

Abstract
Carboxy-terminal domain (CTD) small phosphatase like 2 (CTDSPL2), also known as SCP4 or HSPC129, is a new member of the small CTD phosphatase (SCP) family and its role in cancers remains unclear. Here, we used a Phos-tag technique to screen a series of phosphatases and identified CTDSPL2 as a mitotic regulator. We demonstrated that CTDSPL2 was phosphorylated at T86, S104, and S134 by cyclin-dependent kinase 1 (CDK1) in mitosis. Depletion of CTDSPL2 led to mitotic defects and prolonged mitosis. Resultantly, CTDSPL2 deletion restrained proliferation, migration, and invasion in pancreatic cancer cells. We further confirmed the dominant negative effects of a phosphorylation-deficient mutant form of CTDSPL2, implying the biological significance of CTDSPL2 mitotic phosphorylation. Moreover, RT2 cell cycle array analysis revealed p21 and p27 as downstream regulators of CTDSPL2, and inhibition of p21 and/or p27 partially rescued the phenotype in CTDSPL2-deficient cell lines. Importantly, both CTDSPL2 depletion and phosphorylation-deficient mutant CTDSPL2 hindered tumor growth in xenograft models. Together, our findings for the first time highlight the novel role of CTDSPL2 in regulating cell mitosis, proliferation and motility in pancreatic cancer and point out the implications of CTDSPL2 in regulating two critical cell cycle participants (p21 and p27), providing an alternative molecular target for pancreatic cancer treatment.
Keywords: CTDSPL2, mitosis, phosphorylation, CDK1, pancreatic cancer, p21, p27
1. Introduction
Pancreatic cancer is notorious for its poorest prognosis among all cancer types with a five-year survival rate of only 10% [1]. Such a dismal outcome is due to the limited effectiveness of chemoradiation therapies to prevent recurrence for patients undergoing surgery, as well as to the high toxicity of chemotherapeutic regimens and the emergence of resistance for advanced cases [2]. Therefore, it is imperative that better strategies be explored to improve the treatment and prognosis of this deadly disease. Fortunately, targeted therapies have created therapeutic landscapes for a variety of cancers and promising small molecule inhibitors are being tested [3–5]. Unraveling novel targets in pancreatic cancer will be a crucial step toward improving patient outcomes.
Cancer is increasingly deemed as a cell cycle disease [6, 7]. Hyperactivity of “cell cycle promoters” and hypoactivity of “cell cycle restrictors” account for two hallmarks of cancer — uncontrolled cell proliferation and genomic instability [7, 8]. Based on this feature, people have developed several inhibitors to interfere with cell cycle process in cancers [9, 10], among which PARP inhibitors and CDK4/6 inhibitors have been developed and used clinically. PARP inhibitors are used for “BRCAness” cases to trigger cumulative DNA damage in cancer cells, ultimately resulting in synthetic lethality [11]. CDK4/6 inhibitors arrest cells in G1 to restrict tumor growth in hormone receptor-positive and HER2-negative metastatic breast cancers [12]. To target the mitotic phase of the cell cycle, vinca alkaloids and taxanes are the two main anti-microtubule agents (mitotic poisons) used for cancer treatment [13, 14]. Although highly efficient, the utility of these anti-microtubule agents is limited by unavoidable adverse effects and drug resistance [15]. In increasing specificity, various mitotic regulators have been identified. Until now, specific mitotic inhibitors in clinical trials are either mitotic kinases or motor proteins [13, 15], but mitotic phosphatases are relatively underexplored. Nevertheless, phosphatase dysregulation contributes to cancer development [16] and many phosphatases are promising targets for anti-cancer treatment [17, 18]. For instance, inhibitors against Src homology-2 domain-containing protein tyrosine phosphatase-2 (SHP2) and phosphatase of regenerating liver (PRL) [19] have entered clinical trials. Moreover, mitotic phosphatases play key roles in mitotic entry, sister chromatid cohesion, spindle assembly process, kinetochore-microtubule attachment, and spindle assembly checkpoint (SAC) silencing, as well as mitotic exit [20]. Thus, disclosing critical mitotic phosphatases will offer valuable insight for development of anti-cancer inhibitors.
Carboxy-terminal domain small phosphatase like 2 (CTDSPL2), also named SCP4 or HSPC129, is a metal-dependent serine/threonine phosphatase [21]. CTDSPL2 mainly resides in the nucleus at transcriptionally inactive heterochromatin regions [22–24], yet under certain conditions, CTDSPL2 can translocate from the nucleus to the cytoplasm [23]. Like other small CTD phosphatase (SCP) family members, CTDSPL2 harbors CTD phosphatase activity [21, 23]. CTDSPL2 directly dephosphorylates other targets [22, 25, 26] and interacts with a number of nuclear proteins as well [24]. Functionally, CTDSPL2 dephosphorylates FoxO1/3a to stimulate hepatic gluconeogenesis [26] and muscle proteolysis [27]. Furthermore, CTDSPL2 inhibits BMP-activated osteoblast differentiation by dephosphorylating Smad1/5/8 [22]. In contrast, CTDSPL2 promotes TGFβ-induced epithelial-mesenchymal transition through dephosphorylating Snail [25]. In addition, CTDSPL2 increases the levels of mRNA transcripts from ε- and γ-globin genes in K562 cells and umbilical cord blood-derived CD34+ cells [28]. CTDSPL2 overexpression augments migration and improves survival of chick embryo fibroblasts [29]. However, the role of CTDSPL2 in cancer is obscure.
In this study, we characterized CTDSPL2 as a mitotic phosphatase and investigated the outcome of CTDSPL2 inhibition in pancreatic cancer. We first identified and validated the phosphorylation sites of CTDSPL2 in mitosis. We then demonstrated the important role of CTDSPL2 in regulating pancreatic cancer cell mitosis, proliferation, migration, and invasion. Next, we revealed the dominant negative effects of a phosphorylation-deficient mutant of CTDSPL2. Finally, we unveiled the pivotal targets of CTDSPL2 to complement the hitherto uncompleted interaction web of CTDSPL2.
2. Materials and methods
2.1. Cell culture and transfection
HEK293T, HEK293GP, HeLa, PANC-1, Capan-2, BxPC-3, HPAF-II, Hs766T were purchased from American Type Culture Collection (ATCC). A human pancreatic nestin-expressing cell line transduced with the hTERT gene in a retroviral expression vector (hTERT-HPNE, also known as HPNE) was a generous gift from Dr. Michel Ouellette (University of Nebraska Medical Center). S2.013, Colo-357 and T3M4 were kindly provided by Dr. Michael (Tony) Hollingsworth (University of Nebraska Medical Center). S2.013 and Colo-357 were cultured as described [30]. T3M4 was maintained in DMEM media (Hyclone) supplemented with 10% FBS plus 100 units/ml penicillin and 100 μg/ml streptomycin (Invitrogen). Other cells were cultured following ATCC instructions.
Nocodazole and Taxol (Selleck Chemicals) were used at concentration of 100 ng/mL and 100 nM respectively to arrest cells (at 16 h-24 h) in mitosis. CDK1 inhibitor RO3306 (10 μM) was purchased from ENZO life Sciences. CDK1/2/5 inhibitor purvalanol A (10 μM) and Aurora inhibitor VX680 (2 μM) were from Selleck Chemicals. ATM inhibitor KU55933 (10 μM) and PKA inhibitor H89 (10 μM) were from LC Laboratory. The proteasome inhibitor MG132 (Santa Cruz Biotechnology) was used at concentration of 25 μM to pretreat the cells for 30 min before addition of kinase inhibitors to prevent mitotic exit. Cells were cultured with kinase inhibitors for 1.5 h prior to harvesting for western blotting analysis.
Attractene (Qiagen) was used for transient transfection. HiPerFect (Qiagen) was used for siRNA transfection. VSV.G (Addgene) was used for retroviral packaging. psPAX2 and pMD2.G (Addgene) were used for lentiviral packaging. The transfections were conducted following the manufacturer’s instructions. Virus packaging, infection, and selection process were carried out as previously described [31].
2.2. Expression constructs
pCMV-SPORT6 vector (HsCD00338490) containing the human CTDSPL2 coding sequence (CDS) was purchased from Harvard Medical School Plasmid Repository. The CDS of CTDSPL2 was then subcloned from the pCMV-SPORT6 vector into the pGEM-T vector (Promega) for mutagenesis. Point mutations (T86A, S104A, S134A, and S165A) were generated by the Quik-Change Site-Directed PCR Mutagenesis Kit (Aligent) and verified by Sanger sequencing.
The pSIN4-Flag-IRES-Neo vector was made by inserting an N-terminal Flag-tag with multiple-cloning-site sequences into the empty pSIN4-EF2-ABCG2-IRES-Neo vector (Addgene). Original cDNA (WT) and mutated cDNA (4A) of CTDSPL2 were cloned to the lentiviral pSIN4-Flag-IRES-Neo vector and the retroviral Tet-All (TetOn) vector [32] for establishment of CTDSPL2 stable overexpression and inducible overexpression cell lines, respectively. Cells transduced with Tet-All vectors were cultured with Tet system-approved fetal bovine serum (Clontech Laboratories). pcDNA3-CDK1-AF and GFP-Cyclin B1-R42A were from Addgene.
2.3. RNA interference
The four shRNA constructs were purchased from Millipore-Sigma and the targeting sequences are: shCTDSPL2#A: GCACACAGATTTAATGGATAA; shCTDSPL2#C: GCTCTCAGTTACAATCAATTT; shCDKN1A (shp21): CGCTCTACATCTTCTGCCTTA; shCDKN1B (shp27): GCGCAAGTGGAATTTCGATTT. For TetOn inducible knockdown constructs, oligos were designed, annealed, digested, and inserted to the pLKO-TetOn vector (Addgene) according to manufacturer’s instructions.
2.4. Phos-tag, western blotting, immunoprecipitation, and peptide blocking
Phos-tag gel was made with 10 μM or 20 μM Phos-tag (Wako Pure Chemical Industries) and 100 μM MnCl2 in SDS-acrylamide gel and used as described [33]. Western blotting, lambda phosphatase treatment assays, and immunoprecipitation were done as previously described [31]. Peptide blocking was done following the Abcam website protocol (https://www.abcam.com/protocols/blocking-with-immunizing-peptide-protocol-peptide-competition).
2.5. Recombinant protein purification and in vitro kinase assay
The pGEX-5X1-MCS construct was made by replacing the Flag-WDR5 part of pGEX5X1-Flag-WDR5 (Addgene) with a multiple cloning site sequence. The cDNA of CTDSPL2-WT or CTDSPL2–4A was cloned to pGEX-5X1-MCS for recombinant protein purification as described [34]. Purified GST-CTDSPL2-WT and GST-CTDSPL2–4A were incubated with or without 150 ng Cyclin B1-CDK1 kinase complex (SignalChem) for 45 min at 30°C followed by western blotting analysis.
2.6. Antibodies
Rabbit polyclonal phospho-specific antibodies against CTDSPL2 T86, S134, and S165 were generated and purified by AbMart, Inc. The peptides used for immunizing rabbits were NLITS-pT-PRAGE (T86), NPSSG-pS-PPRTT (S134), and TSGSD-pS-PGQAV (S165). The corresponding non-phosphorylated peptides were also synthesized and used for blocking assays. The p-S104 CTDSPL2 antibody was from Cell Signaling Technology (CST). Anti-Cyclin B1, β-actin, CDC27, ASPP1, ASPP2, iASPP, i2PP2A/SET, and RSK1 were purchased from Santa Cruz Biotechnology. Anti-CTDP1, PP1A, PP1B, PPP1CC, PPP4C, PPP5C, PPP6C, PPP4R1, PPP4R2, PPP4R3A and GST antibodies were from Bethyl Laboratories. Anti-CTDSPL2, PP2A-B55, PPP2R2A, PNUTS, PTEN, Rpb1 NTD/Total CTD, p-CTD S2, p-CTD S5, p-CTD S7, p-T320 PP1α, p-Akt T308, Akt, p-ERK1/2 T202/Y204, ERK1/2, p-YAP S127, YAP, p-GSK3 S9, p-β-catenin S675, β-catenin, p-eIF2α S51, eIF2α, p-STAT3 S727, STAT3, p-SMAD3 S423/425, SMAD3, p-IKKα/β S176/180, IKKβ, p-NF-κB S536, NF-κB, CDK1, p21 and p27 were from CST. Anti-PP2Ac, Flag and p-RSK S380 were from Millipore-Sigma. The anti-α-tubulin antibody was obtained from Abcam Inc. and the anti-GSK3β antibody was from BD Transduction Laboratories. Antibodies were used at 1:500–10000 dilutions.
2.7. Cell proliferation, migration, and invasion
For the cell proliferation assay, 10000 PANC-1, 5000 S2.013, 15000 Capan-2 and 2500 HPNE cells were seeded and doxycycline was added at concentration of 2 μg/mL right after the cells were split. Cells were mixed with trypan blue (HyClone) and counted by the cell counter — Countess II (Invitrogen) at indicated time points.
For the wound healing assay, doxycycline was added at a concentration of 2 μg/mL for 5 days to knock down CTDSPL2. Then, confluent monolayered cells in 6-well plates were scratched by a sterile 200 μL pipette tip, washed with PBS, and then cultured for the indicated time.
For migration and invasion assays, cells were first treated with doxycycline (2 μg/mL) for 5 days to knockdown CTDSPL2. For migration assays, 105 S2.013, 105 PANC-1 (both knockdown and overexpression sets) and 2×105 Capan-2 cells were seeded on BD Falcon Cell Culture Transwell chambers (Corning) for 24 h, 24 h, and 48 h, respectively. For invasion assays, 2×105 S2.013, 5×104 PANC-1 (knockdown groups), 105 PANC-1 (overexpression groups), and 3×105 Capan-2 cells were seeded on BioCoat Matrigel invasion chambers (BD Biosciences) for 24 h, 72 h, 48 h, and 48 h, respectively. The experiments were conducted following the manufacturers’ instructions. The migratory and invasive cells were fixed and stained with 0.4% crystal violet in methanol for 35 min and washed by water. The upper layer of cells was removed using cotton swab and the cells in the lower layer were counted manually.
2.8. Immunofluorescence staining, confocal microscopy, and live-cell imaging
The pHIV-H2B-mRFP plasmid (Addgene) was transfected into HeLa cells for immunofluorescence (IF) staining and PANC-1 cells for live-cell imaging. For IF staining, RFP-HeLa cells were fixed on a slide with 100% methanol for 15 min at room temperature and the slide was treated with mounting media (Vector Laboratories). PANC-1 cells were fixed with 4% paraformaldehyde for 20 min at 37°C and the slide was mounted with DAPI (Invitrogen). Other steps followed the previous protocol [35]. Live-cell imaging was conducted as described [36].
2.9. RNA isolation, reverse transcription, and qRT-PCR
RNA was extracted [36] and reverse transcribed [31] into cDNA as described. RT2 cell cycle array (Qiagen) was used for screening targets of CTDSPL2 following the manufacturer’s instructions. The RT primers for p21 (CDKN1A) were: Forward: AGTCAGTTCCTTGTGGAGCC; Reverse: GACATGGCGCCTCCTCTG. The RT primers for p27 (CDKN1B) were: Forward: GACCTGCAACCGACGATTC; Reverse: CGTTTGACGTCTTCTGAGGC. The qRT-PCR procedure was carried out as described [31].
2.10. Xenograft mouse model
Seven-week-old male athymic nude mice were purchased from Jackson Laboratory. S2.013 cells (i-shControl and i-shCTDSPL2, 3×106) were suspended with PBS and injected subcutaneously into either flank of the mouse. Five animals were used per group. Five days after inoculation, doxycycline (1 mg/mL in 5% sucrose water) was added into the diet. Tumor size was measured by a caliper every three days and tumor volume (V) was calculated by the formula: V=0.5×length×width2 [37]. Mice were euthanized by CO2 inhalation at the end of the experiments and the tumors were resected and dissected for further analysis. The animals were housed in pathogen-free facilities. All animal experiments were approved by the University of Nebraska Medical Center Institutional Animal Care and Use Committee.
2.11. Statistical analysis
Statistical significance was assessed using two-tailed, Student’s t test. P < 0.05 was considered statistically significant.
3. Results
3.1. CTDSPL2 is phosphorylated during anti-tubulin drug-induced mitotic arrest
Cell cycle progression is driven by periodic phosphorylation and dephosphorylation events [10]. To identify potential mitotic phosphatases, we arrested HeLa cells in mitosis using anti-microtubule agents (nocodazole and Taxol), for overnight treatment and tested the phosphorylation status of phosphatases with a Phos-tag system. As shown in Fig. 1A–C, the phosphatases (iASPP [38], PP1A [39], PP1B [39] and i2PP2A [40]) that were previously reported to undergo mitotic phosphorylation were confirmed by the Phos-tag system, and other mitotic arrest-induced hyperphosphorylated phosphatases were also identified (ASPP1, ASPP2, CTDP1, CTDSPL2, PPP4R2, PPP4R3A and PNUTS). Here, we focused on delineating the role of CTDSPL2, an understudied CTD phosphatase. The up-shifted bands of CTDSPL2 were reversed to normal state upon λ-phosphatase treatment (Fig. 1D), suggesting that the retarded movement was caused by phosphorylation modification. To identify the kinase responsible for mitotic phosphorylation of CTDSPL2, we treated the arrested cells with a series of kinase inhibitors and noticed that RO3306 (CDK1 inhibitor) and purvalanol A (CDK1/2/5 inhibitor) successfully blocked mobility upshift of CTDSPL2 (Fig. 1E), signifying the possibility that CDK1 phosphorylates CTDSPL2 in mitosis.
Fig. 1. CTDSPL2 is phosphorylated during mitotic arrest.
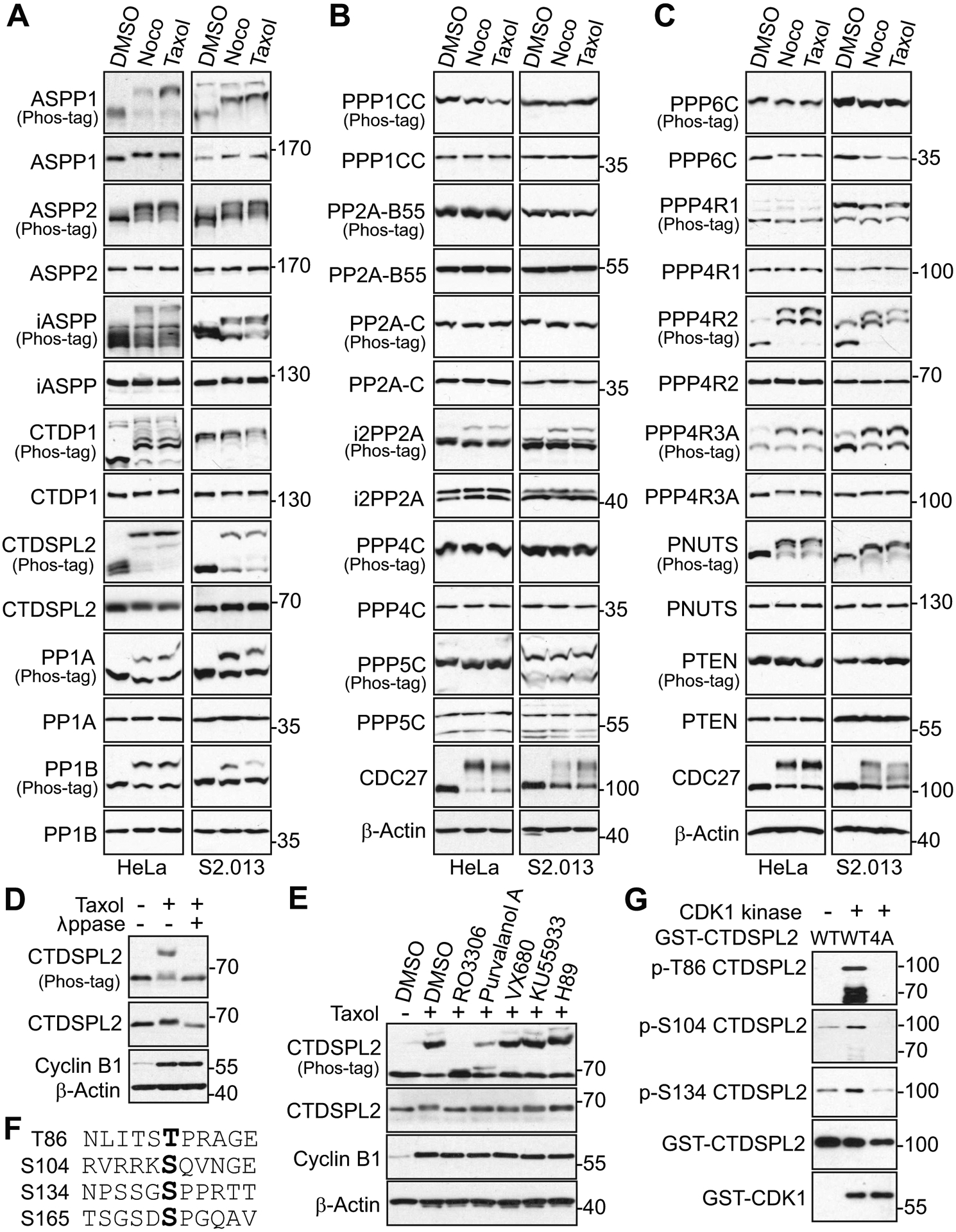
(A-C) HeLa and S2.013 cells were treated with DMSO (control), nocodazole (Noco, 100 ng/mL), or Taxol (100 nM) for 16 h and 24 h, respectively. Total cell lysates were electrophoresed on regular and Phos-tag SDS polyacrylamide gels and probed with the indicated phosphatase antibodies. Upshifted CDC27 marks the mitotic cells. (D) HeLa cells were treated with nocodazole (Noco) as indicated and total cell lysates were further treated with (+) or without (−) lambda phosphatase (λ ppase, NEB). Increased cyclin B1 marks the mitotic cells. (E) HeLa cells were treated with nocodazole together with or without various kinase inhibitors as indicated. MG132 was added 30 min prior to addition of kinase inhibitors to prevent mitotic exit. Increased cyclin B1 confirms the cells in mitosis. (F) Illustration of mitotic phosphorylation sites of CTDSPL2. (G) Bacterially produced and purified recombinant GST-CTDSPL2-WT and GST-CTDSPL2–4A proteins were used for in vitro kinase assays with purified CDK1/cyclin B1 complex for detection of specific phospho-sites. WT: wild type. 4A: T86A/S104A/S134A/S165A.
3.2. CTDSPL2 is phosphorylated at T86, S104, and S134 by CDK1 in mitosis
With the support of a previous study [24] identifying CDK1 (cdc2) as one of the nuclear proteins that interacts with CTDSPL2, we next sought to assess whether CTDSPL2 is a direct substrate of CDK1 and to identify its phosphorylation sites. According to database analysis (www.phosphosite.org), T86, S104, S134, and S165 are potential phosphorylation sites of CTDSPL2 during mitosis. Among these four sites, T86, S134, and S165 (Fig. 1F) satisfy the criteria for the consensus sequence (S/TP) targeted by CDK1 for phosphorylation [41]. In contrast, S104 (serine followed by glutamine, Fig. 1F) does not conform to the pattern of S/TP.
Facilitated by the use of phospho-specific antibodies that we have made against T86, S134, and S165, along with a commercially available phospho-antibody for S104, we first performed in vitro kinase assays to determine whether CDK1 could phosphorylate CTDSPL2 at these sites. As indicated in Fig. 1G, CDK1 robustly phosphorylated GST-tagged wild type (WT) CTDSPL2 at T86, S104, and S134, but failed to phosphorylate a GST-tagged phosphorylation deficient mutant of CTDSPL2 (CTDSPL2–4A: T86A/S104A/S134A/S165A). The phospho-antibody against S165 was unideal for western blot analysis, so phosphorylation of S165 could not be validated. Next, we examined if CDK1 could phosphorylate CTDSPL2 in cells. We transfected Flag-tagged CTDSPL2 into HEK293T cells and arrested the cells with nocodazole and Taxol treatments. As shown in Fig. 2A, T86, S104, and S134 were phosphorylated in the total cell lysates and in the immunoprecipitated samples. To further confirm the specificity of the T86 and S134 phospho-antibodies, we carried out peptide blocking experiments. Incubation with site-specific phospho-peptide, but not regular non-phospho-peptide, completely blocked the phospho-signals (Fig. 2B and 2C), indicating these antibodies precisely detected the phosphorylation of CTDSPL2. We then explored whether knockdown (KD) of CTDSPL2 would abolish endogenous phospho-signals of CTDSPL2 in PANC-1 (Fig. 2D) and S2.013 (Fig. 2E). Unsurprisingly, most (if not all) mitotic arrest-induced phospho-signals were abrogated in CTDSPL2 KD cell lines depending on the KD efficiency of CTDPSL2 (Fig. 2D and 2E). Besides, mutating these sites to non-phosphorylatable alanines largely blocked phosphorylation of T86, S104, and S134 (Fig. 2F), further supporting the specificity of these phospho-antibodies. All these results suggest that CTDSPL2 is phosphorylated at T86, S104, and S134 during mitotic arrest.
Fig. 2. CTDSPL2 is phosphorylated at T86, S104, and S134 by CDK1.
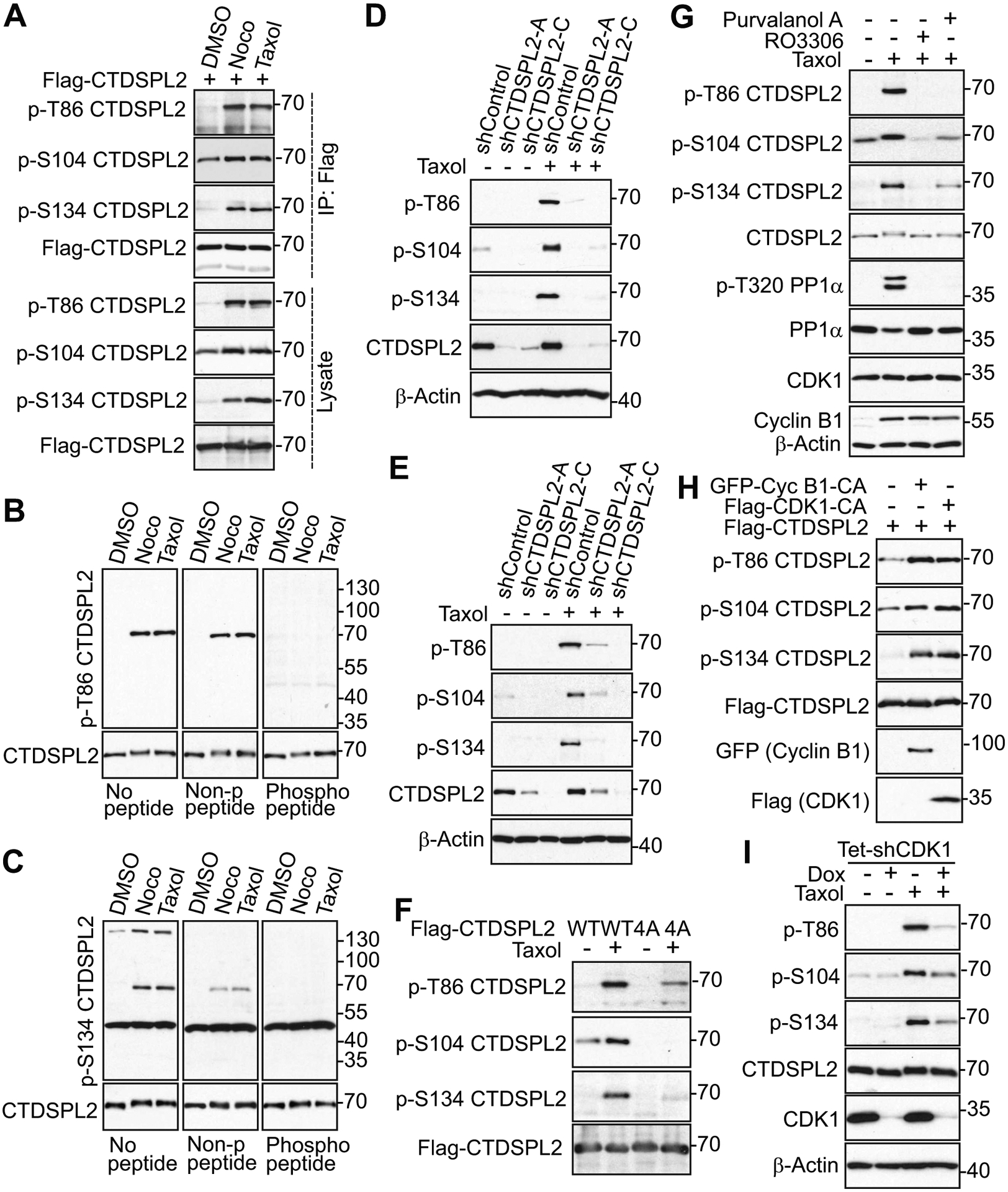
(A) HEK293T cells were transfected with Flag–CTDSPL2 and treated with nocodazole or Taxol for 16 h. Indicated phospho-sites were tested in total cell lysates and immunoprecipitated (IP) samples. (B, C) S2.013 cells were treated with nocodazole or Taxol for 24 h. Lysates were electrophoresed on SDS polyacrylamide gels with non-phospho-peptide or specific phospho-peptide blocking and probed with phospho-antibodies. (D, E) Control and CTDSPL2-knockdown PANC-1 (D) and S2.013 (E) cells were subjected to Taxol treatment for 24 h. Total cell lysates were probed with the indicated phospho-antibodies. (F) HEK293T cells were transfected with CTDSPL2-WT or CTDSPL2–4A plasmid. Total cell lysates were probed with the indicated phospho-antibodies. WT: wild type. 4A: T86A/S104A/S134A/S165A. (G) HeLa cells were treated with Taxol alone or together with RO3306 or Purvalanol A and lysates were harvested for western blotting analysis. Increased phospho-T320 PP1α and Cyclin B1 levels mark the cells in mitosis. PP1α (p-T320) is a well-known substrate of CDK1 and used to measure the CDK1 activity. (H) HEK293T cells were transfected with indicated expression constructs and total cell lysates were analyzed by western blotting. GFP-Cyc B1-CA: GFP-Cyclin B1-R42A (a nondegradable/constitutive active mutant). Flag-CDK1-CA: Flag-CDK1-T14A/Y15A (non-phosphorylatable/constitutive active CDK1). (I) TetOn inducible CDK1 knockdown HeLa cells were treated as indicated and lysates were harvested for western blotting analysis.
To confirm that phosphorylation of T86, S104, and S134 is mediated by CDK1, we tested phosphorylation of these sites under the condition of either pharmacological inhibition of CDK1 or genetic manipulation of CDK1. CDK1 inhibitors dramatically ablated phospho-signals of CTDSPL2 (Fig. 2G). Enhanced expression of constitutively active CDK1 (T14A and Y15F) or cyclin B1 (R42A) was sufficient to stimulate CTDSPL2 phosphorylation at T86, S104, and S134 in a similar pattern to anti-microtubule agents-induced phosphorylation of CTDSPL2 (Fig. 2H). Consistently, inducible KD of CDK1 abolished phosphorylation of CTDSPL2 as well (Fig. 2I). Together, these observations suggest that CTDSPL2 is phosphorylated at T86, S104, and S134 by CDK1 during mitosis.
3.3. CTDSPL2 depletion contributes to mitotic defects, proliferation inhibition, and impaired cell motility
The hyperphosphorylated status of CTDSPL2 in mitosis leads us to postulate that CTDSPL2 is a mitotic regulator and CTDSPL2 ablation will result in mitotic defects. To start, we depleted CTDSPL2 in RFP-H2B-HeLa (Fig. 3A) and PANC-1 (Fig. 3B) cell lines using TetOn-inducible system. Then, we checked the incidence of mitotic defects in these cells using immunofluorescence staining followed by confocal microscopy. In all, mitotic defects could be classified into three categories: 1. spindle defects, including monopolar spindles, multipolar spindles, abnormal spindle length, spindle misorientation and aberrant spindle shape; 2. chromosome distribution defects, also known as chromosome misalignments or unaligned chromosomes; 3. chromosome segregation defects, including lagging chromosome, acentric fragment and chromosome bridge [42]. In our cases, we observed more events of chromosome misalignment, lagging chromosome, multipolar spindle, and misoriented spindle/bent spindle in CTDSPL2-depleted HeLa cells (Fig. 3C and 3D). We also witnessed a higher rate of chromosome misalignment and multipolar spindle in CTDSPL2 knockdown PANC-1 cells (Fig. 3E and 3F). Since aberrant mitotic events could be associated with absence of microtubules’ attachment to the kinetochore or lack of tension at the kinetochore, which triggers hyperactivation of spindle assembly checkpoint to cause mitotic arrest [43], we hypothesized that CTDSPL2 abrogation-induced mitotic defects would give rise to prolonged mitotic length. Indeed, footage from live-cell imaging revealed that unaligned chromosomes in CTDSPL2-depleted cells resulted in drastic delay of anaphase onset and accounted for longer length of mitosis, measured from nuclear envelope breakdown (NEBD) to the end of telophase (Fig. 3G and 3H). These data suggest CTDSPL2 is a bona fide mitotic regulator necessary for the precise and organized progression of mitosis.
Fig. 3. CTDSPL2 depletion contributes to mitotic defects and prolonged mitosis.
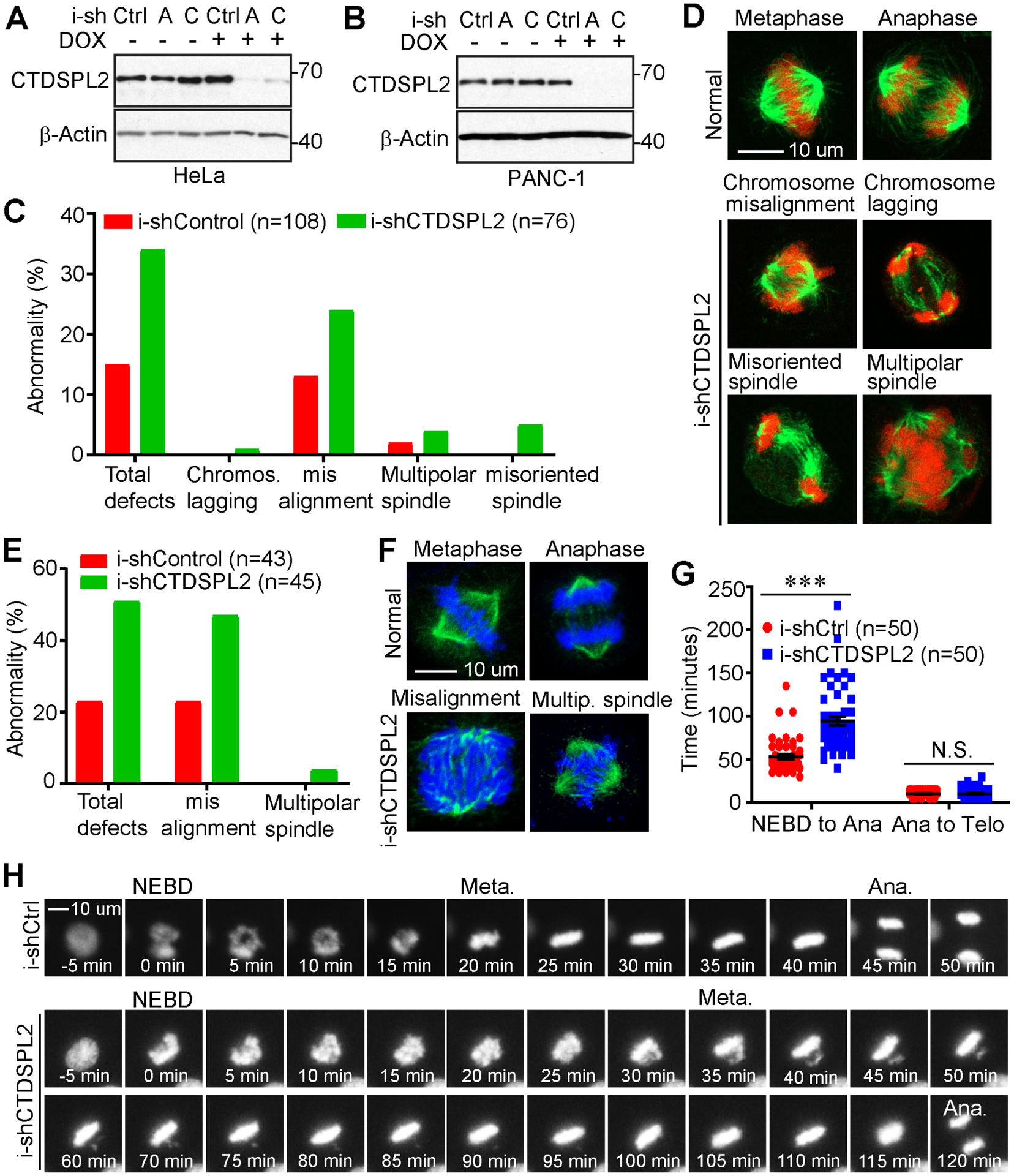
(A, B) Establishment of cell lines with TetOn inducible knockdown of CTDSPL2 in HeLa (A) and PANC-1 (B) cells. (C-F) CTDSPL2 deletion induced mitotic abnormality. Quantification of mitotic defects in control and CTDSPL2-depleted HeLa (C) and PANC-1 (E) cell lines from A and B. Representative mitotic images of TetOn inducible control and CTDSPL2-depleted HeLa (D) and PANC-1 (F) cells. (G) Quantification of mitotic length of control and CTDSPL2-depleted PANC-1 cells using a live-cell imaging system. ***: p=3.7E-10; N.S., not significant. (H) Representative live-cell images of RFP–H2B tagged PANC-1 cells with or without depletion of CTDSPL2.
Next, we investigated whether inducible knockdown of CTDSPL2 (Fig. 4A and 4B) would affect proliferation, migration, and invasion in pancreatic cancer cell lines. As illustrated in Fig. 4C and 4D, cell proliferation was restricted upon CTDSPL2 downregulation. Cells lacking CTDSPL2 migrated significantly slower in wound healing (Fig. 4E–H) and Transwell migration assays (Fig. 4I–K) and exhibited impaired invasive ability in Matrigel invasion assay (Fig. 4L–N). These results indicate that CTDSPL2 deletion restrains proliferation and motility in pancreatic cancer cells.
Fig. 4. CTDSPL2 knockdown leads to impaired cell proliferation, migration, and invasion.
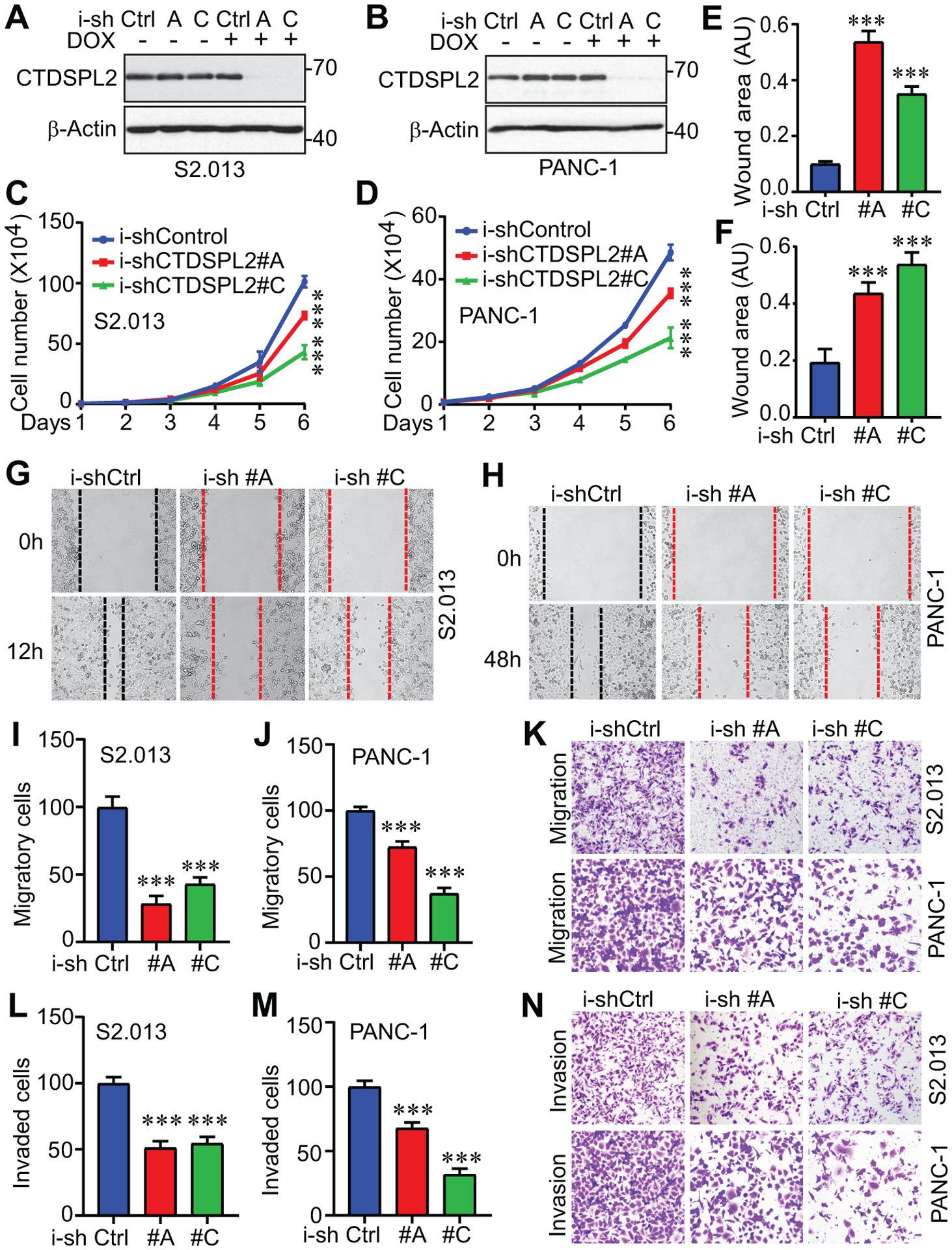
(A, B) Establishment of cell lines with TetOn inducible knockdown of CTDSPL2 in S2.013 (A) and PANC-1 (B) cells. (C, D) Cell proliferation assays in S2.013 (C) and PANC-1 (D) cells from A and B. ***: p=0.0002 (shRNA#A vs Control) and 7.3E-06 (shRNA#C vs Control) in panel C. ***: p=0.0003 (shRNA#A vs Control) and 2.1E-05 (shRNA#C vs Control) in panel D (Student’s t test). (E-H) Quantification of wound healing assays in established S2.013 (E) and PANC-1 (F) cells. ***: p=4.8E-05 (shRNA#A vs Control) and 9.0E-05 (shRNA#C vs Control) in panel E. ***: p=1.8E-04 (shRNA#A vs Control) and 3.2E-05 (shRNA#C vs Control) in panel F (Student’s t test). Representative images of wound healing assays in S2.013 (G) and PANC-1 (H) cells were shown. (I-N) CTDSPL2 knockdown inhibited migration and invasion in S2.013 (I, K, L) and PANC-1 (J, M, N) cells. ***: p=1.2E-05 (shRNA#A vs Control) and 6.4E-05 (shRNA#C vs Control) in panel I. ***: p=6.1E-05 (shRNA#A vs Control) and 1.1E-06 (shRNA#C vs Control) in panel J. ***: p=0.0003 (shRNA#A vs Control) and 0.0005 (shRNA#C vs Control) in panel L. ***: p=5.0E-05 (shRNA#A vs Control) and 6.6E-07 (shRNA#C vs Control) in panel M (Student’s t test). Data were expressed as the mean ± SD of three or four independent experiments.
3.4. Phosphorylation-deficient mutant CTDSPL2 exerts dominant negative effects
Based on the observation that CTDSPL2 undergoes CDK1 mediated phosphorylation modification during mitosis, and phosphorylation modification usually acts as a molecular switch for targets to activate or deactivate, we would like to investigate whether mitotic phosphorylation is required for CTDSPL2 to regulate cell proliferation and migration. For this, we established TetOn-inducible CTDSPL2-WT or -4A (4A: T86A/S104A/S134A/S165A) overexpressed immortalized (but non-malignant) pancreatic HPNE cell lines (Fig. 5A) as well as stably overexpressed pancreatic cancer cell lines (Fig. 5B and 5C). Interestingly, the CTDSPL2–4A mutant retarded the growth of both immortalized pancreatic HPNE cells (Fig. 5D) and pancreatic cancer cells (Fig. 5E and 5F). Cancer cells are characterized by multiple centrosomes and chromosome instability, rendering them more susceptible to mitotic disturbance [44]. This explains the delayed response of HPNE compared to the pancreatic cancer cells under the mitotic disruption caused by the 4A mutant (Fig. 5D–F).
Fig. 5. Phosphorylation-deficient mutant CTDSPL2 has dominant negative effects.
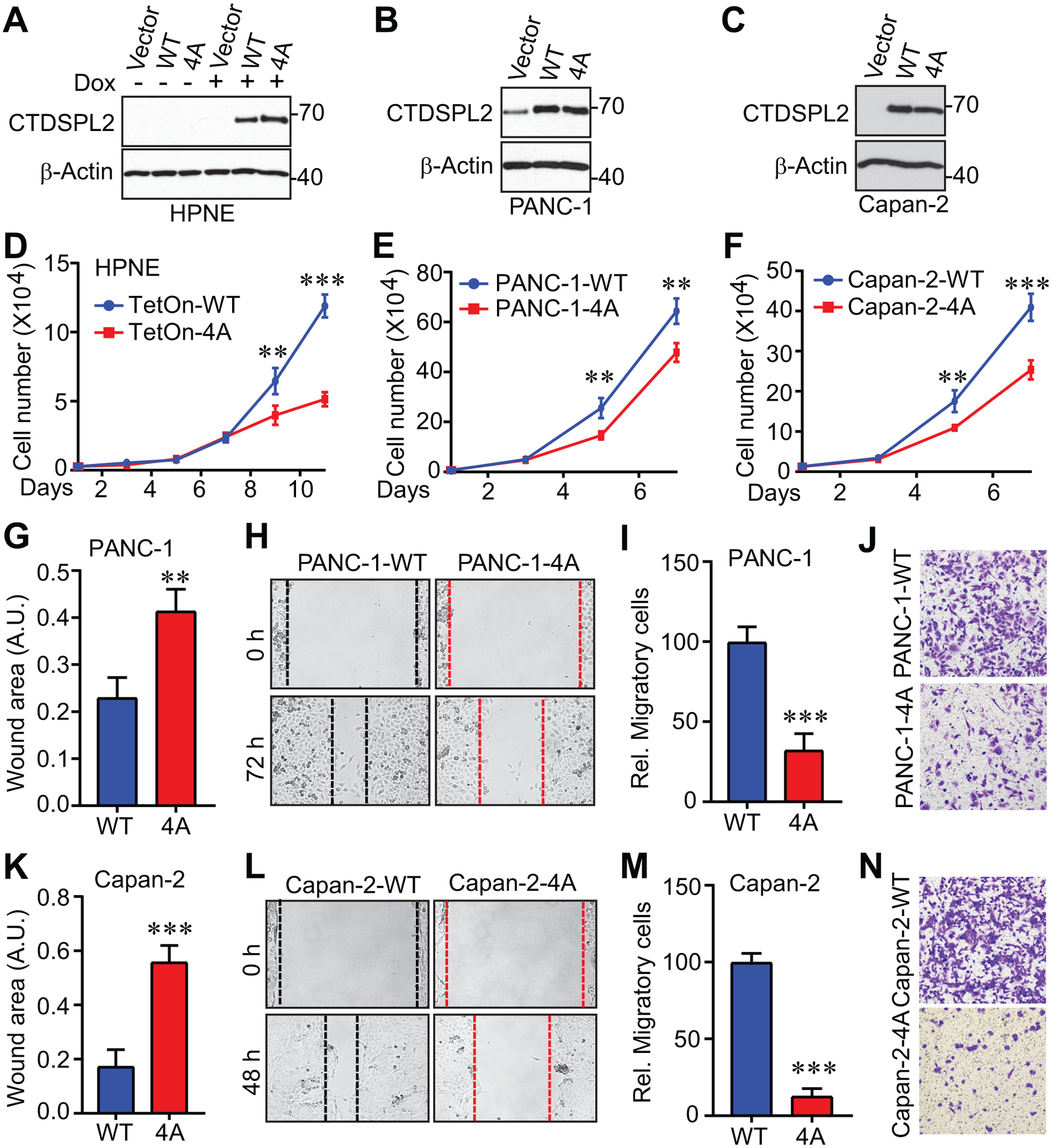
(A) Expression of CTDSPL2 in TetOn inducible CTDSPL2 overexpression HPNE cells. WT: wild type. 4A: T86A/S104A/S134A/S165A. (B, C) Establishment of cell lines with CTDSPL2 overexpression in PANC-1 (B) and Capan-2 (C) cells. (D-F) Cell proliferation assays in HPNE (D) cells from A, PANC-1 (E) cells from B and Capan-2 (F) cells from C. Data were expressed as the mean ± SD of three independent experiments. **: p=0.006; ***: p=9.1E-06 in panel D. **: p=0.002 in panel E. **: p=0.003; ***: p=0.0003 in panel F (Student’s t test). (G-N) Expression of CTDSPL2–4A impaired migration in PANC-1 (G-J) and Capan-2 (K-N) cells. Both wound healing (G, H, K, L) and Transwell (I, J, M, N) assays were used to measure the cell motility. Data were expressed as the mean ± SD of three or four independent experiments. **: p=0.001 in panel G. ***: p=6.4E-05 in panel I. **: p=0.0001 in panel K. ***: p=3.9E-07 in panel M (Student’s t test).
To investigate if, like CTDSPL2 depletion, the presence of the CTDSPL2–4A mutant also hampers cell motility, we conducted the similar sets of experiments in pancreatic cancer cells. As anticipated, cells expressing CTDSPL2–4A spread much more slowly and displayed hindered transmembrane migratory ability (Fig. 5G–N). Therefore, the CTDSPL2–4A mutant exerts dominant negative effects on both cell proliferation and motility.
3.5. CTDSPL2 deletion impedes tumor growth in vivo
We next tested the influence of CTDSPL2 on tumor growth using xenograft mouse models. We subcutaneously inoculated control and CTDSPL2 knockdown cells into the mice and doxycycline was added into the diet on day 5 to achieve CTDSPL2 deletion. We observed decreased growth of tumors initiated by injection of CTDSPL2 knockdown cells compared with control cells (Fig. 6A–C). The expression of CTDSPL2 was indeed reduced in the i-shCTDSPL2 tumors (Fig. 6D).
Fig. 6. CTDSPL2 ablation restrains tumor growth in vivo.
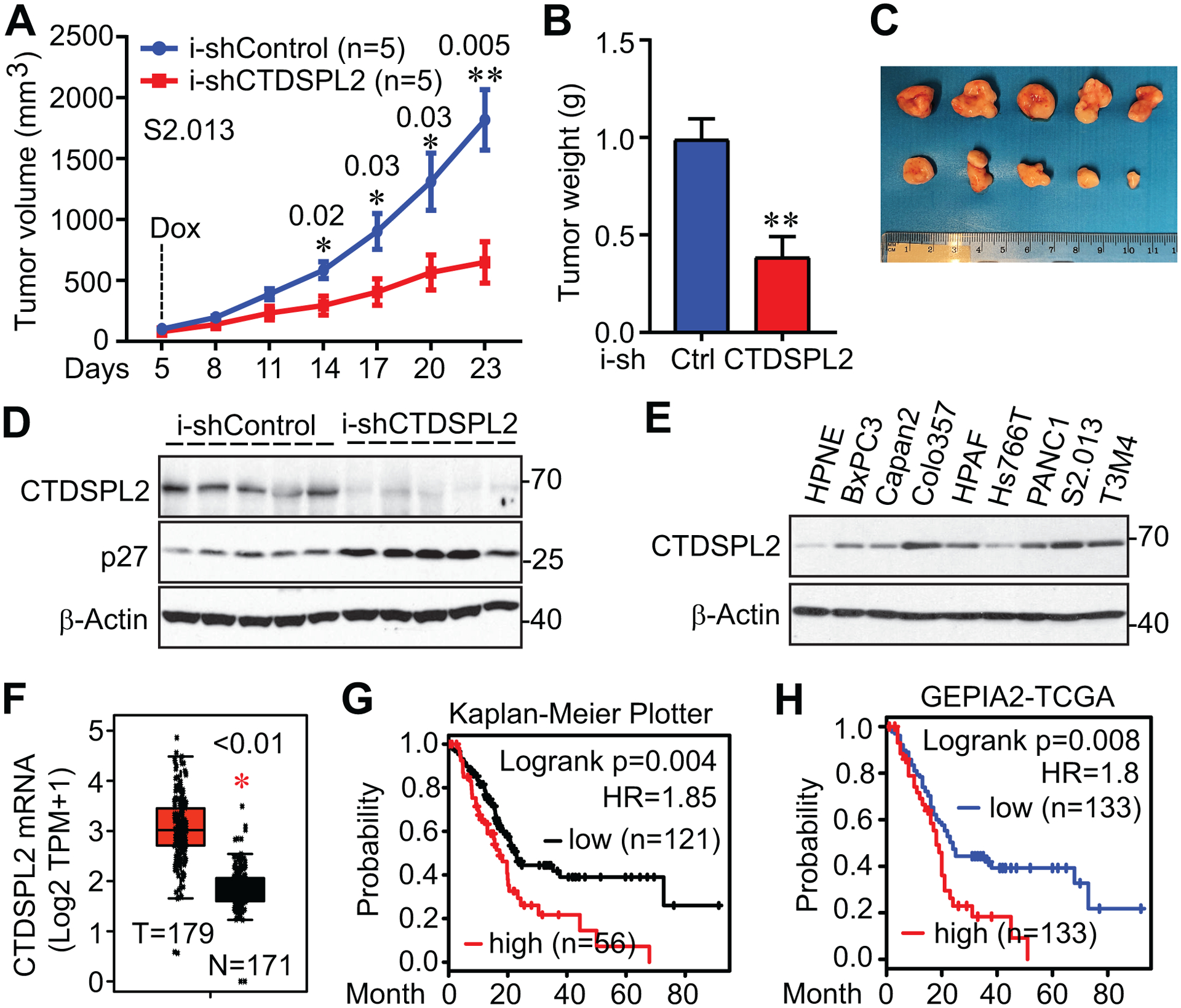
(A) Tumor growth curve of S2.013 cells with/without CTDSPL2 knockdown. Cells were subcutaneously inoculated into athymic nude mice (five mice each group). Doxycycline (1 mg/mL) was used on day 5. The tumor volume was measured at the indicated points. Data were expressed as the mean ± SEM. The p values were also shown (Student’s t test). (B) End-point tumor weight was measured. Data were expressed as the mean ± SD. **: p=0.003 (Student’s t test). (C, D) Tumors were excised and photographed at the endpoint (C) and lysed for western blotting analysis (D). (E) CTDSPL2 is overexpressed in pancreatic cancer cells. Western blotting analysis of CTDSPL2 expression in immortalized pancreatic (HPNE) and pancreatic ductal adenocarcinoma (PDAC) cell lines. (F) CTDSPL2 mRNA levels are increased in PDAC patients of TCGA datasets. The mRNA levels of CTDSPL2 in normal pancreas and pancreatic tumor samples were analyzed using an online tool (GEPIA2). (G, H) CTDSPL2 expression negatively correlated with overall survival in PDAC patients. Data were extracted from Kaplan-Meier plotter (G) and GEPIA2 (H). The cutoff values are 848 in G and 50% in H.
To explore the clinical relevance of CTDSPL2 in pancreatic cancer, we analyzed the expression of CTDSPL2 in HPNE and pancreatic cancer cell lines and found CTDSPL2 was consistently overexpressed in pancreatic cancer cell lines compared to the non-malignant HPNE pancreatic cell line control (Fig. 6E). Congruously, mRNA levels of CTDSPL2 were significantly upregulated in pancreatic tumor samples compared with normal pancreas in the TCGA database (Fig. 6F) [45]. Moreover, CTDSPL2 expression negatively correlated with overall survival in pancreatic cancer patients (Fig. 6G and 6H). Altogether, CTDSPL2 downregulation undermines tumor growth and is associated with poor prognosis in pancreatic cancer.
3.6. p21 and p27 are downstream regulators of CTDSPL2
Since CTDSPL2 is reported to have CTD phosphatase activity [21, 23], we extrapolate that CTDSPL2 functions in pancreatic cancer by dephosphorylating CTD of RNA polymerase II. Unexpectedly, phosphorylation of S2, S5, and S7, which are three sites of CTD that are subject to phosphorylation modification during transcription [46], remained unaffected upon CTDSPL2 knockdown in both S2.013 and PANC-1 cells (Fig. 7A). Alternatively, we screened a series of cancer-related signaling pathways and noted IKKα/β, a key player in the NF-κB pathway, was hyper-phosphorylated upon CTDSPL2 ablation (Fig. 7B and 7C). Combining our finding with the previous discovery that NF-κB binds to the promoter region of CTDSPL2 and stimulates its transcription [27], it was suggested that there might exist a positive feedback loop between CTDSPL2 and the NF-κB pathway.
Fig. 7. p21 and p27 are downstream regulators of CTDSPL2.
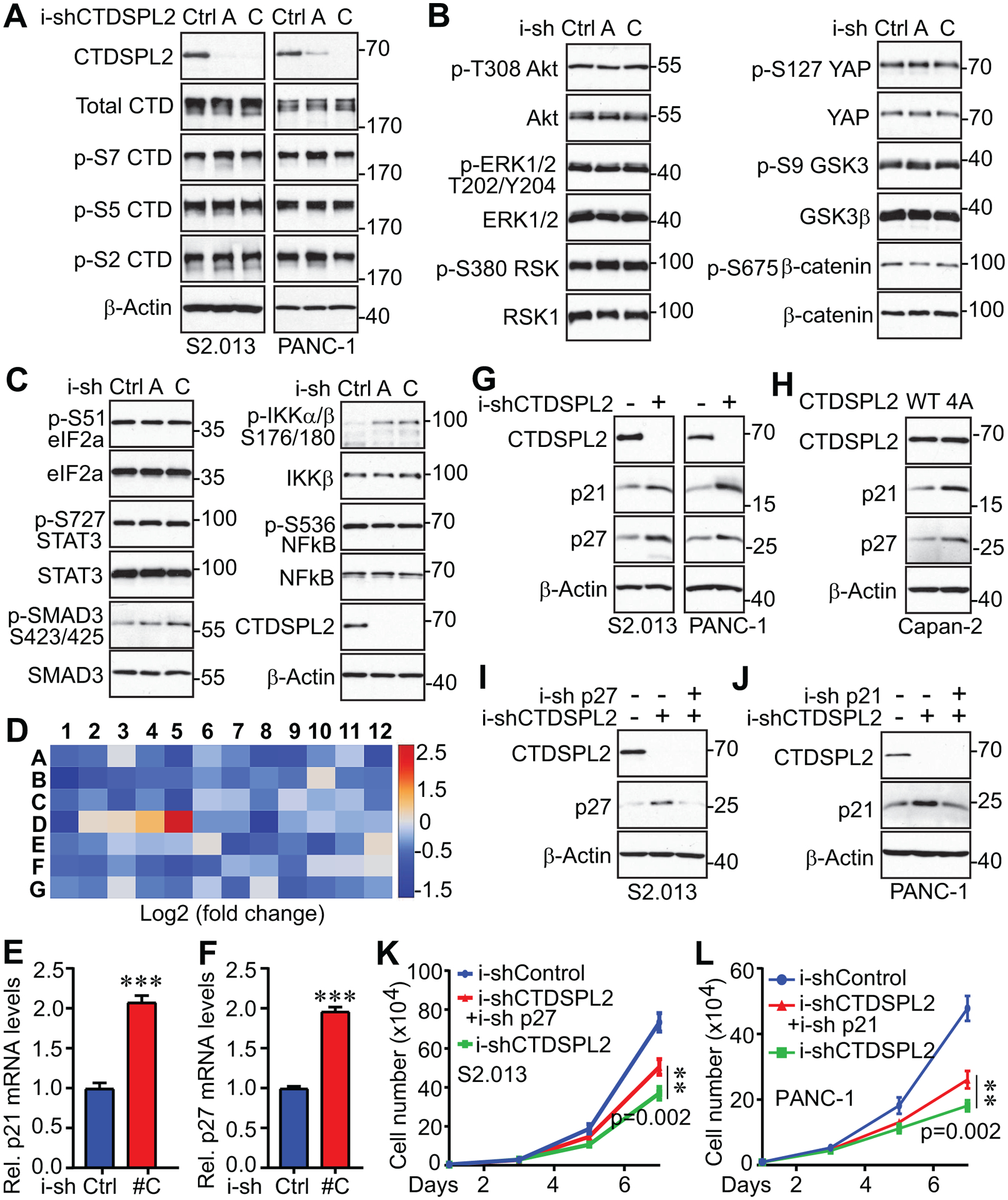
(A) Western blotting analysis with the indicated antibodies in TetOn inducible control and CTDSPL2 knockdown S2.013 and PANC-1 cell lines. (B, C) Western blotting analysis of cancer signaling molecules in control and CTDSPL2 knockdown PANC-1 cell lines. (D) CTDSPL2 deletion increased p21 and p27 expression levels. RT2 cell cycle array analysis in control and CTDSPL2 knockdown PANC-1 cells. (E-F) The qRT-PCR detected elevated p21 (E) and p27 (F) in CTDSPL2-depleted S2.013 cells. Data were expressed as the mean ± SD from three independent experiments. ***: p=5.2E-05 and 6.5E-06 in panel E and F, respectively (Student’s t test). (G) Western blotting analysis detected increased p21 and p27 expression levels in CTDSPL2 knockdown S2.013 and PANC-1 cells. (H) Expression of the CTDSPL2–4A mutant increased p21 and p27 in Capan-2 cells. (I, J) Establishment of inducible knockdown of p27 in S2.013 and inducible knockdown of p21 in PANC-1 cells. (K, L) Proliferation assay in cell lines from panels I and J. Data were expressed as the mean ± SD from three independent experiments. **: p=0.002 in panels K and L (double knockdown vs CTDSPL2 knockdown alone (Student’s t test).
To further explore the downstream signaling of CTDSPL2 in cell cycle progression, we compared mRNA levels of cell cycle participants in i-shControl and i-shCTDSPL2 PANC-1 cells and detected a more than two-fold increase in p21 (CDKN1A) and p27 (CDKN1B) mRNA levels in CTDSPL2-KD cells (Fig. 7D). We then verified the increase of p21 and p27 in CTDSPL2-depleted S2.013 cells (Fig. 7E and 7F). As expected, CTDSPL2 silencing led to enhanced protein levels of p21 and p27 (Fig. 7G). It is worth mentioning that there could exist functional redundancy between p21 and p27, given that the intense increase of p27 was accompanied by only a slight increase of p21 in S2.013, and vice versa for PANC-1 (Fig. 7G). We also noticed that p21 and p27 were boosted in CTDSPL2 phosphorylation-deficient mutant (Fig. 7H), which explains the comparable phenotype between CTDSPL2 knockdown and CTDSPL2–4A overexpression. In view of the results from Fig. 7G, we considered that p27 was the major actor mediating the loss of CTDSPL2 in S2.013, as was p21 in PANC-1. Hence, we downregulated p27 in i-shCTDSPL2 S2.013 cells (Fig. 7I) and blocked p21 in i-shCTDSPL2 PANC-1 cells (Fig. 7J) with an inducible knockdown system. In consequence, p27 knockdown partially recovered retarded growth in CTDSPL-ablated S2.013 (Fig. 7K) as p21 knockdown did in PANC-1 (Fig. 7L).
4. Discussion
Despite belonging to the small phosphatase family (CTDSP1/SCP1, CTDSP2/SCP2, CTDSPL/SCP3, CTDSPL2/SCP4), CTDSPL2 differs from other SCP members in that it harbors an exclusively longer part at its N-terminus [23], which is highly modified by phosphorylation, acetylation, ubiquitination, and others (www.phosphosite.org). As our research interest lies in unveiling novel mitotic phosphatases, we specifically focus on its phosphorylation modification during mitosis. The four identified sites (T86, S104, S134, and S165) all position at the unique N-terminus of CTDSPL2, suggesting the mitotic phosphorylation is not conserved among SCPs, but is a distinct feature for CTDSPL2. Indeed, none of the other SCPs has been reported to have any role in mitotic regulation. This study validates that T86, S104, and S134 of CTDSPL2 are phosphorylated by CDK1 in mitosis. Intriguingly, while S104 applies to the consensus sequence (SQ) of ATM, ATR, and DNA-PK [47], the phosphorylation could not be triggered by carboplatin- or gemcitabine-mediated DNA damage (data not shown). Instead, S104 phosphorylation could be achieved by anti-microtubule agents (nocodazole and Taxol) in a CDK1-dependent manner, indicating S104 is indeed phosphorylated by CDK1 in mitosis (Figs. 1 and 2). A previous report reveals that ATM is phosphorylated and activated at S1403 by Aurora B in mitosis to activate spindle assembly checkpoint [48]. Thus, our current work presents another possible phosphorylation mechanism for S104 — by the Aurora B-ATM-CTDSPL2 axis. Future work is needed to verify this possibility and to explore if phosphorylation of S104 affects cells differently from the other three typical CDK1-targeted sites (T86, S134, and S165).
In our endeavor to reveal how CTDSPL2 deficiency contributes to dampened growth and motility in pancreatic cancer, we discovered that p21 and p27 are the vital players. The p21 and p27 levels are markedly increased upon CTDSPL2 ablation and knockdown of p21 or p27 partially rescues the phenotype caused by CTDSPL2 deletion (Fig. 7K and 7L). p21CIP1 and p27KIP1 are the pan-cell cycle “brakes” that inhibit cyclin E-CDK2, cyclin D-CDK4/6, cyclin A-CDK2, cyclin B-CDK1 throughout cell cycle [7, 49–51]. Importantly, by inhibiting cyclin E-CDK2, p21 and p27 prevent retinoblastoma protein (pRb) hyperphosphorylation-mediated full activation of E2 factor (E2F), a key driver that pushes cells through the restriction point (R point) and ensures the accomplishment of the cell cycle [49, 52]. From a translational standpoint, targeting CTDSPL2 may benefit certain CDK4/6-resistant populations with ectopic activation of cyclin E [53], because CTDSPL2 depletion mediated elevation of p21 and p27 would counteract cyclin E-CDK2 to overcome cyclin E hyperactivation-induced drug resistance. It is very surprising that lack of CTDSPL2, supposedly a mitotic regulator, could restrict cell cycle progression in other phases by activating p21 and p27. A new question to be answered lies ahead: what are the intermediates between CTDSPL2 and p21/p27? As we observe the increase of p21 and p27 at the mRNA level, the activation of p21 and p27 could be either at the transcriptional level or due to post transcriptional modification. p21 is transcriptionally activated by p53 in response to DNA damage [54] and various other factors in p53-independent ways or in other biological conditions [55]. In comparison, p21 is transcriptionally repressed by c-Myc, p53 inhibitory molecules, competitors of TATA-binding protein and others [56]. How p27 is regulated on transcriptional level is less clear. Current knowledge indicates that p27 is transcriptionally induced by Forkhead box O proteins (FOXOs) and MENIN [57], while p27 is transcriptionally inhibited by MYC, the PIM kinase family and the Activator Protein-1 (AP-1) family [57, 58]. Enhanced p21 and p27 could be caused by activation of their transcription factors/transcriptional activators or inhibition of the transcriptional repressors. Given the possible overlap in transcriptional regulation shared by p21 and p27, it’s unknown whether p21 and p27 are induced simultaneously by the same regulators or independently by different mechanisms. ChIP-Seq analysis, RNA-Seq analysis, epigenetic studies and mass spectrometry-based proteomics will assist in the disclosure of this mystery.
Another interesting finding of our study is that phosphorylation of IKKα/β is enhanced upon CTDSPL2 knockdown without activating agents. IKKα/β are the catalytic subunits of the IKK complex that mediates phosphorylation-induced degradation of IκB inhibitors, resulting in activation of the NF-κB pathway [59]. NF-κB signaling is a master regulator of the inflammation response [60] and is activated by various internal and external stress stimuli, including reactive oxygen intermediates, DNA damage, infections, cytokines, antigen receptors, etc. [61, 62]. It is not clear whether phosphorylation of IKKα/β is caused by CTDSPL2 deprivation-mediated internal stress stimuli, direct interaction of CTDSPL2 with IKKα/β, or crosstalk between NF-κB signaling and other CTDSPL2-related pathways. The chronic inflammation induced by NF-κB activation may have tumor-promoting effects [8], but it also invigorates anti-tumor immune responses via facilitating the cross-presentation of tumor antigens [63, 64]. With the previous study reporting that NF-κB transcriptionally activates CTDSPL2 [27] and our current discovery that CTDSPL2 depletion enhances IKKα/β phosphorylation, the potential positive loop between NF-κB and CTDSPL2 needs further investigation.
In summary, our study has identified a new phosphatase (CTDSPL2) in the cell cycle machinery. Our findings provide a standpoint that targeting CTDSPL2 could be a plausible strategy for restricting cell motility and tumor growth in pancreatic cancer. The role of CTDSPL2 in pancreatic cancer development and progression will be further tested using pancreas-specific knockout of CTDSPL2 in genetically engineered pancreatic cancer mouse models. Given its enzymatic activity, pharmacological inhibitors against CTDSPL2 will be developed to fuel the bench-to-bedside translation for pancreatic cancer treatment in the future.
Highlights.
CTDSPL2 is phosphorylated in mitosis by CDK1 at T86, S104, and S134.
Depletion of CTDSPL2 results in mitotic defects and inhibition of cell proliferation, migration, invasion, and tumor growth.
Phosphorylation-deficient form CTDSPL2 exerts dominant negative effects.
p21 and p27 are downstream regulators of CTDSPL2.
Acknowledgements
We thank Dr. Xiao-long Yang (Queen’s University) for the TetOn-shCDK1 HeLa cell line. We thank Dr. Ying Yan (University of Nebraska Medical Center) for PP2A antibodies. Research in the Dong laboratory is supported by the Fred & Pamela Buffett Cancer Center Support Grant (P30 CA036727) and grant R01 GM109066 from the National Institutes of Health (NIH). We are very grateful to Dr. Joyce Solheim for critical reading and comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interests
The authors declare no conflict of interests.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Siegel RL, Miller KD, Fuchs HE, Jemal A, Cancer Statistics, 2021, CA: A Cancer Journal for Clinicians, 71 (2021) 7–33. [DOI] [PubMed] [Google Scholar]
- [2].Adel N, Current treatment landscape and emerging therapies for pancreatic cancer, The American Journal of Managed Care, 25 (2019) S3–s10. [PubMed] [Google Scholar]
- [3].Talwar V, Pradeep Babu KV, Raina S, An overall review of targeted therapy in solid cancers, Current Medicine Research and Practice, 7 (2017) 99–105. [Google Scholar]
- [4].Shimada A, Hematological malignancies and molecular targeting therapy, Eur J Pharmacol, 862 (2019) 172641. [DOI] [PubMed] [Google Scholar]
- [5].Zhong L, Li Y, Xiong L, Wang W, Wu M, Yuan T, Yang W, Tian C, Miao Z, Wang T, Yang S, Small molecules in targeted cancer therapy: advances, challenges, and future perspectives, Signal Transduction and Targeted Therapy, 6 (2021) 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bartek J, Lukas J, Bartkova J, Perspective: defects in cell cycle control and cancer, The Journal of Pathology, 187 (1999) 95–99. [DOI] [PubMed] [Google Scholar]
- [7].Malumbres M, Carnero A, Cell cycle deregulation: a common motif in cancer, Progress in Cell Cycle Research, 5 (2003) 5–18. [PubMed] [Google Scholar]
- [8].Hanahan D, Robert A Weinberg, Hallmarks of Cancer: The Next Generation, Cell, 144 (2011) 646–674. [DOI] [PubMed] [Google Scholar]
- [9].Sherr CJ, Bartek J, Cell Cycle–Targeted Cancer Therapies, Annual Review of Cancer Biology, 1 (2017) 41–57. [Google Scholar]
- [10].Suski JM, Braun M, Strmiska V, Sicinski P, Targeting cell-cycle machinery in cancer, Cancer Cell, 39 (2021) 759–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rose M, Burgess JT, O’Byrne K, Richard DJ, Bolderson E, PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance, Frontiers in Cell and Developmental Biology, 8 (2020) 564601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Whittaker SR, Mallinger A, Workman P, Clarke PA, Inhibitors of cyclin-dependent kinases as cancer therapeutics, Pharmacology & Therapeutics, 173 (2017) 83–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tischer J, Gergely F, Anti-mitotic therapies in cancer, Journal of Cell Biology, 218 (2018) 10–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Škubník J, Jurášek M, Ruml T, Rimpelová S, Mitotic Poisons in Research and Medicine, Molecules (Basel, Switzerland), 25 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chan KS, Koh CG, Li HY, Mitosis-targeted anti-cancer therapies: where they stand, Cell Death Dis, 3 (2012) e411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Stebbing J, Lit LC, Zhang H, Darrington RS, Melaiu O, Rudraraju B, Giamas G, The regulatory roles of phosphatases in cancer, Oncogene, 33 (2014) 939–953. [DOI] [PubMed] [Google Scholar]
- [17].Scott LM, Lawrence HR, Sebti SM, Lawrence NJ, Wu J, Targeting protein tyrosine phosphatases for anticancer drug discovery, Current Pharmaceutical Design, 16 (2010) 1843–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kiyokawa H, Ray D, In vivo roles of CDC25 phosphatases: biological insight into the anti-cancer therapeutic targets, Anticancer Agents Med Chem, 8 (2008) 832–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Vintonyak VV, Antonchick AP, Rauh D, Waldmann H, The therapeutic potential of phosphatase inhibitors, Current Opinion in Chemical Biology, 13 (2009) 272–283. [DOI] [PubMed] [Google Scholar]
- [20].Moura M, Conde C, Phosphatases in Mitosis: Roles and Regulation, Biomolecules, 9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Qian H, Ji C, Zhao S, Chen J, Jiang M, Zhang Y, Yan M, Zheng D, Sun Y, Xie Y, Mao Y, Expression and characterization of HSPC129, a RNA polymerase II C-terminal domain phosphatase, Molecular and Cellular Biochemistry, 303 (2007) 183–188. [DOI] [PubMed] [Google Scholar]
- [22].Zhao Y, Xiao M, Sun B, Zhang Z, Shen T, Duan X, Yu PB, Feng XH, Lin X, C-terminal domain (CTD) small phosphatase-like 2 modulates the canonical bone morphogenetic protein (BMP) signaling and mesenchymal differentiation via Smad dephosphorylation, The Journal of biological chemistry, 289 (2014) 26441–26450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wani S, Sugita A, Ohkuma Y, Hirose Y, Human SCP4 is a chromatin-associated CTD phosphatase and exhibits the dynamic translocation during erythroid differentiation, Journal of Biochemistry, 160 (2016) 111–120. [DOI] [PubMed] [Google Scholar]
- [24].Kang N, Koo J, Wang S, Hur SJ, Bahk YY, A systematic study of nuclear interactome of C-terminal domain small phosphatase-like 2 using inducible expression system and shotgun proteomics, BMB Reports, 49 (2016) 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhao Y, Liu J, Chen F, Feng XH, C-terminal domain small phosphatase-like 2 promotes epithelial-to-mesenchymal transition via Snail dephosphorylation and stabilization, Open Biology, 8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cao J, Yu Y, Zhang Z, Chen X, Hu Z, Tong Q, Chang J, Feng XH, Lin X, SCP4 Promotes Gluconeogenesis Through FoxO1/3a Dephosphorylation, Diabetes, 67 (2018) 46–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Liu X, Yu R, Sun L, Garibotto G, Lin X, Wang Y, Thomas SS, Li R, Hu Z, The nuclear phosphatase SCP4 regulates FoxO transcription factors during muscle wasting in chronic kidney disease, Kidney International, 92 (2017) 336–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ma YN, Zhang X, Yu HC, Zhang JW, CTD small phosphatase like 2 (CTDSPL2) can increase ε- and γ-globin gene expression in K562 cells and CD34+ cells derived from umbilical cord blood, BMC Cell Biology, 11 (2010) 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Winans S, Flynn A, Malhotra S, Balagopal V, Beemon KL, Integration of ALV into CTDSPL and CTDSPL2 genes in B-cell lymphomas promotes cell immortalization, migration and survival, Oncotarget, 8 (2017) 57302–57315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yang S, Zhang L, Purohit V, Shukla SK, Chen X, Yu F, Fu K, Chen Y, Solheim J, Singh PK, Song W, Dong J, Active YAP promotes pancreatic cancer cell motility, invasion and tumorigenesis in a mitotic phosphorylation-dependent manner through LPAR3, Oncotarget, 6 (2015) 36019–36031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Xiao L, Chen Y, Ji M, Dong J, KIBRA regulates Hippo signaling activity via interactions with large tumor suppressor kinases, The Journal of Biological Chemistry, 286 (2011) 7788–7796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yang S, Ji M, Zhang L, Chen Y, Wennmann DO, Kremerskothen J, Dong J, Phosphorylation of KIBRA by the extracellular signal-regulated kinase (ERK)-ribosomal S6 kinase (RSK) cascade modulates cell proliferation and migration, Cellular Signalling, 26 (2014) 343–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhou J, Zeng Y, Cui L, Chen X, Stauffer S, Wang Z, Yu F, Lele SM, Talmon GA, Black AR, Chen Y, Dong J, Zyxin promotes colon cancer tumorigenesis in a mitotic phosphorylation-dependent manner and through CDK8-mediated YAP activation, Proceedings of the National Academy of Sciences of the United States of America, 115 (2018) E6760–e6769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Stauffer S, Zeng Y, Zhou J, Chen X, Chen Y, Dong J, CDK1-mediated mitotic phosphorylation of PBK is involved in cytokinesis and inhibits its oncogenic activity, Cellular Signalling, 39 (2017) 74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhang L, Iyer J, Chowdhury A, Ji M, Xiao L, Yang S, Chen Y, Tsai MY, Dong J, KIBRA regulates aurora kinase activity and is required for precise chromosome alignment during mitosis, The Journal of biological chemistry, 287 (2012) 34069–34077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Stauffer S, Zeng Y, Santos M, Zhou J, Chen Y, Dong J, Cyclin-dependent kinase 1-mediated AMPK phosphorylation regulates chromosome alignment and mitotic progression, Journal of Cell Science, 132 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D, Elucidation of a universal size-control mechanism in Drosophila and mammals, Cell, 130 (2007) 1120–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lu M, Breyssens H, Salter V, Zhong S, Hu Y, Baer C, Ratnayaka I, Sullivan A, Brown NR, Endicott J, Knapp S, Kessler BM, Middleton MR, Siebold C, Jones EY, Sviderskaya EV, Cebon J, John T, Caballero OL, Goding CR, Lu X, Restoring p53 function in human melanoma cells by inhibiting MDM2 and cyclin B1/CDK1-phosphorylated nuclear iASPP, Cancer cell, 23 (2013) 618–633. [DOI] [PubMed] [Google Scholar]
- [39].Qian J, Beullens M, Huang J, De Munter S, Lesage B, Bollen M, Cdk1 orders mitotic events through coordination of a chromosome-associated phosphatase switch, Nature communications, 6 (2015) 10215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yin L, Zeng Y, Xiao Y, Chen Y, Shen H, Dong J, Cyclin-dependent kinase 1-mediated phosphorylation of SET at serine 7 is essential for its oncogenic activity, Cell Death & Disease, 10 (2019) 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Nigg EA, Cellular substrates of p34(cdc2) and its companion cyclin-dependent kinases, Trends in cell biology, 3 (1993) 296–301. [DOI] [PubMed] [Google Scholar]
- [42].Baudoin NC, Cimini D, A guide to classifying mitotic stages and mitotic defects in fixed cells, Chromosoma, 127 (2018) 215–227. [DOI] [PubMed] [Google Scholar]
- [43].Musacchio A, Salmon ED, The spindle-assembly checkpoint in space and time, Nature reviews. Molecular cell biology, 8 (2007) 379–393. [DOI] [PubMed] [Google Scholar]
- [44].Janssen A, Medema RH, Mitosis as an anti-cancer target, Oncogene, 30 (2011) 2799–2809. [DOI] [PubMed] [Google Scholar]
- [45].Tang Z, Kang B, Li C, Chen T, Zhang Z, GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis, Nucleic acids research, 47 (2019) W556–W560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Egloff S, Murphy S, Cracking the RNA polymerase II CTD code, Trends in Genetics : TIG, 24 (2008) 280–288. [DOI] [PubMed] [Google Scholar]
- [47].Kim ST, Lim DS, Canman CE, Kastan MB, Substrate specificities and identification of putative substrates of ATM kinase family members, The Journal of Biological Chemistry, 274 (1999) 37538–37543. [DOI] [PubMed] [Google Scholar]
- [48].Yang C, Tang X, Guo X, Niikura Y, Kitagawa K, Cui K, Wong ST, Fu L, Xu B, Aurora-B mediated ATM serine 1403 phosphorylation is required for mitotic ATM activation and the spindle checkpoint, Molecular Cell, 44 (2011) 597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sherr CJ, The Pezcoller lecture: cancer cell cycles revisited, Cancer Research, 60 (2000) 3689–3695. [PubMed] [Google Scholar]
- [50].Sherr CJ, Roberts JM, Living with or without cyclins and cyclin-dependent kinases, Genes & Development, 18 (2004) 2699–2711. [DOI] [PubMed] [Google Scholar]
- [51].Abukhdeir AM, Park BH, P21 and p27: roles in carcinogenesis and drug resistance, Expert Reviews in Molecular Medicine, 10 (2008) e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Bracken AP, Ciro M, Cocito A, Helin K, E2F target genes: unraveling the biology, Trends in Biochemical Sciences, 29 (2004) 409–417. [DOI] [PubMed] [Google Scholar]
- [53].Álvarez-Fernández M, Malumbres M, Mechanisms of Sensitivity and Resistance to CDK4/6 Inhibition, Cancer Cell, 37 (2020) 514–529. [DOI] [PubMed] [Google Scholar]
- [54].Wood A, Shilatifard A, Transcriptional blackjack with p21, Genes & Development, 20 (2006) 643–647. [DOI] [PubMed] [Google Scholar]
- [55].Gartel AL, Tyner AL, Transcriptional regulation of the p21((WAF1/CIP1)) gene, Experimental Cell Research, 246 (1999) 280–289. [DOI] [PubMed] [Google Scholar]
- [56].Gartel AL, Radhakrishnan SK, Lost in Transcription: p21 Repression, Mechanisms, and Consequences, Cancer Research, 65 (2005) 3980. [DOI] [PubMed] [Google Scholar]
- [57].Hnit SS, Xie C, Yao M, Holst J, Bensoussan A, De Souza P, Li Z, Dong Q, p27(Kip1) signaling: Transcriptional and post-translational regulation, The International Journal of Biochemistry & Cell Biology, 68 (2015) 9–14. [DOI] [PubMed] [Google Scholar]
- [58].Khattar E, Kumar V, Mitogenic regulation of p27(Kip1) gene is mediated by AP-1 transcription factors, The Journal of Biological Chemistry, 285 (2010) 4554–4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Israël A, The IKK complex, a central regulator of NF-kappaB activation, Cold Spring Harbor perspectives in biology, 2 (2010) a000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Liu T, Zhang L, Joo D, Sun S-C, NF-κB signaling in inflammation, Signal Transduction and Targeted Therapy, 2 (2017) 17023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Tilstra JS, Clauson CL, Niedernhofer LJ, Robbins PD, NF-κB in Aging and Disease, Aging and Disease, 2 (2011) 449–465. [PMC free article] [PubMed] [Google Scholar]
- [62].Oeckinghaus A, Ghosh S, The NF-kappaB family of transcription factors and its regulation, Cold Spring Harbor perspectives in biology, 1 (2009) a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Taniguchi K, Karin M, NF-κB, inflammation, immunity and cancer: coming of age, Nature Reviews Immunology, 18 (2018) 309–324. [DOI] [PubMed] [Google Scholar]
- [64].Grivennikov SI, Greten FR, Karin M, Immunity, inflammation, and cancer, Cell, 140 (2010) 883–899. [DOI] [PMC free article] [PubMed] [Google Scholar]