

Abstract
Objective:
Capillary malformation (CM) occurs sporadically and is associated with Sturge-Weber syndrome (SWS). The somatic mosaic mutation in GNAQ (c.548G>A, p.R183Q) is enriched in endothelial cells (EC) in skin CM and SWS brain CM. Our goal was to investigate how the mutant G-protein α-q subunit (Gαq) alters EC signaling and disrupts capillary morphogenesis.
Approach and Results:
We used lentiviral constructs to express p.R183Q or wild-type GNAQ in normal human endothelial colony forming cells (EC-R183Q and EC-WT respectively). EC-R183Q constitutively activated phospholipase C β3 (PLCβ3), a downstream effector of Gαq. Activated PLCβ3 was also detected in human CM tissue sections. Bulk RNA-seq analyses of mutant versus wild-type EC indicated constitutive activation of protein kinase C (PKC), NF-κB and calcineurin signaling in EC-R183Q. Increased expression of downstream targets in these pathways, Angiopoetin-2 (ANGPT2) and Down Syndrome Critical Region Protein 1.4 (DSCR1.4) were confirmed by qPCR and immunostaining of human CM tissue sections. The Gαq inhibitor YM-254890 as well as siRNA targeted to PLCβ3 reduced mRNA expression levels of these targets in EC-R183Q while the pan-PKC inhibitor AEB071 reduced ANGPT2 but not DSCR1.4. EC-R183Q formed enlarged blood vessels in mice, reminiscent of those found in human CM. shRNA knockdown of ANGPT2 in EC-R183Q normalized the enlarged vessels to sizes comparable those formed by EC-WT.
Conclusion:
Gαq-R183Q, when expressed in ECs, establishes constitutively active PLCβ3 signaling that leads to increased ANGPT2 and a pro-angiogenic, pro-inflammatory phenotype. EC-R183Q are sufficient to form enlarged CM-like vessels in mice, and suppression of ANGPT2 prevents the enlargement. Our study provides the first evidence that endothelial Gαq-R183Q is causative for CM and identifies ANGPT2 as a contributor to CM vascular phenotype.
Keywords: Sturge-Weber syndrome, capillary malformation, GNAQ, endothelial cell, Angiopoietin-2, PKC, PLC, DSCR1
INTRODUCTION
Capillary malformation (CM), also termed “port wine stain”, occurs sporadically in 0.3% of newborns 1 and is a slow-flow vascular malformation. CMs consist of clusters of abnormal capillary/venule-like blood vessels below the surface of skin that are often enlarged 2. Sturge-Weber syndrome (SWS) is a rare congenital neurocutaneous disease characterized by CMs in the leptomeninges in addition to facial and extra-craniofacial CMs 3. Patients with SWS are at risk for seizures, stroke-like episodes, and cognitive impairments; symptomatic treatments are available, but to date there are no targeted therapies for the underlying malformation. In SWS, CMs can also occur in the choroidal vessels of the eye, increasing the risk of glaucoma 4,5. A somatic, activating mutation in GNAQ (c.548G>A; p.R183Q) was found in 92% of non-syndromic skin CMs and 88% of SWS skin and brain specimens 6. We subsequently showed that GNAQ p.R183Q is enriched in endothelial cells (ECs) in skin CMs and SWS brain CMs 7,8. A somatic missense mutation (c.547C>T; p.R183C) in GNA11, a GNAQ homologue, was reported in 3 patients with diffuse CM on extremities 9 and a somatic missense mutation in GNB2 (c.232A>G; p.K78E), the β subunit of the heterotrimeric G protein, was recently reported in one patient with SWS 10.
GNAQ encodes Gαq, an alpha subunit of the heterotrimeric G protein that links G-protein coupled receptors (GPCR) to activation of phospholipase Cβ (PLCβ). The latter hydrolyzes phosphatidylinositol 4,5 biphosphate (PIP2) to generate diacylglycerol (DAG) and inositol 1, 4, 5 triphosphate (IP3), two potent signaling molecules. Several endothelial GPCRs are known to signal through Gαq, and in many cell types mitogenic signaling through GPCRs is conducted primarily through Gαq 11. Gαq has inherent guanosine-5’-triphosphate hydrolase (GTPase) activity. Loss of arginine (R) at position 183, located in the conserved GTP-binding pocket, impairs GTPase activity and increases GTP-bound Gαq, which promotes constitutive activity 12.
The GNAQ p.Q209L mutation in uveal melanoma has a similar but stronger effect on Gαq GTPase activity 13,14. As such, the p.Q209L mutation provides a template for understanding the p.R183Q mutation, although computational modeling studies suggest the two mutations may alter Gαq signaling in distinct ways 15. In uveal melanoma, a small GTPase ADP-ribosylation factor 6 (ARF6) functions as an immediate downstream effector of Gαq-Q209L to induce PLC/protein kinase-C(PKC) and Rho/Rac signaling, which in turn activates ERK, p38, YAP and other signaling molecules 16–20. Because the endothelial cellular context is distinct from the melanocytes, we sought to apply an unbiased approach to discover how the p.R183Q mutation alters endothelial Gαq signaling and determine if endothelial p.R183Q is sufficient to drive CM formation in vivo.
Towards this goal, we used lentiviral constructs to express either p.R183Q or wildtype GNAQ in normal human endothelial colony forming cells (ECFC) isolated from umbilical cord blood 21,22, designated EC-R183Q and EC-WT, respectively. In vitro results were corroborated by immunostaining human CM tissue specimens from sporadic CM and SWS cases. Our results show activation of PLCβ3 and increased expression of angiopoietin-2 (ANGPT2), a blood vessel destabilizer, and Down Syndrome Critical Region Protein (DSCR) 1.4, an inhibitor of calcineurin that is induced in ECs by vascular endothelial growth factor (VEGF)-A. We tested blood vessel-forming ability by injecting EC-R183Q or EC-WT subcutaneously into immunodeficient mice using a well-established model for building human microvascular networks in vivo 23–25. EC-R183Q formed enlarged ANGPT2+ blood vessels, reminiscent of the enlarged ANGPT2+ vessels seen in human CM 2. shRNA knockdown of ANGPT2 in EC-R183Q reduced the size of the blood vessels, indicating ANGPT2 contributes to the enlarged vascular phenotype. Taken together, our study uncovers potential pathways and molecules that might provide therapeutic targets to reverse the effects of endothelial Gαq-R183Q on capillary morphogenesis.
MATERIALS AND METHODS
The data that support the findings of this study are available from the corresponding author upon reasonable request. RNA-seq data are publicly available at the Gene Expression Omnibus (GEO) archive at the National Center for Biotechnology Information under GEO accession number GSE186998 (https://www.ncbi.nlm.nih.gov/geo/).
Generation and Culture of EC-WT and EC-R183Q
Human umbilical cord blood-derived ECFCs were isolated, characterized and maintained in our laboratory 24,25 under an IRB-approved protocol. Human GNAQ WT and R183Q cDNA cloned into the pcDNA3.1+ plasmid at Kpnl (5’) and Xho I (3’) were kindly provided by Dr. Kun-Liang Guan 20. Subsequently, WT and R183Q cDNA were inserted into the lentiviral vector CSCW-Fluc-IRES-GFP (Massachusetts General Hospital (MGH) Vector Core) at Nhe l (5’) and Xho I (3’) to replace the Fluc gene. The lentivirus was generated by using 2nd generation packaging system at the MGH or Boston Children’s Hospital vector core facility. The transduction was conducted followed by green fluorescent protein (GFP) sorting. The GFP+ ECs were harvested, expanded, and validated for mutant allelic frequency (MAF) by ddPCR and for Gαq expression by western blot. Lentiviral-engineered ECFCs that express full-length WT or R183Q GNAQ cDNA were designated as EC-WT and EC-R183Q respectively. For expansion, EC-WT and EC-R183Q were plated on fibronectin (0.1 μg/cm2)-coated tissue culture dishes at a density of 15,000 – 20,000 cells/cm2 in Endothelial Cell Growth Medium-2 (Lonza) which consists of Endothelial Cell Growth Basal Medium-2 (EBM-2), SingleQuot supplements (all except hydrocortisone), 10% heat-inactivated fetal bovine serum (FBS, Hyclone) and 1X Glutamine-penicillin-streptomycin solution (GPS, ThermoFisher). To promote quiescence in vitro, ECs were plated at 20,000 cells/cm2 for 30 – 32 hours, then switched to 2% FBS- EBM2 without SingleQuot supplements for 16 hours.
siRNA and shRNA Knockdown
EC-WT and EC-R183Q were transfected with siRNA using INTERFERin® siRNA transfection reagent (Polyplus-transfection®) following the manufacturer’s instruction. 10nM GeneSolution siRNA targeted to PLCB1 and PLCB3 (Qiagen) and control non-target siRNA (Qiagen) were used. Cells were lysed for RT-PCR at 48 hours after transfection and the conditioned media were collected for ELISA at 72 hours after transfection.
EC-R183Q were transduced with ANGPT2 shRNA lentivirus (TRCN0000059223-TRCN000005922325, Sigma) or control lentivirus (Sigma) followed by puromycin (1 μg/mL) selection for 72 hours. Afterwards EC-R183Q sh-ANGPT2 and EC-R183Q sh-CTRL were maintained in full media without puromycin. Knockdown efficacy was confirmed by RT-qPCR and ELISA.
RNA Sequencing
Lentiviral engineered EC-WT and EC-R183Q, and human umbilical vein endothelial cells (HUVEC) transduced in the same manner to create HUVEC-WT and HUVEC-R183Q, were cultured in 2% FBS-EBM2 without SingleQuots for 16 hours to promote quiescence. N=3 biological replicates for each cell type. Total RNA was isolated using the Qiagen RNeasy Plus Mini Kit (Qiagen). RNA Integrity Number was assessed by the Agilent 2100 Bioanalyzer system (BCH IDDRC Molecular Genetics Core Facility) to ensure more than 1μg total RNA with RNA integrity number more than 9.0 was submitted for sequencing. The mRNA library preparation (by Illumina TruSeq Strandard Total RNA with Ribo-Zero kit) and RNA Sequencing (by HiSeq 2500 Rapid, 100 PE Sequencing Throughput) was conducted by Psomagen, Inc (fee for service). Sequencing quality control was performed on raw FASTQ files with FastQC (0.11.4). No samples failed quality thresholds, mean PHRED quality score less than 5 or median PHRED less than 20 for any base on either paired-end read. FASTQ files were aligned with HiSAT2 (2.0.4) to the human reference (hg19) 26. Using GENCODE (release 19) annotations, gene coverage was calculated with StringTie (1.2.4) and coerced into a gene count matrix by Ballgown (2.1.1). Library size normalization was performed using trimmed mean of M-values (TMM) in edgeR (3.26.8). Differential gene expression analysis was conducted on gene-level annotations with edgeR (3.26.8), filtering genes with less than 5 counts in at least one diagnosis group, zero count genes, and removing chromosome Y and mitochondrial genes. Genes were considered differentially expressed with false discovery rate (FDR) less than 0.10. Differentially expressed genes were analyzed for upstream regulators using Ingenuity Pathway Analysis (IPA, Qiagen) to identify concordantly regulated effectors between R183Q mutants and WT samples. Gene Ontology (GO) analyses were performed using the PANTHER Overrepresentation Test (Released 20200407) ( http://geneontology.org), using known, concordant, differentially expressed genes having FDR < 0.00005. Regulated pathways had an FDR correction <0.05. The raw and processed data files are available at the Gene Expression Omnibus (GEO) at the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/gds/).
Histology and Immuno-staining
Human neonatal foreskin, infantile hemangioma, skin CMs (n=6) and SWS brain CM specimens (n=6) (Table 1) were obtained under human subject protocols (IRB-P00003505, S08–01-0013 and 09–02-0043) approved by the Committee on Clinical Investigation at Boston Children’s Hospital. Informed consent was obtained for hemangioma, skin CM and SWS brain lesions according to the Declaration of Helsinki. Foreskin was de-identified, normally discarded tissue. Formalin-fixed, paraffin-embedded (FFPE) tissue sections (5μm) were deparaffinized and either directly stained with hematoxylin and eosin (H&E) or immersed in an antigen retrieval solution (Citrate-EDTA buffer: 10mM Citric Acid, 2mM EDTA, 0.05% Tween-20, pH 6.2) for 20 minutes at 95°C to 99°C. Sections were subsequently blocked for 30 minutes in TNB Blocking buffer (PerkinElmer) followed by incubation with primary antibodies or the plant lectin Ulex europaeus agglutinin-I (UEA I; Vector). UEA I was fluorescently labeled with either DyLight 488, 594 or 649 and used to stain human EC. Anti–human p-PLCβ3(Ser537), ANGPT2, DSCR1, p-ERK1/2 and Ki67 primary antibodies were used (Major Resources Table). Purified class- and species-matched IgGs (Vector Lab) were used as controls to assess non-specific antibody staining. Sections were next incubated with appropriate biotinylated secondary antibodies followed by DyLight 488 or 594 Streptavidin. All slides were mounted using DAPI (Molecular Probe) to visualize nuclei. Images were acquired by a Zeiss Airyscan LSM 880 Fast confocal microscope (Zeiss) and analyzed by Fiji ImageJ software (NIH). A limitation of the immunostaining studies is the small number of skin CM and brain CM available for study, as seen in Table 1 below.
Table 1.
Human Tissue Specimens
| Skin CM | Sex | Age | MAF | p-PLCβ3 | ANGPT2 | DSCR1 | p-ERK1/2 | Ki67 |
|---|---|---|---|---|---|---|---|---|
| 1 | M | 12y | 7.4 | √ | √ | √ | √ | |
| 2 | M | 15y | 5.3 | √ | √ | √ | √ | |
| 3 | M | 28y | 11.1 | √ | √ | √ | √ | |
| 4 | M | 16y | 6.4 | √ | ||||
| 5 | F | 72y | 7.1 | √ | ||||
| 6 | M | 12y | 7.4. | |||||
| SWS brain CM | ||||||||
| 1 | M | 10m | 8.7 | √ | ||||
| 2 | M | 12y | 6.2 | √ | ||||
| 3 | F | 2y | 5.6 | √ | √ | |||
| 4 | M | 11m | 5.0 | √ | ||||
| 5 | M | 10m | 5.6 | √ | ||||
| 6 | M | 2y | 0.1 | √ | ||||
| Hemangioma | ||||||||
| 1 | F | 9m | n.d | √ | √ | √ | √ | |
| Foreskin | ||||||||
| 1 | M | 2d | n.d | √ | √ | √ | √ | √ |
MAF: mutant allelic frequency
n.d.: not done.
RNA Isolation and Quantitative Reverse Transcriptase PCR (RT-qPCR)
Cells were starved in 2% FBS-EBM2 with and without inhibitors for 16 hours. Total cellular RNA was extracted with a RNeasy Micro extraction kit (Qiagen). Reverse transcriptase reactions were performed using an iScript™cDNA synthesis kit (BioRad). qPCR was performed using iTaq Universal SYBR Green Supermix (BioRad). Amplification was carried out in an ABI 7500 (Applied Biosystems). A relative standard curve of each gene amplification was generated to determine the amplification efficiency, with greater than 90% considered acceptable. Gene expression was normalized to GNAQ 27. ATP5B and HPRTI were used as housekeeping genes. Quantitative analysis was performed according to delta CT based Pfaffl’s method 28. Primer sequences are shown in Table 2.
Table 2.
qPCR Primers 5’-3’
| Gene | Forward | Reverse |
|---|---|---|
| ANGPT2 | actcagctaaggaccccactgttg | tgtccacccgcctcctccag |
| ATP5B | ccactaccaagaagggatctatca | gggcagggtcagtcaagtc |
| CD31 | cacctggcccaggagtttc | agtacacagccttgttgccatgt |
| DSCR1.4 | tgcgaccccagtcataaact | gctgcgtgcaattcatacttt |
| E-selectin | cacatctcagggacaatggacaga | gcttgaacattttaccacttggca |
| G-CSF | tcccaccttggacacactg | gggccattcccagttctt |
| GNAQ | aaaatcatgtattcccatctagtcg | ggtccacgaacatcttcagaat |
| GNA11 | gcatccaggaatgctacgac | ggtcaacgtcggtcaggtag |
| GRK2 | tgatggccatggagaagag | catgacactgcggatgct |
| GRK5 | ggtgggcactgttggcta | ctcaggccgtacctctggt |
| HPRT1 | tgaccttgatttattttgcatacc | cgagcaagacgttcagtcct |
| IL-1B | tacctgtcctgcgtgttgaa | tctttgggtaatttttgggatct |
| VE-Cad | ccttgggtcctgaagtgacct | cagggccttccttctgcaa |
Western Blot
Cells were starved 16 hours in 2% FBS-EBM2 with or without inhibitors, lysed in a urea-based lysis buffer (4M Urea, 5mM EDTA, 0.5% SDS, 0.5% NP-40, 100mM Tris, pH7.4) with protease and phosphatase inhibitors (Roche). A nuclear extract kit (Signosis) was used to fractionate nuclear from cytoplasmic proteins. Cell extracts were electrophoresed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), transferred to nitrocellulose, and probed with antibodies (Major Resources Table). All signals detected by enhanced chemiluminescence. The densitometric analysis was conducted using Image Lab software (BioRad) or Fiji ImageJ software.
Enzyme-linked Immunosorbent Assay (ELISA)
Endothelial monolayers were washed with PBS twice, incubated in 0.5% BSA-EBM2 for 24 hours and supernatants collected. ANGPT2 in the media was measured with a commercial human Angiopoietin-2 Quantikine ELISA Kit (R&D systems). An ANGPT2 standard curve was run in parallel.
In Vivo Murine Model for Human Blood Vessel Formation
Animal experiments were conducted according to a protocol approved by the Institutional Animal Care and Use Committee at Boston Children’s Hospital. 0.8×106 ECs (EC-WT or EC-R183Q or EC-R183Q sh-ANGPT2 or EC-R183Q sh-CTRL) were combined with 1.2 ×106 bone marrow mesenchymal progenitor cells (bmMPCs), suspended in 200μL Matrigel™ (Corning) adjusted with 1μg/ml basic fibroblast growth factor (FGF2) and 1μg/ml erythropoietin (EPO) (ProSpec) and injected sub-cutaneously into anesthetized 6-week-old male athymic nu/nu mice 23. Male nude mice were used because they are more docile and easier to handle than female nude mice, which increased safety during animal handling. N=8 for EC-WT, N=6 for EC-R183Q, N=10 for EC-R183Q sh-ANGPT2 and N=12 for EC-R183Q sh-CTRL. After 12 days, mice were euthanized, implants removed and analyzed by H&E staining and immuno-staining. Blood vessels (indicated by the luminal structures containing one or more red blood cells) and UEA I+ human vessels were counted in fields of 0.18 mm2, 8 fields/section, 2 sections/implant. The perimeter of 150 UEA I+ vessels/implant in EC-WT group, 100 UEA I+ vessels/implant in EC-R183Q group, 75 UEA I+ vessels/implant in EC-R183Q sh-CTRL group and 50 UEA I+ vessels/implant in EC-R183Q sh-ANGPT2 group was measured with Fiji ImageJ software.
ANGPT2 RNAscope
Human ANGPT2 specific RNAscope probe Hs-ANGPT2-No-XMn-C1 was designed by Advanced Cell Diagnostics (ACD). RNAscope multiplex fluorescent v2 assay in combination with anti-human VE-Cadherin (R&D) immuno-staining was conducted in FFPE Matrigel implant sections to detect ANGPT2 mRNA expression in human vessels. RNA-Protein Co-Detection Ancillary kit (ACD) and Opal 570 reagent kit (Akoya Biosciences) were used, and the detailed manufacturer’s instruction was followed for co-detection assay. The human RNAscope 3-plex positive control probe (ACD) and RNAscope 3-plex negative control probe (ACD) were run in parallel. All slides were counterstained with DAPI and mounted. Images were acquired by a Zeiss Airyscan LSM 880 Fast confocal microscope (Zeiss) and analyzed by Fiji ImageJ software (NIH).
Statistical analysis
Data were analyzed and plotted by using GraphPad Prism v.8 (GraphPad Software). Results were expressed as mean ± the standard error for the study variables. The normality was not tested due to the small sample size. Data have been analyzed for equal variance across groups or samples by ANOVA and Student-t test unless adjustment was needed. The gene expression differences between EC-R183Q and EC-WT were analyzed by two-tailed Student-t test. For experiments in which cells were treated with inhibitors at multiple concentrations or different siRNA, the differences were assessed by one-way ANOVA followed by Dunnett’s test for multiple comparisons. For experiments in which cells were treated with inhibitors at two concentrations (ELISA), the differences were analyzed by one-way ANOVA followed by Tukey’s test for multiple comparisons. For experiments in which time-dependent response to VEGF-A stimulation was measured, the differences were analyzed by one-way ANOVA followed by Tukey’s test for multiple comparisons. Significant differences were set at P<0.05.
The comparison of vessel density between EC-R183Q and EC-WT and between EC-R183Q sh-ANGPT2 and sh-CTRL was evaluated by two-tailed Student-t test. The vessel circumference distribution was analyzed by Mann-Whitney U test. Statistical significance with Bonferroni adjustment was set as P<0.017.
RESULTS
Characterization of EC-R183Q and EC-WT
Lentiviral engineered EC-R183Q and EC-WT that express p.R183Q or WT GNAQ cDNA were generated (Online Figure I A) and used for all experiments. GFP was co-expressed as a selection marker. GFP-sorted EC-R183Q showed a MAF of 32%, which is consistent with one mutant GNAQ allele in >96% of cells (Online Figure I B). Western blots showed Gαq is over-expressed in EC-R183Q and EC-WT compared to non-transduced EC (Online Figure I C), which was confirmed by qPCR (Online Figure I D). The closely related gene, GNA11, and G protein coupled receptor kinases GRK2 and GRK5 showed similar expression in EC, EC-WT and EC-R183Q. The expression levels of endothelial adhesion molecules VE-cadherin and CD31 were similar between EC-WT and EC-R183Q but increased approximately 3-fold compared to non-transduced EC (Online Figure I D). These data provide characteristics of the EC-WT and EC-R183Q and demonstrate similar levels of GNAQ, GNA11, GRK2, GRK5, VE-cadherin and CD31.
Gαq-R183Q leads to constitutive activation of PLCβ3
Gαq activates PLCβ but not PLCγ 29–32, and VEGF-A stimulation of PLCβ3 and PLCγ1 have been rigorously studied in ECs 30,31,33. To determine if PLCβ3 is constitutively active in response to Gαq-R183Q, EC-WT and EC-R183Q were assayed after the culture for 16 hours in EBM-2 with 2% FBS only, i.e., without exogenous angiogenic factors that are commonly used to expand primary human ECs. We examined PLCβ3 activation in the quiescent cells by western blot detection of its phosphorylation at Ser537 (activation site) and Ser1105 (inhibitory site) 34–36. p-PLCβ3 (Ser537) was significantly increased in EC-R183Q compared to EC-WT while p-PLCβ3 (Ser1105) was significantly decreased (Figure 1A–B), indicating constitutive activation. In contrast, the activity of PLCγ1, measured by the phosphorylation of Ser1248 37,38 was similar in EC, EC-WT and EC-R183Q (Online Figure I E–F). To verify the increased p-PLCβ3 (Ser537) was due to Gαq-R183Q, we treated EC-WT and EC-R183Q cells with 0, 10, 50 or 100nM YM-254890, a specific Gαq/11 inhibitor 39. YM-254890 at 10nM was sufficient to reduce p-PLCβ3 (Ser537) to the basal level (Figure 1C–D). Although there is some evidence that 30nM YM-254890 can inhibit other Gα proteins 40, the potent effect seen with 10nM strongly suggests the activation of PLCβ3 is Gαq-R183Q dependent. We next measured the level of PIP2 (the substrate of PLCβ3) in the quiescent EC-WT and EC-R183Q. As seen in Online Figure I G–H, there was increased PIP2 in EC-R183Q compared to EC-WT, suggesting the PIP2 levels in the mutant ECs may increase to adapt to constitutively activated PLCβ3 – that is, as more substrate is consumed, more is generated.
Figure 1. Gαq-R183Q leads to constitutive activation of PLCβ3.
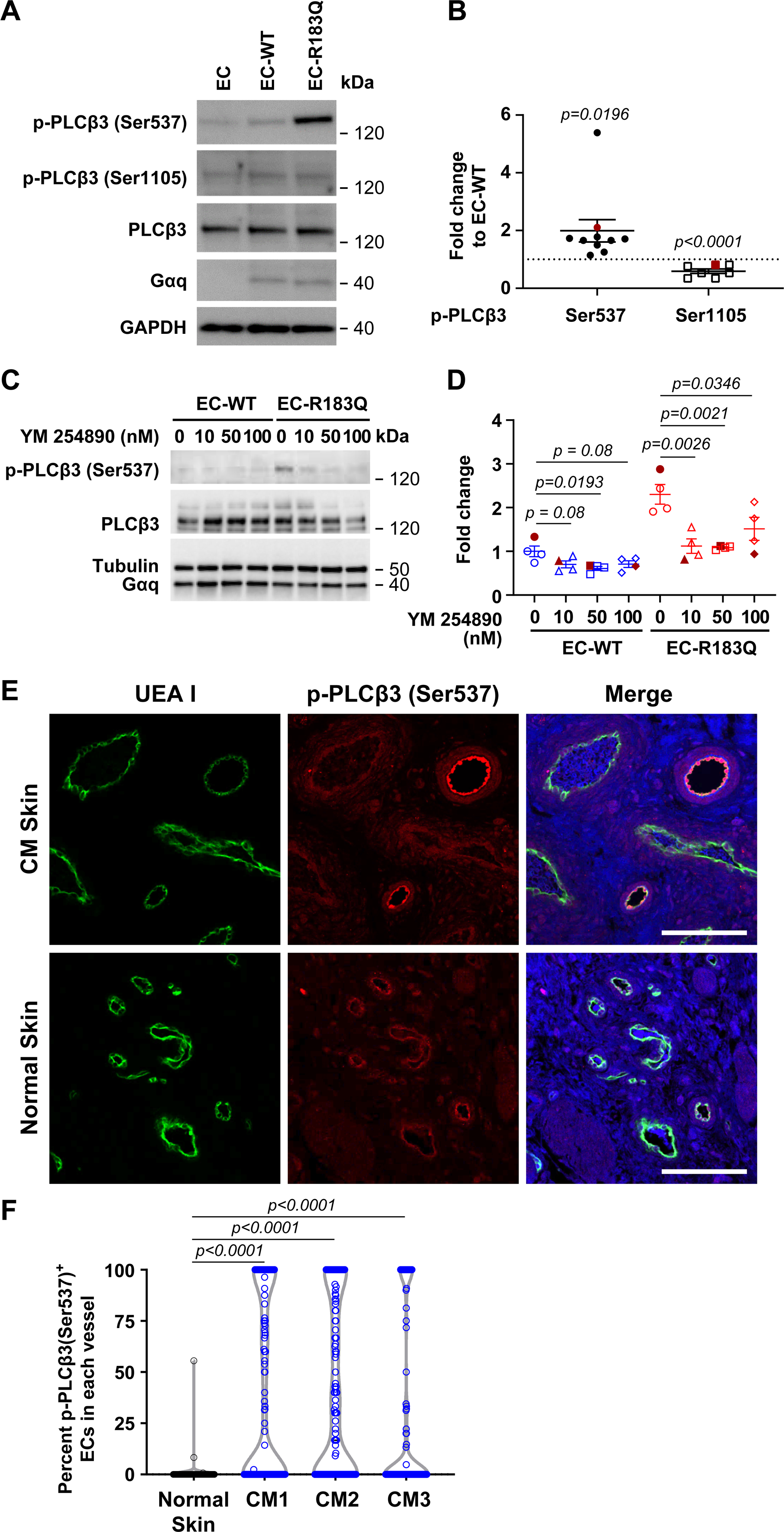
(A) p-PLCβ3 (Ser537) and p-PLCβ3 (Ser1105), total PLCβ3 and Gαq detected by western blot of non-transduced human EC and lentiviral engineered ECs. GAPDH used as the loading control. (B) Levels of p-PLCβ3 (Ser537) and p-PLCβ3 (Ser1105) in EC-R183Q shown as the fold change compared to EC-WT. N=10 for p-PLCβ3 (Ser537) and N=7 for the p-PLCβ3 (Ser1105). Bands in the western blot in (A) are highlighted in red in (B). Dash line marks EC-WT=1. P-values calculated by unpaired two-tailed, Student-t test. (C-D): EC-WT and EC-R183Q treated with increasing concentrations of the Gq/11 inhibitor YM-254890. (C) p-PLCβ3 (Ser537), PLCβ3, Gαq and Tubulin detected by western blot. (D) p-PLCβ3 (Ser537) band intensities, expressed as the fold change to EC-WT without YM-254890 treatment (N=4). Data from western blot shown in C are highlighted by filled red symbols. One-way ANOVA showed P=0.0372 for EC-WT and P=0.0025 for EC-R183Q. Dunnett’s multiple comparison showed significant reduction of p-PLCβ3 (Ser537) in EC-WT at 50nm YM-254890, and in EC-R183Q at all concentrations tested. (E) UEA I (green, human EC) and anti-p-PLCβ3 (Ser537) (red) staining of CM skin tissue (upper panel, CM1 in Table 1) and normal human skin (lower panel). Nuclei counterstained with DAPI (blue). Scale bar, 100 μm. (F) The p-PLCβ3 (Ser537)-positive ECs and total ECs in individual vessels were counted and the percent p-PLCβ3 (Ser537)-positive EC/vessel were calculated and plotted. 100–150 vessels from 4–5 sections of each CM specimen (n=3, Table 1) and from 2 sections of normal skin were analyzed. One-way ANOVA showed P<0.0001 and Dunnett’s multiple comparison showed significant increase in percent p-PLCβ3 (Ser537)-positive ECs in each CM compared to normal skin.
Next, endothelial p-PLCβ3 (Ser537) was examined in vivo in CM skin sections by double-labeling with anti-p-PLCβ3 (Ser537) and the human endothelial marker UEA I 41. p-PLCβ3 (Ser537)+ endothelial cells in CM sections were readily detected in some but not all vessels (Figure 1E). Normal human skin was analyzed in parallel for comparison. The violin plots show the percent p-PLCβ3 (Ser537)+ ECs in individual blood vessels in normal skin and in three different patient CM tissue specimens (Figure 1F). The CM vessels showed a bimodal distribution: some vessels had a high percentage of p-PLCβ3 (Ser537)+ EC and others were negative for p-PLCβ3 (Ser537)+ EC. In normal skin, most blood vessels were p-PLCβ3 (Ser537) negative. Whether the p-PLCβ3 (Ser537)+ EC in CM vessels contain the R183Q allele or non-mutant ECs express active PLCβ3 when in proximity to mutant ECs is not known and will require further investigation. Taken together, these results demonstrate Gαq-R183Q causes constitutive activation of PLCβ3 in ECs.
Gαq-R183Q increases PKC and calcineurin signaling and confers an inflammatory phenotype
To analyze the changes in endothelial gene expression caused by Gαq-R183Q, we performed bulk RNA-Seq on EC-WT and EC-R183Q cultured for 16 hours in EBM-2 with 2% FBS and without growth factors, as described above. We also generated HUVECs expressing WT or R183Q GNAQ, as described in Online Figure 1, and carried out bulk RNA-Seq on these cells in parallel. Ingenuity Pathway Analysis (IPA) of the RNA-Seq data (n=3 biological replicates for each cell type) indicated PKC, NF-κB and calcineurin signaling pathways are activated in EC-R183Q and HUVEC-R183Q (Online Figure II A). An expression heatmap clustered wildtype and mutant HUVEC as well as ECs (with a single outlier, EC_R183Q_2) in Online Figure II B. The connections among these molecules and Gαq is shown in the schematic (Figure 2A). DAG and IP3, products of PLCβ3, activate PKC and calcineurin respectively. PKC can activate NF-kB while calcineurin activates nuclear factor in activated T cells (NFAT); both transcription factors are expressed in ECs and implicated in inflammatory and angiogenic responses. Downstream targets of NF-kB and NFAT that were increased in the bulk RNA-Seq data set and increased in a SOMAscan proteomic assay (data not shown) were further analyzed by qPCR, which showed ANGPT2, DSCR1.4, E-selectin, granulocyte colony-stimulating factor (G-CSF) and interleukin-1β (IL1β) were variably increased in EC-R183Q versus EC-WT (Figure 2B). Gene ontology (GO) analysis partially recapitulated IPA results, with significant enrichment of the NF-kB pathway (GO:0043123; Online Table I).
Figure 2. Gαq-R183Q alters downstream signaling of PLCβ3.

(A) Schematic of Gαq downstream signaling. PKC, NF-kB and calcineurin, highlighted in yellow, were suggested to be increased in EC-R183Q by bulk RNA-Seq. (B) qPCR showed the increased ANGPT2, DSCR1.4 and the NF-kB target genes E-selectin, G-CSF and IL-1B in EC-R183Q compared to EC-WT. Data from 13 independent experiments were plotted. P-values calculated by unpaired two-tailed, Student-t tests. (C) EC-WT and EC-R183Q treated with increasing concentrations of Gαq inhibitor YM-254890 showed reduced ANGPT2 and DSCR1.4 (shown by qPCR, N=5). P-values calculated by one-way ANOVA followed by Dunnett’s multiple comparison. In YM-254890-treated EC-WT, one-way ANOVA showed P=0.0160 for ANGPT2 and P=0.1961 for DSCR1.4. In YM-254890-treated EC-R183Q, one-way ANOVA showed P<0.0001 for ANGPT2 and P=0.0006 for DSCR1.4. (D) ELISA (N=4) showed the increased ANGPT2 protein in EC-R183Q compared to EC-WT. ANGPT2 protein level was reduced by 100 nM YM-254890 both in EC-WT and EC-R183Q. P-values calculated by one-way ANOVA followed by Tukey’s test. One-way ANOVA showed P<0.0001. (E) EC-WT and EC-R183Q treated with 3 different PLCB3-siRNA as well as non-targeted control siRNA. ANGPT2 and DSCR1.4 in EC-R183Q were reduced (shown by qPCR, N=3). P-values calculated by one-way ANOVA followed by Dunnett’s multiple comparison. In EC-WT, one-way ANOVA showed P=0.0773 for ANGPT2 and P=0.0873 for DSCR1.4; in EC-R183Q one-way ANOVA showed P=0.0028 for ANGPT2 and P=0.0502 for DSCR1.4. (F) ELISA showed three different PLCB3-siRNA reduced ANGPT2 protein in both EC-WT and EC-R183Q. P-values calculated by one-way ANOVA followed by Dunnett’s multiple comparison. One-way ANOVA showed P=0.0001 for EC-WT and P=0.0002 for EC-R183Q. (G) EC-WT and EC-R183Q treated with increasing concentrations of the PKC inhibitor AEB071; levels of ANGPT2 was decreased while DSCR1.4 was not (shown by qPCR, N=4). P-values calculated by one-way ANOVA followed by Dunnett’s multiple comparison. In AEB071-treated EC-WT, one-way ANOVA showed P=0.0415 for ANGPT2 and P=0.1489 for DSCR1.4; in AEB071-treated EC-R183Q, one-way ANOVA showed P<0.0001 for ANGPT2 and P=0.8156 for DSCR1.4. (H) ELISA showed increased ANGPT2 protein in EC-R183Q compared to EC-WT; 5μM AEB071 reduced ANGPT2 protein in both EC-WT and EC-R183Q. P-values calculated by one-way ANOVA followed by Tukey’s test. One-way ANOVA showed P<0.0001.
The Gαq inhibitor YM-254890 was again used to determine which transcripts were increased due to Gαq activity (Figure 2C). YM-254890 at 10nM significantly reduced ANGPT2 and DSCR1.4 but had no effect on E-selectin, G-CSF and IL1β (data not shown). YM-254890 reduced ANGPT2 protein significantly (Figure 2D). Cells treated with siRNAs targeted to PLCβ3 showed significantly decreased ANGPT2 and DSCR1.4 mRNA (Figure 2E) and ANGPT2 protein (Figure 2F). In contrast, when PLCβ1 was knocked down by siRNA, there was no effect on ANGPT2 and DSCR1.4 expression (Online Figure IIIA). Additionally, PLCβ3 knockdown did not change PLCβ1 expression, and vice versa, (Online Figure III A–B) and neither affected Gαq expression (Online Figure III A–B). The pan-PKC inhibitor AEB071 42 reduced ANGPT2, in a dose-dependent manner but had no effect on DSCR1.4 (Figure 2G). ANGPT2 protein was reduced by AEB071 (Figure 2H). The calcineurin inhibitor Cyclosporine A (CsA) 43,44 at 1μM reduced DSCR1.4 but not ANGPT2 (Online Figure III C). These results show activation of PLCβ3 and its downstream signaling is specifically due to Gαq-R183Q. The findings also demonstrate roles for endothelial PKC and calcineurin in the pro-inflammatory/pro-angiogenic phenotype induced by Gαq-R183Q and delineate the DAG/PKC/NF-κB from the IP3/calcineurin/NFAT pathway.
NF-κB, a critical regulator of inflammatory gene expression that increases expression of ANGPT2 45, E-selectin, G-CSF and IL1β 46 was examined by western blot of nuclear and cytosolic fractions of EC-WT and EC-R183Q. Canonical and non-canonical NF-κB proteins were increased in EC-R183Q versus EC-WT, in both nuclear and cytoplasmic fractions (Online Figure IV A–B). However, as measured, there did not appear to be consistent, preferential activation of the NF-kB pathway in EC-R183Q, which would be indicated by increased nuclear localization of P50 and P65 (canonical pathway) or P52 and RelB (non-canonical pathway). Functionally blocking NF-κB with the proteasome inhibitor MG132 for 4 hours significantly decreased the elevated ANGPT2 in EC-R183Q and the basal ANGPT2 level in EC-WT (Online Figure IV C), with the caveat that proteosome inhibition may affect other pathways in addition to NF-κB.
DSCR1.4 and VEGF-A signaling in EC-R183Q
Endothelial Gαq has been reported to play essential roles in VEGF-A induced angiogenesis 30,31,33,47. DSCR1.4, which is induced by VEGF-A 48,49 has also been implicated in angiogenesis 50,51. Given the increased DSCR1.4 mRNA in EC-R183Q (Figure 2B), we investigated DSCR1.4 and DSCR1.1 (a constitutively expressed isoform of DSCR1) by western blot. Figure 3A–B show DSCR1.4 protein, not DSCR1.1, was significantly increased in EC-R183Q compared to EC-WT. DSCR1.4 was strongly induced in both EC-R183Q and EC-WT one hour after VEGF-A treatment, with high levels sustained for up to 4 hours (Figure 3C–D); this shows VEGF-A induction of DSCR1.4 is operative in both EC-WT and EC-R183Q, confirmed at the mRNA level by qPCR (Figure 3E). In summary, these experiments show DSCR1.4 protein is expressed in quiescent EC-R183Q and it can be induced by VEGF-A in both EC-R183Q and EC-WT.
Figure 3. Gαq-R183Q does not alter endothelial response to VEGF-A.
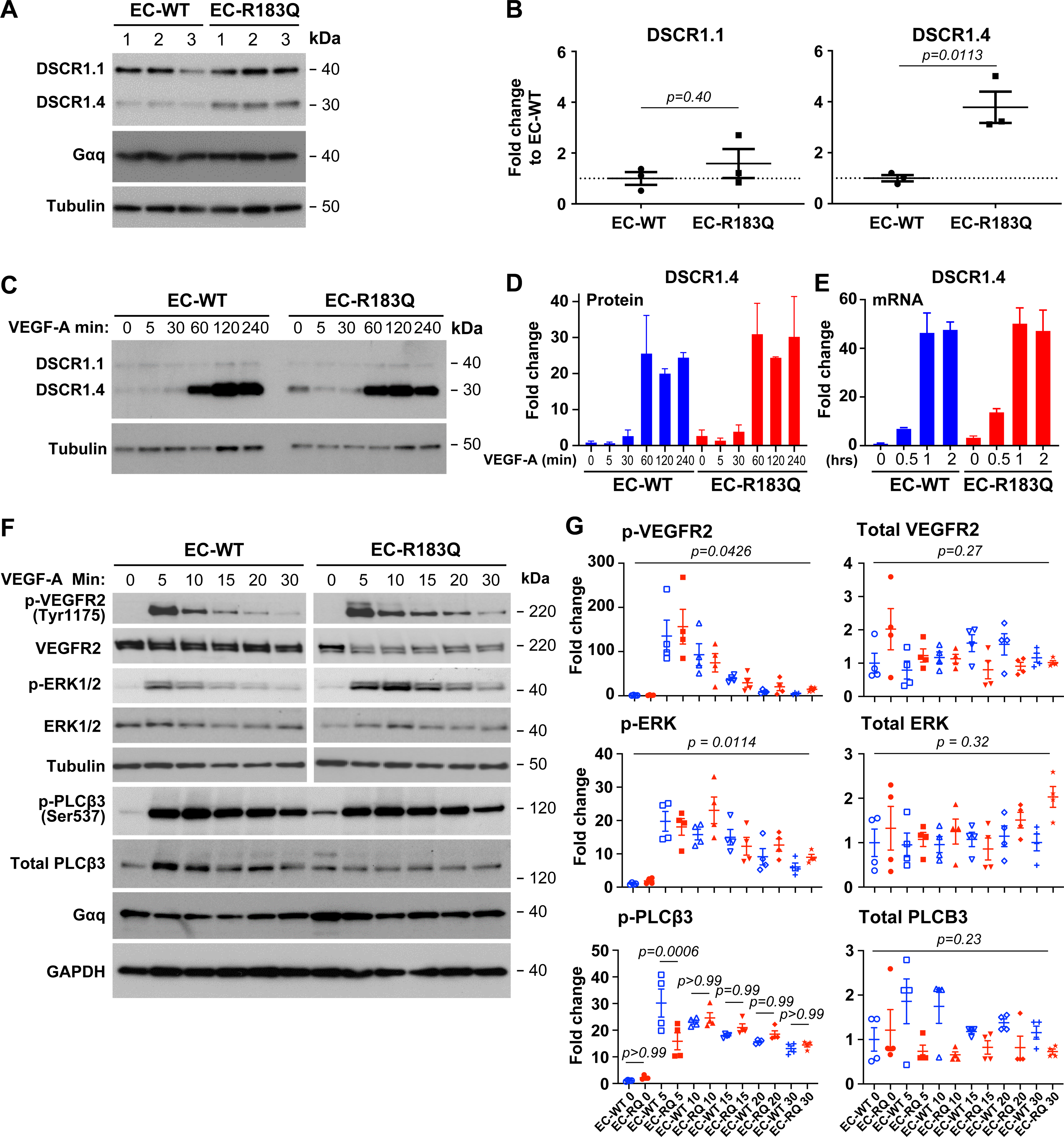
(A-B) Western blot shows inducible DSCR1.4 increased in EC-R183Q compared to EC-WT whereas DSCR1.1 is similar between the two. N=3, P-value calculated by unpaired, two tailed Student-t test. (C-E) Time course of VEGF-A effect on DSCR1.4 and DSCR1.1 analyzed by western blot (C-D) and qPCR (E). Results from two independent experiments. (F-G) Western blot shows p-VEGFR2 (Tyr1175), p-ERK1/2 and p-PLCβ3 (Ser537) in response to VEGF-A over 30 minutes. The band intensities, expressed as the fold change to EC-WT at Time 0 are shown in (G), N=4. P-values calculated by one-way ANOVA followed by Tukey’s multiple comparisons. One-way ANOVA showed P=0.0426 for p-VEGFR2, P=0.0114 for p-ERK1/2, P=0.0171 for p-PLCβ3 and no significant difference for total VEGFR2, ERK1/2 and PLCβ3 over the 30-minute VEGF-A treatment. There were no significant differences between EC-WT and EC-R183Q at each time point during the 30 minutes of VEGF-A-induced p-VEGFR2 (Tyr1175) and p-ERK1/2. P-PLCβ3 was significantly increased in EC-WT at 5 minutes but no significant differences seen at other time points.
VEGF-A caused rapid and robust phosphorylation of VEGFR2 at Tyr1175 in both EC-R183Q and EC-WT (Figure 3F). VEGF-A also caused robust activation of extracellular signal related kinase (ERK1/2) and PLCβ3 in both EC-R183Q and EC-WT, as expected given the strong VEGFR2-(Tyr1175) phosphorylation (Figure 3F–G). Western blots from four biological replicates showed no constitutive activation of VEGFR2 or ERK1/2 in EC-R183Q, i.e. at time=0 before VEGF-A addition, and did not reveal significant differences in the time course or intensity of VEGF-A-induced phosphorylation of VEGFR2 (Tyr1175) and ERK1/2. VEGF-induced phosphorylation of PLCβ3 (Ser537) was similar between EC-R183Q and EC-WT except at the 5 min time point wherein p-PLCβ3 was higher in EC-WT (Figure 3G). Increased expression of DSCR1.4 and p-PLCβ3 (Ser537) at time=0 in EC-R183Q was seen Figure 3C and 3F respectively, consistent with what was observed in Figure 1A and Figure 3A. Taken together, these data show that Gαq-R183Q does not alter the canonical endothelial response to VEGF-A.
ANGPT2 and DSCR1 detected in CM vessels in vivo
To determine if pro-inflammatory/pro-angiogenic molecules seen increased in EC-R183Q are expressed in CM vessels in vivo, we stained CM tissue specimens with the endothelial marker UEA I and anti-ANGPT2 or -DSCR1 antibodies (Figure 4A–B). Although immunofluorescence intensity is not strictly correlated with protein levels, it can be appreciated that skin CM vessels were strongly positive for ANGPT2 compared to vessels in normal human skin (Figure 4A). Anti-DSCR1, which recognizes both 1.1 and 1.4 isoforms of DSCR1, also showed strongly positive vessels in CM compared to normal skin (Figure 4B). Infantile hemangioma, a vascular tumor of infancy displaying proangiogenic properties, served as a positive control for both ANGPT2 52 and DSCR1. Isotype-matched control antibodies for anti-ANGPT2 and DSCR1 are shown in Online Figure V B–C. The mean fluorescence intensity (MFI) of anti-ANGPT2 and DSCR1 stained vessels grouped by increasing circumference was measured in normal skin and patient CM skin specimens (Online Figure V D–E). The violin plots show increased expression of ANGPT2 and DSCR1 in CM vessels compared to similarly sized vessels in normal skin. Note the paucity of small ANGPT2+ and DSCR1+ vessels (<50um in circumference) in CM specimens. Additionally, we measured vessel circumferences in skin CM and compared to those in normal human skin (Online Figure V F); the results were strongly consistent with those reported by Tan and colleagues 2. Collectively, the ANGPT2+ and DSCR1+ blood vessels in human CM sections confirm our in vitro studies on EC-R183Q versus EC-WT and provide further support for increases in PKC and calcineurin signaling revealed by the RNA-Seq study.
Figure 4. Characterization of vessels in CM skin and SWS brain.

(A-B) UEA I staining of human ECs (red) and anti-ANGPT2 (A, green) and anti-DSCR1 (B, green) staining of CM skin tissue sections (upper panels), normal skin sections (middle panels) and hemangioma sections (a positive control, lower panels). (C) UEA I (red) and the anti-p-ERK1/2 (green) staining of CM skin tissue sections (upper left), SWS brain CM (upper right), normal skin (lower left) and a positive control (hemangioma, lower right). The p-ERK1/2-positive ECs and total ECs in each vessel were counted and the percent p-ERK1/2-positive EC/vessel was plotted in (D). One-way ANOVA showed P<0.0001. Dunnett’s multiple comparisons showed no difference between CM specimens and normal skin except brain CM1 (Table 1 shows sex, age and MAF for CM specimens). (E) UEA I (red) and anti-Ki67 (green) staining of skin CM (upper left), SWS brain CM (upper right), normal skin (lower left) and a positive control (hemangioma, lower right). The Ki67-positive ECs and total ECs in each vessel were counted and plotted in (F). P-values calculated by one-way ANOVA (P<0.0001) followed by Dunnett’s multiple comparisons. For the anti-p-ERK1/2 and Ki67 staining, total 50 vessels from 4–5 sections of each skin CM specimen were analyzed, as were 2–3 sections of each SWS brain CM lesion and 2 sections of normal skin. Nuclei counterstained with DAPI (blue). Scale bar, 100 μm.
CM sections from skin and SWS brain, and normal skin were further analyzed for p-ERK1/2 and the proliferating cell marker Ki67 to gain insight into the endothelial proliferative status in CM. Infantile hemangioma again served as a positive control for both p-ERK1/2 and Ki67 53,54. Skin CMs and SWS brain CMs showed endothelial p-ERK1/2 staining, with some vessels containing nearly 100% p-ERK1/2+ ECs and others with very low percentage of p-ERK1/2+ ECs, a pattern akin to what was detected in normal human skin vessels (Figure 4C–D). This finding confirms previous work that shows endothelial p-ERK1/2 in port wine stain and SWS vessels 55 and SWS brain 56 but does not indicate a substantial difference from endothelial p-ERK1/2 in normal skin. Sections were stained with anti-Ki67 to detect and quantify proliferating endothelial cells in CMs from skin and brain (Figure 4E–F). Compared to the positive control and normal skin, CM vessels were largely devoid of proliferating Ki-67+ ECs. This suggests that on-going endothelial proliferation is not a feature of established CM.
EC-R183Q form enlarged, CM-like blood vessels in vivo
We previously showed GNAQ p.R183Q is enriched in ECs in CM from skin and SWS brain 7,8. To test whether endothelial GNAQ p.R183Q is sufficient to drive formation of CM-like vessels, we suspended EC-R183Q or EC-WT with bone marrow mesenchymal progenitor cells (bmMPC) in Matrigel and injected the cell/Matrigel mixture subcutaneously on the flanks of nude mice, as described 23–25. After 12 days, the implants were removed, photographed, and sectioned for histology and IF staining. Both EC-WT and EC-R183Q implants appeared highly vascularized (Figure 5A, upper panels). H&E staining revealed numerous red blood cell-filled vessels, indicating connection to the mouse circulation, however EC-R183Q implants contained enlarged vessels (Figure 5A, middle panel). To identify human endothelial-lined vessels, sections were stained with UEA I, a plant lectin that binds avidly to human endothelial cells but not to murine endothelial cells 41. As expected, many vessels were stained with UEA I, indicating the vessels were lined with human EC, and the UEA I+ vessels appeared enlarged in EC-R183Q implants compared to those formed by EC-WT (Figure 5A, lower panel). Counting total red blood cell-filled lumens in H&E-stained sections from each implant showed that vessel density was reduced in EC-R183Q sections compared to EC-WT (Figure 5C, left), which is consistent with the enlarged vessels observed in EC-R183Q sections. Human vessel density was also reduced in EC-R183Q sections (Figure 5C, right), likely due to the enlarged size of the human vessels. UEA I+ vessel circumference was measured in EC-WT and EC-R183Q sections using ImageJ; the percentage of vessels in each circumference range is shown in Figure 5E. There is a significant decrease in the smallest vessels (<100μm circumference) and a significant increase in larger vessels (101–300μm circumference) in EC-R183Q compared to EC-WT (Figure 5E). A similar shift from small to large vessels is seen in Online Figure V F, confirming findings reported by Tan and colleagues where they show increased vessel circumferences in skin CM, aka port wine stain, compared to normal human skin 2. Thus, the enlarged vessels formed by EC-R183Q are reminiscent of CM in patients.
Figure 5. ANGPT2 contributes to enlarged CM-like vessels.
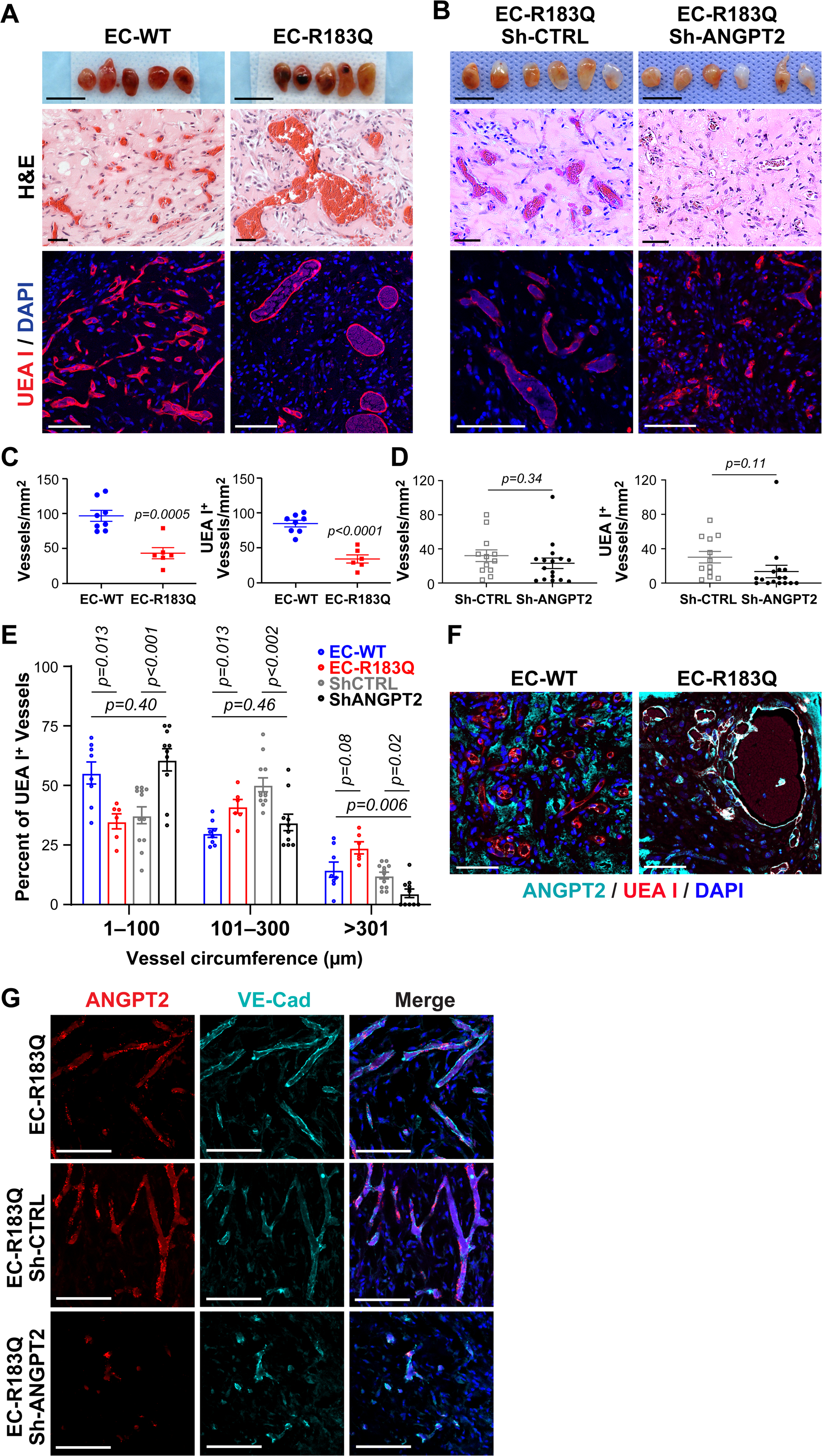
(A) EC-WT and EC-R183Q as well as (B) EC-R183Q sh-CTRL and EC-R183Q sh-ANGPT2 were suspended in Matrigel and injected into nude mice. Implants harvested after 12 days are in the top panels. Scale bar, 10mm. H&E staining showed blood vessels throughout the implants (middle panels) and appearance of enlarged vessels in EC-R183Q implants compared to EC-WT (A) and in EC-R183Q sh-CTRL compared to sh-ANGPT2 (B). Scale bar, 50μm. UEA I (red) staining confirmed enlarged human blood vessels formed in EC-R183Q (A) and EC-R183Q sh-CTRL implants (B) (lower panels). Nuclei counterstained with DAPI (blue). Scale bar, 100μm. Vessel density based on H&E staining (C, left panel) and UEA I+ human vessel density (C, right panel) was lower in EC-R183Q versus EC-WT sections. There was no difference in vessel density between EC-R183Q sh-CTRL and sh-ANGPT2 implants (D, left panel) or in UEA I+ human vessel density (D, right panel). P-values calculated by unpaired, two-tailed Student-t test. N=8 for EC-WT, N=6 for EC-R183Q, N=10 for EC-R183Q sh-ANGPT2 and N=12 for EC-R183Q sh-CTRL. The circumference of each UEA I+ vessel was measured, categorized, and plotted in (E). 150 vessels/implant from EC-WT group, 100 vessels/implant from EC-R183Q group, 75 vessels/implant from EC-R183Q sh-CTRL group and 50 vessels/implant from EC-R183Q sh-ANGPT2 group were analyzed. Mann-Whitney U test with Bonferroni adjustment was applied and P<0.017 set as statistical significance as we focused on 3 comparisons: EC-WT vs. EC-R183Q, EC-R183Q sh-CTRL vs. EC-R183Q sh-ANGPT2 and EC-WT vs. EC-R183Q sh-ANGPT2. (F) Anti-human ANGPT2 (cyan) and UEA I (red) staining of sections from EC-R183Q and EC-WT implants. Nuclei counterstained with DAPI (blue). Scale bar, 100 μm. (G) Sections from implants EC-R183Q (upper), EC-R183Q sh-CTRL (middle) and EC-R183Q sh-ANGPT2 (lower) were stained with human ANGPT2 RNAscope probe (red) and anti-human VE-Cad (cyan) to confirm ANGPT2 expression in human vessels. Nuclei counterstained with DAPI (blue). Scale bar, 100 μm.
We next tested whether shRNA knockdown of ANGPT2 would affect the enlarged vascular phenotype. EC-R183Q were transduced with ANGPT2 shRNA (sh-ANGPT2) lentivirus or control lentivirus (sh-CTRL). QPCR confirmed that ANGPT2 mRNA level was decreased while DSCR1.4 was unchanged (Online Figure VI A). ANGPT2 protein level was also reduced (Online Figure VI B). When mixed with bmMPC in Matrigel and injected into nude mice as described above, both EC-R183Q sh-ANGPT2 and EC-R183Q sh-CTRL formed vessels (Figure 5B). H&E and UEA I staining revealed no significant difference in vessel formation in EC-R183Q sh-ANGPT2 compared to EC-R183Q sh-CTRL (Figure 5B and 5D). Notably, EC-R183Q sh-ANGPT2 implants contained smaller vessels (Figure 5B, lower panel).
UEA I+ vessel circumference was measured in EC-R183Q sh-ANGPT2 and sh-CTRL sections; the percentage of vessels in each circumference range is shown in Figure 5E. There is a significant increase in the smallest vessels (<100μm circumference) and a significant decrease in larger vessels (101–300μm circumference) in EC-R183Q sh-ANGPT2 compared to sh-CTRL (Figure 5E). Indeed, the vessel circumference distribution in EC-R183Q sh-ANGPT2 (black) is similar to EC-WT (blue) (1–100 and 101–300μm circumference, Figure 5E).
To examine ANGPT2 in EC-R183Q vessels, we double-stained cell/Matrigel sections with anti-ANGPT2 and UEA I (Figure 5F). Both large and small UEA I+ vessels were positive for ANGPT2 in the EC-R183Q sections. In contrast, UEA I+ vessels in the EC-WT sections did not stain positive for ANGPT2. Note: the anti-ANGPT2 showed considerable staining of Matrigel only implants, that is implants without cells at the time of subcutaneous injection (Online Figure V G–H). This background staining in the Matrigel sections is clearly delineated from the UEA-1+ human vessels.
We next visualized human ANGPT2 in the cell/Matrigel implants using a RNAscope probe that does not cross-react with mouse Angpt2. Anti-human VE-cadherin staining was conducted directly after the RNAscope. ANGPT2 mRNA was detected in the human VE-cadherin+ vessels in EC-R183Q formed vessels (Figure 5G, top panel), in EC-R183Q-sh-CTRL formed vessels (Figure 5G, middle panel) but was greatly diminished in EC-R183Q-sh-ANGPT2 formed vessels (Figure 5G, bottom panel). Hence, knockdown of the increased ANGPT2 in EC-R183Q normalized the enlarged CM-like vessels, suggesting ANGPT2 is a potential therapeutic target. In summary, the cell/Matrigel implant model revealed that EC-R183Q are sufficient to form blood vessels in vivo that display two key features of CM: enlarged vessels 2 and increased ANGPT2.
DISCUSSION
In this study, we demonstrate that endothelial-expressed Gαq-R183Q causes constitutive activation of PLCβ3, which in turn increases the expression of ANGPT2, DSCR1.4, and other inflammatory mediators. This establishes for the first time how Gαq-R183Q alters endothelial signaling and gene expression. In addition, we show that endothelial Gαq-R183Q is sufficient to form blood vessels in vivo that display enlarged lumens and increased ANGPT2, two features of CM. shRNA knock down of ANGPT2 in the mutant endothelial cells resulted in vessel sizes akin to that seen in normal skin.
ANGPT2 is not essential during embryonic development yet its over-expression in mice causes poorly formed vessels and lethality 57. In adult life, ANGPT2 function is endothelial context dependent. It is expressed at low levels in quiescent EC and is increased in response to inflammatory mediators, which leads to permeable and destabilized blood vessels 58–60. ANGPT2 acts as an antagonist in most settings by blocking its related family member, Angiopoietin-1, from binding to TIE2, an endothelial receptor tyrosine kinase. Angiopoeitin-1 binding to TIE2 activates PI3K/AKT, a pathway that must be properly titrated in endothelium: too much TIE2-PI3K-AKT signaling causes venous malformations, too little can reduce endothelial survival 61. At high concentrations (800 ng/mL), ANGPT2 can activate TIE2, seen by increased phosphorylation of AKT at Ser473, yet lower ANGPT2 concentrations (50–400 ng/ml) had no agonistic effect 62. In EC-R183Q, ANGPT2 is most likely acting as an antagonist because ANGPT2 levels measured by ELISA were less than 30–40 ng/ml and p-AKT (Ser473) was not elevated (Online Figure IV D–E). ANGPT2 has also been shown to destabilize vessels via β1 integrin binding, indicating a TIE2-independent mode of action 63. TIE2 protein levels were comparable between EC-R183Q and EC-WT but p-TIE2 (Tyr992) levels were below limits of western blot detection (data not shown), which precluded our ability to determine if increased ANGPT2 suppresses the phosphorylation status of TIE2. Despite these limitations, our in vitro and in vivo data suggest that pro-inflammatory/pro-angiogenic ANGPT2 is a driver of CM and could therefore be a therapeutic target to prevent CM progression in both cutaneous and intracranial CMs. In support of this speculation, recent literature connects Gαq to ANGPT2 via shear stress: Gαq is involved in EC sensing of shear stress 64; Gαq activation by disturbed shear stress activates NF-κB 65; and NF-κB increases endothelial ANGPT2 45.
DSCR1, also known as regulator of calcineurin 1 (RCAN1), is expressed in two forms – constitutive DSCR1.1 and calcineurin-activated DSCR1.4. Calcineurin is activated by calcium, which can be sourced from endoplasmic reticulum in response to IP3, a product of PLCβ3 (Figure 2A). We speculate DSCR1.4 is elevated in EC-R183Q and CM specimens because of ongoing activation of calcineurin-NFAT pathway by constitutively active PLCβ3. In normal ECs, DSCR1.4 is induced by VEGF-A-mediated calcineurin signaling, which can increase sprouting and migration 50, two features of angiogenic ECs. Once induced by VEGF-A, DSCR1.4 inhibits its own expression by binding to and inhibiting calcineurin phosphatase activity. We speculate that increased DSCR1.4 due to Gq-R183Q is insufficient to inhibit calcineurin because the latter is constantly being activated by the IP3-released calcium. Inhibition of constitutive DSCR1.4 levels with Gαq inhibitor YM-254890 (Figure 2C), PLCB3 siRNA (Figure 2E) and calcineurin inhibitor CsA (Online Figure III C) is consistent with PLCβ3 and calcineurin playing a role in the elevated DSCR1.4.
The inflammatory factors IL1β, E-selectin and G-CSF were also increased in EC-R183Q under quiescent condition. IL-1β is a well-known cytokine that activates NF-κB, and in turn IL-1β transcription is increased by activated NF-κB, yielding a self-amplification loop. E-selectin mediates rolling of neutrophils and monocytes along inflamed endothelium; it is transiently expressed within hours after endothelial exposure to inflammatory agents such as IL1β 66. E-selectin has also been implicated in angiogenesis. The increased levels in NF-κB proteins in EC-R183Q (Online Figure IV A–B) is consistent with increased levels of IL1β, E-selectin and G-CSF (a growth factor used to mobilize bone-marrow to collect stem cells for hematopoietic stem cell transplants). If and how these molecules contribute to CM will require further investigation.
Mitogen-activated protein kinase (MAPK) signaling integrates endothelial responses to growth factor stimulation. A prototype is VEGF-A activation of VEGFR2 that results in transiently increased phosphorylation of ERK1/2, which in turn regulates endothelial proliferation, migration, and vessel integrity 67. While EC-R183Q and EC-WT do not exhibit constitutive activation of p-VEGFR2 (Tyr1175) or p-ERK1/2, both respond rapidly and robustly to VEGF-A (Figure 3F–G), which indicates the initial steps of VEGFR2 signaling are not altered by Gαq-R183Q. There was an expectation that Gαq-R183Q would cause increased levels of p-ERK1/2 6,68 based on what was known about Gαq-Q209L in uveal melanoma. Previous studies showed p-ERK1/2 staining in EC of port-wine stains 55 and SWS brain 56. In this study, we detected nuclear-localized p-ERK1/2 in a set of 3 skin CM and 3 brain CM from SWS patients at levels similar to normal human skin (Figure 4C–D) and we saw no constitutive p-ERK1/2 in EC-R183Q. This is in contrast to the increased p-ERK1/2 seen in Gαq-Q209L-driven uveal melanoma 69,70, suggesting that the two different gain-of-function mutations in Gαq (R183Q versus Q209L) affect cell-specific signaling in distinct ways, which has been suggested by computational studies 15. We also found minimal endothelial proliferation in CM compared to normal infant skin or infantile hemangioma (positive control). This suggests that at the time CM specimens were obtained, p-ERK1/2 and endothelial proliferation were not prominent in CM. This does not rule out the possibility of increased MAPK signaling and/or proliferation in nascent CM.
We tested how Gαq-R183Q affects blood vessel formation in vivo using a well-established human xenograft model pioneered by our lab 25 and that proved useful for studies on venous malformations caused by somatic activating mutation in TIE2 71. EC-R183Q and EC-WT formed perfused human vessels when implanted into nude mice, yet the EC-R183Q implants had fewer vessels/mm2 compared to EC-WT, and this appears to be due to enlargement of some vessels (Figure 5A and 5C). Measuring vessel circumference revealed the percentage of small-sized vessels was reduced and that of larger-sized vessels was increased in EC-R183Q versus EC-WT implants (Figure 5E). This is similar to the enlargement of CM vessels versus those in normal skin 2 (Online Figure V F). Thus, EC-R183Q form vessels with a CM characteristic. Implants were removed from the mice at day 12 and it is possible that further enlargement would occur over time, which could be tested in a time course study. Human vessels formed by EC-R183Q stained positive for ANGPT2 whereas no endothelial ANGPT2 was detected in the EC-WT implants (Figure 5F); this indicates the EC-R183Q-formed vessels display a second characteristic of CM. The anti-ANGPT2 binds to the Matrigel used to suspend the cells prior to sub-cutaneous injection (Online Figure V G–H) and this likely accounts for the non-endothelial staining seen in Figure 5F. We therefore developed an RNAscope assay with a human specific ANGPT2 probe that does not cross-react with murine Angpt2. ANGPT2 was robustly detected in human vessels formed by EC-R183Q (Figure 5G upper panel) but not in the EC-WT implants or in Matrigel (Online Figure VI E).
We examined whether knockdown ANGPT2 in EC-R183Q by shRNA can restore the normal capillary morphology. We confirmed that ANGPT2-shRNA decreased ANGPT2 expression at mRNA and protein levels in EC-R183Q (Online Figure VI A–B) as well as in vivo at mRNA level in the cell/Matrigel implants (Figure 5G lower panel). ANGPT2 knockdown did not reduce the number of EC-R183Q vessels but did shift the vessel size distribution to smaller vessels similar to those formed by EC-WT cells or found in normal skin. These findings indicate ANGPT2 drives at least one aspect of the CM phenotype and thus could be targeted to restore a normal capillary phenotype in CMs.
There are several limitations to this model. An important one is that the human ECs are suspended in an artificial environment that does not reflect tissue microenvironments relevant for CM. Yet, the simplicity of the model allows us to determine what is sufficient to form CM-like vessels in vivo. A second limitation is the lentiviral expression of Gαq-R183Q in human ECFCs. These highly proliferative ECs are isolated from umbilical cord blood and thus may not represent the type of endothelial cell in which the somatic R183Q mutation arises. SWS brain EC isolated and expanded in the culture show GNAQ p.R183Q mutant allelic frequencies of 15–21% 8, i.e. the cultures are a mix mutant and non-mutant endothelial cells, which to date we have not been able to separate. Thus, we lack brain ECs that would provide the appropriate control for brain ECs with the R183Q allele. In contrast, human EC engineered to express either WT or R183Q GNAQ provide, prior to lentiviral transduction, genetically-matched cells, which allows one to identify alterations driven by Gαq-R183Q. Lentiviral-transduced human EC were used previously to show 1) endothelial-expressed TIE2 L914F, a somatic mutation found in sporadic venous malformations 72, was sufficient to recapitulate venous malformations in mice, 2) TIE2 L914F increases PI3K/AKT signaling and 3) blocking this signaling prevented and regressed venous malformations 71. These findings have been expanded upon in subsequent studies 73–75 and provided the rationale for clinical trials to treat difficult venous malformations 71,76. Based on this precedence, the enlarged, ANGPT2+ neovascular networks formed in nude mice by EC-R183Q provide a starting point for functional studies and drug testing.
CM have much in common with other types of vascular malformations, which can occur in capillary, venous, arterial, and lymphatic vessels throughout the body. Vascular malformations can be familial or sporadic: CMs are sporadic. Vascular malformations can result from loss of function mutations or gain of function mutations: CMs are caused by a gain-of-function mutation in GNAQ. The mutations that cause vascular malformations affect endothelial signaling, gene expression and behavior 61 as is shown here for Gαq-R183Q. Furthermore, ANGPT2 is implicated in many types of vascular malformations. Hereditary hemorrhagic telangiectasia (HHT), an inherited disorder characterized by fragile blood vessels prone to hemorrhaging and arteriovenous malformation (AVM), is caused by germline loss of function mutations in TGFβ signaling genes including SMAD4. Crist and colleagues recently showed Smad4 represses Angpt2 transcription, such that loss of endothelial Smad4 results in increased Angpt2 and AVMs. An inhibitor specific for mouse Angpt2 prevented and even reversed AVM formation, and as well reduced enlarged vessels that were prominent in the mouse model 77. Consistent with a role in AVM formation, ANGPT2 was found increased in human brain AVM extracts, measured by ELISA 78. ANGPT2 has also been implicated in cerebral cavernous malformations (CCMs); these are initiated by bi-allelic inactivating mutations in one of three genes, CCM1, CCM2 or CCM3, resulting in capillary-venous vessels with reduced endothelial barrier function and ectasia, which makes them prone to hemorrhage and focal neurological insults. Zhou and colleagues reported CCM3 deficiency facilities exocytosis of ANGPT2 from Weibel-Palade bodies, and CCM3 lesions exhibit enlarged vascular lumens, destabilized EC junctions and disrupted pericyte coverage. An Angpt2 neutralizing antibody blunted CCM progression in Ccm3ECKO mice 79. Another example of potential ANGPT2 involvement is the increased serum levels found in kaposiform lymphangiomatosis (KLA) patients and kaposiform hemangioendothelioma (KHE) patients with Kasabach-Merritt phenomenon coagulopathy compared to normal controls. KLA and KHE patients treated with the mTOR inhibitor sirolimus showed decreased levels of serum ANGPT2 compared to their baselines 12 months earlier 80. In summary, increased ANGPT2 is functionally implicated in cellular and animal models of AVM and CCM wherein it causes ecstatic blood vessels with impacts on blood flow, permeability and function of the underlying tissue or organ. This underscores the importance of strict regulation of ANGPT2 to maintain vessel homeostasis throughout life.
In conclusion, our study demonstrates that endothelial Gαq-R183Q causes constitutive activation of PLCβ3 that leads to a pro-inflammatory/pro-angiogenic phenotype, and we provide the first evidence that endothelial Gαq-R183Q is causative for CM, at least partially driven by increased ANGPT2. At present, targeted drug therapies do not exist for CM and SWS patients. Our discovery of key pathways altered by Gαq-R183Q, the mutation that occurs in ~90% of non-syndromic and SWS CMs, provide a starting point for deeper studies into cellular mechanisms and drug testing that we hope will begin to address the tremendous clinical needs of these patients.
Supplementary Material
HIGHLIGHTS.
Human endothelial cells (EC) engineered to express the GNAQ p.R183Q mutation show constitutive activation of PLCβ3.
ANGPT2, a blood vessel destabilizer, and DSCR1.4, an angiogenic factor, are increased in the mutation-expressing EC.
Endothelial-expressed p.R183Q is sufficient to drive the formation of enlarged blood vessels, reminiscent of what is observed in CM.
shRNA knockdown of ANGPT2 prevents the enlarged vascular phenotype.
Acknowledgments
We thank Boston Children’s Hospital and Harvard Stem Cell Institute flow cytometry research facility for cell sorting, Boston Children’s Hospital viral core and Massachusetts General Hospital vector core (P30 - NS045776) for lentivirus generation, BCH IDDRC Molecular Genetics Core Facility (P50 - HD105351) for evaluating RNA Integrity Number of RNA samples prepared for RNA sequencing and providing technique support for ddPCR. Boston Children’s Hospital Histology research laboratory and Dana-Farber/Harvard Cancer Center Pathology Core for preparation of FFPE sections; Kristin Johnson for preparing figures.
Sources of Funding
Research reported in this manuscript was supported by the National Heart, Lung, and Blood Institute, part of the National Institutes of Health, under Award Number R01 HL127030 (J.B., A.K.G). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The research was also supported by a Sturge-Weber Foundation Lisa’s Fellowship Grant (L.H.), a Translational Neuroscience Center Pilot Study Award, Boston Children’s Hospital (J.B., A.K.G); the Translational Investigator Program, Boston Children’s Hospital (A.K.G.) and a Swiss National Science Foundation Postdoc Mobility Grant (C.B.).
Funding:
Sturge-Weber Foundation Lisa’s Fellowship Grant (L.H.); NIH R01 HL127030 (J.B., A.K.G); Translational Neuroscience Center Pilot Study Award (J.B., A.K.G); Boston Children’s Hospital; Translational Investigator Program Boston Children’s Hospital (A.K.G.) and Swiss National Science Foundation Postdoc.Mobility Grant (C.B.)
ABBREVIATIONS
- ANGPT2
Angiopoietin 2
- CM
Capillary Malformation
- DSCR1
Down Syndrome Critical Region Protein 1
- G-CSF
Granulocyte colony-stimulating factor
- PKC
Protein kinase C
- PLC
Phospholipase C
- SWS
Sturge-Weber syndrome
Footnotes
Competing interests: The authors declare no completing interests.
Disclosure
none
REFERENCE
- 1.Kanada KN, Merin MR, Munden A, Friedlander SF. A prospective study of cutaneous findings in newborns in the united states: Correlation with race, ethnicity, and gestational status using updated classification and nomenclature. J Pediatr. 2012;161:240–245 [DOI] [PubMed] [Google Scholar]
- 2.Tan W, Wang J, Zhou F, Gao L, Yin R, Liu H, et al. Coexistence of eph receptor b1 and ephrin b2 in port-wine stain endothelial progenitor cells contributes to clinicopathological vasculature dilatation. Br J Dermatol. 2017;177:1601–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zallmann M, Mackay MT, Leventer RJ, Ditchfield M, Bekhor PS, Su JC. Retrospective review of screening for sturge-weber syndrome with brain magnetic resonance imaging and electroencephalography in infants with high-risk port-wine stains. Pediatr Dermatol. 2018;35:575–581 [DOI] [PubMed] [Google Scholar]
- 4.Mantelli F, Bruscolini A, La Cava M, Abdolrahimzadeh S, Lambiase A. Ocular manifestations of sturge-weber syndrome: Pathogenesis, diagnosis, and management. Clin Ophthalmol. 2016;10:871–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bichsel CA, Goss J, Alomari M, Alexandrescu S, Robb R, Smith LE, et al. Association of somatic gnaq mutation with capillary malformations in a case of choroidal hemangioma. JAMA Ophthalmol. 2019;137:91–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shirley MD, Tang H, Gallione CJ, Baugher JD, Frelin LP, Cohen B, et al. Sturge-weber syndrome and port-wine stains caused by somatic mutation in gnaq. N Engl J Med. 2013;368:1971–1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Couto JA, Huang L, Vivero MP, Kamitaki N, Maclellan RA, Mulliken JB, et al. Endothelial cells from capillary malformations are enriched for somatic gnaq mutations. Plast Reconstr Surg. 2016;137:77e–82e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang L, Couto JA, Pinto A, Alexandrescu S, Madsen JR, Greene AK, et al. Somatic gnaq mutation is enriched in brain endothelial cells in sturge-weber syndrome. Pediatr Neurol. 2017;67:59–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Couto JA, Ayturk UM, Konczyk DJ, Goss JA, Huang AY, Hann S, et al. A somatic gna11 mutation is associated with extremity capillary malformation and overgrowth. Angiogenesis. 2017;20:303–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fjaer R, Marciniak K, Sundnes O, Hjorthaug H, Sheng Y, Hammarstrom C, et al. A novel somatic mutation in gnb2 provides new insights to the pathogenesis of sturge-weber syndrome. Hum Mol Genet. 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rozengurt E Mitogenic signaling pathways induced by g protein-coupled receptors. J Cell Physiol. 2007;213:589–602 [DOI] [PubMed] [Google Scholar]
- 12.Kimple AJ, Bosch DE, Giguere PM, Siderovski DP. Regulators of g-protein signaling and their galpha substrates: Promises and challenges in their use as drug discovery targets. Pharmacol Rev. 2011;63:728–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O’Brien JM, et al. Frequent somatic mutations of gnaq in uveal melanoma and blue naevi. Nature. 2009;457:599–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, et al. Mutations in gna11 in uveal melanoma. N Engl J Med. 2010;363:2191–2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martins L, Giovani PA, Reboucas PD, Brasil DM, Haiter Neto F, Coletta RD, et al. Computational analysis for gnaq mutations: New insights on the molecular etiology of sturge-weber syndrome. J Mol Graph Model. 2017;76:429–440 [DOI] [PubMed] [Google Scholar]
- 16.Feng X, Degese MS, Iglesias-Bartolome R, Vaque JP, Molinolo AA, Rodrigues M, et al. Hippo-independent activation of yap by the gnaq uveal melanoma oncogene through a trio-regulated rho gtpase signaling circuitry. Cancer Cell. 2014;25:831–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vaque JP, Dorsam RT, Feng X, Iglesias-Bartolome R, Forsthoefel DJ, Chen Q, et al. A genome-wide rnai screen reveals a trio-regulated rho gtpase circuitry transducing mitogenic signals initiated by g protein-coupled receptors. Mol Cell. 2013;49:94–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu X, Li J, Zhu M, Fletcher JA, Hodi FS. Protein kinase c inhibitor aeb071 targets ocular melanoma harboring gnaq mutations via effects on the pkc/erk1/2 and pkc/nf-kappab pathways. Mol Cancer Ther. 2012;11:1905–1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoo JH, Shi DS, Grossmann AH, Sorensen LK, Tong Z, Mleynek TM, et al. Arf6 is an actionable node that orchestrates oncogenic gnaq signaling in uveal melanoma. Cancer Cell. 2016;29:889–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu FX, Luo J, Mo JS, Liu G, Kim YC, Meng Z, et al. Mutant gq/11 promote uveal melanoma tumorigenesis by activating yap. Cancer Cell. 2014;25:822–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ingram DA, Caplice NM, Yoder MC. Unresolved questions, changing definitions, and novel paradigms for defining endothelial progenitor cells. Blood. 2005;106:1525–1531 [DOI] [PubMed] [Google Scholar]
- 22.Ingram DA, Mead LE, Tanaka H, Meade V, Fenoglio A, Mortell K, et al. Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood. 2004;104:2752–2760 [DOI] [PubMed] [Google Scholar]
- 23.Lin RZ, Melero-Martin JM. Bioengineering human microvascular networks in immunodeficient mice. J Vis Exp. 2011:e3065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Melero-Martin JM, Bischoff J. Chapter 13. An in vivo experimental model for postnatal vasculogenesis. Methods Enzymol. 2008;445:303–329 [DOI] [PubMed] [Google Scholar]
- 25.Melero-Martin JM, De Obaldia ME, Kang SY, Khan ZA, Yuan L, Oettgen P, et al. Engineering robust and functional vascular networks in vivo with human adult and cord blood-derived progenitor cells. Circ Res. 2008;103:194–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of rna-seq experiments with hisat, stringtie and ballgown. Nat Protoc. 2016;11:1650–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kankanamge D, Tennakoon M, Weerasinghe A, Cedeno-Rosario L, Chadee DN, Karunarathne A. G protein alphaq exerts expression level-dependent distinct signaling paradigms. Cell Signal. 2019;58:34–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pfaffl MW. A new mathematical model for relative quantification in real-time rt-pcr. Nucleic Acids Res. 2001;29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325 [DOI] [PubMed] [Google Scholar]
- 30.Rhee SG, Choi KD. Regulation of inositol phospholipid-specific phospholipase c isozymes. J Biol Chem. 1992;267:12393–12396 [PubMed] [Google Scholar]
- 31.Sternweis PC, Smrcka AV. Regulation of phospholipase c by g proteins. Trends Biochem Sci. 1992;17:502–506 [DOI] [PubMed] [Google Scholar]
- 32.Litosch I Decoding galphaq signaling. Life Sci. 2016;152:99–106 [DOI] [PubMed] [Google Scholar]
- 33.Mukhopadhyay D, Zeng H. Involvement of g proteins in vascular permeability factor/vascular endothelial growth factor signaling. Cold Spring Harb Symp Quant Biol. 2002;67:275–283 [DOI] [PubMed] [Google Scholar]
- 34.Strassheim D, Law PY, Loh HH. Contribution of phospholipase c-beta3 phosphorylation to the rapid attenuation of opioid-activated phosphoinositide response. Mol Pharmacol. 1998;53:1047–1053 [PubMed] [Google Scholar]
- 35.Strassheim D, Williams CL. P2y2 purinergic and m3 muscarinic acetylcholine receptors activate different phospholipase c-beta isoforms that are uniquely susceptible to protein kinase c-dependent phosphorylation and inactivation. J Biol Chem. 2000;275:39767–39772 [DOI] [PubMed] [Google Scholar]
- 36.Xia C, Bao Z, Yue C, Sanborn BM, Liu M. Phosphorylation and regulation of g-protein-activated phospholipase c-beta 3 by cgmp-dependent protein kinases. J Biol Chem. 2001;276:19770–19777 [DOI] [PubMed] [Google Scholar]
- 37.Wang Y, Wu J, Wang Z. Akt binds to and phosphorylates phospholipase c-gamma1 in response to epidermal growth factor. Mol Biol Cell. 2006;17:2267–2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park DJ, Min HK, Rhee SG. Inhibition of cd3-linked phospholipase c by phorbol ester and by camp is associated with decreased phosphotyrosine and increased phosphoserine contents of plc-gamma 1. J Biol Chem. 1992;267:1496–1501 [PubMed] [Google Scholar]
- 39.Takasaki J, Saito T, Taniguchi M, Kawasaki T, Moritani Y, Hayashi K, et al. A novel galphaq/11-selective inhibitor. J Biol Chem. 2004;279:47438–47445 [DOI] [PubMed] [Google Scholar]
- 40.Peng Q, Shen J. Ym-254890 is a general inhibitor of g proteins. The FASEB Journal. 2019;33:503.507–503.507 [Google Scholar]
- 41.Holthofer H, Virtanen I, Kariniemi AL, Hormia M, Linder E, Miettinen A. Ulex europaeus i lectin as a marker for vascular endothelium in human tissues. Lab Invest. 1982;47:60–66 [PubMed] [Google Scholar]
- 42.Leonard B, McCann JL, Starrett GJ, Kosyakovsky L, Luengas EM, Molan AM, et al. The pkc/nf-kappab signaling pathway induces apobec3b expression in multiple human cancers. Cancer Res. 2015;75:4538–4547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schreiber SL, Crabtree GR. The mechanism of action of cyclosporin a and fk506. Immunol Today. 1992;13:136–142 [DOI] [PubMed] [Google Scholar]
- 44.Wiederrecht G, Lam E, Hung S, Martin M, Sigal N. The mechanism of action of fk-506 and cyclosporin a. Ann N Y Acad Sci. 1993;696:9–19 [DOI] [PubMed] [Google Scholar]
- 45.Kim B, Jang C, Dharaneeswaran H, Li J, Bhide M, Yang S, et al. Endothelial pyruvate kinase m2 maintains vascular integrity. J Clin Invest. 2018;128:4543–4556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heller RA, Schena M, Chai A, Shalon D, Bedilion T, Gilmore J, et al. Discovery and analysis of inflammatory disease-related genes using cdna microarrays. Proc Natl Acad Sci U S A. 1997;94:2150–2155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sivaraj KK, Li R, Albarran-Juarez J, Wang S, Tischner D, Grimm M, et al. Endothelial galphaq/11 is required for vegf-induced vascular permeability and angiogenesis. Cardiovasc Res. 2015;108:171–180 [DOI] [PubMed] [Google Scholar]
- 48.Baggott RR, Alfranca A, Lopez-Maderuelo D, Mohamed TM, Escolano A, Oller J, et al. Plasma membrane calcium atpase isoform 4 inhibits vascular endothelial growth factor-mediated angiogenesis through interaction with calcineurin. Arterioscler Thromb Vasc Biol. 2014;34:2310–2320 [DOI] [PubMed] [Google Scholar]
- 49.Holmes K, Chapman E, See V, Cross MJ. Vegf stimulates rcan1.4 expression in endothelial cells via a pathway requiring ca2+/calcineurin and protein kinase c-delta. PLoS One. 2010;5:e11435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alghanem AF, Wilkinson EL, Emmett MS, Aljasir MA, Holmes K, Rothermel BA, et al. Rcan1.4 regulates vegfr-2 internalisation, cell polarity and migration in human microvascular endothelial cells. Angiogenesis. 2017;20:341–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Minami T, Jiang S, Schadler K, Suehiro J, Osawa T, Oike Y, et al. The calcineurin-nfat-angiopoietin-2 signaling axis in lung endothelium is critical for the establishment of lung metastases. Cell Rep. 2013;4:709–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu Y, Varughese J, Brown LF, Mulliken JB, Bischoff J. Increased tie2 expression, enhanced response to angiopoietin-1, and dysregulated angiopoietin-2 expression in hemangioma-derived endothelial cells. Am J Pathol. 2001;159:2271–2280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boscolo E, Mulliken JB, Bischoff J. Vegfr-1 mediates endothelial differentiation and formation of blood vessels in a murine model of infantile hemangioma. Am J Pathol. 2011;179:2266–2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kraling BM, Razon MJ, Boon LM, Zurakowski D, Seachord C, Darveau RP, et al. E-selectin is present in proliferating endothelial cells in human hemangiomas. Am J Pathol. 1996;148:1181–1191 [PMC free article] [PubMed] [Google Scholar]
- 55.Tan W, Chernova M, Gao L, Sun V, Liu H, Jia W, et al. Sustained activation of c-jun n-terminal and extracellular signal-regulated kinases in port-wine stain blood vessels. J Am Acad Dermatol. 2014;71:964–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wellman RJ, Cho SB, Singh P, Tune M, Pardo CA, Comi AM, et al. Galphaq and hyper-phosphorylated erk expression in sturge-weber syndrome leptomeningeal blood vessel endothelial cells. Vasc Med. 2019;24:72–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, et al. Angiopoietin-2, a natural antagonist for tie2 that disrupts in vivo angiogenesis. Science. 1997;277:55–60 [DOI] [PubMed] [Google Scholar]
- 58.Akwii RG, Sajib MS, Zahra FT, Mikelis CM. Role of angiopoietin-2 in vascular physiology and pathophysiology. Cells. 2019;8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Parikh SM. Angiopoietins and tie2 in vascular inflammation. Curr Opin Hematol. 2017;24:432–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Scholz A, Plate KH, Reiss Y. Angiopoietin-2: A multifaceted cytokine that functions in both angiogenesis and inflammation. Ann N Y Acad Sci. 2015;1347:45–51 [DOI] [PubMed] [Google Scholar]
- 61.Nguyen HL, Boon LM, Vikkula M. Vascular anomalies caused by abnormal signaling within endothelial cells: Targets for novel therapies. Semin Intervent Radiol. 2017;34:233–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim I, Kim JH, Moon SO, Kwak HJ, Kim NG, Koh GY. Angiopoietin-2 at high concentration can enhance endothelial cell survival through the phosphatidylinositol 3’-kinase/akt signal transduction pathway. Oncogene. 2000;19:4549–4552 [DOI] [PubMed] [Google Scholar]
- 63.Hakanpaa L, Sipila T, Leppanen VM, Gautam P, Nurmi H, Jacquemet G, et al. Endothelial destabilization by angiopoietin-2 via integrin beta1 activation. Nat Commun. 2015;6:5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wettschureck N, Strilic B, Offermanns S. Passing the vascular barrier: Endothelial signaling processes controlling extravasation. Physiol Rev. 2019;99:1467–1525 [DOI] [PubMed] [Google Scholar]
- 65.Albarran-Juarez J, Iring A, Wang S, Joseph S, Grimm M, Strilic B, et al. Piezo1 and gq/g11 promote endothelial inflammation depending on flow pattern and integrin activation. J Exp Med. 2018;215:2655–2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pospieszalska MK, Zarbock A, Pickard JE, Ley K. Event-tracking model of adhesion identifies load-bearing bonds in rolling leukocytes. Microcirculation. 2009;16:115–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Simons M, Gordon E, Claesson-Welsh L. Mechanisms and regulation of endothelial vegf receptor signalling. Nat Rev Mol Cell Biol. 2016;17:611–625 [DOI] [PubMed] [Google Scholar]
- 68.O’Hayre M, Degese MS, Gutkind JS. Novel insights into g protein and g protein-coupled receptor signaling in cancer. Curr Opin Cell Biol. 2014;27:126–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen X, Wu Q, Tan L, Porter D, Jager MJ, Emery C, et al. Combined pkc and mek inhibition in uveal melanoma with gnaq and gna11 mutations. Oncogene. 2014;33:4724–4734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Moore AR, Ran L, Guan Y, Sher JJ, Hitchman TD, Zhang JQ, et al. Gna11 q209l mouse model reveals rasgrp3 as an essential signaling node in uveal melanoma. Cell Rep. 2018;22:2455–2468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Boscolo E, Limaye N, Huang L, Kang KT, Soblet J, Uebelhoer M, et al. Rapamycin improves tie2-mutated venous malformation in murine model and human subjects. J Clin Invest. 2015;125:3491–3504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Limaye N, Wouters V, Uebelhoer M, Tuominen M, Wirkkala R, Mulliken JB, et al. Somatic mutations in angiopoietin receptor gene tek cause solitary and multiple sporadic venous malformations. Nat Genet. 2009;41:118–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Limaye N, Kangas J, Mendola A, Godfraind C, Schlogel MJ, Helaers R, et al. Somatic activating pik3ca mutations cause venous malformation. Am J Hum Genet. 2015;97:914–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Castillo SD, Tzouanacou E, Zaw-Thin M, Berenjeno IM, Parker VE, Chivite I, et al. Somatic activating mutations in pik3ca cause sporadic venous malformations in mice and humans. Sci Transl Med. 2016;8:332ra343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Castel P, Carmona FJ, Grego-Bessa J, Berger MF, Viale A, Anderson KV, et al. Somatic pik3ca mutations as a driver of sporadic venous malformations. Sci Transl Med. 2016;8:332ra342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Seront E, Van Damme A, Boon LM, Vikkula M. Rapamycin and treatment of venous malformations. Curr Opin Hematol. 2019;26:185–192 [DOI] [PubMed] [Google Scholar]
- 77.Crist AM, Zhou X, Garai J, Lee AR, Thoele J, Ullmer C, et al. Angiopoietin-2 inhibition rescues arteriovenous malformation in a smad4 hereditary hemorrhagic telangiectasia mouse model. Circulation. 2019;139:2049–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hashimoto T, Wu Y, Lawton MT, Yang GY, Barbaro NM, Young WL. Coexpression of angiogenic factors in brain arteriovenous malformations. Neurosurgery. 2005;56:1058–1065; discussion 1058–1065 [PubMed] [Google Scholar]
- 79.Zhou HJ, Qin L, Zhang H, Tang W, Ji W, He Y, et al. Erratum: Endothelial exocytosis of angiopoietin-2 resulting from ccm3 deficiency contributes to cerebral cavernous malformation. Nat Med. 2016;22:1502. [DOI] [PubMed] [Google Scholar]
- 80.Le Cras TD, Mobberley-Schuman PS, Broering M, Fei L, Trenor CC 3rd, Adams DM. Angiopoietins as serum biomarkers for lymphatic anomalies. Angiogenesis. 2017;20:163–173 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.