

Abstract
Purpose of the review.
Total ceramide levels in cardiac tissue relate to cardiac dysfunction in animal models. However, emerging evidence suggests that the fatty acyl chain length of ceramides also impacts their relationship to cardiac function. This review explores evidence regarding the relationship between ceramides and left ventricular (LV) dysfunction and heart failure (HF). It further explores possible mechanisms underlying these relationships.
Recent findings.
In large, community-based cohorts, a higher ratio of specific plasma ceramides, C16:0/C24:0, related to worse LV dysfunction. Increased LV mass correlated with plasma C16:0/C24:0, but this relationship became non-significant after adjustment for multiple comparisons. Decreased left atrial (LA) function and increased LA size also related to C16:0/C24:0. Furthermore, increased incident HF, overall CVD mortality, and all-cause mortality were associated with higher C16:0/C24:0 (or lower C24:0/C16:0). Finally, a number of possible biological mechanisms are outlined supporting the link between C16:0/C24:0 ceramides, ceramide signaling, and CVD.
Summary.
High cardiac levels of total ceramides are noted in HF. In the plasma, C16:0/C24:0 ceramides may be a valuable biomarker of pre-clinical LV dysfunction, remodeling, HF, and mortality. Continued exploration of the mechanisms underlying these profound relationships may help develop specific lipid modulators to combat cardiac dysfunction and HF.
Keywords: ceramides, LV dysfunction, heart failure, mortality
Introduction
Heart failure (HF) remains a leading cause of morbidity and mortality, affecting 6.5 million Americans, and its prevalence is expected to rise with the aging of the US population [1]. Given the substantial and rising burden of HF and cardiac dysfunction, ongoing efforts are needed to uncover novel mechanistic pathways that may underlie disease pathogenesis and progression. One area of recent interest involves the potential role of specific lipids in the development of heart failure. Data from humans and animal models suggest that complex lipids such as ceramides may be related to the pathogenesis of cardiac dysfunction, especially in the absence of coronary heart disease and myocardial infarction-induced ischemic cardiomyopathy.
Human data linking excessive fatty acid uptake and myocardial ceramides with cardiac dysfunction
In humans, metabolic conditions such as obesity, type 2 diabetes, and other pathologies linked with ectopic triglyceride deposition, such as lipodystrophy, and infection with human immunodeficiency virus (HIV) treated with antiretroviral therapy, are all associated with an increased risk of cardiac dysfunction and/or heart failure, even in the absence of coronary heart disease [2]. Increased fatty acid uptake is usually accompanied by increased fatty acid oxidation [3]. However, the delivery of fatty acids to the myocardium can outstrip even the heart’s prodigious capacity for oxidizing lipids, thus leading to lipid deposition [4]. Several pathology and magnetic resonance spectroscopy (MRS) studies have linked excessive triglyceride deposition with human cardiac dysfunction [5] in a process termed, “lipotoxicity”. However, some data suggest that triglyceride deposition itself is not toxic, but rather that excessive reactive oxygen species generation and/or other bioactive lipids such as ceramides may have detrimental effects on cardiac function [6, 7]. This is supported by a study of LV assist device (LVAD) core samples that noted less triglyceride – but more diacylglycerol and total ceramides – from patients with HF and insulin resistance pre-implantation, as compared with normal controls [8]. Moreover, after LVAD explantation, these changes were reversed [8]. A follow-up study, using untargeted lipidomics, found that particular ceramide species (C16:0, C16:1, C20:1, and C24:1) were increased in the myocardium of patients with HF as compared with normal controls, and the levels of these specific ceramides all decreased after LVAD removal (and improvement in LV function)[9]. In this same study, total serum ceramides were not higher in patients with HF compared with controls, but several distinct species (C16:0, C18:0, C20:1, C20:0, C22:1, and C24:1) were higher in the HF patients. After myocardial unloading and improvement of LV function, these particular serum ceramide levels decreased, and serum C14:0 and C24:0 increased. In sum, specific lipid species – and more precisely specific ceramide species – appear to be related to the pathobiology of heart failure, and tissue and serum ceramide species changes are present in overt, end-stage HF are similar but not exactly the same.
Plasma/serum ceramides and pre-clinical left ventricular dysfunction.
Overt HF in metabolic conditions associated with lipotoxicity is often preceded by asymptomatic alterations in cardiac structure and function. For example, in young women, those who were obese had worse diastolic and systolic function and higher LV mass than age-matched controls [10]. Moreover, increasing body mass index also independently predicted decreased LV relaxation and systolic function, and increased LV concentric remodeling, a precursor to LV concentric hypertrophy [10] confers a high risk for incident HF [11*]. Hence, substantial efforts have been dedicated to identifying clinical measures that can be readily assessed as predictors of preclinical LV dysfunction and used to monitor and prevent progression into overt, symptomatic HF.
Given the histologic data linking ceramides to lipotoxic pathways, and the interest in developing a ‘liquid biopsy’ for those at risk of developing HF, recent studies have aimed to link plasma/serum ceramides to preclinical LV dysfunction and remodeling. In one large study of the Framingham Heart Offspring Cohort, targeted lipidomics were used to evaluate the relationship between the ratio of C16:0 and C24:0 (one of the most abundant ceramides in the plasma) and LV structure and function in patients without overt, prevalent heart failure. In this study, C16:0/C24:0 was directly associated with preclinical LV systolic dysfunction, (i.e., lower ejection fraction and worse [less negative] global circumferential strain) [11*]. Multivariable models also show that C16:0/C24:0 was independently related to worse ejection fraction and global circumferential strain [11*]. These data jibe well with that from the aforementioned study in overt heart failure, in which higher serum C16:0 was again associated with worse function and higher C24:0 was associated with improved LV function [8].
In humans without prevalent HF, left atrial (LA) size and function and LV remodeling also related to plasma C16:0/C24:0. Specifically, an increase in this ratio independently related to worse left atrial (LA) emptying fraction and larger LA volume even after adjustment for multiple variables and multiple comparisons. The ratio also related directly with increased in LV mass [11*]. Interestingly, measures of diastolic dysfunction were not significantly related to plasma C16:0/C24:0 in this study [11*]. In contrast, in a study of the effect of triglyceride lowering in patients with type 2 diabetes, decreased in plasma C24:0/C16:0 (or an increase in C16:0/C24:0) was associated with worsened LV relaxation [12]. Taken together, the plasma ratio of C16:0/C24:0 appears to predict preclinical LV and LA dysfunction, which is often a harbinger of increased risk of HF. The ratio may be an effective tool to monitor dynamic changes in LV function that result from interventions.
Plasma/serum ceramides and incident heart failure, mortality, and HF classification.
Recent data point to particular plasma ceramides as a potential powerful new tool for incident HF risk and mortality prediction as well as preclinical LV dysfunction. In a meta-analysis of data from over 5,000 subjects in the Framingham Heart Study Offspring cohort and the large Study of Health in Pomerania (SHIP) cohort, the ratio of plasma C24:0/C16:0 related inversely (and C16:0/C24:0 related directly) with incident HF [13]. Specifically, for each 3-unit increase in the plasma ratio, there was a 22% lower risk of incident HF (Figure 1). It should be noted that this ceramide ratio also predicted freedom from coronary heart disease, which may have contributed to the decrease in incident HF [13]. This particular ratio of plasma ceramides, as assessed using targeted lipidomics, also related to total cardiovascular disease mortality, non-cardiovascular mortality, and all-cause mortality even after adjustment for multiple cardiovascular risk factors, lipid-lowering medication status and antihypertensive medication status [13]. The reduction in risk for each 3-unit increase in C24:0/C16:0 was a striking 40% decrease in all-cause mortality (95% confidence bounds 0.56 – 0.65) in the meta-analysis (Figure 1) [13]. Moreover, C24:0 alone was associated with reduced all-cause mortality, while C16:0 alone was associated with increased all-cause mortality [13]. In a study of Canadian patients with known HF, total plasma ceramide levels were associated with a significant 31% increase in all-cause mortality, even after multivariable adjustment [14]. Adding total ceramide levels improved the net reclassification index for the prediction of mortality in these patients with HF even after including N-terminal proBNP, LV ejection fraction, body mass index, atrial fibrillation, hemoglobin, and high-sensitivity troponin I [15]. Lastly, increasing total ceramides correlated directly with worsening New York Heart Association (NYHA) classification [15]. In sum, ceramides are related to human HF, but in evaluating the potential pathobiologic effects of ceramides, it is important to distinguish between total ceramides and particular ceramides with specific acyl chain lengths.
Figure 1.
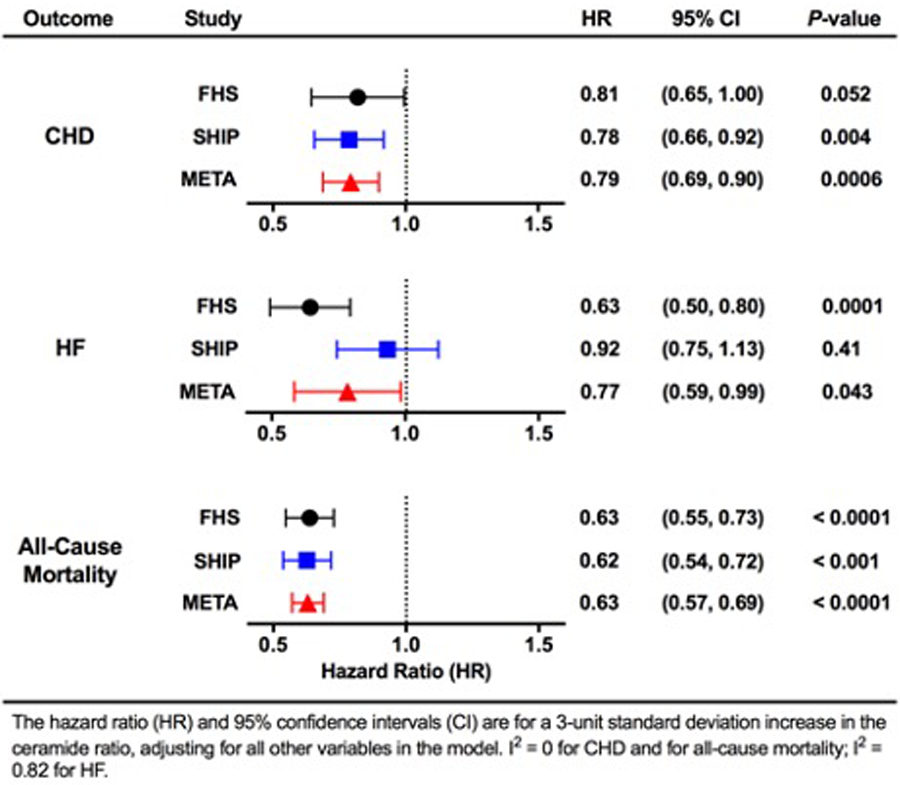
Risk of incident coronary heart disease (CHD), heart failure (HF), and all-cause mortality by the plasma C24:0/C16:0 ceramide ratio. Hazard ratios (HRs) for CHD, HF, and all-cause mortality are reported with 95% confidence intervals (CIs) for each 3-unit increase in the plasma C24:0/C16:0 ceramide ratio (average of SDs between FHS [Framingham Heart Study] and SHIP [Study of Health in Pomerania]), adjusting for all other variables in the model. Data are shown from analysis of subjects in FHS, SHIP, and the combined meta-analysis (META). I2=0 for CHD and for all-cause mortality; I2=0.81 for HF. Reproduced with permission from reference #13.
Hypothesized biological mechanisms linking differing ceramide ratios to cardiac dysfunction and increased mortality.
Ceramide synthesis and regulation of cardiac function
Ceramides are produced by three pathways: de novo pathway, the salvage pathway, and the sphingomyelinase pathway [16] (Figure 2). Total and very long–chain ceramides increase in myocardium and in the circulation following both acute and chronic ischemic injury (myocardial infarction)[9, 17**, 18*], largely via activation of the de novo synthesis pathway [9, 18*]. In the de novo pathway, palmitoyl coenzyme A and serine are catalyzed by the serine palmitoyl transferase (SPT) complex to produce ceramides (Figure 2), which is considered a rate limiting step. Curiously, inhibition of SPT by myriocin reduces the level of C16:0, C24:1, and C24:0 ceramides, inhibits maladaptive cardiac remodeling, and improves cardiac function [9]. In the degradation pathway, ceramidase (CDase) hydrolyzes ceramides to generate sphingosine. Sphingosine is then phosphorylated by sphingosine kinase (SK) to produce the pro-survival molecule sphingosine-1-phosphate (S1P). This counteracts the negative effects of elevated ceramides and promotes cell survival, which may improve cardiac function after myocardial infarction (MI) [17**]. Apart from the de novo pathway, sphingomyelinase pathway also participates in the synthesis of ceramide in heart. Inhibition of acid sphingomyelinase (SMase) reduces cardiac ceramide accumulation in the post-ischemic heart, however this did not affect heart function [19].
Figure 2.
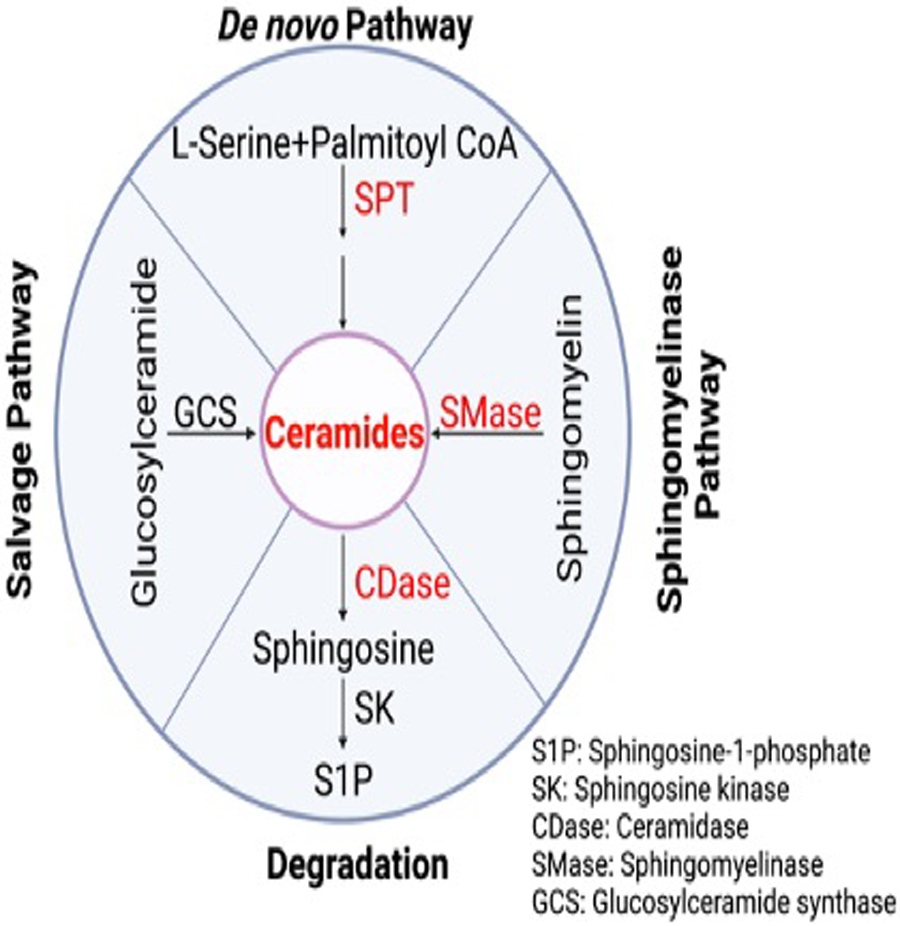
Representation of ceramide synthesis and degradation. The salvage pathway, De novo pathway, and sphingomyelinase synthesis pathway are show along with the degradation pathway. The protein abbreviations and full names are listed in the bottom right corner.
Ceramide metabolism and apoptosis in cardiac dysfunction
Ceramides have been shown to induce apoptosis in the myocardium in mice, rats and humans [17**, 20–22]. Bielawska et al. reported that total ceramides accumulate in the ischemic area in the rat myocardial ischaemia/reperfusion (I/R) model [20]. In vitro, synthetic cell permeable C2 ceramide induces apoptotic death of rat neonatal cardiomyocytes, and ischemia (oxygen/serum/glucose deprivation) leads to a progressive accumulation of total ceramide in cardiomyocytes [20]. These data indicate that ceramide signaling may be involved in myocardial ischemia/reperfusion injury in vitro. A recent study further reported that the increased myocardial ceramide (C16:0, C20:0, C20:1 and C24) during acute MI is associated with exacerbated myocardial injury and deterioration of cardiac function [17**]. Mechanistically, both short and very long chain ceramides induce cardiomyocyte apoptosis via increased cleavage of caspase 3 [22, 23] (Figure 3A). Indeed, cardiomyocyte apoptosis represents a final common pathway in response ceramide signaling via mitochondrial oxidative stress, inflammation, and altered ion channel function [9, 20, 22, 24].
Figure 3.
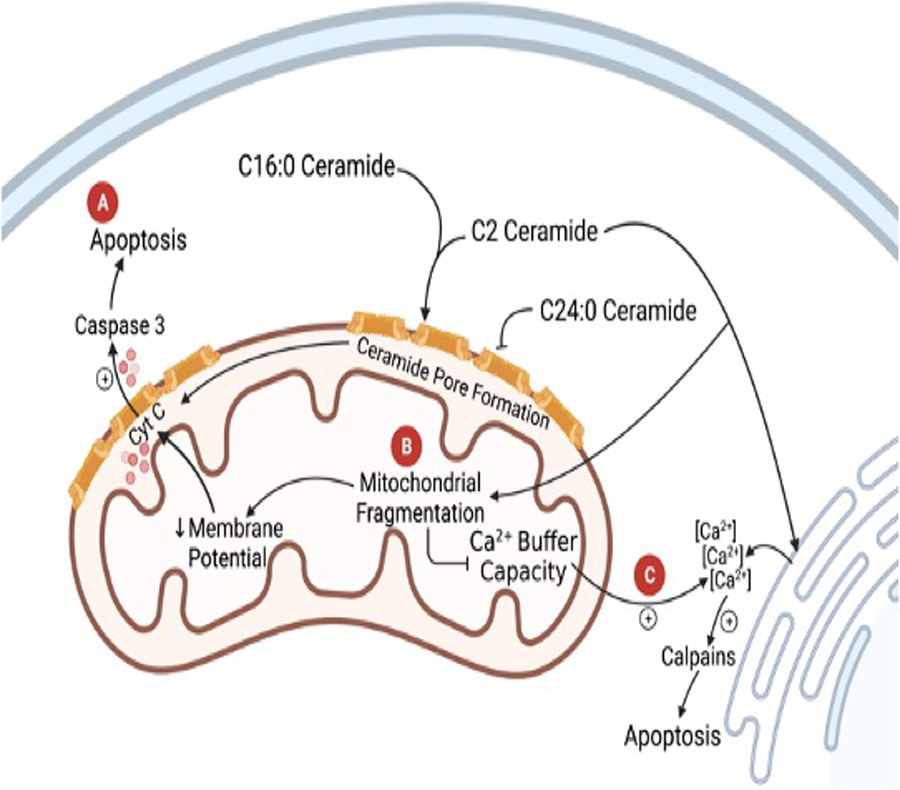
Diagram of ceramide signaling in the mitochondria in cardiomyocytes. (A) shows the release of cyt c through ceramide channels in the mitochondrial outer membrane leading to an increase in caspase 3 and apoptosis. (B) C24:0 ceramide inducing mitochondrial fragmentation and leading to a decrease in inner mitochondrial membrane potential and an inhibition of mitochondrial Ca2+ buffer capacity. (C) Increase in cytoplasmic Ca2+ from C24:0 mediated Ca2+ release from the ER and loss of Ca2+ buffer capacity in the mitochondria. The Ca2+ flux induces apoptosis through calpains.
Ceramide metabolism and mitochondrial dysfunction in cardiac dysfunction
It has been reported in many cell types that mitochondria are an important target in ceramide-mediated apoptosis [25–27]. Consistent with its pro-apoptotic effects, ceramides affect multiple mitochondrial events in cardiomyocytes. Parra et al. reported that C2 ceramide causes mitochondrial fission, which increases mitochondrial outer membrane permeability, decreases mitochondrial inner membrane potential, and decreases mitochondrial Ca2+ buffer capacity in isolated cardiomyocytes [22, 28]. Furthermore, doxorubicin stimulates mitochondrial fission (or mitochondrial fragmentation, Figure 3B) and induces cardiomyopathy and apoptosis through ceramide generation in cardiomyocytes [22]. In addition, it is postulated that C2 and C16:0 ceramides can form channels in the mitochondrial outer membrane. These channels would increase the permeability of the outer membrane and allow the release of cytochrome c into the cytoplasm causing apoptosis through caspase 3 activation [29, 30] (Figure 3A). This would contribute to cardiomyocyte death and ventricular dysfunction. Paola et al. [31] compared the interaction of C2 ceramide and C16:0 ceramide with the respiratory chain of rat heart mitochondria. They showed that both ceramides inhibit pyruvate + malate and succinate oxidation to generate reactive oxygen species, produced at the level of either complex I or complex III, and contributes to cardiomyocyte apoptosis, and myocardial injury.
The chain length of biologically active ceramides serves as an important regulatory factor in mitochondrial ceramide channel formation and permeabilization. Specifically, ceramides with different fatty acyl chain lengths can form different pore sizes in mitochondria [32]. Also, it has been shown that C24:0 ceramide counteracts C16:0 ceramide mediated cytochrome c release in a dose-dependent manner, possibly by interfering with mitochondrial outer membrane channel formation and reducing membrane permeability [30, 33]. As our clinical data reveal that C24:0 ceramide are protective while C16:0 ceramide impair cardiac function [34, 35], one putative mechanistic link is that the presence of C24:0 ceramide can interfere with C16:0-mediated mitochondrial permeabilization [30, 36], whereby these two ceramide species may exist in a counter balancing, yin/yang fashion (Figure 3). In line with this notion, in a stress response in vitro model (serum deprivation), cellular C16:0 ceramide is upregulated while C24:0 ceramide is downregulated [37]. Additionally, selective knockdown of ceramide synthase-2 (CerS2), the enzyme responsible for synthesizing very long chain ceramides, results in near elimination of C24:0 and consequently higher C16:0 ceramide levels[38]. On the other hand, it has been reported that cardiomyocyte overexpression of CerS2 and associated increases in long acyl chain (C20:0-C24:0) ceramides worsen cardiac function by reducing basal oxygen consumption, ATP turnover, and respiration capacity in a high fat diet induced diabetic cardiomyopathy model [23]. Overall, given the critical role of cardiac mitochondrial function to cardiac performance, these biologic mechanisms support the scientific premise that C24:0, C16:0 and their ratios may indeed be significantly linked HF, and adverse cardiac outcomes [11, 13, 34, 35, 39]*.
Ceramide metabolism and Inflammation in cardiac dysfunction
Inflammation and immune cell infiltration into damaged cardiac tissue are crucial factors in heart failure [21]. Krown et al. reported that the inflammatory cytokine TNFα stimulates production of sphingosine and its second messenger molecules: C2 ceramide and S1P, which implicates that C2 ceramide signaling is involved in TNFα mediated cardiomyocyte apoptosis [40]. Likewise, elevated ceramide levels can lead to neutrophil recruitment and activation [41, 42]. Yoav et al. reported that ceramide (C16:0, C20:0, C20:1, and C24:0) levels significantly increase 24 hours after MI [17]**. Acid ceramidase (catalyzes ceramide hydrolysis to free fatty acids and sphingosine) overexpression counteracts the negative effects of elevated ceramide (C20:0, C22:0) by decreasing injurious neutrophil activity, attenuating inflammation, promoting cell survival, and thereby providing a protective effect after MI [17**]. Lastly, Treatment of the Jurkat T cells with C16:0 ceramide resulted in the clustering of Kv1.3 within ceramide-enriched membrane platforms and inhibition of the channel’s activity [43], which may mediate macrophage migration in atherosclerosis [44] (Figure 4A). Apart from neutrophils, the mechanisms underlying ceramide-mediated effects on other immune cells, including cardiac resident macrophages remain unclear. Further research on the association between ceramides, immune cells and inflammation is warranted.
Figure 4.
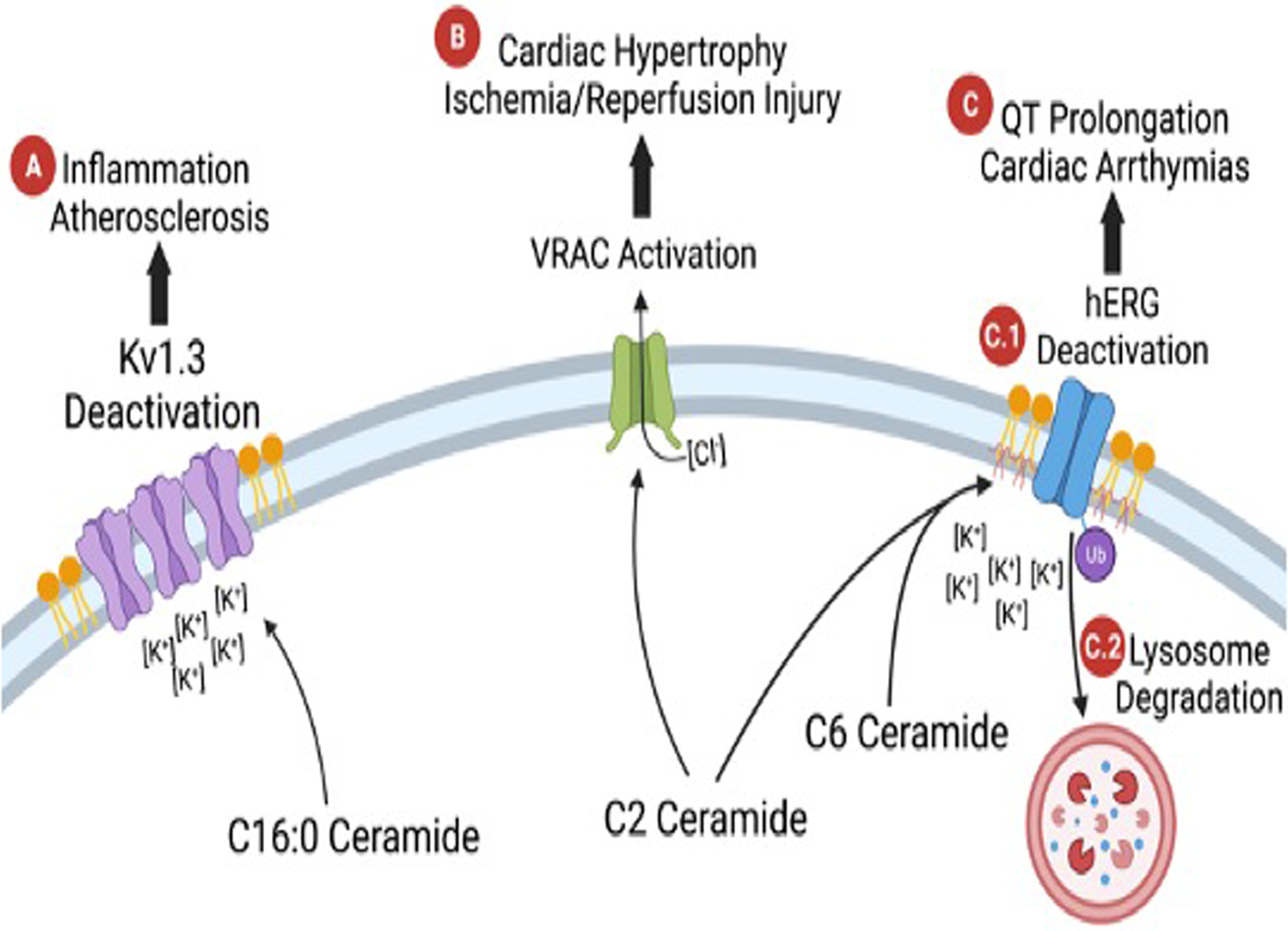
Diagram of ceramides’ interaction with ion channels. (A) C16:0 ceramide closing the Kv1.3 channel and contributing to inflammation and atherosclerosis. (B) C2 ceramide activating VRAC and contributing to cardiac hypotrophy and Ischemia-reperfusion injury. (C) Shows two different mechanisms of C2 and C6 ceramides effects on hERG. (C.1) C2 and C6 ceramides deactivate hERG, closing the channel and (C.2) cause ubiquitin mediated degradation in the lysosome. The decrease in hERG activity impaired action potential repolarization, leading to QT prolongation and cardiac arrhythmia.
Ceramides, intracellular calcium and cardiac contractile function
Intracellular Ca2+ transients play a critical role in regulating cardiac contractility [45]. Liu et al. and Relling et al. reported that exogenous application of C2 ceramide on isolated rat cardiomyocytes has a positive inotropic effect associated with alterations in both intracellular calcium transients and calcium myofilament sensitivity [46, 47]. Moreover, it has been reported that C2 ceramide induces apoptosis and necrosis in cultured cardiomyocytes by a mechanism involving increased Ca2+ influx from the ER and loss of the mitochondrial Ca2+ buffer capacity [28]. The increase in cytosolic Ca2+ triggers the activation of calpains and causes cardiomyocyte apoptosis (Figure 3C) [28] – contributing to cardiac dysfunction. In addition, Simona et al. reported that acute treatment of isolated cardiomyocytes with C6 ceramide depresses cardiac contractility by activating calcium independent isoform PKCε and increasing subsequent phosphorylation of myofilament proteins cardiac troponin I (cTnI) and cardiac myosin binding protein-C (cMyBP-C) [48]. The differences between the effects of C2 and C6 ceramides might be due to their potency and specificity. Further study of the influence of acute ceramide application on membrane ion channels, intracellular Ca2+-regulating proteins and signaling in ventricular myocytes is required to understand the precise role of C16:0 and C24:0 ceramide on cardiac contractile function.
Ceramide and ion channels in cardiac dysfunction
As described above, ceramides are postulated to form channels in phospholipid and cholesterol containing membranes [32, 49, 50] that alter membrane potential. Siskind et al. detected a dose-dependent increase in overall membrane conductance after application of either C2 or C16:0 ceramide in the planar phospholipid membranes and proposed that ceramides form large stable pores in phospholipid membranes [49]. These ceramide containing pores may influence plasma membrane potential directly via their intrinsic conductance, or indirectly by altering the electrophysiological characteristics of ion channels such as K+ channels and the volume-sensitive chloride current (ICl,swell) channels [43, 51–53*, 54,55]. Ceramides have been shown to impact cardiac K+ channels, which in turn regulate the resting membrane potential, the frequency of pacemaker cells, and the shape and duration of the cardiac action potential [56]. Specifically, C6 ceramide down-regulates the surface protein expression within caveolin-enriched lipid rafts of the cardiac Kv channel hERG (a rapid component of the delayed rectifier current (I(kr)) and is associated with ubiquitin-mediated lysosomal degradation [51] (Figure 4C.2). Furthermore, C2 or C6 ceramide application significantly accelerated the fast component of deactivation at more hyperpolarized voltages kinetically and inhibited hERG channel K+ current [51–54]*, which may cause QT prolongation and contribute to adverse cardiac outcomes [57, 58] (Figure 4C).
The swell-activated Cl− current (ICl,swell), or volume-sensitive chloride current (VSOR), or volume regulated anion channel (VRAC) is persistently activated in various cardiac disease including congestive heart failure, ischemia/reperfusion and cardiac hypertrophy [59–61]. Moreover, VRAC is involved in a variety of physiological processes including the cell volume regulation, autophagy, and apoptosis in cardiomyocytes [61–63]. In ventricular cardiomyocytes, exogenous C2 ceramide or endogenous long-chain ceramides generated by SMase have been shown to activate VRAC, were partially inhibited by hyperosmotic shrinkage, suppressed by the ROS scavengers, and fully inhibited by DCPIB (a specific inhibitor of VRAC) and tamoxifen [55]. Activation of VRAC plays a critical role in C2 ceramide induced cardiomyocyte shrinkage and apoptotic volume decrease in apoptosis. Interestingly, VRAC has been reported to both shorten action potential duration and cause depolarization of the resting membrane potential during cell swelling in cardiac myocytes [64], due to the predicted reversal potential of chloride of −40 to −20 mV. It is therefore conceivable that C16:0 and C24:0 ceramide could regulate VRAC and in turn influence cardiac repolarization and indirectly cardiac excitation-contraction coupling [65, 66] to impact contractility, cardiac hypertrophy and ischemia-reperfusion injury (Figure 4B). More studies are required to demonstrate the link between cardiac ceramide accumulation and VRAC activation in regulating cardiomyocyte electrophysiologic characteristics and cardiac function in the context of various cardiac disease.
Conclusion
Heart failure in humans appears to be driven by an increase in specific ceramide species and is directly related to elevated total ceramide levels in the plasma and myocardial tissues. Specifically, the ratio of two ceramide species, C24:0/C16:0, are inversely related to the increased risk of cardiovascular death, total mortality, and incident heart failure. Additionally, the plasma ratio of C24:0/C16:0 relates inversely and independently to worsened LV function (ejection fraction, global longitudinal strain) and LA function (LA emptying fraction) even before frank HF is diagnosed. When taken together, these observations indicate the potential importance of ceramide species, rather than total ceramide levels, present in cardiac tissue and plasma. Current evidence suggests that the relationship between ceramides and cardiac dysfunction may be mediated by the ability of ceramides to alter cardiomyocyte mitochondrial function, oxidative stress, and calcium signaling. Additionally, evidence indicated that ceramides may mediate cardiac dysfunction via interference with ion channel function, negatively impacting apoptosis and contributing to enhanced inflammation.
Key points.
HF in humans is related to increased total ceramide levels in plasma and myocardial tissue. Upon further inspection, it appears that this increase in total ceramides in HF is driven by an increase in specific ceramide species. The increase in these particular ceramide levels are reversed after LVAD therapy and improvement in LV ejection fraction. Specific serum ceramide levels are also altered in association with prevalent end-stage HF.
The ratio of two specific plasma ceramides — C24:0/C16:0 — is inversely related to the risk of incident heart failure, as well as cardiovascular death and total mortality.
The plasma C24:0/C16:0 also relates inversely and independently to worse LV function (ejection fraction, global longitudinal strain) and LA function (LA emptying fraction) even before frank HF is diagnosed. (Plasma C16:0/C24:0 relates directly to worse pre-clinical LV and LA function).
Mechanistically, there is in vitro and in vivo evidence that ceramides may contribute to cardiac dysfunction by altering cardiomyocyte mitochondrial function, oxidative stress, calcium signaling, ion channel function to impact apoptosis and inflammatory pathways.
Acknowledgements
The authors acknowledge support from the following funding: from the National Institutes of Health (NIH, Bethesda, Maryland) 1R34HL138253 (LRP), R21 HL145217 (LRP), R01 AG060499–01 (LRP), 5T32HL130357–05 (LKP, VGB), the Barnes-Jewish Hospital Foundation (LRP). NIH NIDDK R01DK106009 (R.S.), R01DK126068 (R.S.), R01DK127080 (R.S.), NIH VA Merit grant I01BX005072 (R.S.) and the Roy J. Carver Trust grant (R.S.). The authors have no relevant conflicts of interest concerning this article.
Financial support and sponsorship
This work was supported by funding from the National Institutes of Health (NIH), Bethesda Maryland (grants # R21 HL145217–01, R01 AG060499–01, R34HL138253–01 and from the Barnes-Jewish Hospital foundation. NIH NIDDK R01DK106009 (R.S.), R01DK126068 (R.S.), R01DK127080 (R.S.), NIH VA Merit grant I01BX005072 (R.S.) and the Roy J. Carver Trust grant (R.S.).
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
* of special interest
** of outstanding interest
- 1.Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, et al. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation 2018;137(12):e67–e492. [DOI] [PubMed] [Google Scholar]
- 2.Sanon VP, Handelsman Y, Pham SV, Chilton R. Cardiac Manifestations of Congenital Generalized Lipodystrophy. Clin Diabetes 2016;34(4):181–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peterson LR, Herrero P, Schechtman KB, Racette SB, Waggoner AD, Kisrieva-Ware Z, et al. Effect of obesity and insulin resistance on myocardial substrate metabolism and efficiency in young women. Circulation 2004;109(18):2191–6. [DOI] [PubMed] [Google Scholar]
- 4.Sletten AC, Peterson LR, Schaffer JE. Manifestations and mechanisms of myocardial lipotoxicity in obesity. J Intern Med 2018;284(5):478–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, et al. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. Faseb j 2004;18(14):1692–700. [DOI] [PubMed] [Google Scholar]
- 6.Lin CH, Kurup S, Herrero P, Schechtman KB, Eagon JC, Klein S, et al. Myocardial oxygen consumption change predicts left ventricular relaxation improvement in obese humans after weight loss. Obesity (Silver Spring) 2011;19(9):1804–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Listenberger LL, Han X, Lewis SE, Cases S, Farese RV Jr., Ory DS, et al. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A 2003;100(6):3077–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chokshi A, Drosatos K, Cheema FH, Ji R, Khawaja T, Yu S, et al. Ventricular assist device implantation corrects myocardial lipotoxicity, reverses insulin resistance, and normalizes cardiac metabolism in patients with advanced heart failure. Circulation 2012;125(23):2844–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ji R, Akashi H, Drosatos K, Liao X, Jiang H, Kennel PJ, et al. Increased de novo ceramide synthesis and accumulation in failing myocardium. JCI Insight 2017;2(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peterson LR, Waggoner AD, Schechtman KB, Meyer T, Gropler RJ, Barzilai B, et al. Alterations in left ventricular structure and function in young healthy obese women: assessment by echocardiography and tissue Doppler imaging. J Am Coll Cardiol 2004;43(8):1399–404. [DOI] [PubMed] [Google Scholar]
- 11.*Nwabuo CC, Duncan M, Xanthakis V, Peterson LR, Mitchell GF, McManus D, et al. Association of Circulating Ceramides With Cardiac Structure and Function in the Community: The Framingham Heart Study. J Am Heart Assoc 2019;8(19):e013050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang H, Hsu FF, Farmer MS, Peterson LR, Schaffer JE, Ory DS, et al. Development and validation of LC-MS/MS method for determination of very long acyl chain (C22:0 and C24:0) ceramides in human plasma. Anal Bioanal Chem 2013;405(23):7357–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peterson LR, Xanthakis V, Duncan MS, Gross S, Friedrich N, Völzke H, et al. Ceramide Remodeling and Risk of Cardiovascular Events and Mortality. J Am Heart Assoc 2018;7(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu J, Pan W, Shi R, Yang T, Li Y, Yu G, et al. Ceramide is upregulated and associated with mortality in patients with chronic heart failure. Can J Cardiol 2015;31(3):357–63. [DOI] [PubMed] [Google Scholar]
- 15.Pan W, Yu J, Shi R, Yan L, Yang T, Li Y, et al. Elevation of ceramide and activation of secretory acid sphingomyelinase in patients with acute coronary syndromes. Coron Artery Dis 2014;25(3):230–5. [DOI] [PubMed] [Google Scholar]
- 16.Kitatani K, Idkowiak-Baldys J, Hannun YA. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell Signal 2008;20(6):1010–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. **Hadas Y, Vincek AS, Youssef E, Zak MM, Chepurko E, Sultana N, et al. Altering Sphingolipid Metabolism Attenuates Cell Death and Inflammatory Response After Myocardial Infarction. Circulation 2020;141(11):916–30. This paper found the de novo synthesis pathway participated in the production of ceramide in acute MI. Moreover, overexpression of acid ceramidase promotes the degradation of ceramide, decreases cardiac cell death and attenuates the detrimental neutrophil infiltration in the LV, which finally improve cardiac function and lead to improved survival after MI in mice.
- 18. *Hoffman M, Palioura D, Kyriazis ID, Cimini M, Badolia R, Rajan S, et al. Cardiomyocyte Kruppel-Like Factor 5 Promotes De Novo Ceramide Biosynthesis and Contributes to Eccentric Remodeling in Ischemic Cardiomyopathy. Circulation 2021;143(11):1139–56. This study demonstrated the role of the de novo ceramide biosynthesis pathway in cardiac ceramide accumulation in human heart failure and the therapeutic potential of SPT (the key synthesis limit enzyme) inhibition for eccentric remodeling in acute and chronic heart failure. More importantly, they identified cardiomyocyte KLF5 as a pro-hypertrophic factor and knockout of cardiomyocyte KLF5 decreased ceramide biosynthesis and improved cardiac function in heart failure.
- 19.Klevstig M, Stahlman M, Lundqvist A, Scharin Tang M, Fogelstrand P, Adiels M, et al. Targeting acid sphingomyelinase reduces cardiac ceramide accumulation in the post-ischemic heart. J Mol Cell Cardiol 2016;93:69–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bielawska AE, Shapiro JP, Jiang L, Melkonyan HS, Piot C, Wolfe CL, et al. Ceramide is involved in triggering of cardiomyocyte apoptosis induced by ischemia and reperfusion. Am J Pathol 1997;151(5):1257–63. [PMC free article] [PubMed] [Google Scholar]
- 21.Adamo L, Rocha-Resende C, Prabhu SD, Mann DL. Reappraising the role of inflammation in heart failure. Nat Rev Cardiol 2020;17(5):269–85. [DOI] [PubMed] [Google Scholar]
- 22.Parra V, Eisner V, Chiong M, Criollo A, Moraga F, Garcia A, et al. Changes in mitochondrial dynamics during ceramide-induced cardiomyocyte early apoptosis. Cardiovasc Res 2008;77(2):387–97. [DOI] [PubMed] [Google Scholar]
- 23.Law BA, Liao X, Moore KS, Southard A, Roddy P, Ji R, et al. Lipotoxic very-long-chain ceramides cause mitochondrial dysfunction, oxidative stress, and cell death in cardiomyocytes. FASEB J 2018;32(3):1403–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.d’Anglemont de Tassigny A, Souktani R, Henry P, Ghaleh B, Berdeaux A. Volume-sensitive chloride channels (ICl,vol) mediate doxorubicin-induced apoptosis through apoptotic volume decrease in cardiomyocytes. Fundam Clin Pharmacol 2004;18(5):531–8. [DOI] [PubMed] [Google Scholar]
- 25.Bernardi P, Scorrano L, Colonna R, Petronilli V, Di Lisa F. Mitochondria and cell death. Mechanistic aspects and methodological issues. Eur J Biochem 1999;264(3):687–701. [DOI] [PubMed] [Google Scholar]
- 26.Crompton M The mitochondrial permeability transition pore and its role in cell death. Biochem J 1999;341 (Pt 2):233–49. [PMC free article] [PubMed] [Google Scholar]
- 27.Kroemer G, Dallaporta B, Resche-Rigon M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol 1998;60:619–42. [DOI] [PubMed] [Google Scholar]
- 28.Parra V, Moraga F, Kuzmicic J, Lopez-Crisosto C, Troncoso R, Torrealba N, et al. Calcium and mitochondrial metabolism in ceramide-induced cardiomyocyte death. Biochim Biophys Acta 2013;1832(8):1334–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J, Zhen L, Klug MG, Wood D, Wu X, Mizrahi J. Involvement of caspase 3- and 8-like proteases in ceramide-induced apoptosis of cardiomyocytes. J Card Fail 2000;6(3):243–9. [DOI] [PubMed] [Google Scholar]
- 30.Stiban J, Perera M. Very long chain ceramides interfere with C16-ceramide-induced channel formation: A plausible mechanism for regulating the initiation of intrinsic apoptosis. Biochim Biophys Acta 2015;1848(2):561–7. [DOI] [PubMed] [Google Scholar]
- 31.Di Paola M, Cocco T, Lorusso M. Ceramide interaction with the respiratory chain of heart mitochondria. Biochemistry 2000;39(22):6660–8. [DOI] [PubMed] [Google Scholar]
- 32.Colombini M Membrane channels formed by ceramide. Handb Exp Pharmacol 2013(215):109–26. [DOI] [PubMed] [Google Scholar]
- 33.Siskind LJ, Kolesnick RN, Colombini M. Ceramide channels increase the permeability of the mitochondrial outer membrane to small proteins. J Biol Chem 2002;277(30):26796–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tarasov K, Ekroos K, Suoniemi M, Kauhanen D, Sylvänne T, Hurme R, et al. Molecular lipids identify cardiovascular risk and are efficiently lowered by simvastatin and PCSK9 deficiency. J Clin Endocrinol Metab 2014;99(1):E45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laaksonen R, Ekroos K, Sysi-Aho M, Hilvo M, Vihervaara T, Kauhanen D, et al. Plasma ceramides predict cardiovascular death in patients with stable coronary artery disease and acute coronary syndromes beyond LDL-cholesterol. Eur Heart J 2016;37(25):1967–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stiban J, Fistere D, Colombini M. Dihydroceramide hinders ceramide channel formation: Implications on apoptosis. Apoptosis 2006;11(5):773–80. [DOI] [PubMed] [Google Scholar]
- 37.Fekry B, Jeffries KA, Esmaeilniakooshkghazi A, Szulc ZM, Knagge KJ, Kirchner DR, et al. C16-ceramide is a natural regulatory ligand of p53 in cellular stress response. Nat Commun 2018;9(1):4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sassa T, Suto S, Okayasu Y, Kihara A. A shift in sphingolipid composition from C24 to C16 increases susceptibility to apoptosis in HeLa cells. Biochim Biophys Acta 2012;1821(7):1031–7. [DOI] [PubMed] [Google Scholar]
- 39.Peterson LR, Jiang X, Chen L, Goldberg AC, Farmer MS, Ory DS, et al. Alterations in plasma triglycerides and ceramides: links with cardiac function in humans with type 2 diabetes. J Lipid Res 2020;61(7):1065–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krown KA, Page MT, Nguyen C, Zechner D, Gutierrez V, Comstock KL, et al. Tumor necrosis factor alpha-induced apoptosis in cardiac myocytes. Involvement of the sphingolipid signaling cascade in cardiac cell death. J Clin Invest 1996;98(12):2854–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teichgraber V, Ulrich M, Endlich N, Riethmuller J, Wilker B, De Oliveira-Munding CC, et al. Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat Med 2008;14(4):382–91. [DOI] [PubMed] [Google Scholar]
- 42.Corriden R, Hollands A, Olson J, Derieux J, Lopez J, Chang JT, et al. Tamoxifen augments the innate immune function of neutrophils through modulation of intracellular ceramide. Nat Commun 2015;6:8369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bock J, Szabo I, Gamper N, Adams C, Gulbins E. Ceramide inhibits the potassium channel Kv1.3 by the formation of membrane platforms. Biochem Biophys Res Commun 2003;305(4):890–7. [DOI] [PubMed] [Google Scholar]
- 44.Kan XH, Gao HQ, Ma ZY, Liu L, Ling MY, Wang YY. Kv1.3 potassium channel mediates macrophage migration in atherosclerosis by regulating ERK activity. Arch Biochem Biophys 2016;591:150–6. [DOI] [PubMed] [Google Scholar]
- 45.Eisner DA, Caldwell JL, Kistamas K, Trafford AW. Calcium and Excitation-Contraction Coupling in the Heart. Circ Res 2017;121(2):181–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu SJ, Kennedy RH. Positive inotropic effect of ceramide in adult ventricular myocytes: mechanisms dissociated from its reduction in Ca2+ influx. Am J Physiol Heart Circ Physiol 2003;285(2):H735–44. [DOI] [PubMed] [Google Scholar]
- 47.Relling DP, Hintz KK, Ren J. Acute exposure of ceramide enhances cardiac contractile function in isolated ventricular myocytes. Br J Pharmacol 2003;140(7):1163–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Simon JN, Chowdhury SA, Warren CM, Sadayappan S, Wieczorek DF, Solaro RJ, et al. Ceramide-mediated depression in cardiomyocyte contractility through PKC activation and modulation of myofilament protein phosphorylation. Basic Res Cardiol 2014;109(6):445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Siskind LJ, Colombini M. The lipids C2- and C16-ceramide form large stable channels. Implications for apoptosis. J Biol Chem 2000;275(49):38640–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bollinger CR, Teichgraber V, Gulbins E. Ceramide-enriched membrane domains. Biochim Biophys Acta 2005;1746(3):284–94. [DOI] [PubMed] [Google Scholar]
- 51.Chapman H, Ramstrom C, Korhonen L, Laine M, Wann KT, Lindholm D, et al. Downregulation of the HERG (KCNH2) K(+) channel by ceramide: evidence for ubiquitin-mediated lysosomal degradation. J Cell Sci 2005;118(Pt 22):5325–34. [DOI] [PubMed] [Google Scholar]
- 52.Ganapathi SB, Fox TE, Kester M, Elmslie KS. Ceramide modulates HERG potassium channel gating by translocation into lipid rafts. Am J Physiol Cell Physiol 2010;299(1):C74–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. *Miranda WE, Guo J, Mesa-Galloso H, Corradi V, Lees-Miller JP, Tieleman DP, et al. Lipid regulation of hERG1 channel function. Nat Commun 2021;12(1):1409. This paper demonstrated that ceramides are nonconventional inhibitors of hERG1, the electrophysical characteristics are similar to drug-induced QT prolongation. Mechanistically, C6-ceramide promotes hERG1 channel deactivation via a conformational selection mechanism and acts as a gating modulator rather than direct blocker of the hydrophilic permeation pathway.
- 54.Bai Y, Wang J, Shan H, Lu Y, Zhang Y, Luo X, et al. Sphingolipid metabolite ceramide causes metabolic perturbation contributing to HERG K+ channel dysfunction. Cell Physiol Biochem 2007;20(5):429–40. [DOI] [PubMed] [Google Scholar]
- 55.Raucci FJ Jr., Wijesinghe DS, Chalfant CE, Baumgarten CM. Exogenous and endogenous ceramides elicit volume-sensitive chloride current in ventricular myocytes. Cardiovasc Res 2010;86(1):55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tamargo J, Caballero R, Gomez R, Valenzuela C, Delpon E. Pharmacology of cardiac potassium channels. Cardiovasc Res 2004;62(1):9–33. [DOI] [PubMed] [Google Scholar]
- 57.Chiamvimonvat N, Chen-Izu Y, Clancy CE, Deschenes I, Dobrev D, Heijman J, et al. Potassium currents in the heart: functional roles in repolarization, arrhythmia and therapeutics. J Physiol 2017;595(7):2229–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Recanatini M, Poluzzi E, Masetti M, Cavalli A, De Ponti F. QT prolongation through hERG K(+) channel blockade: current knowledge and strategies for the early prediction during drug development. Med Res Rev 2005;25(2):133–66. [DOI] [PubMed] [Google Scholar]
- 59.Clemo HF, Stambler BS, Baumgarten CM. Swelling-activated chloride current is persistently activated in ventricular myocytes from dogs with tachycardia-induced congestive heart failure. Circ Res 1999;84(2):157–65. [DOI] [PubMed] [Google Scholar]
- 60.Huo C, Liu Y, Li X, Xu R, Jia X, Hou L, et al. LRRC8A contributes to angiotensin II-induced cardiac hypertrophy by interacting with NADPH oxidases via the C-terminal leucine-rich repeat domain. Free Radic Biol Med 2021;165:191–202. [DOI] [PubMed] [Google Scholar]
- 61.Xia Y, Liu Y, Xia T, Li X, Huo C, Jia X, et al. Activation of volume-sensitive Cl- channel mediates autophagy-related cell death in myocardial ischaemia/reperfusion injury. Oncotarget 2016;7(26):39345–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang L, Shen M, Guo X, Wang B, Xia Y, Wang N, et al. Volume-sensitive outwardly rectifying chloride channel blockers protect against high glucose-induced apoptosis of cardiomyocytes via autophagy activation. Sci Rep 2017;7:44265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Okada Y, Sato K, Numata T. Pathophysiology and puzzles of the volume-sensitive outwardly rectifying anion channel. J Physiol 2009;587(Pt 10):2141–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vandenberg JI, Bett GC, Powell T. Contribution of a swelling-activated chloride current to changes in the cardiac action potential. Am J Physiol 1997;273(2 Pt 1):C541–7. [DOI] [PubMed] [Google Scholar]
- 65.Sah R, Ramirez RJ, Backx PH. Modulation of Ca(2+) release in cardiac myocytes by changes in repolarization rate: role of phase-1 action potential repolarization in excitation-contraction coupling. Circ Res 2002;90(2):165–73. [DOI] [PubMed] [Google Scholar]
- 66.Sah R, Ramirez RJ, Oudit GY, Gidrewicz D, Trivieri MG, Zobel C, et al. Regulation of cardiac excitation-contraction coupling by action potential repolarization: role of the transient outward potassium current (I(to)). J Physiol 2003;546(Pt 1):5–18. [DOI] [PMC free article] [PubMed] [Google Scholar]