosomes were displaced from the metaphase plate and instead formed a rosette around the ER-MT spindle (Figures 6E and 6F, middle row). These cells failed to divide. The TAOK2 MT-binding domain, when expressed in TAOK2 KO HEK293T cells, localized to the mitotic spindle and not the ER as expected and also did not cause the ER to collapse on the spindle. The number of normal bipolar cells increased from 49.3% in KO cells to 99% on expression of WT-TAOK2 in the TAOK2 KO cells. Only 5.8% of TAOK2 KO cells expressing the abnormal tether construct TAOK2 (941–1,235) had a normal bipolar spindle, while 78.3% of cells expressing the TAOK2 MT-binding domain (1,196–1,235) had a bipolar spindle. Moreover, expression of the TAOK2 MT-binding domain (1,196–1,235) failed to rescue the defect in ER spindle-pole association observed in TAOK2 KO cells (Figure 6F). These data collectively show that TAOK2 mediates ER tethering to the spindle poles during cell division. The association of TAOK2 to the spindle poles is important for ER accumulation at the poles during mitosis. Further, the dissociation of TAOK2 from the spindle MTs is critical for the disengagement of ER from the mitotic spindle.
Figure 6. TAOK2 localizes to the mitotic spindle poles and regulates mitotic ER remodeling.
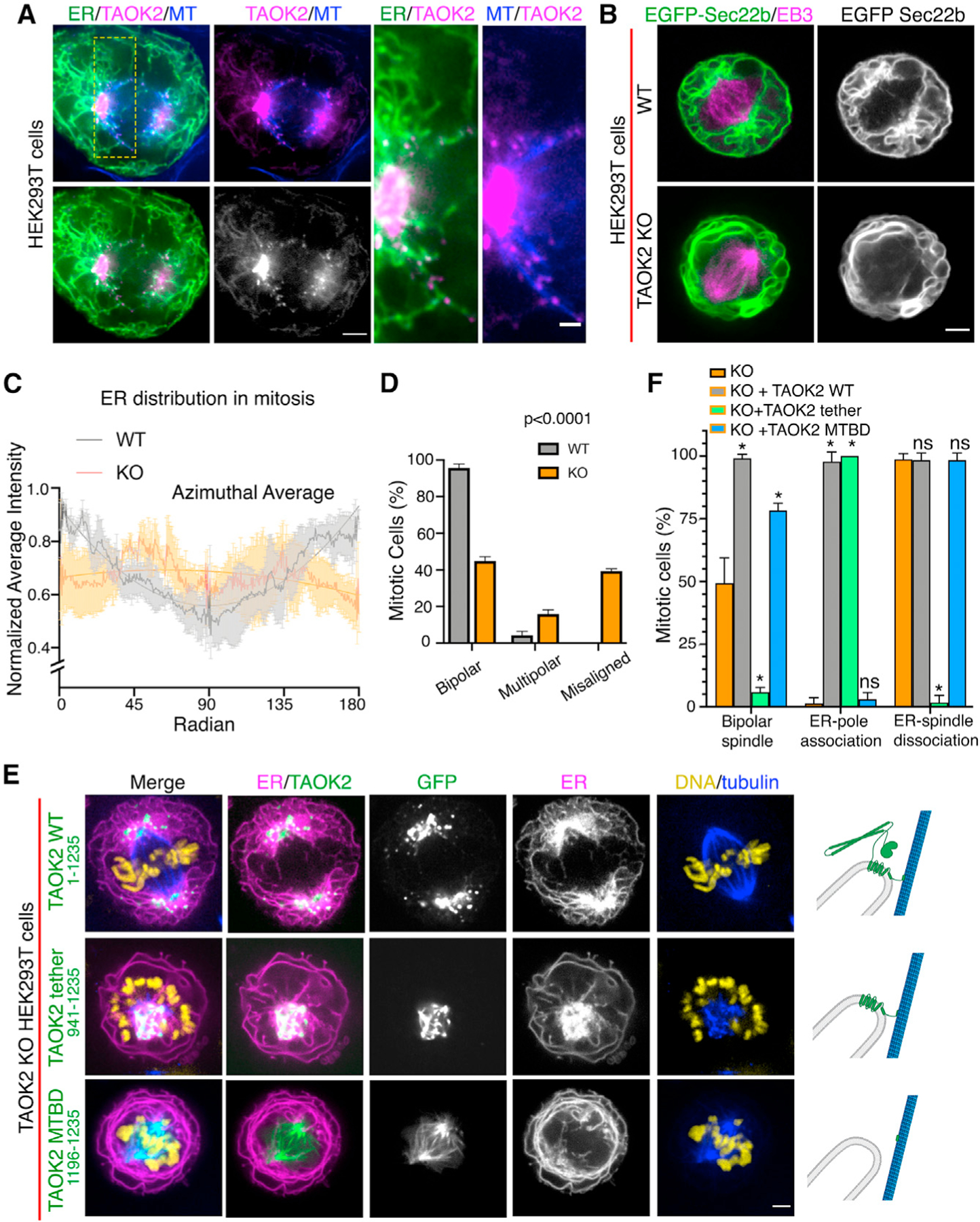
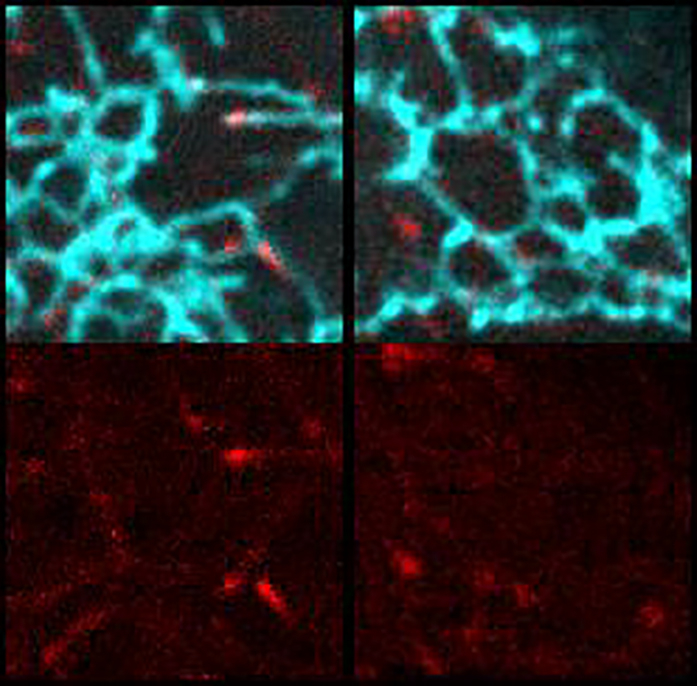
(A) SoRa confocal image of mitotic cell expressing GFP-TAOK2 (magenta) and ER-mRFP (green) and stained with MT dye (blue); scale bar: 2μm. Magnified view of the ER-MT tethering sites; scale bar: 0.5 μm.
(B) WT and TAOK2 KO mitotic cells transfected with EGFP-Sec22b (green) and mCherry-EB3 (magenta). ER morphology in WT and TAOK2 KO cells is shown in grayscale. Scale bar: 5 μm.
(C) Azimuthal average of normalized fluorescence intensity of ER markers (where 0 and 180 correspond to the spindle poles) is plotted for WT (gray) and TAOK2 KO (orange) cells. Values indicate mean ± SEM; n = 3 cells from 3 experiments.
(D) Percent mitotic WT (gray) and TAOK2 KO (orange) cells that exhibit bipolar, multipolar, or misaligned mitotic spindles. Values indicate mean ± SEM; n > 45 mitotic cells from 3 experiments; p < 0.0001 one-way ANOVA.
(E) Mitotic TAOK2 KO cells expressing indicated TAOK2 constructs (green), ER-mRFP (magenta), DNA (yellow), and MT (blue). Localization of TAOK2 constructs and their effect on ER morphology is shown in grayscale. Scale bar: 5 μm. Schematic (right) depicts topology of GFP-TAOK2 deletion constructs.
(F) Percentage of mitotic TAOK2 KO cells expressing the indicated constructs were scored as (1) exhibiting normal bipolar spindle, (2) ER association with spindle poles, and (3) ER dissociation from mitotic spindle. Values indicate mean ± SEM; n = 75 cells from 3 experiments. *p < 0.05.
Catalytic autoregulation of TAOK2-mediated ER-MT coupling
We next tested whether the kinase activity of TAOK2 regulates its MT association. We introduced a kinase-dead mutation K57A, which disrupts the autophosphorylation of TAOK2 at the critical residue S181 in the activation loop (Moore et al., 2000), and renders TAOK2 catalytically dead (Figure 7A). We expressed either GFP-tagged TAOK2 WT or TAOK2-K57A along with ER-mRFP in TAOK2 KO cells to determine how catalytic activity might impact MT tethering and ER motility. Comparative analysis of overlap between ER membrane and MT in cells expressing WT and kinase-dead TAOK2 in TAOK2 KO cells, revealed that the loss of kinase activity increased the association of ER with MT from 42.9% to 75.4% while the peripheral ER density was unaffected (Figures 7B–7D). To test if increased association with MT resulted in stronger ER-MT tethering and hence reduced motility, we measured ER motility in TAOK2 KO cells expressing kinasedead TAOK2-K57A. A substantial decrease in ER-membrane motility was found in cells expressing TAOK2-K57A as opposed to TAOK2-WT (Figures 7E and 7F). The ER-motility index calculated from mean normalized pixel differences over time, was found to decrease from 0.35 ± 0.02 in TAOK2-WT to 0.26 ± 0.03 (n = 9, p = 0.0019) in K57-TAOK2-expressing cells (Figures 7E and 7F). Further, we found that TAOK2 kinase activity was important for MT growth rates. EB3 velocity averaged at 0.21 μm/s in TAOK2-WT expressing KO cells and decreased to 0.14 μm/s in KO cells expressing kinase-dead TAOK2 (Figure 7G). Next, we used a pharmacological TAOK kinase inhibitor CP43 (Giacomini et al., 2018), to acutely inhibit the kinase activity of TAOK2 and test its effect on ER-MT coupling. TAOK2-GFP incubated with 500 nM CP43 in an in vitro kinase assay led to a reduction in its kinase activity (Figure S5A). We measured the ER-motility index in HeLa cells transfected with EGFP-Sec22b incubated with dimethyl sulfoxide (DMSO) or 250 nM CP43 and found that ER motility was decreased from 0.27 ± 0.02 to 0.16 ± 0.05 (n = 9, p = 0.002) in the presence of CP43 (Figure S5B). Additionally, measurement of EB3 velocity in HeLa cells showed that velocity was decreased from 0.33 ± 0.017 μm/s in DMSO-treated cells to 0.24 ± 0.04 μm/s upon the addition of CP43 inhibitor (Figure S5C). These data indicate that catalytic activity of TAOK2 regulates its ER-MT tethering function.
Figure 7. ER-MT tethering is regulated by catalytic activity of TAOK2.
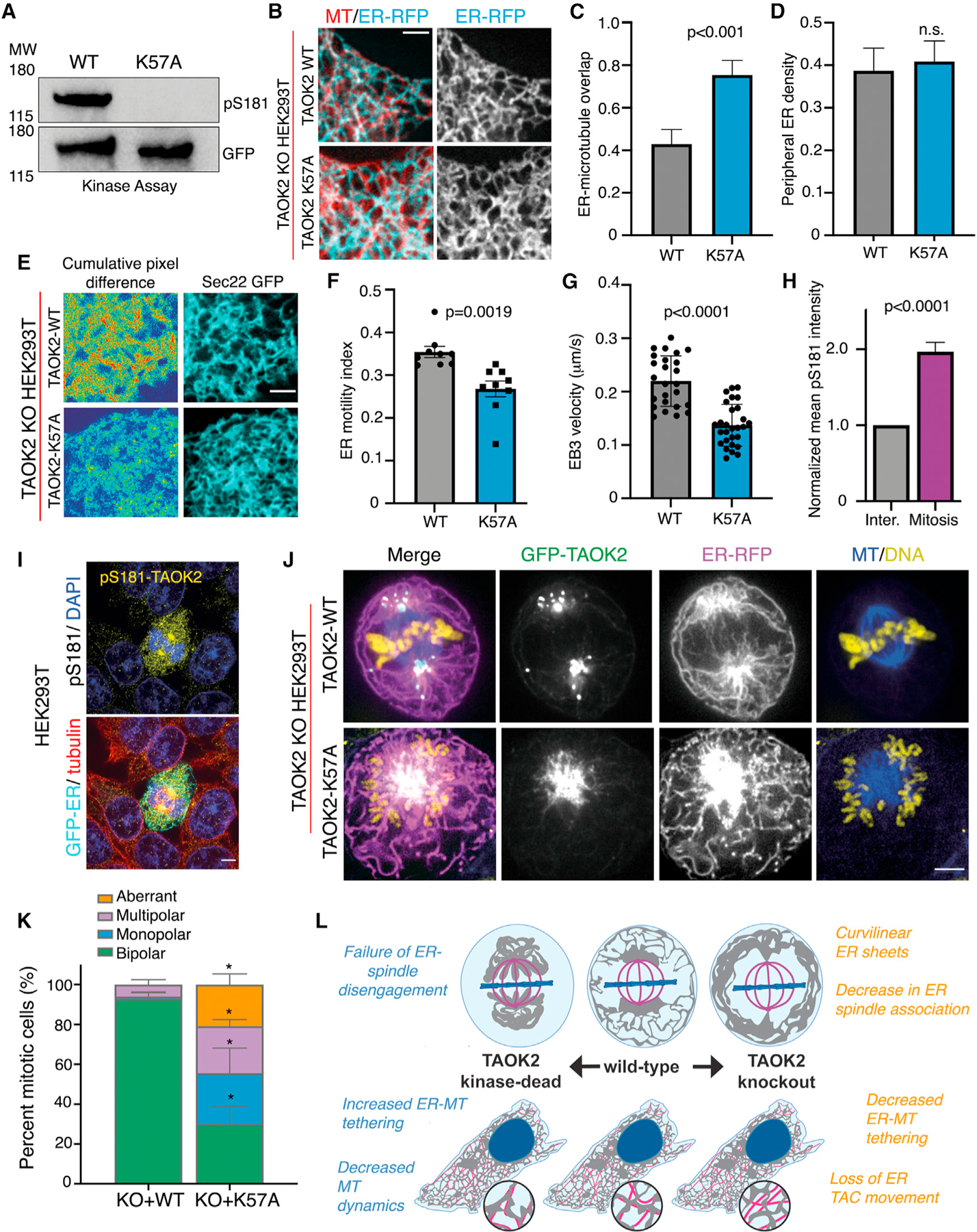
(A) Western blot probed with phospho-S181 antibody to measure kinase activity of GFP-TAOK2 WT and GFP-TAOK2 K57A.
(B) TAOK2 KO cells expressing TAOK2-WT or TAOK2-K57A along with ER-mRFP (cyan) and live-stained with MT dye (red). Scale bar: 3 μm.
(C) Ratio of peripheral ER in contact with MT in a 100-μm2 region in cell periphery is plotted for TAOK2 KO cells expressing either TAOK2-WT or TAOK2-K57A. Values indicate mean ± SEM; n = 6 from 3 experiments; t test with Welch’s correction.
(D) ER density is calculated by measuring the fraction of ER area in a 100-μm2 region in the cell periphery. Values indicate mean ± SEM; n = 6 from 3 experiments; t test with Welch’s correction.
(E) Confocal images of peripheral ER in TAOK2 KO cells expressing TAOK2 WT or TAOK2 K57A along with EGFP-Sec22b at a single time point (left column). Sum slice projection of cumulative pixel difference in successive frames over 30 s; images were acquired every 3 s are shown. Fluorescence intensity is pseudo-colored, red regions representing increased ER motility. Scale bar: 3 μm.
(F) ER-motility index is plotted; error bars indicate SEM; n = 9 cells from 3 experiments; two-tailed t test.
(G) EB3 velocity is plotted; values indicate mean ± SEM; n = 5 cells from 3 experiments with 5 comets/cell; two-tailed t test.
(H) Normalized average intensity of phospho-S181 in mitotic cells is plotted. Error bars indicate SEM, n = 10 cells from 3 experiments; t test with Welch correction.
(I) Confocal images of mitotic and interphase cells stained with phospho-S181 and tubulin antibodies.
(J) Confocal images of TAOK2 KO mitotic cells expressing either GFP-TAOK2 WT or GFP-TAOK2 K57A (green) along with ER-mRFP (magenta) and stained with DNA (yellow) and MT dyes (blue). Scale bar: 5 μm.
(K) Mitotic defects in TAOK2 KO cells expressing either GFP-TAOK2 WT or GFP-TAOK2 K57A. Values indicate mean ± SEM; n = 85 cells from 3 experiments. *p< 0.05.
(L) Representation of pleiotropic function of TAOK2 in maintaining ER-MT tethering, MT dynamics, and ER remodeling during interphase and mitosis. See also Figure S5.
Next, we assessed whether TAOK2 catalytic activity was regulated during mitosis and found that TAOK2 was highly activated during mitosis as has been reported previously (Koo et al., 2017; Wojtala et al., 2011). Immunostaining interphase and mitotic cells with phospho-S181 antibody revealed a 2-fold increase in mitotic cells compared with interphase (Figure 7I). Since this antibody recognizes all isoforms of TAO kinases, we confirmed this result by western blot analyses whereby we can distinguish different TAO proteins based on their molecular weight. Western blot analysis showed a high ratio of pS181-TAOK2/TAOK2 in lysates from synchronized mitotic cells compared to asynchronous cell lysate (Figure 7H). Next, we tested if perturbation of TAOK2 kinase activity would impact either mitosis or ER segregation during cell cycle. We expressed in TAOK2 KO cells either the TAOK2-WT or TAOK2-K57A construct, and then performed four-color imaging of TAOK2, ER membrane, chromosomes, and mitotic MT spindles. We found that in TAOK2-WT-expressing KO cells, both TAOK2 and the ER membranes were enriched at the spindle poles, and a majority of the ER membrane was dissociated from the spindle MTs (Figure 7J). However, in KO cells expressing TAOK2-K57A kinase-dead mutant, TAOK2 association with the spindle MT was markedly increased and ER membranes were extensively associated with the mitotic spindle (Figure 7J). A failure of ER membranes to disengage from the spindle MT led to severe mitotic defects in K57A expressing cells including monopolar (25.5%), multipolar (23.5%), and aberrant spindles (20.8%). The percentage of mitotic cells exhibiting normal bipolar spindles deceased from 92.8% in TAOK2 KO cells expressing TAOK2-WT to 30% in TAOK2-K57A-expressing cells (Figure 7K). These data demonstrate that the increased kinase activity of TAOK2 is important for dynamic regulation of ER-MT tethering during cell division.
DISCUSSION
Our study identifies TAOK2 as an ER-resident kinase that functions as a molecular tether linking the ER membranes to the MT cytoskeleton. We show that distinct structural domains within TAOK2 confer its catalytic activity, ER localization, EB1 interaction, and MT association. Importantly, we demonstrate that the catalytic activity of TAOK2 negatively regulates ER-MT tethering. KO of TAOK2 and expression of kinase-dead TAOK2 have opposing effects on ER-MT tethering and dynamics in both interphase and mitotic cells (Figure 7L). We provide two key pieces of evidence in support of bidirectional autoregulation of tethering by TAOK2. First, we show that the kinase-dead TAOK2 is a stronger ER-MT tether. Second, TAOK2 kinase activity is increased in mitosis, which correlates with a dramatic decrease in the tethering of ER membranes with the mitotic spindle MTs. Perturbation of TAOK2 kinase activity in interphase cells causes increased ER-MT interaction and inhibiting kinase activity in mitotic cells leads to failure of ER disengagement from the mitotic spindle.
While it is well established that the structure of the ER is intimately linked to the MT cytoskeleton, our data provide evidence that the ER can also impact MT dynamics. Increased tethering of ER membranes to the MT cytoskeleton reduces MT growth dynamics as measured by EB3 velocity. Further, enhanced tethering leads to greater MT stability as measured by level of acetylated tubulin. During cell division, aberrant tethering of ER to the mitotic spindle disrupts the dynamics of mitotic spindle, causing mitotic defects. Therefore, our study shows that the ER can regulate MT dynamics both in interphase and mitosis and that TAOK2 as a tethering linker between ER and MT regulates these processes through its catalytic activity.
Our study also revealed another function of TAOK2 in regulating ER-membrane tracking on growing MT plus tips. TAOK2 contains a conserved four-residue motif SxIP (1192TRIP1195), which allows interaction with MT plus end proteins EB (end binding). The SxIP motifs are typically embedded within basic and proline/serine-rich sequence regions and are characteristic features of other +TIPs, such as APC, MACF, CLASP, STIM1, and MCAK (Honnappa et al., 2009). Similarly, we found that SxIP motif within TAOK2 was part of the C-terminal intrinsically disordered region and was surrounded by basic and serine residues. Proteome-wide identification of mammalian S3IP-containing +-TIPs reported several uncharacterized EB partners that have the capacity to accumulate at the growing MT ends. TAOK2 was identified bioinformatically in this study as an EB-binding protein based on its SxIP motif and was also detected in a biotinylation pulldown assay as an EB1 and EB3 (but not EB2) binding partner but was not validated or further characterized (Jiang et al., 2012). Unlike STIM1 which has not been shown to bind MT directly, TAOK2 has binding affinities both for the MT plus tip through association with EB1 and for the MT lattice through association with tubulin, and remarkably these two can be dissociated from each other.
Several ER proteins interact directly or indiretly with microtubules (Klopfenstein et al., 1998; Nikonov et al., 2007; Orso et al., 2009; Pavez et al., 2019; Roll-Mecak and Vale, 2008), how each of them uniquely contributes to cellular physiology is understudied. As a kinase highly expressed in neurons, and one critical for neurodevelopment, the role of TAOK2 as an autoregulated ER-MT tether is likely to serve specific physiological functions in neurons. Neuronal development, connectivity, and plasticity are dependent on the presence of ER membranes within fine neuronal processes such as spines, axons, and dendrites where they are transported along with and tethered to MT. Elucidating TAOK2 function in specialized cells types such as neurons that respond to physiological stimuli by remodeling ER-MT tethering is likely to expand on our current understanding of the importance and dynamics of communication between cellular organelles and the cytoskeleton.
Limitations of the study
Our study shows that TAOK2 is an ER-associated kinase that tethers the ER membrane to MT. We show that TAOK2 is localized within specific subdomains of ER membranes and that many of these are sites of contact with MT. TAOK2 localization in these subdomains is dependent on its amphipathic region and independent of MT binding. Functional relevance of these ER subdomains is unknown. It is also unclear from our results whether these subdomains correspond to domains of specific lipid composition, organelle contact sites, or protein-protein interactions. Finally, while kinase activity of TAOK2 negatively regulates ER-MT tethering, mechanisms that underlie kinase regulation of ER-MT tethering, TAC movement, and EB1-binding properties of TAOK2 were not explored in this study and present interesting avenues to further understand regulation of the dynamic interactions between organelles and cytoskeleton.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Smita Yadav (smitay@uw.edu)
Materials availability
Plasmids and cell lines generated in this study will be shared upon reasonable request.
Data and code availability
Original western blot images and microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture and maintenance
All experiments were performed in HEK293T and HeLa cells, which were grown in DMEM media (Thermo Fisher, Gibco) with 10% fetal bovine serum (Axenia BioLogix) and 1% Pen-Strep (Invitrogen). Cells were maintained at 5% CO2 and 37°C and passaged every 3–4 days.
METHODS DETAILS
Molecular cloning
Full-length human TAOK2 was PCR amplified from TAOK2 cDNA (Origene, isoform alpha), and inserted in vector sfGFP-C1 (Addgene #54579) using restriction sites HindIII and MfeI. Domain dissection mutants were subcloned from sfGFP-TAOK2 using restriction enzymes HindIII and MfeI (New England Biolabs). All resultant plasmids were verified by sequencing. GST-TAOK2-(1187–1235) was subcloned from the sfGFP-TAOK2 into the pGEX4T1 vector using sites SalI and NotI. BFP-STIM1 was created by subcloning BFP from BFP-Rab5 (#49147) and inserting into STIM-YFP (#19754) using restriction sites SalI and NotI.
CRISPR/Cas9 genome editing
Two independent TAOK2 knockout cell lines were generated using CRISPR/Cas9 genome editing in HEK293T cells. Four guides were designed using Synthego guide design tool (https://www.synthego.com) to target coding exon 2. Guides were cloned into plasmid CrisprV2pSpCas9(BB)-2A-Puro (PX459) V2.0 (Addgene Plasmid #62988) which has Puromycin resistance. Cells were passaged in single cell suspension and plated at 50% confluence. Cultures were then transfected with lipofectamine 2000 reagent (Invitrogen 11668–030) and 2mg of each of the 2 guides used per KO line. Cells were then selected with Puromycin for 2 days to select for transfected cells. Non-Homology End Joining (NHEJ) repair created a deletion around the gRNA cutting site. Edited cells were then passaged into single cells and expanded into single cell colonies. Genomic DNA was extracted and the region around the cutting site was PCR amplified to send for sequencing. Knockout of the gene TAOK2 was confirmed by Sanger sequencing analysis, and absence of encoded protein was validated using western blot.
Immunofluorescence and Western blotting
Cells were fixed with 4% paraformaldehyde and 4% sucrose for 20 min at room temperature, followed by 3 washes with phosphate-buffered saline (PBS). One-hour incubation with blocking buffer (200mM Glycine pH 7.4, 0.25% TritonX-100, 10% Normal Donkey Serum, in PBS) was followed by overnight incubation with primary antibody at a 1:1000 dilution in blocking buffer. After three 5min washes in PBS, cells were incubated with secondary antibody at 1:1000 dilution in blocking buffer for 3hr. Coverslips were washed and then mounted onto slides with FluoromontG. Endogenous TAOK2 staining was performed similarly, except cells were fixed with cold methanol incubated on ice for 20 min instead of PFA. Endogenous EB1 staining was adapted from (Grigoriev et al., 2008) with specified changes. Briefly, cells were fixed with −20C methanol on ice for 15 min and subsequently 4% PFA with 4% Sucrose for 15 min. Coverslips were washed with PBS and permeabilized with PBS and 1% TritonX-100. Blocking, washing and antibody dilution steps were performed using PBS with 1% bovine serum albumin and 0.15% Tween-20. Samples for western blot analysis were treated with 4X LDS Sample Buffer (Thermo Fisher) with 125 mM DTT and subsequently heated for 10 min at 95C. Samples were electrophoresed on NuPAGE 4–12% Bis-Tris Polyacrylamide gels (Thermo Fisher) with NuPAGE MOPS running buffer (Thermo Fisher). Western blot transfer to ImmobilonP PVDF membrane (Millipore-Sigma) with Transfer Buffer (25mM Tris, 192mM Glycine, 20% (v/v) Methanol, 0.05% SDS) at 100V for 60min. Resultant blot was blocked in 5% milk or BSA blocking buffer, and subjected to primary antibody and HRP conjugated secondary antibody before visualization with Pierce™ ECL Western Blotting Substrate (Thermo Fisher). Western blot images were obtained using the ChemiDoc Imager (BioRad).
Differential centrifugation assay
HEK293T cells were grown to confluence in four 10cm dishes using DMEM media with 10% fetal bovine serum and 1% Pen-Strep. Cells were washed once with Dulbecco’s PBS, collected in ice cold PBS and pelleted by centrifugation at 200g. Pellet was resuspended in 2 mL of homogenization buffer (250mM sucrose, 10mM HEPES, 1mM EDTA, protease inhibitors (Roche), 1mM PMSF, and 1mM DTT) and homogenized with a 25-gauge syringe needle. Homogenate was subsequently spun at 800g to pellet nuclear fraction. Post nuclear supernatant (S1) was diluted in homogenization buffer to split between two 2mL ultracentrifuge tubes. Heavy membrane fraction (P2) was obtained by centrifuging S1 at 27,000g for 30 min at 4°C. Light membrane fraction (P3) was obtained by centrifuging S2 at 100,00g for 30 min. P3 was resuspended in either 200uL: homogenization buffer (control), or detergent buffer (1% NP-40, 1% TritonX-100, 0.1% SDS) and incubated on ice for 30 min before spinning at 200,000g for 60 min at 4 °C. Resultant high speed pellets (P4) were resuspended in 4x sample buffer with 125mM DTT. Resultant supernatants S4 and cytosolic supernatant fraction S3 were precipitated with ice cold 10% trichloroacetic acid by incubating on ice for 15 minutes followed by centrifugation at 21,000g. Precipitates were washed with ice-cold acetone, and pelleted at 21,000g for 5 min and resuspended in 4x sample buffer with 125 mM DTT. All samples were run on SDS-PAGE gels and transferred to PVDF membrane for western blot analysis.
ER membrane isolation
HEK293T cells were grown to confluence in four 10cm dishes. Cells were washed once with Dulbecco’s PBS, collected in ice cold PBS and pelleted by centrifugation at 200g. Pellet was resuspended in 1mL of isolation buffer (225mM mannitol, 75mM sucrose, 30mM Tris-HCl pH 7.4, 0.1mM EGTA and protease inhibitors (Roche), and homogenized with a 25G needle at 4°C. The homogenate was subsequently subjected to a series of spins at 4°C, retaining the pellet from each and continuing with supernatant to the next spin. The centrifugation schema was as follows (adapted from Hoyer et al., 2018): 2 spins at 600g for 5 min to pellet both cell debris and nuclei (P1 and P2), 3 spins at 7000g for 5 min each to pellet mitochondria (P3, P4, and P5), and a 20,000g spin for 20 min to pellet the crude ER fraction (P6). The supernatant from the last spin yielded the cytosol, and P6 was washed with isolation buffer devoid of EGTA and subject to a 20,000g spin for 15 min at 4°C to re-pellet. P1–6 were resuspended in resuspension buffer (50mM HEPES, 2.5mM MgCl2, 200mM KCl, 5% glycerol, 1% TritonX-100). Protein concentrations of resuspended P1–6 and cytosolic fraction were quantified by BCA assay, and subsequently 4X sample buffer with 0.125M DTT was added. This method of crude organellar separation was adapted from Hoyer et al. (2018). Normalized samples were analyzed by western blot.
Microscopy
Superresolution imaging was performed using the Nikon-CSU-W1 Spinning Disk equipped with a microlensed SoRa emission disk that achieves Super Resolution by Optical Pixel Reassignment with a xy resolution of 120nm. Images were acquired on an inverted Nikon Eclipse Ti2 microscope (Nikon Instruments) attached to a Yokogawa spinning disk unit (CSU-W1 SoRa, Yokogawa Electric) using a 1.49 100x Apo TIRF oil immersion objective lens. Images were captured by an Andor Sona 4.2B-11 camera using the 2.8x SoRa relay, resulting in an effective pixel size of ~40 nm. 405, 488, and 561 nm laser lines were used for excitation. All other live and fixed cell imaging was performed on a Nikon Ti2 Eclipse-CSU-X1 confocal spinning disk microscope equipped with four laser lines 405nm, 488nm, 561nm and 670nm and an sCMOS Andor camera for image acquisition. The microscope was caged within the OkoLab environmental control setup enabling temperature and CO2 control during live imaging. Imaging was performed using Nikon 1.49 100x Apo 100X or 60X oil objectives. Live imaging for ER motility and EB3 comet velocity was performed on fibronectin coated MatTek dishes (MatTek, P35G-1.5–14-C), and images at a single confocal z frame were captured every 2sec. Fixed cell image acquisition was performed as a z stack of images with z distance of 0.3micron. Viafluor MT live imaging dyes (Biotum, #70064, #70063) were used to visualize MTs during live imaging. Cells were incubated for 30 min at 37 degrees C with (1:2000) dye in culture media. Subsequently, dye treated media is replaced with live imaging media with (1:10000) tubulin dye. DNA was stained with NucBlue™ Live ReadyProbes™ Reagent (Hoechst 33342) (Invitrogen), two drops/mL live imaging media incubated for 15 minutes at room temperature before imaging.
Protein purification
TAOK2 C-terminal amino acids 1187–1235 were cloned into pGEX4T1 vector and transformed into BL21 E. coli to bacterially express the GST-tagged TAOK2 1187–1235. A 25ml starter culture grown from a single colony overnight was used to inoculate 1L culture, which was allowed to grow to an O.D, of 0.6 at 37°C. Protein expression was induced by IPTG at a final concentration of 0.4mM for 18 h at 18°C. Bacteria were collected by a 15min spin at 5000g, washed with ice cold PBS, and the pellet was resuspended in ice cold lysis buffer (50mM Tris pH 8.0, 5mM EDTA, 150mM NaCl, 20% glycerol, 5mM DTT, protease inhibitors and PMSF). Addition of 4mg lysozyme (Sigma) was followed by 30min incubation with 0.5% TritonX-100 and sonication on ice. The supernatant was collected after a 60min spin at 25,000g, and incubated with prewashed GST beads (Thermo Fisher) for 1 h. Beads were washed with wash buffer (PBS + 1mM DTT + 0.1% tween 20) followed by wash buffer without detergent. Bound protein was eluted and collected in fractions by glutathione elution buffer at pH8.0 (50mM Tris pH8.0, 250mM KCl, 1mM DTT, 10% glycerol and 30mM glutathione).
MT co-sedimentation assay
MTs were prepared by polymerizing porcine tubulin (Cytoskeleton inc.) in general tubulin buffer (80mM PIPES pH 6.9, 2mM MgCl2, 0.4mM EGTA, Roche Protease Inhibitors) in the presence of 1mM GTP for 20 min at 35C and then diluted further. To prevent depolymerization, MTs were treated with 40μM Taxol. MTs and 5μg purified protein were incubated at room temperature, and pelleted at 100,000g over a 60% glycerol cushion buffer (80mM PIPES pH 7.0, 1 mM MgCl2, 1 mM EGTA, 60% Glycerol, protease inhibitor). The supernatant (top layer above cushion) and the pellet were removed and treated with 4X Sample Buffer with 250mM DTT and 5% beta-mercaptoethanol. Resultant samples were subject to SDS-PAGE and colloidal Coomassie blue staining (Invitrogen).
Mitotic cell lysate
HEK293T cells were synchronized by treatment with 1.67μM nocodazole for 12–16 h. Rounded cells were dislodged by shaking and collected with media. Concurrently, untreated asynchronously growing HEK293T cells were scraped and collected in DPBS. Both tubes of cells were separately pelleted, washed in DPBS, and lysed in HKT buffer (25mM HEPES pH7.2, 150mM KCl, 1% Triton X-100, 1mM DTT, 1 mM EDTA, Protease Inhibitors (Roche, Complete), Halt Phosphatase Inhibitors (Thermo Fisher). Lysate was cleared of cell debris by centrifugation and protein concentrations were determined via BCA assay (Thermo Fisher). Sample Buffer with 125mM DTT was added to equalized amounts of protein and subject to western blot analysis as described above.
Immunoprecipitation kinase assay
HEK293T cells transfected with sfGFP-TAOK2 WT and sfGFP-TAOK2 K57A were lysed with HKT buffer (25mM HEPES pH7.2, 150mM KCl, 1% Triton X-100, 1mM DTT, 1 mM EDTA, Protease Inhibitors (Roche, Complete)). Lysate was precleared with Pierce ProteinG Agarose (Thermo Fisher), and immunoprecipitated with Roche anti-GFP Mouse antibody bound on Pierce protein G agarose. Beads were washed twice with HKT, once for 10 min with HKT with 1mM NaCl, and finally washed with HK buffer (25mM HEPES pH 7.2, 150mM KCl, 1mM DTT, 1 mM EDTA, Protease Inhibitors (Roche, Complete EDTA free)). Beads were washed once with the Kinase Buffer (25mM Tris pH 7.5, 10mM MgCl2, 1mM DTT) prior to the in vitro kinase assay. Kinase assay was then performed by incubating with 0.5mM ATP and Halt protease/phosphatase inhibitors (Thermo Fisher) for 45 min at 30°C on a shaking heat block. Samples were then subjected to western blot analysis detailed above to detect autophosphorylation of TAOK2 at S181 using the rabbit antibody (R&D Systems, PPS037).
CP43 Kinase Assay: Endogenous TAOK2 was immunoprecipitation from untransfected HeLa cells using the TAOK2 Sigma antibody. CP43 (500nM) was added to kinase assay along with 0.5mM ATP, Halt protease/phosphatase inhibitors (Thermo Fisher) and GST-Septin 7 C-terminal coiled coil tail (321–438 amino acids) [Purified as described in Yadav et al., 2017)] as a substrate for 45 min at 30°C on a shaking heat block.
Immunoprecipitation of endogenous EB1 was adapted from (Hoogenraad et al., 2000). Briefly, HeLa cells were lysed with HKT buffer (25mM HEPES pH7.2, 100mM KCl, 0.5% Triton X-100, 1mM DTT, 1 mM EDTA, Protease Inhibitors (Roche, Complete)). Lysate was precleared with Pierce Protein-G Agarose (Thermo Fisher), and immunoprecipitated with anti-GFP Mouse (control), anti-EB1 mouse and anti-STIM1 antibodies separately bound on Pierce protein G agarose. Beads were washed thrice with HKT and washed thrice with HK buffer (25mM HEPES pH 7.2, 100mM KCl, 1mM DTT, 1 mM EDTA) then treated with 4X Sample Buffer with 250mM DTT and 5% beta-mercaptoethanol. Resultant samples were subject to SDS-PAGE.
QUANTIFICATION AND STATISTICAL ANALYSES
Image analysis and quantification
MT growth analysis was performed using the ImageJ plugin for manually tracking objects (Manual Tracking). Single frame time lapse image stacks were processed to measure the distance traveled by EB3 comets in each frame. Velocity was calculated by dividing the total distance traveled by the time taken. Curvature was calculated by dividing the minimum linear path (from beginning to end) by the length of the actual distance the EB3 comet traveled. Time paused was calculated by measuring the number of times the EB3 comet did not change coordinates multiplied by time between each frame (2s). Percent time paused is calculated by divided by the total time the comet was tracked. TAC - EB3 comet quantification was done by counting the number of EB3 comets showing different EB and STIM1 properties during first 10 frames of images acquired every 2 seconds apart. ‘‘Stim1-TAC’’ contacts was counted as percent of EB3 that were positive for STIM1 and where the ER/STIM1 extended along EB3. ‘‘Contact with STIME’’ showed initial EB3-STIM1 contact but no extension of the ER occurred. ‘‘No contact with STIM1’’ represents EB3 comets that did not colocalize with the STIM1/ER during the studied frames. Number of EB3 comets was measured by counting comets in images obtained as z-stacks with 0.1 micron slicing whole cell expressing EB3-mcherry and dividing by the area of the cell. Endogenous EB1 comet number was measured by counting comets in a 100-micron ROI in cells immunostained for EB1.
ER motility was measured from single-z frame image stacks acquired from imaging the ER markers (EGFP-Sec22 or ER-mRFP) using ImageJ. Substacks were created corresponding to frames 6s apart and pixel differences every 6s were calculated using the Stack Difference function to determine a change in fluorescence (ΔF). ΔF was then divided by the mean fluorescence (F) of the earliest time point frame from which it was derived. (i.e., ΔF between frames 3 and 4 would be divided by frame 3) These ΔF/F values were taken for each time point and averaged over 2 min to determine ER motility for each cell. To obtain the cumulative pixel difference the substack obtained from using the Stack Difference function was z-projected with the ‘sum slices’ option, and then pseudocolored using the physics LUT. This method to calculate ER motility index is an adaptation from Dong et al. (2018) with specified changes.
Manders’ Overlap Coefficient (MOC) (Manders et al., 1993) was measured using Just Another Colocalization Plugin (JACoP) (Bolte and Cordelières, 2006). TAOK2 punctae overlap with EB1 and STIM1 was analyzed using a single frame 100-micron ROI in the cell periphery by counting independent and overlapping punctae. The ratio of acetylated tubulin to total a-tubulin was measured from images of immunostained cells acquired as z-stacks with 0.1micron z-steps. Maximum z-projections were thresholded and analyzed for area of acetylated tubulin and a-tubulin respectively. Ratios were determined by dividing the area of acetylated tubulin by that of a-tubulin in each cell.
ER-MT overlap was measured using single frames of images cropped to a 100-micron ROIs at the cell periphery, split into respective channels and thresholded. Total area of ER and MTs was measured using ‘‘analyze particles’’ option on imageJ. Overlapping area was determined by using the ‘image calculator’ AND function on thresholded images of ER and MTs; resultant area was measured using ‘‘analyze particles.’’ Overlap was measured as the ratio of area of overlap to the area of the ER. ER density was subsequently measured as the area of the ER divided by the area of the ROI. These analyses were performed on WT and TAOK2 KO HEK293T cells expressing mEmerald-ensconsin and ER-mRFP, as well as TAOK2 KO HEK293T cells expressing ER-mRFP, TAOK2 WT and K57A respectively, stained with Viafluor MT live imaging dyes (Biotum, #70064, #70063). To assess mitotic defects, four color images were acquired as z-stacks with 0.3micron spacing such that the entire mitotic cell was captured. Mitotic defects were scored manually by visualizing the entire z stack, based on the spindle morphology, ER morphology and chromosomal localization.
Statistics
All statistics were performed in GraphPad software Prism9.0. Multiple groups were analyzed using ANOVA, while two group comparisons were made using unpaired t test unless otherwise stated. Statistically p value less that 0.05 was considered significant. All experiments were done in triplicate, and experimental sample size and p values are indicated with the corresponding figures.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
KEY RESOURCES TABLE
| Reagent or resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
|
| ||
| Mouse monoclonal anti-alpha-Tubulin | Sigma-Aldrich | Cat# T9026; RRID: AB_477593 |
| Mouse monoclonal anti-GAPDH | Invitrogen | Cat# MA5–15738; RRID: AB_10977387 |
| Mouse monoclonal anti-Calreticulin | Abcam | Cat# ab22683; RRID: AB_447253 |
| Rabbit polyclonal anti-TAOK2 | Sigma-Aldrich | Cat# HPA010650; RRID: AB_1080204 |
| Rabbit polyclonal anti-Rtn3a | ProteinTech | Cat# 12055–2-AP; RRID: AB_2301357 |
| Mouse monoclonal anti-Acetylated alpha Tubulin | Sigma-Aldrich | Cat# T6793; RRID: AB_477585 |
| Mouse monoclonal anti-GST | Invitrogen | Cat# 13–6700; RRID: AB_2533028 |
| Mouse monoclonal anti-GM130 | BD Labs | Cat# 610822; RRID: AB_398141 |
| Rabbit polyclonal anti-TAOK2-Cterm | This paper | N/A |
| Mouse monoclonal anti-Stim1 | Santa Cruz Biotech | sc-166840; RRID: AB_2198006 |
| Mouse anti-GFP | Roche | Cat# 11814460001; RRID: AB_390913 |
| Rabbit polyclonal anti-Phospho-TAO2 (S181) | R&D Systems | Cat# PPS037; RRID: AB_2255678 |
| Mouse monoclonal anti-Tom20 | Santa Cruz Biotech | Cat# sc-17764; RRID: AB_628381 |
| Mouse monoclonal anti-EB1 | Santa Cruz Biotech | Cat# sc-47704; RRID: AB_2141631 |
| Rabbit anti-Alpha Tubulin | Cell Signaling | Cat# 2144; RRID: AB_2210548 |
|
| ||
| Bacterial strains | ||
|
| ||
| BL21 (DE3) E. coli | Invitrogen | Cat #EC0114 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Lipofectamine 2000 reagent | Invitrogen | Cat#11668–030 |
| Purified tubulin (>99%) Porcine Brain | Cytoskeleton Inc. | Cat# T240-A |
| Halt Phosphatase Inhibitors | Thermo Fisher | Cat# 78428 |
| CP43 | Tocris | Cat#6558, CAS 850467–66-2 |
| GST-TAOK2C (1187–1235) | This paper | N/A |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| HEK293T | ATCC | Cat# CRL-3216 |
| HEK TAOK2 KO1 | This paper | N/A |
| HEK TAOK2 KO2 | This paper | N/A |
| HeLa | ATCC | Cat# CCL-2 |
|
| ||
| Oligonucleotides | ||
|
| ||
| TAOK2 KO gRNA 1: GAAATGGCAAGACATCATCA |
This paper | N/A |
| TAOK2 KO gRNA 2: ATGGCAAGACATCATCAAGG |
This paper | N/A |
| TAOK2 KO gRNA 3: TGCGGTTCTTACAGAAGCTC |
This paper | N/A |
| TAOK2 KO gRNA 4: AGTACCGGGGCTGTTACCTG |
This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| mCh-Sec61 beta | Addgene | Cat# 49155 |
| sfGFP-C1 | Addgene | Cat# 54579 |
| ER-mRFP | Addgene | Cat# 62236 |
| EGFP-Sec22b | Addgene | Cat# 101918 |
| BFP-Sec61 | Addgene | Cat# 49154 |
| Stim1-YFP | Addgene | Cat# 19754 |
| EB3-mcherry | Addgene | Cat# 55037 |
| mito-BFP | Addgene | Cat# 49151 |
| mTagBFP2-Lysosomes20 | Addgene | Cat# 55308 |
| Rab5-BFP | Addgene | Cat# 49147 |
| mEmerald Ensconsin-C-18 | Addgene | Cat# 62753 |
| sfGFP-TAOK2 | This paper | Origene CAT#: RC214297 |
| GST-TAOK2-(1187–1235) | This paper | N/A |
| BFP-STIM1 | This paper | From Cat# 19754 |
|
| ||
| Software and algorithms | ||
|
| ||
| GraphPad software | Prism 9.0 | https://www.graphpad.com/scientific-software/prism/ |
| TMHMM2.0 | Krogh et al., 2001 | https://services.healthtech.dtu.dk/service.php?TMHMM-2.0 |
| Heliquest | Gautier et al., 2008 | https://heliquest.ipmc.cnrs.fr/ |
| AMPHIPASEEK | Combet et al., 2000 | https://npsa-prabi.ibcp.fr/NPSA/npsa_amphipaseek.html |
| AlphaFold2.0 | Jumper et al., 2021 | https://alphafold.ebi.ac.uk/ |
Highlights.
TAOK2 is an ER kinase that directly binds microtubules, coupling ER to microtubules
Loss of TAOK2 increases ER and microtubule dynamics
TAOK2 associates with EB1 and STIM1 to regulate ER movement on microtubule plus ends
Catalytic activity of TAOK2 drives untethering of ER from the mitotic spindle Nourbakhsh et al., 2021, Developmental Cell 56, 1–13
ACKNOWLEDGMENTS
We are grateful for funding provided by the National Institutes of Health (R00MH108648 and R01MH121674) to S.Y. and the NARSAD Young Investigator Award (27818) to S.Y. K.N. was supported by the NIH predoctoral Pharmacological Sciences Training Program (T32GM007750). We thank Dan Fong (Nikon) for technical assistance on the Nikon CSU-W1 SoRa superresolution microscope. We are grateful to Brian Beliveau (UW), Cole Trapnell (UW), and Jay Shendure (UW, HHMI) for use of their CSU-W1 SoRa superresolution microscope funded by HHMI. Thanks to John D. Scott (UW), Ning Zheng (UW, HHMI), and Shao-En Ong (UW) for comments on the manuscript.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.devcel.2021.11.015.
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science.
REFERENCES
- Carlton JG, Jones H, and Eggert US (2020). Membrane and organelle dynamics during cell division. Nat. Rev. Mol. Cell Biol 21, 151–166. [DOI] [PubMed] [Google Scholar]
- Chen Z, Hutchison M, and Cobb MH (1999). Isolation of the protein kinase TAO2 and identification of its mitogen-activated protein kinase/extracellular signal-regulated kinase kinase binding domain. J. Biol. Chem 274, 28803–28807. [DOI] [PubMed] [Google Scholar]
- Chen Z, Raman M, Chen L, Lee SF, Gilman AG, and Cobb MH (2003). TAO (thousand-and-one amino acid) protein kinases mediate signaling from carbachol to p38 mitogen-activated protein kinase and ternary complex factors. J. Biol. Chem 278, 22278–22283. [DOI] [PubMed] [Google Scholar]
- Combet C, Blanchet C, Geourjon C, and Deléage G (2000). NPS@: network protein sequence analysis. Trends Biochem. Sci 25, 147–150. [DOI] [PubMed] [Google Scholar]
- Cui-Wang T, Hanus C, Cui T, Helton T, Bourne J, Watson D, Harris KM, and Ehlers MD (2012). Local zones of endoplasmic reticulum complexity confine cargo in neuronal dendrites. Cell 148, 309–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Anda FC, Rosario AL, Durak O, Tran T, Gräff J, Meletis K, Rei D, Soda T, Madabhushi R, Ginty DD, et al. (2012). Autism spectrum disorder susceptibility gene TAOK2 affects basal dendrite formation in the neocortex. Nat. Neurosci 15, 1022–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong R, Zhu T, Benedetti L, Gowrishankar S, Deng H, Cai Y, Wang X, Shen K, and De Camilli P (2018). The inositol 5-phosphatase INPP5K participates in the fine control of ER organization. J. Cell Biol 217, 3577–3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farías GG, Fréal A, Tortosa E, Stucchi R, Pan X, Portegies S, Will L, Altelaar M, and Hoogenraad CC (2019). Feedback-driven mechanisms between microtubules and the endoplasmic reticulum instruct neuronal polarity. Neuron 102, 184–201.e8. [DOI] [PubMed] [Google Scholar]
- Friedman JR, Webster BM, Mastronarde DN, Verhey KJ, and Voeltz GK (2010). ER sliding dynamics and ER-mitochondrial contacts occur on acetylated microtubules. J. Cell Biol 190, 363–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier R, Douguet D, Antonny B, and Drin G (2008). HELIQUEST: a web server to screen sequences with specific alpha-helical properties. Bioinformatics 24, 2101–2102. [DOI] [PubMed] [Google Scholar]
- Giacomini C, Koo C-Y, Yankova N, Tavares IA, Wray S, Noble W, Hanger DP, and Morris JDH (2018). A new TAO kinase inhibitor reduces tau phosphorylation at sites associated with neurodegeneration in human tauopathies. Acta Neuropathol. Commun 6, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoriev I, Gouveia SM, van der Vaart B, Demmers J, Smyth JT, Honnappa S, Splinter D, Steinmetz MO, Putney JW, Hoogenraad CC, and Akhmanova A (2008). STIM1 is a MT-plus end-tracking protein involved in remodeling of the ER. Curr. Biol 18, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Li D, Zhang S, Yang Y, Liu J-J, Wang X, Liu C, Milkie DE, Moore RP, Tulu US, et al. (2018). Visualizing intracellular organelle and cytoskeletal interactions at nanoscale resolution on millisecond timescales. Cell 175, 1430–1442.e17. [DOI] [PubMed] [Google Scholar]
- Honnappa S, Gouveia SM, Weisbrich A, Damberger FF, Bhavesh NS, Jawhari H, Grigoriev I, van Rijssel FJA, Buey RM, Lawera A, et al. (2009). An EB1-binding motif acts as a microtubule tip localization signal. Cell 138, 366–376. [DOI] [PubMed] [Google Scholar]
- Hoyer MJ, Chitwood PJ, Ebmeier CC, Striepen JF, Qi RZ, Old WM, and Voeltz GK (2018). A novel class of ER membrane proteins regulates ER-associated endosome fission. Cell 175, 254–265.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Shibata Y, Zhu P-P, Voss C, Rismanchi N, Prinz WA, Rapoport TA, and Blackstone C (2009). A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell 138, 549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang K, Toedt G, Montenegro Gouveia S, Davey NE, Hua S, van der Vaart B, Grigoriev I, Larsen J, Pedersen LB, Bezstarosti K, et al. (2012). A proteome-wide screen for mammalian SxIP motif-containing microtubule plus end tracking proteins. Curr. Biol 22, 1800–1807. [DOI] [PubMed] [Google Scholar]
- Jongsma MLM, Berlin I, and Neefjes J (2015). On the move: organelle dynamics during mitosis. Trends Cell Biol 25, 112–124. [DOI] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klopfenstein DR, Kappeler F, and Hauri HP (1998). A novel direct interaction of endoplasmic reticulum with microtubules. EMBO J 17, 6168–6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo C-Y, Giacomini C, Reyes-Corral M, Olmos Y, Tavares IA, Marson CM, Linardopoulos S, Tutt AN, and Morris JDH (2017). Targeting TAO kinases using a new inhibitor compound delays mitosis and induces mitotic cell death in centrosome amplified breast cancer cells. Mol. Cancer Ther 16, 2410–2421. [DOI] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, and Sonnhammer EL (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol 305, 567–580. [DOI] [PubMed] [Google Scholar]
- Manning G, Plowman GD, Hunter T, and Sudarsanam S (2002). Evolution of protein kinase signaling from yeast to man. Trends Biochem. Sci 27, 514–520. [DOI] [PubMed] [Google Scholar]
- Mitsopoulos C, Zihni C, Garg R, Ridley AJ, and Morris JDH (2003). The prostate-derived sterile 20-like kinase (PSK) regulates microtubule organization and stability. J. Biol. Chem 278, 18085–18091. [DOI] [PubMed] [Google Scholar]
- Moore TM, Garg R, Johnson C, Coptcoat MJ, Ridley AJ, and Morris JD (2000). PSK, a novel STE20-like kinase derived from prostatic carcinoma that activates the c-Jun N-terminal kinase mitogen-activated protein kinase pathway and regulates actin cytoskeletal organization. J. Biol. Chem 275, 4311–4322. [DOI] [PubMed] [Google Scholar]
- Nikonov AV, Hauri H-P, Lauring B, and Kreibich G (2007). Climp-63-mediated binding of microtubules to the ER affects the lateral mobility of translocon complexes. J. Cell Sci 120, 2248–2258. [DOI] [PubMed] [Google Scholar]
- Nourbakhsh K, and Yadav S (2021). Kinase signaling in dendritic development and disease. Front. Cell. Neurosci 15, 624648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orso G, Pendin D, Liu S, Tosetto J, Moss TJ, Faust JE, Micaroni M, Egorova A, Martinuzzi A, McNew JA, and Daga A (2009). Homotypic fusion of ER membranes requires the dynamin-like GTPase atlastin. Nature 460, 978–983. [DOI] [PubMed] [Google Scholar]
- Pavez M, Thompson AC, Arnott HJ, Mitchell CB, D’Atri I, Don EK, Chilton JK, Scott EK, Lin JY, Young KM, et al. (2019). STIM1 is required for remodeling of the endoplasmic reticulum and microtubule cytoskeleton in steering growth cones. J. Neurosci 39, 5095–5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendin D, McNew JA, and Daga A (2011). Balancing ER dynamics: shaping, bending, severing, and mending membranes. Curr. Opin. Cell Biol 23, 435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter M, Murtaza N, Scharrenberg R, White SH, Johanns O, Walker S, Yuen RKC, Schwanke B, Bedürftig B, Henis M, et al. (2019). Altered TAOK2 activity causes autism-related neurodevelopmental and cognitive abnormalities through RhoA signaling. Mol. Psychiatry 24, 1329–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roll-Mecak A, and Vale RD (2008). Structural basis of microtubule severing by the hereditary spastic paraplegia protein spastin. Nature 451, 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaitz A-L, Thompson J, Wong CCL, Yates JR 3rd, and Heald R (2013). REEP3/4 ensure endoplasmic reticulum clearance from metaphase chromatin and proper nuclear envelope architecture. Dev. Cell 26, 315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scorrano L, De Matteis MA, Emr S, Giordano F, Hajnóczky G, Kornmann B, Lackner LL, Levine TP, Pellegrini L, Reinisch K, et al. (2019). Coming together to define membrane contact sites. Nat. Commun 10, 1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth JT, Schoborg TA, Bergman ZJ, Riggs B, and Rusan NM (2015). Proper symmetric and asymmetric endoplasmic reticulum partitioning requires astral microtubules. Open Biol 5, 1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapp EL, Sharma A, Lippincott-Schwartz J, and Hegde RS (2006). Monitoring chaperone engagement of substrates in the endoplasmic reticulum of live cells. Proc. Natl. Acad. Sci. USA 103, 6536–6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasaki M, Chen LB, and Fujiwara K (1986). Microtubules and the endoplasmic reticulum are highly interdependent structures. J. Cell Biol 103, 1557–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasaki M, and Reese TS (1994). Interactions among endoplasmic reticulum, microtubules, and retrograde movements of the cell surface. Cell Motil. Cytoskeleton 29, 291–300. [DOI] [PubMed] [Google Scholar]
- Ultanir SK, Yadav S, Hertz NT, Oses-Prieto JA, Claxton S, Burlingame AL, Shokat KM, Jan LY, and Jan Y-N (2014). MST3 kinase phosphorylates TAO1/2 to enable myosin va function in promoting spine synapse development. Neuron 84, 968–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voeltz GK, Rolls MM, and Rapoport TA (2002). Structural organization of the endoplasmic reticulum. EMBO Rep 3, 944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman-Storer CM, and Salmon ED (1998). Endoplasmic reticulum membrane tubules are distributed by microtubules in living cells using three distinct mechanisms. Curr. Biol 8, 798–806. [DOI] [PubMed] [Google Scholar]
- Wojtala RL, Tavares IA, Morton PE, Valderrama F, Thomas NSB, and Morris JDH (2011). Prostate-derived sterile 20-like kinases (PSKs/TAOKs) are activated in mitosis and contribute to mitotic cell rounding and spindle positioning. J. Biol. Chem 286, 30161–30170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Carvalho P, and Voeltz GK (2018). Here, there, and everywhere: the importance of ER membrane contact sites. Science 361, eaan5835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, and Voeltz GK (2021). Reticulon-3 promotes endosome maturation at ER membrane contact sites. Dev. Cell 56, 52–66.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav S, Oses-Prieto JA, Peters CJ, Zhou J, Pleasure SJ, Burlingame AL, Jan LY, and Jan Y-N (2017). TAOK2 kinase mediates PSD95 stability and dendritic spine maturation through Septin7 phosphorylation. Neuron 93, 379–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original western blot images and microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.