

Abstract
The progression of cancer is an evolutionary process that is challenging to monitor between sampling timepoints. However, investigation of cancer evolution over specific time periods is crucial to the elucidation of key events such as the acquisition of therapeutic resistance and subsequent fatal metastatic spread of therapy-resistant cell populations. Here we apply mutational signature analyses within clinically annotated cancer chronograms to detect and describe the shifting mutational processes caused by both endogenous (e.g. mutator gene mutation) and exogenous (e.g. mutagenic therapeutics) factors between tumor sampling timepoints. In one patient, we find that cisplatin therapy can introduce mutations that confer genetic resistance to subsequent targeted therapy with Erlotinib. In another patient, we trace detection of defective mismatch-repair associated mutational signature SBS3 to the emergence of known driver mutation CTNNB1 S37C. In both of these patients, metastatic lineages emerged from a single ancestral lineage that arose during therapy—a finding that argues for the consideration of local consolidative therapy over other therapeutic approaches in EGFR-positive non-small cell lung cancer. Broadly, these results demonstrate the utility of phylogenetic analysis that incorporates clinical time course and mutational signature deconvolution to inform therapeutic decision making and retrospective assessment of disease etiology.
Keywords: mutational signature, tumor evolution, phylogeny, bottleneck, therapeutic resistance
1. Introduction
Cancer progression is an evolutionary process that frequently enables tumors to evade our best therapeutics and, ultimately, to recur and metastasize. The majority of cancer mortality occurs through the consequent metastatic spread of therapy-resistant cell populations1. The evolution of therapeutic resistance in vivo remains enigmatic in part because it must be reconstructed from patient biopsies whose timing and location are necessarily dictated by patient care, rather than from design to empower discovery. This limitation prevents direct observation of cancer evolution between clinically-appropriate sampling timepoints, and limits investigation of tumor evolution in response to medical intervention. An understanding of the genetic evolution of clinically-important characteristics within incidental sampling intervals could potentially illuminate both the personal and the general etiology of disease progression.
First-line targeted therapies for epidermal growth factor receptor (EGFR)-positive Non-Small Cell Lung Cancer (NSCLC) offer excellent, initially sustained clinical response. Following initial treatment, patients are at risk of developing delayed metastatic disease2, frequently precipitated by EGFR T790M mutation. EGFR T790M arises within a tumor as a consequence of two distinct evolutionary processes: mutation, which leads to its appearance in single cancer-competent cells of the tumor, and selection, which increases its frequency in the tumor in response to treatment. Several in vitro experiments have implicated strong selection for EGFR inhibitor resistance during treatment 3–5. However, determining the cause of the mutation and whether it arises in a single ancestral cell lineage before a metastatic cascade or separately among multiple metastatic lineages has been challenging. Patient care seldom takes the form of a single, monolithic intervention. A better understanding could circumvent clinical trajectories that increase the frequency of resistance, or weigh in on pursuit of local consolidative treatment (LCT) over alternative approaches such as maintenance therapy6.
Here, we reconstruct ancestral states of genetic mutations to infer relationships among clonal lineages and reveal temporal patterns of divergent events in metastatic progression7. We combine Bayesian inference of cancer chronograms with mutational signature analysis to deeply analyze tumor evolution in two patients with metastatic lung adenocarcinoma, providing the first-in-human, clinically correlated tumor phylogenetic analysis of the metastatic cascade in EGFR-mutated NSCLC. Through examination of the phylogenetic topology and branchwise mutational signature analysis, we contextualize the effect of known endogenous and exogenous mutational processes within the evolution of individual patient metastatic cascades. Specifically, we investigate whether exogenous exposure to platinum therapy preceding erlotinib could precipitate T790M mutation. We then examine whether endogenous mutational processes could play a similar role introducing new mutations into a dynamically evolving tumor. Lastly, we test whether such mutations arising in therapy-associated tumor lineage bottlenecks constitute singular lineages that form the basis of multiple therapy-resistant metastatic events. We use these results to evaluate the clinical relevance of shifts in endogenous and exogenous mutational processes and to motivate clinical consideration of potential opportunities for local consolidative treatment.
2. Materials and Methods
2.1. Tumor Sampling and Sequencing
The tissues assessed in this study were obtained from the Yale Pathology Archives based on Yale Human Investigation Committee at Yale University, Protocol no. 0304025173 to Dr. David L. Rimm, which enabled retrieval of tissue from archives that was consented or had been approved for use with waiver of consent. Sample collection and tumor sequencing were performed as described in Zhao et al8. Clinical information was retrospectively obtained for this investigation from electronic medical records after approval from the Yale University Human Investigation Committee (HIC 1508016314).
2.2. Variant Calling
Variant calling was performed using the protocol described in Zhao et al.8 using the same—but updated—SNP reference databases (accessed July 2019). Briefly, this approach involved alignment of whole-exome capture Illumina HiSeq reads to the hg19 human reference genome using Eland. Somatic single-nucleotide variants were then provisionally called using a combination of GATK and freebayes. Polymorphisms within the National Heart, Lung, and Blood Institute Exome Sequencing Project, 1000 Genomes, or Yale Human Exome Database were filtered. The resulting somatic variants were then analyzed for false negatives using the multinomial variant calling approach presented in Zhao et al.8 The resultant Mutational Annotation Format files were converted to multi-fasta formatted alignment files using the paired-normal sequence for reference.
2.3. Phylogenetic Analysis
Evolutionary relationships and divergence times were simultaneously estimated in BEAST v.2.5.2 9 assuming an uncorrelated model of molecular rates with a lognormal distribution (UCLN) and a coalescent exponential population growth branching process prior. Divergence times were calibrated using 1) a lower bound for the most recent common ancestor of the germline lineages at the patients’ year of birth, and 2) a non-contemporaneous tip age for primary tumor sequences at the time of biopsy and contemporaneous tip ages for all metastatic samples taken during autopsy. Each cancer chronogram was inferred by three independent runs of 5.0 × 108 iterations, sampling every 1,000 generations. Sufficient sampling of the posterior distribution for each parameter was evaluated via computation of effective sample size (ESS) values, with ESS values greater than 200 indicating adequate sampling of a target parameter. Independent runs were then assessed for convergence and appropriate levels of burn-in through visual inspection of the marginal posterior probabilities versus the generation state using Tracer v.1.6. Sampled posteriors from these three consistent executions were combined with TreeAnnotator v.2.4 to generate a maximum clade-credibility tree that summarized the posterior distribution of estimated evolutionary relationships and branch lengths.
To ensure the robustness of the tumor tree topology to the choice of alternate phylogenetic inference approaches, we validated the tree topology using maximum-likelihood in IQ-TREE10. We conducted 1000 bootstrap iterations fitting a GTR model to the alignment and found that the bootstrap support corresponded to the posterior support for the topology. Ancestral states of variant nucleotides were then reconstructed via maximum likelihood in FastML11 using the maximum clade-credibility tree under a GTR substitution model specifying a gamma distribution of rates discretized into eight categories. These ancestral states were found to be entirely consistent with those obtained by computing the ancestral states using the empirical-Bayes method in IQ-TREE as well as those obtained by taking the average probability determined by FastML across sampled posterior tree states.
2.4. Tracing of Mutational Processes Associated with COSMIC Mutational Signatures
Mutational signatures were assessed using deconstructSigs (v.1.9.0) 12, referencing only lung-associated endogenous and any known exogenous signatures (COSMIC Exome reference signatures, version 3, May 2019), enforcing a standard minimum signature contribution threshold of 0.05. The proportions of mutations attributable to these mutational signatures were traced to branches within patient chronograms for each set of inferred ancestral-state variants. The results of the mutational signature analysis are located in the Supplemental Materials (Figures S1 and S2)
2.5. Code and Data Availability
Input files for phylogenetic analyses, other code, intermediate files, and diagnostic images are available at https://github.com/Townsend-Lab-Yale/LUAD-PhyLCT. The sequencing reads have been made publicly available through the NCBI Sequence Read Archive, published under the BioProject accession PRJNA674368 on November 3rd, 2020.
3. Results
3.1. Platinum Therapy Preceding Erlotinib Therapy Precipitates T790M Mutation
We applied our Bayesian phylogenetic inference and mutational signature analyses to two patients. Patient 435 received cisplatin, a platinum-based chemotherapeutic, coupled with pemetrexed (Pe), shortly after primary tumor biopsy, then over a year later received the EGFR tyrosine kinase inhibitor erlotinib. Our phylogenetically informed mutational signature inference (Fig. 1) illustrates that the known platinum-associated mutational signature SBS35 (white) was coincident with cisplatin therapy: there was no SBS35 signature attributed to the ancestor of the primary tumor and metastatic lineages, and the highest proportion of mutations attributable to this signature (19.1%) was observed in the inferred ancestor immediately subsequent to cisplatin exposure. The EGFR T790M resistance mutation was coincident with this peak proportion of cisplatin mutations, occurring along the same branch in the phylogeny via the nucleotide substitution c.2369C→T. Signature SBS35 is especially associated with C→T mutations, which constitute 20.3% of mutations associated with the signature. In the mutational landscape of this patient, this contribution from SBS35 corresponds to a 20.1% increase in risk of generating mutations resulting in EGFR T790M. Preceding erlotinib therapy with cisplatin therapy presumably reduces the cancer cell population size. Simultaneously, it induces specific genetic heterogeneity that can include low-frequency, initially undetectable cancer cells with the T790M mutation. While a signal of signature SBS35 persisted until end-of-life, the cessation of cisplatin therapy led to serially smaller average proportions (≤12%) of mutations attributable to SBS35 across the metastatic clade. Mutations arising from other processes increased while mutations from SBS35 had already plateaued as early as three years prior to sampling of the metastases at autopsy.
Figure 1:
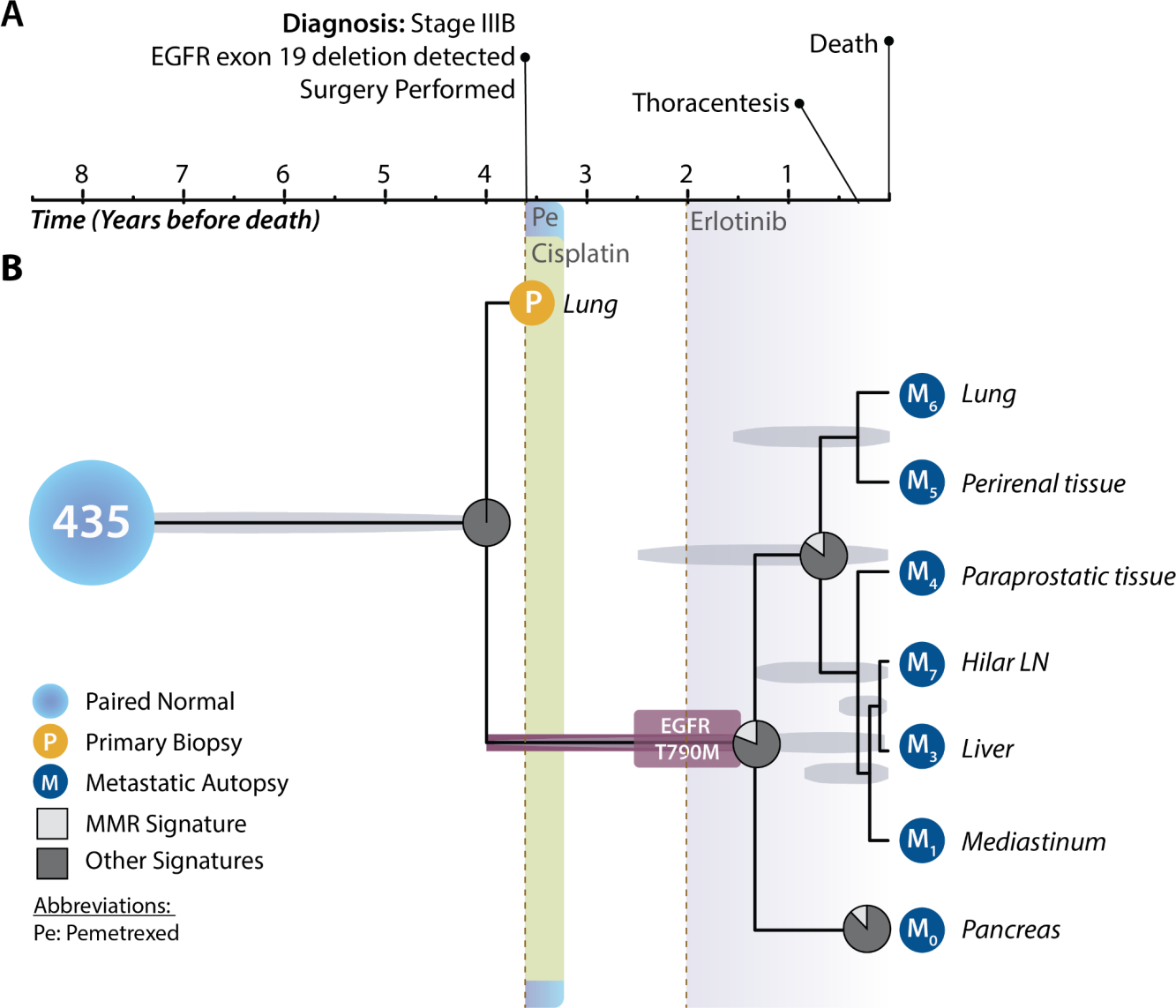
Clinical timeline, phylogeny of primary tumor and metastatic lineages, and shifting mutational processes for patient 435. (A) Clinical timeline designating relevant major events in the medical history of the patient. The patient was a man incidentally diagnosed with stage-IIIB lung cancer after a routine chest x-ray at 79 years of age, just over 3.5 years before death (BD). Resection of the primary tumor of the lung (yellow circle) was attempted upon diagnosis, and initial pathology revealed an EGFR Exon-19 deletion. The patient underwent postoperative chemotherapy with pemetrexed (aqua shading) and cisplatin (beryl green shading). EGFR-targeted therapy had yet to be FDA approved. Fourteen months (1.2 years) after his last cycle of pemetrexed and cisplatin, his disease progressed. Then he received the EGFR-targeted therapy erlotinib (purple-grey shading). Twenty months (1.7 years) after initiation of erlotinib, his disease continued to progress. He developed a malignant pleural effusion. He died of progressive metastatic disease. Metastatic tissues (dark blue circles) were sampled at autopsy from his hilar lymph nodes, liver, lungs, mediastinum, pancreas, as well as perirenal and para-prostatic soft tissue. (B) Tumor chronogram reflecting the temporal evolution of primary and metastatic tissues in relation to disease progression within patient 435 (normal tissue, light blue circle). The divergence time between the primary tumor (yellow circle) and all metastases was estimated at 4 years before death (BD). Proportions of mutations attributable to cisplatin-associated SBS35 (white slice) and to other signatures (dark grey) are indicated at ancestral nodes in the tumor tree. EGFR T790M mutation arose to detectable frequency some time between 1.5–4 years BD (burgundy box), presumably originating at low frequency during cisplatin therapy between 3–3.5 years and rising in frequency in response to treatment with erlotinib 1.5–2 years BD. The length of the truncal branch is truncated so as to provide sufficient space to display treatment, progression, and tumor tree. Light-grey ovals underlying each ancestral node indicate the 95% highest posterior density interval for each node age estimate. The y axis of this chronogram serves only to provide space so that the order of branching events across the x axis can be visualized and thus distances on the y axis have no biological meaning.
3.2. CTNNB1 S37C Mutation Follows Treatment with Bevacizumab and Precipitates Defective Mismatch-Repair
We conducted tumor-tree inference and mutational signature analyses on a second patient. This patient received first-line erlotinib therapy before disease progression at 1 year, prompting a switch to bevacizumab. Later, carboplatin and pemetrexed treatments were added. Following the onset of bevacizumab treatment, a lineage diverged and gave rise to a highly vascular splenic metastatic mass and a distinct metastatic clade with an epithelial-mesenchymal transition- and angiogenesis-associated CTNNB1 S37C mutation. Less than a year before the time of death, lineages within this CTNNB1-mutant metastatic clade diverged genetically and colonized adrenal, lung, kidney, paratracheal lymph node, and liver tissues (Fig. 2). The known defective mismatch repair mutational signature SBS3 (white) followed acquisition of CTNNB1 S37C. There is no detectable SBS3 signature on any lineage lacking the mutation (Fig. 2), including the contemporaneous lesion in the highly vascularized spleen. Unlike the exogenously generated SBS35 signature in patient 435—which attenuated after discontinuation of cisplatin therapy (Fig. 1)—the endogenously generated SBS3 signature only becomes more prominent over time subsequent to the clonal growth of the CTNNB1 S37C lineage and its metastatic diversification. Only 8% of mutations in the common ancestor of the non-splenic metastases were attributable to signature SBS3. In contrast, 18% and 17% of the attributable mutations in the liver lesion and the lineage leading to the other CTNNB1 S37C metastases were attributable to signature SBS3. This signature of DNA mismatch-repair deficiency was specific to the metastatic lineages that arose subsequent to the appearance of the CTNNB1 S37C mutation.
Figure 2:
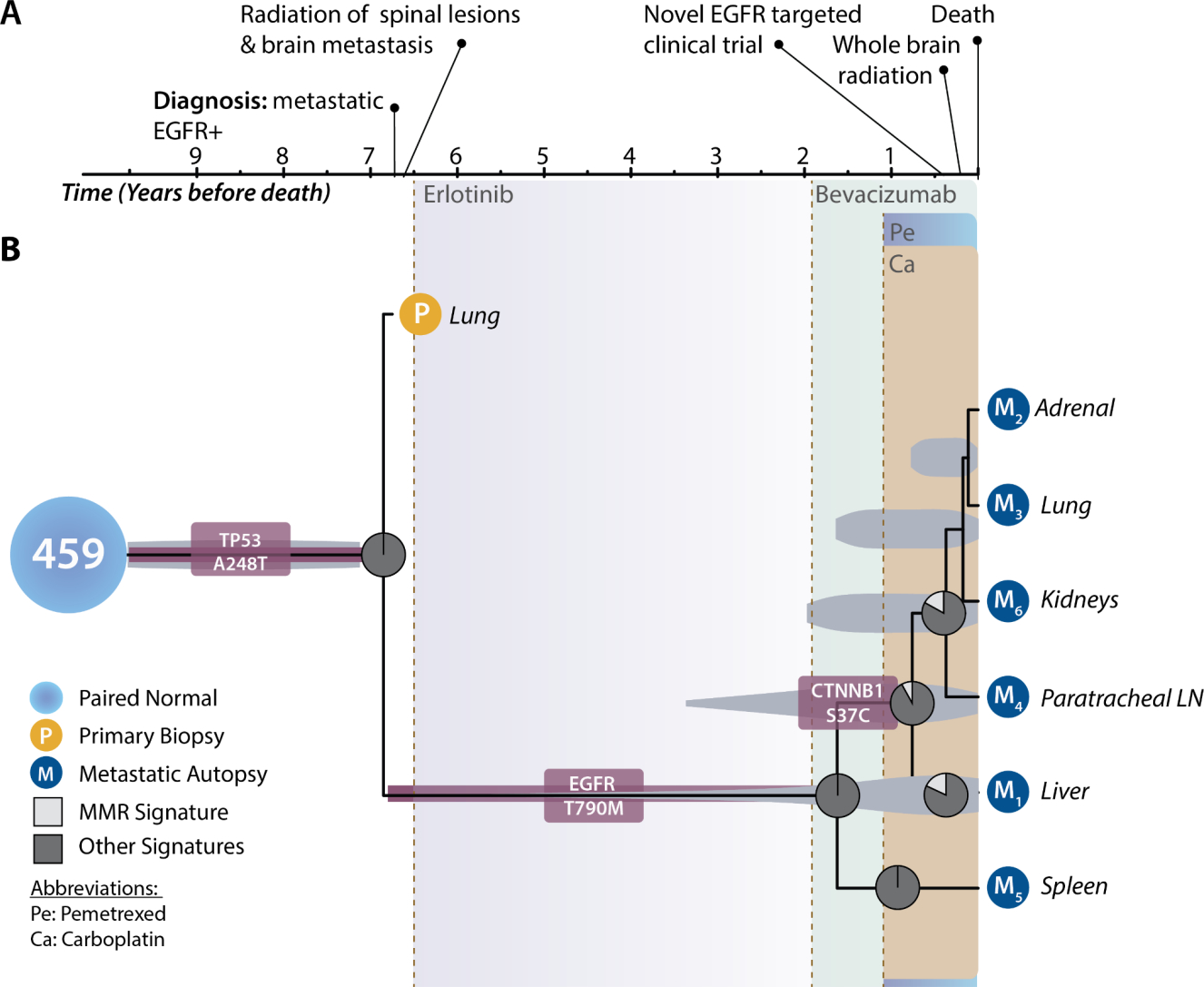
Clinical timeline, phylogeny of primary tumor and metastatic lineages, and shifting mutational processes for patient 459. (A) Clinical timeline designating relevant major events in the medical history of the patient. The patient was a woman diagnosed at 47 years of age with metastatic lung cancer at initial presentation. She subsequently underwent palliative radiotherapy to the spine and brain. Then she received EGFR-targeted therapy (erlotinib, purple-grey shading). Fifty-four months (4.5 years) after initiation of erlotinib, her disease progressed. Her therapeutic regimen was expanded to include bevacizumab (sage shading) along with continued erlotinib. One year after beginning bevacizumab, the disease further progressed. Erlotinib therapy was discontinued. Therapy was expanded with carboplatin (brown shading) and pemetrexed (aqua shading). Due to sustained disease progression, four months BD the patient underwent therapy with a novel EGFR-targeted compound (AP26113) in a clinical trial. Two months and one month prior to her death, additional treatment with whole-brain radiation and re-initiation of erlotinib were elected. She died of progressive metastatic disease. Metastatic tissue (dark blue circles) was sampled at autopsy from her adrenal gland, kidney, liver, lungs, paratracheal lymph nodes (LN), and spleen. (B) Tumor chronogram reflecting the temporal evolution of primary and metastatic tissues in relation to disease progression within patient 459 (normal tissue, light blue circle). A most recent common ancestor of the primary tumor and all metastases was reconstructed at 6.9 years before death (BD) and featured a TP53 mutation (burgundy rounded rectangle). EGFR T790M mutation (burgundy rounded rectangle) arose to detectable frequency some time between 1.6–6.5 years BD. A CTNNB1 mutation (burgundy rounded rectangle) similarly reached detectable frequency in the non-splenic lesions during bevacizumab therapy. Proportions of mutations attributable to defective homologous recombination-based DNA damage repair signature SBS3 (white slice) and attributable to other signatures (dark grey) are indicated at ancestral nodes in the tumor tree. The length of the truncal branch is shortened so as to provide sufficient space to display treatment, progression, and tumor tree. Light grey ovals underlying each ancestral node indicate the 95% highest posterior density interval for each node age estimate. The y axis of this chronogram serves only to provide space so that the order of branching events across the x axis can be visualized and thus distances on the y axis have no biological meaning.
4. Discussion
Here we have demonstrated that placing patient clinical treatment data and disease progression into the context of the molecular evolutionary history of tumor lineages provides clinically relevant insights. In one case, we showed how cisplatin-therapy induced a temporally localized burst of mutations, precipitating the evolution of EGFR T790M resistance to erlotinib. In a second case, a CTNNB1 S37C mutation arose during the administration of Bevacizumab, preceding the rapid diversification of tumor lineages with a signature of DNA mismatch-repair deficiency and leading to a clade of metastases. In both cases, metastatic lineages were found to emerge from a single ancestral lineage arising during therapy. Collectively, these results demonstrate the clinical relevance of shifts in mutational signatures that result from changing endogenous and exogenous mutational processes. They support pursuit of local consolidative treatments in EGFR-driven non-small cell lung cancer. When treatments cause shifts in mutational processes or interact with mutations causing them, the temporal order of treatment matters.
These two cases illustrate that endogenous and exogenous mutational processes provide the raw material for selective evolution enabling the resistance to therapy. The EGFR T790M mutation is known to convey resistance to erlotinib13,14, and our results suggest that CTNNB1 S37C rescued a tumor lineage from the anti-angiogenic selection pressure applied by therapy with bevacizumab 15–17. However, confirmation of these roles necessitates estimation of the strength of selection for resistance and rescue mutations18. Estimation of resistance and rescue effect sizes within distinct branches of tumor trees that are contemporaneous with clinical treatment will require a larger cohort with samples of normal, primary, and recurrent or metastatic tumor tissue. Future clinical trials or other studies incorporating metachronous sampling that apply phylogenetic and mutation signature analyses can illuminate the clinical implications of serial therapeutic treatments on progression.
It might be counterintuitive that a CTNNB1 mutation could be considered a rescue mutation from treatment with bevacizumab, when treatment with bevacizumab is clinically indicated by the presence of CTNNB1. However, both bevacizumab and CTNNB1 operate—at least in part—by shifting the tumor in opposing directions along a phenotypic axis of angiogenesis via modulation of the VEGF pathway. CTNNB1 S37C is a putative gain-of-function mutation associated with activation of the Wnt pathway, which is associated with loss of mismatch-repair function19–21. Through its role in Wnt signaling, CTNNB1 increases VEGF expression by binding its gene promoter22. The VEGF promoter features seven confirmed consensus binding sites for the beta-catenin/TCF complex, so that CTNNB1 S37C stimulates angiogenesis via VEGF23,24. Treatment with bevacizumab inhibits VEGF function. In the context of a tumor with CTNNB1 mutation, treatment with bevacizumab abrogates the increase in angiogenesis and consequent benefits to tumor growth and proliferation of CTNNB1 mutation. In the context of a CTNNB1 wildtype tumor, bevacizumab decreases angiogenesis, throttling the tumor, and CTNNB1 mutation rescues angiogenesis, benefitting tumor growth and proliferation. Our results lend support to the hypothesis that CTNNB1 S37C can act as a rescue mutation from VEGF-inhibitors.
Patient 459 exhibited no detectable SBS31 or SBS35 signature, despite receiving a short end-of-life treatment with carboplatin. It is possible that these mutation signatures were not successfully deconvolved by machine-learning oriented approaches to deconvolve mutational signatures within tumors25–31. However, the presence of SBS35 in all extant and inferred tumor samples subsequent to cisplatin therapy in patient 435 supports the consistency and specificity of signature extraction from tree branches, as does the absence of SBS3 on the early diverging spleen lineage in patient 459, which lacks mutations associated with defective homologous recombination repair. Instead, this lack of immediate detection following treatment is more likely attributable to the lag time before mutations that arise in single cells are selected or drift to clonal fixation32. Mutations require time to reach detectable prominence in the tumor via sustained replication and clonal growth.
Prevention of the emergence of resistant subclones prior to divergence of a metastatic lineage within the primary tumor, could result in significant therapeutic benefit. The observation of a clade of metastases featuring a single common ancestor with the primary tumor implies a strong selective bottleneck associated with erlotinib therapy. Consequent to targeted therapy, low-frequency mutations can form the basis of evolving resistance. The therapy-induced bottleneck suggests an opportunity for local consolidative treatment (LCT) to prevent later metastatic disease. For instance, compared to utilizing first-line treatment in isolation, multi-modality management (such as high-precision ablative radiotherapy or surgery) could alter the trajectory of disease and provide a survival benefit. Notably, recent randomized controlled phase-II trials have confirmed that patients with oligometastatic EGFR-positive NSCLC experience prolonged overall survival and progression-free survival with LCT 6,33,34. This prolonged survival suggests that early intervention with LCT might extinguish the reservoir of drug-tolerant cells. Considering these results in the context of anticipatory treatment of evolving cancers, suggests that such considerations could improve clinical outcomes. For example, less-than-ablative radiation doses have an unintended potential to generate variation enabling tumor escape from targeted therapy. Further investigation of whether ablative radiation doses should be used rather than less-than-ablative doses is warranted.
Shifts in mutational processes driven by therapeutic intervention and by genic aberration can be deconvolved using mutational signature analyses. These analyses provide deeper insights into the tumor evolution than the same analyses devoid of phylogenetic information. Resulting insights have clinical implications regarding the timing of DNA-damaging therapy and targeted cancer treatment for which acceleration of mutational heterogeneity may be detrimental. The presence of SBS35 subsequent to cisplatin therapy in patient 435 in particular has clinical implications regarding the sequencing of chemotherapeutics that increase mutational loads and targeted treatment. If radiation doses are used that are unlikely to be ablative, the same can be said for pre-systemic therapy or concurrent radiation treatment. Our study serves as a template for future evolutionary analyses that seek to reveal the etiology of metastases. Such revelations are essential to defining the general rules that shape the evolutionary trajectory of cancer. Developing such an understanding of how dynamic mutational processes evolve and result in the diversification of metastatic lineages can inform clinical decision making, guide the design of treatment paradigms, and improve patient outcomes.
Supplementary Material
Acknowledgements:
JPT acknowledges the Elihu endowment and the Notsew Orm Sands Foundation, and JPT and DR acknowledge Gilead Sciences, Inc, and NIH NIDCR 1P50DE030707 for funding supporting this research. JNF acknowledges NIH NLM 5T15LM007056-32, NIH NCI 1F31CA257288-01 for financial support to conduct research, and NIH NCI T32CA19300 for training. We also thank the Yale Center for Research Computing for computational resources and support.
Footnotes
Conflicts of Interest: Sanjay Aneja reports personal fees for work as an associate editor from JCO Clinical Cancer Informatics. Joseph N. Contessa reports a research agreement with Spring Bank Pharmaceuticals. David Rimm reports his work as a consultant, advisor, or position on a Scientific Advisory Board for Amgen, Astra Zeneca, Agendia, Biocept, BMS, Cell Signaling Technology, Cepheid, Daiichi Sankyo, GSK, Merck, NanoString, Perkin Elmer, PAIGE, Sanofi, and Ultivue. He has received research funding from Gilead Sciences, Inc, Astra Zeneca, Cepheid, Nanostring, Navigate/Novartis, NextCure, Lilly, Ultivue, and Perkin Elmer. James Yu reports consulting and speaker fees from Augmenix / Boston Scientific. Jeffrey Townsend reports research funding from Gilead Sciences, Inc and consulting with Black Diamond, Agios, and Servier.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data and code availability:
All data and code used in the analysis is referenced within the Methods and Supplementary Tables.
References
- 1.Dillekås H, Rogers MS & Straume O Are 90% of deaths from cancer caused by metastases? Cancer Med. 8, 5574–5576 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stewart EL, Tan SZ, Liu G & Tsao M-S Known and putative mechanisms of resistance to EGFR targeted therapies in NSCLC patients with EGFR mutations-a review. Transl Lung Cancer Res 4, 67–81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ortiz-Cuaran S et al. Heterogeneous Mechanisms of Primary and Acquired Resistance to Third-Generation EGFR Inhibitors. Clin. Cancer Res. 22, 4837–4847 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Bolan PO et al. Genotype-Fitness Maps of EGFR-Mutant Lung Adenocarcinoma Chart the Evolutionary Landscape of Resistance for Combination Therapy Optimization. Cell Syst 10, 52–65.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foggetti G et al. Genetic determinants of EGFR-Driven Lung Cancer Growth and Therapeutic Response In Vivo. Cancer Discov. (2021) doi: 10.1158/2159-8290.CD-20-1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gomez DR et al. Local Consolidative Therapy Vs. Maintenance Therapy or Observation for Patients With Oligometastatic Non-Small-Cell Lung Cancer: Long-Term Results of a Multi-Institutional, Phase II, Randomized Study. J. Clin. Oncol. 37, 1558–1565 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Somarelli JA et al. Molecular Biology and Evolution of Cancer: From Discovery to Action. Mol. Biol. Evol. 37, 320–326 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao Z-M et al. Early and multiple origins of metastatic lineages within primary tumors. Proc. Natl. Acad. Sci. U. S. A. 113, 2140–2145 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bouckaert R et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 15, e1006650 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Minh BQ et al. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 37, 1530–1534 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ashkenazy H et al. FastML: a web server for probabilistic reconstruction of ancestral sequences. Nucleic Acids Res. 40, W580–4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosenthal R, McGranahan N, Herrero J, Taylor BS & Swanton C DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 17, 31 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pao W et al. Acquired Resistance of Lung Adenocarcinomas to Gefitinib or Erlotinib Is Associated with a Second Mutation in the EGFR Kinase Domain. PLoS Med. 2, e73 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Demuth C et al. The T790M resistance mutation in EGFR is only found in cfDNA from erlotinib-treated NSCLC patients that harbored an activating EGFR mutation before treatment. BMC Cancer 18, 1–5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Culy C Bevacizumab: antiangiogenic cancer therapy. Drugs Today 41, 23–36 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Haibe Y et al. Resistance Mechanisms to Anti-angiogenic Therapies in Cancer. Front. Oncol. 10, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.von Felden J et al. Mutations in circulating tumor DNA predict primary resistance to systemic therapies in advanced hepatocellular carcinoma. Oncogene 40, 140–151 (2021). [DOI] [PubMed] [Google Scholar]
- 18.Cannataro VL, Gaffney SG & Townsend JP Effect Sizes of Somatic Mutations in Cancer. J. Natl. Cancer Inst. 110, 1171–1177 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chmara M et al. Multiple pilomatricomas with somatic CTNNB1 mutations in children with constitutive mismatch repair deficiency. Genes Chromosomes Cancer 52, 656–664 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Serebryannyy LA, Yemelyanov A, Gottardi CJ & de Lanerolle P Nuclear α-catenin mediates the DNA damage response via β-catenin and nuclear actin. J. Cell Sci. 130, 1717–1729 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castiglia D et al. Concomitant activation of Wnt pathway and loss of mismatch repair function in human melanoma. Genes Chromosomes Cancer 47, 614–624 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Easwaran V et al. beta-Catenin regulates vascular endothelial growth factor expression in colon cancer. Cancer Res. 63, 3145–3153 (2003). [PubMed] [Google Scholar]
- 23.Pate KT et al. Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J. 33, 1454–1473 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olsen JJ et al. The Role of Wnt Signalling in Angiogenesis. Clin. Biochem. Rev. 38, 131–142 (2017). [PMC free article] [PubMed] [Google Scholar]
- 25.Bayati M et al. CANCERSIGN: a user-friendly and robust tool for identification and classification of mutational signatures and patterns in cancer genomes. Sci. Rep. 10, 1286 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Afsari B et al. Supervised mutational signatures for obesity and other tissue-specific etiological factors in cancer. Elife 10, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lyu X, Garret J, Rätsch G & Lehmann K-V Mutational signature learning with supervised negative binomial non-negative matrix factorization. Bioinformatics 36, i154–i160 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thutkawkorapin J, Eisfeldt J, Tham E & Nilsson D pyCancerSig: subclassifying human cancer with comprehensive single nucleotide, structural and microsatellite mutational signature deconstruction from whole genome sequencing. BMC Bioinformatics 21, 128 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gulhan DC, Lee JJ-K, Melloni GEM, Cortés-Ciriano I & Park PJ Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat. Genet. 51, 912–919 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Huang P-J et al. mSignatureDB: a database for deciphering mutational signatures in human cancers. Nucleic Acids Res. 46, D964–D970 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baez-Ortega A & Gori K Computational approaches for discovery of mutational signatures in cancer. Brief. Bioinform. 20, 77–88 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Foo J, Leder K & Michor F Stochastic dynamics of cancer initiation. Phys. Biol. 8, 015002 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iyengar P et al. Consolidative Radiotherapy for Limited Metastatic Non-Small-Cell Lung Cancer: A Phase 2 Randomized Clinical Trial. JAMA Oncol 4, e173501 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Palma DA et al. Stereotactic ablative radiotherapy versus standard of care palliative treatment in patients with oligometastatic cancers (SABR-COMET): a randomised, phase 2, open-label trial. Lancet 393, 2051–2058 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Input files for phylogenetic analyses, other code, intermediate files, and diagnostic images are available at https://github.com/Townsend-Lab-Yale/LUAD-PhyLCT. The sequencing reads have been made publicly available through the NCBI Sequence Read Archive, published under the BioProject accession PRJNA674368 on November 3rd, 2020.
All data and code used in the analysis is referenced within the Methods and Supplementary Tables.