

Abstract
Antigen specific immunotherapy is a long term goal for the treatment of autoimmune diseases, however developing a means of therapeutically targeting autoimmune T cells in an antigen-specific manner has been difficult. Through the engineering of an HLA-DR1 chimeric antigen receptor (CAR), we have produced CD8+ CAR T cells that target CD4+ T cells in an antigen specific manner, and tested their ability to inhibit the development of autoimmune arthritis in a mouse model. The DR1 CAR molecule was engineered to contain CD3ζ activation and CD28 signaling domains and a covalently linked autoantigenic peptide from type II collagen (DR1-CII) to provide specificity for targeting the autoimmune T cells. Stimulation of the DR1-CII CAR T cells by an anti-DR antibody induced cytokine production indicating that the DR1-CAR functions as a chimeric molecule. In vitro CTL assays using cloned CD4+ T cells as target cells demonstrated that the DR1-CII CAR T cells efficiently recognize and kill CD4+ T cells that are specific for the CII autoantigen. The CTL function was highly specific, as no killing was observed using DR1-restricted CD4+ T cells that recognize other antigens. When B6.DR1 mice, in which autoimmune arthritis had been induced, were treated with the DR1-CII CAR T cells, the CII-specific autoimmune CD4+ T cell response was significantly decreased, autoantibody production was suppressed, and the incidence and severity of the autoimmune arthritis was diminished. These data demonstrate that HLA-DR CAR T cells have the potential to provide a highly specific therapeutic approach for the treatment of autoimmune disease.
Introduction
Autoimmune diseases are serious health problems, affecting more than 4% of the population (1). Therapeutic modalities for autoimmune diseases have been largely based on non-specific immunosuppression that can lead to significant long-term side effects. Although the biologic drugs currently used for autoimmune therapy are an improvement, immunosuppressive effects are still a health concern for many patients (2). While understanding the origins of autoimmune pathogenesis remains a complex problem, it is clear that most are associated with the expression of specific HLA class II alleles (3). Implicit in this association is a role for CD4+ T cells, and the mechanism by which MHC class II molecules stimulate CD4+ T cells is well defined. Class II molecules bind peptides and the resulting MHC:peptide complexes serve as ligands for the T cell receptor (TCR) expressed by CD4+ T cells. Peptide binding is dependent on the interaction of its amino acid side chains with five pockets located in the cleft of the class II binding groove (4), and these pockets are the primary locations of the class II allelic polymorphisms. Thus, allele specific binding of peptides by disease associated HLA class II is likely a basis for the susceptibility to autoimmunity. One example is rheumatoid arthritis (RA) in which susceptibility is associated with select DR alleles, including DR1 (DRB1*01:01) and DR4 (DRB1*04:01, *04:04, and*04:05) (5) that share a stretch of amino acids at positions 70 to 74 in the DRB1 chains (6), termed the shared epitope. This observation suggests that collectively these RA-associated alleles bind and present the same autoantigenic peptide(s) to CD4+ T cells that promote the autoimmune disease.
Targeting the pathogenic CD4+ T cells that recognize the autoantigens presented by RA-associated HLA-DR alleles would be a highly effective means of treating the autoimmune disease and would avoid the creation of a state of general immunosuppression that is commonly associated with current therapies for RA. The difficulty in designing antigen specific modalities for treating autoimmune diseases is determining which antigen(s) is driving the autoimmune T cell response and how to use this information to target the CD4+ T cells that are mediating the pathogenesis of the disease. While advances have been made at identifying autoantigens for a number of autoimmune diseases, a means of therapeutically targeting the pathogenic CD4+ T cells that are stimulated by these autoantigens remains elusive.
Recent advancements in cancer immunotherapy have demonstrated an effective means of targeting and eliminating cancer cells in an antigen specific manner. This immunotherapy is based on the use of CD8+ chimeric antigen receptor (CAR) T cells in which the CAR molecules are genetically engineered proteins. In most cases, the CAR extracellular domains are composed of antibody single chain variable fragment (scFv) that provide the specificity for an antigen expressed by the cancer cell, and the CAR intracellular domains are derived from TCR signaling proteins, e.g. CD28 and CD3ζ, that link the CAR with the signaling pathway used by endogenous TCR (7). cDNA constructs encoding these anti-cancer CAR are inserted into CD8+ T cells using replication deficient viral vectors that have been extensively developed for gene therapy (8). Essentially these engineered CD8+ T cells are re-targeted CTL that use the specificity of the scFv CAR to identify the target cell and the endogenous lytic mechanism of the CTL. Once the scFv CAR binds to the target cell, the CTL is activated via the CAR intracellular activation and signaling domains and the CTL is stimulated and lyses the cancer cell. Several of these scFv CAR T cell therapies for cancer are now FDA approved, and many more are in clinical trials.
The challenge to applying the scFv CAR approach to the treatment of autoimmune diseases is defining a cellular target that is specific for the pathogenic CD4+ T cells. To circumvent this issue, we have developed a novel CAR therapeutic approach for treating autoimmunity that targets only the pathogenic CD4+ T cells that are mediating the pathology of the autoimmune disease, and targets them in a manner that is dependent on the antigen specificity of their TCR. To achieve this, we designed CAR molecules based on HLA-DRB1*01:01 (DR1) that incorporates a model autoantigen as part of its molecular structure, and linked both the HLA-DRB1 and DRA1 chains to CD28 and CD3ζ activation and signaling domains in the transmembrane and intracellular portion of each DR1 chain. Upon transduction, the resulting DR1 CAR T cells lyse CD4+ T cells in an antigen specific manner, i.e. only CD4+ T cells expressing a TCR restricted to DR1 and specific for the antigenic peptide in the DR1 CAR are lysed. Using our HLA-DRB1*01:01 humanized mouse model of RA in which autoimmunity is induced by immunization with type II collagen (CII), our studies demonstrate that DR1-CII CAR T cells that include the immunodominant peptide from the autoantigen CII identify and lyse CII-specific CD4+ T cells in vivo, and effectively inhibit the autoimmune T cell response. In addition, following autoimmune disease induction, the treatment of the B6.DR1 mice with DR1-CII CAR CD8+ T cells significantly reduced both the B cell autoantibody response and inhibited the severity and incidence of the autoimmune arthritis. In all, these data demonstrate the potential utility of MHC class II based CAR T cells in treating autoimmune diseases in an antigen specific manner.
Materials & Methods
Mice.
The generation of DRB1*0101 transgenic mice (DR1) has been previously described (9). The DRA1 and DRB1 transgenes were established in (C57BL/6 x SJL/J) F2 mice, backcrossed to the B6 background, and the I-Ab molecule expressed by the B6 mice was genetically deleted by backcrossing with the B6.129-H2dlAb1-Ea knockout strain (stock number 003584, Jackson Laboratories). All mice used in these studies were bred in our facility, maintained in micro-isolators in a pathogen-free environment, and were fed standard rodent chow (Ralston Purina) and water ad libitum. All animal studies were approved by the institutional animal care and use committee.
CAR T cells.
The functional design of the DR1 CAR molecules is shown in Figure 1A. cDNA encoding the HLA-DR1*01:01 CAR was cloned into the MSGV1-28z mut1-3 (MSGV) retroviral expression vector (gift of Dr. James Kochenderfer, NIH) (8) using In-Fusion cloning (Takara Bio). The DR1B and DR1A chains were separated by a ribosome skipping T2A sequence allowing both chains to be expressed off the same promoter in a single mRNA transcript that is translated as two separate peptide chains (Figure 1B) (10). The endogenous DR1 transmembrane and cytoplasmic domains were removed and replaced with the corresponding domains from CD28 and CD3ζ encoded in MSGV (Figure 1). The DR1 cDNA used is also chimeric for mouse class II by replacement of the second domains of DRA1 and DRB1 with corresponding domains derived from mouse I-E to enable interaction of the DR1 molecule with murine CD4 (9). To load the DR1 molecule with a covalently linked antigenic peptide (Figure 1), cDNA encoding the peptide sequences for the CII peptide (GIAGFKGEQGPKGEP; pMSGV1-DR1-CII-28z mut1-3) or the control HA peptide (PKYVKQNTLKLAT; pMSGV1-DR1-HA-28z mut1-3) were added to the amino-terminus of DRB1 chain as previously described (11).
Figure 1. Molecular design of the HLA-DR1 CAR molecule for targeting CD4+ T cells.
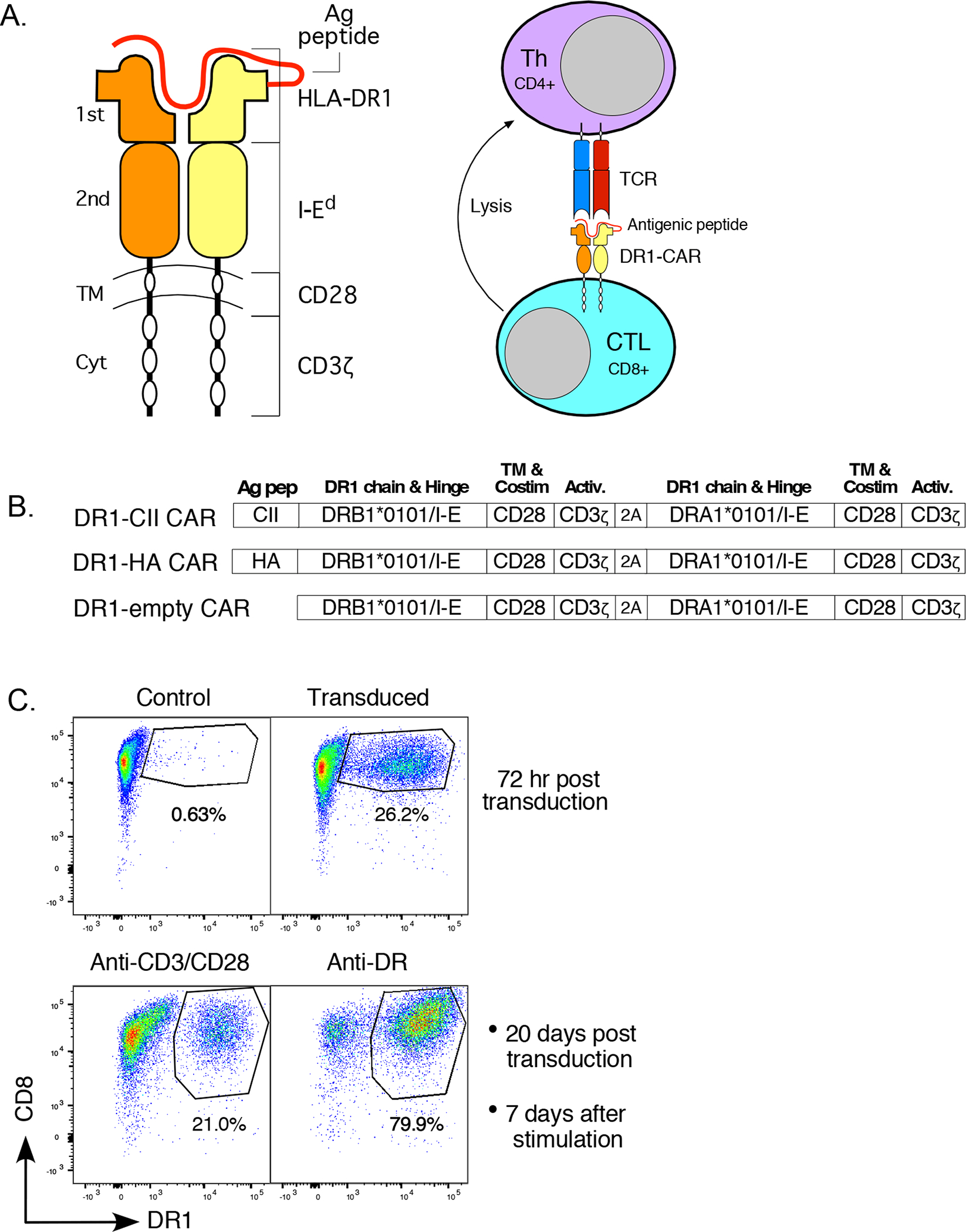
A. The HLA-DR1 CAR molecule consists of HLA-DR 1st domains, an antigenic peptide covalently linked to the DRB1 chain, second domains from I-Ed to enable murine CD4 interaction, and CD28 trans-membrane/costimulatory and CD3ζ activation domains to deliver signals to the CD8+ CTL. The first domains of the HLA-DR1-peptide complex serves as the ligand for the TCR expressed by CD4+ autoimmune T cell targets. Once these cells interact, the CTL is activated by signaling through the CD28/CD3ζ domain, and the attached CD4+ T cell is lysed. B. The cDNA construct of the HLA-DR1 CAR is based on the construct expressed by our DR1 Tg mouse that enables mouse CD4 interaction. In constructs 1 and 2, cDNA encoding an antigenic peptide and linker was added to the DRB1 chain. Transmembrane (TM) and cytoplasmic (Cyt) domains of the DRB1 and DRA1 chains are replaced with corresponding CD28 and CD3ζ domains enabling intracellular signaling through the DR1 molecule. The DRB1 and DRA1 chains are separated by a ribosome skipping T2A sequence allowing both chains to be expressed off the same promoter as a single mRNA that is translated as two separate peptide chains. C. For expression of DR1 CAR in T cells, purified CD8+ T cells were transduced with the MSGV vector encoding the chimeric DR1 construct or mock transduced (control). Data shown are from the DR1-CII construct, and similar results were observed with transduction of the DR1-HA and DR1 empty constructs. Seven days after transduction, cells were stimulated with either antibodies to CD3 and CD28, or with an antibody to DR (LB3.1). Cells were stained with anti-CD8-FITC and anti-DR-Alexa 647 (antibody L243) and analyzed by flow cytometry. Stimulation of the CAR T cells in culture through the CAR molecule using the anti-DR antibody significantly enriched for CAR expressing cells.
Replication deficient retrovirus encoding the DR1 CAR molecule was produced using 293T cells co-transfected with pEQ-pam 3 (-E), pCAG-Eco env (gifts from Dr. Stephen Gottschalk, St Jude Children’s Research Hospital), and pMSGV-DR1. 293T cells, grown in DMEM + 10% heat inactivated FBS and 2 mM L-glutamine, were transfected with 10 μg of total DNA in a 15:15:10 ratio (pEQ-pam 3(-E):pMSGV-DR1:pCAG-Eco env, respectively) using GeneJuice Transfection Reagent (Novagen), following the manufacturer’s protocol. Viral supernatants were collected 48 and 72 hours after transfection, filtered with a 0.45 μm syringe filter and snap frozen for future use. 72 hr after transfection, 293T cells were stained with an anti-DR antibody (LB3.1) and analyzed by flow cytometry to confirm expression of DR1.
DR1 CAR T cells were produced by transduction of mouse CD8+ T cells using retronectin-coated plates (TaKaRa Bio) as follows. CD8+ T cells were purified from the spleen and lymph nodes of B6.DR1 mice using anti-CD8a magnetic beads and an LS column (Miltenyi), according to manufacturer’s protocol. After purification, the CD8+ T cells were stimulated overnight by incubation in 24-well plates coated with anti-CD3 and anti-CD28 (1 ug/ml) in RPMI 1640 complete media containing 10% heat inactivated FBS (Hyclone), 2 mM L-glutamine (Gibco), 100 μg/ml streptomycin (Gibco), 100 U/ml penicillin (Gibco), 50 μM β-mercaptoethanol (Gibco), 1 mM Na-pyruvate (Gibco), 1x non-essential amino acids (Gibco), 1 mM HEPES (Gibco), and 20 U of IL-2/ml (PeproTech). The following day, the CD8+ cells were washed and resuspended at 1×106 cells/ml in RPMI 1640 complete media containing 5 μM β-mercaptoethanol and 40 U of IL-2/ml.
Prior to transduction, wells of 24 well plates (non-treated tissue culture plate, Falcon) were coated overnight with retronectin (0.5 ml/well of 10 ug/ml) at 4°C. The retronectin-coated wells were then blocked with 0.5 ml of 2% BSA in sterile PBS for 30 min at room temperature and washed once with PBS. 0.5 ml of virus supernatant was then added to each well and the plate centrifuged for 2 hr at 2000 × g, 32°C. After centrifugation, 1 ml of the stimulated, washed CD8+ T cells were centrifuged onto the virus-retronectin coated wells for 2 hr at 1000 × g, 32°C. After centrifugation, the plates containing the cells were cultured overnight at 37°C.
Three days after transduction, DR1 expression by the transduced CAR T cells was analyzed by flow cytometry using a fluorescent labeled anti-DR antibody (LB3.1) to determine transduction efficiency. For maintenance in culture, the CAR T cells were seeded at 1 × 106 cells/ml in complete RPMI 1640 with 20 U/ml of IL-2, and the cell concentration was adjusted daily. The cells were re-stimulated weekly on anti-CD3 and anti-CD28 antibody (1 ug/ml) coated plates, and in some cases the first re-stimulation was done with anti-DR (LB3.1, 2 μg/ml) coated plates. For some experiments purified, non-transduced (NT) CD8+ T cells were used as a control, and these cells were maintained in culture using the same anti-CD3/CD28 stimulation protocol.
Immunizations and autoimmune arthritis induction.
For arthritis induction and T cell proliferation assays, mice were immunized subcutaneously at the base of the tail with 100 μg of peptide or bovine CII emulsified in equal volumes of CFA consisting of 85% heavy paraffin oil (Fisher Scientific), 15% mannide mono-oleate (Sigma), and 4 mg/ml of heat killed mycobacterium (H37Ra, Difco) (12). For arthritis studies, mice were examined 3 times per week starting at day 18 after immunization and the presence of arthritis, number of affected limbs, and the severity were assessed. Severity of disease was evaluated by visual inspection and assigned a score using a scale of 0 to 4 based on the degree of inflammation, as described (12).
T cell proliferation assays.
Eleven or 12 days after immunization, T cells were recovered from draining lymph nodes of CII-immunized mice. T-cell proliferation assays were performed in 96-well micro-titer plates in a total volume of 300 μl containing 4 × 105 lymph node cells and various concentrations of peptide in complete HL-1 medium. Cell cultures were maintained at 37° C in 5% humidified CO2 for 4 days. On day 3 of culture, 1 μCi of [3H]-thymidine was added, and on day 4 the plates were harvested onto filter plates. After the filter plates were dried, scintillation fluid was added and [3H]-thymidine incorporation measured using a Hidex Sense Microplate Reader (Lab Logic).
T-cell hybridomas.
T-cell hybridomas were established by polyethylene glycol fusion (Boehringer Mannheim) of lymph node cells with BW5147 thymoma cells (TCR α-/β-) as previously described (13). Lymph node cells were obtained from DR1 (DRB1*0101) transgenic mice (9) immunized 10 days previously with antigen/CFA. Prior to fusion, lymph node T cells were stimulated with antigenic peptide for four days, followed by IL-2 stimulation for three days. Resulting hybridomas were screened for their ability to recognize the antigenic peptide presented by DR1.
Antigen presentation assays.
Antigen presentation assays were performed in 96-well microtiter plates in a total volume of 0.3 ml containing 5 × 104 antigen presenting cells (APC), 5 × 104 T-hybridoma cells, and 10 μg of synthetic peptide (RS Synthesis). Assays were performed in HL-1 medium (Bio-Whittaker) supplemented to 2 mM L-glutamine (Gibco), 50 units/ml penicillin, 50 μg/ml streptomycin (Gibco), and 50 μM β-mercaptoethanol (Gibco). Assay cultures were incubated at 37° C in 5% humidified CO2 for 20 to 24 hours. After this time, culture supernatants were harvested and IL-2 production by the T cell hybridomas was measured in a bioassay using the IL-2 addicted cell line HT-2 (9). HT-2 cell viability was assessed by cleavage of MTT (Sigma) (14, 15). IL-2 titers were quantified by the reciprocal of the highest two-fold serial dilution maintaining HT-2 cell viability greater than two-fold over negative control cultures. Results are presented as units of IL-2 per ml as described by Kappler et a. (16).
Autoantibody Measurement.
CII-specific antibody levels in plasma of mice immunized with CII/CFA were quantitated using a bead based ELISA and a MagPix instrument (Bio-Rad). The CII-beads for the ELISA were produced by incubation of purified bovine type II collagen with Bio-Plex Pro magnetic COOH beads (Bio-Rad). The magnetic beads were activated with EDAC/S-NHS and the collagen was coupled using an Amine Coupling Kit (BioRad) according to manufactures protocol. For the ELISA, the CII-coupled beads were incubated with various dilutions of mouse plasma starting at 1:4000 and the quantity of mouse IgG bound detected by the addition of a biotinylated anti-mouse IgG (Sigma) followed by PE-Streptavidin (Invitrogen). The quantity of anti-CII antibody was calculated using a curve generated using purified CII-specific antibody as the standard.
Cytokine assays.
Cytokines produced by CAR T cells after stimulation with anti-DR or anti-CD3/CD28 antibodies, were measured using a multiplexed bead assay (Bio-Rad) and analyzed using a Bio-Rad MagPix. 1 × 106 CAR T cells were cultured in a 48 well plate in which the wells had been coated with 250 μl of an anti-DR antibody (LB 3.1, 2 μg/ml), or anti-CD3 and anti-CD28 antibodies (1 μg/ml each), or PBS. After 18 hr of stimulation, 500 ul of supernatant was collected and frozen @ −20° C prior to the cytokine ELISA. Supernatants were assayed for expression of IFNγ, TNFα, IL-10, IL-17A, and IL-6. Standard curves were used to calculate cytokine concentration, and data are based on duplicate samples and representative of two independent experiments.
CTL assays.
A flow cytometry based assay was used to measure the cytolytic activity of the CAR T cells. 105 DR1-CII or DR1-HA CAR T cells were co-cultured in 300 μl RPMI medium in a 96 well microtiter plate with 104 cloned DR1-restricted T cells for a 10:1 effector:target (E:T) ratio. Targets consisted of T cell hybridomas specific for either the CII peptide (GIAGFKGEQGPKGEP) or the HA peptide (PKYVKQNTLKLAT). After 4 hr at 37° C, cells were collected from the wells and stained with anti-CD8-APC-Cy7 (clone 53-6.7, BD), anti-BV14-FITC (clone 14.2, BD), anti-CD4-PerCP-Cy5.5 (clone RM4-5, Tonbo), anti-BV8-PE (clone F23.1, BD), anti-HLA-DR-Pacific Orange (clone LB 3.1), and DAPI (Sigma) was added just prior to analysis for exclusion of dead cells. Data analyses were focused on live cells (DAPI negative), HLA-DR1 negative, CD8 negative, and CD4+/BV14+ (HA specific) or CD4+/BV8+ (CII-specific) T cells.
Results
Genetic engineering and expression of HLA-DR1 CAR.
A chimeric receptor for expression in CD8+ T cells was designed using HLA-DRB1*01:01 and HLA-DRA1:01:01 cDNA in which the transmembrane and cytoplasmic domains were replaced with corresponding domains from CD28 and CD3ζ, respectively, to provide activation and signaling to the CD8+ T cell (Figure 1A). In addition, the second domains of the HLA-DR chains were replaced with corresponding domains from the murine I-E α and β chains to enable interaction with murine CD4 (9), and cDNA encoding an antigenic peptide was inserted near the amino terminus of the DRB1 chain (Figure 1B) (11). This design was selected to provide the highest potential affinity between the DR1 CAR molecule and the TCR expressed by CD4+ T cells, and to provide antigen specificity to the CAR T cell targeting and subsequent lysis of the CD4+ T cells.
Using replication deficient retrovirus produced from the MSGV1-28z mut1-3 (MSGV) expression vector containing the DR1 CAR, mouse CD8+ T cells from B6.DR1 mice were transduced and expression of the DR1-CII CAR molecule was assessed by flow cytometry (Figure 1C). Transduction results were similar for the DR1-HA and the DR1-empty CAR constructs (Figure 1B, and data not shown), and overall efficiencies ranged from 25 to 50%. Given the dependency of the anti-DR antibody on tertiary structure, these data indicated that the DR1 CAR molecule was folded in a native state. While anti-CD3/CD28 stimulation of the DR1 CAR T cells allowed us to maintain the CAR T cells and to expand their number (Figure 1C, lower panels), we found that stimulation directly through the CAR molecule with an anti-DR antibody selectively enriched for the CAR T cells. These data support the concept that intracellular signaling is occurring through the CAR to the CD8+ T cell, however subsequent re-stimulation of the CAR T cells with anti-DR antibody alone was insufficient to maintain the CAR T cell line over prolonged periods (data not shown). Consequently for the remainder of our experiments, the first stimulation post transduction was done with anti-DR antibody to enrich for the CAR T cells, and all subsequent stimulations were done with anti-CD3/CD28 antibodies to expand the cells for experimental use.
Specificity of the DR1 CAR molecule and functional activity of the DR1 CAR T cell.
To demonstrate that the DR1 CAR molecule was recognized by DR1-restricted TCR, antigen presentation assays were performed with T cell hybridomas specific for the immunodominant determinant of type II collagen, CII(259–273), or a hemagglutinin peptide, HA(306–318), presented by DR1. 293T cells that had been transfected with the DR1 CAR molecules were mixed with the T cell hybridomas and IL-2 production by the T cells was measured as an indication of TCR recognition of the CAR molecule. As shown in Figure 2, DR1 CAR molecules that have the CII peptide covalently attached to the DRB1 chain (DR1-CII CAR) are recognized by DR1-restricted CII-specific T cells but not HA-specific T cells in the absence of exogenous added antigen. CAR molecules that are “empty” of an antigenic peptide (DR1-CAR) are not recognized by these T cells, however, the addition of exogenous CII peptide to the cells expressing the DR1-CAR enabled stimulation of the CII-specific T cells. Thus these data demonstrate that the DR1 CAR molecule is in a native conformation that is recognized by CD4+ T cells in an antigen specific manner.
Figure 2. The DR1-CAR expressed by transfected cells is recognized by CD4+ T cells in an MHC restricted, antigen specific manner.
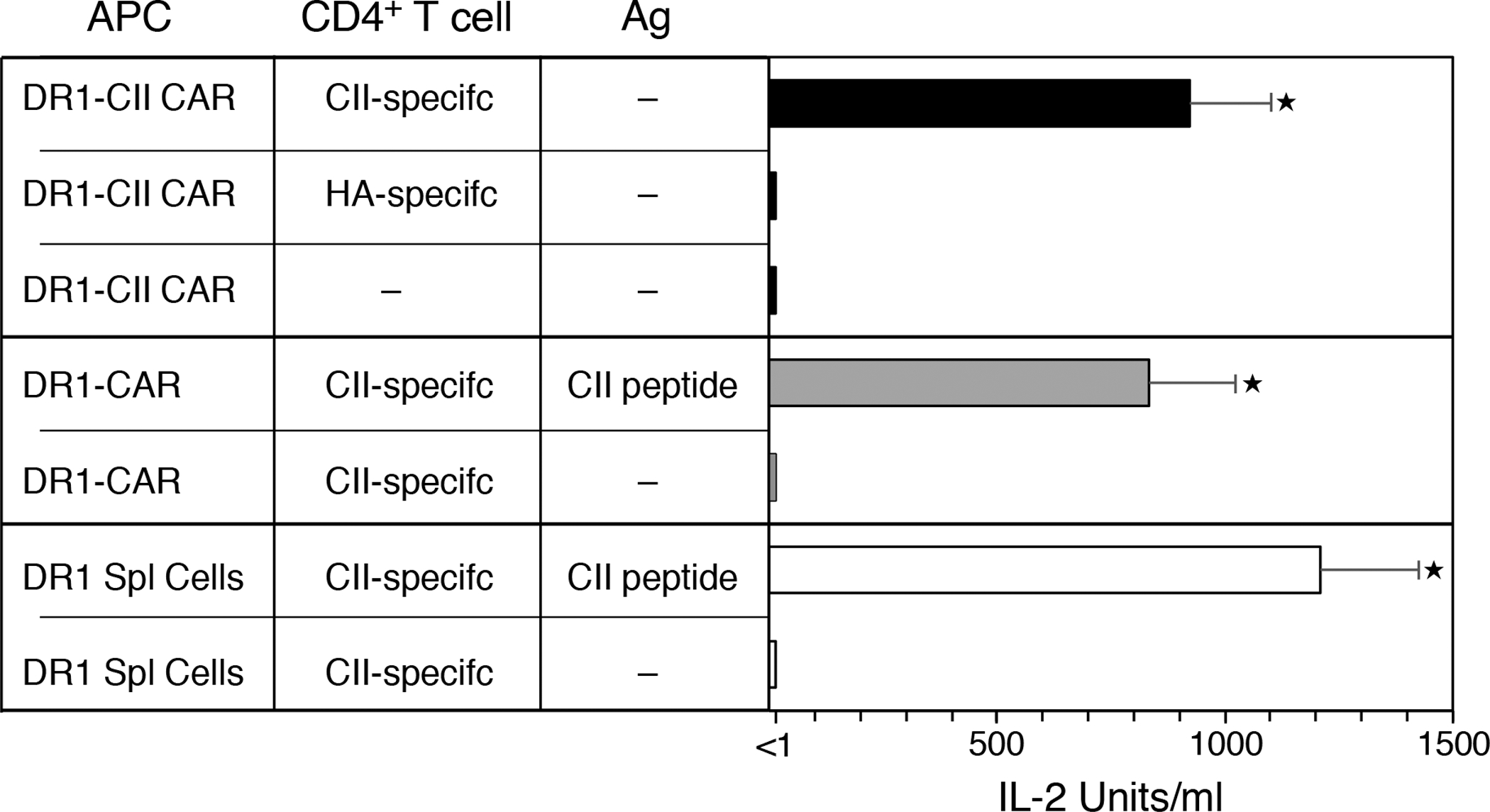
The chimeric DR1 molecules were transfected into 293T cells and tested for their ability to be recognized as antigen specific ligands using T cell hybridomas specific for CII or HA peptide. The DR1-CII chimeric molecule was recognized by the CII-specific CD4+ T cells (■ bars) as indicated by the production of IL-2, but not by the HA-specific T cells. CAR T-cells expressing empty DR1 (DR1-CAR) did not stimulate the CII-specific T cells unless the CII peptide was added exogenously to the cultures (■ bars). In both instances, the positive response of the two CAR T-cells were similar to that observed using spleen cells as APC from B6.DR1 mice (☐ bars). These data indicate that the chimeric DR1-CII-CD28-CD3 CAR molecule functions as an MHC restriction element. Experiment was repeated at least 3 times; error bars indicate standard error of the mean (SEM); ★ indicates p value < 0.0001.
When the DR1 CAR CD8+ T cells were tested for cytolytic function, the CAR T cells were found to be active CTL and lysed CD4+ T cells in antigen specific manner. Using a flow cytometric assay, CII-specific (TCR-BV8+) and HA-specific (TCR-BV14+) T cells were incubated with either DR1-CII or DR1-HA CAR T cells at a 10:1 E:T ratio for 4 hours, and the cultures were then analyzed for CD8 negative, DR1 negative, and BV14+ and BV8+ cells (Figure 3). The CII-specific and HA-specific target cells were mixed at a 1:1 ratio, and after 4 hr in the absence of CAR T cells, were still found to be present in roughly equal numbers (Figure 3A). While the addition of non-transduced (NT) CD8+ T cells to the cultures had no effect on either the BV8+ (CII-specific) or BV14+ (HA-specific) target cell populations (Figure 3B), the addition of the CAR T cells resulted in the elimination of the target cells in an antigen specific manner. The DR1-CII CAR T cells effectively lysed the BV8+ CII-specific T cells and not the BV14+ HA-specific T cells (Figure 3C), while the DR1-HA CAR T cells lysed the HA-specific T cell but not the CII-specific T cells (Figure 3D). Similar results were observed using 51Cr-release cytolytic assays (data not shown). Summaries of these data and their statistical analyses are shown in Figures 3E and 3F. In all, these data demonstrate the lytic functionality of the DR1 CAR T cells and the specificity of the DR1 CAR in targeting CD4+ T cells in a manner that reflects the TCR specificity of the CD4+ target cells.
Figure 3. DR1-CII and DR1-HA CAR T cells lyse CD4+ T cells in an antigen specific manner.
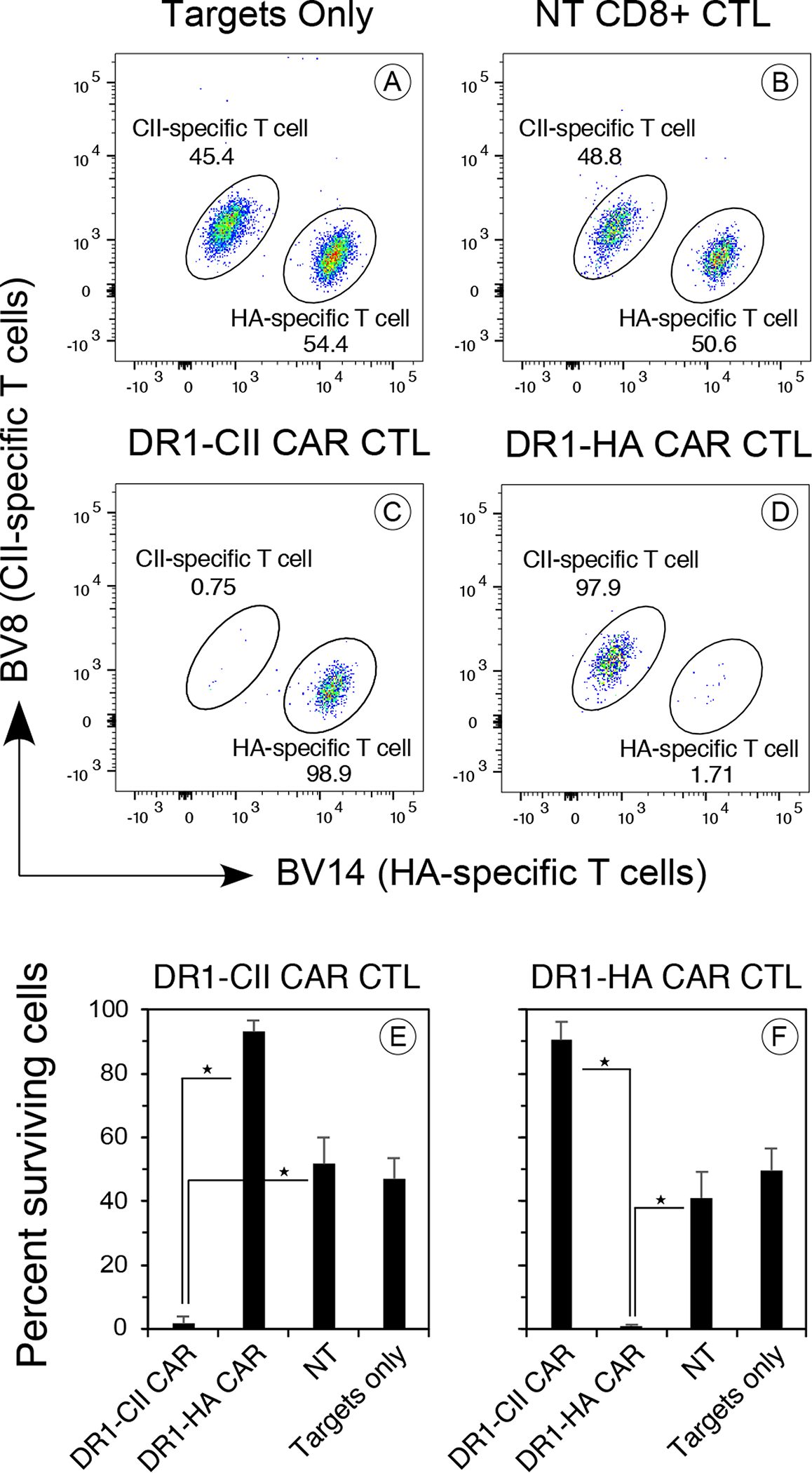
CD8+ T cells were transduced with vectors encoding the DR1-CII or DR1-HA CAR and the resulting T cells were incubated with a mixture of CII-specific and HA-specific T cell hybridoma clones at 10:1 effector:target ratio. After 4 hours, the cells were labeled with fluorochrome labeled antibodies specific for CD8, DR1, TCR-BV8, and TCR-BV14, stained with DAPI, and analyzed by flow cytometry. A. Target cells in the absence of CAR T cells, showing roughly an even mix of target cells in the assay. The CII-specific T cells express TCR-BV8, while the HA-specific T-cells express BV14. B. Target cells mixed with non-transduced CD8+ T cells. No lysis of target cells was observed. C. DR1-CII CAR T-cells lyse the T cells specific for CII (0.75% BV8+), but not the HA-specific T cells (98.9% BV14+). D. DR1-HA CAR T cells lyse the HA-specific T cells (1.7% BV14+), but not the CII-specific T cells (97.9% BV8+). E and F. Summary of at least 6 experiments each for the cytolytic efficacy and specificity of the DR1-CII and DR1-HA CAR T cells. Each CAR T cell was highly effective in lysing its appropriate target cell (★ indicates p value <0.00001), whereas NT CD8+ T cells had no effect on the targets. Error bars indicate standard deviation. Data are based on DAPI negative, CD8 negative, and DR negative gates in the flow cytometry data.
In vivo activity of the DR1 CAR T cells.
Given that the in vitro CTL data indicated the therapeutic potential of these CAR T cells, we tested their ability to inhibit an antigen specific T cell response in vivo. B6.DR1 mice were immunized with either CII or the HA peptide, treated with 2 × 106 DR1-CII, or DR1-HA CAR T cells, or NT CD8+ T cells as control, on day 4 and 7 after immunization. On day 11, T cells from draining lymph node were recovered and tested for their ability to be re-stimulated by antigen in an in vitro proliferation assay. As shown in Figure 4A, mice immunized with CII and treated with DR1-CII CAR T cells had a significantly reduced T cell proliferative response (p < 0.03). While the effect varied among the CAR T cell treated mice, with some mice having a 50% reduction to others a near total reduction, all showed a definitive therapeutic effect on the autoimmune CII T cell response. Similar data was obtained from DR1 CAR T cells that had been in culture for 6 days or 76 days (data not shown). The efficacy of this CAR T cell approach was not limited to autoimmune T cell responses. Treatment of HA immunized B6.DR1 mice with DR1-HA CAR T cells also effectively reduced the T cell response to this foreign antigen (Figure 4B, p < 0.004). While in both of these experiments some T cell responses were still observed in the DR1 CAR T cell treated mice (Figures 4A and 4B), increasing the number of DR1 CAR T cells administered did not enhance the overall effect (Figure 4C). While all 3 doses of DR1-CII CAR T cells significantly reduced the in vivo T cell response to CII in comparison to the NT control cells (p < 0.05), treating mice twice within 10 days with the highest number of DR1-CII CAR T cells (6 × 106) did not result in a complete elimination of the CII-specific T cell response in the DR1 mice. The small proliferative response still detectable indicated that not all CD4+ CII-specific T cells had been eliminated in vivo by the DR1 CAR T cells.
Figure 4. DR1 CAR T cells suppress CD4+ T cell responses in vivo.
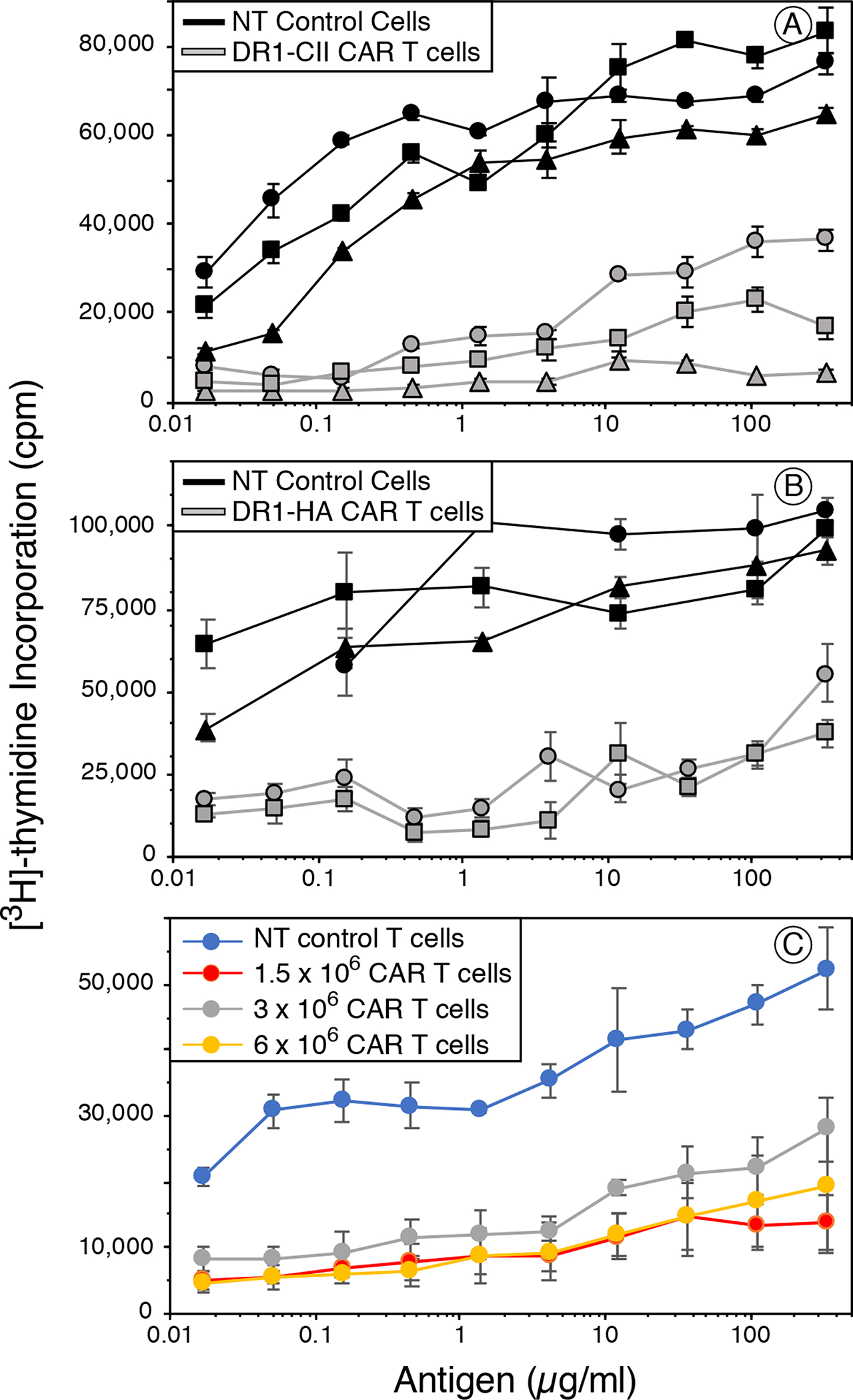
B6.DR1 mice were immunized with CII/CFA or HA/CFA, and on days 4 and 7 mice were treated with 2 × 106 DR1-CII or DR1-HA CAR T-cells or non-transduced CD8+ T cells as the negative control. Four days after the last treatment, draining lymph node cells were collected, and tested for their ability to be re-stimulated in vitro with various concentrations of CII or HA peptide in a T cell proliferation assay. A. Mice immunized with CII/CFA and treated with DR1-CII CAR T cells. Mice receiving the NT control CD8+ T cells developed a strong T cell proliferative response when re-stimulated with the CII peptide (black lines, 3 individual mice). In comparison, mice treated with the DR1-CII CAR T-cells had significantly reduced proliferative responses (grey lines, 3 different mice, p < 0.03 for 11 highest antigen concentrations in comparison to the NT control cells). B. Mice immunized with HA/CFA and treated with DR1-HA CAR T cells. Similar to panel A, HA - immunized mice treated with DR1-HA CAR T cells (grey lines) had a significantly reduced T cell response to HA in comparison to NT controls (black lines, p < 0.004 for 8 highest antigen concentrations in comparison to the NT control cells). C. Dose effect of CAR T cells on the suppression of the T cell response. Mice were immunized with CII/CFA and treated with various numbers of CAR T cells on days 5 and 10. On day 12 draining lymph node cells were collected, and tested for their ability to be re-stimulated with CII peptide in vitro. While the T cell response was suppressed in mice receiving all 3 dose of CAR T cells (p < 0.05 for each dose in comparison to NT control cells), no significant differences were observed among the 1.5 × 106, 3 × 106, and 6 × 106 CAR T cell doses. Data from these experiments are representative of 2 or 3 experiments each. Each experiment was repeated a minimum of 2 times, and data are based on quadruplicate repeats for each point. Errors bars indicate standard error of the means.
Inhibition of autoimmune arthritis by DR1-CII CAR T cells.
Given the ability of the DR1-CII CAR T cells to downregulate an autoimmune T cell response, we tested their ability to prevent the development of autoimmune arthritis in the B6.DR1 mouse model of rheumatoid arthritis (9). Mice were immunized with CII, and on days 7, 14 and 21 after disease induction they were treated with either 2 × 106 DR1-CII CAR T cells, or NT CD8+ T cells as a control, or left untreated. Mice were then examined 3 times per week for the presence and severity of arthritis in both fore and hind limbs. In comparison to the ≥ 80% disease incidence in the 2 control groups (NT CD8+ T cells and untreated, Figure 5A), mice receiving the DR1-CAR T cells had a 50% reduction in the incidence of autoimmune arthritis that was statistically significant (p < 0.05 by Fisher Exact test) at nearly every time point (Figure 5). For the DR1-CII CAR treated mice that did develop disease, the onset was delayed in comparison to controls. While the effect of the DR1-CII CAR T cells in this model was statistically significant, 40% of the mice still developed disease by the later time point (Figure 5A). However the arthritis severity in these DR1-CII CAR T cell treated mice was generally lower than that of the arthritic mice in the untreated and NT CD8+ T cell control groups (Figure 5B). Given that the CAR T cell treatment ended as the arthritis first began to develop in the CAR treated mice, these data imply that the DR1-CII CAR T cells may have a short half-life in vivo. As noted for the T cell proliferation studies, similar arthritis incidence data were obtained using CAR T cells that were maintained in culture for short periods (23 days) or long periods (> 70 days).
Figure 5. DR1-CII CAR T cells inhibit the development reduce the severity of autoimmune arthritis in B6.DR1 mice.
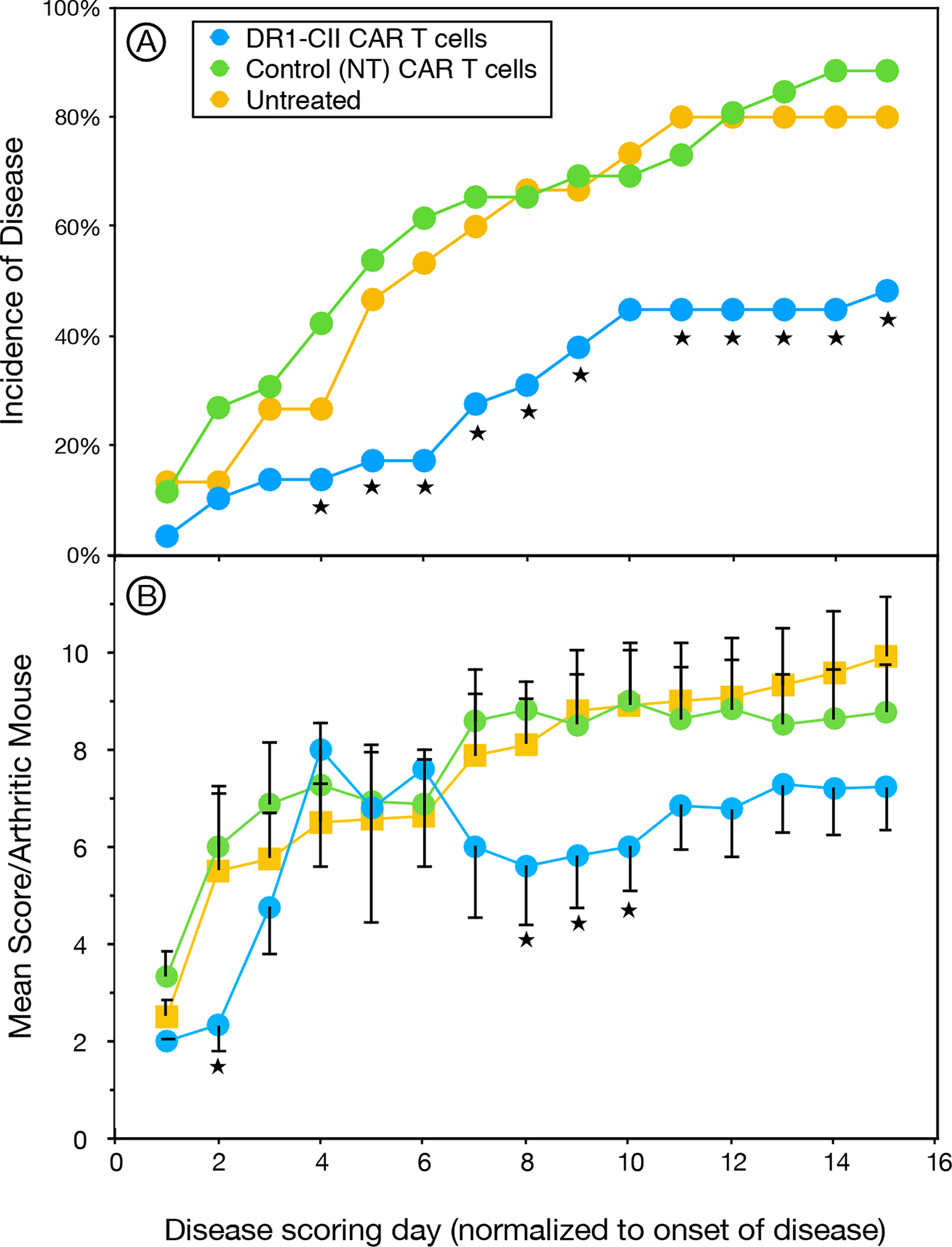
B6.DR1 mice were immunized with CII/CFA and treated on days 7, 14 and 21 following disease induction with 2 × 106 DR1-CII CAR T cells, or non-transduced (NT) CD8+ T cells, or left untreated. Mice were observed 3 times per week for the presence of arthritis in fore and hind limbs. A. Mice receiving DR1-CII CAR T cells had a statistically significantly lower incidence of disease than both control groups (★ indicates p < 0.05 by Fisher Exact test, p values ranged from 0.04 to 0.0015), and had a later onset. B. In mice that developed arthritis, the mean severity of the disease was decreased in comparison to control groups (NT CAR T cells and untreated). Data indicate mean severity score of the sum of all 4 limbs of only mice that developed disease. Error bars indicate standard error of the mean (SEM), and ★ indicates statistical significance (p ≤ 0.05 by student’s t-test). The last day of CAR cell treatment corresponds to the first day of disease scoring. Data are a compilation of 3 independent experiments, n = 15 for the untreated group, n = 29 for the NT treated group, and n = 30 for the CAR T cell treated group.
Autoimmune arthritis in the B6.DR1 mouse model is dependent on both a CD4+ T cell response to the autoantigen, CII, as well as a B cell response in the production of pathogenic autoantibody to CII (9, 17). Consistent with the inhibition of both the autoimmune T cell response (Figure 4) and the inhibition of autoimmune arthritis (Figure 5), the autoantibody levels in the mice treated with the DR1-CII CAR T cells were also significantly reduced in comparison to the control groups (Figure 6). At all 3 time points measured, the anti-CII autoantibody response was statistically reduced (p ≤ 0.02) by more than 50% in comparison to both the untreated mice and those treated with the control NT CD8+ T cells (Figure 6). In all, these data demonstrate that CAR T cells expressing HLA-DR as a chimeric receptor have the potential to be a highly specific therapeutic modality for the treatment of autoimmune disorders.
Figure 6. B6.DR1 mice treated with DR1-CII CAR T cells produce significantly less autoantibody.
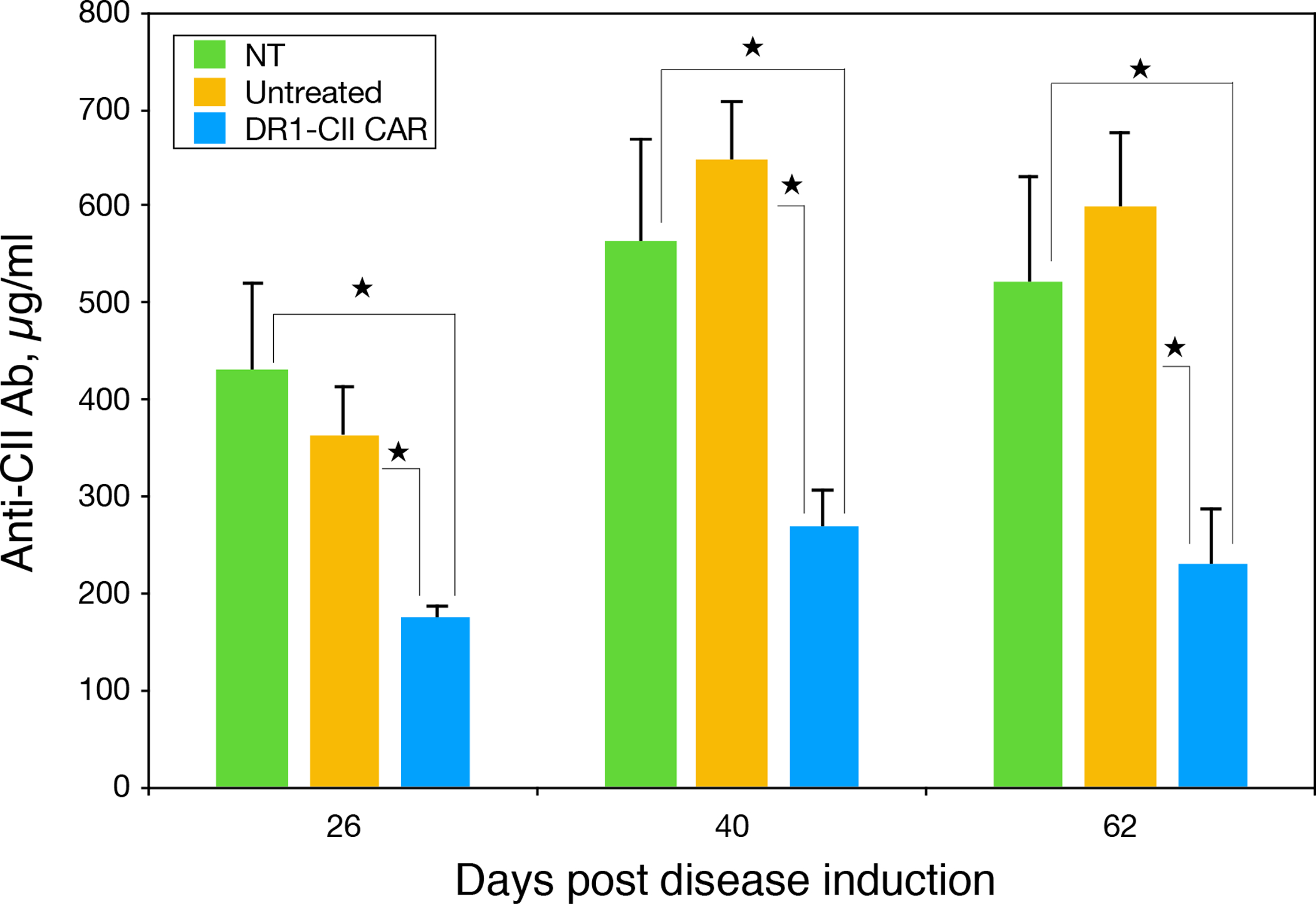
Plasma was collected from mice in the arthritis experiment in Figure 5 and tested for the presence of anti-CII autoantibody. All mice were immunized with CII/CFA to induce autoimmunity to CII, and then treated with DR1-CII CAR T cells or control NT CD8+ T cells, or left untreated as indicated. A bead based ELISA using CII coupled Luminex beads was used to quantitate the antibody. A standard curve produced using affinity purified anti-CII antibody was used to calculate the quantity of autoantibody measured. ★ indicates p ≤ 0.02 by Student’s t-test.
While the DR1-CAR T cells were effective in reducing autoimmune disease manifestations, the CAR T cells did not appear to undergo significant expansion in vivo. Enumeration of the CAR T cells by flow cytometry after therapeutic administration indicated that CAR T cells were only detectable a short time after passive transfer. As shown in Figure 7, 4 days after adoptive transfer, the DR1-CII-CAR T cells could be detected in lymph nodes (4.9% DR1+/CD8+ T cells) and in the spleen (2.1%), and possibly in the blood at a very small percentage. However by day 10, they were no longer detectable by this approach in any of these tissues. These data imply that the DR1-CII CAR T cells are not strongly stimulated to expand in vivo, as is necessary for successful therapy with CAR T cells used to treat cancer (18, 19). This observation is consistent with the concept that the CD4+ CII-specific T cells that are the targets for the DR1-CII CAR T cells are in low frequency (11, 20), thus it is likely that only a small number of these CAR T cells undergo expansion in vivo.
Figure 7. Detection of CAR T cells in lymphoid tissue 4 days after adoptive transfer.
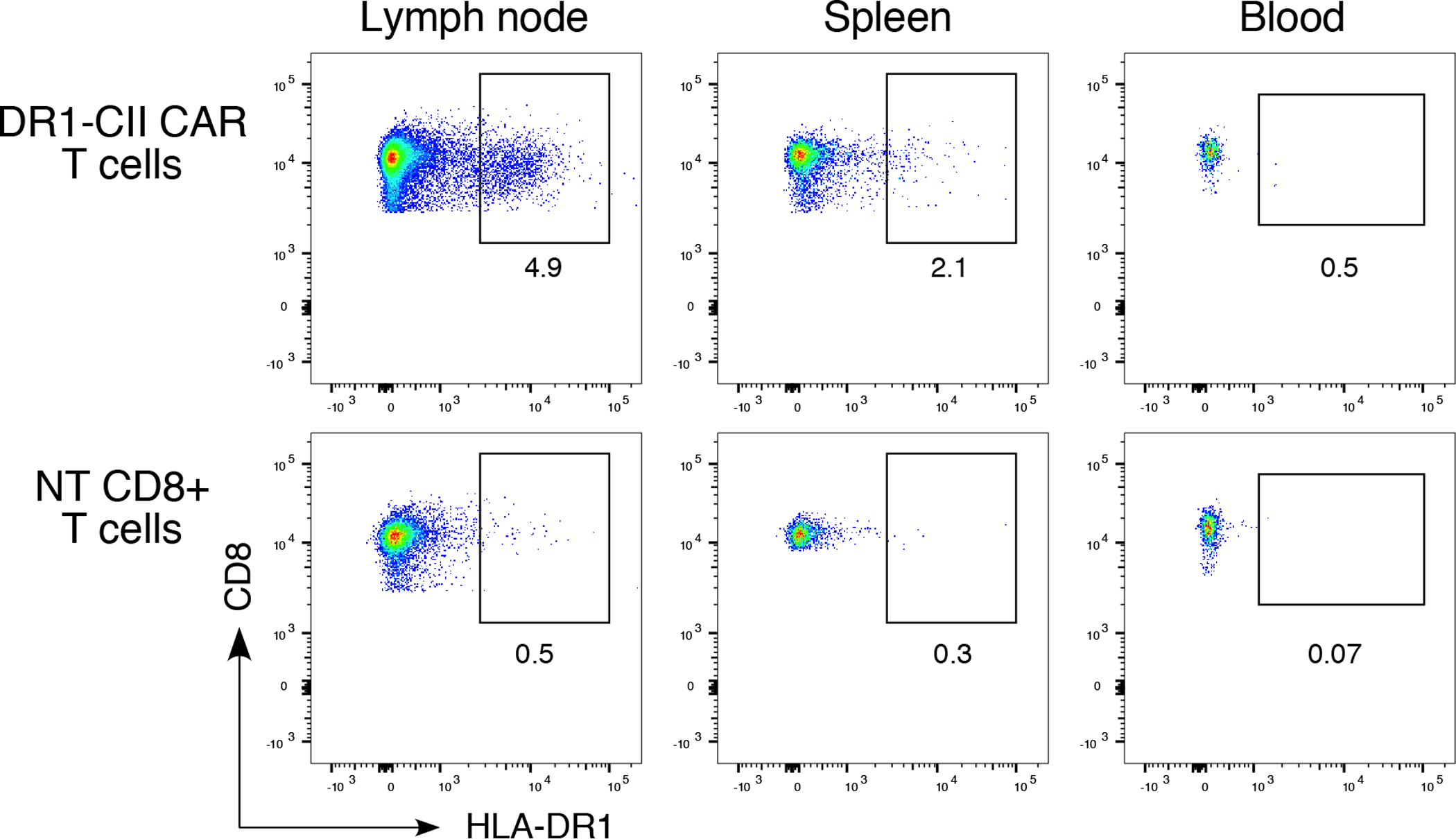
B6.DR1 mice were immunized with CII/CFA, and 7 days later, mice were treated with 2 × 106 DR1-CII CAR T-cells or NT CD8+ T cells (negative control). Four days after treatment, cells from draining lymph nodes, peripheral blood, and spleens were collected, incubated with fluorochrome labeled antibodies specific for CD3, CD19, CD4, CD8, and DR, stained with DAPI, and analyzed by flow cytometry. Data shown were generated from a single mouse and are representative of 3 experiments. Flow data were gated on CD3+/CD19−, and CD4−/CD8+, and are based on a minimum of 10,000 cells analyzed.
Functional phenotype of the HLA-DR1 CAR T cells.
The structure of our CAR differs substantially from those used in cancer therapy in that the DR1-CAR is a two chain molecule, each with its own activation and costimulatory domains (Figure 1). To analyze how this affects the cell signaling through the DR1-CAR in these T cells, we analyzed cytokine production following simulation of these cells through their CAR molecule compared to pan stimulation with anti-CD3 and anti-CD28 antibodies. As shown in Table 1, cytokine production via stimulation through DR1-CII CAR was very similar to anti-CD3/CD28 stimulation in terms of the production of TNFα, IFNγ, IL-6, IL-10, and IL-17A, although some differences were observed with the stimulation of DR1-HA CAR T cells using these two stimulations, particularly TNFα production. It is unclear why these two CARs would differ in cytokine production, given that both are identical with the exception of the nature of the peptide bound by the DR 1st domains. In all, these data indicate that the signaling domains are functional, and that stimulation of these T cells through their CAR does not appear to differ from that generated through CD3/CD28 stimulation, at least when antibody is used to stimulate. However, given that the DR1-CAR cell lines could not be maintained by repetitive stimulation through the CAR with an anti-DR antibody indicates that, despite the cytokine production, some signaling aspect is different when initiated through the CAR molecule in comparison to signaling through the endogenous CD3 and CD28 cell membrane proteins.
Table 1.
Cytokines produced by DR1 CAR T cells following stimulation with antibody to HLA-DR1 or antibodies to CD3 and CD28.
| Cytokine, pg/ml | ||||||
|---|---|---|---|---|---|---|
| CAR T cell | Stimulation | TNF α | IFNγ | IL-6 | IL-10 | IL-17A |
| DR1-CII | anti-DR1 | 5583.9 ± 1431.5 | >15,000 | 54.5 ± 8.3 | 132.5 ± 35.3 | 2.6 ± 0.7 |
| DR1-CII | anti-CD3/CD28 | 5650.2 ± 538.7 | >15,000 | 70.7 ± 6.1 | 184.2 ± 37.1 | 2.7 ± 0.6 |
| DR1 CII | none | 29.0 ± 14.8 | 13.4 ± 1.9 | –* | 34.6 ± 18.7 | – * |
| DR1-HA | anti-DR1 | 145.0 ± 20.2 | >15,000 | 8.5 ± 0.9 | 122.9 ± 23.4 | 2.5 ± 0.5 |
| DR1-HA | anti-CD3/CD28 | 3381.0 ± 1008.5 | >15,000 | 40.4 ± 3.2 | 366.7 ± 31.1 | 3.8 ± 0.4 |
| DR1-HA | none | 106.6 ± 99.4 | 29.9 ± 12.2 | 4.3 ± 3.8 | 133.0 ± 127.2 | 3.5 ± 2.6 |
Below detection level
To determine if the expression of exhaustion markers was playing a negative role in the function of the DR1-CAR T cells (21), CAR T cell expression of PD1, Tim-3, and CD69 was measured at several time points after these cell lines were established. Like most other CAR T cells, the expression of these exhaustion markers by our DR1-CII CAR cells increases over time in culture (Figure 8). By day 10 post transduction, 34% of the cells have acquired Tim-3+ expression, and by day 23 nearly all of the cells express Tim-3 and continue to express this marker through at least day 65 (Figure 8A). Only a small percentage of these cells express PD-1 through day 23, and reach 50% by day 37 and maintain this level through day 65. Similarly, more than 50% of these CAR T cells express CD69 by day 10 and stay at that level until day 37 after which nearly all of the cells are CD25+/CD69+. The memory phenotype of these CAR T cells initially included CD62L+ stem cell type (TSCM), but this was quickly lost in culture as the phenotype became predominantly CD44+/CD62L negative T effector (TEFF) by Day 17.
Figure 8. Phenotype of DR1 CAR T cells maintained in culture.
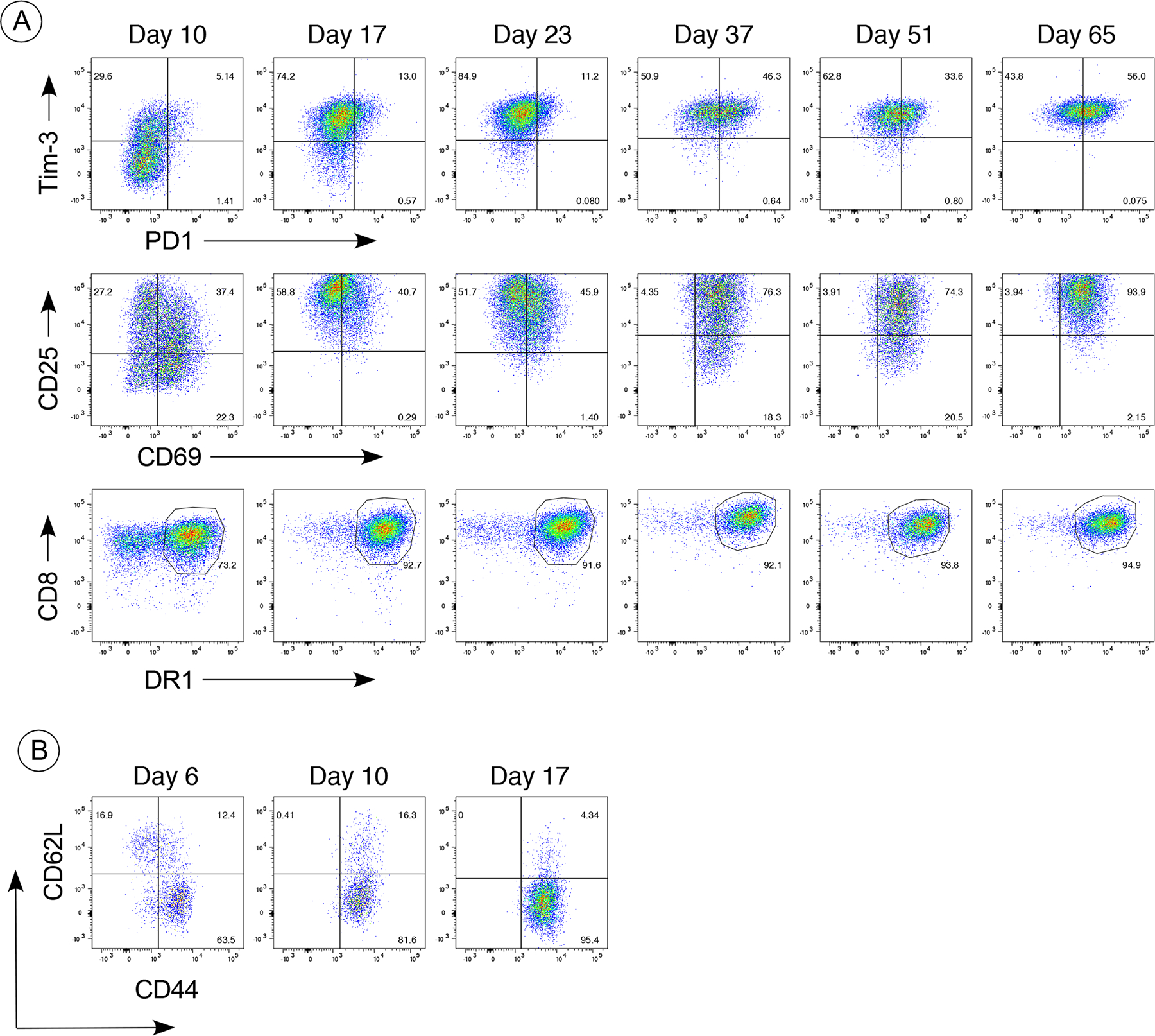
Expression of exhaustion markers PD1, CD69, and Tim-3 (panel A) and CTL memory markers (panel B) were measured from day 10 post transduction through day 65 in culture. For in vitro maintenance, the CAR T cells were seeded at 1 × 106 cells/ml in complete RPMI 1640 with 20 U/ml of IL-2 and were re-stimulated weekly by culture in tissue culture plates coated with anti-CD3 and anti-CD28 antibody (1 ug/ml). Cells were labeled with fluorochrome-conjugated antibodies specific for CD3, CD4, CD8, DR1, Tim-3, PD1, CD25, CD69 (panel A), or CD3, CD4, CD8, DR1, CD62L, and CD44 (panel B). Data shown were gated on CD3+, CD4−, CD8+, and DR+ and are based on a minimum of 10,000 cells analyzed. Data are representative of 6 different CAR T cell lines.
Despite the expression of these exhaustion and CTL phenotypic markers there was no loss of their cytolytic function using either CD4+ CII-specific T cell lines (Figure 9) or naïve CII-specific T cells from a TCR transgenic mouse (data not shown) (22) as targets. Short term cultured cells lysed CD4+ target T cells with similar efficiency as long term cultured CAR T cells that had now acquired exhaustion marker expression, and similar data was observed when comparing efficacy of short term and long term cultured cells in preventing the development of autoimmune arthritis. Our analysis of these target cells revealed that they do not express PD-L1, the ligand for PD-1, indicating that this exhaustion marker is likely not inhibiting the activity of our CAR T cells in vivo, unlike cancer therapy where PD-1/PD-L1 interactions play a significant inhibitory role in CAR T cell efficacy in vivo (23).
Figure 9. CTL activity of DR1-CII CAR T cells maintained in culture.
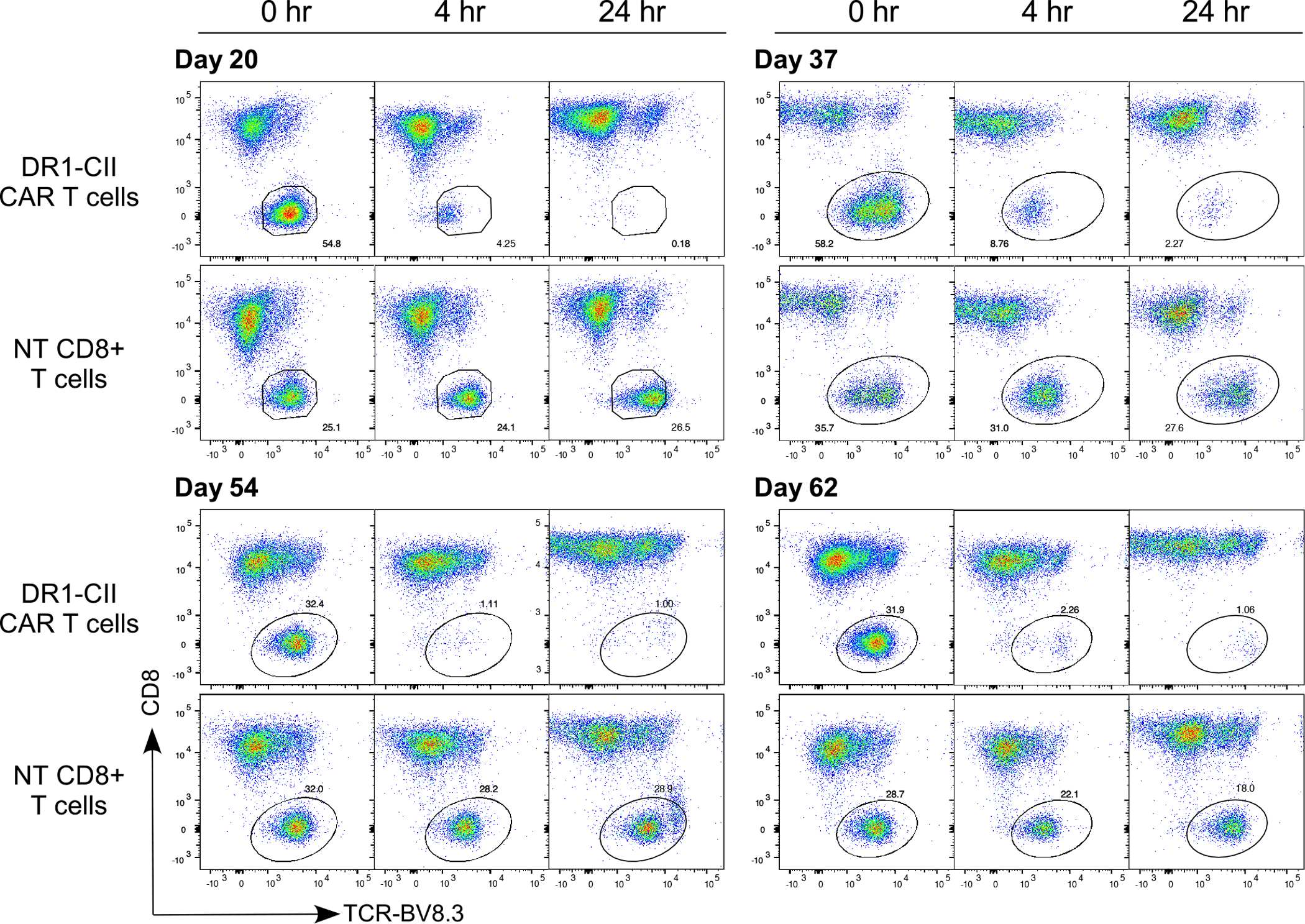
CTL activity of the CAR T cells was measured using a flow cytometry based assay. CAR T cells and target cells (CII-specific T cell hybridoma) were mixed at a 2:1 effector to target ratio, and CTL lysis measured at 4 and 24 hrs. Despite the expression of exhaustion markers, DR1-CII CAR T cells were active as CTL in the lysis of CD4+ T cells to similar levels from day 20 through day 62 in culture. To measure lysis, cells were labeled with fluorochrome conjugated antibodies specific for TCR-BV8.3 (marker for CII-specific T cell targets), CD8, and DR, and data are based on gates with a minimum of 10,000 cells analyzed. Data are representative of a minimum of 3 different CAR T cell lines.
Discussion
The results of these studies demonstrate the potential utility of CAR T cells in treating autoimmune disorders. Using a humanized mouse model of RA, we have shown that chimeric MHC class II molecules can be used to construct CAR T cells that target and lyse CD4+ T cells in an antigen specific manner in vivo. By encoding the immunodominant peptide for CII in the HLA-DR1 CAR molecule, the DR1 CAR T cells downregulated both the CII-specific CD4+ T cell response and the CII-specific autoantibody response, resulting in a significantly reduced incidence of autoimmune arthritis in the B6.DR1 mouse model of RA. The antigen specificity of the CAR T cell was devised by encoding an antigenic peptide as part of the DRB1 chain construct (24), and as was demonstrated using a second antigenic peptide HA(306–318), the specificity of the CAR T cell was easily reprogramed by changing this peptide sequence. Based on the cytokines produced by ligation of the DR1 CAR molecule with an anti-DR antibody, our CAR construct with CD28-CD3ζ transmembrane and cytoplasmic costimulatory domains is capable of generating TCR-like signaling events in CD8+ T cells. While cytokines characteristically expressed by CD8+ T cells were produced by this approach, periodic anti-CD3/CD28 stimulation in vitro was required to maintain the DR1 CAR T cells in culture, as repeated signaling through the DR1 CAR was insufficient to maintain the DR1 CAR T cells. In vitro stimulation of the CAR T cells with a combination of anti-CD3/CD28 did drive the expression of Tim-3, PD-1, and CD69, markers associated with CD8 T cell exhaustion, although we saw little to no effect of the expression of these markers on the function of the DR1 CAR T-cells. DR1 CAR T cells maintained in culture for > 90 days were as effective in inhibiting both the autoimmune response and the development of arthritis in vivo as CAR T cells maintained for only 6 to 25 days, and we have seen no loss of their cytolytic function in vitro with a variety of target cells after their expression of these markers. In all, these data demonstrate the therapeutic potential for an antigen specific therapy using engineered HLA based CAR to target and lyse autoimmune CD4+ T cells.
Our HLA-DR CAR T cell therapy differs in a number of ways from the anti-CD19 CAR T cells used in cancer therapy. CAR molecules designed for cancer therapy are primarily based on genetically engineered antibodies, scFv single chain molecules composed of the heavy and light chain variable regions (8). These scFv have only 1 transmembrane and cytoplasmic domain hence a single CD28/CD3ζ activation and costimulatory complex in the CAR molecule. HLA-DR molecules are composed of two chains, an α and β, and each chain has its own transmembrane and cytoplasmic (TM/Cyto) domains. The constructs used in our studies replaced the endogenous TM/Cyto in each of these chains with corresponding CD28-CD3ζ signaling domains. How this affects the function of our DR1 CAR in comparison to the scFv CARs is currently unknown. While it might be anticipated that such a design would double the signaling strength upon engagement of the DR1 CAR by the TCR, we have seen no evidence of this in our preliminary signaling studies measuring ZAP-70 phosphorylation (ZAP-70p, K. Whittington and E. Rosloniec, unpublished observation). Although we have CD28/CD3 signaling domains on both the DR1 chains, it is not clear if both are participating in the generation of ZAP-70p. It is also possible that inclusion of signaling domains on both chains generates steric hindrance for the phosphorylating enzymes and limits the CD28-CD3 ζ signaling events in the DR1 CAR CD8+ T cells. Assessing the signaling events that occur through the DR1 CAR will be paramount to optimizing therapeutic use of these CAR T cells. Given several differences between the DR1 CAR and the scFv CAR including affinity and membrane density, it is possible that the TM/Cyto signaling domains required for optimal CD8+ T cell activation through the DR1 CAR will be different than the CD28-CD3ζ signaling domains often used with scFv CAR.
CAR T cell therapy in cancer is dependent on three primary outcomes in vivo – activation, expansion, and persistence via maturation of the CD+8 T cell to a stem cell memory phenotype (TSCM) (25, 26). While our DR1 CAR T cells demonstrated function in vivo, we were unable to detect evidence of expansion implying that persistence to a memory phenotype did not occur. Two factors that are likely playing a role in this outcome are frequency of the target cells and affinity of the DR1 CAR for the TCR on the target cells. In terms of frequency, antigen stimulated CD4+ T cells comprise a very small percentage of total CD4+ T cells in the mouse, even during an active autoimmune disease. Our previous studies using a DR1-CII tetramer to analyze the T cell response in B6.DR1 mice with autoimmune arthritis indicated that only 1 to 4% of CD4+ T cells are specific for the immunodominant determinant of the CII autoantigen, and these comprise 1% or less of the total lymphoid population (11). In terms of the number of target cells for the CAR T cells in vivo, this is a target frequency that is orders of magnitude less than the target frequency of the anti-CD19 CAR T cells used for cancer therapy. Second, the affinity of the DR1 CAR for its target cells is likely significantly lower than the affinity of the antibody-based scFv CARs for their ligand. The affinity of MHC class II and class I binding by TCR is considered to be 10 to 100 fold lower than the affinity of antibody for antigen (27, 28), and how this difference in affinity affects the ability of the CAR to initiate T cell stimulation and function is unknown. Recent studies of CAR signaling focused on protein phosphorylation by Salter et al. have indicated that signal strength is a key determinant in the fate of CAR T cells after adoptive transfer (29). While sufficient stimulation of the CAR T cell is required for its cytolytic activity, too much stimulation tends to drive the CAR T cell to an exhaustion phenotype with less than optimal persistence for long term efficacy. To address this issue, new 2nd and 3rd generation signaling domains have been developed for CARs to moderate signal strength for enhanced clinical efficacy (30), and some have been found to improve clinical CAR T cell expansion and persistence (31). Investigation of these new domains with our DR1 CAR may prove beneficial for its function, although given the lower affinity of our CAR with its receptor it is likely that enhanced signaling through these new domains will be the goal. This concept is supported by our observation that continuous stimulation of the DR1 CAR T cell through the DR1 CAR was insufficient to maintain these cells in culture over extended periods.
The development of CAR T cells for the treatment of other autoimmune diseases has been described by several investigators with a variety of results (32). For the treatment of pemphigus vulgaris, a CAR T cell expressing a construct encoding the autoantigen desmoglein fused to CD137-CD3ζ signaling domains was designed to target B cells that produce the autoantibody for desmoglein (33). A high degree of success was achieved with the desmoglein CAR T cells in treating a mouse model of pemphigus, although the model is based on CAR T cell targeting of passively transferred B cell hybridomas, and not endogenous autoimmune B cells. Two CAR T cell studies also have been described for the treatment of autoimmune diabetes. In one study, a CAR based on an antibody that recognizes an MHC class II:peptide complex linked to CD28-CD137-CD247 signaling domains was used to target the antigen presenting cells involved in stimulating autoimmune CD4+ T cells in diabetes (34). In a second approach, Fishman et al., developed CAR T cells that target autoimmune CD8+ T cells in diabetes using an MHC class I CAR linked to antigenic-peptide/β2m/CD3-ζ domains (35). Similar to our studies, both of these CAR T cells significantly reduced the incidence of autoimmune disease, but neither generated a durable or curative therapy. As with our CAR T cells used to treat autoimmune arthritis, the target cells for the diabetes CAR T cells are very small in number, and reside predominantly within lymphoid tissue. The combination of the low frequency of the target cells and their location in solid tissue may result in the inability of CAR T cells to achieve the activation, expansion, and persistence necessary for high therapeutic efficacy. This problem is similar to the use of CAR T cells in cancer therapy when the target frequency is low and/or the targets are in solid tissue (36, 37). Both in our CAR studies and in the diabetes CAR studies, expansion and persistence appear to be lacking, suggesting that different costimulatory domains for these CARs may be needed to achieve these functional goals. Recent studies of costimulatory domains used in CAR T cells for cancer therapy suggest that the use of ICOS and 4–1BB domains (38) or a mutation in the CD28 domain (39) might solve these problems.
While immunodepletion prior to CAR T cell infusion enhances the efficacy of CAR T cell cancer therapy (40), we were not able to include this approach in our studies. Immunodepletion appears to create space for the CAR T cells to take up residence within the lymphoid organs, and promotes activation, expansion, and persistence through the development of a TSCM memory phenotype (41–43). Since immunodepletion in our studies would also interfere with the development of the autoimmune arthritis, it would confound the interpretation of the CAR T cell treatment data. The inability to create this lymphoid space may also explain why infusing more CAR T cells did not enhance the efficacy of the CAR T cell treatment in our autoimmune arthritis model. However, for treatment of patients with autoimmune disorders, immunodepletion could be used prior to CAR T-cell treatment, as an acute immunosuppression in combination with CAR T cell therapy would likely provide significant benefit in treating the disease.
In summary, the ability to target T cells in an antigen specific manner would represent a significant advancement in immunotherapy of autoimmune diseases, and the data presented here demonstrate the feasibility of achieving this goal using HLA-DR engineered CAR T cells. By targeting the CD4+ T cells that drive the autoimmune response in our model of RA, the T cell and B cell components of this autoimmune disease were significantly suppressed as well as the incidence of disease, clearly indicating the therapeutic potential of this approach. Additionally, these data also demonstrate the flexibility of CAR T cell design and the potential utility in treating a variety of diseases. While design of CAR for producing engineered T cells is highly attractive for therapeutic use, the optimal signaling domains to be used are likely to be highly variable based on several factors including target frequency, affinity between the CAR and its ligand on the target cells, and the necessity for long or short term CAR T cell function in vivo. Combining the knowledge of these functional components with recent advancements in the regulation of CAR T cells after adoptive transfer, both in terms of turning them on and off (44–46) or activating them via exogenous stimuli (47, 48), has significant potential in the generation of new, highly effective immunotherapies.
Key Points.
HLA-DR can be engineered to function as a CAR molecule in CD8+ T cells
CD8+ T cells expressing HLA-DR1 CAR molecules efficiently lyse CD4+ T cells in vivo
HLA-DR1:CII-peptide CAR T cells inhibit the development of autoimmune arthritis
Acknowledgments
This work was supported by a grant from NIH-NIAMS AR071633 (E.R.).
References
- 1.Hayter SM, and Cook MC. 2012. Updated assessment of the prevalence, spectrum and case definition of autoimmune disease. Autoimmun Rev 11: 754–765. [DOI] [PubMed] [Google Scholar]
- 2.Boyman O, Comte D, and Spertini F. 2014. Adverse reactions to biologic agents and their medical management. Nat Rev Rheumatol 10: 612–627. [DOI] [PubMed] [Google Scholar]
- 3.Matzaraki V, Kumar V, Wijmenga C, and Zhernakova A. 2017. The MHC locus and genetic susceptibility to autoimmune and infectious diseases. Genome Biol 18: 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown JH, Jardetzky TS, Gorga JC, Stern LJ, Urban RG, Strominger JL, and Wiley DC. 1993. Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature 364: 33–39. [DOI] [PubMed] [Google Scholar]
- 5.Nepom GT, Byers P, Seyfried C, Healey LA, Wilske KR, Stage D, and Nepom BS. 1989. HLA genes associated with rheumatoid arthritis. Identification of susceptibility alleles using specific oligonucleotide probes. Arthritis Rheum. 32: 15–21. [DOI] [PubMed] [Google Scholar]
- 6.du Montcel ST, Michou L, Petit-Teixeira E, Osorio J, Lemaire I, Lasbleiz S, Pierlot C, Quillet P, Bardin T, Prum B, Cornelis F, and Clerget-Darpoux F. 2005. New classification of HLA-DRB1 alleles supports the shared epitope hypothesis of rheumatoid arthritis susceptibility. Arthritis Rheum 52: 1063–1068. [DOI] [PubMed] [Google Scholar]
- 7.Gill S, and June CH. 2015. Going viral: chimeric antigen receptor T-cell therapy for hematological malignancies. Immunol Rev 263: 68–89. [DOI] [PubMed] [Google Scholar]
- 8.Kochenderfer JN, Yu Z, Frasheri D, Restifo NP, and Rosenberg SA. 2010. Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood 116: 3875–3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosloniec EF, Brand DD, Myers LK, Whittington KB, Gumanovskaya M, Zaller DM, Woods A, Altmann DM, Stuart JM, and Kang AH. 1997. An HLA-DR1 transgene confers susceptibility to collagen-induced arthritis elicited with human type II collagen. J. Exp. Med 185: 1113–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Donnelly ML, Luke G, Mehrotra A, Li X, Hughes LE, Gani D, and Ryan MD. 2001. Analysis of the aphthovirus 2A/2B polyprotein ‘cleavage’ mechanism indicates not a proteolytic reaction, but a novel translational effect: a putative ribosomal ‘skip’. J Gen Virol 82: 1013–1025. [DOI] [PubMed] [Google Scholar]
- 11.Latham KA, Whittington KB, Zhou R, Qian Z, and Rosloniec EF. 2005. Ex vivo characterization of the autoimmune T cell response in the HLA-DR1 mouse model of collagen-induced arthritis reveals long-term activation of type II collagen-specific cells and their presence in arthritic joints. J Immunol 174: 3978–3985. [DOI] [PubMed] [Google Scholar]
- 12.Rosloniec EF, Cremer M, Kang AH, Myers LK, and Brand DD. 2010. Collagen-induced arthritis. Curr Protoc Immunol Chapter 15: Unit 15 15 11–25. [DOI] [PubMed] [Google Scholar]
- 13.Rosloniec EF, Whittington KB, Zaller DM, and Kang AH. 2002. HLA-DR1 (DRB1*0101) and DR4 (DRB1*0401) use the same anchor residues for binding an immunodominant peptide derived from human type II collagen. J Immunol 168: 253–259. [DOI] [PubMed] [Google Scholar]
- 14.Denizot F, and Rita L. 1986. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods 89: 271–277. [DOI] [PubMed] [Google Scholar]
- 15.Mosmann T 1983. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65: 55–63. [DOI] [PubMed] [Google Scholar]
- 16.Kappler JW, Skidmore B, White J, and Marrack P. 1981. Antigen-inducible, H-2-restricted, interleukin-2-producing T cell hybridomas. Lack of independent antigen and H-2 recognition. J. Exp. Med 153: 1198–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosloniec EF, Brand DD, Myers LK, Esaki Y, Whittington KB, Zaller DM, Woods A, Stuart JM, and Kang AH. 1998. Induction of autoimmune arthritis in HLA-DR4 (DRB1*0401) transgenic mice by immunization with human and bovine type II collagen. J Immunol 160: 2573–2578. [PubMed] [Google Scholar]
- 18.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, Shen A, Teachey DT, Levine BL, June CH, Porter DL, and Grupp SA. 2014. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 371: 1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E, Liu H, Wu MF, Gee AP, Mei Z, Rooney CM, Heslop HE, and Brenner MK. 2011. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 118: 6050–6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qian Z, Latham KA, Whittington KB, Miller DC, Brand DD, and Rosloniec EF. 2010. An autoantigen-specific, highly restricted T cell repertoire infiltrates the arthritic joints of mice in an HLA-DR1 humanized mouse model of autoimmune arthritis. J Immunol 185: 110–118. [DOI] [PubMed] [Google Scholar]
- 21.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, and Venkateshwara VR. 2015. 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nature medicine 21: 581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tang B, Kim S, Hammond S, Cullins DL, Brand DD, Rosloniec EF, Stuart JM, Postlethwaite AE, Kang AH, and Myers LK. 2014. Characterization of T cell phenotype and function in a double transgenic (collagen-specific TCR/HLA-DR1) humanized model of arthritis. Arthritis Res Ther 16: R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, Sadelain M, and Adusumilli PS. 2016. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest 126: 3130–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosloniec EF, Ivey RA 3rd, Whittington KB, Kang AH, and Park HW. 2006. Crystallographic structure of a rheumatoid arthritis MHC susceptibility allele, HLA-DR1 (DRB1*0101), complexed with the immunodominant determinant of human type II collagen. J Immunol 177: 3884–3892. [DOI] [PubMed] [Google Scholar]
- 25.Gattinoni L, Speiser DE, Lichterfeld M, and Bonini C. 2017. T memory stem cells in health and disease. Nat Med 23: 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Biasco L, Izotova N, Rivat C, Ghorashian S, Richardson R, Guvenel A, Hough R, Wynn R, Popova B, and Lopes A. 2021. Clonal expansion of T memory stem cells determines early anti-leukemic responses and long-term CAR T cell persistence in patients. Nature Cancer: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van der Merwe PA, and Davis SJ. 2003. Molecular interactions mediating T cell antigen recognition. Annu Rev Immunol 21: 659–684. [DOI] [PubMed] [Google Scholar]
- 28.Matsui K, Boniface JJ, Reay PA, Schild H, Fazekas de St Groth B, and Davis MM. 1991. Low affinity interaction of peptide-MHC complexes with T cell receptors. Science 254: 1788–1791. [DOI] [PubMed] [Google Scholar]
- 29.Salter AI, Ivey RG, Kennedy JJ, Voillet V, Rajan A, Alderman EJ, Voytovich UJ, Lin C, Sommermeyer D, Liu L, Whiteaker JR, Gottardo R, Paulovich AG, and Riddell SR. 2018. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci Signal 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Subklewe M, von Bergwelt-Baildon M, and Humpe A. 2019. Chimeric Antigen Receptor T Cells: A Race to Revolutionize Cancer Therapy. Transfus Med Hemother 46: 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramos CA, Rouce R, Robertson CS, Reyna A, Narala N, Vyas G, Mehta B, Zhang H, Dakhova O, Carrum G, Kamble RT, Gee AP, Mei Z, Wu MF, Liu H, Grilley B, Rooney CM, Heslop HE, Brenner MK, Savoldo B, and Dotti G. 2018. In Vivo Fate and Activity of Second- versus Third-Generation CD19-Specific CAR-T Cells in B Cell Non-Hodgkin’s Lymphomas. Mol Ther 26: 2727–2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zmievskaya E, Valiullina A, Ganeeva I, Petukhov A, Rizvanov A, and Bulatov E. 2021. Application of CAR-T Cell Therapy beyond Oncology: Autoimmune Diseases and Viral Infections. Biomedicines 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, Di Zenzo G, Lanzavecchia A, Seykora JT, Cotsarelis G, Milone MC, and Payne AS. 2016. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 353: 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang L, Sosinowski T, Cox AR, Cepeda JR, Sekhar NS, Hartig SM, Miao D, Yu L, Pietropaolo M, and Davidson HW. 2019. Chimeric antigen receptor (CAR) T cells targeting a pathogenic MHC class II:peptide complex modulate the progression of autoimmune diabetes. J Autoimmun 96: 50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fishman S, Lewis MD, Siew LK, De Leenheer E, Kakabadse D, Davies J, Ziv D, Margalit A, Karin N, Gross G, and Wong FS. 2017. Adoptive Transfer of mRNA-Transfected T Cells Redirected against Diabetogenic CD8 T Cells Can Prevent Diabetes. Mol Ther 25: 456–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmidts A, and Maus MV. 2018. Making CAR T Cells a Solid Option for Solid Tumors. Front Immunol 9: 2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma S, Li X, Wang X, Cheng L, Li Z, Zhang C, Ye Z, and Qian Q. 2019. Current Progress in CAR-T Cell Therapy for Solid Tumors. Int J Biol Sci 15: 2548–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guedan S, Posey AD Jr., Shaw C, Wing A, Da T, Patel PR, McGettigan SE, Casado-Medrano V, Kawalekar OU, Uribe-Herranz M, Song D, Melenhorst JJ, Lacey SF, Scholler J, Keith B, Young RM, and June CH. 2018. Enhancing CAR T cell persistence through ICOS and 4–1BB costimulation. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guedan S, Madar A, Casado-Medrano V, Shaw C, Wing A, Liu F, Young RM, June CH, and Posey AD Jr. 2020. Single residue in CD28-costimulated CAR-T cells limits long-term persistence and antitumor durability. J Clin Invest 130: 3087–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, Taylor C, Yeh R, Bartido S, Borquez-Ojeda O, Olszewska M, Bernal Y, Pegram H, Przybylowski M, Hollyman D, Usachenko Y, Pirraglia D, Hosey J, Santos E, Halton E, Maslak P, Scheinberg D, Jurcic J, Heaney M, Heller G, Frattini M, and Sadelain M. 2011. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 118: 4817–4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, Robinson MR, Raffeld M, Duray P, Seipp CA, Rogers-Freezer L, Morton KE, Mavroukakis SA, White DE, and Rosenberg SA. 2002. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 298: 850–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muranski P, Boni A, Wrzesinski C, Citrin DE, Rosenberg SA, Childs R, and Restifo NP. 2006. Increased intensity lymphodepletion and adoptive immunotherapy--how far can we go? Nat Clin Pract Oncol 3: 668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wrzesinski C, and Restifo NP. 2005. Less is more: lymphodepletion followed by hematopoietic stem cell transplant augments adoptive T-cell-based anti-tumor immunotherapy. Curr Opin Immunol 17: 195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu BX, Song NJ, Riesenberg BP, and Li Z. 2019. Development of molecular and pharmacological switches for chimeric antigen receptor T cells. Exp Hematol Oncol 8: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Park S, Pascua E, Lindquist KC, Kimberlin C, Deng X, Mak YSL, Melton Z, Johnson TO, Lin R, Boldajipour B, Abraham RT, Pons J, Sasu BJ, Van Blarcom TJ, and Chaparro-Riggers J. 2021. Direct control of CAR T cells through small molecule-regulated antibodies. Nat Commun 12: 710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Viaud S, Ma JSY, Hardy IR, Hampton EN, Benish B, Sherwood L, Nunez V, Ackerman CJ, Khialeeva E, Weglarz M, Lee SC, Woods AK, and Young TS. 2018. Switchable control over in vivo CAR T expansion, B cell depletion, and induction of memory. Proc Natl Acad Sci U S A 115: E10898–E10906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zheng Y, Stephan MT, Gai SA, Abraham W, Shearer A, and Irvine DJ. 2013. In vivo targeting of adoptively transferred T-cells with antibody- and cytokine-conjugated liposomes. J Control Release 172: 426–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tang L, Zheng Y, Melo MB, Mabardi L, Castano AP, Xie YQ, Li N, Kudchodkar SB, Wong HC, Jeng EK, Maus MV, and Irvine DJ. 2018. Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat Biotechnol 36: 707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]