

Summary
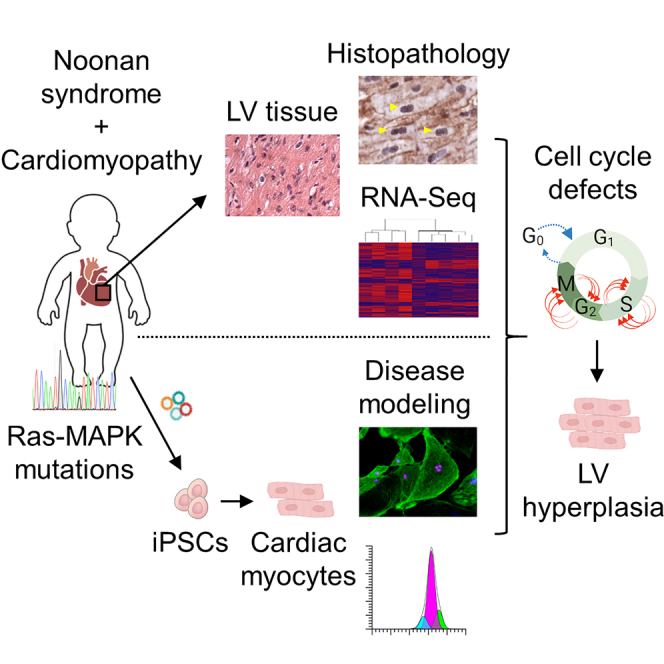
Childhood-onset myocardial hypertrophy and cardiomyopathic changes are associated with significant morbidity and mortality in early life, particularly in patients with Noonan syndrome, a multisystemic genetic disorder caused by autosomal dominant mutations in genes of the Ras-MAPK pathway. Although the cardiomyopathy associated with Noonan syndrome (NS-CM) shares certain cardiac features with the hypertrophic cardiomyopathy caused by mutations in sarcomeric proteins (HCM), such as pathological myocardial remodeling, ventricular dysfunction, and increased risk for malignant arrhythmias, the clinical course of NS-CM significantly differs from HCM. This suggests a distinct pathophysiology that remains to be elucidated. Here, through analysis of sarcomeric myosin conformational states, histopathology, and gene expression in left ventricular myocardial tissue from NS-CM, HCM, and normal hearts complemented with disease modeling in cardiomyocytes differentiated from patient-derived PTPN11N308S/+ induced pluripotent stem cells, we demonstrate distinct disease phenotypes between NS-CM and HCM and uncover cell cycle defects as a potential driver of NS-CM.
Subject areas: Cell biology, Stem cells research, Transcriptomics
Graphical abstract
Highlights
-
•
Cardiomyopathy associated with Noonan syndrome (NS-CM) differs from sarcomeric HCM
-
•
Cell cycle pathways are dysregulated in left ventricular tissue from NS-CM patients
-
•
Cell cycle defects drive cardiomyocyte multinucleation and hyperplasia in NS-CM
Cell biology; Stem cells research; Transcriptomics
Introduction
Left ventricular (LV) hypertrophy and severe cardiomyopathic changes occur in approximately 20% of patients with Noonan syndrome (Colquitt and Noonan, 2014; Shaw et al., 2007), a genetic disorder with a prevalence of 1 in 1,000 to 2,500 live births presenting with varying degrees of facial dysmorphism, short stature, congenital heart defects, hematologic disorders, and mental retardation (Romano et al., 2010; Tartaglia and Gelb, 2005). Noonan syndrome belongs to a spectrum of diseases named “RASopathies” because it is caused by autosomal-dominant mutations in genes of the Ras-mitogen-activated protein kinase (MAPK) signaling pathway, primarily PTPN11, SOS1, KRAS, NRAS, RIT1, and RAF1 (El Bouchikhi et al., 2016; Gelb and Tartaglia, 2006). By contrast, classical hypertrophic cardiomyopathy (HCM), recognized as unexplained LV hypertrophy that is generally found in adults and less frequently in young children (Norrish et al., 2019), is caused by mutations in genes encoding sarcomeric proteins, such as MYH7, MYBPC3, ACTC1, TNNT2, TNNI3, TPM1, MYL2, and MYL3 (reviewed in Seidman and Seidman, 2011). Although the cardiac features of childhood-onset cardiomyopathy associated with Noonan syndrome (NS-CM) and HCM are similar, we and others have reported a significantly younger age at diagnosis and a shorter time of survival without hospitalization for heart failure or cardiac intervention in NS-CM (Calcagni et al., 2018; Kaltenecker et al., 2019; Wilkinson et al., 2012). The fact that myocardial hypertrophy and dysfunction do not progress over time in a subset of NS-CM patients (Kaltenecker et al., 2019; Prendiville et al., 2014) while they typically progress in HCM patients, particularly with early onset (Ho et al., 2018), also hints to distinct disease mechanisms. To investigate the molecular processes accounting for these clinical findings, we performed histopathological analyses of LV myocardial tissue from NS-CM, HCM, and normal hearts, identifying distinguishing features between the two diseases. Whole-transcriptome analysis of NS-CM myocardium and disease modeling in cardiomyocytes derived from induced pluripotent stem cells (iPSCs) of a NS-CM patient carrying a PTPN11N308S/+ mutation further illustrated the phenotypic differences between NS-CM and HCM and revealed cell cycle defects leading to increased cardiomyocyte proliferation as a potential cause for LV hypertrophy in NS-CM.
Results
NS-CM and HCM cause opposite changes in sarcomeric myosin conformations in LV myocardial tissue
We collected LV myocardial tissue during surgical intervention from infants and children diagnosed with Noonan syndrome and cardiomyopathy. These individuals had massive myocardial hypertrophy leading to severe LV outflow tract obstruction (Figures 1A and S1–S8; Tables S1 and S2). For phenotypic comparisons, we collected LV myectomy samples from HCM patients (Table S2). Patients without overt cardiomyopathy or known genetic variants who underwent cardiac surgery for various reasons served as ‘normal’ controls (Table S2).
Figure 1.

Histopathological features of LV myocardial tissue differ between patients with HCM and NS-CM
(A) Exemplary transthoracic echocardiographic imaging of NS-CM patient 5 at the time of presentation for surgical intervention. Marked increase of the end-diastolic interventricular septal diameter (∗), of the end-diastolic left ventricular (LV) posterior wall (#), and reduced end-diastolic LV diameter (§) compared to an unaffected age- and sex-matched infant (CTRL) on parasternal long axis view. See also Figures S1–S8.
(B) Percentage of myosin heads in SRX and DRX conformations in human LV myocardial tissue from control (CTRL, n = 10 replicates from N = 2 patients), HCM (mutated MYH7, n = 9 replicates from N = 2 patients) or NS-CM patients (mutated RAF1, n = 12 replicates from N = 3 patients; mutated PTPN11, n = 12 replicates from N = 2 patients). Box-and-whiskers plot indicating the minimum, 25th percentile, median, 75th percentile, maximum, and all data points; p-values from one-way ANOVA with Tukey's multiple comparisons test.
(C–E) Representative images of LV myocardial tissue from age- and sex-matched control, NS-CM, and HCM patients stained with hematoxylin-eosin. Yellow arrowheads show examples of multinucleated cardiomyocytes (CMs); scale bars = 25 μm (left) and 100 μm (right) (C).
Corresponding percentage of multinucleated CMs (D) and CM density (E). Box-and-whiskers plots indicating the minimum, 25th percentile, median, 75th percentile, maximum, and all data points; p values from one-way ANOVA with Tukey's multiple comparisons test. (CTRL N = 6 patients; NS-CM N = 5 patients; HCM N = 6 patients). See also Figure S9.
The pathophysiology of HCM has been well studied using human cardiac tissue, animal models, and cultured cells (Hooijman et al., 2011; Prendiville et al., 2014; Seidman and Seidman, 2011). In particular, pathogenic HCM-causing MYH7 variants and MYBPC3 truncating variants have been shown to destabilize the myosin interacting heads motif in LV myocardium, leading to an increase in the proportion of myosins in a disordered relaxed state (DRX) reducing the number of myosins occupying the super relaxed state (SRX) when compared to healthy tissue (Toepfer et al., 2019, 2020). Increased DRX myosin populations are associated with hypercontractility, slowed relaxation, and energetic imbalance in HCM cells. Clinical and echocardiographic features of hypercontractility and diastolic dysfunction also occur in NS-CM (Burch et al., 1992; Hudsmith et al., 2006; Kaltenecker et al., 2019), but underlying cellular mechanisms have not yet been described in human NS-CM tissue. Therefore, we examined the proportion of myosins in the DRX/SRX state in LV tissue obtained from NS-CM, thick filament HCM, and unaffected (control) patients using a fluorescent ATP analogue, Mant-ATP, chased with nonfluorescent ATP (see STAR Methods) (Hooijman et al., 2011; Toepfer et al., 2019). The tissue from HCM patients, which carried two different MYH7 variants, had a higher proportion of DRX myosin when compared to control tissue (% DRX 48.2 ± 3.2 in HCM vs 37.9 ± 2.6 in control; p = 0.0013) (Figure 1B). NS-CM samples had the opposite phenotype with a shift towards the SRX state, indicating that NS-CM does not operate through destabilization of the myosin interacting head motifs, and therefore has a distinct pathomechanism from thick filament HCM (% SRX 76.4 ± 6.7 in NS-CM vs 62.1 ± 2.6 in control; p <0.0001) (Figure 1B). This was consistent in NS-CM patients carrying both PTPN11 and RAF1 variants.
Histopathological analysis of left ventricular myocardial tissue points to cell cycle defects in NS-CM
We then compared the histology of NS-CM and HCM tissues. As described previously, NS-CM and HCM tissues share common histopathological hallmarks such as fibrosis and myocardial disarray (Burch et al., 1992; Hudsmith et al., 2006; Kaltenecker et al., 2019). Blinded evaluation of hematoxylin-eosin and dystrophin-stained sections additionally revealed a higher fraction of multinucleated cardiomyocytes in NS-CM tissue compared to age- and sex-matched HCM tissue and ‘control’ tissue from transplanted heart biopsies (multinucleated CMs 14.6 ± 6.2% in NS-CM vs. 4.2 ± 0.7% in HCM, p = 0.0005; 3.9 ± 0.8 in control, p = 0.0004) (Figures 1C, 1D, and S9). This previously undescribed feature suggests that cell cycle regulation may be perturbed in NS-CM. Moreover, we found cardiomyocyte density to be significantly lower in HCM LV tissue than in control tissue, reflecting the cellular hypertrophy characteristic of HCM (CMs/mm2 913.4 ± 144.1 in HCM vs 1,604.5 ± 315.4 in control, p = 0.017) (Figures 1C and 1E). By contrast, the mean cardiomyocyte density in NS-CM tissue was significantly higher than in HCM tissue (CMs/mm2 1,914.5 ± 588.8 in NS-CM vs 913.4 ± 144.1 in HCM, p = 0.0017), suggesting a mechanism of hyperplasia rather than cellular hypertrophy in NS-CM (Figures 1C and 1E).
Whole-transcriptome analysis of left ventricular myocardial tissue by RNA-Seq reveals dysregulation of cell cycle pathways in NS-CM
To better define the underlying molecular processes, we assessed RNA expression in NS-CM LV myocardial tissues using bulk RNA sequencing (RNA-Seq). As age-matched and sex-matched controls, we used LV myocardial tissue samples removed during ventricular septal defect repair surgery in otherwise healthy pediatric patients (Table S2); principal component analysis showed a clear distinction between the groups (Figure S10). We first assessed the top differentially expressed genes in NS-CM, using a log2 fold change ≥1.5 as cutoff (= fold change of 2.8), of which 101 were upregulated and 308 were downregulated (Figure 2A, Table S3). In contrast to what has been described in HCM myocardial tissue and cardiomyocytes differentiated from HCM patient-derived iPSCs, there was no upregulation of genes encoding sarcomeric or mitochondrial proteins (Cohn et al., 2019; Gao et al., 2020). The highest upregulated gene in NS-CM was EREG, which encodes the epidermal growth factor receptor (EGFR) ligand epiregulin (Figure 2B). Among the top 5 upregulated genes were also the transcription factor CUX2 and the growth factor FGF10 (Figure 2B) (Hubert et al., 2018). Top downregulated genes in NS-CM included FOSB, also known as G0/G1 switch regulatory protein 3, and SOST, encoding sclerostin, a negative regulator of the canonical Wnt pathway (Figure 2B). It can be noted that 16 out of the top 20 upregulated genes in NS-CM were not significantly upregulated in LV tissue of two pediatric HCM patients compared to control tissue (with the caveat of limited sample size and poor age-matching), an additional indicator of distinct mechanisms between the two diseases (Table S4).
Figure 2.

Whole-transcriptome analysis of left ventricular myocardial tissue by RNA-Seq reveals dysregulation of cell cycle in NS-CM
(A) Heatmap of genes differentially expressed in NS-CM LV samples (N = 5 patients) compared to control (CTRL; N = 4 patients) (log2 fold change ≥1.5, p <0.05).
(B) Top 5 upregulated (top) and downregulated genes (bottom) in NS-CM compared to CTRL. FC: fold change.
(C) Gene set enrichment analysis (GSEA) indicating GO terms related to biological processes dysregulated in NS-CM compared to control (p < 0.05).
(D) GSEA of gene sets related to cell cycle, with normalized enrichment score (NES) and p value are indicated. See also Figure S10.
We next performed gene set enrichment analysis (GSEA) to identify the functional processes affected in NS-CM. This revealed a significant enrichment of gene ontology terms related to cell cycle regulation (Figures 2C and 2D, leading edge genes in Table S5). Specifically, in NS-CM, there was increased expression of genes controlling DNA replication and cell cycle checkpoints. The dysregulation of genes controlling chromosome segregation, such as the cohesin complex subunits STAG2, SMC3, and SMC1A, pointed to specific defects in mitosis. GSEA also confirmed an overrepresentation of terms linked to cardiac contraction and the regulation of action potentials by ion channels, consistent with functional features of cardiomyopathy (Figure 2C and Table S5). Terms related to extracellular matrix and cell adhesion also emerged, as could be expected from fibrotic myocardial tissue described in NS-CM (Figure 2C, Tables S1 and S5) (Kaltenecker et al., 2019).
Cardiomyocytes derived from PTPN11N308S/+ induced pluripotent stem cells do not display hallmarks of HCM
As patient tissue collected at the time of surgical intervention ultimately represents a fixed and advanced state of the disease, we sought to model early disease development in cardiomyocytes derived from patient-specific induced pluripotent stem cells (iPSC-CMs). Although the more prevalent RAF1-associated NS-CM has been studied in mouse and iPSC-CM models, there is a lack of data on the molecular mechanisms of PTPN11-associated NS-CM (Jaffré et al., 2019; Passariello et al., 2016; Sakai et al., 2018; Wu et al., 2011). We therefore generated iPSCs from a male patient affected by severe neonatal onset cardiomyopathy with spontaneous stagnation of disease progression beyond infancy who harbored a heterozygous 923A > G mutation in PTPN11 leading to a gain-of-function serine substitution in position 308 of the SHP-2 protein (N308S) (referred to as patient 5 in Table S1 and Figure S5). This is a mutational hot spot in the PTP domain of SHP-2 that is affected in roughly 25% of cases of Noonan syndrome and related disorders (Tartaglia and Gelb, 2005). Two clones (#1 and #2) were confirmed to carry the 923A > G mutation in PTPN11 and were fully tested for alkaline phosphatase activity, expression of the pluripotency markers NANOG and TRA1-81, reactivation of endogenous pluripotency genes (OCT4, SOX2, NANOG, TDGF1, and REX1), loss of Sendai viral transgenes, and normal karyotype (Figures S11A–S11E and S11G). Multilineage differentiation potential was confirmed by detection of derivatives of all three germ layers in in vitro differentiated embryoid bodies (Figure S11F). For all experiments, the two PTPN11N308S/+ iPSC clones and two independent healthy control lines (#1: male and #2: female) were differentiated into cardiomyocytes through a Wnt modulation protocol consistently yielding a high percentage of cardiac troponin T (cTnT) positive cells after 15 days (control: 88.6 ± 4.6%; PTPN11N308S/+: 91.9 ± 4.4%) (Figures S12A–S12C).
In cardiomyocytes derived from PTPN11N308S/+ iPSCs, we evaluated phenotypical features that have been consistently reported in iPSC-CM models of HCM, as outlined in a systematic review of the literature (Eschenhagen and Carrier, 2019): increased cardiomyocyte size, sarcomere disorganization, higher NPPA/NPPB and MYH7/MYH6 expression levels, and arrhythmic beating behavior (Eschenhagen and Carrier, 2019). We first measured the cell surface area of cardiomyocytes at day 30 of differentiation via immunofluorescence staining of cTnT combined with the desmosomal protein plakophilin-2 (PKP2) to delimit cell borders. This was done 15 days after dissociation and reseeding of cells at either 15,000, 25,000, or 50,000 cells/cm2 to control for the effect of density on cell size. The lower the seeding density, the higher was the mean cardiomyocyte size reached in both diseased and control settings; however, no significant differences were detected between control and PTPN11N308S/+ cells (Figures 3A and 3B). We next used an immunofluorescence staining of sarcomeric α-actinin (ACTN2) to visualize myofibril organization and found control and PTPN11N308S/+ cardiomyocytes to have fully organized, partially organized, and disorganized sarcomeres in equivalent proportions (Figures 3C and 3D). We also did not observe significant differences in the expression levels of MYH7 relative to MYH6 and NPPA relative to NPPB (Figure 3E). TNNT2 and GATA4, two other genes typically upregulated in HCM, also showed equivalent expression levels in the two groups (Figure 3E). Finally, to evaluate the presence of functional features of HCM, we performed single-cell calcium transient imaging with the fluorescent calcium indicator Fluo-4 on day 30 using cardiomyocytes submitted to field stimulation at different frequencies (0.4, 0.5, or 1 Hz). The transients recorded in PTPN11N308S/+ cells were comparable to control cells with regards to duration to 90% peak decay at any of the tested stimulation frequencies, but showed a significantly shorter time to 50% peak decay at 0.4 and 0.5 Hz as well as reduced amplitude at 0.4 Hz (Figures 3F and 3G). Of note, we did not observe the occurrence of arrhythmic events in PTPN11N308S/+ iPSC-CMs (data not shown).
Figure 3.

PTPN11N308S/+ iPSCs do not display hallmarks of sarcomeric hypertrophic cardiomyopathy
(A and B) Representative images of control and PTPN11N308S/+ iPSC-derived cardiomyocytes (iPSC-CMs) seeded at 15,000, 25,000, or 50,000 cells/cm2 15 days before immunofluorescence staining of cardiac troponin T (cTnT) and plakophilin-2 (PKP2) at day 30 of differentiation (A) and corresponding measurements of mean cell surface area of cTnT+ cells indicated as mean ± SEM (B), p-values from a two-way ANOVA with Tukey's multiple comparisons test. Control #1: N = 2 differentiations, n = 715 cells; #2: N = 2 differentiations, n = 1,039 cells. PTPN11N308S/+ #1: N = 2 differentiations, n = 602 cells; #2: N = 2 differentiations, n = 1,303 cells. Scale bar in (A) = 100 μm.
(C and D) Representative images of control and PTPN11N308S/+ iPSC-CMs stained for sarcomeric α-actinin (ACTN2) at day 30 (C) and corresponding percentage of cardiomyocytes displaying fully organized, partially organized, or disorganized myofibrils indicated as mean ± SD (D). Control #1: N = 4 differentiations, n = 879 cells; #2: N = 3 differentiations, n = 929 cells. PTPN11N308S/+ #1: N = 2 differentiations, n = 543 cells; #2: N = 3 differentiations, n = 481 cells. Scale bar in (C) = 50 μm; in insets = 10 μm.
(E) qRT-PCR analysis of the ratio of expression of β-myosin heavy chain (MYH7) to α-myosin heavy chain (MYH6) and natriuretic peptide A (NPPA) to natriuretic peptide B (NPPB) and level of expression of cardiac troponin T (TNNT2) and GATA-binding protein 4 (GATA4) in day 30 control and PTPN11N308S/+ iPSC-CMs, indicated as mean ± SEM, both groups #1: N = 2 differentiations, #2: N = 2 differentiations.
(F and G) Representative single-cell calcium traces based on Fluo-4 fluorescence imaging on day 30 control and PTPN11N308S/+ iPSC-CMs under 0.5, 1, and 1.5 Hz pacing conditions (F). Calcium transient duration to 50% decay (TD50, left), duration to 90% decay (TD90, middle) and transient amplitude (right) measured by Fluo-4 fluorescence imaging in day 30 control and PTPN11N308S/+ iPSC-CMs under 0.5, 1 or 1.5 Hz field stimulation; dot plots showing all points, the mean and 95% CI (G). Control #1: N = 2 differentiations, n = 135 Ca2+ transients/pacing frequency in 34 cells; #2: N = 1 differentiation, n = 94 Ca2+ transients/pacing frequency in 23 cells (pooled). PTPN11N308S/+ #1: N = 2 differentiations, n = 117 Ca2+ transients/pacing frequency in 30 cells; #2: N = 1 differentiation, n = 99 transients/pacing frequency in 25 cells (pooled). p-values from a two-way ANOVA with Sidak's multiple comparisons test. See also Figures S11 and S12.
Hyperactive cell cycling leads to aberrant multinucleation and increased proliferation in PTPN11N308S/+ iPSC-CMs
Having observed multinucleation in NS-CM patient tissue, we used an immunofluorescence staining of cTnT in combination with PKP2 and Hoechst to quantify the percentage of mononucleated, binucleated, and multinucleated iPSC-CMs, the latter being defined as cardiomyocytes containing three or more nuclei. On day 30, a significantly higher percentage of both binucleation and multinucleation was detected in PTPN11N308S/+ cells compared to controls (binucleated CMs: 10.9 ± 2.8% vs. 6.6 ± 4.3%, p = 0.0359; multinucleated CMs: 3.6 ± 1.1% vs. 0.5 ± 0.5%, p <0.0001) (Figures 4A and 4B). We further evaluated cell cycle activity in PTPN11N308S/+ and control cells to test the hypothesis that cardiomyocyte hyperplasia could be driving LV hypertrophy in NS-CM. A first immunocytochemical evaluation of the cell cycle activity marker Ki67, which is expressed during all phases of an active cell cycle (Scholzen and Gerdes, 2000), showed a significantly higher proportion of Ki67+ PTPN11N308S/+ cardiomyocytes compared to controls (18.9 ± 0.6% vs. 11.7 ± 2.5%, p = 0.0002) (Figures 4C and 4D), with 2.8 ± 0.13% of PTPN11N308S/+ CMs containing two or more Ki67+ nuclei (data not shown). To better analyze cell cycle progression, we then dissociated and purified day 30 iPSC-CMs, extracted their nuclei, and performed propidium iodide staining to measure DNA content by flow cytometry (Figures S12D and S12E). This revealed a significantly lower percentage of nuclei in G0/G1 phase and a higher percentage in S and G2/M phases in PTPN11N308S/+ cells compared to control cells, confirming an increase in cell cycle activity (Figure 4E). We also noted a rare occurrence of polyploid PTPN11N308S/+ nuclei with a DNA content superior to 4n, virtually absent in the control population (0.6 ± 0.3% vs. 0.1 ± 0.1%, p = 0.013), although the presence of confounding doublets cannot be entirely excluded (Figure 4E). Lastly, we assessed the proportion of cardiomyocytes completing full division cycles including cytokinesis by loading day 15 cardiomyocytes with the cell tracing dye CFSE and analyzing the percentage of cTnT+ cells in successive daughter generations 96 h later by flow cytometry (Figure 4F). This analysis revealed that during the same period of time, PTPN11N308S/+ iPSC-CMs had predominantly completed three cycles of division while the majority of control iPSC-CMs had completed two, resulting in a significantly higher proliferation index in PTPN11N308S/+ iPSC-CMs than in the control (6.1 ± 0.7% vs. 4.7 ± 0.9%, p = 0.018) (Figures 4G and 4H).
Figure 4.

Hyperactive cell cycling leads to aberrant multinucleation and increased proliferation in PTPN11N308S/+ iPSC-CMs
(A and B) Representative images of control and PTPN11N308S/+ iPSC-CMs stained for plakophilin-2 (PKP2) and cardiac troponin T (cTnT) at day 30 of differentiation (A) and corresponding percentage of binucleated cardiomyocytes (left) and multinucleated cardiomyocytes containing 3 or more nuclei (right) (B). Box-and-whiskers plot indicating the minimum, 25th percentile, median, 75th percentile, maximum, and all data points; p-value from an unpaired two-tailed t test; Control #1: N = 4 differentiations, n = 681 cells, #2: N = 4 differentiations, n = 1,170 cells. PTPN11N308S/+ #1: N = 4 differentiations, n = 858 cells, #2: N = 4 differentiations, n = 947 cells. Scale bar in (A) = 100 μm; full arrowheads show examples of binucleated CMs, empty arrowheads show examples of CMs containing 3 nuclei.
(C and D) Representative images of control and PTPN11N308S/+ iPSC-CMs stained for cTnT and Ki67 at day 30 of differentiation (C) and corresponding percentage of Ki67+ CMs (D), indicated as mean ± SEM, p-value from an unpaired two-tailed t-test. Control #1: N = 3 differentiations, n = 591 cells; #2: N = 3 differentiations, n = 793 cells. PTPN11N308S/+ #1: N = 3 differentiations, n = 706 cells; #2: N = 2 differentiations, n = 656 cells. Scale bar in (C) = 100 μm, arrowheads show Ki67+ nuclei.
(E) Percentage of nuclei isolated from day 30 purified control and PTPN11N308S/+ iPSC-CMs in G0/G1, G2/M, S phases or a polyploid state as measured by flow cytometry analysis of propidium iodide staining. Box-and-whiskers plots indicating the minimum, 25th percentile, median, 75th percentile, maximum, and all data points; p values from unpaired two-tailed t-tests. Control #1 and #2 both N = 3 differentiations. PTPN11N308S/+ #1: N = 3 differentiations; #2: N = 4 differentiations.
(F–H) Representative plots of proliferation analysis of control and PTPN11N308S/+ iPSC-CMs loaded with CFSE and seeded at day 15 (t0) and collected after 96 h (t96) (F). Cells were stained for cTnT before flow cytometry analysis and the percentage of cTnT+ CFSE+ cells in successive daughter generations (Gen1-4) at t96 was calculated relative to the parent generation (Gen0) at t0 using ModFit LT v. 5.0.9 (G). A proliferation index was calculated in ModFit LT, representing the average number of cells that one original cell gave rise to (H). Box-and-whiskers plots indicating the minimum, 25th percentile, median, 75th percentile, maximum, and all data points; p values from a two-way ANOVA with Sidak's multiple comparisons test (G) and an unpaired two-tailed t-test (H). Control #1: N = 4 differentiations; #2: N = 3 differentiations. PTPN11N308S/+ #1 and #2 both N = 3 differentiations. CFSE = carboxyfluorescein succinimidyl ester. See also Figures S11 and S12.
Discussion
In this study, we describe histopathological, transcriptomic, and functional features in LV tissue and patient-specific iPSC-CMs that point to distinct disease mechanisms between childhood-onset NS-CM and HCM. We additionally identify cell cycle defects leading to increased cardiomyocyte proliferation as a potential cause for LV hypertrophy in NS-CM.
Dysfunctional cardiac contraction triggers the development of heart failure in cardiomyopathy. This was apparent in our whole-transcriptome analysis of LV tissue from NS-CM patients, which showed important dysregulation of gene categories associated with cardiac conduction and contraction. However, it is not known if the same mechanisms are at play in NS-CM and HCM. To address this, we evaluated the conformational state of myosins in LV myocardial tissue of NS-CM and HCM patients compared to healthy controls as an indicator of contractile function. Thick filament mutations causing HCM have been shown to induce a shift towards myosins in DRX conformation in murine and human ventricular myocardium (McNamara et al., 2016, 2017; Schmid and Toepfer, 2021; Toepfer et al., 2019, 2020). By contrast, we found that destabilization of the myosin interacting head motifs does not seem to contribute to the disease pathomechanism of NS-CM, which argues against the use of the cardiac myosin inhibitor Mavacamten in this disease entity (Olivotto et al., 2020). Moreover, LV myocardial tissue of NS-CM patients did not show the upregulation of sarcomeric protein and mitochondrial genes typical of HCM (Cohn et al., 2019; Ren et al., 2016).
To further investigate the mechanisms underlying NS-CM, we generated iPSCs from a NS-CM patient carrying a gain-of-function PTPN11N308S/+ mutation. There have only been few other studies in iPSC-CM models of NS-CM so far, and none of NS-CM associated with a mutation in PTPN11 (Hanses et al., 2020; Jaffré et al., 2019; Sakai et al., 2018). The molecular mechanisms of HCM have however been extensively studied in iPSC-CMs, allowing the identification of hallmark phenotypical features that we evaluated in PTPN11N308S/+ iPSC-CMs (Eschenhagen and Carrier, 2019). We found neither increased size nor sarcomere disorganization in these cells compared to two independent control lines. PTPN11N308S/+ iPSC-CMs also did not have a transcriptional profile typical of HCM. Similar results were obtained by Hanses et al. in iPSC-CMs from a NS-CM patient carrying a biallelic mutation in LZTR1, which, although they had higher cell size, showed neither increased sarcomere disorganization nor upregulation of hypertrophic markers (Hanses et al., 2020). By contrast, Jaffré et al. reported a phenotype similar to HCM in iPSC-CMs from a NS-CM patient carrying a RAF1S275L/+ mutation, including increased cardiomyocyte size and sarcomere disorganization (Jaffré et al., 2019). However, in our study, myocardial tissue from patients carrying RAF1 mutations (including RAF1S275L/+) or PTPN11 mutations had equivalent histopathological features and did not segregate according to the affected gene in RNA-Seq analysis, suggesting common pathophysiology.
Analysis of the electrophysiological function of PTPN11N308S/+ iPSC-CMs by single-cell calcium transient imaging revealed a significantly shorter duration to 50% peak decay compared to control cells, which was also reported in LZTR1-mutated iPSC-CMs (Hanses et al., 2020). Although data on calcium abnormalities in iPSC-CM models of HCM are not consistent – either because of mutation-specific effects or differences in experimental conditions (Eschenhagen and Carrier, 2019), an opposite phenotype of increased decay duration has been described in iPSC-CMs of equivalent age carrying a MYH7R442G/+ mutation (Han et al., 2014). PTPN11N308S/+ iPSC-CMs also did not show arrhythmic events in calcium transients, one of the only unifying features of HCM iPSC-CMs reported in up to 20% of cardiomyocytes (Han et al., 2014; Lan et al., 2013; Mosqueira et al., 2018). Arrhythmic events were also not mentioned in any previous NS-CM iPSC-CM model (Hanses et al., 2020; Jaffré et al., 2019; Sakai et al., 2018).
The main cellular process we found dysregulated in both NS-CM patient tissue and PTPN11N308S/+ iPSC-CMs was cell cycle activity. LV myocardial tissue of NS-CM patients showed significant upregulation of cell cycle pathways, including genes controlling mitotic processes. A key finding was the highly elevated expression of FGF10. In addition to promoting the differentiation and proliferation of second heart field cells contributing to the right ventricle during development, it was shown in mice that the overexpression of Fgf10 triggered cell cycle re-entry in adult cardiomyocytes of both ventricles (Hubert et al., 2018; Rochais et al., 2014). It is therefore plausible that an aberrant increase of FGF10 expression is driving left ventricular cardiomyocyte proliferation in NS-CM. Similarly, the upregulation of EREG and downregulation of FOSB and SOST that we observed in NS-CM have previously been associated with increased cell proliferation in a number of physiological and cancerous processes (Cao et al., 2013; Kyotani et al., 2013; Morato et al., 2021; Nishioka et al., 2020; White et al., 2010; Wu et al., 2017; Xu et al., 2017; Zhang et al., 2020; Zou et al., 2017). As an upstream component of the Ras-MAPK pathway, the EGFR ligand EREG is a particularly interesting candidate for further studies of the mechanisms of NS-CM. The observed increase in CUX2 expression may also be related to Ras-MAPK hyperactivation, as CUT domain proteins have been shown to accelerate DNA repair and prevent senescence in cells with elevated reactive oxygen species (ROS) levels because of increased Ras signaling (Ramdzan et al., 2014, 2021). We also found high multinucleation in NS-CM LV cardiomyocytes, which was recapitulated in PTPN11N308S/+ iPSC-CMs. Although increased multinucleation has also been reported in some iPSC-CM models of HCM (Lan et al., 2013; Mosqueira et al., 2018), the presence of high multinucleation in HCM patient tissue is still debated and was not confirmed in our samples. In PTPN11N308S/+ iPSC-CMs, we found cell cycle hyperactivity to increase not only acytokinetic nuclear division but also cytokinetic cell division, suggesting that cardiomyocyte hyperplasia rather than hypertrophy could be the leading cause of left ventricular hypertrophy and cardiomyocyte multinucleation in NS-CM. This was supported by the higher cardiomyocyte density observed in NS-CM LV myocardial tissue compared to HCM. One can further speculate that the exit of cardiomyocytes from the cell cycle during the early postnatal period could explain why ventricular wall thickness does not progress further in a subset of affected children (Ahuja et al., 2007; Kaltenecker et al., 2019). This was the case in the patient carrying the PTPN11N308S/+ mutation from which we report iPSC-derived CMs results. Jaffré et al. did not observe increased proliferation in RAF1S275L/+ iPSC-CMs, though interestingly complete inactivation of RAF1 significantly decreased proliferation in these cells (Jaffré et al., 2019). Data on potential cell cycle defects in other NS-CM iPSC-CM models is lacking. However, cardiomyocyte hyperplasia was recently described in a neonatal patient who died from cardiomyopathy associated with Noonan syndrome with multiple lentigines (NSML, formerly LEOPARD syndrome) caused by a loss-of-function mutation in PTPN11 (Nakagama et al., 2018), providing additional evidence for cardiomyocyte cell cycle dysfunction in the wider spectrum of RASopathies.
Overall, our study supports the recognition of NS-CM and HCM as distinct disease entities, calling for differential treatment of syndromic cardiomyopathy patients. However, it is not yet clear to what extent our findings are common to other mutations causing NS-CM. Refining genotype-phenotype correlations will be needed to realize the potential of small-molecule therapies targeting specific components of the Ras-MAPK pathway, which have shown encouraging results in RASopathy mouse models (Marin et al., 2011; Schramm et al., 2012; Wang et al., 2017) and in a few case reports (Andelfinger et al., 2019; Hahn et al., 2015).
Limitations of the study
The limited availability of patient myocardial tissue samples precluded a statistically meaningful comparison between NS-CM patients carrying PTPN11 and RAF1 mutations, which may provide information about gene-specific effects, as well as adequate RNA-Seq analysis of age-matched HCM tissue. In addition, because of the lack of myocardial control samples from age-matched healthy children, left ventricular samples from patients with ventricular septal defects were used as controls that do not constitute absolutely unaffected patients. The robustness of the in vitro analysis of hiPSC-derived cardiomyocytes would be enhanced by increasing the number of patient lines as well as by comparing NS-CM cells with genetically engineered isogenic control cells rather than with cells derived from unrelated healthy controls.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Cardiac Troponin T | Invitrogen | Cat# MA5-12960; RRID: AB_11000742 |
| Rabbit monoclonal anti-Cardiac Troponin T | Abcam | Cat# ab92546; RRID: AB_10585593 |
| Human monoclonal anti-Cardiac Troponin T-VioBlue | Miltenyi Biotec | Cat# 130-120-402; RRID: AB_2783891 |
| Guinea pig polyclonal anti-Plakophilin 2 | Origene | Cat# AP09554SU-N; RRID: AB_10556427 |
| Mouse monoclonal anti-Ki67 | BD Biosciences | Cat# 556003; RRID: AB_396287 |
| Mouse monoclonal anti-α-actinin | Sigma-Aldrich | Cat# A7811; RRID: AB_476766 |
| Rabbit polyclonal anti-Dystrophin | Zytomed | Cat# 504-2664 |
| Biological samples | ||
| Left ventricular myocardium from NS-CM, HCM and unaffected patients: see Table S2 | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| CHIR99021 | Axon Medchem | Cat# 1386 |
| IWR1 | Tocris | Cat# 3132 |
| Critical commercial assays | ||
| TruSeq Stranded mRNA Library Prep | Illumina | Cat# 20020594 |
| CellTrace CFSE | Invitrogen | Cat# C34554 |
| Fluo-4 AM | Thermo Fisher | Cat# F14201 |
| Deposited data | ||
| Raw bulk RNA-Seq data | This paper | GEO: GSE188238 |
| Experimental models: Cell lines | ||
| Healthy male human iPSCs | Moretti et al., 2020 | Human Pluripotent Stem Cell Registry, ID: MRIi003-A |
| Healthy female human iPSCs | Krane et al., 2021 | Human Pluripotent Stem Cell Registry, ID: MRIi001-A |
| Male NS-CM patient iPSCs | This paper | Human Pluripotent Stem Cell Registry, ID: MRIi025-A |
| Oligonucleotides | ||
| See Table S7 for PCR and qPCR primer sequences | ||
| Software and algorithms | ||
| ImageJ | National Institutes of Health, Bethesda, Maryland, USA | https://imagej.nih.gov/ij/ |
| RStudio | Integrated Development for R. RStudio, Inc., Boston, Massachusetts, USA | http://www.rstudio.com/ |
| ModFit LT version 5.0.9 | Verity Software House | https://www.vsh.com/products/mflt/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Alessandra Moretti (amoretti@mytum.de).
Materials availability
The newly generated PTPN11N308S/+ patient iPSCs will be shared upon request.
Experimental model and subject details
Human subjects
This study used left ventricular myocardium samples obtained from 8 NS-CM patients (4 male, 4 female), 9 HCM patients (6 male, 3 female), and 12 unaffected control patients (7 male, 5 female). Details of the clinical characteristics of NS-CM patients are listed in Table S1; details of the age, sex, and known genotype of all patients are listed in Table S2.
All recruitment and consenting procedures were done under institutional review board-approved protocols of Technical University of Munich. Written informed consent was obtained from all patients or families. Human subjects participating in Sarcomeric Human Cardiomyopathy Registry (SHaRe) were recruited after obtaining written signed consent using protocols that were reviewed and approved by each institution of the participating SHaRe investigators, as previously described (Ho et al., 2018).
Cell lines
Two healthy human induced pluripotent stem cell (iPSC) lines were used: one male (#1) and one female (#2). iPSCs were reprogrammed from a male NS-CM patient carrying a PTPN11N308S/+ mutation and two clones were used in experiments (#1 and #2) (see iPSC reprogramming). iPSCs were maintained in standard culture conditions (37°C, 5% CO2) in Essential 8 (E8) medium (Gibco A1517001) on Geltrex-coated plates (Gibco A1413301) and passaged every 4 days with 0.5 mM EDTA (Gibco 15575020).
Method details
Human tissue samples
Tissue samples from patients with Noonan syndrome presenting with a diagnosis of hypertrophic cardiomyopathy and from patients with non-syndromic sarcomeric hypertrophic cardiomyopathy were gained at the time of surgical septal myectomy for treatment of refractory symptomatic patients with a left ventricular outflow tract gradient of ≥50 mm Hg at rest or with provocative maneuvers, associated with New York Heart Association (NYHA) functional classes/Ross ≥ III despite maximum medical management (Kühn et al., 2013; Maron et al., 2003a, 2003b; Nishimura and Holmes, 2004; Weidemann et al., 2002). The septal myectomy operation in the present cohort was performed as previously described (Maron et al., 1998; Morrow and Brockenbrough, 1961). Molecular genetic DNA-based analysis was performed in the clinical setting as recommended by European and North-American guidelines (Ackerman et al., 2011; Gersh et al., 2011) and in accredited laboratories only and criteria for assessing variant pathogenicity were based on the variant type, variant database, literature review, frequency in the general population, and in silico analysis according to the ACMG (American College of Medical Genetics and Genomics) guidelines (Richards et al., 2015). Control left ventricular tissue was gained from transplanted donor hearts at the time of first myocardial biopsy after transplantation for histopathological analysis and from patients undergoing surgical ventricular septal defect repair for RNA-sequencing analysis (Table S2). For Mant-ATP assays, control tissue consisted of left ventricular tissue samples from non-failing hearts discarded during surgical procedures at the Brigham and Women's Hospital, obtained through the SHaRe registry (Table S2).
Mant-ATP assay
Mant-ATP assays were performed on snap frozen human myectomy samples prepared as previously described (Toepfer et al., 2019, 2020). Briefly, tissue samples were thawed and permeabilized in permeabilization buffer consisting of 100 mM NaCl, 8 mM MgCl2, 5 mM EGTA, 5 mM K2HPO4, 5 mM KH2PO4, 3 mM NaN3, 5 mM ATP, 1 mM dithiothreitol (DTT), 20 mM 2,3-butanedione monoxime (BDM), and 0.1% Triton X100 (pH 7.0). Once samples were permeabilized, they were placed in a glycerinating solution consisting of 120 mM K acetate, 5 mM Mg acetate, 2.5 mM K2HPO4, 2.5 mM KH2PO4, 50 mM Mops, 5 mM ATP, 20 mM BDM, 2 mM DTT, and 50% (v/v) glycerol (pH 6.8) to allow storage (-20°C for 2 days) or immediate use. Before fluorescence acquisition, sections of tissue were pinned in a flow chamber constructed out of a microscope slide and coverslip and each chamber was flushed with an ATP buffer consisting of 120 mM K acetate, 5 mM Mg acetate, 2.5 mM K2HPO4, 2.5 mM KH2PO4, 4 mM ATP, 50 mM Mops, and 2 mM DTT (pH 6.8) to remove glycerol. This buffer was replaced with rigor buffer consisting of 120 mM K acetate, 5 mM Mg acetate, 2.5 mM K2HPO4, 2.5 mM KH2PO4, 50 mM Mops, and 2 mM DTT (pH 6.8). For fluorescence acquisition, a Nikon TE2000-E was used with a Nikon 20x/0.45 NA objective using a Hamamatsu C9100 electron multiplying charge-coupled device. Frames were acquired every 10 s with a 20-ms acquisition and exposure time using a 4’,6-diamidino-2-phenylindole filter set. Initial fluorescence acquisition was simultaneous with the addition of rigor buffer containing 250 μm Mant-ATP to visualize fluorescent Mant-ATP wash in. At the end of a 15-min acquisition, ATP buffer (rigor buffer and 4 mM ATP) was added to the chamber with simultaneous acquisition of the Mant-ATP chase. After experimental procedures, three regions of each chamber were sampled for fluorescence decay analyses in ImageJ (National Institutes of Health, Bethesda, MD) as previously described (McNamara et al., 2016; Toepfer et al., 2019).
Histopathology
The percentage of multinucleated cardiomyocytes was assessed on hematoxylin-eosin stained histopathologic slides. Five slides were evaluated per patient and nuclei from 100 cardiomyocytes were counted for each slide by a cardiovascular pathologist (KK) blinded to the groups. Cardiomyocyte density was determined by quantifying the number of cardiomyocytes per mm2 using ImageJ (National Institutes of Health, Bethesda, MD). Selected slides were additionally stained for dystrophin (Zytomed 504-2664, 1:1000) to highlight cardiomyocyte membranes.
RNA sequencing
Strand specific, polyA-enriched RNA sequencing was performed as previously described (Haack et al., 2013). Briefly, RNA was isolated from whole-cell lysates using the AllPrep RNA Kit (Qiagen) and RNA integrity number (RIN) was determined with the Agilent 2100 BioAnalyzer (RNA 6000 Nano Kit, Agilent). For library preparation, 1 μg of RNA was poly(A) selected, fragmented, and reverse transcribed with the Elute, Prime, Fragment Mix (Illumina). A-tailing, adaptor ligation, and library enrichment were performed as described in the TruSeq Stranded mRNA Sample Prep Guide (Illumina). RNA libraries were assessed for quality and quantity with the Agilent 2100 BioAnalyzer and the Quant-iT PicoGreen dsDNA Assay Kit (Life Technologies). RNA libraries were sequenced as 100 bp paired-end runs on an Illumina HiSeq4000 platform. The STAR aligner (version 2.4.2a)(Dobin et al., 2013) with modified parameter settings (--twopassMode=Basic) was used for split-read alignment against the human genome assembly hg19 (GRCh37) and UCSC knownGene annotation. To quantify the number of reads mapping to annotated genes we used HTseq-count (version 0.6.0) (Anders et al., 2015). FPKM (Fragments Per Kilobase of transcript per Million fragments mapped) values were calculated using custom scripts. Differential expression analysis was performed using the R Bioconductor package DESeq2 (Love et al., 2014). For pathway- and gene set enrichment analysis we used the R Bioconductor packages GAGE (Luo et al., 2009) plus pathview (Luo and Brouwer, 2013) and GOseq (Young et al., 2010), respectively. All gene set files for this analysis were obtained from GSEA algorithm implemented in R software (R Core Team, 2021). Enrichment score (ES) and False discovery rate (FDR) value were applied to sort pathways enriched. Furthermore, to assess statistical significance, we randomized our data set by permuting gene sets 1000 times and considered only gene sets with a p-value ≤ 0.05.
iPSC reprogramming
The healthy iPSC lines were reprogrammed with the CytoTune-iPS 2.0 Sendai Reprogramming kit (Invitrogen A16517) using the peripheral blood mononuclear cells (PBMCs) of male (#1: Human Pluripotent Stem Cell Registry ID MRIi003-A) and female (#2: ID MRIi001-A) volunteers, as previously described (Krane et al., 2021; Moretti et al., 2020). PTPN11N308S/+ iPSCs (Human Pluripotent Stem Cell Registry ID MRIi025-A) were reprogrammed with the same protocol from the PBMCs of a male Noonan syndrome patient carrying a heterozygous 923A→G mutation in the PTPN11 gene leading to a p.N308S substitution in the protein tyrosine phosphatase domain of the SHP-2 protein. For two clones (#1 and #2), pluripotency was assessed after reprogramming via alkaline phosphatase staining (Roche 11681451001), immunofluorescence analysis of the pluripotency markers Nanog and TRA-1-81 (all antibodies listed in Table S6) and qRT-PCR analysis of the pluripotency markers OCT4, SOX2, NANOG, REX1 and TDGF-1 (all primers listed in Table S7). Trilineage differentiation potential was tested via spontaneous embryoid body differentiation in DMEM/F12 medium (Gibco 11320033) containing 20% FCS, 50 μg/mL L-ascorbic acid (Sigma-Aldrich A5960), 1% L-glutamine, 1% non-essential amino acids (Gibco 11140050) and 0.5% Penicillin-Streptomycin for 21 days followed by qRT-PCR analysis of markers of endoderm (SOX7, AFP, PDX1), mesoderm (CD31, DES, ACTA2, SCL, CDH5) and ectoderm (KRT14, NCAM1, TH, GABRR2) using GAPDH as an endogenous control. Loss of Sendai virus was confirmed via immunofluorescence analysis and RT-PCR of the Sendai vector and viral transgenes OCT4, SOX2, KLF4 and c-MYC using GAPDH as an endogenous control. Karyotyping was performed by the Institute of Human Genetics of the Klinikum rechts der Isar, Technical University of Munich.
iPSC-cardiomyocyte culture
Cardiomyocytes were differentiated from iPSCs using a protocol previously described by Lian and colleagues, with some modifications (Lian et al., 2013). Briefly, iPSCs were seeded at a 1:8 ratio in Geltrex-coated 12-well plates in E8 medium containing 10 μM Y27632 (Calbiochem 688000). E8 medium was replaced every day for 4-5 days until the cells reached 100% confluence, at which point cardiac differentiation was initiated by adding basal cardiac differentiation medium consisting of RPMI1640 (Gibco 21875091) with B27 minus insulin (Gibco A1895601) supplemented with 5 μM CHIR99021 (Axon Medchem 1386). After 48 hours, medium was replaced with basal cardiac differentiation medium supplemented with 5 μM IWR1 (Tocris 3132). The cells were then maintained in basal cardiac differentiation medium for another 10 days before beating areas were mechanically transferred to plates coated with 2 μg/cm2 fibronectin (Sigma-Aldrich F1141) and cultured in cardiomyocyte maintenance medium consisting of DMEM/F12 with 2% FCS, 1% non-essential amino acids, 1% Penicillin-Streptomycin-Glutamine and 0.1 mM β-mercaptoethanol.
Single cell dissociation of cardiomyocytes for re-seeding or analysis was performed using a papain-based protocol as previously described (Fischer et al., 2018). Briefly, cells were washed twice with 2 mM EDTA in DPBS with Ca2+ and Mg2+ (Gibco 14040133) before incubation in a solution containing 20 U/mL papain (Worthington Biochemical Corporation LS003124) and 1 mM L-cysteine (Sigma-Aldrich C6852) in DPBS for 30 minutes at 37°C. The reaction was stopped by addition of one volume of DPBS containing 1 mg/mL trypsin inhibitor (Sigma-Aldrich T9253) and 40 μg/mL DNAse I (Sigma-Aldrich DN25). Single cardiomyocytes were seeded at the indicated densities onto fibronectin-coated plates in cardiomyocyte maintenance medium with a concentration of FCS increased to 20% to promote survival. Cells were then cultured in cardiomyocyte maintenance medium replaced every 3 days until analysis.
For experiments using purified cardiomyocytes, cells were dissociated with papain as described above and non-cardiomyocytes present in the culture were depleted via magnetic-activated cell sorting using the human PSC-derived cardiomyocyte isolation kit (Miltenyi 130-110-188).
Immunofluorescence analysis
For immunofluorescence staining, cells were washed with DPBS with Ca2+ and Mg2 and fixed with 4% PFA for 15 min at room temperature (RT). After washing 3 times with DPBS, cells were permeabilized with 0.25% Triton X-100 in DPBS for 15 min at RT. After washing another 3 times with DPBS, cells were blocked with 3% BSA in DPBS for at least 1 hour at RT. Primary antibodies (Table S6) were then added at the indicated dilution in 0.5% BSA in DPBS and incubated overnight at 4°C. After washing 3 times for 5 min with PBST (0.05% Tween 20 in DPBS), appropriate secondary antibodies (Table S6) diluted in 0.5% BSA in DPBS were added for 1 hour at RT, protected from light. After washing 3 times for 5 min with PBST, Hoechst 33258 (Sigma-Aldrich, 94403) was added at a final concentration of 5 μg/mL in DPBS for 15 min at RT, protected from light. After washing once with DPBS, cells were then covered with fluorescence mounting medium (Dako S3023) and stored at 4°C until imaging with an inverted microscope (DMI6000B, Leica Microsystems, Wetzlar, Germany). The area of cTnT+ cells was measured manually using ImageJ (National Institutes of Health, Bethesda, MD).
DNA content analysis in isolated iPSC-cardiomyocyte nuclei
For nuclei isolation, 30 day-old iPSC-cardiomyocytes were dissociated to single cells and purified as described above. Purified cells were resuspended in ice-cold nuclei extraction buffer (320 mM sucrose, 5 mM MgCl2, 10 mM HEPES, 1% Triton X-100 in H2O) at 106 cells/mL, gently vortexed for 10 s and placed on ice for 10 min. The nuclei were then pelleted by centrifugation at 2,000 g for 8 min at 4°C and washed twice with nuclei wash buffer (320 mM sucrose, 5 mM MgCl2, 10 mM HEPES in H2O). The nuclei were resuspended in 100 μL flow buffer (320 mM sucrose, 5 mM MgCl2, 10 mM HEPES, 1% BSA in H2O) and treated with RNAse A (Qiagen 158922) at 100 U/mL for 1 hour at 37°C on a shaker at 400 rpm, adding propidium iodide (Sigma-Aldrich P4864) at 50 μg/mL after 30 min. Flow cytometry data was acquired on a Gallios flow cytometer (Beckman Coulter) and analyzed with Kaluza software version 1.2.
Proliferation analysis of iPSC-cardiomyocytes
For proliferation analysis, 15 day-old iPSC-cardiomyocytes were dissociated to single cells as described above and loaded with 4 μM CellTrace CFSE (Invitrogen C34554) according to manufacturer’s instructions. After loading, a t0 sample was collected for flow cytometry and the rest of the cells was seeded on fibronectin-coated plates at 25,000 cells/cm2 in cardiomyocyte maintenance medium containing 20% FCS and 10 μM Y27632 (Calbiochem 688000) to promote survival, replaced with cardiomyocyte medium containing 2% FCS the next day. After 96 hours (t96), the cells were collected by papain dissociation as described above. For flow cytometry analysis, the cells were fixed with 4% PFA for 10 min at RT, then washed 3 times with 2% FCS in DPBS with Ca2+ and Mg2 and blocked in 10% FCS, 0.1% Triton-X, 0.1% saponin in DPBS for 1 hour at RT protected from light. To label cardiomyocytes, an antibody against cTnT (Miltenyi Biotec 130-120-402) was then added at 2 μL per 106 cells in 100 μL 1% FCS, 0.1% Triton-X, 0.1% saponin in DPBS and incubated overnight at 4°C. The cells were then washed 3 times with antibody diluent and resuspended in 2% FCS in DPBS. Flow cytometry data was acquired on a Gallios flow cytometer (Beckman Coulter) and the proportion of single cTnT+ cells in successive daughter generations at t96 relative to the t0 parent generation as well as the proliferation index were calculated with ModFit LT version 5.0.9.
Calcium imaging in iPSC-cardiomyocytes
For calcium imaging, day 15 iPSC-CMs were seeded at 25,000 cells/cm2 in 3.5 cm glass bottom cell culture microdishes (MatTek Corporation P35G-1.5 14-C) coated with 2 μg/cm2 fibronectin. At day 30, calcium imaging was performed using the calcium indicator Fluo-4 AM (Thermo Fisher F14201) at a concentration of 1 μM in Tyrode’s solution supplemented with Ca2+ as previously described (Moretti et al., 2020). Briefly, field stimulation electrodes (RC-37FS, Warner Instruments, Hamden, CT, USA) were connected to a stimulus generator (HSE Stimulator P, Hugo Sachs Elektronik, March-Hugstetten, Germany) providing depolarizing pulses (50 V, 5 ms duration) at the frequencies indicated. ImageJ (National Institutes of Health, Bethesda, MD) was used to quantify fluorescence over single cells and background regions. Subsequent analysis was performed in RStudio (RStudio Team (2015). RStudio: Integrated Development for R. RStudio, Inc., Boston, MA) using custom-written scripts. After subtraction of background fluorescence, the time course of Fluo-4 fluorescence was normalized to the initial value (F/F0). After manual selection of the starting points and peaks of the calcium transients, the transient duration at 50% decay (TD50) and 90% decay (TD90) was automatically determined by the script. The amplitude of calcium transients was calculated by subtracting the basal fluorescence value from the peak value.
PCR and real-time qPCR
Total RNA was extracted from iPSCs or purified day 30 iPSC-cardiomyocytes using the Absolutely RNA Microprep kit (Agilent 400805) and cDNA was prepared with the High Capacity cDNA RT kit (Applied Biosystems 4368814) according to the manufacturers’ instructions. PCR was performed with the FIREPol DNA Polymerase (Solis Biodyne 01-01-00500) and primers listed in Table S7. Real-time qPCR was performed with a 7500 Fast Real-time PCR System (Applied Biosystems, Germany) using the Power SYBR Green PCR Master Mix (Applied Biosystems 4367659) and the primers listed in Table S7. Data was analyzed using the 2-ΔCt method with normalization to GAPDH expression.
Quantification and statistical analysis
Statistical analysis was performed with GraphPad Prism version 8.4.0. (La Jolla California, USA). Statistical details of each experiment including the statistical tests used and the exact value and definition of n are indicated in figure legends. Results are presented as box-and-whiskers plots indicating the minimum, 25th percentile, median, 75th percentile, maximum and all data points unless otherwise indicated. Results cited in the text are given as mean ± standard deviation. Normally distributed data from two groups were compared by two-tailed t-test. Normally distributed data from more than two experimental groups were compared using one-way or two-way analysis of variance (ANOVA) with appropriate multiple comparison tests as indicated. A p-value < 0.05 was considered statistically significant.
Acknowledgments
We would like to thank the patients and families who participated in this study. This work was supported by grants from: the European Research Council, ERC 788381 (to A.M.), the German Research Foundation, Transregio Research Unit 152 (to A.M., K-L.L.) and 267 (to A.M., K-L.L.), the DZHK (German Centre for Cardiovascular Research) and the Deutsche Herzstiftung e.V. Gerd-Killian Award (to C.M.W.) and the Sir Henry Wellcome Fellowship 206466/Z/17/Z (to C.N.T.). The graphical abstract was created with BioRender.com.
Author contributions
CMW and AM designed the study and reviewed the data. ABM, CMW, AM, and JGS wrote the paper. CMW and JC developed the clinical sample cohort. AG, PE, JGS, CES, and KLL contributed to study conception and design. MS and CNT performed the Mant-ATP experiments and analyzed the data. GS, TS, EM, and RB performed the bioinformatics analysis. KK performed the histological analysis. ABM, SRM, and HR analyzed the data.
Declaration of interests
Dr. Wolf is a consultant for Day One Therapeutics. All other authors declare no competing interests.
Published: January 21, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103596.
Contributor Information
Alessandra Moretti, Email: amoretti@mytum.de.
Cordula M. Wolf, Email: wolf@dhm.mhn.de.
Supplemental information
Data and code availability
Bulk RNA-Seq data have been deposited at GEO and are publicly available as of the date of publication. The accession number is listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper will be shared by the lead contact upon request.
References
- Ackerman M.J., Priori S.G., Willems S., Berul C., Brugada R., Calkins H., Camm A.J., Ellinor P.T., Gollob M., Hamilton R., et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies. Heart Rhythm. 2011;8:1308–1339. doi: 10.1016/j.hrthm.2011.05.020. [DOI] [PubMed] [Google Scholar]
- Ahuja P., Sdek P., MacLellan W.R. Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol. Rev. 2007;87:521–544. doi: 10.1152/physrev.00032.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andelfinger G., Marquis C., Raboisson M.-J., Théoret Y., Waldmüller S., Wiegand G., Gelb B.D., Zenker M., Delrue M.-A., Hofbeck M. Hypertrophic cardiomyopathy in Noonan syndrome treated by MEK-inhibition. J. Am. Coll. Cardiol. 2019;73:2237–2239. doi: 10.1016/j.jacc.2019.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S., Pyl P.T., Huber W. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Bouchikhi I., Belhassan K., Moufid F.Z., Iraqui Houssaini M., Bouguenouch L., Samri I., Atmani S., Ouldim K. Noonan syndrome-causing genes: Molecular update and an assessment of the mutation rate. Int. J. Pediatr. Adolesc. Med. 2016;3:133–142. doi: 10.1016/j.ijpam.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burch M., Mann J.M., Sharland M., Shinebourne E.A., Patton M.A., Mckenna W.J., Mann J.M., Sharland M., Patton M.A., Mckenna W.J. Myocardial disarray in Noonan syndrome. Br. Heart J. 1992;68:586–594. doi: 10.1136/hrt.68.12.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcagni G., Adorisio R., Martinelli S., Grutter G., Baban A., Versacci P., Digilio M.C., Drago F., Gelb B.D., Tartaglia M., et al. Clinical presentation and natural history of hypertrophic cardiomyopathy in RASopathies. Heart Fail. Clin. 2018;14:225–235. doi: 10.1016/j.hfc.2017.12.005. [DOI] [PubMed] [Google Scholar]
- Cao Y., Xia D.S., Qi S.R., Du J., Ma P., Wang S.L., Fan Z.P. Epiregulin can promote proliferation of stem cells from the dental apical papilla via MEK/Erk and JNK signalling pathways. Cell Prolif. 2013;46:447–456. doi: 10.1111/cpr.12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn R., Thakar K., Lowe A., Ladha F.A., Pettinato A.M., Romano R., Meredith E., Chen Y.S., Atamanuk K., Huey B.D., et al. A contraction stress model of hypertrophic cardiomyopathy due to sarcomere mutations. Stem Cell Rep. 2019;12:71–83. doi: 10.1016/j.stemcr.2018.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquitt J.L., Noonan J.A. Cardiac findings in Noonan syndrome on long-term follow-up. Congenit. Heart Dis. 2014;9:144–150. doi: 10.1111/chd.12102. [DOI] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eschenhagen T., Carrier L. Cardiomyopathy phenotypes in human-induced pluripotent stem cell-derived cardiomyocytes—a systematic review. Pflügers Arch. - Eur. J. Physiol. 2019;471:755–768. doi: 10.1007/s00424-018-2214-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer B., Meier A., Dehne A., Salhotra A., Tran T.A., Neumann S., Schmidt K., Meiser I., Neubauer J.C., Zimmermann H., et al. A complete workflow for the differentiation and the dissociation of hiPSC-derived cardiospheres. Stem Cell Res. 2018;32:65–72. doi: 10.1016/j.scr.2018.08.015. [DOI] [PubMed] [Google Scholar]
- Gao J., Collyer J., Wang M., Sun F., Xu F. Genetic dissection of hypertrophic cardiomyopathy with myocardial RNA-seq. Int. J. Mol. Sci. 2020;21:3040. doi: 10.3390/ijms21093040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelb B.D., Tartaglia M. Noonan syndrome and related disorders: Dysregulated RAS-mitogen activated protein kinase signal transduction. Hum. Mol. Genet. 2006;15:R220–R226. doi: 10.1093/hmg/ddl197. [DOI] [PubMed] [Google Scholar]
- Gersh B.J., Maron B.J., Bonow R.O., Dearani J.A., Fifer M.A., Link M.S., Naidu S.S., Nishimura R.A., Ommen S.R., Rakowski H., et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: Executive summary. Circulation. 2011;124:2761–2796. doi: 10.1161/CIR.0b013e318223e230. [DOI] [PubMed] [Google Scholar]
- Haack T.B., Kopajtich R., Freisinger P., Wieland T., Rorbach J., Nicholls T.J., Baruffini E., Walther A., Danhauser K., Zimmermann F.A., et al. ELAC2 mutations cause a mitochondrial RNA processing defect associated with hypertrophic cardiomyopathy. Am. J. Hum. Genet. 2013;93:211–223. doi: 10.1016/j.ajhg.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn A., Lauriol J., Thul J., Behnke-Hall K., Logeswaran T., Schänzer A., Böğürcü N., Garvalov B.K., Zenker M., Gelb B.D., et al. Rapidly progressive hypertrophic cardiomyopathy in an infant with Noonan syndrome with multiple lentigines: Palliative treatment with a rapamycin analog. Am. J. Med. Genet. A. 2015;167:744–751. doi: 10.1002/ajmg.a.36982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L., Li Y., Tchao J., Kaplan A.D., Lin B., Li Y., Mich-Basso J., Lis A., Hassan N., London B., et al. Study familial hypertrophic cardiomyopathy using patient-specific induced pluripotent stem cells. Cardiovasc. Res. 2014;104:258–269. doi: 10.1093/cvr/cvu205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanses U., Kleinsorge M., Roos L., Yigit G., Li Y., Barbarics B., El-Battrawy I., Lan H., Tiburcy M., Hindmarsh R., et al. Intronic CRISPR repair in a preclinical model of Noonan syndrome-associated cardiomyopathy. Circulation. 2020;142:1059–1076. doi: 10.1161/CIRCULATIONAHA.119.044794. [DOI] [PubMed] [Google Scholar]
- Ho C.Y., Day S.M., Ashley E.A., Michels M., Pereira A.C., Jacoby D., Cirino A.L., Fox J.C., Lakdawala N.K., Ware J.S., et al. Genotype and lifetime burden of disease in hypertrophic cardiomyopathy insights from the sarcomeric human cardiomyopathy registry (SHaRe) Circulation. 2018;138:1387–1398. doi: 10.1161/CIRCULATIONAHA.117.033200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooijman P., Stewart M.A., Cooke R. A new state of cardiac myosin with very slow ATP turnover: A potential cardioprotective mechanism in the heart. Biophys. J. 2011;100:1969–1976. doi: 10.1016/j.bpj.2011.02.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert F., Payan S.M., Rochais F. FGF10 signaling in heart development, homeostasis, disease and repair. Front. Genet. 2018;9:599. doi: 10.3389/fgene.2018.00599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudsmith L.E., Petersen S.E., Francis J.M., Robson M.D., Watkins H., Neubauer S. Hypertrophic cardiomyopathy in Noonan syndrome closely mimics familial hypertrophic cardiomyopathy due to sarcomeric mutations. Int. J. Cardiovasc. Imaging. 2006;22:493–495. doi: 10.1007/s10554-005-9034-y. [DOI] [PubMed] [Google Scholar]
- Jaffré F., Miller C.L., Schänzer A., Evans T., Roberts A.E., Hahn A., Kontaridis M.I. iPSC-derived cardiomyocytes reveal aberrant ERK5 and MEK1/2 signaling concomitantly promote hypertrophic cardiomyopathy in RAF1-associated Noonan syndrome. Circulation. 2019;140:207–224. doi: 10.1161/CIRCULATIONAHA.118.037227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltenecker E., Schleihauf J., Meierhofer C., Shehu N., Mkrtchyan N., Hager A., Kühn A., Cleuziou J., Klingel K., Seidel H., et al. Long-term outcomes of childhood onset Noonan compared to sarcomere hypertrophic cardiomyopathy. Cardiovasc. Diagn. Ther. 2019;9:S299–S309. doi: 10.21037/cdt.2019.05.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krane M., Dreßen M., Santamaria G., My I., Schneider C.M., Dorn T., Laue S., Mastantuono E., Berutti R., Rawat H., et al. Sequential defects in cardiac lineage commitment and maturation cause hypoplastic left heart syndrome. Circulation. 2021;144:1409–1428. doi: 10.1161/CIRCULATIONAHA.121.056198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühn A., De Pasquale Meyer G., Müller J., Petzuch K., Fratz S., Röhlig C., Hager A., Schreiber C., Hess J., Vogt M. Tricuspid valve surgery improves cardiac output and exercise performance in patients with Ebstein’s anomaly. Int. J. Cardiol. 2013;166:494–498. doi: 10.1016/j.ijcard.2011.11.033. [DOI] [PubMed] [Google Scholar]
- Kyotani Y., Ota H., Itaya-Hironaka A., Yamauchi A., Sakuramoto-Tsuchida S., Zhao J., Ozawa K., Nagayama K., Ito S., Takasawa S., et al. Intermittent hypoxia induces the proliferation of rat vascular smooth muscle cell with the increases in epidermal growth factor family and erbB2 receptor. Exp. Cell Res. 2013;319:3042–3050. doi: 10.1016/j.yexcr.2013.08.014. [DOI] [PubMed] [Google Scholar]
- Lan F., Lee A.S., Liang P., Sanchez-Freire V., Nguyen P.K., Wang L., Han L., Yen M., Wang Y., Sun N., et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell. 2013;12:101–113. doi: 10.1016/j.stem.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian X., Zhang J., Azarin S.M., Zhu K., Hazeltine L.B., Bao X., Hsiao C., Kamp T.J., Palecek S.P. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat. Protoc. 2013;8:162–175. doi: 10.1038/nprot.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W., Brouwer C. Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics. 2013;29:1830–1831. doi: 10.1093/bioinformatics/btt285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W., Friedman M.S., Shedden K., Hankenson K.D., Woolf P.J. GAGE: Generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics. 2009;10:161. doi: 10.1186/1471-2105-10-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin T.M., Keith K., Davies B., Conner D.A., Guha P., Kalaitzidis D., Wu X., Lauriol J., Wang B., Bauer M., et al. Rapamycin reverses hypertrophic cardiomyopathy in a mouse model of LEOPARD syndrome-associated PTPN11 mutation. J. Clin. Invest. 2011;121:1026–1043. doi: 10.1172/JCI44972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maron B.J., Nishimura R.A., Danielson G.K. Pitfalls in clinical recognition and a novel operative approach for hypertrophic cardiomyopathy with severe outflow obstruction due to anomalous papillary muscle. Circulation. 1998;98:2505–2508. doi: 10.1161/01.CIR.98.23.2505. [DOI] [PubMed] [Google Scholar]
- Maron B.J., McKenna W.J., Danielson G.K., Kappenberger L.J., Kuhn H.J., Seidman C.E., Shah P.M., Spencer W.H., Spirito P., Ten Cate F.J., et al. American college of cardiology/European society of cardiology clinical expert consensus document on hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2003;42:1687–1713. doi: 10.1016/S0735-1097(03)00941-0. [DOI] [PubMed] [Google Scholar]
- Maron M.S., Olivotto I., Betocchi S., Casey S.A., Lesser J.R., Losi M.A., Cecchi F., Maron B.J. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N. Engl. J. Med. 2003;348:295–303. doi: 10.1056/NEJMoa021332. [DOI] [PubMed] [Google Scholar]
- McNamara J.W., Li A., Smith N.J., Lal S., Graham R.M., Kooiker K.B., van Dijk S.J., Remedios C.G.do., Harris S.P., Cooke R. Ablation of cardiac myosin binding protein-C disrupts the super-relaxed state of myosin in murine cardiomyocytes. J. Mol. Cell. Cardiol. 2016;94:65–71. doi: 10.1016/j.yjmcc.2016.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara J.W., Li A., Lal S., Bos J.M., Harris S.P., Van Der Velden J., Ackerman M.J., Cooke R., Dos Remedios C.G. MYBPC3 mutations are associated with a reduced super-relaxed state in patients with hypertrophic cardiomyopathy. PLoS One. 2017;12:e0180064. doi: 10.1371/journal.pone.0180064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morato A., Martignani E., Miretti S., Baratta M., Accornero P. External and internal EGFR-activating signals drive mammary epithelial cells proliferation and viability. Mol. Cell. Endocrinol. 2021;520:111081. doi: 10.1016/j.mce.2020.111081. [DOI] [PubMed] [Google Scholar]
- Moretti A., Fonteyne L., Giesert F., Hoppmann P., Meier A.B., Bozoglu T., Baehr A., Schneider C.M., Sinnecker D., Klett K., et al. Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy. Nat. Med. 2020;26:206–214. doi: 10.1038/s41591-019-0738-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow A.G., Brockenbrough E.C. Surgical treatment of idiopathic hypertrophic subaortic stenosis: Technic and hemodynamic results of subaortic ventriculomyotomy. Ann. Surg. 1961;154:181–189. doi: 10.1097/00000658-196108000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosqueira D., Mannhardt I., Bhagwan J.R., Lis-Slimak K., Katili P., Scott E., Hassan M., Prondzynski M., Harmer S.C., Tinker A., et al. CRISPR/Cas9 editing in human pluripotent stem cell-cardiomyocytes highlights arrhythmias, hypocontractility, and energy depletion as potential therapeutic targets for hypertrophic cardiomyopathy. Eur. Heart J. 2018;39:3879–3892. doi: 10.1093/eurheartj/ehy249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagama Y., Inuzuka R., Ichimura K., Hinata M., Takehara H., Takeda N., Kakiuchi S., Shiraga K., Asakai H., Shindo T., et al. Accelerated cardiomyocyte proliferation in the heart of a neonate with LEOPARD syndrome-associated Fatal cardiomyopathy. Circ. Hear. Fail. 2018;11:1–3. doi: 10.1161/CIRCHEARTFAILURE.117.004660. [DOI] [PubMed] [Google Scholar]
- Nishimura R.A., Holmes D.R. Clinical practice. Hypertrophic obstructive cardiomyopathy. N. Engl. J. Med. 2004;350:1320–1327. doi: 10.1056/NEJMcp030779. [DOI] [PubMed] [Google Scholar]
- Nishioka S., Wu P.-H., Yakabe T., Giaccia A.J., Le Q.-T., Aoyama H., Shimizu S., Shirato H., Onodera Y., Nam J.-M. Rab27b contributes to radioresistance and exerts a paracrine effect via epiregulin in glioblastoma. Neuro-Oncology Adv. 2020;2:vdaa091. doi: 10.1093/noajnl/vdaa091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norrish G., Field E., McLeod K., Ilina M., Stuart G., Bhole V., Uzun O., Brown E., Daubeney P.E.F., Lota A., et al. Clinical presentation and survival of childhood hypertrophic cardiomyopathy: A retrospective study in United Kingdom. Eur. Heart J. 2019;40:986–993. doi: 10.1093/eurheartj/ehy798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivotto I., Oreziak A., Barriales-Villa R., Abraham T.P., Masri A., Garcia-Pavia P., Saberi S., Lakdawala N.K., Wheeler M.T., Owens A., et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2020;396:759–769. doi: 10.1016/S0140-6736(20)31792-X. [DOI] [PubMed] [Google Scholar]
- Passariello C.L., Martinez E.C., Thakur H., Cesareo M., Li J., Kapiloff M.S. RSK3 is required for concentric myocyte hypertrophy in an activated Raf1 model for Noonan syndrome. J. Mol. Cell. Cardiol. 2016;93:98–105. doi: 10.1016/j.yjmcc.2016.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prendiville T.W., Gauvreau K., Tworog-Dube E., Patkin L., Kucherlapati R.S., Roberts A.E., Lacro R.V., Prendiville T. Cardiovascular disease in Noonan syndrome. Arch. Dis. Child. 2014;99:629–634. doi: 10.1136/archdischild-2013-305047. [DOI] [PubMed] [Google Scholar]
- R Core Team . R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2021. https://www.R-project.org/ [Google Scholar]
- Ramdzan Z.M., Vadnais C., Pal R., Vandal G., Cadieux C., Leduy L., Davoudi S., Hulea L., Yao L., Karnezis A.N., et al. RAS Transformation requires CUX1-dependent repair of oxidative DNA damage. PLoS Biol. 2014;12:e1001807. doi: 10.1371/journal.pbio.1001807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramdzan Z.M., Vickridge E., Faraco C.C.F., Nepveu A. Cut domain proteins in DNA repair and cancer. Cancers (Basel) 2021;13:2953. doi: 10.3390/cancers13122953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren C.W., Liu J.J., Li J.H., Li J.W., Dai J., Lai Y.Q. RNA-seq profiling of mRNA associated with hypertrophic cardiomyopathy. Mol. Med. Rep. 2016;14:5573–5586. doi: 10.3892/mmr.2016.5931. [DOI] [PubMed] [Google Scholar]
- Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E., et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and Genomics and the association for molecular pathology. Genet. Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochais F., Sturny R., Chao C.-M., Mesbah K., Bennett M., Mohun T.J., Bellusci S., Kelly R.G. FGF10 promotes regional foetal cardiomyocyte proliferation and adult cardiomyocyte cell-cycle re-entry. Cardiovasc. Res. 2014;104:432–442. doi: 10.1093/cvr/cvu232. [DOI] [PubMed] [Google Scholar]
- Romano A.A., Allanson J.E., Dahlgren J., Gelb B.D., Hall B., Pierpont M.E., Roberts A.E., Robinson W., Takemoto C.M., Noonan J.A. Noonan syndrome: Clinical features, diagnosis, and management guidelines. Pediatrics. 2010;126:746–759. doi: 10.1542/peds.2009-3207. [DOI] [PubMed] [Google Scholar]
- Sakai T., Naito A.T., Kuramoto Y., Ito M., Okada K., Higo T., Nakagawa A., Shibamoto M., Yamaguchi T., Sumida T., et al. Phenotypic screening using patient-derived induced pluripotent stem cells identified Pyr3 as a candidate compound for the treatment of infantile hypertrophic cardiomyopathy. Int. Heart J. 2018;59:1096–1105. doi: 10.1536/ihj.17-730. [DOI] [PubMed] [Google Scholar]
- Schmid M., Toepfer C.N. Cardiac myosin super relaxation (SRX): A perspective on fundamental biology, human disease and therapeutics. Biol. Open. 2021;10 doi: 10.1242/bio.057646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholzen T., Gerdes J. The Ki-67 protein: From the known and the unknown. J. Cell. Physiol. 2000;182:311–322. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Schramm C., Fine D.M., Edwards M.A., Reeb A.N., Krenz M. The PTPN11 loss-of-function mutation Q510E-Shp2 causes hypertrophic cardiomyopathy by dysregulating mTOR signaling. Am. J. Physiol. - Heart Circ. Physiol. 2012;302:231–243. doi: 10.1152/ajpheart.00665.2011. [DOI] [PubMed] [Google Scholar]
- Seidman C.E., Seidman J.G. Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: A personal history. Circ. Res. 2011;108:743–750. doi: 10.1161/CIRCRESAHA.110.223834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw A.C., Kalidas K., Crosby A.H., Jeffery S., Patton M.A. The natural history of Noonan syndrome: A long-term follow-up study. Arch. Dis. Child. 2007;92:128–132. doi: 10.1136/adc.2006.104547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia M., Gelb B.D. Noonan syndrome and related disorders: Genetics and Pathogenesis. Annu. Rev. Genomics Hum. Genet. 2005;6:45–68. doi: 10.1146/annurev.genom.6.080604.162305. [DOI] [PubMed] [Google Scholar]
- Toepfer C.N., Wakimoto H., Garfinkel A.C., McDonough B., Liao D., Jiang J., Tai A.C., Gorham J.M., Lunde I.G., Lun M., et al. Hypertrophic cardiomyopathy mutations in MYBPC3 dysregulate myosin. Sci. Transl. Med. 2019;11:eaat1199. doi: 10.1126/scitranslmed.aat1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toepfer C.N., Garfinkel A.C., Venturini G., Wakimoto H., Repetti G., Alamo L., Sharma A., Agarwal R., Ewoldt J.F., Cloonan P., et al. Myosin sequestration regulates sarcomere function, cardiomyocyte energetics, and metabolism, informing the pathogenesis of hypertrophic cardiomyopathy. Circulation. 2020;141:828–842. doi: 10.1161/CIRCULATIONAHA.119.042339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Chandrasekhar V., Abbadessa G., Yu Y., Schwartz B., Kontaridis M.I. In vivo efficacy of the AKT inhibitor ARQ 092 in Noonan syndrome with multiple lentigines-associated hypertrophic cardiomyopathy. PLoS One. 2017;12:e0178905. doi: 10.1371/journal.pone.0178905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidemann F., Eyskens B., Jamal F., Mertens L., Kowalski M., D’Hooge J., Bijnens B., Gewillig M., Rademakers F., Hatle L., et al. Quantification of regional left and right ventricular radial and longitudinal function in healthy children using ultrasound-based strain rate and strain imaging. J. Am. Soc. Echocardiogr. 2002;15:20–28. doi: 10.1067/mje.2002.116532. [DOI] [PubMed] [Google Scholar]
- White G.E., Tan T.C.C., John A.E., Whatling C., McPheat W.L., Greaves D.R. Fractalkine has anti-apoptotic and proliferative effects on human vascular smooth muscle cells via epidermal growth factor receptor signalling. Cardiovasc. Res. 2010;85:825–835. doi: 10.1093/CVR/CVP341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson J.D., Lowe A.M., Salbert B.A., Sleeper L.A., Colan S.D., Cox G.F., Towbin J.A., Connuck D.M., Messere J.E., Lipshultz S.E. Outcomes in children with Noonan syndrome and hypertrophic cardiomyopathy: A study from the pediatric cardiomyopathy registry. Am. Heart J. 2012;164:442–448. doi: 10.1016/j.ahj.2012.04.018. [DOI] [PubMed] [Google Scholar]
- Wu T., Wang L.N., Tang D.R., Sun F.Y. SOST silencing promotes proliferation and invasion and reduces apoptosis of retinoblastoma cells by activating Wnt/β-catenin signaling pathway. Gene Ther. 2017;24:399–407. doi: 10.1038/gt.2017.31. [DOI] [PubMed] [Google Scholar]
- Wu X., Simpson J., Hong J.H., Kim K.-H., Thavarajah N.K., Backx P.H., Neel B.G., Araki T. MEK-ERK pathway modulation ameliorates disease phenotypes in a mouse model of Noonan syndrome associated with the Raf1(L613V) mutation. J. Clin. Invest. 2011;121:1009–1025. doi: 10.1172/JCI44929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., Chen W., Leng S., Padilla M.T., Saxton B., Hutt J., Tessema M., Kato K., Kim K.C., Belinsky S.A., et al. Muc1 knockout potentiates murine lung carcinogenesis involving an epiregulin-mediated EGFR activation feedback loop. Carcinogenesis. 2017;38:604–614. doi: 10.1093/CARCIN/BGX039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young M.D., Wakefield M.J., Smyth G.K., Oshlack A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010;11:R14. doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R., Li X., Liu Z., Wang Y., Zhang H., Xu H. EZH2 inhibitors-mediated epigenetic reactivation of FOSB inhibits triple-negative breast cancer progress. Cancer Cell Int. 2020;20:175. doi: 10.1186/s12935-020-01260-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J., Zhang W., Li X.L. Effects of SOST gene silencing on proliferation, apoptosis, invasion, and migration of human osteosarcoma cells through the Wnt/β-catenin signaling pathway. Calcif. Tissue Int. 2017;100:551–564. doi: 10.1007/s00223-016-0231-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bulk RNA-Seq data have been deposited at GEO and are publicly available as of the date of publication. The accession number is listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper will be shared by the lead contact upon request.