

Abstract

Asymmetric hydroalkoxylation of alkenes constitutes a redox-neutral and 100% atom-economical strategy toward enantioenriched oxygenated building blocks from readily available starting materials. Despite their great potential, catalytic enantioselective additions of alcohols across a C–C multiple bond are particularly underdeveloped, especially compared to other hydrofunctionalization methods such as hydroamination. However, driven by some recent innovations, e.g., asymmetric MHAT methods, asymmetric photocatalytic methods, and the development of extremely strong chiral Brønsted acids, there has been a gratifying surge of reports in this burgeoning field. The goal of this review is to survey the growing landscape of asymmetric hydroalkoxylation by highlighting exciting new advances, deconstructing mechanistic underpinnings, and drawing insight from related asymmetric hydroacyloxylation and hydration. A deep appreciation of the underlying principles informs an understanding of the various selectivity parameters and activation modes in the realm of asymmetric alkene hydrofunctionalization while simultaneously evoking the outstanding challenges to the field moving forward. Overall, we aim to lay a foundation for cross-fertilization among various catalytic fields and spur further innovation in asymmetric hydroalkoxylations of C–C multiple bonds.
1. Introduction
Hydrofunctionalization of a C–C multiple bond provides an atom- and step-economical strategy to introduce structural and stereochemical complexity toward value-added chemicals and medicinally germane compounds.1−5 Given its widespread synthetic utility, translating this approach into asymmetric hydrofunctionalization methods has garnered significant attention from the catalysis community.6 In particular, stereoselective additions of alcohols, carboxylic acids, and water across C–C multiple bonds (i.e., hydroalkoxylation, hydroacyloxylation, and hydration) are attractive redox-neutral tools for generating stereoenriched ethers, acetals, esters (including lactones), and alcohols.7−9 Given the prevalence of such motifs in bioactive compounds,10 stereoselective additions of O–H bonds across C–C multiple bonds have enabled access to key chemical synthons en route to natural products or derivatives thereof (Figure 1).11−18 However, catalyst-controlled enantioselective methods have remained relatively scarce compared to other asymmetric hydrofunctionalizations (e.g., hydroamination).19 We attribute the dearth of asymmetric methods to intrinsic challenges associated with such transformations rather than a lack of compelling interest to efficiently access the corresponding synthetically important functionalities. For example, akin to hydroamination reactions,20 while additions of O–H bonds to C–C double bonds tend to be thermodynamically feasible or thermoneutral (e.g., ΔG° = −4.1 kcal/mol for the hydration of 2-butene in H2O21), they are typically impeded by relatively high kinetic barriers. Further, garnering reactivity with relatively weak oxygen nucleophiles under reaction conditions conducive to asymmetric induction is challenging (N (MeNH2) = 15.19 in MeCN;22N (MeOH) = 6.86 in MeCN23), particularly with weakly Lewis basic alkenes.
Figure 1.

A survey of natural products or related structures prepared via stereoselective hydroalkoxylation or hydroacyloxylation.
Gratifyingly, as a result of key developments in a broad range of catalytic fields (transition metal catalysis, photocatalysis, organocatalysis, enzyme catalysis, heterogeneous catalysis, Lewis base catalysis), enantioselective additions of O–H bonds to unsaturated molecules have evolved considerably in recent years (Scheme 1). Building upon these successes, we find it timely to examine the current state-of-the-art research in catalyst-controlled stereoselective hydroalkoxylation while also considering potential growth areas for the future. In this review, we specifically aim to provide readers with a broad and encompassing overview of the diverse assortment of catalytic strategies employed in this developing field. Given the highly relevant mechanistic overlap, we also include several examples of asymmetric hydroacyloxylations and hydrations as well as cycloisomerizations involving the addition of an O–H group across a polyene scaffold. While we cover transformations of a broad range of C–C multiple bonds (alkenes, alkynes, allenes, and enol ethers), additions to alkenes bearing an electron-withdrawing group (Michael additions) are beyond the scope of this review. We encourage interested readers to consult more focused reviews on that topic.24,25
Scheme 1. General Overview of the Topics Discussed in This Review.

Central to the organization of the review, we categorize the reported literature based on the underlying reaction mechanism/catalytic activation mode. As such, we have broadly classified these reactions in seven different categories (sections 2–8). In each subfield, we provide a general overview of the methodology, focusing on the general principles governing regio- and/or stereoselectivity as well as specific challenges encountered for a given approach. Further, we have placed a special emphasis on the accompanied physical-organic and theoretical analysis that has played an important role in providing molecular level details into the mechanism and identify the existing bottlenecks that warrant further developments. Despite our earnest effort to categorize these reactions based on the underlying mechanism, overlap between subfields and/or a lack of conclusive experimental evidence can convolute unambiguous assignments. When relevant, we note such gray areas throughout our discussions. We conclude this review by highlighting some outstanding challenges and identifying potential areas of improvement that could provide an inspiration for future studies.
2. Transition Metal Catalysis
Owing to the versatile reactivity of metal π complexes, transition-metal catalysis has provided innumerable platforms for hydrofunctionalization reactions of C–C multiple bonds, including hydroamination, hydroformylation, and hydroboration reactions, among others.1−5,26 In recent years, considerable attention has been devoted to the employment of chiral ligand scaffolds and/or chiral anions to effect asymmetric variants of such processes. Interestingly, however, relative to other hydrofunctionalization strategies, transition metal-catalyzed asymmetric additions of oxygen nucleophiles to C–C multiple bonds are starkly underdeveloped.
It has been suggested that asymmetric hydroalkoxylations are particularly challenging for transition-metal systems due to hidden Brønsted acid catalysis, i.e., the propensity of metal complexes to release competent Brønsted acids that are responsible for nonasymmetric activity.27 In fact, it has been demonstrated that common conditions used for metal-catalyzed hydroalkoxylation lead to the formation of trifluoromethanesulfonic acid (triflic acid, TfOH), a strong Brønsted acid (pKa = −14.3 ± 2.0 in DMSO28) known to effect both intra- and intermolecular hydroalkoxylations.29,30 For example, the Hintermann group has shown that heating AgOTf in chlorinated solvents irreversibly forms TfOH (detected by 19F NMR spectroscopy), which the authors demonstrate as the active catalytic species in a phenol–isoprene cyclization (Scheme 2).27 The authors provide additional strong evidence that an early example of an asymmetric hydroalkoxylation using Cp*RuCl2 in the presence of AgOTf and a chiral bisphosphine ligand in toluene31 is catalyzed by in situ formed TfOH and disclose that the reported enantioselectivities are not reproducible (not depicted).
Scheme 2. Hidden Brønsted Acid Catalysis.

As such, it is imperative that hidden acid catalysis be considered during the development of asymmetric hydroalkoxylations and complexes bearing more basic ligands/anions might be preferred over triflates. As Hintermann points out, control reactions that simply replace metal triflates with equal loadings of TfOH and draw conclusions based on quantitative or qualitative differences in reactivity profiles can lead to a false impression that Brønsted acids are not involved in the main pathway of catalysis.27 Oftentimes, hidden acids can have advantages over pure TfOH and therefore such experiments can be misleading. For example, in a hydroalkoxylation reaction of dicyclopentadiene using Cu(OTf)2, the [Cu] species suppresses polymerization of the nucleophilic partner (2-hydroxyethyl methacrylate), and therefore the control experiment with pure TfOH resulted in gelation of the reaction mixture and poor overall yields of the desired product.32 Nevertheless, the authors provide strong evidence that TfOH is indeed the catalytically active species, underscoring the importance of well-designed control experiments.
To this end, protocols developed by Hintermann that deliberately generate hidden acids are encouraged as benchmark control experiments in reactions involving metal triflates.27 Additionally, one of the strongest arguments that a transition-metal complex is responsible for catalytic reactivity is the induction of high levels of enantioselectivity when employing a chiral ligand scaffold. We will herein delineate transition-metal-catalyzed methods that achieve enantioselectivity in hydroalkoxylation/acyloxylation reactions. When relevant, we will discuss possible hidden acid catalysis and highlight relevant mechanistic probes.
This section has been divided into two mechanistically distinct subclasses: (1) metal complexes that proceed through an inner-sphere mechanism (Scheme 3, left) and (2) those proceeding via an outer-sphere nucleophilic attack by the oxygenated species (Scheme 3, right).7 Whether a process follows an inner- or outer-sphere pathway can have important implications on the stereochemical outcome of a reaction and is dependent on a number of factors, including the nature of the unsaturated substrate, the transition metal involved and its corresponding coordination geometry, the ligand scaffold, the nucleophilic species, and the general reaction conditions. Recent studies have revealed relatively low barriers of migratory insertion of alkenes into M–OR bonds and have suggested that some methods previously considered outer-sphere might in fact proceed through inner-sphere mechanisms.33 This distinction can be especially ambiguous in Pd-catalyzed transformations. Hence, we recognize the ever-evolving nature of mechanistic postulation and have organized these methods based on current proposals.
Scheme 3. Two Mechanistic Subclasses of Transition Metal-catalyzed Hydroalkoxylation.

2.1. Inner-Sphere Mechanism
2.1.1. Alkenes
Metal-catalyzed additions of O–H bonds across C–C π bonds commonly proceed through an inner-sphere mechanism in the presence of electron-rich metals (e.g., Rh, Ir).7 Herein, we will consider two general mechanistic scenarios that govern hydroalkoxylations/acyloxylations of alkenes proceeding through nucleophilic activation (Scheme 4).34 Type I describes a redox neutral cycle initiated by protonolysis of a M–X bond to yield an alkoxy/acyloxy metal complex (M–OR, 1). Alkene coordination and subsequent 1,2-insertion into the M–OR bond results in the branched product following protonolysis. Alternatively, type II mechanisms proceed via oxidative addition into the O–H bond to form an alkoxy/acyloxy-hydrido metal complex (H–M–OR, 1′). Alkene coordination and subsequent 1,2-migratory insertion into the M–OR bond provides the terminal alkyl-hydrido metal complex (3′), which undergoes reductive elimination to form the Markovnikov adduct and regenerate the active catalyst. In either case, the regioselectivity of migratory insertion is typically dictated by the formation of the kinetically favored metal–alkyl complex (3 or 3′), in which the metal resides at the less encumbered carbon. Alternatively, from complex 2′, migratory insertion into the M–H bond could also be envisaged; however, insertion of alkenes into the M–H bonds of such complexes are rare and, to the best of our knowledge, are so far limited to examples proceeding through kinetically favored metallocycles (vide infra).8 As a result, transition-metal mediated hydroalkoxylation/acyloxylation of alkenes generally provides the branched adduct in high regioselectivities and pathways to linear adducts prove challenging and largely undiscovered. Notably, M–H insertions have been demonstrated in an anti-Markovnikov hydroalkoxylation of porphyrin-based Rh-alkyl substrates; however, these reactions are stoichiometric in Rh.35
Scheme 4. Two General Processes Describing Inner-sphere Mechanisms of Hydroalkoxylation.

In 2015, Ohmiya and Sawamura et al. reported an (R)-DTBM-Segphos-based Cu(I)-catalyzed asymmetric cyclization of 2,2-diphenylpent-4-en-1-ol to form the corresponding tetrahydrofuran with an er up to 85.5:14.5 in relatively low yield (Scheme 5).36 The authors propose a catalytic cycle consistent with type I, in which mesityl-Cu(I) undergoes protonolysis to form an alkoxy Cu(I) complex (and mesitylene), which proceeds through subsequent migratory insertion and finally protonolysis of the corresponding alkyl–M bond to form the cyclized product. Asymmetric induction is demonstrated with a single substrate and reactivity is generally limited to β,β-disubstituted alkenols containing a terminal alkene.
Scheme 5. Cu-Catalyzed Hydroalkoxylation of 2,2-Diphenylpent-4-en-1-ol.

Similarly, the Nishumura group developed an Ir-catalyzed hydroacyloxylation of geminally disubstituted alkenoic acids using (R)-DTBM-Segphos to yield γ-lactones with good levels of enantioselectivity (Scheme 6).37 Stoichiometric studies of the precatalyst in the presence of substrate revealed the formation of a hydrido iridium (Ir–H) complex, providing evidence that the reaction proceeds via an oxidative addition pathway. However, in contrast to the depicted type II mechanism, the authors propose a 2,1-insertion into the M–H bond to form the kinetically favored 6-membered iridacycle (4), ultimately yielding the Markovnikov adduct following reductive elimination. Notably, in each of the above examples, chiral ligands derived from 3,5-di-tert-butyl-4-methoxyphenyl (DTBM)-substituted bisphosphines prove dramatically superior at enantioinduction compared to other examined ligand scaffolds.
Scheme 6. Ir-Catalyzed Hydroacyloxylation toward γ-Lactones.

More recently, Chemler and co-workers described an asymmetric cyclization of a range of 4- and 5-substituted alkenols catalyzed by a chiral Cu(II)-bis(oxazoline) (BOX) complex to yield the corresponding 5- and 6-membered cyclic ethers with moderate to high levels of enantioselectivity (Scheme 7).38 The authors suggest that the catalytic cycle commences with anion exchange of the Cu–OTf precatalyst with the alkenol followed by ligand dissociation to form a cationic alkoxy Cu intermediate. An enantiodetermining oxycupration subsequently provides an alkyl Cu species (7) that undergoes homolytic C–Cu bond cleavage to render an alkyl radical (8). A subsequent hydrogen atom transfer (HAT) from 1,4-cyclohexadiene yields the product and a stoichiometric oxidant (MnO2 or Ag2CO3) regenerates the active Cu(II) species. The addition of K2CO3 seemingly prevents the formation of TfOH, suppressing hidden acid catalysis and enabling high levels of enantioinduction. However, the enantioselectivity is essentially unaffected without added K2CO3 (using either MnO2 or air as the oxidant), albeit with reduced yields. This experiment suggests that, if TfOH does form, the Cu complex outcompetes the acid pathway in the formation of the product (though the authors do not comment on side reactivity).
Scheme 7. Cu(BOX)-Catalyzed Hydroalkoxylation.

While each of the above methods have provided access to a number of enantioenriched cyclic ethers and lactones, there is a clear pattern of substrate engineering tailored to the Thorpe–Ingold effect, such that the reactivity scope of transition-metal catalyzed intramolecular hydroalkoxylations remains inherently limited. Efforts toward the development of asymmetric methods employing simpler alkenols/alkenoic acids are encouraged. For example, Marks and co-workers have reported organolanthanide-catalyzed intramolecular Markovnikov hydroalkoxylations of a diverse range of alkenols, including the cyclization of simple pent-4-en-1-ol to 2-methyltetrahydrofuran.39 While a free TfOH-catalyzed process as a major pathway has tentatively been ruled out, asymmetric variants of these methods have not been reported.
Another significant challenge in the development of transition metal-catalyzed asymmetric hydroalkoxylations and hydroacyloxylations is their extension to intermolecular systems. The relatively low basicity of simple C–C double bonds renders the formation of metal π complexes with alkenes extremely challenging and reactivity typically depends on either high alkene loadings or Lewis basic groups tethered to the alkenic unit.40,41 In 2013, the Hartwig group impressively showed that the combination of Ir and (S)-DTBM-Segphos catalyzes the addition of phenols to structurally simple alkenes (solvent quantities) in good yields and poor to moderate enantioselectivities (Scheme 8).19 A number of experiments (including the use of chiral ligands and measurable enantioinduction with all substrates) provide strong evidence that the reaction is indeed catalyzed by the metal complex and that hidden Brønsted acid catalysis is either limited or completely suppressed. The kinetic profile of the reaction is consistent with a type II redox mechanism and a turnover-limiting oxymetalation step via an alkoxy-hydrido Ir(III) complex. As with the aforementioned intramolecular examples, this method highlights the privileged nature of the DTBM-derived ligand scaffolds, as all other evaluated ligand classes provided little to no reactivity.
Scheme 8. Ir-Catalyzed Intermolecular Hydroalkoxylation of Simple Alkenes.

This method reveals two additional challenges facing transition-metal-catalyzed hydroalkoxylations. First, alkene isomerization leads to unreactive internal C–C double bonds, further necessitating high loadings of the starting terminal alkene. Notably, alkene isomerization is more significant in this hydroalkoxylation with phenol than in a similar Ir-catalyzed hydroamination published by the same authors.42 In this hydroalkoxylation case, the phenol unit is not basic enough to stabilize the Ir complex and, consequently, an 18 electron Ir(III) allyl hydride complex is the catalyst resting state (Scheme 8, structure 9), which leads to increased isomerization. A second challenge is that the alkyl Ir complex following oxymetalation is prone to β-hydride elimination and the corresponding enol ethers are observed in moderate quantities. Further, because the alkene is the terminal reductant for the enol ether side product, the corresponding saturated alkanes are also observed. This represents a general challenge in similar hydroamination strategies, as analogous enamines are typically observed.42,43 Despite these drawbacks, this method demonstrates the robust capacity of transition-metal systems to engage weakly basic and unfunctionalized alkenes and effect asymmetric hydroalkoxylation reactions. We anticipate improved chemo- and enantioselectivities in future developments.
2.1.2. Allenes and Alkynes
The increased Lewis basic nature of allenes and alkynes compared to alkenes increases the propensity of these functionalities to form metal π complexes and subsequently participate in organometallic reactions. Additionally, the degree of unsaturation that remains following metal-catalyzed reactions of allenes and alkynes has been shown to stabilize metal complexes through π-coordination and accelerate catalytic processes.44 We will herein describe transition-metal-catalyzed asymmetric hydroalkoxylations and hydroacyloxylations of allenes and alkynes involving inner-sphere attack of the nucleophile, a field so far dominated by Rh catalysis. For the methods described in this section, alkynes and allenes proceed through a common catalytic intermediate, and therefore we will discuss these functionalities in parallel.
The seminal report on intermolecular asymmetric hydroalkoxylations of allenes was published by Nishimura and Hayashi et al. in 2009 and describes a DTBM-Segphos-based Rh-catalyzed addition of phenols to diphenylphosphinylallenes to yield vinyl ethers in high yields and enantioselectivities (Scheme 9).45 On the basis of 1H and 31P NMR studies, the authors propose that the reaction proceeds via protonolysis of the Rh precatalyst to form a phenoxorhodium species that reacts with the allene substrate to concomitantly forge a C–O bond and generate a π-allylrhodium intermediate (10). Protonation of the π-allyl system with phenol regenerates the active catalyst and yields the enantioenriched enol ether product. While this method provides precedence for an enantioselective intermolecular Rh-catalyzed hydroalkoxylation of allenes, the dependency on diphenylphosphoryl substituents imposes limitations on its synthetic applicability and elicits the development of more general catalytic solutions.
Scheme 9. Intermolecular Rh-catalyzed Hydroalkoxylation of Diphenylphosphinylallenes.

To this end, Trost and Yamamoto published a series of methods throughout the 1990s on Pd-catalyzed additions of nucleophiles (including carboxylic acids and alcohols) to alkyl-substituted allenes and alkynes.46−53 A key mechanistic feature of these reactions is the formation of a Pd π-allyl intermediate, which undergoes a regioselective outer-sphere nucleophilic attack to yield linear allylic products. A few enantioselective Pd-catalyzed hydroalkoxylations and hydroacyloxylations have been developed based on this work and will be discussed in section 2.2.1. Alternatively, Evans and others have shown that Rh-catalyzed allylic substitution reactions (which proceed through analogous Rh π-allyl intermediates) result in high levels of regioselectivity toward branched allylic compounds.54,55 As such, Breit recognized that Rh-catalyzed nucleophilic additions to alkynes and allenes could offer a platform for asymmetric syntheses of branched allylic compounds, avoiding stoichiometric byproducts and offering complementary regioselectivity to related Pd-catalyzed transformations.56
Accordingly, in 2011, Breit and co-workers described a highly enantioselective Rh-catalyzed hydroacyloxylation of terminal allenes to form allylic esters using (R,R)-DIOP as a chiral ligand (Scheme 10, from allene).57 Notably, the method is tolerant of free alcohols and could be used to form a quaternary stereocenter with excellent enantioselectivity. Additionally, the Breit group has shown the robustness of this method with the formation of various macrocyclic scaffolds, as well as key intermediates en route to several natural products.58−63
Scheme 10. Rh-Catalyzed Hydroacyloxylations of Allenes and Alkynes.

The authors propose that the reaction proceeds via an oxidative addition into the carboxylic acid to generate acyloxy-hydrido rhodium complex 11, which, following allene coordination, undergoes a hydrometalation to generate Rh π-allyl intermediate 13. An ensuing reductive elimination yields the branched allylic ester and regenerates the Rh(I) catalyst (Scheme 11, cycle I). However, the authors have not ruled out an outer-sphere nucleophilic attack.
Scheme 11. Mechanistic Proposal for Rh-Catalyzed Hydroacyloxylations of Allenes and Alkynes.
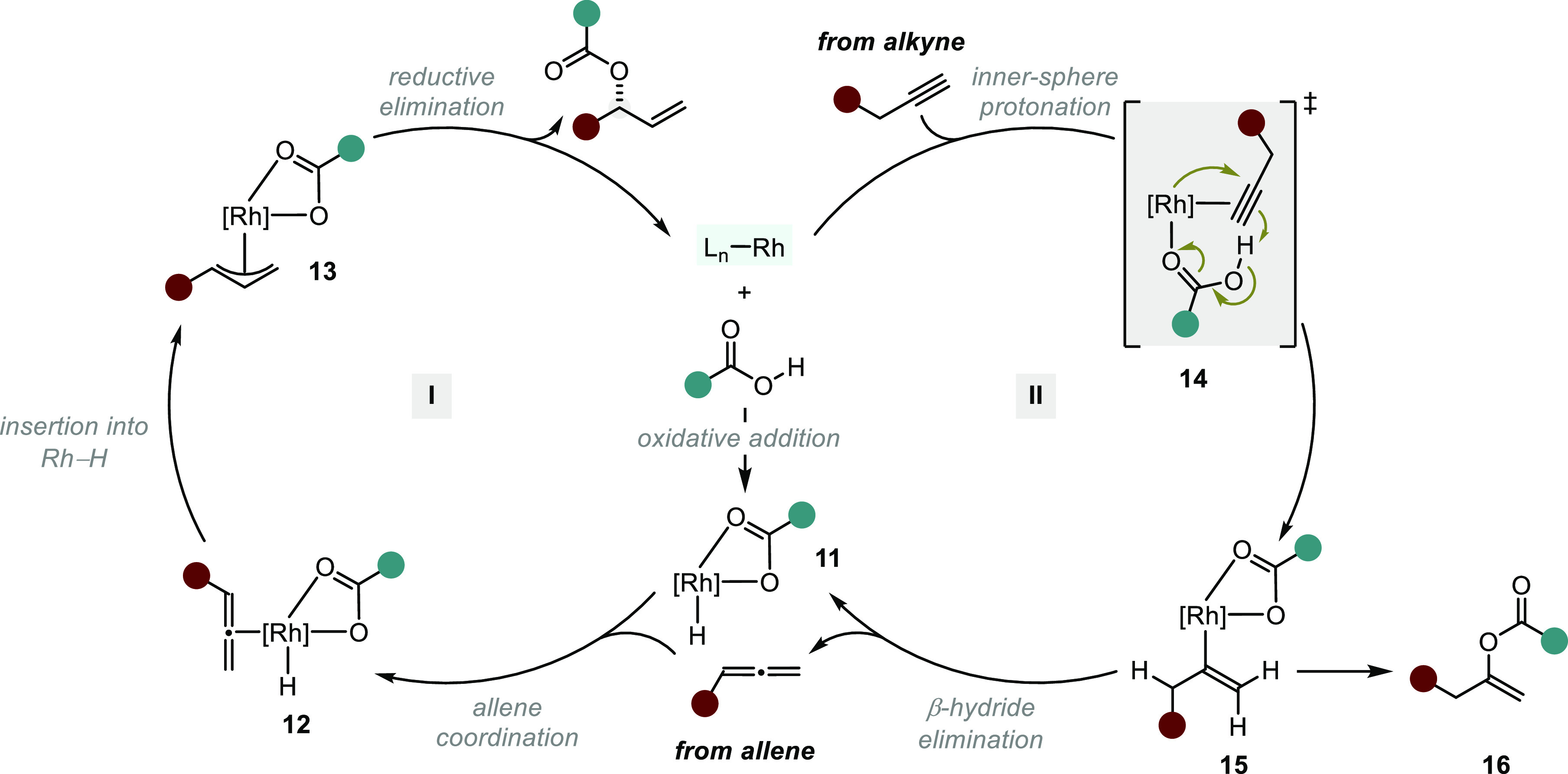
Breit and co-workers later expanded this methodology by capitalizing on the well-established proclivity of alkynes to isomerize to allenes in the presence of Rh–H complexes.64 On the basis of their previous work using an achiral phosphine ligand,65 Breit et al. developed a Rh(I)/(R,R)-Cp-DIOP-catalyzed enantioselective hydroacyloxylation of alkynes (Scheme 10, from alkyne).66 Mechanistic studies, including DFT calculations and extensive experimental investigations, suggest that the isomerization pathway proceeds via an inner-sphere protonation of the terminal alkyne by a Rh-coordinated carboxylic acid, leading to the formation of 15 (Scheme 11, cycle II).67 This is mechanistically distinct from related Pd-catalyzed isomerizations, which are thought to proceed through hydrometalation pathways. From complex 15, β-hydride elimination releases an allene, and concurrently generates the acyloxy-hydrido complex (11) and enters the aforementioned catalytic cycle I. Alternatively, reductive elimination of complex 15 yields gem-enol ester 16, a side product observed in relatively low quantities.
In 2016, the same group demonstrated a Rh(I)/diphenyl phosphoric acid-catalyzed enantioselective hydroalkoxylation of allenes and internal Me-substituted alkynes (Scheme 12).68 A wide range of simple and functionalized alcohols were tolerated, including a later addition of N-hydroxyphthalimides,69 which undergo facile cleavage to furnish enantioenriched allylic alcohols. The mechanism is similar to that proposed for the addition of carboxylic acids; however, in this case, the phosphoric acid is used to generate the Rh–H intermediate and subsequently form the electrophilic Rh π-allyl species 18. Anion exchange yields the alkoxy Rh π-allyl intermediate 19, followed by reductive elimination to furnish the branched allylic ether product. The authors can again not rule out the possibility of an outer-sphere nucleophilic attack of the alcohol on the Rh π-allyl species. We wonder if enantioinduction would be observed using a chiral phosphoric acid in combination with an achiral phosphine ligand, which could probe the inner- vs outer-sphere nature of this step.
Scheme 12. Rh-Catalyzed Hydroalkoxylation of Allenes and Alkynes.

2.2. Outer-Sphere Mechanism
Most transition metals, by virtue of their low-lying vacant d-orbitals, are able to bind carbon–carbon multiple bonds as π-Lewis acids. In contrast to inner-sphere hydrofunctionalizations that proceed via migratory insertion of a coordinated π-bond into a metal-alkoxide, nucleophilic addition can occur from outside the ligand sphere of the metal; an elementary step referred to as outer-sphere attack (Scheme 13). Following addition, the newly formed alkyl-metal bond is cleaved through protonolysis to regenerate the active catalyst and yield the hydroalkoxylation product. While enantioselectivity can be induced by a chiral neutral metal-bound ligand, an intrinsic challenge for asymmetric induction originates from the distal nucleophilic attack, a factor that can be increasingly problematic depending on the coordination geometry of the metal complex (vide infra). Alternatively, asymmetric counteranion directed catalysis (ACDC) has emerged as a powerful tool for enantioselective hydrofunctionalizations. In this section, we will elaborate on both concepts through the aid of selected examples. Here, we will exclusively discuss examples where the newly formed C–O bond is part of a carbon stereocenter. Additionally, numerous examples of desymmetrizing hydroalkoxylations and hydroacyloxylations can be found in the literature that render the transformation asymmetric only by creation of a stereocenter distal to the reactive site.70−73 These transformations therefore lie outside the objective of this review.
Scheme 13. General Outer-sphere Mechanism Governing Lewis Acid-catalyzed Hydroalkoxylations.

2.2.1. Chiral Metal–Ligand Scaffolds
In this section, we will highlight selected hydrofunctionalizations that proceed via outer-sphere nucleophilic attack, where stereoselectivity is induced by a chiral, neutral, metal-bound ligand. We will discuss the examples in order of the corresponding Lewis acidic metal and will start with a mechanistically distinct type of catalysis.
Similar to previously discussed inner-sphere Rh-catalyzed hydroalkoxylations and hydroacyloxylations of alkynes and allenes (section 2.1.2), outer-sphere hydrofunctionalization can be accomplished through Pd catalysis (Scheme 14).46 A key mechanistic feature is the formation of a palladium hydride 20 upon oxidative addition into an acidic O–H bond (alcohol or carboxylic acid). A series of hydropalladation and β-hydride elimination reactions lead to allyl-Pd complex 23 via an intermediate vinyl-palladium 21 and Pd–allene complex 22. Ultimately, outer-sphere nucleophilic attack generates the product and, together with simultaneous or subsequent proton transfer, the initial Pd-hydride 20. Asymmetric intramolecular alkoxide additions to allyl–palladium intermediates generated from allylic systems have also been reported in an overall oxidative fashion. We will not explicitly elaborate on these examples, as the overall transformation does not classify as a hydrofunctionalization.74
Scheme 14. Mechanism of Palladium(0) Catalyzed Intramolecular Hydroalkoxylation of Alkynes.

On the basis of this concept, Yamamoto and co-workers achieved an intramolecular asymmetric hydroalkoxylation of alkynes (Scheme 15).75 Through the use of a chiral bisphosphine ligand, a palladium(0) source, and benzoic acid, an alkyne is converted to the corresponding allyl–palladium complex, which is ultimately intercepted by the alcohol. The authors were thus able to obtain furans, pyrans, as well as isochromanes in moderate to excellent yields and good enantioselectivity. Importantly, the origin of stereoinduction could not be unambiguously disclosed by the authors, as neither an allene, nor a 1,3-diene, both well-known precursors to Pd(II)-allyl complexes, could be reacted with high enantioselectivity. Computations suggest that the initial alkyne complexation already determines the enantioselectivity and that the corresponding Lewis acidic enantiopure palladium(II) complex remains closely associated to the π-cloud throughout the reaction mechanism.
Scheme 15. Palladium-catalyzed Intramolecular Hydroalkoxylation of Alkynes via Intermediate Allyl–Pd Complexes.

An intermolecular palladium-catalyzed hydroalkoxylation via allyl–Pd intermediates was realized by Rhee et al. in 2014 (Scheme 16).76 Starting from terminal alkoxyallenes and simple enantiopure secondary alcohols, using Trost ligand 22, the authors obtained acetals 23 that were in situ cyclized via olefin metathesis to the corresponding dihydropyrans. Under the influence of either enantiomer of the chiral ligand, opposite diastereomers of the products could be isolated with good selectivity, highlighting the catalyst control over diastereomeric reaction pathways. Notably, the products could be further elaborated to valuable glycoside building blocks by dihydroxylation. Additionally, Overman and co-workers later employed this method to access an enantiopure 3-chloro-5-alkoxybutenolide en route to several natural products.16,77 Further, Cao and co-workers later disclosed an asymmetric addition of phenols to alkoxyallenes using a Trost ligand in the presence of Pd(0) to yield acyclic O,O-acetals.78 On the basis of experimental evidence, the authors propose that the enantiodetermining step might be the insertion of the Pd–H bond into the allene system.
Scheme 16. Palladium-catalyzed Asymmetric Intermolecular Hydroalkoxylation of Alkoxyallenes.

In a purely π-Lewis-acidic activation mode, an early and rare example of an intermolecular asymmetric hydroalkoxylation was reported by Katsuki and Nagano in 2002 (Scheme 17).79 The authors were able to achieve high stereoselectivity in the acetalization reaction of electronically biased primary or secondary alcohols with dihydrofuran under the influence of a chiral Ru(II)–salen complex. Notably, the reaction of either enantiomer of the starting material provided the THF-protected compounds in good diastereoselectivity, showcasing efficient catalyst-control even though a small matched–mismatched effect was observed. Mechanistically, catalyst 24 releases nitrous oxide upon irradiation,80 providing the active Lewis acidic ruthenium(III) catalyst with an open coordination site for substrate binding.
Scheme 17. Ruthenium-catalyzed Asymmetric Intermolecular Acetal Formation.

Through the use of highly Lewis acidic platinum(II) pincer complexes, Gagné and Nguyen achieved a catalytic enantioselective cycloisomerization of polyenic compounds (Scheme 18).81 The Pt-bis(oxazoline)pyridine (PyBOX) precatalyst 25 is activated by silver tetrafluoroborate to generate a dicationic Pt(II) catalyst with noncoordinating anions enabling strong substrate binding and consequently high reactivity. In the reaction of several linear aromatic and aliphatic substrates, the authors were able to obtain cycloisomerized products in good to excellent yields, however only with poor enantioselectivities. The authors propose a catalytic cycle that begins with Lewis acidic activation of the least substituted C=C bond via coordination to the (PyBOX)Pt2+.
Scheme 18. Platinum-catalyzed Asymmetric Polyene Cyclizations.

After the cyclization cascade, the former OH group transfers a proton which rapidly protodemetalates the in situ generated alkyl-platinum intermediate and thus releases the catalyst. The putative mechanism is supported by deuterium labeling studies and other control experiments. It is of importance to state that these types of polyene cyclizations do not formally represent a hydroalkoxylation, as an OH moiety is not added across a single multiple bond. However, the reaction can be perceived as an extended hydroalkoxylation across multiple double bonds, resulting in ring formations. We therefore include polyene cyclizations in this discussion for their mechanistic relevance and as a means to highlight catalytic approaches that could enable future progress in the field.
In another report showcasing the ability of platinum to activate C–C double bonds, Xu and co-workers recently described a platinum(II)-catalyzed intramolecular hydroalkoxylation of alkenes (Scheme 19).82 The author’s approach relies on the design a of “donor–acceptor” type bifunctional Pt(II)-catalyst, where the monodentate phosphine ligand contains a basic imidazole unit that can engage in hydrogen bonding with the nucleophilic alcohol to form a well-defined TS 27. Thus, with additional aid of a Thorpe–Ingold effect in the substrate, a chiral tetrahydrofuran was obtained in moderate yield, albeit with poor enantioselectivity. Interestingly, removal of the basic functionality in the catalyst lead to complete erosion of enantioselectivity, providing some proof for the mechanistic hypothesis. However, it should be noted that the reaction conditions employed by the authors (AgOTf in DCE at 50 °C) have been demonstrated by Hintermann to lead to the formation of TfOH via decomposition of the solvent (see Scheme 2). Although the authors describe a set of control experiments to rule out hidden acid catalysis, we believe the results are not conclusive and a more rigorous analysis would be adequate. While the observed enantioinduction does indeed imply the active involvement of platinum complex in the hydroalkoxylation, due to the low level of selectivity, nonasymmetric background reactivity cannot fully be ruled out. An additional role of the basic functionality in the catalyst might be the capture and deactivation of small amounts of triflic acid rather than the active involvement in the stereodetermining cyclization step.
Scheme 19. Platinum-catalyzed Asymmetric Intramolecular Hydroalkoxylation of Alkenes.

Lewis acidic gold complexes have been widely employed in hydrofunctionalizations of allenes and alkynes. Some important features in Au(I) catalysts that define their reactivity include: (a) the propensity to coordinate to C–C multiple bonds with a strong kinetic preference to react with alkynes, (b) a well-defined and modular catalyst structure with a strong metal–ligand bond, (c) “aurophilic” behavior, i.e., a stabilizing Au–Au interaction with the magnitude of a hydrogen bond, and (d) the linear bidentate coordination geometry of Au(I) complexes (which consequently mandates outer-sphere attack) (Scheme 20).83 Thus, a chiral ligand is placed opposite to the outer-sphere approach of a nucleophile, rendering enantiocontrol especially difficult. Additionally, the reactivity of Au(I)-catalysts is largely defined by the properties of the corresponding counteranion, with noncoordinating ions dissociating faster from the metal center and thus facilitating substrate binding. As a general observation, Au-catalyzed asymmetric transformations have been most successful with chiral binuclear catalysts of the type L(AuX)2. Additionally, the prominent strategy to in situ activate Au precatalysts by chloride abstraction using silver salts introduces a second transition metal in the reaction mixture, leading to potential oligomerization and loss of the well-defined catalyst structure due to the well-known silver effect.84
Scheme 20. Coordination Sphere of Au(I) Complexes and the Associated Difficult Stereoinduction by the Ligand.

Seminal contributions to ligand controlled asymmetric Au catalysis were made by the Widenhoefer et al., who documented the first enantioselective hydroalkoxylation of allenes catalyzed by a dinuclear Au(I) complex, formed upon activation of precatalyst (S)-28(AuCl)2 with AgOTs (Scheme 21).85 The transformation gives rise to chiral THFs and THPs in good to excellent yield and moderate to excellent selectivity; however, a significant drop in enantioselectivity was observed when employing a substrate bearing no substituents along the tether. Control experiments were conducted using a chiral AgI–phosphine complex or a chiral phosphonium salt to rule out potential background catalysis. Similar asymmetric transformations have since been reported, although no high enantioselectivity was observed for the examined ligand systems.86,87
Scheme 21. First Gold(I)-catalyzed Asymmetric Intramolecular Hydroalkoxylation of Allenes.

To move away from dinuclear Au complexes, Fürstner et al. designed and synthesized a series of TADDOL-related phosphoramidite ligands, one of which was successfully employed in an asymmetric cyclization of a hydroxyallene compound to a substituted tetrahydrofuran in excellent yield and enantioselectivity (Scheme 22).88 Notably, the authors observe a pseudo C3 symmetric ligand sphere around the Au(I) cation in the solid state of the catalyst. It was rationalized that an extended arene would allow the ligand by virtue of attractive π interactions to reach to the opposite site of the metal center and thus enable enantioinduction in an outer-sphere nucleophilic attack. A striking feature of these reactions is that the sense of asymmetric induction in the cyclization can be inverted solely by changing the solvent or the temperature or the escorting counteranion (e.g., from BF4– to CF3CO2–). To gain insights into this intriguing behavior, the authors subsequently conducted experimental as well as computational mechanistic studies.89 Key findings include an apparent change of the stereodetermining step at reduced temperatures as well as a strong entropic contribution to the ΔΔG⧧ of the diastereomeric transition states, leading to pronounced temperature dependency. Additionally, the proton-transfer and protodeauration steps seem to be highly dependent on the choice of solvent and counteranion, with protic solvents and coordinating ions (CF3CO2–) serving as proton shuttles. Remarkably, this study sheds light on the importance of entropic changes along the reaction coordinate that remain underappreciated in asymmetric catalysis.
Scheme 22. Phosphoramidite Gold(I)-catalyzed Asymmetric Intramolecular Hydroalkoxylation of Allenes.

In an impressive display of the ability of chiral phosphine–gold(I) complexes to activate alkynes, Toste and co-workers reported in 2010 enantioselective polyene cyclization cascade reactions, leading to overall hydrophenoxylations and hydroacyloxylations (Scheme 23).90 While analogous reactions involving the initial activation of an alkene were reported, the initiation of an asymmetric polyene cyclization by Lewis acid activation of an alkyne had remained elusive. The authors postulate that the coordination of the chiral biphenylphosphine-based Au(I) catalyst to the triple bond initiates a 6-exo-dig cyclization, while enantioselectivity is translated by preorganization of the polyene throughout the whole cyclization cascade according to the Stork–Eschenmoser postulate (via 30).91 Consequently, cis-fused decaline systems could also be obtained from the corresponding (Z)-alkenes, albeit with reduced enantioselectivity. The reaction conditions are suitable for terminal as well as internal alkynes (even though higher temperature and longer reaction times are necessary for the latter). Impressively, high enantioselectivity was not only obtained in bicyclization reactions but also in the homologated tricyclization process toward tetracyclic scaffolds.
Scheme 23. Gold(I)-catalyzed Asymmetric Polyene Cyclizations.

A rationally designed bifunctional chiral Au(I) complex was developed by Zhang et al. in 2019 and utilized in an asymmetric isomerization of an alkyne to the corresponding chiral allene and a subsequent stereospecific intramolecular hydroalkoxylation to yield 2,5-dihydrofurans in good yield and stereoselectivity (Scheme 24).92 Importantly, starting from enantiopure secondary alcohols, the diastereomeric ratio in the cyclization product was controlled by the catalyst and not by the substrate. To achieve efficient asymmetric isomerization, the authors deliberately introduced a basic tertiary amine into the ligand that deprotonates the substrate in the propargylic position upon Lewis acid activation by the gold complex. Interestingly, while the precatalyst [(R)-31]AuCl exists as a mixture of atropisomers that were isolated and analyzed by single crystal structure analysis, anion exchange to a noncoordinating tetraarylboronate at elevated temperatures lead to quantitative equilibration of the complex to a single isomer with (Ra,R)-configuration. With this in situ generated catalyst isomer, the authors developed a stereochemical model where a syn-periplanar Au(I)-assisted deprotonation and subsequent protodeauration lead to enantioenriched Au complex 32, from which stereospecific cyclization provides the desired product.
Scheme 24. Gold(I)-catalyzed Asymmetric Alkyne Isomerization and Intramolecular Hydroalkoxylation.

2.2.2. Asymmetric Counteranion Directed Catalysis
In the previously discussed section, neutral chiral ligands coordinate tightly to the metal, and the stereoselectivity is induced during the interaction of these complexes with the substrates. However, complexes with cationic metal centers are necessarily associated with anions or anionic ligands. When those anions are chiral and enantiomerically pure, the corresponding reactions can be broadly classified as asymmetric counteranion directed catalysis (ACDC). In this context, chiral anions, for example, BINOL derived phosphates, in addition to neutralizing charge or acting as a (weak) ligand, are capable of engaging in interactions such as hydrogen bonding and deprotonation. There exists a continuum of possibilities, where selectivity is dependent with varying extent on electrostatic, noncovalent, and covalent interactions. A detailed discussion of ACDC is beyond the scope of this review, and interested readers are advised to consult the literature.93,94
The area of gold catalysis has offered a fertile breeding ground for the development of ACDC, as often the use of traditional ligand scaffolds result in poor selectivity due to the linear coordination geometry around the metal center (Scheme 20). On the other hand, the counteranion in Au(I) catalysis necessarily needs to dissociate from the metal center to allow substrate coordination. Thus, it is possible to create a chiral ion pair with the metal-bound substrate and guide nucleophilic approach from outside the ligand sphere (Scheme 25). It has moreover been observed that a chiral counteranion can be combined additively with a chiral ligand to enable asymmetric transformations. In this section, we will focus on examples where π-Lewis acidic metals are employed in the context of asymmetric counteranion directed hydroalkoxylation. A separate section related to ACDC within the realm of chiral Brønsted acids can be found in section 5.
Scheme 25. Coordination Sphere of Au(I) Complexes under the Influence of a Chiral Anion.

Pioneering synthetic method development toward asymmetric chiral counteranion directed hydroalkoxylation can be attributed to Toste and co-workers (Scheme 26).95 Their first example of an intramolecular asymmetric hydroalkoxylation of an allenol can be perceived as further development of the seminal chiral ligand-controlled contribution by Widenhoefer et al. (Scheme 21). The authors found that a Au precatalyst with an achiral ligand scaffold (dppm) could be employed in the presence of (R)-TRIP as a chiral counteranion to effect high levels of enantioinduction in intramolecular cyclizations. The method was found to be compatible with a variety of allenol substrates, including α- or β-substituted alcohols of varying tether linkage. Notably, the synergistic nature of the ligand scaffold and chiral counteranion can be appreciated in the cyclization to form THF 33. In this case, the anion alone induces 80% ee (er = 90:10) when the reaction is performed with dppm; however, when employing a chiral-at-phosphorus ligand (DIPAMP) together with the (R)-TRIP counteranion, enantioinduction is improved to 92% ee (er = 96:4). Control experiments rule out the possibility of background catalysis by the AgTRIP-salt or the corresponding CPA alone. Consistent with an ion-pair model, it was observed that nonpolar solvents gave optimal enantioselectivity while more coordinating solvents lead to erosion of selectivity due to attenuation of crucial electrostatic effects (solvent-separated ion pair). Furthermore, the authors were able to extend the concept to asymmetric hydroacyloxylations of allenes by employing (S)-BINAP as a chiral ligand and (R)-TRIP as a chiral counteranion. These reactions exhibit a strong matched–mismatched effect: When (R)-Ag-TRIP is combined with the antipodal (R)-BINAP(AuCl)2, the product is formed in a near-racemic fashion. Such behavior has previously also been described in organocatalytic reactions involving ion pair catalysts consisting of chiral ammonium cations with chiral phosphate counteranions.96 Further, this methodology was successfully applied to intramolecular asymmetric hydroamination and has since also been demonstrated in intramolecular asymmetric aminoxylations of allenes.97 Additionally, hydroxyl-allenecycloisomerizations have been reported under ACDC using a slightly different catalytic system.98
Scheme 26. Gold(I)-catalyzed Asymmetric Hydroalkoxylation of Allenes via ACDC.

A desymmetrization of allene-containing 1,3-diols has been contributed by Toste and co-workers using a purely asymmetric counteranion directed protocol with an achiral dppe-ligand and (R)-Ag-C8-TRIP (Scheme 27).99 Under optimized reaction conditions, tetrahydrofurans were formed in good yield and with high selectivity. However, the homologated tetrahydropyrans displayed reduced enantioselectivity. Mechanistically, the authors did not observe a nonlinear correlation between the silver salt’s ee and the product ee, suggesting the influence of only a single chiral counteranion in the enantiodetermining step. The structure of the active dinuclear catalyst therefore remains of interest, especially with regard to the ongoing aspiration to design more efficient catalyst scaffolds.
Scheme 27. Gold(I)-catalyzed Asymmetric Desymmetrizing Hydroalkoxylation of Allene-Containing Diols.

Brimble and co-workers have leveraged a chiral counterion-based strategy to achieve an asymmetric dihydroalkoxylation of suitably crafted alkyne-diols toward valuable spiroacetals (Scheme 28).100 Under the influence of a Au(I)-catalyst possessing both a chiral bisphospine ligand as well as a chiral phosphate anion, separable regioisomeric mixture of spiroacetals (34 and 35) were obtained in quantitative yield but with poor regioisomeric ratio and moderate enantioselectivity. The intermediacy of a putative enol ether has been verified by a control experiment, in which subjecting the separately prepared intermediate to the reaction condition leads to the experimentally observed product with identical selectivity. A later example by Rexit and Mailikezati adopted a modified approach to successfully catalyze an asymmetric dihydroxylation to yield spiroacetals in good yields and enantioselectivities.101 As will be discussed in section 5.1, highly enantioselective spiroacetalizations have been achieved under the influence of confined organic Brønsted acid catalysts.102
Scheme 28. Gold(I)-catalyzed Asymmetric Dihydroalkoxylation of Alkynes toward Spiroacetals.

While it is very prominent within Au(I) catalysis, the use of chiral counteranions to induce enantioselectivity has also been extended to other π-Lewis acidic metals. We will conclude this section by highlighting some of these examples. Whereas chiral silver-phosphates play a crucial role in generating an active Au(I) catalyst, Ag(I) complexes can also display catalytic activity themselves. In 2012, Hong and co-workers reported a chiral silver(I)-catalyzed kinetic resolution of racemic allenic secondary alcohols toward the cycloisomerized products (Scheme 29).103 Using a BINOL-derived silver phosphate, good selectivity (s up to 189) was observed for a wide range of substrates, although high catalyst loading was required. To rationalize the observed stereochemical outcome, the authors propose a geometric model in which the allene coordinates to the silver to form an η2 complex. The different stability of the diastereomeric complexes formed upon complexation of either enantiomer of the starting material might account for the cyclization rate difference required for the kinetic resolution. On the basis of the steric contour of the four schematic quadrants created by the chiral phosphate anion, the (R)-enantiomer of the starting material will preferentially coordinate to the metal, as the sterically demanding substituent will be able to reside in the unhindered area. Additional support for this model arises from the observation that both tertiary as well as C2-substituted allenic alcohols cyclize with drastically reduced selectivities, presumably due to steric clash of the additional substituent with the phosphate anion. An additional mechanistic possibility includes hydrogen bonding of the phosphate anion to the free OH to increase its nucleophilicity and create a well-defined transition state.
Scheme 29. Silver(I)-catalyzed Kinetic Resolution of Allenic Alcohols.

In 2012, Hii and co-workers disclosed an asymmetric silver(I)-catalyzed intramolecular hydroalkoxylation and hydroacyloxylation of allenes (Scheme 30) under the influence of chiral phosphate anions.104 Using the silver salts of 6-phospha-2,4,8-trioxa-adamantane (β-CgPO2Ag, 37) or TADDOL derived phosphoric acid ((R,R)-TADDOL-PO2Ag, 38), the authors were able to obtain cycloisomerized products in quantitative yield but only with poor enantioselectivity. Additionally, the protocol relies on the use of geminal disubstituted starting materials, a setback that was already disentangled by Au(I) catalysis (Scheme 26).
Scheme 30. Silver(I)-catalyzed Hydroacyloxylations and Hydroalkoxylations of Allenes.

Similar to what was already discussed within the realm of Au(I) catalysis, a desymmetrization of allenic 1,3-diols has been disclosed by Cao and Zheng via Pd(II) and CPA cocatalysis.105 Under the influence of catalytic amounts of Pd(OAc)2 and SPINOL-derived phosphoric acid (R)-STRIP, substituted dihydrofurans could be obtained in excellent yields and moderate enantioselectivities (Scheme 31). On the basis of literature precedence, the authors propose that the phosphate serves as both an anionic ligand to the metal center and as the base to simultaneously activate one of the alcohols, thus inducing stereoselectivity (see complex 39).
Scheme 31. Palladium(II)-CPA-cocatalyzed Asymmetric Desymmetrizing Hydroalkoxylation of Allenic Diols.

3. Metal-Hydride Hydrogen Atom Transfer
An emerging strategy in asymmetric hydrofunctionalization chemistry capitalizes on transition-metal hydrides (M–H) that react with alkenes via hydrogen atom transfer (HAT).106−109 The ensuing formation of a solvent-caged metallo/organic radical pair offers a versatile platform for functionalization with a variety of coupling partners (Scheme 32). Such catalytic systems are common with first-row transition metals, including Fe, Co, and Mn, and proceed under mild reaction conditions with high levels of chemoselectivity. Additionally, the intermediacy of carbon centered radicals dictates the regioselectivity of MHAT hydrofunctionalizations and leads to Markovnikov adducts (detailed mechanistic discussion follows). The robustness of this methodology has manifested in its use in a myriad of natural product syntheses, with the Mukayaima hydration being a model example.106 However, because of the inherent challenge of controlling the facial differentiation of putative alkyl radical intermediates, stereoselective processes have largely been limited to auxiliary-controlled reactions.110,111 Only in the past few years have highly enantioselective MHAT reactions been developed, with asymmetric hydroalkoxylation reactions being among the first examples in the field.
Scheme 32. MHAT Hydrofunctionalization of an Alkene.

Namely, in 2019, Pronin and co-workers reported the first example of a highly asymmetric HAT-initiated hydrofunctionalization, demonstrating an intramolecular cyclization of tertiary allylic alcohols to furnish the corresponding epoxides with good to excellent levels of enantioselectivity (Scheme 33).112 The reaction employs methylphenylsilane as reductant and an N-fluoropyridinium oxidant (41(F)) and is catalyzed by chiral Co salen complex 40 containing dibenzofuran units bound to the ethylenediamine-derived fragment, which prove to play an crucial role in enantioinduction (vide infra). High levels of asymmetric induction were observed with heterocycle-derived allylic alcohols (e.g., tetrahydropyrans or piperidine derivatives) as well as cyclohexanes bearing heteroatoms. Alternatively, substrates derived from simple cyclohexanes, including those containing alkyl substituents, resulted in decreased enantioselectivities. Further, employment of an acyclic substrate yields the desired epoxide, however, with nearly complete loss of enantioinduction.
Scheme 33. Co-catalyzed Intramolecular Hydroalkoxylation of Allylic Alcohols to Enantioenriched Epoxides.

Alternatively, in 2016, Shigehisa and co-workers reported a single example of an intramolecular asymmetric hydroalkoxylation of 2,2-diphenylpent-4-en-1-ol to yield the corresponding tetrahydrofuran with moderate enantioselectivity (er = 64:36).113 In 2020, the same research group showed that incorporation of chiral binaphthyl units into the Co salen scaffold dramatically improves the observed enantioinduction (Scheme 34).114 The authors demonstrated a moderate scope of alkenols, however, all substrates contain bulky substituents along the tether. Surprisingly, the steric nature of the employed silane has a striking effect on the enantioselectivity of the reaction. In particular, relatively nonsterically hindered secondary silanes, e.g., diethylsilane, selectively form the (S)-enantiomer of the product. Alternatively, more sterically encumbered silanes, e.g., diisopropylsilane and tetramethyldisiloxane, result in the formation of the product antipode.
Scheme 34. Co-catalyzed Intramolecular Asymmetric Hydroalkoxylation toward Tetrahydrofurans.

A general mechanistic proposal for Co-catalyzed HAT-initiated hydroalkoxylation is depicted in Scheme 35. While it is broadly accepted that such transformations proceed through metal hydrides, the mechanism to form the requisite M–H is not yet well understood, particularly with Co salen complexes. Shigeshi and Hiroya have suggested that the catalytic cycle commences with the oxidation of two equivalents of a Co(II) precatalyst with an N-fluoropyridinium salt (41(F)) to concomitantly form a cationic Co(III) complex and a Co(III)–F intermediate (as well as 2,4,6-collidine).113,115 The Co(III)–F species subsequently reacts with a silane (R3Si–H) via ligand exchange to furnish the catalytically active Co(III)–H species, thermodynamically driven by the formation of a strong (and chemically inert) Si–F bond (Scheme 35a). However, in a recent mechanistic investigation into a Co/Ni dual-catalyzed hydroarylation, Shenvi et al. disclosed that the reaction of a Co(II) salen complex with an N-fluoropyridinium triflate does not lead to the isolation of a Co(III)–F complex but rather exclusive isolation of a cationic Co(III) species with an outer-sphere triflate counteranion.116 The authors suggest that Co–H formation could thereafter result from hydride delivery from a pentavalent silicate intermediate to the cationic Co(III) species, still leading to the formation of a Si–F bond.108
Scheme 35. A General Mechanistic Proposal for Asymmetric MHAT Hydroalkoxylation.

While detailed investigations are still necessary to disentangle the mechanistic possibilities toward M–H formation with Co salen complexes, a key feature undoubtedly includes oxidation to Co(III), as the Co(II) precatalyst is inactive in MHAT reactions.116 Further, a compatible reductant (typically a silane) is required and the uphill formation of a highly reactive (and weak) M–H bond must be compensated by the concomitant formation of a strong bond (typically a Si–F or Si–O bond).108 This represents the enabling driving force of MHAT catalysis. Recent methods, such as those described herein, employ N-fluoro salts, as these are not only capable oxidants but further provide the fluoride required for Si–F bond formation.
In the presence of an alkene, HAT from the transient Co–H forms a metal radical/carbon radical cage pair (Scheme 35b, 43).117,118 Subsequent reactivity can proceed via three pathways: (1) a hydrogen atom abstraction by the metal can occur to reform the starting materials (and can also lead to alkene isomerization119), (2) the radical pair can undergo solvent cage escape, generating a free alkyl radical (44), or (3) the radical pair can collapse to form a diastereomeric mixture of the corresponding organometallic intermediate (45).116,120,121 In an earlier example of a nonasymmetric intermolecular hydroalkoxylation, Shigehisa and Hiroya proposed that a free alkyl radical escapes from the cage and is subsequently oxidized to form a carbocation, which is then captured by an alcohol.115 However, this scenario cannot explain the enantioselectivity observed in the presence of chiral Co–salen complexes. Alternatively, in 2018, Pronin and co-workers reported a catalyst-controlled chemodivergent functionalization of tertiary allylic alcohols to yield either the above-mentioned epoxides (with low ee in the model system using a chiral Co–salen complex not optimized for enantioinduction) or semipinacol rearrangement products.122 The striking influence of the salen scaffold on both the chemoselectivity, as well as the diastereoselectivity of the semipinacol rearrangements, led Pronin et al. to suggest that these reactions involve alkyl–Co(IV) complexes as electrophilic intermediates, rather than free carbocations. In particular, the authors propose a radical polar crossover (RPC) process in which complex 45 undergoes a single electron oxidization to furnish a reactive cationic alkyl–Co(IV)intermediate 46, thus enabling a stereoinvertive nucleophilic displacement. Concomitant deprotonation yields the corresponding epoxide. This elementary step regenerates the Co(II) precatalyst, necessitating the stoichiometric use of oxidant and silane. These postulations are supported by pioneering work by Halpern and others describing the formation123−128 and ensuing reactivity129−131 of alkyl–Co(IV) complexes.
In their 2019 report (Scheme 33), Pronin et al. propose that the observed enantioselectivity may result from reversible epimerization of the stereocenter bearing the homolytically labile Co–C bond in 46 (blue dashed arrows), followed by an enantiodetermining displacement via a dynamic kinetic resolution. Eyring analysis using sterically and electronically differentiated salen complexes shows that the enantioselectivity of this transformation is enthalpically controlled and positively correlates to the polarizability and quadrupole moment of the arene ring in the backbone of the salen complex. Hence, the authors suggest that cation−π interactions between the radical cation of the Co(IV)–salen complex, and the electron-rich dibenzofuran motifs play an integral role in stabilizing the transition state, leading to the major enantiomer (Scheme 35c, 47).132 Alternatively, the authors do not rule out the possibility of an enantiodetermining radical capture by a cationic Co(III) complex to give a scalemic mixture of the cationic Co(IV) intermediate (maroon dashed arrow). In either scenario, a stereospecific displacement of (S)-46 predominates and renders the formation of the observed (R)-epoxide.
The profound effect of the employed silane on the observed enantioselectivity in the asymmetric hydroalkoxylation reported by Shigehisa et al. indicates that at least two competing enantiodetermining steps are operating. The authors propose that, in the presence of sterically unencumbered secondary silanes, an extensive radical chain reaction between 45 and a diffused radical (44) results in a diastereomeric enrichment of (R)-45 (teal dashed arrows). The subsequent oxidation would therefore furnish a scalemic mixture of 46, favoring (R)-46, from which stereospecific displacement yields enantioenriched (S)-product. On the other hand, when employing sterically bulky silanes, the authors suggest that this radical chain reaction is suppressed and propose a dynamic kinetic resolution analogous to Pronin to yield the (R)-enantiomer. The authors provide computational evidence for favorable CH−π interactions between the catalyst and substrate that stabilize either the favored alkyl–Co(III) intermediate or the transition state of nucleophilic displacement.
Given that the mechanistic scenarios for asymmetric induction involve, to varying extents, cationic intermediates in the enantiodetermining step, it might be interesting to study the effects of chiral anions on the enantioselectivity of these reactions. To the best of our knowledge, chiral anions have not yet been employed in MHAT reactions. Further, while an intermolecular asymmetric hydroamination has been recently described,133 intermolecular MHAT hydroalkoxylation reactions have so far not been rendered asymmetric. In a recent perspective, Holland and Shenvi et al. suggest that analogous differentiation of Fe- or Mn-based diastereomeric organometallic intermediates may be untenable; however, the authors offer other potential approaches to asymmetric MHAT method development.108
4. Photocatalysis
Photocatalysis has emerged as a powerful tool used to enable new activation modes and offer complementary reactivity to thermally catalyzed methodologies. Similar to MHAT strategies, photocatalysis offers a mild and highly chemoselective approach to the generation of reactive radical intermediates. However, in contrast to MHAT, photocatalytic hydrofunctionalization methods provide linear adducts, a notable aspect because anti-Markovnikov products are otherwise challenging to obtain. As mentioned in section 3, the intermediacy of highly reactive radical intermediates presents challenges in inducing enantioselectivity, and further, nonstereoselective background reactivity can hamper light-driven processes. Nevertheless, two general approaches to enantioselective photocatalysis have been developed. Type I describes the excitation of a substrate within a preassembled chiral environment, whereas in type II, the enantioselective bond formation is catalyzed by a “conventional” chiral catalyst and occurs separately from the photochemical reaction (this strategy is mainly used in photoredox catalysis).134−136 This section will include a detailed discussion of the photocatalytic methods developed for asymmetric hydroalkoxylation and will consider future directions within this subfield.
One of the earliest reports of an enantioselective intermolecular hydroalkoxylation was disclosed in 1993 by the Inoue group and describes a photocatalytic anti-Markovnikov hydroalkoxylation of stilbene derivatives.137 In a later development, naphthalene dicarboxylate catalyst 48 containing chiral saccharide-based motifs was employed to enable improved enantioselectivities (Scheme 36).218 The authors propose that an arene exciplex is formed between the catalyst and alkene substrate and that the polar nature of the auxiliary units facilitates electron transfer from the alkene to furnish a chiral radical ion pair, of which dissociation is disfavored in relatively nonpolar reaction media (Et2O). Hence, the olefin radical cation is contained within a chiral environment, and facial differentiation can therefore be achieved upon addition of an alcohol (type I). The regioselective preference is dictated by the favored nucleophilic attack to the less-substituted carbon of the olefin cation radical, ultimately giving rise to the anti-Markovnikov adduct (Scheme 37).138 While moderate levels of enantioselectivity are observed, the poor reactivity of this method restricts its overall applicability.
Scheme 36. Chiral Photosensitizer-catalyzed Intermolecular Hydroalkoxylation of Stilbene Derivatives.

Scheme 37. General Regioselectivity Observed for the Addition of Nucleophiles to Olefin Radical Cations.

Since 2012, the Nicewicz group has published a range of reports on anti-Markovnikov selective hydrofunctionalizations of alkenes using an acridinium-derived photocatalyst139 in the presence of a hydrogen atom transfer reagent.140 As with the previous example, these reactions proceed through the intermediacy of an olefin radical cation and are therefore highly selective toward linear products. Nicewicz and Hamilton initially demonstrated the success of this methodology with an intramolecular hydroalkoxylation reaction of alkenols to form a wide range of tetrahydrofurans and pyranes using 2-phenylmalononitrile as an H atom donor.141 In 2017, Luo and co-workers recognized that the cationic nature of the photocatalyst, as well as the intermediacy of cationic species in this transformation, revealed an opportunity to induce enantioselectivity via ion-pairing catalysis, specifically asymmetric counteranion directed catalysis. Indeed, the authors demonstrated that in the presence of a chiral ion pair photoredox organocatalyst, derived from Mes-Acr-Me+ and a chiral BINOL-based phosphate anion (49–), moderate levels of enantioselectivity could be achieved (Scheme 38).142
Scheme 38. Asymmetric Hydroalkoxylation of Alkenols Using Acridinum-based Photocatalyst.

In alignment with thorough mechanistic studies performed by Nicewicz and Romero,143 the authors propose a catalytic cycle entailing a photoinduced SET from the alkenol to the excited Mes-Acr-Me–49 to yield radical cation 50, followed by an enantiodetermining C–O bond-forming cyclization/proton transfer step to provide 51 (type II). Finally, a hydrogen atom transfer event regenerates the chiral anion and furnishes the desired product (Scheme 39). Further mechanistic studies by Luo et al. revealed that the chiral phosphate anion plays three critical roles in the catalytic process: (1) to increase the lifetime of the chiral photocatalyst’s triplet state, (2) to induce asymmetry in the cyclization step, and (3) to facilitate the proton shuttle in the cyclization/proton transfer process through H-bonding. Given the poor to moderate enantioselectivities observed, coupled with the known effect of confined organocatalysts (see section 5.1), the use of imidodiphosphate anions could potentially render this reaction highly enantioselective. Additionally, while Nicewicz has demonstrated intermolecular reactivity with methanol, so far this strategy has not been extended to an intermolecular asymmetric hydroalkoxylation and is still limited to alkenols benefiting from the Thorpe–Ingold effect.
Scheme 39. Proposed Mechanism for the Intramolecular Hydroalkoxylation with an Acridiunium-based Photocatalyst and an HAT Reagent in the Presence of a Chiral Phosphate Anion.

Knowles and co-workers recently reported a fundamentally unique approach to photocatalyzed hydroalkoxylation reactions, in which homolytic activation of an O–H bond initiates an intramolecular addition to variously substituted alkenes (Scheme 40).144 In particular, a phosphate anion promotes a proton-coupled electron transfer (PCET) activation of the O–H bond in the presence of an IrIII-based visible light photo-oxidant and subsequently mediates the ring-closing event via complex 52. This work has, however, not yet been rendered asymmetric.
Scheme 40. Hydroalkoxylation via Proton-coupled Electron Transfer (PCET) Activation of Alcohol O–H Bond.

5. Bro̷nsted Acid Catalysis
Inspired by the impeccable enantioselectivities observed in Brønsted acid–mediated hydroalkoxylations using enzymes (section 7), coupled with the challenges of hidden acid catalysis in transition metal systems (section 2), chiral small molecule Brønsted acid catalysis stands out as a promising approach to developing asymmetric hydroalkoxylation methods. The formation of the branched (i.e., Markovnikov) adduct of acid-mediated hydrofunctionalizations with aliphatic alkenes is both kinetically and thermodynamically favored, and therefore such reactions typically proceed with high levels of regioselectivity.1 However, there exists a number of important challenges associated with this approach to asymmetric hydroalkoxylation and hydroacyloxylation. Namely, alkenes are weakly basic145 functionalities and therefore protonating such motifs could require very strong acids and/or harsh reaction conditions, both of which might lead to issues with functional group tolerance. Further, controlling the facial selectivity of nucleophilic attack onto intermittent carbocations is notoriously challenging146,147 and moreover, controlling such intermediates via ion-pairing is not well-explored. Nevertheless, a few small-molecule acids have effected moderate to excellent levels of enantioinduction in hydroalkoxylations of C–C multiple bonds. These systems can be divided into three general categories: (a) chiral Brønsted acid catalysis, (b) Lewis acid-assisted chiral Brønsted acid (LBA) catalysis, and (c) chiral Lewis base-assisted Brønsted acid (LBBA) catalysis (Scheme 41). This section will briefly introduce each of these strategies and specifically focus on their applications in asymmetric hydroalkoxylation. Similar to discussion in section 2.2.2, for gold-catalyzed reactions proceeding through ACDC, there exists a continuum of noncovalent interactions that dictate stereoselectivity, i.e., ion pairing and/or hydrogen bonding. When relevant and elucidated by the authors, such distinctions will be made.
Scheme 41. General Approaches to Brønsted Acid-catalyzed Hydroalkoxylation.

5.1. Chiral Brønsted Acid Organocatalysis
Since seminal reports in 2004 by Akiyama148 and Terada149,150 on Mannich-type reactions, BINOL-based chiral phosphoric acids (CPAs) and their derivatives have proven to be privileged motifs in asymmetric catalysis.151−153 In sections 2.2.2 and 4, we delineated the effective use of chiral phosphates (e.g., TRIP) as anions in asymmetric transformations of C–C multiple bonds, in which either a metal or photocatalyst activates the substrate and the chiral phosphate provides enantioinduction through ion pairing with cationic intermediates. Alternatively, the direct use of CPAs as chiral Brønsted acids to protonate C–C multiple bonds and subsequently control facial selectivity of nucleophilic attack on carbocation intermediates presents an elegant approach to asymmetric hydroalkoxylation. However, the relatively weakly acidic nature of CPAs (pKa = 12.5–14 in MeCN154) is inherently limiting and has rendered aliphatic alkenes out of reach. Strategies to overcome this limitation have involved the use of electronically activated alkenes (e.g., enol ethers) or, more recently, the design of very strong and confined Brønsted acids that are capable of protonating unactivated alkenes and imparting stereocontrol on corresponding carbocation intermediates. This section will provide an overview of each of these approaches.
Upon the basis of our group’s previous endeavors in asymmetric spiroacetalizations,155 in 2012, we described a catalytic enantioselective spiroacetalization reaction of hydroxyenol ethers (Scheme 42).102 The success of this method was enabled by the development of a new class of highly confined C2-symmetric imidodiphosphates (IDPs), which maintain the bifunctional nature of CPAs while providing an enzyme-inspired geometrically constrained reaction site (pKa = 11.5 in MeCN,156 when 3,3′ substituents = −C6H5). The employed IDP catalyst 53 is tolerant of a range of substrates to yield the corresponding spiroacetals with low catalyst loading (as low as 0.1 mol %), including in a highly enantioselective synthesis of (S)-olean, a sex pheromone of female olive fruit flies. Impressively, with chiral nonracemic hydroxyenol ether starting materials, the catalyst displays exquisite stereocontrol, overriding the thermodynamic preference of the substrate and providing a range of nonthermodynamic spiroacetals with good levels of diastereoselectivity.
Scheme 42. IDP-catalyzed Hydroalkoxylation of Enol Ethers toward Enantioenriched Spiroacetals.

Soon after, the Nagorny group published a conceptually similar approach with a highly stereoselective spiroacetalization using (S)-TRIP.157 The substrates employed do show dependence on Thorpe–Ingold substituents along the tether, perhaps a consequence of the more open active site of CPA catalysts in comparison to the IDP catalyst employed in the above example from our group. Alternatively, the authors additionally employed a range of d-glycal derivatives and observed nonthermodynamic spiroacetals in high yields and good to excellent diastereoselectivities (Scheme 43a).
Scheme 43. (S)-TRIP-catalyzed Asymmetric Hydroalkoxylation of Enol Ethers.

Accompanied mechanistic and computational studies were conducted in order to differentiate between potential SN1-like, SN2-like, and covalent phosphate intermediate-based mechanisms.158 In particular, the authors employed deuterium-labeled hydroxyenol ether 58 and concluded that the TRIP-catalyzed spiroacetalization proceeds via a syn-selective addition of the O–H group across the C–C double bond (Scheme 43b). Further, a significant inverse secondary kinetic isotope effect was observed for this substrate, suggesting that rehybridization occurs in the rate-determining step (RDS). Additionally, a Hammett analysis conducted with aromatic enol ethers is consistent with the buildup of positive charge in the transition state of the RDS, although the absolute value suggests a concerted pathway rather than the formation of a fully charged oxocarbenium ion. Taken together, and in combination with in-depth computational studies (including use of the growing string method and molecular dynamics), the authors suggest that these TRIP-catalyzed spiroacetalizations occur via a concerted, though asynchronous mechanism in which a fully formed oxocarbenium ion is not implied. Instead, the bifunctional nature of the CPA is exploited to initially effect protonation of the enol ether followed by deprotonation of the alcohol appendage and simultaneous spiroacetalization via TS 61 (Scheme 43c).
While the above examples achieve reactivity with phosphoric acid-derived catalysts, the dependency on electronically activated C–C multiple bonds limits their generality in asymmetric hydroalkoxylation reactions. Alternatively, the use of electronically unbiased alkenes have eluded chiral Brønsted acid catalysis since seminal reports, and the design and synthesis of stronger BINOL-derived chiral acids has been at the center of research efforts in asymmetric organic Brønsted acid catalysis for nearly two decades. To this end, our group recently disclosed imidodiphosphorimidate (IDPi) catalysts,159 which not only maintain the confined reaction pocket of the above-mentioned IDP’s but additionally offer significantly increased acid strength due to Yagupolskii-type160 substitution of P=O double bonds with P=NTf motifs (pKa = 4.5 in MeCN,156 when 3,3′ substituents = −C6H5).
With such scaffolds in hand, our group disclosed an intramolecular hydroalkoxylation of simple alkenols to provide highly sought-after enantioenriched tetrahydrofurans and tetrahydropyrans in high yields (41–95%) and excellent enantioselectivities (er = 92:8 to 98.5:1.5) using IDPi 62 (Scheme 44).17 Importantly, opposed to the intramolecular examples described throughout section 2.1.1, this method does not require Thorpe–Ingold substituents along the tether of the alkenol to induce cyclization. In fact, a substrate bearing a dimethyl unit along the chain proved significantly less reactive, underscoring the sensitivity of the confined active site. To probe the mechanism and origin of enantioselectivity for this transformation, a combination of computational and experimental investigations were carried out. Analogous to the previous example by Nagorny and Zimmerman et al., density functional theory (DFT) studies (B3LYP/def2- TZVP/D3(BJ)/CPCM) suggest that the reaction proceeds through a concerted, though asynchronous mechanism in which the reaction is triggered by protonation of the alkene followed by enantiodetermining C–O bond formation. Further, an intramolecular competitive Hammett analysis with a series of styrene derivatives was performed. Plotting log(kX/kH) against substituent parameter σ+ results in a linear correlation with a negative slope (ρ= −2.08 ± 0.04), corroborating the buildup of carbocationic character at the internal position of the double bond.
Scheme 44. IDPi-catalyzed Asymmetric Intramolecular Hydroalkoxylation of Simple Alkenols.

The generality of this method was further showcased by the encouraging enantioselectivity (er 76.5:23.5) observed during the initial screening of highly challenging intermolecular hydroalkoxylation (Scheme 45).17 This elegant design demonstrates the truly unique capabilities of small-molecule catalysts to exert enzyme-like sterocontrol while simultaneously opening the door to explore new reactivity that were previously beyond the reach of the traditional organocatalysts.
Scheme 45. IDPi-catalyzed Asymmetric Intermolecular Hydroalkoxylation of Styrene.

5.2. Lewis-Acid Assisted Chiral Brønsted Acid Catalysis
In 1994, Yamamoto and co-workers demonstrated that the combination of an achiral Lewis acid and a chiral Brønsted acid results in a conformationally rigidified chiral Brønsted acid with increased strength, termed by the authors as a Lewis acid-assisted chiral Brønsted acid (chiral LBA).161−163 Chiral LBAs are typically generated in situ by combining optically active binaphthyl derivatives with equal or excess amounts of Lewis acid at room temperature to form solution-stable coordinated complexes. Early studies highlighted the use of LBAs in asymmetric protonations of silyl enols ethers with the first catalytic variant reported in 1996.164 In 1999, Yamamoto and co-workers published a breakthrough report on small-molecule mediated asymmetric polyene cyclizations using chiral LBAs.165,166 Substrates containing a free hydroxy group (polyolefinic phenols and alcohols) require stoichiometric amounts of the chiral LBA, likely a consequence of the −OH group competitively binding to the Lewis acid and impeding reactivity. Alternatively, enantioselective cyclizations of geranyl aryl ethers proceed smoothly with catalytic quantities of chiral LBA 64 to provide the cyclized products with moderate enantioselectivity and high degrees of diastereoselectivity (Scheme 46). The authors suggest that this reaction proceeds through a [1,3] rearrangement (abnormal Claisen rearrangement) to provide polyolefinic phenol 65 that subsequently undergoes cyclization. On the basis of these successes, Yamamoto and co-workers published a series of reports throughout the early 2000s, demonstrating the robust ability of chiral LBAs to mediate a range polyene cyclizations en route to natural products, including syntheses of (−)-ambrox,13 (−)-chromazonarol,12 and (−)-caparrapi oxide,167 although these examples require stoichiometric quantities of a chiral LBA. In 2009, Bhat et al. employed chiral LBA-mediated polyene cyclizations in the total syntheses of (+)-sclareolide and (+)-tetrahydroactinidiolide, starting from (3R,6E)-nerolidol and (R)-linalool, respectively.168 In this case, carboxylic acids were used as terminating groups and the cyclizations proceeded with high levels of enantioinduction with substoichiometric amounts of the chiral BINOL derivative (in the presence of stoichiometric quantities of SnCl4).
Scheme 46. Enantioselective Cyclization of Geranyl Aryl Ethers via Abnormal Claisen.

More recently, in 2015, the Hintermann group showed that an in situ generated enantiopure titanium-derived complex catalyzes the cyclization of 2-allylphenols to yield enantioenriched 2-methylcourmarans (Scheme 47).169 The chiral catalyst is prepared by combining an enantiopure 1,1′-binaphthyl-2-carboxylic acid (BINA-Cox), Ti(Oi-Pr)4, and a cocatalytic quantity of water in a 1:1:1 ratio (5 mol % each). Despite requiring very high temperatures (240 °C, μW), the intramolecular cyclizations proceed with moderate enantioselectivites.
Scheme 47. Intramolecular Hydroalkoxylation of 2-Allylphenols to 2-Methylcourmarans.

Recent efforts by the authors have focused on expanding the limited reactivity scope by preparing and testing a diverse library of BINA–Cox ligands (>30).170 While incremental improvements to the substrate scope were achieved, this method has so far not been successfully applied to aliphatic alkenols. The authors suggest that a multinuclear titanium-μ-oxo species is in situ generated and acts as the active catalytic species, although they have not yet confirmed the structure of the formed complex. Further, the authors have not yet proposed a mechanism for this transformation, although some insight can be gained in a related nonasymmetric hydroalkoxylation by the authors with Al(i-PrO)3.171
In 2021, Xie and Li reported a highly related intramolecular hydroalkoxylation using a chiral 1,1′-binaphthyl-2,2′-disulfonic acid (BINSA) in the presence of Ti(EHO)4 (EHO = 2-ethylhexyloxy) and water.172 The shown intramolecular cyclizations of 2-allylphenols proceed under comparatively mild reaction conditions (75 °C) and generally improved enantioselectivities are observed, relative to the previous example. In line with Hintermann, the authors propose that such transformations proceed through a multinuclear titanium-μ-oxo species and have tentatively provided a possible mechanism, with the enantiodetermining step involving concomitant protonation of the alkene and C–O bond formation to yield enantioenriched cyclized products (Scheme 48).
Scheme 48. Proposed Transition State in Disulfonic Acid/Ti/H2O-Catalyzed Cyclization of 2-Allylphenols.

In 2020, Zhao, Zhao, and co-workers capitalized on the combination of chiral N-Tf-phosphoramide 69 and TiCl4 to effect an asymmetric hydroalkoxylation of alkenols with moderate levels of enantioselectivity (Scheme 49).173 Similar to aforementioned examples using transition metals, the reactivity of this system is highly dependent on bulky substituents along the chain to promote substrate cyclization through the Thorpe–Ingold effect. Replacement of the aromatic rings with either methyl groups or hydrogens resulted in either a near or a complete loss of reactivity, respectively. Notably, the N-Tf-phosphoramide Brønsted acid cannot alone promote the cyclization, likely a consequence of inadequate Brønsted acidity. While the authors propose a structure for the Ti(VI)-(R)-69 complex, there is currently insufficient evidence to distinguish whether this complex acts a chiral Lewis acid or chiral Brønsted acid. Despite this mechanistic ambiguity, it is herein categorized under LBA catalysis for consistency of reaction conditions (combination of a chiral Brønsted acid and a strong Lewis acid) and because a key step of the reaction mechanism likely includes protonation of the C–C double bond.
Scheme 49. Chiral N-Triflyl Phosphoramide/TiCl4-catalyzed Intramolecular Hydroalkoxylation.

5.3. Chiral Lewis Base Assisted Brønsted Acid Catalysis
Complementary to previously introduced LBAs, the combination of an achiral Brønsted acid with a chiral enantiopure Lewis base is coined Lewis base–assisted Brønsted acid (LBBA) catalysis. Notably, while the design of LBAs relies on the increased Brønsted acidity upon Lewis acid activation, LBBAs show diminished acidity with regard to the original acid. Therefore, an excess of Lewis base catalyst with regard to the acid is usually required to suppress unselective background reactivity. Also, a significant difference in pKa between Lewis base and Brønsted acid is crucial for quantitative consumption of the achiral acid. The first LBBAs introduced by Ishihara and co-workers in 2011 fulfill these criteria by incorporating a sufficiently Lewis basic chiral BINOL-derived phosphonate and a strong Brønsted acid (Scheme 50).174
Scheme 50. Generalized Concept of LBBAs.

Chiral Lewis bases similar to 70 were previously employed in asymmetric halopolycyclizations using N-iodosuccinimide (NIS) by Ishihara and co-workers.175 Because of the intermediacy of an ionic (R3P-I+) species that acts as an electrophilic activator to induce high enantioselectivity in the cyclization, the authors speculated that the replacement of iodine by a simple proton would result in a similarly structured Brønsted acid that might be able to induce a biomimetic polyene cyclization. In their seminal report, Ishihara and co-workers thus utilized LBBAs for this purpose (Scheme 51).174 The authors were able to obtain trans-fused tricyclic frameworks with good yield and high diastereo- and enantioselectivity from a variety of substituted 2-geranylphenols. Importantly, sterically nondemanding Brønsted acids like fluorosulfonic acid showed generally improved enantioselectivity. Additionally, fine-tuning of the electronic and steric properties of the Lewis basic phosphine turned out to be crucial for high yield, enantioselectivity, and reduced catalyst decomposition under the reaction conditions. With a related catalytic principle, the same group in 2013 reported the LBBA-catalyzed kinetic resolution of racemic 2-substituted carboxylic acids via intramolecular hydroacyloxylation.176
Scheme 51. Asymmetric LBBA-catalyzed Polyene Cyclization.

6. Lewis Base Catalysis
As was discussed in the previous section, chiral phosphines can act cooperatively with achiral acids to form Lewis base assisted Brønsted acid catalysts. In a distinct activation mode, phosphines can also engage with activated alkynes, related to the well-explored Morita–Baylis–Hillman reaction, as nucleophilic Lewis bases. In 1994, Trost and co-workers established that in the presence of 1,3-bis(diphenylphosphino)propane (DPPP), an intramolecular hydroalkoxylation of ω-hydro-alkynoates can be achieved.177 As outlined in Scheme 52, the mechanism commences with the 1,4-addition of a chiral phosphine to an alkynoate to give zwitterionic intermediate 71. Subsequently, a series of proton transfers leads to the key electrophilic cationic intermediate 73 that, after nucleophilic addition, generates ylide intermediate 74. Further proton transfer generates an enolate 75 that releases the catalyst upon product formation. Notably, the overall reaction can be understood as a γ-umpolung of alkynoates, with a formal oxidation in the γ-position, while the alkyne is reduced to an alkene. Akin to some π-Lewis acidic metals (see section 2), these transformations are compatible with both alkynes and allenes, as both proceed via a common catalytic intermediate.178
Scheme 52. General Mechanism of Phosphine-catalyzed Hydroalkoxylations of Propiolate Derivatives.

Because of the covalent nature of the catalytic intermediates formed between the Lewis basic catalyst and the substrate, a reasonable advancement was the development of chiral phosphines for asymmetric hydroalkoxylations of alkynoates. The first asymmetric variant of this reaction was reported in 2009 by Fu and co-workers (Scheme 53).179 Using spiroindane-derived phosphepine 76 as catalyst, the authors achieved an asymmetric intramolecular hydroalkoxylation to furnish THFs, THPs, and (iso)chromanes in good yields and enantioselectivities. Notably, the use of benzoic acid derivatives as additives had a profound effect on reactivity and selectivity. Control experiments show that the added carboxylic acid does not protonate the phosphine. Possibly, it is instead involved in the proton transfer reactions as well as in the stereodetermining step, as an engineered bromobenzoic acid was required to achieve high enantioselectivity for phenolic substrates. Additionally, spectroscopic analysis of the reaction mixture identified the resting state of the catalyst as the free phosphine rather than a phosphonium adduct with the substrate.
Scheme 53. Phosphine-catalyzed Asymmetric Intramolecular Hydroalkoxylation of Alkynoates.

Fu and co-workers further demonstrated the generality of phosphephine catalyst 76 in an intermolecular hydroalkoxylation of benzylic alkynoates (Scheme 54).180 A wide array of aromatic substrates as well as alcohols are well tolerated, furnishing the ethereal products with generally high yield and selectivity. Remarkably, the catalyst control was further highlighted in the reaction with enantiomerically pure α-methyl benzyl alcohols, where high diastereomeric control was imparted regardless of the nucleophiles absolute configuration. Despite the impressive stereochemical outcome, the transformation still suffers from several drawbacks. First, the transformation relies on aromatic substrates. Second, sterically demanding secondary alcohols like cyclohexanol are not well tolerated, giving significantly diminished product yield. Further, the utilization of water as nucleophile was impossible under the reported reaction conditions. Improvements in these directions would represent a significant leap toward general protecting group free functionalization of alkynoates.
Scheme 54. Phosphine-catalyzed Asymmetric Intermolecular Hydroalkoxylation of Alkynoates.

7. Enzyme Catalysis
Given the vast ubiquity of hydrofurans and hydropyrans in natural products, Nature’s chemical toolkit includes several highly stereoselective methods for the formation of cyclic ethers. Known biosyntheses of these motifs include general acid- and general base-catalyzed additions of alcohols to epoxides, carbonyls, and Michael acceptors.181 Despite the efficiency and versatility of hydroalkoxylation processes, known enzymatic asymmetric additions of alcohols to C–C π bonds remain relatively rare, and to the best of our knowledge, are limited to intramolecular reactions. In this section, we will delineate enzyme-catalyzed asymmetric hydroalkoxylations and discuss recent advances in hydration reactions of alkenes.
In the early 1990s, Woggon and co-workers identified a previously undiscovered enzyme responsible for the asymmetric cyclization of 77 to γ-tocopherol, a key step in the biosynthetic route toward the vitamin E family (Scheme 55).182 Labeling studies revealed that the enzyme-catalyzed cyclization proceeds through Si-protonation of the double bond and concomitant Re-attack of the phenolic oxygen to furnish γ-tocopherol.183 Further, the authors discovered three critical substrate-enzyme recognition sites: (1) the OH group at C(1) of the hydroquinone, (2) the (E)-configuration of the double bond, and (3) the length of the lipophilic side chain. Variation of these components, i.e., acylation of the alcohol, use of the (Z)-isomer, or shortening of the side chain, results in either significantly reduced yields or no observed cyclization.184 Alternatively, mimicking the stereocontrol of asymmetric cyclizations toward this chromane framework using strong chiral Brønsted acids has proven to be a formidable challenge.185 As such, the development of small-molecule catalysts capable of achieving such reactivity with high levels of enantioinduction and a potentially increased reactivity scope is warranted.
Scheme 55. Enzyme-catalyzed Hydroalkoxylation Toward γ-Tocopherol.

More recently, Garg, Houk, and Tang et al. identified a dedicated enzyme that catalyzes an enantioselective hydroalkoxylation reaction in the biosynthesis of herqueinone, a fungal metabolite. In particular, the authors discovered a 149aa protein encoded from the gene PhnH that is responsible for the intramolecular addition of a phenol to the terminal alkene of a reverse prenyl group (Scheme 56).186 DFT calculations (using acetic acid as a general acid model), coupled with enzymatic modeling and docking studies, suggest that the reaction is initiated by deprotonation of the phenol group, followed by C–O bond formation and a concerted proton transfer from a nearby glutamate residue (via TS 78).187 Notably, the authors do not rule out an alternative mechanism in which the glutamate residue positions a water molecule as a specific acid in the protonation step. Unfortunately, attempts to translate this chemistry to a simplified and non-natural substrate, 2-allyl resorcinol, resulted in no rate acceleration of the hydroalkoxylation adduct. To our knowledge, promiscuous reactivity of engineered analogues of this enzyme with non-natural substrates has yet to be studied, but could offer meaningful solutions to the field.
Scheme 56. An Intramolecular Asymmetric Hydroalkoxylation in the Biosynthesis of Herqueinone.

While enantioselective enzymatic hydroalkoxylations are still largely undiscovered, several enzymes are known to catalyze intermolecular asymmetric hydrations of electronically unactivated C–C double bonds. For example, fatty acid hydratases convert isolated alkenes of unsaturated fatty acids to the corresponding chiral alcohols.188 Additionally, linalool dehydratase/isomerase catalyzes an asymmetric hydration reaction in the anaerobic degradation of the monoterpene mercene to (S)-linalool.189 In general, however, wild-type variants of hydratases display limited promiscuity, and therefore the synthetic applicability of these enzymes has been severely restricted.190
In 2013, Glueck and Faber et al. serendipitously discovered “hydratase activity” of a variety of phenolic acid decarboxylases and demonstrated formal additions of water to hydroxystyrene derivatives in the presence of carbonate buffer with moderate levels of enantioselectivity (Scheme 57).191 The dependency on hydroxystyrenes is explained by the proposed p-quinone methide intermediate, an electronically activated motif that undergoes a stereoselective 1,6-addition of water (via TS 79), a mechanistic feature that severely limits the applicability of the method. Further, the addition of aromatic and alkyl alcohols were later attempted in the presence of ferulic acid decarboxylase from Enterobacter sp.; however, none of the tested alcohol nucleophiles underwent addition and only competitive hydration was observed.192
Scheme 57. Promiscuous Catalytic Hydration of Hydroxystyrenes with Phenolic Acid Decarboxylates.

In 2018, the Hauer group published a groundbreaking study in which they described the promiscuous reactivity of fatty acid hydratases for the hydration of structurally simple C–C double bonds (Scheme 58). In particular, the authors discovered that wild-type oleic acid hydratase (OAH) from Elizabethkingia meningoseptica, in the presence of a carboxylic acid decoy molecule catalyzes an asymmetric hydration of 1-decene to form (S)-2-decanol in moderate yield (44% isolated, 0.5 mmol) and excellent enantioselectivity (er >99.5:0.5).193 Interestingly, the activity of the wild-type enzyme was nearly completely diminished in the presence of shorter chain alkenes, e.g., 1-octene and 1-heptene. To overcome the substrate specificity displayed by the wild type, the authors engineered enzymatic variants capable of the asymmetric hydration of a variety of aliphatic alkenes, including cis- and trans-internal alkenes and the relatively bulky 4-phenyl-1-butene. Impressively, excellent levels of enantioselectivity were observed in all cases. A current drawback of the method is relatively low enzyme activity; however, this discovery nevertheless represents significant progress in the field and shows tremendous promise for future developments in asymmetric additions of oxygen nucleophiles to simple alkenes using engineered enzymes.
Scheme 58. Enzyme-catalyzed Asymmetric Hydration of Simple Alkenes.

8. Heterogeneous and Supramolecular Catalysis
Heterogeneous and supramolecular catalysis offer unique possibilities for sustainable chemistry in terms of catalyst recyclability and/or the choice of solvent. In addition to these practical considerations, immobilized or encapsulated active sites can profit from confinement effects that are potentially crucial for product selectivity.194 Significant effort has been devoted to the design of heterogeneous conditions that allow catalytic reactivity that is beyond the scope of homogeneous catalysis. Herein we wish to highlight recent endeavors in heterogeneous and supramolecular catalysis within the context of asymmetric hydroalkoxylation and hydroacyloxylation.
An application of nanomicelles in asymmetric gold catalysis has been published by Lipshutz and co-workers (Scheme 59). The authors utilized the hydrophobic environment within the spontaneously aggregated surfactant TPGS-750-M to enhance tight ion pairing within a cationic (R,R)–Au(I) complex consisting of a chiral BIPHEP-ligand 28 and an enantiopure TRIP-counteranion to achieve asymmetric intramolecular hydroacyloxylations of allenes.195 Notably, the reaction is performed in water at room temperature to give enantioenriched lactones in good yield and enantioselectivity, while an α-substituted racemic allene could be converted to diastereomerically enriched material with high selectivity. Under optimal reaction conditions, additional small amounts of organic solvent (DMSO, benzene, or toluene) were required to achieve short reaction times. As both the catalyst and the surfactant can be recycled, the associated E-factor, a measure of the sustainability of a reaction protocol, is comparably low (4.9). DFT calculations suggest a change in mechanism with regard to the analogous homogeneous gold-catalyzed cyclization of allenic alcohols.196 The computed energies for the cyclization mechanism in a model system with PH3 as ligand show that the traditional 5-exo-trig cyclization to a protonated lactone is substantially disfavored in comparison to a base-assisted pathway, where initial deprotonation of the carboxylic acid allows almost barrierless product formation. Such change in mechanism gathers further support by the observed rate acceleration in the presence of trimethylamine as additive.
Scheme 59. Gold(I)-catalyzed Intramolecular Hydroacyloxylation of Allenes within Nanomicelles.

In addition to their pioneering work in homogeneous gold catalysis, the Toste group has developed heterogeneous conditions for the asymmetric intramolecular hydroacyloxylation of allenes (Scheme 60).197 Utilizing a mesoporous silica support material (SBA-15) for cationic gold complexes together with chiral BIPHEP-ligand 78, the authors were able to obtain lactones in high yield and enantioselectivity. The catalyst design relies on acidic hydroxyl groups within the heterogeneous catalyst to facilitate the crucial protodeauration step that is suggested to be turnover limiting. Therefore, significant rate and selectivity enhancement was observed for the supported catalyst with the regard to corresponding homogeneous conditions. Control experiments with Au catalyst on nonporous support material (pores occupied with cetyltrimethylammonium bromide) suggests that the confined space within the silica pores only plays a minor role in controlling the selectivity. Rather, cooperative hydrogen bonding with the acidic OH groups on the surface is crucial for enantiofacial discrimination. Remarkably, the catalysts could be easily recovered and recycled up to 11 times with no significant loss of selectivity. Stability studies using coupled plasma optical emission spectroscopy (ICP-OES) revealed a total of 3.2% leaching of the gold complex. Further, FTIR spectroscopy shows that a total of 63% molecular catalyst was left after 11 cycles.
Scheme 60. Silica-supported Gold(I)-catalyzed Intramolecular Hydroacyloxylation of Allenes.

A typical observation in gold catalysis is the undesired formation of Au nanoparticles (NP). As for the silica-supported Au catalyst described in Scheme 60, formation of Au-NPs has been observed by the authors after multiple cycles of catalyst recovery. The Toste group therefore attempted to extend the concept of heterogeneous gold catalysis to the purposeful utilization of nanoparticles. Notably, NP-containing catalysts are often unstable toward aggregation or leaching. To circumvent these deleterious effects, the authors designed a catalyst consisting of SBA-15 as mesoporous support material and PAMAM G4OH to form dendrimer-encapsulated metal clusters (DEMCs) together with a chiral NHC ligand 79 (Scheme 61).198 The solid catalyst thus obtained consists of highly active aggregation-stable Au-NPs that seemingly possess a surface oxidation state of Au(I) with bound NHC ligands responsible for high activity and induction of stereoselectivity. Despite the poor enantioinduction, this is the first demonstration of stereoselective catalysis by NHC-ligated AU NPs as well as by AU DEMCs. Notably, Au species remaining in solution also show catalytic activity, albeit with no substantial enantioinduction. Additionally, the analogous homogeneous NHC–Au-Cl complex does not catalyze the hydrolactonization while chloride abstraction with AgBF4 leads to decomposition and precipitation of Au(0).
Scheme 61. Silica-supported DEMC-catalyzed Intramolecular Hydroacyloxylation of Allenes.

A unique approach to combine transition metal and enzyme catalysis has been pioneered by Toste and co-workers.199 The supramolecular encapsulation of cationic phosphine–Au(I) complexes in gallium clusters prevents diffusion of the metal into the reaction solution, where it could bind to amino acid residues of the protein such as cysteine, histidine, or asparagine, thus compromising the enzymes activity. The authors where able to demonstrate a tandem enzymatic kinetic resolution and gold-catalyzed intramolecular hydroalkoxylation of allenes with excellent enantioselectivity. However, achiral Me3PAu(I) complexes were utilized, consequently resulting in a substrate-controlled hydroalkoxylation with poor diastereomeric selectivity. Yet, the work demonstrates that metal encapsulation can prevent adverse interactions and catalyst deactivation pathways. The extension of the concept to truly asymmetric hydroalkoxylations is anticipated.
9. Future Directions and Outlook
The past decade has witnessed a remarkable surge in asymmetric hydroalkoxylation of C–C multiple bonds. The quickening pace of progress in this field using a range of catalytic strategies, aided by its profound synthetic utility, assures the continued vibrancy of this emerging research area in the years to come. Our goal of this section is to identify gaps in the current knowledge and stimulate collective thinking on alternative approaches, which might present avenues for further research.
In particular, asymmetric Markovnikov hydroalkoxylations of unactivated tri- and tetra-substituted alkenes remain out of reach, although reactivity promises to be challenging. Along these lines, considering the wide range of examples shown throughout this review, there is a significant dearth of methods describing asymmetric intermolecular hydroalkoxylations. In fact, only three catalytic methods have displayed this reactivity, each with moderate enantioinduction. Efforts to expand reactivity and stereoselectivity to higher-substituted alkenes, as well as intermolecular systems, is highly warranted. Our group’s work in Brønsted acid-catalyzed intermolecular hydroalkoxylations of styrene has recently opened a new avenues for such reactivity and further developments in this regard can be anticipated.
Despite astounding progress in achieving stereoselectivity for alkenes, asymmetric hydroalkoxylations of cyclopropanes (a well-known alkene surrogate) remain elusive.200 Cyclopropanes are prevalent in drug molecules, and their direct functionalization would enable a new strategy for late state functionalization (LSF) as well as study structure–activity relationships (SAR). Looking ahead, we anticipate significant opportunities lie in designing new intra- and intermolecular asymmetric hydroalkoxylation methods of cyclopropanes, cyclopropenes, and vinylcyclopropanes to further expand the repertoire of hydroalkoxylations.
Furthermore, asymmetric anti-Markovnikov additions are still a work in progress. In this regard, harnessing open-shell selectivity presents untold prospects for reaction discovery. Controlling absolute stereochemistry in radical intermediates presents a long-standing challenge, although contemporary efforts have bridled some of the key challenges.201 Among different radical-based approaches of designing novel asymmetric anti-Markovnikov hydroalkoxylation, employing photoredox catalysis perhaps holds promise. A significant body of work, spearheaded by Nicewicz and co-workers, leading to the anti-Markovnikov hydrofunctionalization of alkenes have been reported, and initial efforts by Luo and co-workers are highly promising.140,142 Similarly, designing new enantioselective PCET-based asymmetric hydrofunctionalization represents another emerging frontier. Given the recent reports of PCET-based hydroalkoxylation144 and asymmetric hydroamination202 by Knowles and co-workers, we believe the stage is well set for designing new PCET-based asymmetric hydroalkoxylation.
Electrocatalysis represents another enabling tool for taming radical intermediates for asymmetric catalysis. One unique feature of electrochemistry that can be particularly effective in the current context is its ability to dial in precise potential, which allows excellent chemoselectivity among various redox active functionalities. A recent report by Lin and co-workers leveraged electrochemistry to accomplish highly enantioselective hydrocyanation,203 highlighting the prospect of these methods to successfully unleash similar hydroalkoxylation reactions. Perhaps a suitable chiral HAT reagent, a chiral photocatalysts, or a chiral mediator could render the above-mentioned open-shell processes asymmetric.
In regard to controlling the stereoselectivity of radical-like intermediates, seminal work by the Pronin112 and Shigehisa113,114 groups in asymmetric MHAT hydroalkoxylation holds significant promise for the future development of general and highly enantioselective Markovnikov-selective methods. Beyond the practical developments already achieved in this area, the fundamental questions regarding the mechanistic underpinnings of enantioinduction are highly stimulating and offer opportunities for interdisciplinary investigations. We look forward to continued work in this area.
Going beyond the use of a traditional single catalytic activation approach, successful integration of different activation modes has shown to be effective for asymmetric alkene functionalization. Related studies by Melchiorre and co-workers using photo-organocatalytic enantioselective radical cascade reactions (leading to an enantioselective difunctionalization),204 and a tandem difunctionalization205 by Knowles and co-workers provide a compelling background to design a similar asymmetric hydroalkoxylation using synergistic catalysis.
Another avenue to achieve an anti-Markovnikov hydrofunctionalization is the use of chiral bulky super base. Seminal work by Bandar et al. has shown the successful implementation of super bases in achieving anti-Markovnikov selectivity.206 Designing chiral superbase catalysts for the development of asymmetric anti-Markovnikov hydroalkoxylation is therefore ripe for development.
Finally, the synergy among different fields can be judicially harnessed for mechanism-driven reaction development.207 As alluded before, the acidity of a given catalyst has remained one of the key considerations in Brønsted acid-catalyzed alkenes activations. The experimental measurement of acidity can inform a suitable catalyst choice for a given transformation.208 Acidity of these Brønsted acids can also be computed.209 On a similar note, DFT-based methods can also provide valuable impetus in understanding the cooperativity among different catalyst motifs and to facilitate more definitive studies of structure–activity relationship.210
While the recent surge in biocatalytic alkene activation has been impressive, its success has suffered due to substrate specificity. With the help of advanced protein engineering techniques such as site-selective mutagenesis, installing new residues has shown to enhance the capabilities of natural enzymes. Theory and machine learning-based methods can play a crucial role in guiding and expediting such processes.211−213
Additionally, it seems inevitable that the push to apply site-selective supramolecular catalysis and design asymmetric heterogeneous surface will cause further rapid progress. In this regard, the recent discovery by Tiefenbacher and co-workers using self-assembled resorcin[4]arene hexamer perhaps suits best for an asymmetric development.214 We expect that a closer interaction among theory, physical organic study and nanoengineering can play an important role in making these processes more robust.215−217 Empowered by mechanistic understanding coupled with increased awareness, more synthetic chemists will likely adopt these unconventional techniques with growing enthusiasm.
By highlighting and conceptualizing many of the recent developments in state-of-the-art asymmetric hydroalkoxylations, we hope that this review will knit together the broad cross-section of computational and synthetic chemists active across various areas of asymmetric catalysis and nucleate new efforts to explore the unknown avenues for further research in this burgeoning area.
Acknowledgments
Generous support by the Max-Planck-Society, the Horizon 2020 Marie Sklodowska-Curie Postdoctoral Fellowship (to R.M, grant agreement no. 897130), the Studienstiftung des deutschen Volkes (doctoral scholarship to M.J.S.), and the European Research Council (Advanced Grant “C–H Acids for Organic Synthesis, CHAOS” to B.L) is gratefully acknowledged.
Glossary
Abbreviations
- CPA
chiral phosphoric acid
- BIPHEP
bis(phosphanyl)biphenyl
- BINOL
1,1′-bi-2-naphthol
- DTBM
3,5-di-tert-butyl-4-methoxyphenyl
- IDP
imidodiphosphate
- IDPi
imidodiphosphorimidate
- LBBA
Lewis base assisted Brønsted acid
- LBA
Lewis acid assisted Brønsted acid
- NHC
nitrogen heterocyclic carbenes
- PMB
p-methoxybenzyl
- SPINOL
1,1′-spirobiindane-7,7′-diol
- TADDOL
α,α,α′,α′-tetraaryl-1,3-dioxolane-4,5-dimethanol
- THF
tetrahydrofuran
- THP
tetrahydropyran
Biographies
Jennifer L. Kennemur attended Texas Tech University, where she conducted her undergraduate research with Prof. Michael Mayer and earned a B.S. in Chemistry in 2013. She then moved to the University of Illinois at Urbana—Champaign and studied Rh- and Pd-catalyzed regio- and enantioselective functionalizations of alkenes in the laboratory of Prof. Kami Hull, earning a M.S. in Chemistry in 2016. In 2017, she moved to Mülheim an der Ruhr, Germany, and began her Ph.D. studies with Prof. Dr. Benjamin List at the Max-Planck-Institut für Kohlenforschung. Her work in the List Lab involves the design and synthesis of strong chiral Brønsted acids and their utility in asymmetric organocatalysis.
Rajat Maji was born in West Bengal, India, and obtained M.S. degrees from the IIT-Kharagpur. During this time, he worked on synthetic carbohydrates under the tutelage of Prof. T. Pathak and also carried out a project in the research group of Prof. J. J. Vittal at the National University of Singapore (NUS). Subsequently, he spent one year as a CSIR Fellow with Prof. S. Hajra, working on asymmetric thiourea organocatalysis. Rajat earned his Ph.D. in 2018 from the Texas A & M University, under the supervision of Prof. Steven E Wheeler, where he honed his knowledge of organic structure and reactivity through the lens of state-of-the-art computations. In April 2019, Rajat resumed his training by undertaking a postdoctoral position supported by Marie Curie fellowship in the group of Prof. Benjamin List, where he is developing new asymmetric olefin activation strategy, harnessing the synergy of experiment and theory.
Manuel J. Scharf was born in 1995 in Hannover, Germany, where he also conducted his Bachelors and Masters studies at the Leibniz Universität Hannover. During a short stay with Prof. Barry Trost at Stanford University supported by a DAAD fellowship, he worked on Zn-prophenol-catalyzed direct asymmetric Mannich reactions. He ultimately received his M.Sc. degree in Medicinal and Natural Product Chemistry in 2018. He subsequently went on to the Max-Planck-Institut für Kohlenforschung in Mülheim an der Ruhr, Germany, to pursue a Ph.D. under the supervision of Prof. Dr. Benjamin List as a fellow of the Studienstiftung des deutschen Volkes. His graduate studies focus on the design and application of strong and confined Brønsted acids in the catalytic asymmetric synthesis of alkaloid natural products.
Benjamin List was born in 1968 in Frankfurt, Germany. He graduated from the Freie University Berlin (1993) and received his Ph.D. (1997) from the University of Frankfurt under the supervision of Prof. J. Mulzer. After postdoctoral studies (1997–1998) as a Feodor Lynen Fellow of the Alexander von Humboldt foundation at The Scripps Research Institute with Prof. R. A. Lerner, he became a Tenure Track Assistant Professor there in January 1999. In 2003, he moved to the Max-Planck-Institut für Kohlenforschung, where he has been a director since 2005. From 2012 until 2014, he has been the managing director of the institute. Since 2004, he has served as an honorary professor at the University of Cologne, and since 2020 he has been a Specially Appointed Professor at Hokkaido University, Japan. His research interests are new catalysis concepts and chemical synthesis in general. He has developed several concepts, including aminocatalysis, enamine catalysis, and asymmetric counteranion-directed catalysis (ACDC). He was recently announced as a recipient of the 2021 Nobel Prize in Chemistry for his pioneering work in asymmetric organocatalysis.
Author Contributions
† J.L.K. and R.M. contributed equally
Open access funded by Max Planck Society.
The authors declare no competing financial interest.
References
- Beller M.; Seayad J.; Tillack A.; Jiao H. Catalytic Markovnikov and anti-Markovnikov Functionalization of Alkenes and Alkynes: Recent Developments and Trends. Angew. Chem., Int. Ed. 2004, 43, 3368–3398. 10.1002/anie.200300616. [DOI] [PubMed] [Google Scholar]
- Blieck R.; Taillefer M.; Monnier F. Metal-Catalyzed Intermolecular Hydrofunctionalization of Allenes: Easy Access to Allylic Structures via the Selective Formation of C–N, C–C, and C–O Bonds. Chem. Rev. 2020, 120, 13545–13598. 10.1021/acs.chemrev.0c00803. [DOI] [PubMed] [Google Scholar]
- Beletskaya I. P.; Najera C.; Yus M. Catalysis and Regioselectivity in Hydrofunctionalization Reactions of Unsaturated Carbon Bonds. Part I. Russ. Chem. Rev. 2020, 89, 250–274. 10.1070/RCR4916. [DOI] [Google Scholar]
- Beletskaya I. P.; N C.; Yus M. Catalysis and Regioselectivity in Hydrofunctionalization Reactions of Unsaturated Carbon Bonds. Part II. Hydroamination. Russ. Chem. Rev. 2020, 89, 1074–1114. 10.1070/RCR4953. [DOI] [Google Scholar]
- Beletskaya I. P.; Najera C.; Yus M. Catalysis and Regioselectivity in Hydrofunctionalization Reactions of Unsaturated Carbon Bonds. Part III. Russ. Chem. Rev. 2021, 90, 70–93. 10.1070/RCR4983. [DOI] [Google Scholar]
- Chen J.; Lu Z. Asymmetric Hydrofunctionalization of Minimally Functionalized Alkenes via Earth Abundant Transition Metal Catalysis. Org. Chem. Front. 2018, 5, 260–272. 10.1039/C7QO00613F. [DOI] [Google Scholar]
- Abbiati G.; Beccalli E. M.; Rossi E., Groups 9 and 10 Metals-Catalyzed O–H Bond Addition to Unsaturated Molecules. In Hydrofunctionalization; Ananikov V. P., Tanaka M., Eds.; Springer: Berlin, Heidelberg, 2011; pp 231–290. [Google Scholar]
- Hintermann L., Recent Developments in Metal-Catalyzed Additions of Oxygen Nucleophiles to Alkenes and Alkynes. In C–X Bond Formation; Vigalok A., Ed.; Springer: Berlin, Heidelberg, 2010; pp 123–155. [Google Scholar]
- Xie W.-B.; Li Z. Asymmetric Synthesis of Ethers by Catalytic Alkene Hydroalkoxylation. Synthesis 2020, 52, 2127–2146. 10.1055/s-0039-1690874. [DOI] [Google Scholar]
- McGrath N. A.; Brichacek M.; Njardarson J. T. A Graphical Journey of Innovative Organic Architectures That Have Improved Our Lives. J. Chem. Educ. 2010, 87, 1348–1349. 10.1021/ed1003806. [DOI] [Google Scholar]
- Haydl A. M.; Breit B. The Total Synthesis of Epothilone D as a Yardstick for Probing New Methodologies. Chem. - Eur. J. 2017, 23, 541–545. 10.1002/chem.201605011. [DOI] [PubMed] [Google Scholar]
- Ishibashi H.; Ishihara K.; Yamamoto H. A New Artificial Cyclase for Polyprenoids: Enantioselective Total Synthesis of (−)-Chromazonarol, (+)-8-epi-Puupehedione, and (−)-11‘-Deoxytaondiol Methyl Ether. J. Am. Chem. Soc. 2004, 126, 11122–11123. 10.1021/ja0472026. [DOI] [PubMed] [Google Scholar]
- Ishihara K.; Ishibashi H.; Yamamoto H. Enantio- and Diastereoselective Stepwise Cyclization of Polyprenoids Induced by Chiral and Achiral LBAs. A New Entry to (−)-Ambrox, (+)-Podocarpa-8,11,13-triene Diterpenoids, and (−)-Tetracyclic Polyprenoid of Sedimentary Origin. J. Am. Chem. Soc. 2002, 124, 3647–3655. 10.1021/ja0124865. [DOI] [PubMed] [Google Scholar]
- Lee S.; Rhee Y. H. Synthesis of Deoxyaminosugar Cyclohexyl-l-callipeltose and Its Diastereomer Using Pd-Catalyzed Asymmetric Hydroalkoxylation. J. Org. Chem. 2019, 84, 9353–9357. 10.1021/acs.joc.9b01059. [DOI] [PubMed] [Google Scholar]
- Schmidt J. P.; Breit B. Rhodium-Catalyzed Cyclization of Terminal and Internal Allenols: An Atom Economic and Highly Stereoselective Access Towards Tetrahydropyrans. Angew. Chem., Int. Ed. 2020, 59, 23485–23490. 10.1002/anie.202009166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slutskyy Y.; Jamison C. R.; Zhao P.; Lee J.; Rhee Y. H.; Overman L. E. Versatile Construction of 6-Substituted cis-2,8-Dioxabicyclo[3.3.0]octan-3-ones: Short Enantioselective Total Syntheses of Cheloviolenes A and B and Dendrillolide C. J. Am. Chem. Soc. 2017, 139, 7192–7195. 10.1021/jacs.7b04265. [DOI] [PubMed] [Google Scholar]
- Tsuji N.; Kennemur J. L.; Buyck T.; Lee S.; Prévost S.; Kaib P. S. J.; Bykov D.; Farès C.; List B. Activation of Olefins Via Asymmetric Brønsted Acid Catalysis. Science 2018, 359, 1501–1505. 10.1126/science.aaq0445. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Song S.; Jiang F.; Fu C.; Ma S. Efficient Syntheses of Traumatic Lactone and Rhizobialide. Chem. - Eur. J. 2019, 25, 9948–9958. 10.1002/chem.201901210. [DOI] [PubMed] [Google Scholar]
- Sevov C. S.; Hartwig J. F. Iridium-Catalyzed, Intermolecular Hydroetherification of Unactivated Aliphatic Alkenes with Phenols. J. Am. Chem. Soc. 2013, 135, 9303–9306. 10.1021/ja4052153. [DOI] [PubMed] [Google Scholar]
- Johns A. M.; Sakai N.; Ridder A.; Hartwig J. F. Direct Measurement of the Thermodynamics of Vinylarene Hydroamination. J. Am. Chem. Soc. 2006, 128, 9306–9307. 10.1021/ja062773e. [DOI] [PubMed] [Google Scholar]
- Dey J.; O’Donoghu A. C.; More O’Ferrall R. A. Equilibrium Constants for Dehydration of Water Adducts of Aromatic Carbon–Carbon Double Bonds. J. Am. Chem. Soc. 2002, 124, 8561–8574. 10.1021/ja0126125. [DOI] [PubMed] [Google Scholar]
- Nigst T. A.; Antipova A.; Mayr H. Nucleophilic Reactivities of Hydrazines and Amines: The Futile Search for the α-Effect in Hydrazine Reactivities. J. Org. Chem. 2012, 77, 8142–8155. 10.1021/jo301497g. [DOI] [PubMed] [Google Scholar]
- Kumara M. N.; Nakahara T.; Kobayashi S.; Fujio M.; Mishima M. Nucleophilicities of Alcohols and Water in Acetonitrile Based on Reactivities of Benzhydrylium Ions. Bull. Chem. Soc. Jpn. 2018, 91, 523–530. 10.1246/bcsj.20170360. [DOI] [Google Scholar]
- Nising C. F.; Bräse S. The Oxa-Michael Reaction: From Recent Developments to Applications in Natural Product Synthesis. Chem. Soc. Rev. 2008, 37, 1218–1228. 10.1039/b718357g. [DOI] [PubMed] [Google Scholar]
- Nising C. F.; Bräse S. Recent Developments in the Field of Oxa-Michael Reactions. Chem. Soc. Rev. 2012, 41, 988–999. 10.1039/C1CS15167C. [DOI] [PubMed] [Google Scholar]
- Müller T. E.; Hultzsch K. C.; Yus M.; Foubelo F.; Tada M. Hydroamination: Direct Addition of Amines to Alkenes and Alkynes. Chem. Rev. 2008, 108, 3795–3892. 10.1021/cr0306788. [DOI] [PubMed] [Google Scholar]
- Dang T. T.; Boeck F.; Hintermann L. Hidden Brønsted Acid Catalysis: Pathways of Accidental or Deliberate Generation of Triflic Acid from Metal Triflates. J. Org. Chem. 2011, 76, 9353–9361. 10.1021/jo201631x. [DOI] [PubMed] [Google Scholar]
- Trummal A.; Lipping L.; Kaljurand I.; Koppel I. A.; Leito I. Acidity of Strong Acids in Water and Dimethyl Sulfoxide. J. Phys. Chem. A 2016, 120, 3663–3669. 10.1021/acs.jpca.6b02253. [DOI] [PubMed] [Google Scholar]
- Rosenfeld D. C.; Shekhar S.; Takemiya A.; Utsunomiya M.; Hartwig J. F. Hydroamination and Hydroalkoxylation Catalyzed by Triflic Acid. Parallels to Reactions Initiated with Metal Triflates. Org. Lett. 2006, 8, 4179–4182. 10.1021/ol061174+. [DOI] [PubMed] [Google Scholar]
- Li Z.; Zhang J.; Brouwer C.; Yang C.-G.; Reich N. W.; He C. Brønsted Acid Catalyzed Addition of Phenols, Carboxylic Acids, and Tosylamides to Simple Olefins. Org. Lett. 2006, 8, 4175–4178. 10.1021/ol0610035. [DOI] [PubMed] [Google Scholar]
- Ohta T.; Kataoka Y.; Miyoshi A.; Oe Y.; Furukawa I.; Ito Y. Ruthenium-Catalyzed Intramolecular Cyclization of Hetero-Functionalized Allylbenzenes. J. Organomet. Chem. 2007, 692, 671–677. 10.1016/j.jorganchem.2006.05.059. [DOI] [Google Scholar]
- Tschan M. J.-L.; Thomas C. M.; Strub H.; Carpentier J.-F. Copper(II) Triflate as a Source of Triflic Acid: Effective, Green Catalysis of Hydroalkoxylation Reactions. Adv. Synth. Catal. 2009, 351, 2496–2504. 10.1002/adsc.200800750. [DOI] [Google Scholar]
- Hanley P. S.; Hartwig J. F. Migratory Insertion of Alkenes Into Metal-Oxygen and Metal-Nitrogen Bonds. Angew. Chem., Int. Ed. 2013, 52, 8510–8525. 10.1002/anie.201300134. [DOI] [PubMed] [Google Scholar]
- Ishii A.; Nakata N., The Mechanism for Transition-Metal-Catalyzed Hydrochalcogenation of Unsaturated Organic Molecules. In Hydrofunctionalization; Ananikov V. P., Tanaka M., Eds.; Springer: Berlin, Heidelberg, 2013; pp 21–50. [Google Scholar]
- Sanford M. S.; Groves J. T. Anti-Markovnikov Hydrofunctionalization of Olefins Mediated by Rhodium-Porphyrin Complexes. Angew. Chem., Int. Ed. 2004, 43, 588–590. 10.1002/anie.200351941. [DOI] [PubMed] [Google Scholar]
- Murayama H.; Nagao K.; Ohmiya H.; Sawamura M. Copper(I)-Catalyzed Intramolecular Hydroalkoxylation of Unactivated Alkenes. Org. Lett. 2015, 17, 2039–2041. 10.1021/acs.orglett.5b00758. [DOI] [PubMed] [Google Scholar]
- Nagamoto M.; Nishimura T. Iridium-Catalyzed Asymmetric Cyclization of Alkenoic Acids Leading to γ-Lactones. Chem. Commun. 2015, 51, 13466–13469. 10.1039/C5CC05393E. [DOI] [PubMed] [Google Scholar]
- Chen D.; Berhane I. A.; Chemler S. R. Copper-Catalyzed Enantioselective Hydroalkoxylation of Alkenols for the Synthesis of Cyclic Ethers. Org. Lett. 2020, 22, 7409–7414. 10.1021/acs.orglett.0c01691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzudza A.; Marks T. J. Efficient Intramolecular Hydroalkoxylation/Cyclization of Unactivated Alkenols Mediated by Lanthanide Triflate Ionic Liquids. Org. Lett. 2009, 11, 1523–1526. 10.1021/ol8029559. [DOI] [PubMed] [Google Scholar]
- Hoveyda A. H.; Evans D. A.; Fu G. C. Substrate-Directable Chemical Reactions. Chem. Rev. 1993, 93, 1307–1370. 10.1021/cr00020a002. [DOI] [Google Scholar]
- Ickes A. R.; Ensign S. C.; Gupta A. K.; Hull K. L. Regio- and Chemoselective Intermolecular Hydroamination of Allyl Imines for the Synthesis of 1,2-Diamines. J. Am. Chem. Soc. 2014, 136, 11256–11259. 10.1021/ja505794u. [DOI] [PubMed] [Google Scholar]
- Sevov C. S.; Zhou J.; Hartwig J. F. Iridium-Catalyzed Intermolecular Hydroamination of Unactivated Aliphatic Alkenes with Amides and Sulfonamides. J. Am. Chem. Soc. 2012, 134, 11960–11963. 10.1021/ja3052848. [DOI] [PubMed] [Google Scholar]
- Beller M.; Trauthwein H.; Eichberger M.; Breindl C.; Herwig J.; Müller T. E.; Thiel O. R. The First Rhodium-Catalyzed Anti-Markovnikov Hydroamination: Studies on Hydroamination and Oxidative Amination of Aromatic Olefins. Chem. - Eur. J. 1999, 5, 1306–1319. . [DOI] [Google Scholar]
- Yu Z.-X.; Cheong P. H.-Y.; Liu P.; Legault C. Y.; Wender P. A.; Houk K. N. Origins of Differences in Reactivities of Alkenes, Alkynes, and Allenes in [Rh(CO)2Cl]2-Catalyzed (5 + 2) Cycloaddition Reactions with Vinylcyclopropanes. J. Am. Chem. Soc. 2008, 130, 2378–2379. 10.1021/ja076444d. [DOI] [PubMed] [Google Scholar]
- Kawamoto T.; Hirabayashi S.; Guo X.-X.; Nishimura T.; Hayashi T. Rhodium-Catalyzed Asymmetric Hydroalkoxylation and Hydrosulfenylation of Diphenylphosphinylallenes. Chem. Commun. 2009, 3528–3530. 10.1039/b900976k. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Brieden W.; Baringhaus K. H. Intermolecular Additions and Cycloisomerizations by a Pd-Catalyzed Sequence of an Intramolecular Redox Reaction and an Addition. Angew. Chem., Int. Ed. Engl. 1992, 31, 1335–1336. 10.1002/anie.199213351. [DOI] [Google Scholar]
- Yamamoto Y.; Al-Masum M.; Asao N. Palladium-Catalyzed Addition of Activated Methylene and Methyne Compounds to Allenes. J. Am. Chem. Soc. 1994, 116, 6019–6020. 10.1021/ja00092a083. [DOI] [Google Scholar]
- Trost B. M.; Gerusz V. J. Palladium-Catalyzed Addition of Pronucleophiles to Allenes. J. Am. Chem. Soc. 1995, 117, 5156–5157. 10.1021/ja00123a020. [DOI] [PubMed] [Google Scholar]
- Al-Masum M.; Yamamoto Y. Palladium-Catalyzed Hydrocarboxylation of Allenes. J. Am. Chem. Soc. 1998, 120, 3809–3810. 10.1021/ja974223+. [DOI] [Google Scholar]
- Kadota I.; Shibuya A.; Gyoung Y. S.; Yamamoto Y. Palladium/Acetic Acid Catalyzed Allylation of Some Pronucleophiles with Simple Alkynes. J. Am. Chem. Soc. 1998, 120, 10262–10263. 10.1021/ja981299c. [DOI] [Google Scholar]
- Patil N. T.; Nawaz Khan F.; Yamamoto Y. Microwave-Enhanced Pd(0)/Acetic Acid Catalyzed Allylation Reactions of C, N, and O-Pronucleophiles With Alkynes. Tetrahedron Lett. 2004, 45, 8497–8499. 10.1016/j.tetlet.2004.09.099. [DOI] [Google Scholar]
- Patil N. T.; Pahadi N. K.; Yamamoto Y. Pd(0)-PhCOOH Catalyzed Addition of Oxygen Pronucleophiles to Allenes and Internal Alkynes. Can. J. Chem. 2005, 83, 569–573. 10.1139/v05-043. [DOI] [Google Scholar]
- Patil N. T.; Kadota I.; Shibuya A.; Gyoung Y. S.; Yamamoto Y. Allylation of Carbon Pronucleophiles with Alkynes in the Presence of Palladium/Acetic Acid Catalyst. Adv. Synth. Catal. 2004, 346, 800–804. 10.1002/adsc.200404046. [DOI] [Google Scholar]
- Evans P. A.; Leahy D. K. Regio- and Enantiospecific Rhodium-Catalyzed Allylic Etherification Reactions Using Copper(I) Alkoxides: Influence of the Copper Halide Salt on Selectivity. J. Am. Chem. Soc. 2002, 124, 7882–7883. 10.1021/ja026337d. [DOI] [PubMed] [Google Scholar]
- Hayashi T.; Okada A.; Suzuka T.; Kawatsura M. High Enantioselectivity in Rhodium-Catalyzed Allylic Alkylation of 1-Substituted 2-Propenyl Acetates. Org. Lett. 2003, 5, 1713–1715. 10.1021/ol0343562. [DOI] [PubMed] [Google Scholar]
- Koschker P.; Breit B. Branching Out: Rhodium-Catalyzed Allylation with Alkynes and Allenes. Acc. Chem. Res. 2016, 49, 1524–1536. 10.1021/acs.accounts.6b00252. [DOI] [PubMed] [Google Scholar]
- Koschker P.; Lumbroso A.; Breit B. Enantioselective Synthesis of Branched Allylic Esters via Rhodium-Catalyzed Coupling of Allenes with Carboxylic Acids. J. Am. Chem. Soc. 2011, 133, 20746–20749. 10.1021/ja210149g. [DOI] [PubMed] [Google Scholar]
- Ganss S.; Breit B. Enantioselective Rhodium-Catalyzed Atom-Economical Macrolactonization. Angew. Chem., Int. Ed. 2016, 55, 9738–9742. 10.1002/anie.201604301. [DOI] [PubMed] [Google Scholar]
- Brosowsky J.; Lutterbeck M.; Liebich A.; Keller M.; Herp D.; Vogelmann A.; Jung M.; Breit B. Syntheses of Thailandepsin B Pseudo-Natural Products: Access to New Highly Potent HDAC Inhibitors via Late-Stage Modification. Chem. - Eur. J. 2020, 26, 16241–16245. 10.1002/chem.202002449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steib P.; Breit B. Concise Total Synthesis of (−)-Vermiculine through a Rhodium-Catalyzed C2-Symmetric Dimerization Strategy. Chem. - Eur. J. 2019, 25, 3532–3535. 10.1002/chem.201900216. [DOI] [PubMed] [Google Scholar]
- Steib P.; Breit B. Enantioselective Rhodium-Catalyzed Dimerization of omega-Allenyl Carboxylic Acids: Straightforward Synthesis of C2 -Symmetric Macrodiolides. Angew. Chem., Int. Ed. 2018, 57, 6572–6576. 10.1002/anie.201803369. [DOI] [PubMed] [Google Scholar]
- Haydl A. M.; Breit B. Atom-Economical Dimerization Strategy by the Rhodium-Catalyzed Addition of Carboxylic Acids to Allenes: Protecting-Group-Free Synthesis of Clavosolide A and Late-Stage Modification. Angew. Chem., Int. Ed. 2015, 54, 15530–15534. 10.1002/anie.201506618. [DOI] [PubMed] [Google Scholar]
- Schotes C.; Ostrovskyi D.; Senger J.; Schmidtkunz K.; Jung M.; Breit B. Total Synthesis of (18S)- and (18R)-Homolargazole by Rhodium-Catalyzed Hydrocarboxylation. Chem. - Eur. J. 2014, 20, 2164–2168. 10.1002/chem.201303300. [DOI] [PubMed] [Google Scholar]
- Wolf J.; Werner H. Basic metals. Part 61. Synthesis of [(C5H5)Rh(η3-1-MeC3H4)[P(CHMe2)3]PF6 from (C5H5)Rh(MeC.tplbond.CMe)[P(CHMe2)3]. The Mechanism of Conversion of an Alkyne Into an Allyl Ligand via an Allene Intermediate. Organometallics 1987, 6, 1164–1169. 10.1021/om00149a005. [DOI] [Google Scholar]
- Lumbroso A.; Koschker P.; Vautravers N. R.; Breit B. Redox-Neutral Atom-Economic Rhodium-Catalyzed Coupling of Terminal Alkynes with Carboxylic Acids Toward Branched Allylic Esters. J. Am. Chem. Soc. 2011, 133, 2386–2389. 10.1021/ja1108613. [DOI] [PubMed] [Google Scholar]
- Koschker P.; Kähny M.; Breit B. Enantioselective Redox-Neutral Rh-Catalyzed Coupling of Terminal Alkynes with Carboxylic Acids Toward Branched Allylic Esters. J. Am. Chem. Soc. 2015, 137, 3131–3137. 10.1021/jacs.5b01131. [DOI] [PubMed] [Google Scholar]
- Gellrich U.; Meißner A.; Steffani A.; Kähny M.; Drexler H.-J.; Heller D.; Plattner D. A.; Breit B. Mechanistic Investigations of the Rhodium Catalyzed Propargylic CH Activation. J. Am. Chem. Soc. 2014, 136, 1097–1104. 10.1021/ja411204d. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Breit B. Rhodium-Catalyzed Enantioselective Intermolecular Hydroalkoxylation of Allenes and Alkynes with Alcohols: Synthesis of Branched Allylic Ethers. Angew. Chem., Int. Ed. 2016, 55, 8440–8443. 10.1002/anie.201603538. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Breit B. Rhodium-Catalyzed Regio- and Enantioselective Addition of N-Hydroxyphthalimide to Allenes: A Strategy To Synthesize Chiral Allylic Alcohols. Org. Lett. 2018, 20, 300–303. 10.1021/acs.orglett.7b03709. [DOI] [PubMed] [Google Scholar]
- Sridharan V.; Fan L.; Takizawa S.; Suzuki T.; Sasai H. Pd(II)–SDP-Catalyzed Enantioselective 5-Exo-Dig Cyclization of γ-Alkynoic Acids: Application to the Synthesis of Functionalized Dihydofuran-2(3H)-ones Containing a Chiral Quaternary Carbon Center. Org. Biomol. Chem. 2013, 11, 5936–5943. 10.1039/c3ob41103f. [DOI] [PubMed] [Google Scholar]
- Ali M.; Li C. Desymmetrization Construction of Chiral Lactones by Synergistic Cu(II) Complex and Organic Base. Tetrahedron Lett. 2020, 61, 152106. 10.1016/j.tetlet.2020.152106. [DOI] [Google Scholar]
- Sota Y.; Yamamoto M.; Murai M.; Uenishi J. i.; Uemura M. Gold(I)-Catalyzed Asymmetric Desymmetrization of meso-Alkynyl Diols and Kinetic Resolution of the Corresponding dl-Diols: Effects of Celite Filtration and Silver Salts. Chem. - Eur. J. 2015, 21, 4398–4404. 10.1002/chem.201405889. [DOI] [PubMed] [Google Scholar]
- Zheng Y.; Guo L.; Zi W. Enantioselective and Regioselective Hydroetherification of Alkynes by Gold-Catalyzed Desymmetrization of Prochiral Phenols with P-Stereogenic Centers. Org. Lett. 2018, 20, 7039–7043. 10.1021/acs.orglett.8b02982. [DOI] [PubMed] [Google Scholar]
- Wang P.-S.; Liu P.; Zhai Y.-J.; Lin H.-C.; Han Z.-Y.; Gong L.-Z. Asymmetric Allylic C–H Oxidation for the Synthesis of Chromans. J. Am. Chem. Soc. 2015, 137, 12732–12735. 10.1021/jacs.5b08477. [DOI] [PubMed] [Google Scholar]
- Patil N. T.; Lutete L. M.; Wu H.; Pahadi N. K.; Gridnev I. D.; Yamamoto Y. Palladium-Catalyzed Intramolecular Asymmetric Hydroamination, Hydroalkoxylation, and Hydrocarbonation of Alkynes. J. Org. Chem. 2006, 71, 4270–4279. 10.1021/jo0603835. [DOI] [PubMed] [Google Scholar]
- Lim W.; Kim J.; Rhee Y. H. Pd-Catalyzed Asymmetric Intermolecular Hydroalkoxylation of Allene: An Entry to Cyclic Acetals with Activating Group-Free and Flexible Anomeric Control. J. Am. Chem. Soc. 2014, 136, 13618–13621. 10.1021/ja508587f. [DOI] [PubMed] [Google Scholar]
- Garnsey M. R.; Slutskyy Y.; Jamison C. R.; Zhao P.; Lee J.; Rhee Y. H.; Overman L. E. Short Enantioselective Total Syntheses of Cheloviolenes A and B and Dendrillolide C via Convergent Fragment Coupling Using a Tertiary Carbon Radical. J. Org. Chem. 2018, 83, 6958–6976. 10.1021/acs.joc.7b02458. [DOI] [PubMed] [Google Scholar]
- Jiang L.; Jia T.; Wang M.; Liao J.; Cao P. Pd-Catalyzed Enantioselective Hydroalkoxylation of Alkoxyallenes with Phenol for Construction of Acyclic O,O-Acetals. Org. Lett. 2015, 17, 1070–1073. 10.1021/acs.orglett.5b00146. [DOI] [PubMed] [Google Scholar]
- Nagano H.; Katsuki T. Stereocontrolled OH Protection: Asymmetric Tetrahydrofuranylation. Chem. Lett. 2002, 31, 782–783. 10.1246/cl.2002.782. [DOI] [Google Scholar]
- Works C. F.; Ford P. C. Photoreactivity of the Ruthenium Nitrosyl Complex, Ru(salen)(Cl)(NO). Solvent Effects on the Back Reaction of NO with the Lewis Acid RuIII(salen)(Cl)1. J. Am. Chem. Soc. 2000, 122, 7592–7593. 10.1021/ja000137p. [DOI] [Google Scholar]
- Nguyen H.; Gagné M. R. Enantioselective Cascade Cyclization/Protodemetalation of Polyenes with N3Pt2+ Catalysts. ACS Catal. 2014, 4, 855–859. 10.1021/cs401190c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Xu X.; Sun H.; Tao G.; Chang X.-Y.; Xing X.; Chen B.; Xu C. Development of Highly Efficient Platinum Catalysts for Hydroalkoxylation and Hydroamination of Unactivated Alkenes. Nat. Commun. 2021, 12, 1953. 10.1038/s41467-021-22287-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorin D. J.; Toste F. D. Relativistic Effects in Homogeneous Gold Catalysis. Nature 2007, 446, 395–403. 10.1038/nature05592. [DOI] [PubMed] [Google Scholar]
- Homs A.; Escofet I.; Echavarren A. M. On the Silver Effect and the Formation of Chloride-Bridged Digold Complexes. Org. Lett. 2013, 15, 5782–5785. 10.1021/ol402825v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Widenhoefer R. A. Gold(I)-Catalyzed Intramolecular Enantioselective Hydroalkoxylation of Allenes. Angew. Chem., Int. Ed. 2007, 46, 283–285. 10.1002/anie.200603260. [DOI] [PubMed] [Google Scholar]
- Malik G.; Ferry A.; Guinchard X. When Phosphosugars Meet Gold: Synthesis and Catalytic Activities of Phostones and Polyhydroxylated Phosphonite Au(I) Complexes. Molecules 2015, 20, 21082–21093. 10.3390/molecules201219755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S.; Nanko M.; Shinozaki T.; Kojima M.; Aikawa K.; Mikami K. Dynamic Chirality Control of tropos DPCB-digold Skeleton by Chiral Binaphthyldicarboxylate. Chem. - Asian J. 2016, 11, 823–827. 10.1002/asia.201501326. [DOI] [PubMed] [Google Scholar]
- Teller H.; Corbet M.; Mantilli L.; Gopakumar G.; Goddard R.; Thiel W.; Fürstner A. One-Point Binding Ligands for Asymmetric Gold Catalysis: Phosphoramidites with a TADDOL-Related but Acyclic Backbone. J. Am. Chem. Soc. 2012, 134, 15331–15342. 10.1021/ja303641p. [DOI] [PubMed] [Google Scholar]
- Ilg M. K.; Wolf L. M.; Mantilli L.; Farès C.; Thiel W.; Fürstner A. A Striking Case of Enantioinversion in Gold Catalysis and Its Probable Origins. Chem. - Eur. J. 2015, 21, 12279–12284. 10.1002/chem.201502160. [DOI] [PubMed] [Google Scholar]
- Sethofer S. G.; Mayer T.; Toste F. D. Gold(I)-Catalyzed Enantioselective Polycyclization Reactions. J. Am. Chem. Soc. 2010, 132, 8276–8277. 10.1021/ja103544p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stork G.; Burgstahler A. W. The Stereochemistry of Polyene Cyclization. J. Am. Chem. Soc. 1955, 77, 5068–5077. 10.1021/ja01624a038. [DOI] [Google Scholar]
- Cheng X.; Wang Z.; Quintanilla C. D.; Zhang L. Chiral Bifunctional Phosphine Ligand Enabling Gold-Catalyzed Asymmetric Isomerization of Alkyne to Allene and Asymmetric Synthesis of 2,5-Dihydrofuran. J. Am. Chem. Soc. 2019, 141, 3787–3791. 10.1021/jacs.8b12833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahlau M.; List B. Asymmetric Counteranion-Directed Catalysis: Concept, Definition, and Applications. Angew. Chem., Int. Ed. 2013, 52, 518–533. 10.1002/anie.201205343. [DOI] [PubMed] [Google Scholar]
- Phipps R. J.; Hamilton G. L.; Toste F. D. The Progression of Chiral Anions From Concepts to Applications in Asymmetric Catalysis. Nat. Chem. 2012, 4, 603–614. 10.1038/nchem.1405. [DOI] [PubMed] [Google Scholar]
- Hamilton G. L.; Kang E. J.; Mba M.; Toste F. D. A Powerful Chiral Counterion Strategy for Asymmetric Transition Metal Catalysis. Science 2007, 317, 496–499. 10.1126/science.1145229. [DOI] [PubMed] [Google Scholar]
- Martin N. J. A.; List B. Highly Enantioselective Transfer Hydrogenation of α,β-Unsaturated Ketones. J. Am. Chem. Soc. 2006, 128, 13368–13369. 10.1021/ja065708d. [DOI] [PubMed] [Google Scholar]
- LaLonde R. L.; Wang Z. J.; Mba M.; Lackner A. D.; Toste F. D. Gold(I)-Catalyzed Enantioselective Synthesis of Pyrazolidines, Isoxazolidines, and Tetrahydrooxazines. Angew. Chem., Int. Ed. 2010, 49, 598–601. 10.1002/anie.200905000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aikawa K.; Kojima M.; Mikami K. Synergistic Effect: Hydroalkoxylation of Allenes through Combination of Enantiopure BIPHEP-Gold Complexes and Chiral Anions. Adv. Synth. Catal. 2010, 352, 3131–3135. 10.1002/adsc.201000672. [DOI] [Google Scholar]
- Zi W.; Toste F. D. Gold(I)-Catalyzed Enantioselective Desymmetrization of 1,3-Diols through Intramolecular Hydroalkoxylation of Allenes. Angew. Chem., Int. Ed. 2015, 54, 14447–14451. 10.1002/anie.201508331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quach R.; Furkert D. P.; Brimble M. A. Synthesis of Benzannulated Spiroacetals Using Chiral Gold–Phosphine Complexes and Chiral Anions. Tetrahedron Lett. 2013, 54, 5865–5868. 10.1016/j.tetlet.2013.08.077. [DOI] [Google Scholar]
- Rexit A. A.; Mailikezati M. Asymmetric Synthesis of Optically Active Spiroacetals From Alkynyl Glycols Catalyzed by Gold Complex/Brønsted Acid Binary System. Tetrahedron Lett. 2015, 56, 2651–2655. 10.1016/j.tetlet.2015.03.007. [DOI] [Google Scholar]
- Čorić I.; List B. Asymmetric Spiroacetalization Catalysed by Confined Brønsted Acids. Nature 2012, 483, 315–319. 10.1038/nature10932. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zheng K.; Hong R. Chiral Silver Phosphate-Catalyzed Cycloisomeric Kinetic Resolution of α-Allenic Alcohols. J. Am. Chem. Soc. 2012, 134, 4096–4099. 10.1021/ja300453u. [DOI] [PubMed] [Google Scholar]
- Arbour J. L.; Rzepa H. S.; Contreras-García J.; Adrio L. A.; Barreiro E. M.; Hii K. K. Silver-Catalysed Enantioselective Addition of O-H and N-H Bonds to Allenes: A New Model for Stereoselectivity Based on Noncovalent Interactions. Chem. - Eur. J. 2012, 18, 11317–11324. 10.1002/chem.201200547. [DOI] [PubMed] [Google Scholar]
- Cao K.-S.; Zheng W.-H. Enantioselective Desymmetrization of Prochiral Allenic Diols via Cooperative Catalysis Of Pd(OAc)2 and a Chiral Phosphoric Acid. Tetrahedron: Asymmetry 2015, 26, 1150–1155. 10.1016/j.tetasy.2015.08.010. [DOI] [Google Scholar]
- Crossley S. W. M.; Obradors C.; Martinez R. M.; Shenvi R. A. Mn-, Fe-, and Co-Catalyzed Radical Hydrofunctionalizations of Olefins. Chem. Rev. 2016, 116, 8912–9000. 10.1021/acs.chemrev.6b00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green S. A.; Crossley S. W. M.; Matos J. L. M.; Vásquez-Céspedes S.; Shevick S. L.; Shenvi R. A. The High Chemofidelity of Metal-Catalyzed Hydrogen Atom Transfer. Acc. Chem. Res. 2018, 51, 2628–2640. 10.1021/acs.accounts.8b00337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevick S. L.; Wilson C. V.; Kotesova S.; Kim D.; Holland P. L.; Shenvi R. A. Catalytic Hydrogen Atom Transfer to Alkenes: A Roadmap for Metal Hydrides and Radicals. Chem. Sci. 2020, 11, 12401–12422. 10.1039/D0SC04112B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenvi R. A.; Matos J. L. M.; Green S. A., Hydrofunctionalization of Alkenes by Hydrogen-Atom Transfer. In Organic Reactions; Denmark S. E., Ed.; John Wiley & Sons: Hoboken NJ, 2020; pp 383–470. [Google Scholar]
- Sato M.; Gunji Y.; Ikeno T.; Yamada T. Stereoselective Preparation of α-Hydroxycarboxamide by Manganese Complex Catalyzed Hydration of α,β-Unsaturated Carboxamide with Molecular Oxygen and Phenylsilane. Chem. Lett. 2004, 33, 1304–1305. 10.1246/cl.2004.1304. [DOI] [Google Scholar]
- Sato M.; Gunji Y.; Ikeno T.; Yamada T. Stereoselective α-Hydrazination of α,β-Unsaturated Carboxylates Catalyzed by Manganese(III) Complex with Dialkylazodicarboxylate and Phenylsilane. Chem. Lett. 2005, 34, 316–317. 10.1246/cl.2005.316. [DOI] [Google Scholar]
- Discolo C. A.; Touney E. E.; Pronin S. V. Catalytic Asymmetric Radical–Polar Crossover Hydroalkoxylation. J. Am. Chem. Soc. 2019, 141, 17527–17532. 10.1021/jacs.9b10645. [DOI] [PubMed] [Google Scholar]
- Shigehisa H.; Hayashi M.; Ohkawa H.; Suzuki T.; Okayasu H.; Mukai M.; Yamazaki A.; Kawai R.; Kikuchi H.; Satoh Y.; Fukuyama A.; Hiroya K. Catalytic Synthesis of Saturated Oxygen Heterocycles by Hydrofunctionalization of Unactivated Olefins: Unprotected and Protected Strategies. J. Am. Chem. Soc. 2016, 138, 10597–10604. 10.1021/jacs.6b05720. [DOI] [PubMed] [Google Scholar]
- Ebisawa K.; Izumi K.; Ooka Y.; Kato H.; Kanazawa S.; Komatsu S.; Nishi E.; Shigehisa H. Catalyst- and Silane-Controlled Enantioselective Hydrofunctionalization of Alkenes by Cobalt-Catalyzed Hydrogen Atom Transfer and Radical-Polar Crossover. J. Am. Chem. Soc. 2020, 142, 13481–13490. 10.1021/jacs.0c05017. [DOI] [PubMed] [Google Scholar]
- Shigehisa H.; Aoki T.; Yamaguchi S.; Shimizu N.; Hiroya K. Hydroalkoxylation of Unactivated Olefins with Carbon Radicals and Carbocation Species as Key Intermediates. J. Am. Chem. Soc. 2013, 135, 10306–10309. 10.1021/ja405219f. [DOI] [PubMed] [Google Scholar]
- Shevick S. L.; Obradors C.; Shenvi R. A. Mechanistic Interrogation of Co/Ni-Dual Catalyzed Hydroarylation. J. Am. Chem. Soc. 2018, 140, 12056–12068. 10.1021/jacs.8b06458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg D. C.; Norton J. R. Hydrogen-Atom Transfer Reactions of Transition-Metal Hydrides. Isr. J. Chem. 1991, 31, 55–66. 10.1002/ijch.199100006. [DOI] [Google Scholar]
- Sweany R. L.; Halpern J. Hydrogenation of α-Methylstyrene by Hydridopentacarbonylmanganese (I). Evidence for a Free-Radical Mechanism. J. Am. Chem. Soc. 1977, 99, 8335–8337. 10.1021/ja00467a045. [DOI] [Google Scholar]
- Crossley S. W. M.; Barabé F.; Shenvi R. A. Simple, Chemoselective, Catalytic Olefin Isomerization. J. Am. Chem. Soc. 2014, 136, 16788–16791. 10.1021/ja5105602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green S. A.; Matos J. L. M.; Yagi A.; Shenvi R. A. Branch-Selective Hydroarylation: Iodoarene–Olefin Cross-Coupling. J. Am. Chem. Soc. 2016, 138, 12779–12782. 10.1021/jacs.6b08507. [DOI] [PubMed] [Google Scholar]
- Ungvary F.; Marko L. Reaction of HCo(CO)4 and CO with Styrene. Mechanism of (α-Phenylpropionyl)- and (β-Phenylpropionyl)cobalt Tetracarbonyl Formation. Organometallics 1982, 1, 1120–1125. 10.1021/om00069a003. [DOI] [Google Scholar]
- Touney E. E.; Foy N. J.; Pronin S. V. Catalytic Radical–Polar Crossover Reactions of Allylic Alcohols. J. Am. Chem. Soc. 2018, 140, 16982–16987. 10.1021/jacs.8b12075. [DOI] [PubMed] [Google Scholar]
- Abley P.; Dockal E. R.; Halpern J. Oxidative Cleavage of Cobalt-Carbon Bonds In Organobis(dimethylglyoximato)cobalt Compounds. J. Am. Chem. Soc. 1972, 94, 659–660. 10.1021/ja00757a070. [DOI] [Google Scholar]
- Halpern J.; Chan M. S.; Hanson J.; Roche T. S.; Topich J. A. Detection and Characterization of Radical Cations Produced by One-Electron Chemical And Electrochemical Oxidations of Organocobalt Compounds. J. Am. Chem. Soc. 1975, 97, 1606–1608. 10.1021/ja00839a072. [DOI] [Google Scholar]
- Halpern J.; Topich J.; Zamaraev K. I. Electron Paramagnetic Resonance Spectra and Electronic Structures of Organobis(dimethylglyoximato)cobalt(IV) Complexes. Inorg. Chim. Acta 1976, 20, L21–L24. 10.1016/S0020-1693(00)94064-7. [DOI] [Google Scholar]
- Topich J.; Halpern J. Organobis(dioximato)cobalt(IV) Complexes: Electron Paramagnetic Resonance Spectra and Electronic Structures. Inorg. Chem. 1979, 18, 1339–1343. 10.1021/ic50195a036. [DOI] [Google Scholar]
- Halpern J.; Chan M. S.; Roche T. S.; Tom G. M.; Hoyer E.; Spiridonov V. P.; Strand T. G. Redox Chemistry of Organobis(dimethylglyoximato)cobalt Complexes. Acta Chem. Scand. 1979, 33a, 141–148. 10.3891/acta.chem.scand.33a-0141. [DOI] [Google Scholar]
- Lande S. S.; Kochi J. K. Formation and Oxidation of Alkyl Radicals by Cobalt(III) Complexes. J. Am. Chem. Soc. 1968, 90, 5196–5207. 10.1021/ja01021a023. [DOI] [Google Scholar]
- Anderson S. N.; Ballard D. H.; Chrzastowski J. Z.; Dodd D.; Johnson M. D. Inversion of Configuration in the Nucleophilic Displacement of Cobalt From Alkylcobalt(IV) Complexes and Its Relevance to the Halogenation of the Corresponding Alkylcobalt(III) Complexes. J. Chem. Soc., Chem. Commun. 1972, 685–686. 10.1039/c39720000685. [DOI] [Google Scholar]
- Magnuson R. H.; Halpern J.; Levitin I. Y.; Vol’pin M. E. Stereochemistry of the Nucleophilic Cleavage of Cobalt–Carbon Bonds in Organocobalt(IV) Compounds. J. Chem. Soc., Chem. Commun. 1978, 44–46. 10.1039/C39780000044. [DOI] [Google Scholar]
- Vol’pin M. E.; Levitin I. Y.; Sigan A. L.; Halpern J.; Tom G. M. Reactivity of Organocobalt(IV) Chelate Complexes Toward Nucleophiles: Diversity of Mechanisms. Inorg. Chim. Acta 1980, 41, 271–277. 10.1016/S0020-1693(00)88468-6. [DOI] [Google Scholar]
- Knowles R. R.; Lin S.; Jacobsen E. N. Enantioselective Thiourea-Catalyzed Cationic Polycyclizations. J. Am. Chem. Soc. 2010, 132, 5030–5032. 10.1021/ja101256v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X.; Chen X.; Chen J.; Sun Y.; Cheng Z.; Lu Z. Ligand-Promoted Cobalt-Catalyzed Radical Hydroamination of Alkenes. Nat. Commun. 2020, 11, 783. 10.1038/s41467-020-14459-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brimioulle R.; Lenhart D.; Maturi M. M.; Bach T. Enantioselective Catalysis of Photochemical Reactions. Angew. Chem., Int. Ed. 2015, 54, 3872–3890. 10.1002/anie.201411409. [DOI] [PubMed] [Google Scholar]
- Rigotti T.; Alemán J. Visible Light Photocatalysis – From Racemic to Asymmetric Activation Strategies. Chem. Commun. 2020, 56, 11169–11190. 10.1039/D0CC03738A. [DOI] [PubMed] [Google Scholar]
- Jiang C.; Chen W.; Zheng W.-H.; Lu H. Advances in Asymmetric Visible-Light Photocatalysis, 2015–2019. Org. Biomol. Chem. 2019, 17, 8673–8689. 10.1039/C9OB01609K. [DOI] [PubMed] [Google Scholar]
- Inoue Y.; Okano T.; Yamasaki N.; Tai A. First Photosensitized Enantiodifferentiating Polar Addition: Anti-Markovnikov Methanol Addition to 1,1-Diphenylpropene. J. Chem. Soc., Chem. Commun. 1993, 718–720. 10.1039/c39930000718. [DOI] [Google Scholar]
- Asaoka S.; Wada T.; Inoue Y. Microenvironmental Polarity Control of Electron-Transfer Photochirogenesis. Enantiodifferentiating Polar Addition of 1,1-Diphenyl-1-alkenes Photosensitized by Saccharide Naphthalenecarboxylates. J. Am. Chem. Soc. 2003, 125, 3008–3027. 10.1021/ja028680o. [DOI] [PubMed] [Google Scholar]
- Gassman P. G.; Bottorff K. J. Anti-Markovnikov Addition of Nucleophiles to a Non-Conjugated Olefin via Single Electron Transfer Photochemistry. Tetrahedron Lett. 1987, 28, 5449–5452. 10.1016/S0040-4039(00)96751-0. [DOI] [Google Scholar]
- Fukuzumi S.; Kotani H.; Ohkubo K.; Ogo S.; Tkachenko N. V.; Lemmetyinen H. Electron-Transfer State of 9-Mesityl-10-methylacridinium Ion with a Much Longer Lifetime and Higher Energy Than That of the Natural Photosynthetic Reaction Center. J. Am. Chem. Soc. 2004, 126, 1600–1601. 10.1021/ja038656q. [DOI] [PubMed] [Google Scholar]
- Margrey K. A.; Nicewicz D. A. A General Approach to Catalytic Alkene Anti-Markovnikov Hydrofunctionalization Reactions via Acridinium Photoredox Catalysis. Acc. Chem. Res. 2016, 49, 1997–2006. 10.1021/acs.accounts.6b00304. [DOI] [PubMed] [Google Scholar]
- Hamilton D. S.; Nicewicz D. A. Direct Catalytic Anti-Markovnikov Hydroetherification of Alkenols. J. Am. Chem. Soc. 2012, 134, 18577–18580. 10.1021/ja309635w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.; Li H.; Li S.; Zhang M.-T.; Luo S. A Chiral Ion-Pair Photoredox Organocatalyst: Enantioselective Anti-Markovnikov Hydroetherification of Alkenols. Org. Chem. Front. 2017, 4, 1037–1041. 10.1039/C6QO00806B. [DOI] [Google Scholar]
- Romero N. A.; Nicewicz D. A. Mechanistic Insight into the Photoredox Catalysis of Anti-Markovnikov Alkene Hydrofunctionalization Reactions. J. Am. Chem. Soc. 2014, 136, 17024–17035. 10.1021/ja506228u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui E.; Metrano A. J.; Tsuchiya Y.; Knowles R. R. Catalytic Hydroetherification of Unactivated Alkenes Enabled by Proton-Coupled Electron Transfer. Angew. Chem., Int. Ed. 2020, 59, 11845–11849. 10.1002/anie.202003959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurence C; Gal J.-F.. Thermodynamic and Spectroscopic Scales of Hydrogen-Bond Basicity and Affinity. In Lewis Basicity and Affinity Scales; John Wiley & Sons: Hoboken NJ, 2009; pp 111–227. [Google Scholar]
- Isomura M.; Petrone D. A.; Carreira E. M. Coordination-Induced Stereocontrol over Carbocations: Asymmetric Reductive Deoxygenation of Racemic Tertiary Alcohols. J. Am. Chem. Soc. 2019, 141, 4738–4748. 10.1021/jacs.9b00862. [DOI] [PubMed] [Google Scholar]
- Naredla R. R.; Klumpp D. A. Contemporary Carbocation Chemistry: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 6905–6948. 10.1021/cr4001385. [DOI] [PubMed] [Google Scholar]
- Akiyama T.; Itoh J.; Yokota K.; Fuchibe K. Enantioselective Mannich-Type Reaction Catalyzed by a Chiral Brønsted Acid. Angew. Chem., Int. Ed. 2004, 43, 1566–1568. 10.1002/anie.200353240. [DOI] [PubMed] [Google Scholar]
- Uraguchi D.; Terada M. Chiral Brønsted Acid-Catalyzed Direct Mannich Reactions via Electrophilic Activation. J. Am. Chem. Soc. 2004, 126, 5356–5357. 10.1021/ja0491533. [DOI] [PubMed] [Google Scholar]
- Hatano M.; Moriyama K.; Maki T.; Ishihara K. Which Is the Actual Catalyst: Chiral Phosphoric Acid or Chiral Calcium Phosphate?. Angew. Chem., Int. Ed. 2010, 49, 3823–3826. 10.1002/anie.201000824. [DOI] [PubMed] [Google Scholar]
- Akiyama T. Stronger Brønsted Acids. Chem. Rev. 2007, 107, 5744–5758. 10.1021/cr068374j. [DOI] [PubMed] [Google Scholar]
- Terada M. Chiral Phosphoric Acids as Versatile Catalysts for Enantioselective Transformations. Synthesis 2010, 2010, 1929–1982. 10.1055/s-0029-1218801. [DOI] [Google Scholar]
- Zamfir A.; Schenker S.; Freund M.; Tsogoeva S. B. Chiral BINOL-Derived Phosphoric Acids: Privileged Brønsted Acid Organocatalysts for C–C Bond Formation Reactions. Org. Biomol. Chem. 2010, 8, 5262–5276. 10.1039/c0ob00209g. [DOI] [PubMed] [Google Scholar]
- Kaupmees K.; Tolstoluzhsky N.; Raja S.; Rueping M.; Leito I. On the Acidity and Reactivity of Highly Effective Chiral Brønsted Acid Catalysts: Establishment of an Acidity Scale. Angew. Chem., Int. Ed. 2013, 52, 11569–11572. 10.1002/anie.201303605. [DOI] [PubMed] [Google Scholar]
- Lee J.-W.; List B. Deracemization of α-Aryl Hydrocoumarins via Catalytic Asymmetric Protonation of Ketene Dithioacetals. J. Am. Chem. Soc. 2012, 134, 18245–18248. 10.1021/ja3096202. [DOI] [PubMed] [Google Scholar]
- Schreyer L.; Properzi R.; List B. IDPi Catalysis. Angew. Chem., Int. Ed. 2019, 58, 12761–12777. 10.1002/anie.201900932. [DOI] [PubMed] [Google Scholar]
- Sun Z.; Winschel G. A.; Borovika A.; Nagorny P. Chiral Phosphoric Acid-Catalyzed Enantioselective and Diastereoselective Spiroketalizations. J. Am. Chem. Soc. 2012, 134, 8074–8077. 10.1021/ja302704m. [DOI] [PubMed] [Google Scholar]
- Khomutnyk Y. Y.; Argüelles A. J.; Winschel G. A.; Sun Z.; Zimmerman P. M.; Nagorny P. Studies of the Mechanism and Origins of Enantioselectivity for the Chiral Phosphoric Acid-Catalyzed Stereoselective Spiroketalization Reactions. J. Am. Chem. Soc. 2016, 138, 444–456. 10.1021/jacs.5b12528. [DOI] [PubMed] [Google Scholar]
- Kaib P. S. J.; Schreyer L.; Lee S.; Properzi R.; List B. Extremely Active Organocatalysts Enable a Highly Enantioselective Addition of Allyltrimethylsilane to Aldehydes. Angew. Chem., Int. Ed. 2016, 55, 13200–13203. 10.1002/anie.201607828. [DOI] [PubMed] [Google Scholar]
- Yagupolskii L. M.; Petrik V. N.; Kondratenko N. V.; Sooväli L.; Kaljurand I.; Leito I.; Koppel I. A. The Immense Acidifying Effect of The Supersubstituent -NSO2CF3 on the Acidity of Amides and Amidines of Benzoic Acids in Acetonitrile. J. Chem. Soc., Perkin trans. 2002, 2, 1950–1955. 10.1039/B204172C. [DOI] [Google Scholar]
- Ishihara K.; Kaneeda M.; Yamamoto H. Lewis Acid Assisted Chiral Bronsted Acid for Enantioselective Protonation of Silyl Enol Ethers and Ketene Bis(trialkylsilyl) Acetals. J. Am. Chem. Soc. 1994, 116, 11179–11180. 10.1021/ja00103a052. [DOI] [Google Scholar]
- Ishibashi H.; Ishihara K.; Yamamoto H. Chiral proton donor reagents: tin tetrachloride--coordinated optically active binaphthol derivatives. Chem. Rec. 2002, 2, 177–188. 10.1002/tcr.10020. [DOI] [PubMed] [Google Scholar]
- Yamamoto H.; Futatsugi K. ″Designer Acids″: Combined Acid Catalysis for Asymmetric Synthesis. Angew. Chem., Int. Ed. 2005, 44, 1924–1942. 10.1002/anie.200460394. [DOI] [PubMed] [Google Scholar]
- Ishihara K.; Nakamura S.; Kaneeda M.; Yamamoto H. First Example of a Highly Enantioselective Catalytic Protonation of Silyl Enol Ethers Using a Novel Lewis Acid-Assisted Brønsted Acid System. J. Am. Chem. Soc. 1996, 118, 12854–12855. 10.1021/ja962414r. [DOI] [Google Scholar]
- Ishihara K.; Nakamura S.; Yamamoto H. The First Enantioselective Biomimetic Cyclization of Polyprenoids. J. Am. Chem. Soc. 1999, 121, 4906–4907. 10.1021/ja984064+. [DOI] [Google Scholar]
- Nakamura S.; Ishihara K.; Yamamoto H. Enantioselective Biomimetic Cyclization of Isoprenoids Using Lewis Acid-Assisted Chiral Brønsted Acids: Abnormal Claisen Rearrangements and Successive Cyclizations. J. Am. Chem. Soc. 2000, 122, 8131–8140. 10.1021/ja001165a. [DOI] [Google Scholar]
- Uyanik M.; Ishibashi H.; Ishihara K.; Yamamoto H. Biomimetic Synthesis of Acid-Sensitive (−)-Caparrapi Oxide and (+)-8-Epicaparrapi Oxide Induced by Artificial Cyclases. Org. Lett. 2005, 7, 1601–1604. 10.1021/ol050295r. [DOI] [PubMed] [Google Scholar]
- Upar K. B.; Mishra S. J.; Nalawade S. P.; Singh S. A.; Khandare R. P.; Bhat S. V. Efficient Enantioselective Synthesis of (+)-Sclareolide and (+)-Tetrahydroactinidiolide: Chiral LBA-Induced Biomimetic Cyclization. Tetrahedron: Asymmetry 2009, 20, 1637–1640. 10.1016/j.tetasy.2009.06.020. [DOI] [Google Scholar]
- Schlüter J.; Blazejak M.; Boeck F.; Hintermann L. Asymmetric Hydroalkoxylation of Non-Activated Alkenes: Titanium-Catalyzed Cycloisomerization of Allylphenols at High Temperatures. Angew. Chem., Int. Ed. 2015, 54, 4014–4017. 10.1002/anie.201409252. [DOI] [PubMed] [Google Scholar]
- Helmbrecht S. L.; Schlüter J.; Blazejak M.; Hintermann L. Axially Chiral 1,1’-Binaphthyl-2-Carboxylic Acid (BINA-Cox) as Ligands for Titanium-Catalyzed Asymmetric Hydroalkoxylation. Eur. J. Org. Chem. 2020, 2020, 2062–2076. 10.1002/ejoc.201901895. [DOI] [Google Scholar]
- Schlüter J.; Blazejak M.; Hintermann L. Aluminum-Catalyzed Hydroalkoxylation at Elevated Temperatures: Fast and Simple Access to Coumarans and Other Oxygen Heterocycles. ChemCatChem 2013, 5, 3309–3315. 10.1002/cctc.201300182. [DOI] [Google Scholar]
- Xie W.-B.; Li Z. Bis(μ-oxo)–Dititanium(IV)–Chiral Binaphthyldisulfonate Complexes for Highly Enantioselective Intramolecular Hydroalkoxylation of Nonactivated Alkenes. ACS Catal. 2021, 11, 6270–6275. 10.1021/acscatal.1c01146. [DOI] [Google Scholar]
- Zhao P.; Cheng A.; Wang X.; Ma J.; Zhao G.; Li Y.; Zhang Y.; Zhao B. Asymmetric Intramolecular Hydroalkoxylation of Unactivated Alkenes Catalyzed by Chiral N-Triflyl Phosphoramide and TiCl4†. Chin. J. Chem. 2020, 38, 565–569. 10.1002/cjoc.201900544. [DOI] [Google Scholar]
- Sakakura A.; Sakuma M.; Ishihara K. Chiral Lewis Base-Assisted Brønsted Acid (LBBA)-Catalyzed Enantioselective Cyclization of 2-Geranylphenols. Org. Lett. 2011, 13, 3130–3133. 10.1021/ol201032t. [DOI] [PubMed] [Google Scholar]
- Sakakura A.; Ukai A.; Ishihara K. Enantioselective Halocyclization of Polyprenoids Induced by Nucleophilic Phosphoramidites. Nature 2007, 445, 900–903. 10.1038/nature05553. [DOI] [PubMed] [Google Scholar]
- Sakuma M.; Sakakura A.; Ishihara K. Kinetic Resolution of Racemic Carboxylic Acids through Asymmetric Protolactonization Promoted by Chiral Phosphonous Acid Diester. Org. Lett. 2013, 15, 2838–2841. 10.1021/ol401313d. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Li C.-J. Phosphine-Catalyzed Isomerization-Addition of Oxygen Nucleophiles to 2-Alkynoates. J. Am. Chem. Soc. 1994, 116, 10819–10820. 10.1021/ja00102a071. [DOI] [Google Scholar]
- Guo H.; Fan Y. C.; Sun Z.; Wu Y.; Kwon O. Phosphine Organocatalysis. Chem. Rev. 2018, 118, 10049–10293. 10.1021/acs.chemrev.8b00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung Y. K.; Fu G. C. Phosphine-Catalyzed Enantioselective Synthesis of Oxygen Heterocycles. Angew. Chem., Int. Ed. 2009, 48, 2225–2227. 10.1002/anie.200805377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler D. T.; Fu G. C. Catalytic Enantioselective Carbon–Oxygen Bond Formation: Phosphine-Catalyzed Synthesis of Benzylic Ethers via the Oxidation of Benzylic C–H Bonds. J. Am. Chem. Soc. 2016, 138, 12069–12072. 10.1021/jacs.6b08486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmerling F.; Hahn F. Biosynthesis of Oxygen and Nitrogen-Containing Heterocycles in Polyketides. Beilstein J. Org. Chem. 2016, 12, 1512–1550. 10.3762/bjoc.12.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker A.; Woggon W.-D.; Rüttimann A. Identification of The Tocopherol-Cyclase in the Blue-Green Algae Anabaena variabilis KÜTZING (Cyanobacteria). Helv. Chim. Acta 1993, 76, 1729–1738. 10.1002/hlca.19930760429. [DOI] [Google Scholar]
- Stocker A.; Derungs G.; Woggon W.-D.; Netscher T.; Rüttimann A.; Müller R. K.; Schneider H.; Todaro L. J. The Reaction Mechanism of Chromanol-Ring Formation Catalyzed by tocopherol cyclase from Anabaena variabilis KÜTZING (Cyanobacteria). Helv. Chim. Acta 1994, 77, 1721–1737. 10.1002/hlca.19940770705. [DOI] [Google Scholar]
- Stocker A.; Fretz H.; Frick H.; Rüttimann A.; Woggon W.-D. The Substrate Specificity of Tocopherol Cyclase. Bioorg. Med. Chem. 1996, 4, 1129–1134. 10.1016/0968-0896(96)00125-3. [DOI] [PubMed] [Google Scholar]
- Kaib P. S. J.; List B. Highly Acidic BINOL-Derived Phosphoramidimidates and their Application in the Brønsted Acid Catalyzed Synthesis of α-Tocopherol. Synlett 2015, 27, 156–158. 10.1055/s-0035-1560971. [DOI] [Google Scholar]
- Gao S.-S.; Garcia-Borràs M.; Barber J. S.; Hai Y.; Duan A.; Garg N. K.; Houk K. N.; Tang Y. Enzyme-Catalyzed Intramolecular Enantioselective Hydroalkoxylation. J. Am. Chem. Soc. 2017, 139, 3639–3642. 10.1021/jacs.7b01089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y.; Yu X.; Huang J.-W.; Liu W.; Li Q.; Hu Y.; Yang Y.; Chen Y.; Jin J.; Li H.; Chen C.-C.; Guo R.-T. Crystal Structure and Proposed Mechanism of an Enantioselective Hydroalkoxylation Enzyme from Penicillium herquei. Biochem. Biophys. Res. Commun. 2019, 516, 801–805. 10.1016/j.bbrc.2019.06.100. [DOI] [PubMed] [Google Scholar]
- Engleder M.; Pavkov-Keller T.; Emmerstorfer A.; Hromic A.; Schrempf S.; Steinkellner G.; Wriessnegger T.; Leitner E.; Strohmeier G. A.; Kaluzna I.; Mink D.; Schurmann M.; Wallner S.; Macheroux P.; Gruber K.; Pichler H. Structure-Based Mechanism of Oleate Hydratase from Elizabethkingia meningoseptica. ChemBioChem 2015, 16, 1730–1734. 10.1002/cbic.201500269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling B.; Wang X.; Su H.; Liu R.; Liu Y. Protonation State And Fine Structure of the Active Site Determine the Reactivity of Dehydratase: Hydration and Isomerization of β-Myrcene Catalyzed by Linalool Dehydratase/Isomerase From Castellaniella defragrans. Phys. Chem. Chem. Phys. 2018, 20, 17342–17352. 10.1039/C8CP02362J. [DOI] [PubMed] [Google Scholar]
- Nestl B. M.; Geinitz C.; Popa S.; Rizek S.; Haselbeck R. J.; Stephen R.; Noble M. A.; Fischer M.-P.; Ralph E. C.; Hau H. T.; Man H.; Omar M.; Turkenburg J. P.; van Dien S.; Culler S. J.; Grogan G.; Hauer B. Structural and Functional Insights Into Asymmetric Enzymatic Dehydration of Alkenols. Nat. Chem. Biol. 2017, 13, 275–281. 10.1038/nchembio.2271. [DOI] [PubMed] [Google Scholar]
- Wuensch C.; Gross J.; Steinkellner G.; Gruber K.; Glueck S. M.; Faber K. Asymmetric Enzymatic Hydration of Hydroxystyrene Derivatives. Angew. Chem., Int. Ed. 2013, 52, 2293–2297. 10.1002/anie.201207916. [DOI] [PubMed] [Google Scholar]
- Payer S. E.; Sheng X.; Pollak H.; Wuensch C.; Steinkellner G.; Himo F.; Glueck S. M.; Faber K. Exploring the Catalytic Promiscuity of Phenolic Acid Decarboxylases: Asymmetric, 1,6-Conjugate Addition of Nucleophiles Across 4-Hydroxystyrene. Adv. Synth. Catal. 2017, 359, 2066–2075. 10.1002/adsc.201700247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demming R. M.; Hammer S. C.; Nestl B. M.; Gergel S.; Fademrecht S.; Pleiss J.; Hauer B. Asymmetric Enzymatic Hydration of Unactivated, Aliphatic Alkenes. Angew. Chem., Int. Ed. 2019, 58, 173–177. 10.1002/anie.201810005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitschke B.; Turberg M.; List B. Confinement as a Unifying Element in Selective Catalysis. Chem. 2020, 6, 2515–2532. 10.1016/j.chempr.2020.09.007. [DOI] [Google Scholar]
- Handa S.; Lippincott D. J.; Aue D. H.; Lipshutz B. H. Asymmetric Gold-Catalyzed Lactonizations in Water at Room Temperature. Angew. Chem., Int. Ed. 2014, 53, 10658–10662. 10.1002/anie.201404729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhdanko A.; Maier M. E. The Mechanism of Gold(I)-Catalyzed Hydroalkoxylation of Alkynes: An Extensive Experimental Study. Chem. - Eur. J. 2014, 20, 1918–1930. 10.1002/chem.201303795. [DOI] [PubMed] [Google Scholar]
- Shu X.-Z.; Nguyen S. C.; He Y.; Oba F.; Zhang Q.; Canlas C.; Somorjai G. A.; Alivisatos A. P.; Toste F. D. Silica-Supported Cationic Gold(I) Complexes as Heterogeneous Catalysts for Regio- and Enantioselective Lactonization Reactions. J. Am. Chem. Soc. 2015, 137, 7083–7086. 10.1021/jacs.5b04294. [DOI] [PubMed] [Google Scholar]
- Ye R.; Zhukhovitskiy A. V.; Kazantsev R. V.; Fakra S. C.; Wickemeyer B. B.; Toste F. D.; Somorjai G. A. Supported Au Nanoparticles with N-Heterocyclic Carbene Ligands as Active and Stable Heterogeneous Catalysts for Lactonization. J. Am. Chem. Soc. 2018, 140, 4144–4149. 10.1021/jacs.8b01017. [DOI] [PubMed] [Google Scholar]
- Wang Z. J.; Clary K. N.; Bergman R. G.; Raymond K. N.; Toste F. D. A Supramolecular Approach to Combining Enzymatic and Transition Metal Catalysis. Nat. Chem. 2013, 5, 100–103. 10.1038/nchem.1531. [DOI] [PubMed] [Google Scholar]
- Pirenne V.; Muriel B.; Waser J. Catalytic Enantioselective Ring-Opening Reactions of Cyclopropanes. Chem. Rev. 2021, 121, 227–263. 10.1021/acs.chemrev.0c00109. [DOI] [PubMed] [Google Scholar]
- Proctor R. S. J.; Colgan A. C.; Phipps R. J. Exploiting Attractive Non-Covalent Interactions for the Enantioselective Catalysis of Reactions Involving Radical Intermediates. Nat. Chem. 2020, 12, 990–1004. 10.1038/s41557-020-00561-6. [DOI] [PubMed] [Google Scholar]
- Roos C. B.; Demaerel J.; Graff D. E.; Knowles R. R. Enantioselective Hydroamination of Alkenes with Sulfonamides Enabled by Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2020, 142, 5974–5979. 10.1021/jacs.0c01332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L.; Fu N.; Ernst B. G.; Lee W. H.; Frederick M. O.; DiStasio R. A.; Lin S. Dual Electrocatalysis Enables Enantioselective Hydrocyanation of Conjugated Alkenes. Nat. Chem. 2020, 12, 747–754. 10.1038/s41557-020-0469-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilla P.; Rey Y. P.; Holden C. M.; Melchiorre P. Photo-Organocatalytic Enantioselective Radical Cascade Reactions of Unactivated Olefins. Angew. Chem., Int. Ed. 2018, 57, 12819–12823. 10.1002/anie.201808183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry E. C.; Rono L. J.; Hale M. E.; Matsuura R.; Knowles R. R. Enantioselective Synthesis of Pyrroloindolines via Noncovalent Stabilization of Indole Radical Cations and Applications to the Synthesis of Alkaloid Natural Products. J. Am. Chem. Soc. 2018, 140, 3394–3402. 10.1021/jacs.7b13616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C.; Bandar J. S. Superbase-Catalyzed anti-Markovnikov Alcohol Addition Reactions to Aryl Alkenes. J. Am. Chem. Soc. 2018, 140, 3547–3550. 10.1021/jacs.8b00766. [DOI] [PubMed] [Google Scholar]
- Bandar J. S.; Pirnot M. T.; Buchwald S. L. Mechanistic Studies Lead to Dramatically Improved Reaction Conditions for the Cu-Catalyzed Asymmetric Hydroamination of Olefins. J. Am. Chem. Soc. 2015, 137, 14812–14818. 10.1021/jacs.5b10219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae H. Y.; Höfler D.; Kaib P. S. J.; Kasaplar P.; De C. K.; Döhring A.; Lee S.; Kaupmees K.; Leito I.; List B. Approaching Sub-ppm-Level Asymmetric Organocatalysis of a Highly Challenging and Scalable Carbon–Carbon Bond Forming Reaction. Nat. Chem. 2018, 10, 888–894. 10.1038/s41557-018-0065-0. [DOI] [PubMed] [Google Scholar]
- Yang C.; Xue X.-S.; Jin J.-L.; Li X.; Cheng J.-P. Theoretical Study on the Acidities of Chiral Phosphoric Acids in Dimethyl Sulfoxide: Hints for Organocatalysis. J. Org. Chem. 2013, 78, 7076–7085. 10.1021/jo400915f. [DOI] [PubMed] [Google Scholar]
- Yang C.; Xue X.-S.; Li X.; Cheng J.-P. Computational Study on the Acidic Constants of Chiral Brønsted Acids in Dimethyl Sulfoxide. J. Org. Chem. 2014, 79, 4340–4351. 10.1021/jo500158e. [DOI] [PubMed] [Google Scholar]
- Mazurenko S.; Prokop Z.; Damborsky J. Machine Learning in Enzyme Engineering. ACS Catal. 2020, 10, 1210–1223. 10.1021/acscatal.9b04321. [DOI] [Google Scholar]
- Musil M.; Konegger H.; Hon J.; Bednar D.; Damborsky J. Computational Design of Stable and Soluble Biocatalysts. ACS Catal. 2019, 9, 1033–1054. 10.1021/acscatal.8b03613. [DOI] [Google Scholar]
- Wu Z.; Kan S. B. J.; Lewis R. D.; Wittmann B. J.; Arnold F. H. Machine Learning-Assisted Directed Protein Evolution with Combinatorial Libraries. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 8852–8858. 10.1073/pnas.1901979116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catti L.; Tiefenbacher K. Intramolecular Hydroalkoxylation Catalyzed Inside a Self-Assembled Cavity of an Enzyme-Like Host Structure. Chem. Commun. 2015, 51, 892–894. 10.1039/C4CC08211G. [DOI] [PubMed] [Google Scholar]
- Jorner K.; Tomberg A.; Bauer C.; Sköld C.; Norrby P.-O. Organic reactivity from mechanism to machine learning. Nat. Chem. Rev. 2021, 5, 240–255. 10.1038/s41570-021-00260-x. [DOI] [PubMed] [Google Scholar]
- Santiago C. B.; Guo J.-Y.; Sigman M. S. Predictive and Mechanistic Multivariate Linear Regression Models for Reaction Development. Chem. Sci. 2018, 9, 2398–2412. 10.1039/C7SC04679K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahrt A. F.; Athavale S. V.; Denmark S. E. Quantitative Structure–Selectivity Relationships in Enantioselective Catalysis: Past, Present, and Future. Chem. Rev. 2020, 120, 1620–1689. 10.1021/acs.chemrev.9b00425. [DOI] [PMC free article] [PubMed] [Google Scholar]