

Studies of the pathogenesis of chronic lymphocytic leukemia (CLL) have been hampered by disease heterogeneity, the limited number of CLL cell lines, and the difficulty in manipulating primary B cells to express potential driver genes of interest. In a definitive review, ten Hacken and Wu describe the contribution of mouse models to study the pathogenesis and treatment of CLL, where genetic strategies allow modeling the full range of CLL phenotypes from indolent to transformed disease.
Visual Abstract

Abstract
Rapid advances in large-scale next-generation sequencing studies of human samples have progressively defined the highly heterogeneous genetic landscape of chronic lymphocytic leukemia (CLL). At the same time, the numerous challenges posed by the difficulties in rapid manipulation of primary B cells and the paucity of CLL cell lines have limited the ability to interrogate the function of the discovered putative disease “drivers,” defined in human sequencing studies through statistical inference. Mouse models represent a powerful tool to study mechanisms of normal and malignant B-cell biology and for preclinical testing of novel therapeutics. Advances in genetic engineering technologies, including the introduction of conditional knockin/knockout strategies, have opened new opportunities to model genetic lesions in a B-cell–restricted context. These new studies build on the experience of generating the MDR mice, the first example of a genetically faithful CLL model, which recapitulates the most common genomic aberration of human CLL: del(13q). In this review, we describe the application of mouse models to the studies of CLL pathogenesis and disease transformation from an indolent to a high-grade malignancy (ie, Richter syndrome [RS]) and treatment, with a focus on newly developed genetically inspired mouse lines modeling recurrent CLL genetic events. We discuss how these novel mouse models, analyzed using new genomic technologies, allow the dissection of mechanisms of disease evolution and response to therapy with greater depth than previously possible and provide important insight into human CLL and RS pathogenesis and therapeutic vulnerabilities. These models thereby provide valuable platforms for functional genomic analyses and treatment studies.
Introduction
Despite the flood of knowledge gained from large-scale sequencing studies over the last 20 years,1-11 we still know relatively little about the functional impact of the discovered putative drivers of chronic lymphocytic leukemia (CLL) and how their interplay impacts disease progression and response to therapy. Although notable advances in the clinical management of CLL have been achieved because of the US Food and Drug Administration approvals of novel targeted agents (the BCL2 inhibitor venetoclax and the Bruton’s tyrosine kinase [BTK] inhibitors ibrutinib and acalabrutinib) including in combination with the anti-CD20 antibodies rituximab and obinutuzumab,12-20 CLL remains incurable, and tailored therapeutic interventions are challenged by the high degree of genetic heterogeneity characterizing patients with CLL. Another critical barrier to CLL management is represented by Richter syndrome (RS), an aggressive transformation of CLL primarily into a diffuse large B-cell lymphoma (DLBCL) histology that occurs in up to 10% of patients21 who are largely refractory to existing therapies and exhibit a median overall survival of less than 12 months.
Although early studies of CLL predominantly focused on dissecting the relevance of B-cell receptor (BCR) signaling and microenvironmental interactions,22 CLL genetics have been progressively found to play an important role in patient stratification and clinical outcome.23 Common genomic aberrations [eg, del(13q), del(11q), del(17p), tri 12] were already identified in the 1990s through karyotyping24 and later confirmed by fluorescent in situ hybridization1 (Figure 1). With the introduction of next-generation sequencing approximately a decade ago, the complexity of CLL genetic2,4,5,25-27 and epigenetic7,10,28,29 heterogeneity has been progressively unraveled through a number of studies across the world. Most recently, the analysis of large CLL patient cohorts,8,9 combined together to integrate genetic, transcriptional, and epigenetic information from >1000 patients,11 has allowed further identification of CLL patient subsets with distinct molecular features and clinical outcome. The architecture and evolutionary trajectories of RS have also been progressively unraveled.3,30-33 Here again, these analyses have been based on statistical inference, with limited information available on the functional implications of the so-called identified "drivers."
Figure 1.

Timeline of human vs murine studies highlighting landmark papers and ASH abstracts related to genetic/epigenetic analyses of human CLL and RS and to the generation of genetically engineered mouse models and xenografts.
Functional studies in CLL have long faced many obstacles because of the difficulties in genetic manipulation of primary CLL B cells and the lack of patient-derived cell lines and xenografts.34 The most used mouse model of CLL is the Eμ-TCL1 transgenic mouse,35 which is recognized to develop an aggressive disease variant and thus does not recapitulate the indolent nature of most human CLL. Likewise, because of the relative rarity of RS primary specimens and the limited availability of murine or cellular models, RS pathogenesis has remained largely undefined. Given the wealth of human genetic information afforded by sequencing of CLL, it would be informative to generate models that are reflective of human genetics, and in doing so, create the opportunity to dissect mechanisms of CLL progression and to evaluate therapeutic vulnerabilities of disease subsets with selected genetic makeups (Figure 2). Several transformative advances in genetic engineering have enabled an improved ability to assess the in vivo effects of candidate drivers, including through the use of conditional knock-in/knock-out strategies, and, more recently, the clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 gene editing technology. We provide an overview of recent efforts in generating genetically engineered and xenograft models of CLL and RS and a perspective on novel technological advances and their application to disease modeling.
Figure 2.

Schematic representation of the integration of human genomic analyses into the generation of mouse models recapitulating human disease drivers. Novel models can be exploited for studies of disease pathogenesis and treatment and allow findings to be translated back into personalized treatment interventions for patients with selected genetic makeups.
Early days of CLL mouse modeling: spontaneous development and the TCL1 mouse
For many years, CLL was considered a B-cell tumor derived from accumulation of malignant B cells failing to undergo apoptosis because of prosurvival signaling from microenvironmental components.22 The first mouse model of CLL, the New Zealand Black (NZB) mouse (Figure 1),36 indeed developed disease spontaneously with aging, starting at 12 months, in the absence of any genetic manipulation. Of note, later studies carried out in this mouse strain identified a point mutation in the locus encoding mir16-1,37 a modulator of BCL2 expression,38 suggesting the necessity for constitutive dysregulation of antiapoptotic pathways as the basis for CLL development. In the early 2000s, CLL mouse modeling was revolutionized by the introduction of the Eμ-TCL1 mouse,35 generated through engineering of the T-cell leukemia oncogene TCL1 under the control of the variable heavy-chain promoter -IgH-Eμ enhancer, thus overexpressing TCL1 in all B cells. Eμ-TCL1 mice develop B220+ CD5+ lympho-proliferation at 100% penetrance by 2 months of age, with clonal overt disease by approximately 12 months. Although TCL1 protein is overexpressed by a subset of immunoglobulin heavy-chain variable (IGHV)-unmutated CLL cells,39 and functions as an activator of NF-κB signaling,40 it does not represent a recurrent CLL genetic lesion. TCL1-derived CLL cells are transplantable into secondary recipients,41 providing a platform highly amenable for therapeutic applications. BCRs of TCL1 mice resemble those of aggressive human CLL, showing high structural similarity compatible with recognition of autoantigens and microbial proteins, similar to those of aggressive treatment-refractory CLL.42 Since its generation, the Eμ-TCL1 mouse has been widely used in numerous studies, aimed both at dissecting the relevance of selected proteins or immune components in disease progression and at testing drugs preclinically; these studies have been previously summarized in Simonetti et al,43 Pekarsky et al,44 and Chen et al.45
Novel applications of the TCL1 mouse: models of intrinsic and acquired resistance to therapy
The US Food and Drug Administration approval of targeted therapeutic strategies, including the BTK kinase inhibitor ibrutinib,12,13 and the BCL2 inhibitor venetoclax,14,15 has substantially changed the therapeutic landscape for patients with CLL but has also led the appreciation of novel mechanisms of therapeutic resistance, distinct from those associated with chemotherapeutic agents. Thus, it has become important to develop preclinical models of resistance to novel agents, which could serve as the basis for mechanistically dissecting therapeutic resistance and for testing novel combinatorial strategies. At baseline, the TCL1 mouse appears to be a model reflective of venetoclax resistance.46 Through BH3 profiling, a technique that defines which antiapoptotic proteins are primarily responsible for cell survival in a test sample,47 TCL1 cells were identified to be dependent on MCL1 and BFL-148 proteins for survival rather than BCL2 (the archetypal vulnerability of CLL) and to show reduced sensitivity to venetoclax treatment both in vitro and in transplantable models.46,49 Intrinsic resistance to ibrutinib was identified in a compound Eμ-TCL1 × MYC model, mimicking coexistence of CLL and B-cell lymphoma. In this model, BTK inhibitor resistance was overcome either by inhibition of nuclear export via XPO150 or by the reversible and noncovalent BTK and SRC kinase inhibitor ARQ531.51 More recently, CRISPR-Cas9 gene editing was used to introduce the common BTK resistance mutation C481S and generate a mouse model of resistance to irreversible BTK inhibitors, including ibrutinib, acalabrutinib, and zanubrutinib.52 Although the primary report only demonstrated resistance to such inhibitors in vitro in mouse splenocytes, this model (if crossed with a leukemogenic strain; eg, TCL1, MDR) could represent a useful system to study combinatorial treatments in the context of BTK inhibitor resistance.
Models of acquired resistance have been developed through continuous exposure of TCL1-derived CLL cells to targeted therapy in serial transplant settings. Scheffold et al53 generated a transplantable mouse line displaying acquired resistance to phosphatidylinositol 3-kinase δ (PI3K-δ) inhibition by continuous treatment of serial TCL1 transplants with the PI3K-δ inhibitor GS-649443, identifying IGF1R signaling and the FOXO/GSK3β transcription factors as important mediators (and druggable targets) of PI3Kδ inhibitor resistance. Because of the intrinsic resistance to venetoclax of the TCL1 model, modeling acquired resistance to BCL2 inhibition in this setting is not possible, whereas other models (eg, the MDR mice, characterized by upregulated BCL2 signaling), could be amenable platforms for similar serial treatment studies and induction of BCL2i resistance.
CLL patient–derived xenografts: challenges and successes
Over the years, numerous attempts to xenograft primary CLL from peripheral blood mononuclear cells (PBMCs) have been made (reviewed in Bertilaccio et al34), with overall limited success compared with other B-cell malignancies.54 One of the first successful studies was reported by Durig et al55 using recipient NOD/SCID mice, a strain devoid of B and T cells but retaining functional natural killer (NK) cells. After infusion of approximately 108 CLL PBMCs both IV and intraperitoneally (compared with most B-cell lymphoma xenografts that can be generated from infusion of approximately 1-2 × 106 cells),54 stable recovery of CLL PBMCs was obtained for up to 8 weeks, with Binet stage C PBMCs showing higher engraftment rates compared with more indolent cells derived from patients in Binet stage A. With the introduction of novel immune-deficient strains (ie, NSG mice), increasingly devoid of functional T and NK cells that could mediated graft rejection, more robust xenograft models have been generated,56,57 particularly when CLL PBMCs were coinjected with autologous CD4+ T cells (with higher engraftment efficacy when T cells were preactivated in vitro)58 or allogeneic CD14+ monocytes. The life span of recipient animals in this setting has been reported to be 4 to 12 weeks, a much shorter time frame for analysis compared with that of TCL1 (ie, 9-12 months). Despite the lack of functional B, T, and NK cells, and the only partial functionality of macrophages and dendritic cells characterizing the NSG model, NSG xenografts have been proven effective in recapitulating microenvironmental interactions typical of human disease, such as activation of NF-κB and BCR signaling in peripheral lymphoid organs,59 presumably by interactions of human CLL PBMCs with autologous engrafted immune cells and/or murine macrophages or stroma. Microenvironment-derived prosurvival signals (ie, BCR and Notch2) can be successfully inhibited by treatment with ibrutinib59 or daptomycin,60 confirming the validity of this preclinical model for testing targeted therapies within a limited time frame. An intriguing alternate approach for xenografting CLL cells was explored by Kikushige et al,61 who xeno-transplanted CLL hematopoietic stem cells into NSG mice, with the hypothesis that the propensity to generate clonal B cells is already present at the stem cell level in patients with CLL. These xeno-transplants, performed in newborn NSGs by facial vein injection of CLL hematopoietic stem cells within 48 hours of birth, gave rise to monoclonal B-cell expansions, consistent with monoclonal B-cell lymphocytosis (MBL; ie, the precursor state of human CLL) but never to emergence of full-blown CLL. Moreover, the BCR immunoglobulin heavy-chain variable identity of the clonal B cells emerging after transplant was distinct from the predominant sequence identified in the primary donor cells, suggesting selection of an alternate donor-derived subclone on infusion. Immune-deficient mice have also been amenable models for xenografting cell lines, including the MEC1 cell line,62 OSU-CLL,63 PCL12,64 JVM3,65 and HG3,66 with the caveat that these CLL-derived cell lines are Epstein-Barr virus transformed, generally carry aggressive genetic lesions, and are only partially able to engage with murine microenvironmental components, given the incomplete homology to human receptor–ligand pairs.
Genetically inspired mouse models of CLL: new tools for functional genomic analyses
Advances in genetic engineering technologies, including novel knockin/knockout strategies, have undoubtedly changed the spectrum of opportunities for in vivo genetic modeling, as it has become increasingly feasible to introduce CLL-specific mutations in a cellular background of choice (Table 1). The first example of genetically faithful mouse models are the MDR67 and CDR mice,68 created through either germline or Cd19-restricted deletion of the minimal or common deleted region (MDR and CDR, respectively) of chromosome 14q, corresponding to human chromosome 13q, thus recapitulating the most common genomic aberration of human disease, focal del(13q). Both models develop a wide range of B-cell lympho-proliferations, ranging from MBL to CLL (with a small proportion of other non-Hodgkin B-cell lymphomas), with an approximate 16- to 18-month onset and an approximate 20% to 40% penetrance (depending on zygosity and germline or B cell–restricted deletion), thus recapitulating the indolent nature of human disease. Subsequent studies have taken advantage of similar conditional strategies to model common genetic aberrations of human disease (Figures 1 and 3), including hot spot mutation in the splicing factor Sf3b1, alone or combined with deletion in the DNA damage response gene Atm (ie, the Sf3b1/Atm mice),69 as well as novel disease drivers, such as the B-cell developmental factor Ikzf3,70 the ribosomal protein Rps15,71 and combined mutation in MDR and Sf3b1.72 Given the pervasive locally disordered methylation detected in human CLL and the impact of dysregulated methylation in facilitating disease initiation and promoting clonal evolution in CLL,7 various investigators have dissected the mechanisms of disease progression in mouse models of DNA methyltransferase-3A (Dnmt3a) deletion.73-75 A suite of models recapitulating XPO1 mutations in CLL and lymphomas was also recently generated.76 All together, these models have allowed deep functional investigation of the role of driver lesions in the B-cell lineage and the interpretation of mutation-specific oncogenic mechanisms at the basis of CLL initiation and progression (Figure 4). Furthermore, transplantability of primary murine tumors into secondary recipients has allowed the development of robust platforms for drug treatment studies. Considering the negative prognostic value of SF3B12 and NOTCH16 mutations and the previously reported association between mutations of IKZF3 and RPS15 and fludarabine resistance,8 these models represent important platforms for the study of mechanisms of therapeutic resistance and the development of novel combinatorial treatment strategies tailored to selected genetic contexts.
Table 1.
Genetically inspired mouse models of CLL and RS
| Mouse model | Human genotype/phenotype | Onset (mo) | Penetrance (%) | Disease entities (as reported) | Phenotype | Reference |
|---|---|---|---|---|---|---|
| MDR | del (13q) | 16-18 | 20-40 | MBL, CLL, DLBCL | Increased B-cell proliferation | 67 |
| MDR/Sf3b1 | del (13q) + Sf3b1-K700E | 18-20 | 25 | CLL | Increased mTOR signaling | 72 |
| Sf3b1/Atm | SF3B1-K700E+del(11q) | 18-22 | 10 | CLL | Increased DNA damage, aberrant 3' splice site selection, low BCR responsiveness | 69 |
| Ikzf3 | IKZF3-L162R | 16-18 | 40 | CLL, DLBCL | Increased follicular and germinal center development, high BCR responsiveness | 70 |
| Rps15 (alone or + Trp53) |
RPS15-S138F TP53-mut or del(17p) |
18-20 | 20-30 | CLL | Aberrant MYC signaling | 71 |
| Xpo1 | XPO1-E571K | 6 (SRBC stimulated) | N/A (<100%) | CLL, B cell lymphomas | Aberrant nuclear export | 76 |
| Dnmt3a | Disordered methylation | 10-16 | 60-100 | CLL | Increased B1a-cell survival, aberrant NOTCH signaling | 73-75 |
| CRISPR-multiplexed | LOF in ATM, TP53, CHD2, BIRC3, MGA, SAMHD1, combined with del(13q) | 9-14 | 60-80 | CLL, CLL/RS, RS | Increased DNA damage, MYC signaling | 90 |
N/A, not applicable.
Figure 3.

Comutation plot highlighting disease drivers identified in our latest published dataset of 538 CLLs that were since then modeled in mice. Modeled human gene mutations, name of mouse model, and reference study are indicated in the table. Published dataset of 538 CLLs from Landau et al.8
Figure 4.

B-cell–restricted strains carrying CLL putative disease drivers and functional interrogation of the role of selected gene mutations in CLL pathogenesis.
-
1.
The Sf3b1/Atm mouse. Sf3b1 is among the most commonly deleted genes in CLL, frequently co-occurring with focal deletion in chromosome 11q [del(11q)].8 Through B cell–restricted introduction of Sf3b1-K700E in a mouse model, dysregulated splicing was a result of Sf3b1 mutation, with patterns consistent with SF3B1-mutant human CLL (ie, alternative 3' splice-site selection).77 Importantly, Sf3b1-mutant mouse cells showed features of cellular senescence, a program associated with reduced metabolic activity and cell cycle block. The senescent state could be overcome by presence of co-occurring Atm deletion, which also increased DNA damage. Indeed, only when co-occurring, Sf3b1 and Atm lesions were able to lead to CLL development in elderly mice at approximately 10% penetrance, whereas neither lesion alone was sufficient to generate leukemia. Focal amplifications in chr15 and chr17 (encompassing MAPK and MYC signaling genes) were consistently found in all diseased mice, consistent with the necessity of a “third-hit” to facilitate oncogenesis in these late-onset models. Another key aspect of this study was the identification of diminished BCR signaling in association with Sf3b1 mutation, in both mouse and human SF3B1-mutant CLL, which correlated with differential response to ibrutinib treatment, with SF3B1-mutant CLL patients experiencing a delayed onset of peak absolute lymphocyte counts (ALC), compared with SF3B1 wild type. Thus, this mouse model informed on new and unexpected insights into the biology of CLL cells carrying SF3B1 mutations.
-
2.
The Ikzf3 mouse. Hotspot mutation in Ikzf3 was identified as a novel disease driver in our latest published dataset of 538 CLLs8; occurring in approximately 3% patients and associated with fludarabine-refractoriness, mutation of this B-cell transcription factor was found to be invariably located in a hotspot within the DNA binding domain region. Ikzf3 mutation was demonstrated to potentiate BCR and NF-κB signaling through direct regulation of BCR signaling gene transcription. Ikzf3 as a sole mutation was able to induce CLL generation in approximately 40% of 15- to 18-month-old animals, confirming its role as a CLL driver. Interestingly, the same chr15 amplification (involving the MYC oncogene) found in Sf3b1/Atm CLL mice was also identified in Ikzf3-mutant CLLs, suggesting a key role of MYC pathway dysregulation in both mouse models. In contrast to Sf3b1, Ikzf3-mutation was associated with heightened BCR signaling responses in both preleukemic and leukemic murine CLLs. IKZF3 mutation and/or its overexpression in human primary CLLs correlated with reduced responsiveness to the BTK kinase inhibitor ibrutinib, with implications for IKZF3-mediated resistance to ibrutinib monotherapy in patients, as previously suggested by human studies.78 These results also suggest that IKZF3 overexpression can phenocopy its mutation in patients, an important association given the otherwise relatively low prevalence of IKZF3 mutations in CLL patients.
-
3.
The Rps15 mouse. Ribosomal protein RPS15 was also identified to be a novel disease driver in approximately 5% CLL and frequently co-occurring with mutation in TP53.79 RPS15 point mutants have been modeled in HEK293T cells, already providing evidence of altered translational fidelity and reduced sensitivity to drug treatment,80 although the HEK293T system cannot fully recapitulate the characteristics of a mature B-cell lineage. When modeled in a B cell–restricted fashion, Rps15 mutation associated with strong induction of MYC-associated transcriptional programs and led to leukemia with an approximate 20% penetrance and approximate 20-month disease onset. Studies aimed at deciphering the function of Rps15 mutation (alone and combined with B cell–restricted Trp53 mutation) in both normal and leukemic B-cell biology are currently underway and anticipated to provide insight into the relevance of translatome dysregulation in CLL.
-
4.
The MDR-Sf3b1 mouse. Because the SF3B1 mutation frequently co-occurs with chromosome 13q deletion [del(13q)], Zhang et al72 explored co-occurrence of these 2 lesions (ie, MDR-Sf3b1) in mice and observed approximately 24% MDR-Sf3b1 developing CLL-like disease with an approximate 18- to 20-month onset. Unique to the MDR-Sf3b1 double-mutant CLL is the upregulation of the mTORC1 pathway, an important metabolic signaling pathway characterized by increased activity in poor prognostic CLL subgroups.81 mTORC1 activity in the MDR-Sf3b1 mouse could be selectively inhibited through the mTORC1 inhibitor temsirolimus, alone or combined with splicing inhibition by H3B-8800, proposing a novel dual combinatorial treatment strategy for del(13q)-SF3B1mut patients.
-
5.
The Xpo1 mouse. Mutations in exportin 1 (XPO1) have been identified in a number of mature B-cell lymphomas, including CLL, where they appear as primarily clonal events, persistent at disease relapse.82 Taylor et al76 developed a suite of mouse models of the most common E571 mutation, either introduced alone, in a conditional B-cell–restricted fashion or combined with MYC or BCL2, demonstrating the role of Xpo1 as a driver in CLL and B-cell lymphoma. Mutant Xpo1 acts by altering nuclear/cytoplasmic compartmentalization of proteins involved in inflammation, DNA repair, and chromatin remodeling. Importantly, Xpo1-mutant cells appeared to be preferentially sensitive to Xpo1 inhibition by selinexor, previously shown to be effective in patients with CLL and in the context of ibrutinib resistance.83 More recently, Xpo-E571 mutation was shown to accelerate disease onset and progression in an Eμ-TCL1–based cross,84 confirming the oncogenicity of this mutation.
-
6.
The Dnmt3a mouse. Epigenetic dysregulation is as a hallmark of transforming CLL, with disordered methylation patterns associated to increased cancer cell plasticity and propensity for clonal evolution.7 A few studies have modeled mutations in Dnmt3a in mice,73-75 all converging on dysregulated development of the B1a CD5+ peritoneal B-cell subset, leading to CLL generation with an approximate 10- to 16-month onset and an approximate 60% to 100% penetrance (depending on the zygosity and germline vs conditional expression). Of note, DNMT3A was reported to be downregulated at the protein level in TCL1-overexpressing cells,85 again suggesting the importance of reduced expression of this DNA methyltransferase in leukemia development. A Notch1-related expression signature was unique to Dnmt3a-deleted CLL but not to other previously characterized mouse lines (ie, Sf3b1/Atm, Ikzf3, MDR). As nonmutational Notch activation occurs in human CLL,86 this model may represent a unique platform for the study of constitutive NOTCH1 signaling in CLL.
Modeling RS: transgenic mouse models and xenografts
The first evidence describing occasional transformation of CLL cells into large cell lymphoma comes from a study where NZB-derived cells were engrafted into secondary recipients,36 providing the proof-of-principle that CLL cells can transform to acquire a DLBCL-like histology in vivo. Genetically engineered mouse models of RS are still quite limited. A few recent studies have reported on some cases of RS transformation of TCL1-based aggressive CLL in the context of coexpression of high-risk alterations, such as Trp53 or Akt mutations,87 of AKT protein,88 or of lesions primarily found in lymphomas, such as the MYC oncogene.50 Eμ-TCL1–derived splenocytes engineered to express mutant Trp53 and Cdkn2A/B (lesions frequently co-occurring in human RS)3 via CRISPR-Cas9 gene editing also gave rise to RS-like disease when transplanted into congenic immune-competent recipients, showing BCR dependence and sensitivity to combined BCR and CDK4/6 inhibition.89
The CRISPR-Cas9 technology was recently leveraged to introduce multiplexed combinatorial gene edits in recurrent loss-of-function lesions (ie, Atm, Trp53, Chd2, Birc3, Samhd1, Mga) in hematopoietic stem cells of del(13q) donor mice,90 which, on transplantation into either immune-competent CD45.1 or immune-deficient NSG, gave rise not only to CLL carrying heterogeneous combinatorial traits but also transformed into RS, with approximately 60% to 80% penetrance and approximately 9- to 14-month onset (depending on the recipient strain). This system has allowed identification of recurrent combinatorial lesions typical of transforming disease, such as comutation in Trp53 and in the MYC signaling regulator Mga90 and of increased presence of clonal drivers on CLL transformation into RS, consistent with human disease.32 Importantly RS development was primarily observed in CD45.1 immune-competent hosts, compared with NSG, highlighting the importance of immune microenvironmental selective pressure for lymphoma development.
Xenografting of RS cells is more reliable than CLL PBMCs, although RS primary samples are more difficult to obtain. Moreover, by their very nature (ie, primary RS samples transplanted into immune-deficient hosts), they do not allow studies of disease transformation and/or tumor evolution from the antecedent CLL (Figure 5). RS xenografts have been successfully developed by injecting either peripheral blood– or lymph node–derived cells subcutaneously or IV in NSG mice, with first passage tumors maintaining transcriptional, genetic, and phenotypic stability compared with primary tumors.91 These models are potentially useful for short-term treatment studies, as animals reach survival end points within approximately 20 to 40 days after engraftment in the absence of treatment.92,93 Using 4 different RS xenografts expressing variable surface levels of the receptor tyrosine kinase–like orphan receptor 1 (ROR1), Vaisitti et al94 recently demonstrated efficacy of the novel antibody–drug conjugate VLS-101 (an anti-ROR1 antibody conjugated to an anti-microtubule cytotoxin) in RS xenografts expressing high ROR1 levels compared with lower ROR1 expressors. Recent xenograft-based therapeutic studies demonstrated RS sensitivity to BCR inhibition, alone or combined with BRD4 inhibitors95 or venetoclax,96 further validating RS xenografts as a valuable platform for personalized short-term treatment analyses. As the RS genomic landscape is progressively unraveled,3,30-33 we anticipate a multitude of efforts will be focused on the development of novel RS models in the coming years.
Figure 5.
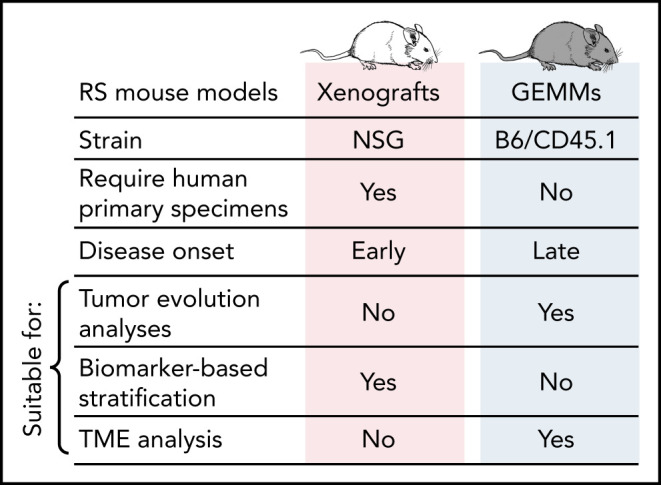
Comparative characteristics of CLL and RS genetically engineered mouse models (GEMMs) and xenografts and their amenability for studies of CLL pathogenesis and treatment.
A perspective on new technologies and applications to CLL and RS modeling
The rapid advances in novel genetic engineering methodologies and single-cell technologies are allowing us to interrogate biological processes at increasingly greater depth. Although the aforementioned efforts have been mostly focused on understanding tumor-intrinsic features of B cells carrying cancer driver mutation, there has been increased interest in linking genetic events with tumor-extrinsic changes in immune microenvironmental composition, which in turn, is impactful in understanding disease progression and response to therapy.97 Although flow cytometry has historically been the golden standard for evaluation of tumor immune subpopulations in mouse models, novel technologies including mass cytometry and single cell RNA-seq (alone or combined with surface molecule detection, ie, cellular indexing of transcriptomes and epitopes by sequencing [CITE-seq]) are progressively emerging as a widely used approach to evaluate the composition of the tumor immune microenvironment, and to analyze cellular states and infer functional immune cell interactomes. A recent mass cytometry–based study, carried out in the Eμ-TCL1 mouse model, identified high PD1 and LAG3 expression in a number of tumor-infiltrating immune populations, including CD8+ T cells, regulatory T cells, macrophages, and NK cells,98 all contributing to immune suppression. More recently, single-cell RNA-seq (scRNA-seq) of splenocytes derived from a TCL1 model of AKT overexpression88 has allowed identification of a unique CD4+ T-cell subset enriched in TCL1 × AKT mice (but not in age-matched Eμ-TCL1), leading to DLL1 overexpression and mediating NOTCH-signaling activation in autologous CLL cells.
scRNA-seq requires single-cell suspensions as input and disruption of tissue integrity, therefore limiting tumor-immune cell proximity analyses, which are fundamental to address the impact of microenvironmental interactions on tumor progression, in particular on resistance to treatment. Slide-Seq, a novel adaptation of scRNA-seq to flash-frozen tissue sections,99 allows in situ definition of gene expression patterns, thus overcoming this obstacle. An increasingly common approach to address immune cell interactions within tumor microenvironments at high resolution is represented by multiplexed imaging techniques, including multiplexed spatial cytometry (codetection by indexing)100 and multiplexed ion beam imaging by time of flight,101 based on fluorescent or mass spectrometry detection, respectively. These techniques allow quantitative characterizations of cellular components in relationship to tissue architecture, allowing detection of ≥30 markers simultaneously, far more than conventional confocal microscopy. In CLL, a few elegant studies have explored microenvironmental interactions via confocal imaging in mice,102-104 but these larger-scale imaging applications are yet to be explored.
A powerful approach in cancer evolution studies is represented by lineage tracing,105 whereby a cellular population (and its progeny) can be identified over time because of expression of trackable markers (usually fluorescent markers or barcodes). Recently, the novel tool ClonMapper was developed,106 which combines lineage tracing with transcriptional profiling, allowing single-cell identification of transcribed barcodes. Another related application is represented by mitochondrial ATAC sequencing, whereby mitochondrial DNA mutations, detectable via single-cell ATAC sequencing, can be used to stably trace clones in primary samples and can be combined with chromatin accessibility profiling.107 Single-cell DNA sequencing at high throughput,108 including in conjunction with surface protein expression,109,110 is also becoming more broadly used, providing opportunities for proteogenomic analysis of tumor evolution. These technologies were thus far applied to the study of mechanisms of clonal evolution of cell lines106 or primary CLL samples111 in response to chemotherapy or targeted treatment, and one could envision adapting them to analyses related to mouse models.
We are increasingly powered to identify more completely the spectrum of disease drivers for CLL via whole-exome and whole-genome sequencing because of the ever-larger size of patient cohorts.11 Multimodal integration of genomic, transcriptomic, epigenetic, and functional information is essential for a more complete patient stratification and the identification of selected vulnerabilities of individual pharmacotypes, which have thus far been difficult to predict. With the discovery of new drivers, our genetic engineering tool kit is also expanding because of the introduction of the novel Cas9 variants (eg, base editors) that allow precise introduction of point mutations at the RNA or DNA level,112 with higher efficacy than the earlier homology-directed repair strategies. We anticipate the application of these novel engineering strategies will expand our capacity of modeling single or combined traits in vitro and in vivo in the near future.
Acknowledgments
The authors thank Romain Guieze and Satyen Gohil for critical reading of the manuscript.
C.J.W. acknowledges support from National Institutes of Health/National Cancer Institute grants P01 CA206978, R01 CA216273, and is a scholar of the Leukemia and Lymphoma Society. E.t.H. is a Scholar of the American Society of Hematology.
Authorship
Contribution: E.t.H. and C.J.W. wrote the manuscript.
Conflict-of-interest disclosure: C.J.W. is an equity holder of Biontech, Inc and receives research funding from Pharmacyclics. E.t.H. declares no competing financial interests.
Correspondence: Catherine J. Wu, 450 Brookline Ave, Boston, MA 02215; e-mail: cwu@partners.org.
REFERENCES
- 1.Döhner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910-1916. [DOI] [PubMed] [Google Scholar]
- 2.Wang L, Lawrence MS, Wan Y, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365(26):2497-2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fabbri G, Khiabanian H, Holmes AB, et al. Genetic lesions associated with chronic lymphocytic leukemia transformation to Richter syndrome. J Exp Med. 2013;210(11):2273-2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Puente XS, Pinyol M, Quesada V, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475(7354):101-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quesada V, Conde L, Villamor N, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2011;44(1):47-52. [DOI] [PubMed] [Google Scholar]
- 6.Rossi D, Rasi S, Fabbri G, et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood. 2012;119(2):521-529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Landau DA, Clement K, Ziller MJ, et al. Locally disordered methylation forms the basis of intratumor methylome variation in chronic lymphocytic leukemia. Cancer Cell. 2014;26(6):813-825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Landau DA, Tausch E, Taylor-Weiner AN, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526(7574):525-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Puente XS, Beà S, Valdés-Mas R, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature. 2015; 526(7574):519-524. [DOI] [PubMed] [Google Scholar]
- 10.Beekman R, Chapaprieta V, Russiñol N, et al. The reference epigenome and regulatory chromatin landscape of chronic lymphocytic leukemia. Nat Med. 2018; 24(6):868-880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Knisbacher BA, Lin Z, Hahn CK, et al. The CLL-1100 project: towards complete genomic characterization and improved prognostics for CLL. Blood. 2020; 136(suppl 1):2-4. [Google Scholar]
- 12.Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burger JA, Tedeschi A, Barr PM, et al. ; RESONATE-2 Investigators . Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med. 2015;373(25):2425-2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jain N, Keating M, Thompson P, et al. Ibrutinib and venetoclax for first-line treatment of CLL. N Engl J Med. 2019; 380(22):2095-2103. [DOI] [PubMed] [Google Scholar]
- 15.Roberts AW, Davids MS, Pagel JM, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374(4):311-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Byrd JC, Harrington B, O’Brien S, et al. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374(4):323-332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shanafelt TD, Wang XV, Kay NE, et al. Ibrutinib-rituximab or chemoimmunotherapy for chronic lymphocytic leukemia. N Engl J Med. 2019;381(5):432-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moreno C, Greil R, Demirkan F, et al. Ibrutinib plus obinutuzumab versus chlorambucil plus obinutuzumab in first-line treatment of chronic lymphocytic leukaemia (iLLUMINATE): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019;20(1):43-56. [DOI] [PubMed] [Google Scholar]
- 19.Seymour JF, Kipps TJ, Eichhorst B, et al. Venetoclax-rituximab in relapsed or refractory chronic lymphocytic leukemia. N Engl J Med. 2018;378(12):1107-1120. [DOI] [PubMed] [Google Scholar]
- 20.Fischer K, Al-Sawaf O, Bahlo J, et al. Venetoclax and obinutuzumab in patients with CLL and coexisting conditions. N Engl J Med. 2019;380(23):2225-2236. [DOI] [PubMed] [Google Scholar]
- 21.Allan JN, Furman RR. Current trends in the management of Richter syndrome. Int J Hematol Oncol. 2019;7(4):IJH09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghia P, Caligaris-Cappio F. The indispensable role of microenvironment in the natural history of low-grade B-cell neoplasms. Adv Cancer Res. 2000;79:157-173. [DOI] [PubMed] [Google Scholar]
- 23.Kipps TJ, Stevenson FK, Wu CJ, et al. Chronic lymphocytic leukaemia [published correction appears in Nat Rev Dis Primers. 2017;3:17008]. Nat Rev Dis Primers. 2017; 3(1):16096.28102226 [Google Scholar]
- 24.Juliusson G, Oscier DG, Fitchett M, et al. Prognostic subgroups in B-cell chronic lymphocytic leukemia defined by specific chromosomal abnormalities. N Engl J Med. 1990;323(11):720-724. [DOI] [PubMed] [Google Scholar]
- 25.Fabbri G, Rasi S, Rossi D, et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med. 2011;208(7):1389-1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rossi D, Fangazio M, Rasi S, et al. Disruption of BIRC3 associates with fludarabine chemorefractoriness in TP53 wild-type chronic lymphocytic leukemia. Blood. 2012;119(12):2854-2862. [DOI] [PubMed] [Google Scholar]
- 27.Rossi D, Rasi S, Spina V, et al. Integrated mutational and cytogenetic analysis identifies new prognostic subgroups in chronic lymphocytic leukemia. Blood. 2013;121(8):1403-1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oakes CC, Claus R, Gu L, et al. Evolution of DNA methylation is linked to genetic aberrations in chronic lymphocytic leukemia. Cancer Discov. 2014;4(3):348-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kulis M, Heath S, Bibikova M, et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet. 2012; 44(11):1236-1242. [DOI] [PubMed] [Google Scholar]
- 30.Rossi D, Spina V, Deambrogi C, et al. The genetics of Richter syndrome reveals disease heterogeneity and predicts survival after transformation. Blood. 2011;117(12):3391-3401. [DOI] [PubMed] [Google Scholar]
- 31.Klintman J, Appleby N, Stamatopoulos B, et al. Genomic and transcriptomic correlates of Richter transformation in Chronic Lymphocytic Leukemia. Blood. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parry E, Guieze R, Leshchiner I, et al. Genetic determinants and evolutionary history of Richter syndrome. Blood. 2020;136(suppl 1):47-48. [Google Scholar]
- 33.Chigrinova E, Rinaldi A, Kwee I, et al. Two main genetic pathways lead to the transformation of chronic lymphocytic leukemia to Richter syndrome. Blood. 2013;122(15):2673-2682. [DOI] [PubMed] [Google Scholar]
- 34.Bertilaccio MT, Scielzo C, Simonetti G, et al. Xenograft models of chronic lymphocytic leukemia: problems, pitfalls and future directions. Leukemia. 2013;27(3):534-540. [DOI] [PubMed] [Google Scholar]
- 35.Bichi R, Shinton SA, Martin ES, et al. Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc Natl Acad Sci USA. 2002;99(10):6955-6960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Phillips JA, Mehta K, Fernandez C, Raveché ES. The NZB mouse as a model for chronic lymphocytic leukemia. Cancer Res. 1992; 52(2):437-443. [PubMed] [Google Scholar]
- 37.Scaglione BJ, Salerno E, Balan M, et al. Murine models of chronic lymphocytic leukaemia: role of microRNA-16 in the New Zealand Black mouse model. Br J Haematol. 2007;139(5):645-657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cimmino A, Calin GA, Fabbri M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2 [published correction appears in Proc Natl Acad Sci USA. 2006;103(7):2464]. Proc Natl Acad Sci USA. 2005;102(39):13944-13949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Herling M, Patel KA, Khalili J, et al. TCL1 shows a regulated expression pattern in chronic lymphocytic leukemia that correlates with molecular subtypes and proliferative state. Leukemia. 2006;20(2):280-285. [DOI] [PubMed] [Google Scholar]
- 40.Pekarsky Y, Palamarchuk A, Maximov V, et al. Tcl1 functions as a transcriptional regulator and is directly involved in the pathogenesis of CLL. Proc Natl Acad Sci USA. 2008;105(50):19643-19648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zanesi N, Aqeilan R, Drusco A, et al. Effect of rapamycin on mouse chronic lymphocytic leukemia and the development of nonhematopoietic malignancies in Emu-TCL1 transgenic mice. Cancer Res. 2006; 66(2):915-920. [DOI] [PubMed] [Google Scholar]
- 42.Yan XJ, Albesiano E, Zanesi N, et al. B cell receptors in TCL1 transgenic mice resemble those of aggressive, treatment-resistant human chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2006; 103(31):11713-11718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simonetti G, Bertilaccio MT, Ghia P, Klein U. Mouse models in the study of chronic lymphocytic leukemia pathogenesis and therapy. Blood. 2014;124(7):1010-1019. [DOI] [PubMed] [Google Scholar]
- 44.Pekarsky Y, Drusco A, Kumchala P, Croce CM, Zanesi N. The long journey of TCL1 transgenic mice: lessons learned in the last 15 years. Gene Expr. 2015; 16(3):129-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen SS, Chiorazzi N. Murine genetically engineered and human xenograft models of chronic lymphocytic leukemia. Semin Hematol. 2014;51(3):188-205. [DOI] [PubMed] [Google Scholar]
- 46.Ten Hacken E, Valentin R, Regis FFD, et al. Splicing modulation sensitizes chronic lymphocytic leukemia cells to venetoclax by remodeling mitochondrial apoptotic dependencies. JCI Insight. 2018; 3(19):121438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Certo M, Del Gaizo Moore V, Nishino M, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006; 9(5):351-365. [DOI] [PubMed] [Google Scholar]
- 48.Yecies D, Carlson NE, Deng J, Letai A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood. 2010;115(16):3304-3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patel VK, Lamothe B, Ayres ML, et al. Pharmacodynamics and proteomic analysis of acalabrutinib therapy: similarity of on-target effects to ibrutinib and rationale for combination therapy. Leukemia. 2018; 32(4):920-930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lucas F, Rogers KA, Harrington BK, et al. Eμ-TCL1xMyc: a novel mouse model for concurrent CLL and B-cell lymphoma. Clin Cancer Res. 2019;25(20):6260-6273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reiff SD, Mantel R, Smith LL, et al. The BTK inhibitor ARQ 531 targets ibrutinib-resistant CLL and Richter transformation. Cancer Discov. 2018;8(10):1300-1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Estupiñán HY, Bouderlique T, He C, et al. Novel mouse model resistant to irreversible BTK inhibitors: a tool identifying new therapeutic targets and side effects. Blood Adv. 2020;4(11):2439-2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scheffold A, Jebaraj BMC, Tausch E, et al. IGF1R as druggable target mediating PI3K-δ inhibitor resistance in a murine model of chronic lymphocytic leukemia. Blood. 2019;134(6):534-547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Townsend EC, Murakami MA, Christodoulou A, et al. The public repository of xenografts enables discovery and randomized phase II-like trials in mice [published correction appears in Cancer Cell. 2016;30(1):P183]. Cancer Cell. 2016;30(1):183. [DOI] [PubMed] [Google Scholar]
- 55.Dürig J, Ebeling P, Grabellus F, et al. A novel nonobese diabetic/severe combined immunodeficient xenograft model for chronic lymphocytic leukemia reflects important clinical characteristics of the disease. Cancer Res. 2007;67(18):8653-8661. [DOI] [PubMed] [Google Scholar]
- 56.Bagnara D, Kaufman MS, Calissano C, et al. A novel adoptive transfer model of chronic lymphocytic leukemia suggests a key role for T lymphocytes in the disease. Blood. 2011;117(20):5463-5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oldreive CE, Skowronska A, Davies NJ, et al. T-cell number and subtype influence the disease course of primary chronic lymphocytic leukaemia xenografts in alymphoid mice. Dis Model Mech. 2015; 8(11):1401-1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Patten PEM, Ferrer G, Chen SS, et al. A detailed analysis of parameters supporting the engraftment and growth of chronic lymphocytic leukemia cells in immune-deficient mice. Front Immunol. 2021; 12:627020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Herman SE, Sun X, McAuley EM, et al. Modeling tumor-host interactions of chronic lymphocytic leukemia in xenografted mice to study tumor biology and evaluate targeted therapy. Leukemia. 2013; 27(12):2311-2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mangolini M, Götte F, Moore A, et al. Notch2 controls non-autonomous Wnt-signalling in chronic lymphocytic leukaemia. Nat Commun. 2018;9(1):3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kikushige Y, Ishikawa F, Miyamoto T, et al. Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell. 2011;20(2):246-259. [DOI] [PubMed] [Google Scholar]
- 62.Bertilaccio MT, Scielzo C, Simonetti G, et al. A novel Rag2-/-gammac-/-xenograft model of human CLL. Blood. 2010;115(8):1605-1609. [DOI] [PubMed] [Google Scholar]
- 63.Hertlein E, Beckwith KA, Lozanski G, et al. Characterization of a new chronic lymphocytic leukemia cell line for mechanistic in vitro and in vivo studies relevant to disease. PLoS One. 2013; 8(10):e76607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Agathangelidis A, Scarfò L, Barbaglio F, et al. Establishment and characterization of PCL12, a novel CD5+ chronic lymphocytic leukaemia cell line. PLoS One. 2015; 10(6):e0130195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Loisel S, Ster KL, Quintin-Roue I, et al. Establishment of a novel human B-CLL-like xenograft model in nude mouse. Leuk Res. 2005;29(11):1347-1352. [DOI] [PubMed] [Google Scholar]
- 66.Quijada-Álamo M, Hernández-Sánchez M, Alonso-Pérez V, et al. CRISPR/Cas9-generated models uncover therapeutic vulnerabilities of del(11q) CLL cells to dual BCR and PARP inhibition. Leukemia. 2020;34(6):1599-1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Klein U, Lia M, Crespo M, et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell. 2010; 17(1):28-40. [DOI] [PubMed] [Google Scholar]
- 68.Lia M, Carette A, Tang H, et al. Functional dissection of the chromosome 13q14 tumor-suppressor locus using transgenic mouse lines. Blood. 2012;119(13):2981-2990. [DOI] [PubMed] [Google Scholar]
- 69.Yin S, Gambe RG, Sun J, et al. A murine model of chronic lymphocytic leukemia based on B cell-restricted expression of Sf3b1 mutation and Atm deletion. Cancer Cell. 2019;35(2):283-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lazarian G, Yin S, Ten Hacken E, et al. A hotspot mutation in transcription factor IKZF3 drives B cell neoplasia via transcriptional dysregulation. Cancer Cell. 2021;39(3):380-393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gutierrez C, Ouspenskaia T, Fu D, et al. RPS15 and TP53 co-mutation drives B cell malignancy through altered translation and MYC activation in a murine model. Blood. 2020;136(suppl 1):28-29. [Google Scholar]
- 72.Zhang B, Iyer P, Jin M, et al. Expression of Sf3b1-K700E accelerates the development of chronic lymphocytic leukemia in a del(13q) murine model. Blood. 2020; 136(suppl 1):4-5.32614961 [Google Scholar]
- 73.Biran A, et al. B cell–restricted depletion of Dnmt3a activates Notch and Myc signaling, generating a consistent murine model of chronic lymphocytic leukemia. Cancer Res. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Peters SL, Hlady RA, Opavska J, et al. Tumor suppressor functions of Dnmt3a and Dnmt3b in the prevention of malignant mouse lymphopoiesis. Leukemia. 2014; 28(5):1138-1142. [DOI] [PubMed] [Google Scholar]
- 75.Haney SL, Upchurch GM, Opavska J, et al. Promoter hypomethylation and expression is conserved in mouse chronic lymphocytic leukemia induced by decreased or inactivated Dnmt3a. Cell Rep. 2016; 15(6):1190-1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Taylor J, Sendino M, Gorelick AN, et al. Altered nuclear export signal recognition as a driver of oncogenesis. Cancer Discov. 2019;9(10):1452-1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang L, Brooks AN, Fan J, et al. Transcriptomic characterization of SF3B1 mutation reveals its pleiotropic effects in chronic lymphocytic leukemia. Cancer Cell. 2016;30(5):750-763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ahn IE, Chen Y-C, Underbayey C, et al. Diverging clonal evolution during sequential therapy with chemoimmunotherapy followed by BTK inhibitors. Blood. 2019; 134(suppl 1):850. [Google Scholar]
- 79.Ljungström V, Cortese D, Young E, et al. Whole-exome sequencing in relapsing chronic lymphocytic leukemia: clinical impact of recurrent RPS15 mutations. Blood. 2016;127(8):1007-1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bretones G, Álvarez MG, Arango JR, et al. Altered patterns of global protein synthesis and translational fidelity in RPS15-mutated chronic lymphocytic leukemia. Blood. 2018;132(22):2375-2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cosimo E, Tarafdar A, Moles MW, et al. AKT/mTORC2 inhibition activates FOXO1 function in CLL cells reducing B-cell receptor-mediated survival. Clin Cancer Res. 2019;25(5):1574-1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Landau DA, Carter SL, Stojanov P, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152(4):714-726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hing ZA, Mantel R, Beckwith KA, et al. Selinexor is effective in acquired resistance to ibrutinib and synergizes with ibrutinib in chronic lymphocytic leukemia. Blood. 2015;125(20):3128-3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Walker JS, Hing ZA, Harrington B, et al. Recurrent XPO1 mutations alter pathogenesis of chronic lymphocytic leukemia. J Hematol Oncol. 2021;14(1):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Palamarchuk A, Yan PS, Zanesi N, et al. Tcl1 protein functions as an inhibitor of de novo DNA methylation in B-cell chronic lymphocytic leukemia (CLL). Proc Natl Acad Sci USA. 2012;109(7):2555-2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fabbri G, Holmes AB, Viganotti M, et al. Common nonmutational NOTCH1 activation in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2017;114(14):E2911-E2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Knittel G, Rehkämper T, Korovkina D, et al. Two mouse models reveal an actionable PARP1 dependence in aggressive chronic lymphocytic leukemia. Nat Commun. 2017;8(1):153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kohlhaas V, Blakemore SJ, Al-Maarri M, et al. Active AKT signaling triggers CLL towards Richter transformation via over-activation of Notch1. Blood. 2020. [DOI] [PubMed] [Google Scholar]
- 89.Chakraborty S, Martines C, Porro F, et al. B cell receptor signaling and genetic lesions in TP53 and CDKN2A/CDKN2B cooperate in Richter transformation. Blood. 2021; 137(5):646-660. [DOI] [PubMed] [Google Scholar]
- 90.Ten Hacken E, Sewastianik T, Redd R, et al. Multiplexed CRISPR in vivo editing of CLL loss-of-function lesions models transformation of chronic lymphocytic leukemia into Richter syndrome. Blood. 2020;136(suppl 1):2-3. [Google Scholar]
- 91.Vaisitti T, Braggio E, Allan JN, et al. Novel Richter syndrome xenograft models to study genetic architecture, biology, and therapy responses. Cancer Res. 2018; 78(13):3413-3420. [DOI] [PubMed] [Google Scholar]
- 92.Arruga F, Bracciamà V, Vitale N, et al. Bidirectional linkage between the B-cell receptor and NOTCH1 in chronic lymphocytic leukemia and in Richter syndrome: therapeutic implications [published correction appears in Leukemia. 2020;34:1721]. Leukemia. 2020;34(2):462-477. [DOI] [PubMed] [Google Scholar]
- 93.Vaisitti T, Gaudino F, Ouk S, et al. Targeting metabolism and survival in chronic lymphocytic leukemia and Richter syndrome cells by a novel NF-κB inhibitor. Haematologica. 2017;102(11):1878-1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vaisitti T, Arruga F, Vitale N, et al. ROR1 targeting with the antibody-drug conjugate VLS-101 is effective in Richter syndrome patient-derived xenograft mouse models. Blood. 2021;137(24):3365-3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fiskus W, Mill CP, Perera D, et al. BET proteolysis targeted chimera-based therapy of novel models of Richter Transformation-diffuse large B-cell lymphoma [published online ahead of print 2 March 2021]. Leukemia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Iannello A, Vitale N, Coma S, et al. Synergistic efficacy of dual PI3K-d/g inhibitor duvelisib with Bcl2 inhibitor venetoclax in Richter syndrome PDX models. Blood. 2021;137(24):3378-3389. [DOI] [PubMed] [Google Scholar]
- 97.Nguyen KB, Spranger S. Modulation of the immune microenvironment by tumor-intrinsic oncogenic signaling. J Cell Biol. 2020; 219(1):e201908224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wierz M, Pierson S, Guyonnet L, et al. Dual PD1/LAG3 immune checkpoint blockade limits tumor development in a murine model of chronic lymphocytic leukemia. Blood. 2018;131(14):1617-1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rodriques SG, Stickels RR, Goeva A, et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science. 2019; 363(6434):1463-1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Goltsev Y, Samusik N, Kennedy-Darling J, et al. Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell. 2018;174(4):968-981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Angelo M, Bendall SC, Finck R, et al. Multiplexed ion beam imaging of human breast tumors. Nat Med. 2014; 20(4):436-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ioannou N, Hagner PR, Stokes M, et al. Triggering interferon signaling in T cells with avadomide sensitizes CLL to anti-PD-L1/PD-1 immunotherapy. Blood. 2021; 137(2):216-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Farinello D, Wozińska M, Lenti E, et al. A retinoic acid-dependent stroma-leukemia crosstalk promotes chronic lymphocytic leukemia progression. Nat Commun. 2018; 9(1):1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Heinig K, Gätjen M, Grau M, et al. Access to follicular dendritic cells is a pivotal step in murine chronic lymphocytic leukemia B-cell activation and proliferation. Cancer Discov. 2014;4(12):1448-1465. [DOI] [PubMed] [Google Scholar]
- 105.Kretzschmar K, Watt FM. Lineage tracing. Cell. 2012;148(1-2):33-45. [DOI] [PubMed] [Google Scholar]
- 106.Gutierrez C, Al’Khafaji A, Brenner E, et al. Multi-functional barcoding enables high resolution study of clonal dynamics. Nat Can. 2021;2;758-772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lareau CA, Ludwig LS, Muus C, et al. Massively parallel single-cell mitochondrial DNA genotyping and chromatin profiling. Nat Biotechnol. 2021;39(4):451-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ten Hacken E, Clement K, Li S, et al. High throughput single-cell detection of multiplex CRISPR-edited gene modifications. Genome Biol. 2020;21(1):266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Miles LA, Bowman RL, Merlinsky TR, et al. Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature. 2020;587(7834):477-482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Demaree B, Delley CL, Vasudevan HN, et al. Joint profiling of DNA and proteins in single cells to dissect genotype-phenotype associations in leukemia. Nat Commun. 2021;12(1):1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Penter L, Gohil SH, Lareau C, et al. Cancer Discov. 2021;candisc.0276.2021. [Google Scholar]
- 112.Rees HA, Liu DR. Base editing: precision chemistry on the genome and transcriptome of living cells [published correction appears in Nat Rev Genet. 2018;19:801]. Nat Rev Genet. 2018;19(12):770-788.30323312 [Google Scholar]