

Abstract
Purposes: Recent studies have suggested that loss-of-function mutations of the tunica intima endothelial receptor tyrosine kinase (TEK) are responsible for approximately 5% of primary congenital glaucoma (PCG) cases in diverse populations. However, the causative role of TEK mutations has not been studied in Chinese PCG patients. Here, we report the mutation spectrum of TEK after screening a large cohort of PCG patients of Chinese Han origin and analyze the identified variants in functional assays.
Methods: TEK-targeted next-generation sequencing (NGS) was performed in 200 PCG patients. Candidate variants were prioritized by mutation type and allele frequency in public datasets. Plasmids containing wild type and identified variants of TEK were constructed and used to assess protein expression, solubility, receptor auto-phosphorylation, and response to ligand stimulation in cell-based assays.
Results: Ten missense and one nonsense heterozygous variants were detected by NGS in 11 families. The clinical features of TEK variants carriers were comparable to that of TEK-mutated patients identified in other populations and CYP1B1-mutated individuals from in-house database. Functional analysis confirmed four variants involving evolutionarily conserved residues to be loss-of-function, while one variant (p.R1003H) located in tyrosine kinase domain seemed to be an activating mutation. However, our results did not support the pathogenicity of the other five variants (p.H52R, p.M131I, p.M228V, p.H494Y, and p.L888P).
Conclusion: We provide evidence for TEK variants to be causative in Chinese PCG patients for the first time. Attention needs to be paid to TEK mutations in future genetic testing.
Keywords: TEK receptor tyrosine kinase, primary congenital glaucoma, next generation sequencing, mutations, functional analysis
Introduction
In healthy eyes, the intraocular pressure (IOP) is maintained by dynamic balance between aqueous humor production via ciliary body and drainage through trabecular and uveoscleral pathway (Johnson et al., 2017). Developmental defects of aqueous outflow structures can give rise to elevated IOP and early-onset glaucoma, causing buphthalmos, cornea stretching, optic disc cupping, and irreversible blindness (Lewis et al., 2017). Affecting children in the first 3 years of life, primary congenital glaucoma (PCG) is the most common form of infantile glaucoma with isolated angle anomalies (Young et al., 2020).
Genetic factors serve a critical role in the pathogenesis of PCG. To date, mutations of CYP1B1, LTBP2, MYOC, FOXC1, TEK, and ANGPT1 have been implicated in PCG patients from diverse populations (Stoilov et al., 1997; Kaur et al., 2005; Ali et al., 2009; Chakrabarti et al., 2009; Souma et al., 2016; Thomson et al., 2017). However, the molecular basis of PCG remains largely unrevealed, especially for patients of Chinese Han origin with CYP1B1 heterozygous and homozygous mutations accounting for only 17.2% of cases and no mutations of LTBP2 being identified (Chen et al., 2008; Chen et al., 2016).
Haploinsufficiency of TEK (Tunica intima endothelial receptor tyrosine kinase, Gene ID: 7010) caused by heterozygous loss-of-function mutations was reported to be causative for 5% of multiethnic PCG patients characterized by autosomal dominant inheritance with decreased penetrance and variable expressivity (Souma et al., 2016). This gene encodes a transmembrane receptor tyrosine kinase (Tie2) involved in cardiovascular development and homeostasis (Saharinen et al., 2017). Activating mutations of TEK have been identified as the genetic causes for most inherited and sporadic cases of venous malformation (VM) (Vikkula et al., 1996; Limaye et al., 2009). Notably, TEK is also expressed in the endothelia of Schlemm’s canal (SC) and collector channels, vital components of conventional aqueous outflow pathway (van Zyl et al., 2020). Conditional knockout of TEK from embryonic day 17.5 caused completely absent SC and significantly increased IOP, while heterozygous TEK deletion led to a severely hypomorphic SC and elevated IOP in mice (Souma et al., 2016). This dose–effect relationship clearly demonstrated that the normal function of Tie2 is indispensable during angle development.
For the first time, we performed TEK-targeted next generation sequencing in a large cohort of Chinese PCG patients aiming to verify the causative role and depict the mutation spectrum of TEK in this population.
Materials and Methods
Study Participants
Two hundred patients along with their parents were enrolled via the Eye and ENT Hospital Biobank from 2004 to 2018. The diagnosis of PCG must agree with the following criteria: (1) expansion of corneal diameter, corneal clouding, or Haab’s striae; (2) IOP >21 mm Hg; (3) optic nerve cupping or asymmetric cup-to-disc ratio; and (4) disease onset before age 3. Other developmental glaucoma or secondary glaucoma, such as Peter’s anomaly, Axenfeld–Rieger syndrome, and aniridia were excluded.
All PCG cases were confirmed by glaucoma specialists before surgery under general anesthesia. Anterior segment examination and corneal diameter measurement were performed with the aid of surgical microscopes. IOP was measured using a Tono-PEN tonometry (Mentor, Norwell, MA, United States). The structures of anterior chamber angle and the optic disc were observed under gonioscopy whenever possible. Five milliliters of venous blood was collected from each PCG patient and his/her parents and preserved in the biobank for genomic DNA extraction.
Next Generation Sequencing and Variants Prioritization
In this study, a customized panel with 58 pairs of primers was designed to cover the coding regions and 50-bp sequences flanking the splice sites of TEK gene (Supplementary Table S1). DNA samples were amplified by multiplex PCR and sequenced by a Hiseq4000 platform (Illumina, San Diego, CA). Sequencing reads were aligned to the human reference genome assembly (hg19). Variants were called by Genome Analysis Toolkit (version 3.70) and annotated by ANNOVAR (Wang et al., 2010). Exonic and splice site (±5 bp) variants with an allele frequency <0.1% in the Genome Aggregation database (gnomAD_all and gnomAD_eas) were validated by Sanger sequencing and selected for further analysis. Additionally, four probands were exome sequenced using a SureSelect Human All Exon Kit (Aglient, Santa Clara, CA, United States) and the aforementioned Hiseq4000 platform. When available, parental DNA samples were also subjected to Sanger sequencing for co-segregation analysis.
In Silico Functional Study
Twelve in silico tools were integrated to predict the pathogenicity of non-synonymous variations, including SIFT (Sim et al., 2012), Polyphen-2 (Adzhubei et al., 2010), LRT (Chun and Fay, 2009), MutationTaster (Schwarz et al., 2014), MutationAssessor (Reva et al., 2011), FATHMM (Shisha et al., 2013), PROVEN (Choi and Chan, 2015), VEST (Carter et al., 2013), Meats (Kim et al., 2017), M-CAP (Jagadeesh et al., 2016), CADD (Kircher et al., 2014), and DANN (Quang et al., 2015). Multiple Tie2 protein sequences from different species were retrieved to evaluate the evolutionary conservation of affected residues. To assess the effect of residue substitution on protein structure and stability, the FoldX plugin for YASARA (Van Durme et al., 2011) was used to calculate the free energy change induced by specific mutations with three repetitions each, and the resulting structures were visualized with YASARA View (Krieger and Vriend, 2014). For variants located within immunoglobulin (Ig)-like domains and the epidermal growth factor (EGF)-like domain, the Tie2 ligand-binding domain crystal structure (PDB ID: 2GY5, 23-445 aa) was utilized as the template (Barton et al., 2006). Additionally, crystal structure of the membrane proximal three fibronectin type III (FNIII) domains of Tie2 (PDB ID: 5UTK, 438-741 aa) and Tie2 kinase domain (PDB: 1FVR, 798-1124 aa) were used as templates for respective missense variants (Moore et al., 2017) (Shewchuk et al., 2000).
Cloning of TEK-Expressing Plasmids
The full-length human TEK cDNA (reference sequence: NM_000459.5, 4,683 bp) with 3x Flag (DYKDDDDK) tag was cloned into the pCDNA3.1-ZsGreen vector (Supplementary Figure S1A). Site-directed mutagenesis was performed to introduce mutations into TEK-expressing vectors. Specifically, two plasmids (Y1024X-Flag and TEK1023-Flag, Supplementary Figures S1B, C) were generated to verify the nonsense mutation p.Y1024*. All plasmids were validated by Sanger sequencing.
Western Blot Analysis
Wild-type (WT) or mutant TEK-Flag plasmids (1 μg) were transfected into subconfluent 293T cells in six-well dishes using Lipofectamine 2000 (Life Technologies, Carlsbad, CA). Twenty-four hours later, cells were lysed by 1% Triton X-100, centrifuged, and separated into supernatant and insoluble pellets. Radioimmunoprecipitation assay (RIPA) buffer was added to the pellets before sonication. Same amount of proteins (15 μg) was separated by electrophoresis and transferred to polyvinylidene fluoride (PVDF) membranes. After blocking with 5% non-fat milk, antibodies against phosphotyrosine (1:3,000; ab179530, Abcam, Cambridge, United Kingdom) and Flag (1:5,000; 20543-1-AP, Proteintech, Rosemont, IL, United States) were used to detect the phosphorylation and expression of Flag-tagged Tie2, respectively. β-Actin (1:5,000; 66009-1-Ig, Proteintech, Rosemont, IL, United States) was used as a loading control. For proteasomal inhibition assay, transfected 293T cells were treated with 5 μM MG132 (MedChemExpress, Shanghai, China) for 24 h before harvest.
Quantitative Real-Time PCR
Total RNA was extracted from 293T cells using EZ-press RNA Purification Kit (EZBioscience, Roseville, MN, United States) 24 h after transfection. One thousand nanograms of RNA was used for reverse transcription and subsequent quantitative PCR in CFX96TM real-time system (Bio-Rad, Hercules, CA, United States). The housekeeping gene ACTB was used as the internal control. All mRNA variants were normalized to WT-TEK using the δ-delta Ct method. The primers are listed below: forward primer, 5′-TCCAGGCAACTTGACTTCGG-3; reverse primer, 5′-CCTTGAACCTTGTAACGGATAG-3.
TEK Localization Assay
Human umbilical vein endothelial cells (HUVECs) were obtained from ScienCell and cultured using endothelial cell medium (ScienCell, Carlsbad, CA, United States). Cells were transfected with either WT or mutant TEK-Flag plasmids by electroporation with Neon transfection system (Thermo Fisher, MA, United States) following the manufacturer’s instructions. The transfected cells were seeded on glass coverslips in 24-well dishes. After 48 h, cells were either directly fixed with 4% paraformaldehyde for 15 min at room temperature or stimulated with 600 ng/ml recombinant human Angiopoietin 1 (R&D systems, Minneapolis, MN, United States) for another 30 min at 37°C before fixation. Cells were then permeabilized and blocked by 5% goat serum and 0.3% TritonX-100 in phosphate-buffered saline (PBS) for an hour at room temperature and incubated overnight at 4°C with anti-Flag antibody (1:200; 66008-3-Ig, Proteintech, Rosemont, IL, United States) and anti-ZO1 antibody (1:500; 21773-1-AP, Proteintech, Rosemont, IL, United States). Cyanine3- and Cyanine5-conjugated secondary antibodies (Thermo Fisher, MA, United States) were used to detect Flag and ZO-1 signals, respectively. Cell nuclei were stained by 4,6-diamidino-2-phenylindole (DAPI) (1 μg/ml; Sigma-Aldrich, St. Louis, MO, United States) for 5 min. Images were taken with a fluorescence microscope (DM4000 B, Leica, Wetzlar, Germany).
Statistical Analysis
Statistical analysis was performed with Stata software (15.1, StataCorp). Continuous variables were compared using Student’s t-test or paired t-test, while Pearson chi-square test was applied to comparing categorical variables. A p value <0.05 was considered statistically significant.
Results
Identification of Rare/Novel TEK Variants
An average read depth of 1,938.3 was achieved in this study, while 99.3% of the targeted regions had 100x coverage per base. In total, 44 TEK variants were detected in 199 samples. Among these, 11 rare or novel heterozygous nonsynonymous variants were identified in 11 unrelated patients (Table 1 and Supplementary Figure S2). Four variants (p.R1003H, p.H52R, p.H494Y, and p.Y1024*) co-segregated with the glaucoma phenotype. Other six variants were inherited from unaffected parents, consistent with the autosomal dominant pattern with decreased penetrance and variable expressivity suggested by Souma and others (Souma et al., 2016).
TABLE 1.
TEK variants identified in 11 families.
| Family ID | Patient ID | Chromosomal position | Exons | DNA change | Amino acid change | gnomAD_all | gnomAD_eas | In silico predictions | Average ddG |
|---|---|---|---|---|---|---|---|---|---|
| 01 | C-9 | 27217702 | exon19 | c.3008G > A | p.R1003H | 0.000012 | 0.000054 | 11|12 | 2.55 |
| 02 | C-56 | 27168521 | exon3 | c.393G > A | p.M131I | 2|12 | 0.81 | ||
| 03 | C-39 | 27192508 | exon11 | c.1511T > C | p.L504P | 8|12 | 4.16 | ||
| 04 | C-64 | 27172667 | exon5 | c.682A > G | p.M228V | 0.000032 | 0.000272 | 0|12 | 0.36 |
| 05 | C-99 | 27209206 | exon16 | c.2663T > C | p.L888P | 10|12 | 2.33 | ||
| 06 | C-151 | 27157931 | exon2 | c.155A > G | p.H52R | 0.000028 | 0 | 0|12 | −0.55 |
| 07 | C-165 | 27172715 | exon5 | c.730C > T | p.P244S | 8|12 | 4.00 | ||
| 08 | C-270 | 27206737 | exon15 | c.2522C > T | p.A841V | 0.000004 | 0 | 11|12 | 1.40 |
| 09 | C-290 | 27190679 | exon10 | c.1480C > T | p.H494Y | 0|12 | −0.55 | ||
| 10 | C-350 | 27173250 | exon6 | c.791G > T | p.C264F | 10|12 | 8.48 | ||
| 11 | C-354 | 27218784 | exon20 | c.3072T > A | p.Y1024X | 3|3 | NA |
Chromosomal positions correspond to GRCh37/hg19 assembly. Reference TEK mRNA, sequence, NM_000459.4. Reference TEK, protein sequence, NP_000450.2. gnomAD_all, allele frequency in all populations from gnomAD, dataset. gnomAD_eas, allele frequency in East Asian populations from gnomAD, dataset. In silico predicitons were made combining 12 programs listed in Methods, and presented as “Number of deleterious predictions | Number of total predictions”. Changes in Gibbs free energy were calculated via FoldX plugin for YASARA (average of three repetitions).
One mutation (p.A841V) was previously identified in a large American pedigree by Young et al. (2020). In addition, a different amino acid change in proline residue 244 (p.P244R) was also reported in the same study. The nonsense mutation (p.Y1024*) was confirmed by Western blotting (Supplementary Figure S1D). The truncated protein lacks tyrosine residues for phosphorylation in C-terminus (e.g., 1,102 and 1,108), which are indispensable in mediating cellular signals. Therefore, it was reasonable to speculate that these variants were detrimental to protein function.
The remaining eight missense variants differed in pathogenicity prediction. Half of them (p.C264F, p.L504P, p.L888P, and p.R1003H) involved evolutionarily conserved residues (Supplementary Figure S3) and evaluated as destabilizing by FoldX (Table 1), whereas the other four variants (p.H52R, p.M131I, p.M228V, and p.H494Y) were predicted to be benign by most in silico tools.
Since family 01-07 were previously confirmed negative for CYP1B1 and LTBP2 mutations, probands from family 08-11 were exome sequenced to explore mutations in other known disease-causing genes. C-354 was found to inherit the nonsense mutation of TEK (p.Y1024*) and a missense variant of CYP1B1 (p.L107V) from his affected father. C-350 carried an additional missense variant of LTBP2 (p.T886K) from her mother compared to her unaffected father. No rare or novel variants of MYOC, FOXC1, and ANGPT1 were detected (Supplementary Table S2).
Clinical Characteristics of TEK-Mutated PCG Patients
In this study, 5.5% of PCG patients carried rare or novel variants of TEK (Table 2). Of the carriers, 13 out of 19 (68.4%) exhibited clinically identifiable glaucoma phenotypes starting from 9.5 months after birth on average. There appeared to be a gender predilection, as most cases were male. In addition, more patients were bilaterally affected. Eight patients were followed up for disease prognosis (Table 3). Most of them (5/8) remained stable after initial trabeculotomy. Two patients underwent secondary angle surgeries, and one was enucleated for ocular components.
TABLE 2.
Comparison of clinical characteristics of TEK-mutated PCG cohorts.
| Souma et al. (2016) | Young et al. (2020) | This study | p-value | |
|---|---|---|---|---|
| Mutation rate (%) | 10/189 (5.3) | NA | 11/200 (5.5) | 0.931 a |
| Penetrance (%) | 14/22 (63.6) | 13/21 (61.9) | 13/19 (68.4) | 0.979 a |
| Sex (male: female) | 9:5 | 6:11 | 8:3 | 0.103 a |
| Bilaterally: unilaterally involvement | 6:6 | 10:4 | 7:4 | 0.470 a |
| Age at onset (month, mean ± SD) | 3.73 ± 7.1 | 7.27 ± 7.2 | 9.55 ± 10.2 | 0.162 b |
Pearson’s chi squared test.
Kruskal–Wallis test.
TABLE 3.
Clinical information for TEK-mutated patients.
| Family ID | Patient ID | Amino acid change | Sex | Age at onset | Eyes | Preoperative IOP (mmHg) | Cornea clouding | Corneal diameter (mm) | Cup–disc ratio | Treatment |
|---|---|---|---|---|---|---|---|---|---|---|
| 01 | C-9 | p.R1003H | M | Birth | OD | 20.6 | No | 12 | 0.5 | Loss to follow-up |
| OS | 20.6 | Haab’s striae | 13 | 0.7 | ||||||
| 02 | C-56 | p.M131I | M | 2y | OD | 20.6 | Macula | 13 | 0.4 | 1 trabeculotomy |
| 03 | C-39 | p.L504P | M | 2y | OD | 31 | Clear | 14.5 | 0.4 | 1 trabecolotomy in both eyes |
| OS | 48.5 | Yes | 16.5 | 0.9 | ||||||
| 04 | C-64 | p.M228V | F | 3 m | OD | 34 | Yes | 14.5 | 0.9 | Initial trabeculotomy and loss to follow-up |
| OS | 25 | Yes | 14.5 | 0.8 | ||||||
| 05 | C-99 | p.L888P | M | 2y | OD | 50.62 | Haab’s striae | 14 | 0.8 | 2 trabeculotomies in both eyes |
| OS | 54.66 | Haab’s striae | 13 | 0.8 | ||||||
| 06 | C-151 | p.H52R | F | Birth | OD | – | – | – | – | 1 trabeculotomy and 1 trabecolectomy in both eyes |
| OS | – | – | – | – | With subsequent tube shunt and enucleation of the left eye | |||||
| 07 | C-165 | p.P244S | M | Birth | OS | 28 | – | 13.5 | 1.0 | 1 trabeculotomy |
| 08 | C-270 | p.A841V | M | 2 m | OS | 42 | Yes | 13 | 1.0 | 1 trabeculotomy |
| 09 | C-290 | p.H494Y | M | 1y | OS | 45 | Yes | 13 | 1.0 | 2 trabeculotomies |
| 10 | C-350 | p.C264F | F | 1y | OD | 28 | Haab’s striae | 13.5 | 1.0 | 1 trabeculotomy in both eyes |
| OS | 30.4 | Haab’s striae | 13.5 | 1.0 | ||||||
| 11 | C-354 | p.Y1024X | M | 4 m | OD | 31.8 | – | 13 | – | Initial trabeculotomy in both eyes and loss to follow-up |
| OS | 29 | – | 12.5 | – |
Sex: M, male; F, female. Age at onset: m, month; y, year. Eyes: OD, right eye; OS, left eye.
There were no statistically significant difference between TEK-mutated PCG groups with regard to clinical features listed in Table 2. Similarly, the clinical presentations were comparable between Chinese patients with TEK mutations and those with CYP1B1 mutations or unidentified causes (Table 4).
TABLE 4.
Comparison of clinical characteristics of PCG subgroups.
| CYP1B1 | TEK | Unknown causes | p-value | |
|---|---|---|---|---|
| Mutation rate (%) | 20/124 (16.1) | 11/200 (5.5) | NA | 0.004 a |
| Sex (Male: Female) | 14:6 | 8:3 | 68:29 | 0.983 a |
| Bilaterally: unilaterally involvement | 12:8 | 7:4 | 46:51 | 0.398 a |
| Age at onset (month, mean ± SD) | 11.18 ± 13.04 | 9.55 ± 10.2 | 11.84 ± 12.49 | 0.647 b |
| Preoperative IOP (mmHg) | 40.94 ± 11.03 | 35.34 ± 5.89 | 35.45 ± 11.73 | 0.143 b |
| Corneal diameter (mm) | 13.26 ± 1.30 | 13.78 ± 1.15 | 13.35 ± 1.00 | 0.587b |
| Age at operation (day) | 972.21 ± 1008.5 | 665.3 ± 564.5 | 718.51 ± 703.3 | 0.876b |
Pearson’s chi squared test.
Kruskal–Wallis test.
Functional Impact of TEK Variants
All PCG-related TEK missense variants but p.A841V were tested in cell-based assays to evaluate their impact on protein expression, solubility, auto-phosphorylation, and response to ligand stimulation. First, WT and mutant TEK plasmids were overexpressed in 293T cells. The transcription levels of missense variants were all comparable to WT (Supplementary Figure S4). Three variants (p.P244S, p.C264F, and p.L504P) located in the ectodomain of Tie2 led to a dramatic reduction in protein expression in both soluble and insoluble fractions (Figure 1). After inhibition of proteosomal activity by MG132, the insoluble fraction of p.P244S and p.C264F mutant was remarkably increased, suggesting significant protein degradation, whereas p.L504P was insensitive to MG132 treatment. P244 together with P243 is critical in the structure of a β-turn (Figure 2). Substitution of proline (non-polar) to a serine (polar) is likely to change protein conformation. As C264 forms a disulfide bond with C255, we inferred that the p.C264F variant might lead to the disruption of the disulfide bridge and misfolding of translation product. L504 locates in the E β-strand of fibronectin type IIIa (FNIIIa) domain, which contributes to a classical antiparallel β-sheet motif (Moore et al., 2017). The replacement of leucine by proline disrupted intra-strand hydrogen bond resulting in an energetically unstable structure.
FIGURE 1.

Functional assessment of variants p.P244S, p.C264F, and p.L504P. The expression of soluble protein was significantly reduced (A) and remained unchanged after MG132 treatment (C). No dramatic change in auto-phosphorylation was detected (A). The expression of insoluble protein was also significantly reduced with increased phosphorylation level (B). In addition, protein degradation was reversed in p.P244S and p.C264F, while p.L504P was unresponsive to proteasomal inhibition (D). Sol, soluble; Ins, insoluble; PY, phosphotyrosine; WT, wild type; MG, MG132.^a
FIGURE 2.
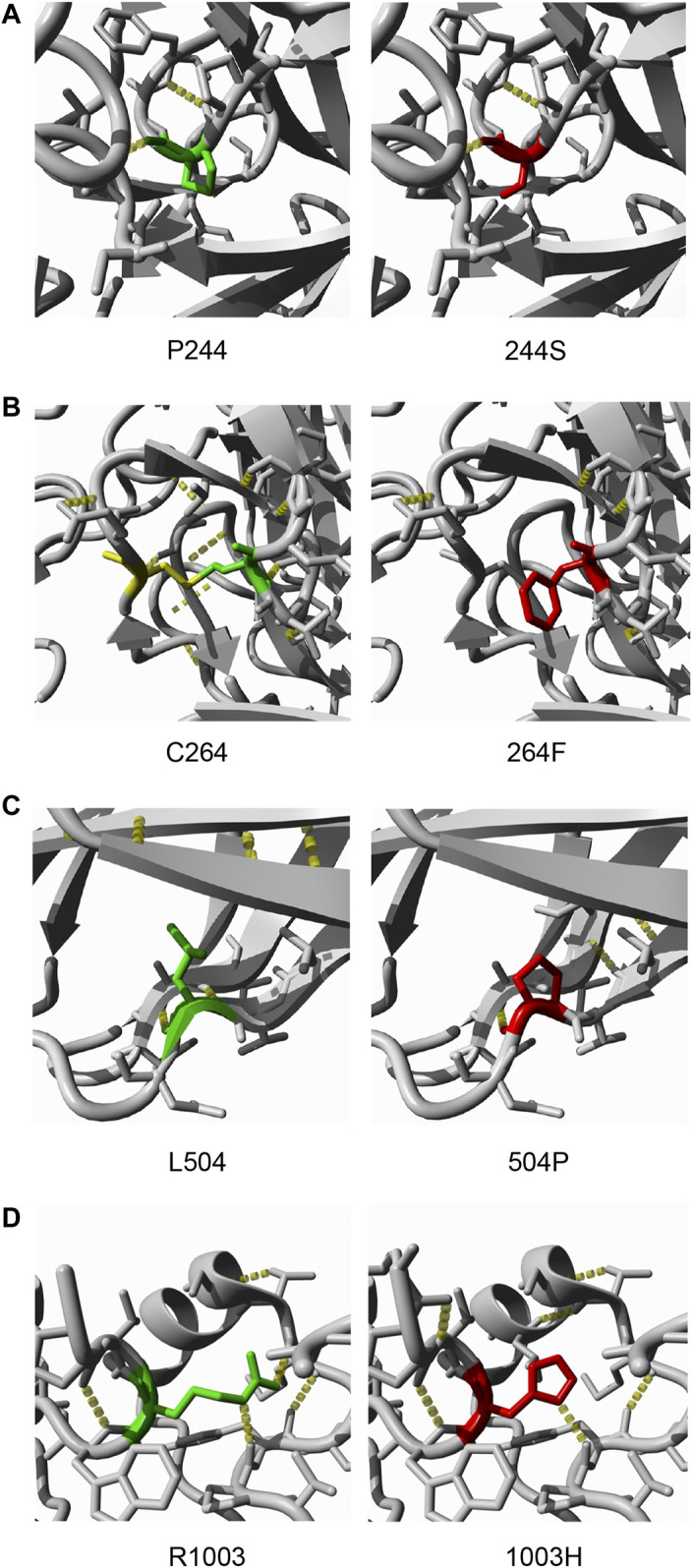
Crystal structures of four pathogenic TEK mutants (A). p.P244S substitutes a nonpolar proline to polar serine, which may influence the structure of a β-turn (B). Cysteine-264 forms a disulfide bond with Cysteine-255. Thus, substitution of C264 was predicted to be destabilizing due to the disruption of a disulfide bond (C). p.L504P is within a β-sheet of the FNIII domain; the substitution is likely to cause loss of hydrogen bonds, as is the case in p.R1003H (D). Green, wild-type residue; red, mutated residue; yellow, cysteine-255; yellow dots, hydrogen bonds.
Both protein expression and auto-phosphorylation levels of the other four variants in the ectodomain (p.H52R, p.M131I, p.M228V, and p.H494Y) were comparable to that of WT (Supplementary Figure S5), consistent with the in silico prediction of pathogenicity and structural analysis (Supplementary Figure S6). Notably, p.R1003H mutant was overexpressed in the insoluble component with increased auto-phosphorylation in the soluble fraction, matching the features of activating mutations (Figure 3). Structural analysis indicated that the de novo variant p.R1003H was likely to cause the disruption a hydrogen bond connecting R1003 with M1024 (Figure 2). However, variant p.L888P could not be distinguished from WT in our cell-based assay (Figure 3).
FIGURE 3.

Functional assessment of cytodomain variants p.L888P and p.R1003H. The protein expression and auto-phosphorylation levels of p.L888P are comparable to that of wild type (A, B). However, p.R1003H is significantly phosphorylated in soluble portion (A) and enriched in insoluble fraction (B), which does not respond to MG132 treatment (C, D). Sol, soluble; Ins, insoluble; PY, phosphotyrosine; WT, wild type; MG, MG132.
The ectodomain of Tie-2 consists of three Ig-like domains and an EGF-like domain, which are responsible for ligand binding (Yu et al., 2013), and a FNIII domain that is involved in ligand-induced receptor multimerization (Moore et al., 2017). Variants in these regions have the potential to weaken or disrupt ligand-mediated signaling. Thus, we transiently expressed all variants in HUVECs and explored their response to stimulation by potent agonist angiopoeitin-1 (ANGPT1). As anticipated, the WT TEK was diffusely expressed on the cytomembrane of HUVECs and relocalized to cell–cell junctions when treated with ANGPT1 (Figure 4). Surprisingly, the expression of variants p.P244S, p.C264F, and p.L504P was greatly reduced, consistent with the results of Western blotting. However, the other seven mutants did not exhibit significant alteration in response to ligand stimulation (Figure 4). A summary of functional analysis results is provided in Supplementary Table S3.
FIGURE 4.

Expression and ligand responsiveness of TEK variants (A). Expression of p.P244S, p.C264F, and p.L504P were greatly decreased in HUVECs. GFP (green) signals indicated cells were successfully transfected. Wild-type Tie2 (anti-Flag, red) was diffusely distributed in cellular membrane of quiescent HUVECs (first row). However, Flag signals for three loss-of-function variants were notably reduced. Scale bars indicate 25 μm. (B) After stimulation by Angiopoietin-1 for 30 min (lower panel), TEK proteins were relocated to cell–cell junctions as indicated by anti-ZO-1 (green). Nuclei were stained by DAPI (blue). Scale bars indicate 25 μm in upper panel and 12.5 μm in lower panel for magnified images. Pictures presented were representative of four biological replicates.
Discussion
In this study, we screened for TEK mutations in a large cohort of Chinese PCG patients for the first time. Deep-targeted NGS yielded 10 rare/novel potentially pathogenic variants and one previously confirmed mutation. Subsequent cell-based functional study was able to classify four variants (p.P244S, p.C264F, p.L504P, and p.Y1024*) as loss-of-function, one (p.R1003H) as gain-of-function, while the remaining 5 (p.H52R, p.M131I, p.M228V, p.H494Y, and p.L888P) appeared to be benign.
The development of the vascular and lymphatic system relies upon the orderly expression of TEK (Dumont et al., 1994; Thomson et al., 2014). As an essential part of aqueous humor drainage pathway, SC shares striking similarities with lymphatic vascular system (Aspelund et al., 2014). Indeed, some studies suggested SC as a specialized hybrid vessel with both lymphatic and vascular features (Ramos et al., 2007; Kizhatil et al., 2014). However, it was not until 2014 that an unintentional discovery linked the unique properties of SC and the pathogenesis of PCG (Thomson et al., 2014). Further studies delineated a clear dose effect between TEK function and SC morphology (Souma et al., 2016). Since then, more loss-of-function TEK mutations were reported in PCG patients with different ethnic backgrounds (Kabra et al., 2017; Young et al., 2020).
Despite being exhaustively investigated, the genetic cause of PCG remained largely unknown for most outbred populations (Mashima et al., 2001; Lim et al., 2013; Mohanty et al., 2013). In China, mutations in confirmed pathogenic genes only accounts for approximately 20% of PCG patients (Chen et al., 2008; Chen et al., 2016). Our results depicted the mutation spectrum of the recently reported causative gene and suggested the necessity of incorporating TEK in targeted panel for genetic diagnosis of PCG.
The incidence of TEK mutations in this study was similar to that of earlier reports (Souma et al., 2016; Kabra et al., 2017; Young et al., 2020). In addition, identified variants were predominantly located in the ectodomain of Tie2 (7/11), a distinctive feature varying from those associated with familial or sporadic VM (Saharinen et al., 2017). Possibly due to the wide coverage of coding sequence (23 exons, 4683bp for longest transcript), disease-associated variants rarely recur. Nevertheless, one of our patients shared a missense mutation (p.A841V) with a multigeneration pedigree reported by Young et al. (2020). This family was unique in terms of high disease penetrance, complex phenotypes, and refractoriness to treatment. A modifier gene, SVEP1, was proposed to explain the remarkable clinical features. Carrying the same TEK mutation, proband from family 8 presented relatively mild symptoms, such as unilateral involvement, limited penetrance, and favorable prognosis. Subsequent WES failed to detect any variants in SVEP1, which possibly explained the phenotypic disparities.
Based on the conservation analysis, missense variants identified in this study can be categorized into two groups. Variants p.H52R, p.M131I, p.M228V, and p.H494Y involve revolutionarily un-conservative residues, while the rest locates in conserved positions. p.H52R and p.H494Y both co-segregated with phenotype; however, in silico predictions and functional studies did not support their causative roles. Previous structural analysis revealed that the ligand–receptor interface is confined to the top of the Tie2 Ig2 domain, including the loops B-C (147-150 aa), C, (158–161 aa), and F–G (195–200 aa), strand C (151–157 aa), and strand C(162–167 aa) (Yu et al., 2013). Thus, amino acid changes in position 52, 131, and 228 are theoretically function sparing. Of note, we identified a rare de novo activating mutation (p.R1003H) in one patient. Unfortunately, we could not confirm whether this patient presented with VM or not since she was lost to follow-up. However, based on previous reports, this variant should not be considered as the cause of PCG. Similarly, there was not enough evidence to classify variant p.L888P to be pathogenic judging from structural and functional analysis. There might be other mechanisms through which those variants could lead to PCG. For instance, variants in other genes involved in ANG-Tie2 pathway could contribute jointly to the phenotype. It is necessary to screen relevant genes in future studies. Another possibility is that those variants could interfere the expression of TEK gene in vivo, which might be revealed using gene-edited animals. Additionally, TEK variants are likely to influence the phosphorylation level of particular amino acid sites (Souma et al., 2016). To this end, technologies of proteomics are required (e.g., mass spectrometry).
Mutant TEK proteins were previously shown to exhibit diminished or reduced interaction with CYP1B1, suggesting a potential digenic inheritance (Kabra et al., 2017). Interestingly, coexistence of variants in different pathogenic genes for PCG was noticed in our patients. The CYP1B1 variants that co-occurred with TEK mutations in this study were also found in healthy controls from Asian populations (Kim et al., 2011; Gong et al., 2015; Do et al., 2016). Nevertheless, how the cytoplasmic receptor Tie2 comes into direct contact with endoplasmic reticulum-targeted enzyme and what is the biological implication of this protein complex require further investigation.
There are some shortcomings to our study. First, limited by sequencing methods, it is outside the scope of this study to illustrate the mutation profile of other known disease-causing genes or uncover new potentially causal genes in this cohort. Considering the critical role of ANG-Tie2 signaling in SC development, it is tempting to screen genes encoding other component involved in this pathway (e.g., ANGPT1, ANGPT2, ANGPT4, and TIE1). In addition, the number of patients identified with TEK mutations was small, which limited the statistic power of comparison between patient groups and precluded any meaningful genotype–phenotype association analysis.
In summary, we confirmed the causative role of TEK mutation in Chinese PCG patients for the first time. The majority of disease-associated mutations are heterozygous missense variants involving evolutionarily conserved amino acid residues. Although TEK mutations account for <5% of total cases, due attention should be given in future genetic testing.
Acknowledgments
The samples used for the analyses described in this manuscript were obtained from the EENT Biobank. We would like to thank all the participants and the staffs for their valuable contribution to this research.
Data Availability Statement
The original contributions presented in the study are publicly available in NCBI under accession number PRJNA762473.
Ethics Statement
The studies involving human participants were reviewed and approved by the Institutional Review Board of Eye and ENT Hospital, Fudan University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
JC, XC, and XS contributed in the conception and design of the study and revised the manuscript. YC and CT recruited the patients. YQ acquired and analyzed the data and drafted the article. All authors approved the manuscript.
Funding
This work was supported by National Natural Science Foundation of China (Grant Numbers 81870661 and 81770968). The funding organization had no role in the design or conduct of this research.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.764509/full#supplementary-material
References
- Adzhubei I. A., Schmidt S., Peshkin L., Ramensky V. E., Gerasimova A., Bork P., et al. (2010). A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 7, 248–249. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali M., Mckibbin M., Booth A., Parry D. A., Jain P., Riazuddin S. A., et al. (2009). Null Mutations in LTBP2 Cause Primary Congenital Glaucoma. Am. J. Hum. Genet. 84, 664–671. 10.1016/j.ajhg.2009.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspelund A., Tammela T., Antila S., Nurmi H., Leppänen V.-M., Zarkada G., et al. (2014). The Schlemm's Canal Is a VEGF-C/VEGFR-3-responsive Lymphatic-like Vessel. J. Clin. Invest. 124, 3975–3986. 10.1172/jci75395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton W. A., Tzvetkova-Robev D., Miranda E. P., Kolev M. V., Rajashankar K. R., Himanen J. P., et al. (2006). Crystal Structures of the Tie2 Receptor Ectodomain and the Angiopoietin-2-Tie2 Complex. Nat. Struct. Mol. Biol. 13, 524–532. 10.1038/nsmb1101 [DOI] [PubMed] [Google Scholar]
- Carter H., Douville C., Stenson P. D., Cooper D. N., Karchin R. (2013). Identifying Mendelian Disease Genes with the Variant Effect Scoring Tool. BMC Genomics 14 (Suppl. 3), S3. 10.1186/1471-2164-14-S3-S3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti S., Kaur K., Rao K. N., Mandal A. K., Kaur I., Parikh R. S., et al. (2009). The Transcription Factor GeneFOXC1Exhibits a Limited Role in Primary Congenital Glaucoma. Invest. Ophthalmol. Vis. Sci. 50, 75–83. 10.1167/iovs.08-2253 [DOI] [PubMed] [Google Scholar]
- Chen X., Chen Y., Fan B. J., Xia M., Wang L., Sun X. (2016). Screening of the LTBP2 Gene in 214 Chinese Sporadic CYP1B1-Negative Patients with Primary Congenital Glaucoma. Mol. Vis. 22, 528–535. [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Jiang D., Yu L., Katz B., Zhang K., Wan B., et al. (2008). CYP1B1 and MYOC Mutations in 116 Chinese Patients with Primary Congenital Glaucoma. Arch. Ophthalmol. 126, 1443–1447. 10.1001/archopht.126.10.1443 [DOI] [PubMed] [Google Scholar]
- Choi Y., Chan A. P. (2015). PROVEAN Web Server: a Tool to Predict the Functional Effect of Amino Acid Substitutions and Indels. Bioinformatics 31, 2745–2747. 10.1093/bioinformatics/btv195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun S., Fay J. C. (2009). Identification of Deleterious Mutations within Three Human Genomes. Genome Res. 19, 1553–1561. 10.1101/gr.092619.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do T., Shei W., Chau P. T. M., Trang D. L., Yong V. H. K., Ng X. Y., et al. (2016). CYP1B1 and MYOC Mutations in Vietnamese Primary Congenital Glaucoma Patients. J. Glaucoma 25, e491–e498. 10.1097/ijg.0000000000000331 [DOI] [PubMed] [Google Scholar]
- Dumont D. J., Gradwohl G., Fong G. H., Puri M. C., Gertsenstein M., Auerbach A., et al. (1994). Dominant-negative and Targeted Null Mutations in the Endothelial Receptor Tyrosine Kinase, Tek, Reveal a Critical Role in Vasculogenesis of the Embryo. Genes Develop. 8, 1897–1909. 10.1101/gad.8.16.1897 [DOI] [PubMed] [Google Scholar]
- Gong B., Qu C., Li X., Shi Y., Lin Y., Zhou Y., et al. (2015). Mutation Spectrum ofCYP1B1in Chinese Patients with Primary Open-Angle Glaucoma. Br. J. Ophthalmol. 99, 425–430. 10.1136/bjophthalmol-2014-306054 [DOI] [PubMed] [Google Scholar]
- Jagadeesh K. A., Wenger A. M., Berger M. J., Guturu H., Stenson P. D., Cooper D. N., et al. (2016). M-CAP Eliminates a Majority of Variants of Uncertain Significance in Clinical Exomes at High Sensitivity. Nat. Genet. 48, 1581–1586. 10.1038/ng.3703 [DOI] [PubMed] [Google Scholar]
- Johnson M., Mclaren J. W., Overby D. R. (2017). Unconventional Aqueous Humor Outflow: A Review. Exp. Eye Res. 158, 94–111. 10.1016/j.exer.2016.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabra M., Zhang W., Rathi S., Mandal A. K., Senthil S., Pyatla G., et al. (2017). Angiopoietin Receptor TEK Interacts with CYP1B1 in Primary Congenital Glaucoma. Hum. Genet. 136, 941–949. 10.1007/s00439-017-1823-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur K., Reddy A., Mukhopadhyay A., Mandal A., Hasnain S., Ray K., et al. (2005). Myocilin Gene Implicated in Primary Congenital Glaucoma. Clin. Genet. 67, 335–340. 10.1111/j.1399-0004.2005.00411.x [DOI] [PubMed] [Google Scholar]
- Kim H. J., Suh W., Park S. C., Kim C. Y., Park K. H., Kook M. S., et al. (2011). Mutation Spectrum of CYP1B1 and MYOC Genes in Korean Patients with Primary Congenital Glaucoma. Mol. Vis. 17, 2093–2101. [PMC free article] [PubMed] [Google Scholar]
- Kim S., Jhong J.-H., Lee J., Koo J.-Y. (2017). Meta-analytic Support Vector Machine for Integrating Multiple Omics Data. BioData Mining 10, 2. 10.1186/s13040-017-0126-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M., Witten D. M., Jain P., O'roak B. J., Cooper G. M., Shendure J. (2014). A General Framework for Estimating the Relative Pathogenicity of Human Genetic Variants. Nat. Genet. 46, 310–315. 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizhatil K., Ryan M., Marchant J. K., Henrich S., John S. W. M. (2014). Schlemm's Canal Is a Unique Vessel with a Combination of Blood Vascular and Lymphatic Phenotypes that Forms by a Novel Developmental Process. Plos Biol. 12. e1001912 10.1371/journal.pbio.1001912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger E., Vriend G. (2014). YASARA View-Molecular Graphics for All Devices-From Smartphones to Workstations. Bioinformatics 30, 2981–2982. 10.1093/bioinformatics/btu426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis C. J., Hedberg-Buenz A., Deluca A. P., Stone E. M., Alward W. L. M., Fingert J. H. (2017). Primary Congenital and Developmental Glaucomas. Hum. Mol. Genet. 26, R28–r36. 10.1093/hmg/ddx205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S.-H., Tran-Viet K.-N., Yanovitch T. L., Freedman S. F., Klemm T., Call W., et al. (2013). CYP1B1, MYOC, and LTBP2 Mutations in Primary Congenital Glaucoma Patients in the United States. Am. J. Ophthalmol. 155, 508–517. 10.1016/j.ajo.2012.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limaye N., Wouters V., Uebelhoer M., Tuominen M., Wirkkala R., Mulliken J. B., et al. (2009). Somatic Mutations in Angiopoietin Receptor Gene TEK Cause Solitary and Multiple Sporadic Venous Malformations. Nat. Genet. 41, 118–124. 10.1038/ng.272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashima Y., Suzuki Y., Sergeev Y., Ohtake Y., Tanino T., Kimura I., et al. (2001). Novel Cytochrome P4501B1 (CYP1B1) Gene Mutations in Japanese Patients with Primary Congenital Glaucoma. Invest. Ophthalmol. Vis. Sci. 42, 2211–2216. [PubMed] [Google Scholar]
- Mohanty K., Tanwar M., Dada R., Dada T. (2013). Screening of the LTBP2 Gene in a north Indian Population with Primary Congenital Glaucoma. Mol. Vis. 19, 78–84. [PMC free article] [PubMed] [Google Scholar]
- Moore J. O., Lemmon M. A., Ferguson K. M. (2017). Dimerization of Tie2 Mediated by its Membrane-Proximal FNIII Domains. Proc. Natl. Acad. Sci. USA 114, 4382–4387. 10.1073/pnas.1617800114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quang D., Chen Y., Xie X. (2015). DANN: a Deep Learning Approach for Annotating the Pathogenicity of Genetic Variants. Bioinformatics 31, 761–763. 10.1093/bioinformatics/btu703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos R. F., Hoying J. B., Witte M. H., Daniel Stamer W. (2007). Schlemm’s Canal Endothelia, Lymphatic, or Blood Vasculature? J. Glaucoma 16, 391–405. 10.1097/ijg.0b013e3180654ac6 [DOI] [PubMed] [Google Scholar]
- Reva B., Antipin Y., Sander C. (2011). Predicting the Functional Impact of Protein Mutations: Application to Cancer Genomics. Nucleic Acids Res. 39, e118. 10.1093/nar/gkr407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saharinen P., Eklund L., Alitalo K. (2017). Therapeutic Targeting of the Angiopoietin-TIE Pathway. Nat. Rev. Drug Discov. 16, 635–661. 10.1038/nrd.2016.278 [DOI] [PubMed] [Google Scholar]
- Schwarz J. M., Cooper D. N., Schuelke M., Seelow D. (2014). MutationTaster2: Mutation Prediction for the Deep-Sequencing Age. Nat. Methods 11, 361–362. 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- Shewchuk L. M., Hassell A. M., Ellis B., Holmes W. D., Davis R., Horne E. L., et al. (2000). Structure of the Tie2 RTK Domain. Structure 8, 1105–1113. 10.1016/s0969-2126(00)00516-5 [DOI] [PubMed] [Google Scholar]
- Shihab H. A., Gough J., Cooper D. N., Stenson P. D., Barker G. L. A., Edwards K. J., et al. (2013). Predicting the Functional, Molecular, and Phenotypic Consequences of Amino Acid Substitutions Using Hidden Markov Models. Hum. Mutat. 34, 57–65. 10.1002/humu.22225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim N.-L., Kumar P., Hu J., Henikoff S., Schneider G., Ng P. C. (2012). SIFT Web Server: Predicting Effects of Amino Acid Substitutions on Proteins. Nucleic Acids Res. 40, W452–W457. 10.1093/nar/gks539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souma T., Tompson S. W., Thomson B. R., Siggs O. M., Kizhatil K., Yamaguchi S., et al. (2016). Angiopoietin Receptor TEK Mutations Underlie Primary Congenital Glaucoma with Variable Expressivity. J. Clin. Invest. 126, 2575–2587. 10.1172/jci85830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoilov I., Akarsu A. N., Sarfarazi M. (1997). Identification of Three Different Truncating Mutations in Cytochrome P4501B1 (CYP1B1) as the Principal Cause of Primary Congenital Glaucoma (Buphthalmos) in Families Linked to the GLC3A Locus on Chromosome 2p21. Hum. Mol. Genet. 6, 641–647. 10.1093/hmg/6.4.641 [DOI] [PubMed] [Google Scholar]
- Thomson B. R., Heinen S., Jeansson M., Ghosh A. K., Fatima A., Sung H.-K., et al. (2014). A Lymphatic Defect Causes Ocular Hypertension and Glaucoma in Mice. J. Clin. Invest. 124, 4320–4324. 10.1172/jci77162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson B. R., Souma T., Tompson S. W., Onay T., Kizhatil K., Siggs O. M., et al. (2017). Angiopoietin-1 Is Required for Schlemm's Canal Development in Mice and Humans. J. Clin. Invest. 127, 4421–4436. 10.1172/jci95545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Durme J., Delgado J., Stricher F., Serrano L., Schymkowitz J., Rousseau F. (2011). A Graphical Interface for the FoldX Forcefield. Bioinformatics 27, 1711–1712. 10.1093/bioinformatics/btr254 [DOI] [PubMed] [Google Scholar]
- Van Zyl T., Yan W., Mcadams A., Peng Y.-R., Shekhar K., Regev A., et al. (2020). Cell Atlas of Aqueous Humor Outflow Pathways in Eyes of Humans and Four Model Species Provides Insight into Glaucoma Pathogenesis. Proc. Natl. Acad. Sci. USA 117, 10339–10349. 10.1073/pnas.2001250117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vikkula M., Boon L. M., Iii K. L. C., 3rd, Calvert J. T., Calvert A. J., Goumnerov B., et al. (1996). Vascular Dysmorphogenesis Caused by an Activating Mutation in the Receptor Tyrosine Kinase TIE2. Cell 87, 1181–1190. 10.1016/s0092-8674(00)81814-0 [DOI] [PubMed] [Google Scholar]
- Wang K., Li M., Hakonarson H. (2010). ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 38, e164. 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young T. L., Whisenhunt K. N., Jin J., Lamartina S. M., Martin S. M., Souma T., et al. (2020). SVEP1 as a Genetic Modifier of TEK-Related Primary Congenital Glaucoma. Invest. Ophthalmol. Vis. Sci. 61, 6. 10.1167/iovs.61.12.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X., Seegar T. C. M., Dalton A. C., Tzvetkova-Robev D., Goldgur Y., Rajashankar K. R., et al. (2013). Structural Basis for Angiopoietin-1-Mediated Signaling Initiation. Proc. Natl. Acad. Sci. 110, 7205–7210. 10.1073/pnas.1216890110 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original contributions presented in the study are publicly available in NCBI under accession number PRJNA762473.