

Abstract
Anthracyclines are associated with risk of significant dose-dependent cardiotoxicity. Conventional heart failure therapies have neither ameliorated declining cardiac function nor addressed the underlying cause. Gene therapy may confer long-term cardioprotection by rendering the heart resistant to anthracyclines after 1 treatment, although the optimal therapeutic target remains to be elucidated. Recombinant adeno-associated virus is now clinically approved for the treatment of lipoprotein lipase deficiency, spinal muscular atrophy, and hereditary transthyretin amyloidosis. High-throughput methods allow selection of recombinant adeno-associated virus capsids that facilitate efficient gene delivery to specific target cells. Vector safety is enhanced by incorporating cardiac-specific promoters into vector design and localizing delivery to reduce off-target risk. Any cardioprotective transgene may bear a degree of risk as they may play as yet unknown roles, which require careful assessment using clinically relevant models. The innovative technologies outlined here make gene therapy a promising proof of principle, with potential further application to nonanthracycline chemotherapeutics.
Key Words: cardioprotection, gene therapy, recombinant adeno-associated virus vector
Abbreviations and Acronyms: AAV, adeno-associated virus; FT, functional transduction; GFP, green fluorescent protein; hiPSC-CM, human induced pluripotent stem cell derived cardiomyocyte; ITR, inverted terminal repeat; rAAV, recombinant adeno-associated virus; RC, replication-competent; SERCA2A, sarcoplasmic/endoplasmic reticulum calcium ATPase isoform 2A
Central Illustration

Highlights
-
•
Protection against anthracycline cardiotoxicity may be achieved by gene delivery to the heart.
-
•
The optimal cardioprotective target gene remains to be identified.
-
•
Targeted gene expression in human myocytes can now be achieved with advances in AAV vectorology.
-
•
It is critical to minimize risk of off-target effects which may impede anthracycline oncotherapy.
Anthracyclines are a major group of chemotherapeutic drugs used to treat a range of cancers. Although effective, long-term exposure to these anticancer drugs causes cardiotoxicity, putting cancer patients at high risk of developing heart failure and other cardiac complications later in life. Myocardial damage is dose dependent, with 9% of adult cancer patients exhibiting cardiotoxicity within the first year after completion of chemotherapy (1). Historically, a heart failure incidence of 4%-36% was reported when the dose of doxorubicin ranged from 500 mg/m2 to >600 mg/m2 (2). However, modern dosing regimens use a lower anthracycline dose of 100-385 mg/m2, which has resulted in a lower mean left ventricular ejection fraction decline (5.4%) compared with 15 years ago (9%-17%) (3). Early echocardiographic surveillance also allows stratification of patients who are at higher risk of developing cardiotoxicity, thus allowing timely intervention to address left ventricular dysfunction, particularly in childhood cancer survivors (1,4).
Current preventative strategies require frequent monitoring of heart function and use of the iron chelating agent dexrazoxane, a U.S. Food and Drug Administration–approved drug recommended by the American Society of Clinical Oncology (5). Dexrazoxane limits the formation of anthracycline-iron complexes, which are believed to cause cardiotoxicity by generating free radicals. However, dexrazoxane administration has been associated with a potential increase in the incidence of secondary primary malignancies in pediatric oncology patients, and may therefore require further evaluation and consideration as a preventative therapy (6,7).
Current treatments for anthracycline-induced cardiotoxicity are heart failure therapies such as angiotensin-converting enzyme inhibitors and β-blockers. However, a meta-analysis of cancer patients who received neurohormonal therapies in conjunction with chemotherapy demonstrated that although higher left ventricular ejection fraction was observed, the decline in LV systolic function was not dramatically improved (8). Therefore, an alternative strategy may be desirable to protect against cardiotoxicity in cancer patients.
The challenge of protecting organs from off-target toxicity by gene therapy is an established paradigm. In cancer cells, the DNA repair enzyme O6-DNA-methyl-guanine-methyl-transferase (MGMT) has been associated with high tumor grade and drug resistance (9,10). Depletion of MGMT activity using O6-benzylguanine before treatment with methylating chemotherapy drugs results in a failure to repair DNA lesions within tumor cells, thus leading to apoptotic cancer cell death (11). Although effective, this strategy inadvertently leads to toxicity in the bone marrow, where endogenous MGMT is already low (12).
Chemoprotection has been demonstrated by using gene therapy to specifically protect the bone marrow compartment from toxicity caused by brain cancer chemotherapy. A strategy was developed to facilitate delivery of a mutated gene for MGMT to the hematopoietic compartment. Due to a single amino acid substitution, the protein is rendered insensitive to depletion by O6-benzyguanine, thus protecting hematopoietic stem cells from chemotoxicity (13). Several studies have shown successful chemoprotection in preclinical models using this approach (14,15).
The advantage of gene therapy is that chemoprotection can be achieved from a single treatment by prevention of toxicity at the molecular level, rather than prolonged management of the downstream pathophysiology. Successful application of gene therapy is dependent on identifying the underlying molecular cause of toxicity. For example, sequence variations in drug transporter genes such as ABCB1, ABCB4, ABCC1 (MRP1), and ABCC2 (MRP2) identified in a subset of patients have been associated with reduction of function and subsequent increased susceptibility to cardiotoxicity (16, 17, 18). Overexpression of human cDNA for the multiple drug resistance (MDR1) gene in the hearts of transgenic mice led to cardioprotection against adriamycin (19). Transgenic mice did not exhibit pathological changes in the heart tissue, as was observed in control mice. By augmenting the drug efflux capacity of the heart, it is possible to increase resistance against anthracycline cardiotoxicity, at least in mice. However, drug transporters may not be the ideal cardioprotective target, because they are able to efflux a range of drugs, which may subsequently lose their effectiveness in patients who have received this therapeutic transgene.
One of the key challenges of developing cardioprotective strategies is to restrict delivery and expression of therapeutic genes to the heart, because inadvertent delivery to cancer cells may impede the anticancer activity of anthracyclines. The potential for unknown unintended effects in the heart must also be studied with care. This review aims to highlight emerging technology that will enable development of strategies for heart targeted delivery of protective genes to prevent cardiotoxicity during chemotherapy (Central Illustration).
Central Illustration.

Potential of Cardiac Gene Therapy to Protect Against Anthracycline Cardiotoxicity
(1) Current therapies do not provide sufficient protection against anthracyclines, and require monitoring of heart function and subsequent intervention throughout chemotherapy. (2) Adeno-associated virus (AAV) mediated gene therapy may provide a way to confer long-term cardioprotection after a single treatment, by targeted delivery of therapeutics to the heart. This can be achieved by development of new capsids with the enhanced ability for (A) cell entry and (B) gene expression in the target cells. (C) Application of the therapeutic vector can then be tested in clinically relevant small animal models to assess cardioprotection and cancer cytotoxicity.
Translation of Cardiac Gene Therapy From Bench to Bedside
Gene therapy has shown promise for targeted gene delivery in treating a variety of diseases. Over the last decade, the field of gene therapy has taken significant strides toward clinical application, driven by improvements in the vector technology and an array of promising preclinical data. Nonviral and viral vectors have been innovatively engineered, modified, and applied in both pre- and clinical settings in an endeavor to treat and prevent cardiac abnormalities (20, 21, 22, 23, 24). With advancements in vector design and delivery, which contributes to improved safety and efficacy, viral vector-based gene therapies are now cutting-edge tools at the forefront of medicine, and are being used in clinical trials for a range of diseases (25,26).
Currently, recombinant vectors based on adeno-associated virus (rAAVs) are one of the most actively investigated vectors for human gene therapy (27). The properties of rAAV that render it favorable as a gene delivery vehicle include its high safety profile, low immunogenicity, longevity of transgene expression, and distinctive capsid-dependent tissue tropism (28,29).
Wildtype AAVs were first discovered in 1965 as contaminants in an adenovirus-infected cell preparation. In its natural form, AAV is a single-stranded DNA parvovirus that requires coinfection with a helper virus to successfully replicate (30, 31, 32, 33). The AAV viral genome is ∼4.8 kb in length and is comprised of 2 genes responsible for viral replication and encoding structural protein: the rep (Replicase) and cap (Capsid). These genes are flanked by viral inverted terminal repeats (ITRs), which are the only elements required in cis for viral genome packaging and replication (Figure 1A).
Figure 1.
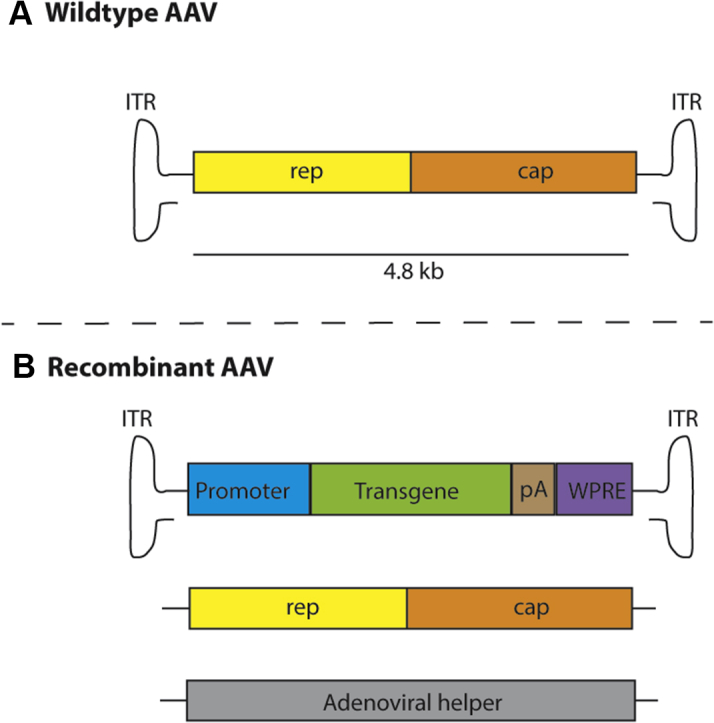
Vectorization of the Wildtype AAV Genome to Produce Recombinant AAV
(A) The 4.8kb wildtype adeno-associated virus (AAV) genome is comprised of replication (rep) and capsid (cap) genes flanked by inverted terminal repeats (ITRs). (B) Vectorization of AAV requires the expression construct containing ITRs in cis flanking the promoter, transgene, pA (polyadenylation signal) and optionally the WPRE (post-transcriptional regulatory element). Also required is a packaging plasmid and adenoviral helper plasmid which provide the required elements in trans for vector packaging.
Vectorization of AAV to produce recombinant vectors (rAAV) involves removal of the wildtype rep and cap open reading frames and replacing them with an expression cassette encoding the therapeutic transgene and all relevant regulatory elements (Figure 1B). Recombinant AAV particles are then produced in packaging cells supplemented with the required genes encoded on separate plasmids. In the most commonly used method, the packaging constructs are divided between 3 plasmids that include: 1) the rAAV genome with ITRs in cis flanking the selected therapeutic gene expression cassette; 2) the packaging plasmid delivering wildtype AAV rep and cap genes in trans; and 3) a helper plasmid encoding necessary adenovirus genes to enable vector production (28) (Figure 1B). Thus, the resulting rAAVs lack the necessary viral coding sequences required to replicate but retain the ability to deliver genomic information to the target cell.
The tropism and immunological characteristics of a rAAV vector are influenced by its capsid, the protein shell which protects the viral genome (34). To date, hundreds of capsids have been identified or generated in laboratories via bioengineering techniques. However, the first 13 naturally-occurring AAVs (AAV1-13) to be identified remain the best characterized. Cellular binding is mediated by primary receptor interactions between the capsid and the target cell surface. Slight differences in capsid amino acid sequences may therefore lead to significant changes in tropism among AAV variants. This provides a robust and extensive toolbox, which allows tailoring of molecular therapies for delivery to the target organ. A vector can be designed for optimal heart-targeted gene delivery simply by packaging the therapeutic construct into a cardiotropic capsid.
In particular, AAV9 capsid has shown efficient gene delivery to the heart when tested in mouse and rat models (35). Therefore, it is one of the more prolifically used serotypes for preclinical cardiac gene therapy studies in rodents. However, AAV1 has been shown to robustly transduce the sheep and pig myocardium, as well as outperform AAV9 in human-induced pluripotent stem cell derived cardiomyocytes (hiPSC-CMs) (36,37).
Thus far, over 200 clinical trials have been conducted using rAAV, leading to 3 market-approved gene therapies in the United States and EU for the treatment of lipoprotein lipase deficiency (Glybera, uniQure), spinal muscular atrophy (Zolgensma, Novartis), and hereditary transthyretin amyloidosis (Luxturna, Spark Therapeutics) (38, 39, 40, 41, 42). The CUPID (Calcium Up-Regulation by Percutaneous Administration of Gene Therapy in Cardiac Disease) clinical trials were the first cardiac gene therapy protocols to employ rAAV (43). In these trials, a naturally occurring cardiotropic AAV1 vector encoding sarcoplasmic/endoplasmic reticulum calcium ATPase 2A isoform (SERCA2A) administered by percutaneous intracoronary delivery was used to treat patients with heart failure. AAV1.SERCA2A exhibited an excellent safety profile as a human cardiac gene therapy vector. In the initial phase 1 trial, patients receiving the maximum dosage of 1013 total DNase-resistant particles presented with improved health outcomes compared with those administered mid- and low-range doses (44,45).
Disappointingly, however, treatment efficacy was not observed when the trial progressed to Phase 2b (45). Assessment of patient cardiac tissue showed that lack of benefit was attributed to marginal uptake of the AAV1.SERCA2a vector (<2%), approximately 1,000 times less than was achieved in preclinical models showing significant overexpression of SERCA2a in the heart (21,45). This hinted at differences in permissiveness of human and porcine cardiomyocytes to the AAV1 capsid and highlights the need for the development of human cardiac-tropic variants to enable clinical development of novel rAAV-based therapies.
The differences between preclinical models and human outcomes are not limited to cardiac gene therapy. This same phenomenon was observed in a study that clearly demonstrated capsid-mediated differences in the transduction efficiency of rAAV between human and mouse cells using a xenograft mouse model with a humanized liver (46). The AAV8 capsid, which is robustly liver tropic in rodents, was shown to transduce human hepatocytes 20 times less efficiently than mouse cells within the same liver. The clinical data obtained with AAV8 provided further evidence of significant misalignment of murine data and clinical outcomes. Therefore, there is accumulating evidence indicating that permissiveness of human cardiomyocytes to AAV transduction is not only capsid-dependent, but also fundamentally different between primary human tissue and nonhuman preclinical models. This has important ramifications for the development pipeline of cardiac gene therapeutics, in terms of capsid design and selection for tropism to human cardiomyocytes.
Methods of Selection for Cardiotropic AAV Vectors
An emerging approach to create vectors with enhanced tropism is the use of directed evolution to screen for cardiotropic AAV capsids (27). In its simplest form, this technique relies on the generation of an extensive library of replication-competent (RC) AAV virions from plasmid library containing randomly mutagenized AAV capsid gene. The capsid mutagenesis can be generated via a number of methods, ranging from DNA shuffling, error-prone polymerase chain reaction, and short peptide insertions, to targeted mutagenesis of selected residues. The final packaged library contains virions enclosing full AAV genomes, ITR-rep-cap-ITR, in which the cap gene is replaced with a unique mutated variant that corresponds to a novel capsid. Libraries can be generated at a complexity of >108 variants (47). Directed evolution allows high throughput screening of these variants for tropism, because positive selection pressure is applied by the model used and allows isolation of capsids which mediate successful cell entry.
As an additional step to improve stringency of cell targeting, negative selection pressure can also be applied to deplete variants which display unwanted characteristics (48,49) (Figure 2A). Negative selection has been successfully employed to generate vectors that can evade the immune system, by rescue of variants that retain infectivity after incubation with human sera containing anti-AAV immunoglobulins (50). It has also been demonstrated in the selection of variants that have improved targeting toward HIV-1 infected compared with uninfected H9 T cells (51).
Figure 2.

Screening AAV Variants for Efficient Entry Into Target Cardiomyocytes
(A) Negative selection by incubating adeno-associated virus (AAV) library with human sera or opposing cells. (B) Collection of AAV-containing media to use for positive selection in target cells. (C) Adenovirus (Ad) mediated replication yields enriched AAV capsid candidates.
In general, positive selection is more commonly used to screen for variants in target cells to isolate capsids that exhibit superior cell entry (Figure 2B). As outlined in the previous text, one such approach uses RC library design. Coinfection of cardiomyocytes with such RC library and human wild-type adenovirus allows for replication of AAV variants that were able to enter the cells and complete intracellular journey to the nucleus, where AAV replication takes place. Following adenovirus-induced cell lysis, the virus containing supernatant is collected and used to infect cardiomyocytes in a subsequent round of selection. Successive rounds of selection in fresh cultures of target cells allow enrichment of tropic variants, which can then be isolated and characterized by sequencing (Figure 2C).
While directed evolution using adenovirus mediated replication can efficiently enrich for cardiomyocyte-tropic AAV variants, nonspecific capsids may inadvertently be enriched if unwanted contaminating cell populations are present in the culture system. Dependency on adenovirus coinfection means that AAV selection may select for features that are not necessary for a gene delivery vector, such as efficient replication or cellular escape. It is therefore not possible to predict whether the enriched capsids would subsequently exhibit functional transduction as a packaged vector in the target cell type (52). Finally, the requirement for adenovirus coinfection limits RC approaches to cells that are permissive to adenoviral infection, significantly restricting the utility of the RC approach.
The functional transduction (FT) platform was developed as an alternative method, which allows simultaneous evaluation of cell entry and transgene expression in the target cell type based on cell sorting (53). The shuffled cap library is cloned into a construct in which a green fluorescent protein (GFP) expression cassette replaces the endogenous rep gene (Figure 3A). The library is then packaged, with rep supplied in trans, to obtain an AAV library which is subsequently used to transduce target cells. Transduction of target cells with this library will result in fluorescent labeling of successfully transduced target cells (Figure 3B). Sorting can then be performed using cell surface markers to isolate cells of interest from the GFP-positive cells (54), from which DNA can then be extracted. The cap gene can subsequently be rescued by polymerase chain reaction amplification, then cloned back into the AAV plasmid backbone for additional rounds of selection. Enrichment of cardiotropic variants would then become evident at the end of the selection process (Figure 3C). The FT platform is highly flexible in that the GFP expression cassette can be replaced with a reporter gene (fluorescent protein or drug resistance gene) under the control of a tissue-restrictive promoter, enabling selection in specific cells without the need for FACS sorting.
Figure 3.

Screening AAV Variants for Entry and Gene Expression Into Cardiomyocytes
(A) Generation of the shuffled adeno-associated virus (AAV) capsid library using the functional transduction (FT) platform. The construct contains both the shuffled cap gene and green fluorescent protein (GFP) under the control of a cardiac specific promoter (CSP). (B) Transduction of cells, followed by sorting to isolate GFP-positive cells. DNA is then extracted and the cap gene recovered. The cap gene is then recloned back into the library and taken for further rounds of sequencing. (C) Enriched AAV capsid candidates at the end of successive rounds of selection. hiPSC-CM = human-induced pluripotent stem cell derived cardiomyocyte; ITR = inverted terminal repeat; PCR = polymerase chain reaction
Although functional transduction requires a longer period of time compared with adenovirus-mediated replication because of the need for cloning steps between each round of selection, it provides an alternative method of capsid selection that is independent of adenovirus-mediated replication, and allows evaluation of both cell entry and gene expression in target cells.
Although each selection method has its advantages and limitations, the choice of selection model used is critical to the successful identification of cardiotropic variants. Because AAV transduction is receptor-mediated, the selection should ideally be performed in the cell type ultimately targeted for therapeutic gene delivery.
Preclinical Models for Testing Gene Transfer to Human Cardiomyocytes
Successful translation of novel gene therapeutics to clinical application is difficult, and preclinical assessment should ideally be done using the most biologically relevant model systems. In choosing models, whether for generation or functional validation of gene therapeutics, a balance of tissue availability, feasibility, and clinical relevance is required. The most clinically relevant model would be to use an intact human heart as this would retain the architecture and heterogeneity of the functional organ. Although it is possible to assess gene delivery ex vivo, as proven in a porcine model of ex vivo adenoviral transduction, it is logistically difficult to maintain the heart, for extended periods of time, under normothermic conditions without the use of specialized equipment that is not readily available (35). Intact human hearts would also be difficult to obtain because these would most likely be prioritized for transplantation, and it would be even more difficult to obtain a whole organ from the target population of interest (Figure 4). An additional challenge would also be the large amount of vector required to transduce the whole organ.
Figure 4.

Models for Assessing Clinical Utility of Cardiac Gene Therapeutics
Pros and cons of each model system as represented by sliding scales, showing a larger value on the wider end and a smaller value on the narrow end. Colors represent favorability of each characteristic, with green being most favorable, and red being the least favorable. hiPSC-CM = human-induced pluripotent stem cell derived cardiomyocyte; rAAV = recombinant adeno-associated virus.
It is also possible to obtain primary ventricular tissue for generating organotypic slices, which can be cultured for up to 5 days for transduction experiments with rAAV (55). This is highly desirable because capsid-dependent gene transfer can be tested using primary cardiac tissue of human origin. The tissue slices retain the cellular complexity of the whole organ, although structural integrity of the intact organ is sacrificed. However, as with the whole heart, it may not be possible to readily source representative heart samples from the same population demographics as the target patients (Figure 4).
Cardiomyocytes derived from hiPSCs may provide an alternative to primary tissues. A stem cell line can be generated by first reprogramming isolated somatic cells (eg, fibroblasts, PBMCs) from patients, then using established protocols to differentiate the multipotent cells into cardiomyocytes (56,57). Due to ready availability of the cells, it would then be possible to establish high throughput workflows to screen for cardiotropic therapeutics (Figure 4). Inherently, the most obvious risk of using hiPSC-CM as the selection model is that these cells are not primary, and their gene expression profile is not identical to bona fide human cardiomyocytes (58). Thus, hiPSC-CM may express receptors that are not present in primary cardiomyocytes, and may therefore inadvertently drive selection of variants that are ultimately unable to bind to primary human myocardium.
Another risk of using hiPSC-CM for positive selection is that there will always be a noncardiomyocyte fraction that may contaminate the target cardiomyocyte population. Thus, it would not be possible to ascertain whether enriched variants were solely cardiomyocyte derived. To address this, it would be necessary to eliminate nontarget cells from the culture. In mammalian cells, glucose is a major source of energy, but fetal cardiomyocytes are also able to use lactate as an alternative (59). By making use of this metabolic pathway, purity of differentiated cells can be enriched by exposure to lactate supplemented glucose depleted culture media (60)
An alternative method would be to label cells with a selectable marker, which would enable temporal control over selection of cells of interest using an antibiotic selection cassette. Although potentially more effective than metabolic selection, this would require modification of cells prior to differentiation, which may subsequently interfere with cardiomyocyte formation.
It is also important to select candidate vectors using cardiomyocytes with a phenotype that is relevant to the target patient population, because maturity can significantly affect transduction efficiency of each AAV variant (61). It is hypothesized that hiPSC-CMs retain a fetal phenotype partly because of preferential use of glycolysis for energy metabolism, as opposed to β-oxidation of fatty acids in mature adult cardiomyocytes (59,62). To model patients from adult populations, maturation of hiPSC-CM can be induced by supplementing culture media with physiological concentrations of the fatty acids palmitic, oleic, and linoleic acid (63). This maturation process is designed to mimic the transition from glycolytic to oxidative metabolism in the developing heart and can therefore be used to generate hiPSC-CM that are closer to an adult phenotype.
High Throughput Screening of AAV Variants to Compare Transduction Efficiencies
Depending on the complexity of the AAV library and the selection model used, the number of enriched variants that will need to be vectorized and validated may vary depending on the stringency of selection. As the number of candidate variants increases, so does the workload, cost, and time required to select the final AAV variant for preclinical and clinical studies. One approach to perform comparison of multiple vectors is to transduce target cell population at the same multiplicity of transduction (MOT) with each of the variants encoding a fluorescent marker and to quantify the number of positive cells using fluorescent readout. Although straightforward, this approach becomes very time consuming and suffers from lower accuracy when large number of vectors are to be tested. To address this technical limitation, a new approach that makes use of next-generation sequencing for high-throughput screening and analysis of rAAV variants for both efficient target cell entry and induction of gene expression has been recently developed and validated (64,65).
Identical AAV cassettes containing green fluorescent protein (GFP) are designed to include short DNA sequences known as a barcode. These barcoded vectors are packaged into multiple AAV vector capsid variants and mixed in equimolar ratio to obtain a mix of packaged AAVs, in which each capsid is uniquely labeled at the DNA level (Figure 5A). The AAV mix is then used to transduce cells of interest, which can subsequently be sorted based on GFP expression (Figure 5B). DNA and RNA can be isolated from the successfully transduced target cells. Analysis of the relative prevalence of barcode sequences from the extracted DNA provides subsequent insight into the efficiency of cell entry for each variant, whereas the RNA/cDNA shows the efficiency of gene expression (Figure 5C). Therefore, variants that show high vector genome copy numbers at both DNA and RNA/cDNA levels are likely to be highly efficient at gene delivery to the cell target.
Figure 5.

Select AAV Variants With Cardiomyocyte Targeted Entry and Gene Expression
(A) An adeno-associated virus (AAV) kit is generated by mixing green fluorescent protein (GFP) expressing vectors with unique barcoding sequence (BC) and optional woodchuck hepatitis virus post-transcriptional regulatory element (WPRE) for each capsid variant. (B) The barcoded AAV library is used to transduce target cells, followed by sorting to isolate GFP-positive target cells only. Nucleic acids (DNA and RNA) are then extracted. (C) Next-generation sequencing (NGS) is then utilized to detect and quantify barcode sequences.
Optimizing Safety by Pre-Emptive Risk Mitigation
The efficiency of gene delivery to the heart greatly affects the dose required for cardioprotective efficacy. This is an important consideration, because nonhuman primates injected systemically with high doses of AAV9 (2 × 1014 GC/kg) were reported to exhibit acute systemic inflammation, coagulation defects, and hepatic toxicity (66). Toxicity was hypothesized to have resulted from hepatocyte damage and activation of systemic inflammation. Although the molecular mechanism is still unclear, the severity of toxicity is clearly dose-dependent. Therefore, by identifying high-performing AAV capsids with optimized cardiomyocyte entry and gene expression in the heart, lower vector doses may be sufficient to reach a therapeutic threshold.
It is also important to screen all vectors for liver transduction because the liver is the natural target for most AAVs (67,68). A liver-specific enhancer-promoter element exists in the 3’ untranslated region between the cap gene and the right hand ITR (Figure 1), which is evidence of a direct evolutionary link between AAV and human liver and provides at least a partial explanation of why AAVs naturally transduce human liver with high efficiency. Therefore, if 2 capsids are otherwise identical in function, clinical development should be granted to the variant with lower liver transduction, to reduce the risk of liver toxicity.
In striving to protect the heart, care must be taken to avoid interference with anthracycline-mediated cancer killing brought on by inadvertent delivery to tumors. Despite stringent selection for cardiotropism, it is not possible to guarantee that the candidate capsid is completely detargeted from tumors, because they are comprised of a heterogeneous population of cancer cells.
Systemic injection of a vector that exhibits homing to the heart would be the least invasive. However, to reduce the risk of gene transfer to cancer cells, the route of vector administration can also be restricted to localize vector delivery. Direct intramyocardial injection can achieve strong gene expression, although this method is relatively more invasive and may not achieve global transduction of the heart because of the focal nature of gene delivery (69). Percutaneous intracoronary injection of AAV in a porcine model was shown to induce strong transgene expression in the heart, with only low levels detectable in the liver (70). This route is minimally invasive, and therefore, would be more clinically applicable.
As a secondary strategy, cardiac muscle-specific promoters can be incorporated into the rAAV vector cassette, such that gene expression is activated in cardiomyocytes only. Proof-of-concept studies have shown that the cardiac sodium-calcium exchanger (NCX1), ventricle-specific myosin light chain-2 (MLC 2-v) promoters can induce targeted expression restricted to cardiomyocytes (22,71,72). By incorporating cell-specific promoters into the vector design, the risk of off-target gene expression is further reduced.
Validation of Novel Therapeutics in a Functioning Heart
Cell culture models allow high-throughput screening of capsid transduction performance and subsequent functional validation of novel AAV variants. However, they do not recapitulate the complex architecture and physiological barriers presented by a whole organ. Therefore, verification of treatment efficacy would require both a functioning whole heart and a quantifiable tumor burden in vivo.
Small animal tumor models have been developed for preclinical validation of novel therapeutics, based on the generation of orthotopic tumors in rodents. An immunodeficient rodent could first be engrafted with human tumor cells (Figure 6). Once the cancer cells are seeded (eg, mammary fat pad to simulate breast cancer) to form the tumor, gene delivery to the heart can then be achieved by tail vein infusion (20). By testing for efficient cardiac gene transfer in vivo, it is then possible to determine whether there are off-target effects related to both the route of administration and inadvertent nonspecific transgene expression. This is vital in ensuring that the putative cardioprotective vector will not interfere with cancer killing activity of anthracyclines.
Figure 6.

Dual-Targeted Raav for Cardioprotective Chemotherapy
Sequence of events for treatment of a hypothetical breast cancer preclinical model. Tumor burden will be established in a rodent model, followed by pretreatment with the cardiac-targeted adeno-associated virus (AAV) to deliver a cardioprotective gene to the heart. The animal model will then be treated with anthracycline at a cardiotoxic dose. Control animals that receive only vehicle or empty vector should exhibit left ventricle dysfunction, whereas animals that receive the therapeutic vector should retain normal heart function while the tumor is eliminated. PBS = phosphate buffered saline.
Limitations of Current Technology and Future Considerations for Vector Development
In this review, we have focused on cardioprotection against anthracyclines as a proof of principle for cardiac gene therapy. This illustrates how cell-targeted therapies can confer protection without interfering with the activity of anticancer drugs. However, cardiac gene therapy is not limited to cardiomyocyte-targeted delivery. Other investigators have pursued targeting the toxic effects of doxorubicin in endothelial cells. An AAV9 vector encoding vascular endothelial growth factor-B was given as a prophylactic therapy to mice 7 days before commencement of doxorubicin treatment (73). This study was promising, because the therapy was shown to reduce doxorubicin-induced cardiac atrophy and whole body wasting. Depending on the molecular strategy, this gene transfer technology could also be adapted to protect against other cardiotoxic anticancer drugs, such as trastuzumab (74). However, careful consideration must be given to the disease-causing mechanism, timing of pathogenesis, and target patient population.
Gene therapy is suited for application to monogenic diseases, because gene addition can restore normal protein levels in defective cells. Gene editing can also be used to directly restore defects in the genes directly. As previously described, sequence variations in drug transporter genes may lead to reduction of efflux function and increased susceptibility to cardiotoxicity (16,17). These patients may therefore benefit from gene correction, which would restore normal transporter function and potentially augment natural resistance to anthracycline cardiotoxicity.
However, we also acknowledge the limitation of lack of specificity regarding efflux of anthracyclines by drug transporters. They may also efflux nonanthracycline cardiac drugs such as statins, which may affect the efficacy of treatment for nonmalignant diseases (75). Drug transporters such as MRP1 may also efflux glutathione conjugates, which may have as yet unknown biological consequences for redox balance within cells with supraphysiological transporter activity (76,77). The long-term potential for side effects of the gene therapy must therefore be considered together with initial cardioprotection during chemotherapy.
Although various cardioprotective genes have been explored in preclinical models, our knowledge of what genes would be most suited for protection against anthracycline-induced cardiotoxicity in humans are as yet unknown. Regardless of the gene that is ultimately used, the long-term risks of vector induced gene expression in the heart need to be acknowledged and screened.
One way to address this limitation is to design an AAV vector that allows transgene expression to be switched off at completion of chemotherapy. A unique “Cre-off” method was tested for switching off transgene expression in the mouse brain in vivo (78). By creating a vector where the open reading frame is flanked by lox sites, it becomes possible to regulate gene expression to ensure that the transgene is not expressed in cells that also express Cre recombinase (Cre). Using this system, it could be possible to switch off the protective transgene expression in a temporally controlled manner by transient induction of Cre expression. To our knowledge, this has not been tested in the context of cardiac gene therapy and would be a novel method of reducing the risk of long-term expression of cardioprotective targets. However, there is then the challenge of not only specifically delivering Cre recombinase to the heart, but also to do so in a strictly transient manner so that neither the Cre nor the original therapeutic transgene will be expressed at the end of cancer therapy.
The rapid evolution of rAAV development may result in vectors that can incorporate both efficiency of cardiac targeting and also mitigation of long-term risks of permanent transgene expression.
Conclusions
Although application of AAV-mediated gene therapy for cardioprotection is still in its research infancy, it will be exciting to see how emerging vector technologies will guide the development of novel therapies. Improvements in AAV vectorology and big data analysis have enabled innovative evolution of this promising gene delivery system to maximize gene delivery and transduction efficiency in target cells. The versatility of this technology allows cardiotropic delivery of any gene that is within the packaging size constraints of the AAV genome. Accumulating evidence of key differences between available preclinical models has highlighted the importance of testing therapies in relevant cell types. Being able to tailor therapies to target the heart while minimizing off-target effects will improve the safety of AAV vectors and increase the feasibility of its use in cardioprotection.
Funding Support and Author Disclosures
Dr Kok is funded by NHMRC Ideas grant APP1184929. Miss Maclean is funded by the Yim Family Foundation Scholarship. Mr Ho was supported by the University of Sydney Honours Scholarship. Assoc Prof Lisowski is supported by NHMRC Project Grants APP1156431 and APP1161583; is supported by a grant from the National Science Centre, Republic of Poland (OPUS 13) (UMO-2017/25/B/NZ1/02790); is a scientific cofounder and holds stocks in LogicBio Therapeutics; is a cofounder and holds shares of Exigen Biotherapeutics, IGS Therapeutics, and Orphinic Scientific; and served as a consultant for AAV technologies discussed in this review. Assoc Prof Kizana is funded by NHMRC Ideas Grants APP1184929 and APP1188348 and by the NSW Health–Cardiovascular Research Capacity Program.
Acknowledgments
Dr Kok thanks Prof Christopher Semsarian for his invaluable advice and mentorship through the Franklin Women Mentoring Program (2020). The figures were drawn by Dr Kok using Adobe Illustrator.
Footnotes
The authors attest they are in compliance with human studies committees and animal welfare regulations of the authors’ institutions and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the Author Center.
References
- 1.Cardinale D., Colombo A., Bacchiani G., et al. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation. 2015;131(22):1981–1988. doi: 10.1161/CIRCULATIONAHA.114.013777. [DOI] [PubMed] [Google Scholar]
- 2.Lefrak E.A., Pitha J., Rosenheim S., Gottlieb J.A. A clinicopathologic analysis of adriamycin cardiotoxicity. Cancer. 1973;32(2):302–314. doi: 10.1002/1097-0142(197308)32:2<302::aid-cncr2820320205>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 3.Jeyaprakash P., Sangha S., Ellenberger K., Sivapathan S., Pathan F., Negishi K. Cardiotoxic effect of modern anthracycline dosing on left ventricular ejection fraction: a systematic review and metaanalysis of placebo arms from randomized controlled trials. J Am Heart Assoc. 2021;10(6) doi: 10.1161/JAHA.120.018802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leerink J.M., van der Pal H.J.H., Kremer L.C.M., et al. Refining the 10-year prediction of left ventricular systolic dysfunction in long-term survivors of childhood cancer. J Am Coll Cardiol CardioOnc. 2021;3(1):62–72. doi: 10.1016/j.jaccao.2020.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Armenian S.H., Lacchetti C., Barac A., et al. Prevention and monitoring of cardiac dysfunction in survivors of adult cancers: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol. 2017;35(8):893–911. doi: 10.1200/JCO.2016.70.5400. [DOI] [PubMed] [Google Scholar]
- 6.Tebbi C.K., London W.B., Friedman D., et al. Dexrazoxane-associated risk for acute myeloid leukemia/myelodysplastic syndrome and other secondary malignancies in pediatric Hodgkin's disease. J Clin Oncol. 2007;25(5):493–500. doi: 10.1200/JCO.2005.02.3879. [DOI] [PubMed] [Google Scholar]
- 7.Salzer W.L., Devidas M., Carroll W.L., et al. Long-term results of the pediatric oncology group studies for childhood acute lymphoblastic leukemia 1984-2001: a report from the children's oncology group. Leukemia. 2010;24(2):355–370. doi: 10.1038/leu.2009.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vaduganathan M., Hirji Sameer A., Qamar A., et al. Efficacy of neurohormonal therapies in preventing cardiotoxicity in patients with cancer undergoing chemotherapy. J Am Coll Cardiol CardioOnc. 2019;1(1):54–65. doi: 10.1016/j.jaccao.2019.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brent T.P., Houghton P.J., Houghton J.A. O6-Alkylguanine-DNA alkyltransferase activity correlates with the therapeutic response of human rhabdomyosarcoma xenografts to 1-(2-chloroethyl)-3-(trans-4-methylcyclohexyl)-1-nitrosourea. Proc Natl Acad Sci U S A. 1985;82(9):2985–2989. doi: 10.1073/pnas.82.9.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bobola M.S., Berger M.S., Ellenbogen R.G., Roberts T.S., Geyer J.R., Silber J.R. O6-methylguanine-DNA methyltransferase in pediatric primary brain tumors: relation to patient and tumor characteristics. Clin Cancer Res. 2001;7(3):613–619. [PubMed] [Google Scholar]
- 11.Gerson S.L. Clinical relevance of MGMT in the treatment of cancer. J Clin Oncol. 2002;20(9):2388–2399. doi: 10.1200/JCO.2002.06.110. [DOI] [PubMed] [Google Scholar]
- 12.Gerson S.L., Phillips W., Kastan M., Dumenco L.L., Donovan C. Human CD34+ hematopoietic progenitors have low, cytokine-unresponsive O6-alkylguanine-DNA alkyltransferase and are sensitive to O6-benzylguanine plus BCNU. Blood. 1996;88(5):1649–1655. [PubMed] [Google Scholar]
- 13.Xu-Welliver M., Kanugula S., Pegg A.E. Isolation of human-alkylguanine-DNA alkyltransferase mutants highly resistant to inactivation by -benzylguanine. Cancer Research. 1998;58(9):1936. [PubMed] [Google Scholar]
- 14.Kramer B.A., Lemckert F.A., Alexander I.E., Gunning P.W., McCowage G.B. Characterisation of a P140K mutant O6-methylguanine-DNA-methyltransferase (MGMT)-expressing transgenic mouse line with drug-selectable bone marrow. J Gene Med. 2006;8(9):1071–1085. doi: 10.1002/jgm.937. [DOI] [PubMed] [Google Scholar]
- 15.Jansen M., Sorg U.R., Ragg S., et al. Hematoprotection and enrichment of transduced cells in vivo after gene transfer of MGMT(P140K) into hematopoietic stem cells. Cancer Gene Ther. 2002;9(9):737–746. doi: 10.1038/sj.cgt.7700490. [DOI] [PubMed] [Google Scholar]
- 16.Visscher H., Ross C.J., Rassekh S.R., et al. Pharmacogenomic prediction of anthracycline-induced cardiotoxicity in children. J Clin Oncol. 2012;30(13):1422–1428. doi: 10.1200/JCO.2010.34.3467. [DOI] [PubMed] [Google Scholar]
- 17.Wojnowski L., Kulle B., Schirmer M., et al. NAD(P)H oxidase and multidrug resistance protein genetic polymorphisms are associated with doxorubicin-induced cardiotoxicity. Circulation. 2005;112(24):3754–3762. doi: 10.1161/CIRCULATIONAHA.105.576850. [DOI] [PubMed] [Google Scholar]
- 18.Bhatia S. Genetics of anthracycline cardiomyopathy in cancer survivors. J Am Coll Cardiol CardioOnc. 2020;2(4):539–552. doi: 10.1016/j.jaccao.2020.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dell'Acqua G., Polishchuck R., Fallon J.T., Gordon J.W. Cardiac resistance to adriamycin in transgenic mice expressing a rat alpha-cardiac myosin heavy chain/human multiple drug resistance 1 fusion gene. Human Gene Therapy. 1999;10(8):1269–1279. doi: 10.1089/10430349950017950. [DOI] [PubMed] [Google Scholar]
- 20.Inagaki K., Fuess S., Storm T.A., et al. Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol Ther. 2006;14(1):45–53. doi: 10.1016/j.ymthe.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greenberg B., Butler J., Felker G.M., et al. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet. 2016;387(10024):1178–1186. doi: 10.1016/S0140-6736(16)00082-9. [DOI] [PubMed] [Google Scholar]
- 22.Konkalmatt P.R., Beyers R.J., O'Connor D.M., Xu Y., Seaman M.E., French B.A. Cardiac-selective expression of extracellular superoxide dismutase after systemic injection of adeno-associated virus 9 protects the heart against post-myocardial infarction left ventricular remodeling. Circ Cardiovasc Imaging. 2013;6(3):478–486. doi: 10.1161/CIRCIMAGING.112.000320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kho C., Lee A., Jeong D., et al. SUMO1-dependent modulation of SERCA2a in heart failure. Nature. 2011;477(7366):601–605. doi: 10.1038/nature10407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zangi L., Lui K.O., von Gise A., et al. Modified mRNA directs the fate of heart progenitor cells and induces vascular regeneration after myocardial infarction. Nat Biotechnol. 2013;31(10):898–907. doi: 10.1038/nbt.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Russell S., Bennett J., Wellman J.A., et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390(10097):849–860. doi: 10.1016/S0140-6736(17)31868-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lowes L.P., Alfano L.N., Arnold W.D., et al. Impact of age and motor function in a phase 1/2A study of infants with SMA type 1 receiving single-dose gene replacement therapy. Pediatr Neurol. 2019;98:39–45. doi: 10.1016/j.pediatrneurol.2019.05.005. [DOI] [PubMed] [Google Scholar]
- 27.Kok C.Y., Alexander I., Lisowski L., Kizana E. Directed evolution of adeno-associated virus vectors in human cardiomyocytes for cardiac gene therapy. Heart, Lung and Circulation. 2018;27(11):1270–1273. doi: 10.1016/j.hlc.2018.08.014. [DOI] [PubMed] [Google Scholar]
- 28.Samulski RJ Muzyczka N. AAV-mediated gene therapy for research and therapeutic purposes. Annu Rev Virol. 2014;1(1):427–451. doi: 10.1146/annurev-virology-031413-085355. [DOI] [PubMed] [Google Scholar]
- 29.Gushchina L.V., Frair E.C., Rohan N.L., et al. Lack of toxicity in non-human primates receiving clinically relevant doses of an AAV9.U7snRNA vector designed to induce DMD exon 2 skipping. Hum Gene Ther. 2021;32(17-18):882–894. doi: 10.1089/hum.2020.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Atchison R., Casto B., Hammon W. Adenovirus-associated defective virus particles. Science. 1965;149(3685):754–756. doi: 10.1126/science.149.3685.754. [DOI] [PubMed] [Google Scholar]
- 31.Buller R.M., Janik J.E., Sebring E.D., Rose J.A. Herpes simplex virus types 1 and 2 completely help adenovirus-associated virus replication. J Virol. 1981;40(1):241–247. doi: 10.1128/jvi.40.1.241-247.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Georg-Fries B., Biederlack S., Wolf J., zur Hausen H. Analysis of proteins, helper dependence, and seroepidemiology of a new human parvovirus. Virology. 1984;134(1):64–71. doi: 10.1016/0042-6822(84)90272-1. [DOI] [PubMed] [Google Scholar]
- 33.McPherson R.A., Rosenthal L.J., Rose J.A. Human cytomegalovirus completely helps adeno-associated virus replication. Virology. 1985;147(1):217–222. doi: 10.1016/0042-6822(85)90243-0. [DOI] [PubMed] [Google Scholar]
- 34.Buning H., Srivastava A. Capsid modifications for targeting and improving the efficacy of AAV vectors. Mol Ther Methods Clin Dev. 2019;12:248–265. doi: 10.1016/j.omtm.2019.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bishawi M., Roan J.-N., Milano C.A., et al. A normothermic ex vivo organ perfusion delivery method for cardiac transplantation gene therapy. Scientific Reports. 2019;9(1):8029. doi: 10.1038/s41598-019-43737-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Asokan A., Samulski R.J. An emerging adeno-associated viral vector pipeline for cardiac gene therapy. Hum Gene Ther. 2013;24(11):906–913. doi: 10.1089/hum.2013.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ambrosi C.M., Sadananda G., Han J.L., Entcheva E. Adeno-associated virus mediated gene delivery: implications for scalable in vitro and in vivo cardiac optogenetic models. Frontiers in Physiology. 2019;10:168. doi: 10.3389/fphys.2019.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oh J.G., Ishikawa K. Recent highlights and advances in cardiac gene therapy. Discov Med. 2019;28(155):229–235. [PMC free article] [PubMed] [Google Scholar]
- 39.Büning H. Gene therapy enters the pharma market: the short story of a long journey. EMBO Molecular Medicine. 2013;5(1):1–3. doi: 10.1002/emmm.201202291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoy S.M. Onasemnogene abeparvovec: first global approval. Drugs. 2019;79(11):1255–1262. doi: 10.1007/s40265-019-01162-5. [DOI] [PubMed] [Google Scholar]
- 41.First systemic CRISPR agent in humans. Nature Biotechnology. 2020;38(12):1364. doi: 10.1038/s41587-020-00773-8. [DOI] [PubMed] [Google Scholar]
- 42.Stroes E.S., Nierman M.C., Meulenberg J.J., et al. Intramuscular administration of AAV1-lipoprotein lipase S447X lowers triglycerides in lipoprotein lipase-deficient patients. Arterioscler Thromb Vasc Biol. 2008;28(12):2303–2304. doi: 10.1161/ATVBAHA.108.175620. [DOI] [PubMed] [Google Scholar]
- 43.Gabisonia K., Recchia F.A. Gene therapy for heart failure: new perspectives. Curr Heart Fail Rep. 2018;15(6):340–349. doi: 10.1007/s11897-018-0410-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jaski B.E., Jessup M.L., Mancini D.M., et al. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J Card Fail. 2009;15(3):171–181. doi: 10.1016/j.cardfail.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jessup M., Greenberg B., Mancini D., et al. Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation. 2011;124(3):304–313. doi: 10.1161/CIRCULATIONAHA.111.022889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lisowski L., Dane A.P., Chu K., et al. Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature. 2014;506(7488):382–386. doi: 10.1038/nature12875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Varadi K., Michelfelder S., Korff T., et al. Novel random peptide libraries displayed on AAV serotype 9 for selection of endothelial cell-directed gene transfer vectors. Gene Therapy. 2012;19(8):800–809. doi: 10.1038/gt.2011.143. [DOI] [PubMed] [Google Scholar]
- 48.Perabo L., Büning H., Kofler D.M., et al. In vitro selection of viral vectors with modified tropism: the adeno-associated virus display. Molecular Therapy. 2003;8(1):151–157. doi: 10.1016/s1525-0016(03)00123-0. [DOI] [PubMed] [Google Scholar]
- 49.Bartel M.A., Weinstein J.R., Schaffer D.V. Directed evolution of novel adeno-associated viruses for therapeutic gene delivery. Gene Therapy. 2012;19(6):694–700. doi: 10.1038/gt.2012.20. [DOI] [PubMed] [Google Scholar]
- 50.Grimm D., Lee J.S., Wang L., et al. In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J Virol. 2008;82(12):5887–5911. doi: 10.1128/JVI.00254-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wooley D.P., Sharma P., Weinstein J.R., Kotha Lakshmi Narayan P., Schaffer D.V., Excoffon K.J.D.A. A directed evolution approach to select for novel Adeno-associated virus capsids on an HIV-1 producer T cell line. J Virol Methods. 2017;250:47–54. doi: 10.1016/j.jviromet.2017.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Westhaus A., Cabanes-Creus M., Rybicki A., et al. High-throughput in vitro, ex vivo, and in vivo screen of adeno-associated virus vectors based on physical and functional transduction. Human Gene Therapy. 2020;31(9-10):575–589. doi: 10.1089/hum.2019.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cabanes-Creus M., Westhaus A., Navarro R.G., et al. Attenuation of heparan sulfate proteoglycan binding enhances in vivo transduction of human primary hepatocytes with AAV2. Mol Ther Methods Clin Dev. 2020;17:1139–1154. doi: 10.1016/j.omtm.2020.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Skelton R.J.P., Costa M., Anderson D.J., et al. SIRPA, VCAM1 and CD34 identify discrete lineages during early human cardiovascular development. Stem Cell Res. 2014;13(1):172–179. doi: 10.1016/j.scr.2014.04.016. [DOI] [PubMed] [Google Scholar]
- 55.Liu Z., Klose K., Neuber S., Jiang M., Gossen M., Stamm C. Comparative analysis of adeno-associated virus serotypes for gene transfer in organotypic heart slices. J Transl Med. 2020;18(1):437. doi: 10.1186/s12967-020-02605-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takahashi K., Tanabe K., Ohnuki M., et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 57.Zwi L., Caspi O., Arbel G., et al. Cardiomyocyte differentiation of human induced pluripotent stem cells. Circulation. 2009;120(15):1513–1523. doi: 10.1161/CIRCULATIONAHA.109.868885. [DOI] [PubMed] [Google Scholar]
- 58.Pavlovic B.J., Blake L.E., Roux J., Chavarria C., Gilad Y. A comparative assessment of human and chimpanzee iPSC-derived cardiomyocytes with primary heart tissues. Sci Rep. 2018;8(1):15312. doi: 10.1038/s41598-018-33478-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fisher D.J., Heymann M.A., Rudolph A.M. Myocardial consumption of oxygen and carbohydrates in newborn sheep. Pediatr Res. 1981;15(5):843–846. doi: 10.1203/00006450-198105000-00003. [DOI] [PubMed] [Google Scholar]
- 60.Tohyama S., Hattori F., Sano M., et al. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell. 2013;12(1):127–137. doi: 10.1016/j.stem.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 61.Rossi A., Dupaty L., Aillot L., et al. Vector uncoating limits adeno-associated viral vector-mediated transduction of human dendritic cells and vector immunogenicity. Sci Rep. 2019;9(1) doi: 10.1038/s41598-019-40071-1. 3631-3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang X., Pabon L., Murry Charles E. Engineering adolescence. Circ Res. 2014;114(3):511–523. doi: 10.1161/CIRCRESAHA.114.300558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang X., Rodriguez M.L., Leonard A., et al. Fatty acids enhance the maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cell Rep. 2019;13(4):657–668. doi: 10.1016/j.stemcr.2019.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Adachi K., Enoki T., Kawano Y., Veraz M., Nakai H. Drawing a high-resolution functional map of adeno-associated virus capsid by massively parallel sequencing. Nat Commun. 2014;5:3075. doi: 10.1038/ncomms4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marsic D., Mendez-Gomez H.R., Zolotukhin S. High-accuracy biodistribution analysis of adeno-associated virus variants by double barcode sequencing. Mol Ther Methods Clin Dev. 2015;2:15041. doi: 10.1038/mtm.2015.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hinderer C., Katz N., Buza E.L., et al. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum Gene Ther. 2018;29(3):285–298. doi: 10.1089/hum.2018.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Logan G.J., Dane A.P., Hallwirth C.V., et al. Identification of liver-specific enhancer–promoter activity in the 3′ untranslated region of the wild-type AAV2 genome. Nat Gen. 2017;49(8):1267–1273. doi: 10.1038/ng.3893. [DOI] [PubMed] [Google Scholar]
- 68.Cabanes-Creus M., Hallwirth C.V., Westhaus A., et al. Restoring the natural tropism of AAV2 vectors for human liver. Sci Transl Med. 2020;12(560) doi: 10.1126/scitranslmed.aba3312. [DOI] [PubMed] [Google Scholar]
- 69.Prasad K.-M.R., Smith R.S., Xu Y., French B.A. A single direct injection into the left ventricular wall of an adeno-associated virus 9 (AAV9) vector expressing extracellular superoxide dismutase from the cardiac troponin-T promoter protects mice against myocardial infarction. J Gene Med. 2011;13(6):333–341. doi: 10.1002/jgm.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kaspar B.K., Roth D.M., Lai N.C., et al. Myocardial gene transfer and long-term expression following intracoronary delivery of adeno-associated virus. J Gene Med. 2005;7(3):316–324. doi: 10.1002/jgm.665. [DOI] [PubMed] [Google Scholar]
- 71.Barth A.S., Kizana E., Smith R.R., et al. Lentiviral vectors bearing the cardiac promoter of the Na+-Ca2+ exchanger report cardiogenic differentiation in stem cells. Mol Ther. 2008;16(5):957–964. doi: 10.1038/mt.2008.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Müller O.J., Leuchs B., Pleger S.T., et al. Improved cardiac gene transfer by transcriptional and transductional targeting of adeno-associated viral vectors. Cardiovasc Res. 2006;70(1):70–78. doi: 10.1016/j.cardiores.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 73.Räsänen M., Degerman J., Nissinen T.A., et al. VEGF-B gene therapy inhibits doxorubicin-induced cardiotoxicity by endothelial protection. Proc Natl Acad Sci U S A. 2016;113(46):13144–13149. doi: 10.1073/pnas.1616168113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ayres L.R., de Almeida Campos M.S., de Oliveira Gozzo T., et al. Trastuzumab induced cardiotoxicity in HER2 positive breast cancer patients attended in a tertiary hospital. Int J Clin Pharm. 2015;37(2):365–372. doi: 10.1007/s11096-015-0070-y. [DOI] [PubMed] [Google Scholar]
- 75.Knauer M.J., Urquhart B.L., Schwabedissen HEMz, et al. Human skeletal muscle drug transporters determine local exposure and toxicity of statins. Circ Res. 2010;106(2):297–306. doi: 10.1161/CIRCRESAHA.109.203596. [DOI] [PubMed] [Google Scholar]
- 76.Leier I., Jedlitschky G., Buchholz U., Cole S.P., Deeley R.G., Keppler D. The MRP gene encodes an ATP-dependent export pump for leukotriene C4 and structurally related conjugates. J Biol Chem. 1994;269(45):27807–27810. [PubMed] [Google Scholar]
- 77.Cole S.P., Deeley R.G. Transport of glutathione and glutathione conjugates by MRP1. Trends Pharmacol Sci. 2006;27(8):438–446. doi: 10.1016/j.tips.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 78.Saunders A., Johnson C.A., Sabatini B.L. Novel recombinant adeno-associated viruses for Cre activated and inactivated transgene expression in neurons. Front Neural Circuits. 2012;6:47. doi: 10.3389/fncir.2012.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]