

Summary
Hepatitis C virus (HCV) is a leading cause of liver-associated disease and liver cancer. Of the major HCV subtypes, patients infected with subtype 1b have been associated with having a higher risk of developing chronic infection and hepatocellular carcinoma. However, underlying reasons for this increased disease severity remain unknown. Here, we provide an evolutionary rationale, based on a comparative study of fitness landscape and in-host evolutionary models of the E2 glycoprotein of HCV subtypes 1a and 1b. Our analysis demonstrates that a higher chronicity rate of 1b may be attributed to lower fitness constraints, enabling 1b viruses to more easily escape antibody responses. More generally, our results suggest that differences in evolutionary constraints between HCV subtypes may be an important factor in mediating distinct disease outcomes. Our analysis also identifies antibodies that appear escape-resistant against both subtypes 1a and 1b, providing directions for designing HCV vaccines having cross-subtype protection.
Subject areas: Hepatology, In silico biology, Systems biology, Virology
Graphical abstract
Highlights
-
•
Comparative analysis of the fitness landscapes of HCV subtypes 1a and 1b
-
•
Subtype 1b evolution is subject to less constraints than 1a
-
•
Subtype 1b appears to evade antibodies more easily compared with 1a
-
•
Antibodies are identified that are difficult to escape for both subtypes 1a and 1b
Hepatology; In silico biology; Systems biology; Virology
Introduction
HCV is a highly mutable single-stranded RNA virus (Rosen, 2011). Approximately 15%–25% of infected people clear the virus spontaneously, while the remaining develop chronic liver disease, commonly leading to cirrhosis and hepatocellular carcinoma (HCC) (Centers for Disease Control and Prevention, 2018). HCV is estimated to cause chronic infection in 58 million people worldwide (World Health Organization, 2021). Although therapeutic treatments for chronic HCV infection have improved since the introduction of direct-acting antivirals (DAAs), these drugs are only administered to a limited number of infected individuals because of high cost (Rosenthal and Graham, 2016) and low diagnostic rate (World Health Organization, 2021). The effectiveness of DAAs also has limitations due to the inability to prevent reinfection (Rossi et al., 2018) and the emergence of drug-resistant variants (Wyles and Luetkemeyer, 2017). Thus, for complete eradication of HCV, developing an effective vaccine is essential.
HCV is classified into eight major genotypes and 90 subtypes (ICTV, 2019). Among the major genotypes, genotype 1 is the most prevalent, representing roughly half of all HCV infections worldwide (Petruzziello et al., 2016). Of the genotype 1 infections for which subtypes have been specified, 99% are either subtype 1a or 1b (Messina et al., 2014), with subtype 1b known to be the most prevalent subtype worldwide (Gower et al., 2014). The dominant HCV subtype however varies across geographical regions, with subtype 1a more prevalent in North America, Tropical Latin America, Northwestern Europe, and Australia, and subtype 1b more prevalent in Asia, Western and Eastern Europe, Southern and Central Latin America, and North Africa (Messina et al., 2014). The most prominent difference between these two highly prevalent HCV subtypes is in terms of disease outcome. Multiple studies have shown that the chronicity rate of subtype 1b infections is about two times higher than that of 1a and other subtypes (Amoroso et al., 1998; Cho et al., 2014; Hwang et al., 2001). Moreover, subtype 1b infections have been reported to increase the risk of developing cirrhosis (Osella et al., 2001), and present almost double the risk of developing HCC compared with 1a and other subtypes (Bruno et al., 2007; Lee et al., 2014; Raimondi et al., 2009; Silini et al., 1996).
Despite these observations, it remains unclear as to why subtype 1b infections may lead to more severe disease outcomes. This may be attributed to various reasons, including differences in viral phenotype, host-specific immune factors, demographics, transmission route (injectable drug use in the case of 1a and blood transfusion in the case of 1b (Magiorkinis et al., 2009)), etc. One plausible explanation is that subtype 1b may evolve under less rigid fitness constraints than other subtypes, allowing it to escape immune responses more easily and to more effectively propagate infection. Evidence of a similar phenomenon has been presented for HIV, for which the fitness of the infecting subtype has been reported to be a factor associated with disease outcome (Venner et al., 2016). For HCV, host-specific factors such as patient's ethnicity, gene composition (e.g., IL28B polymorphisms), alcohol consumption, and co-infection with HIV have been shown to contribute to the disparate disease outcome of infections (Chen and Morgan, 2006; Missiha et al., 2008; Yan and Wang, 2017). However, it is not yet known whether different HCV subtypes exhibit markedly different fitness properties, and if so, whether this could be a significant factor in determining disease outcome.
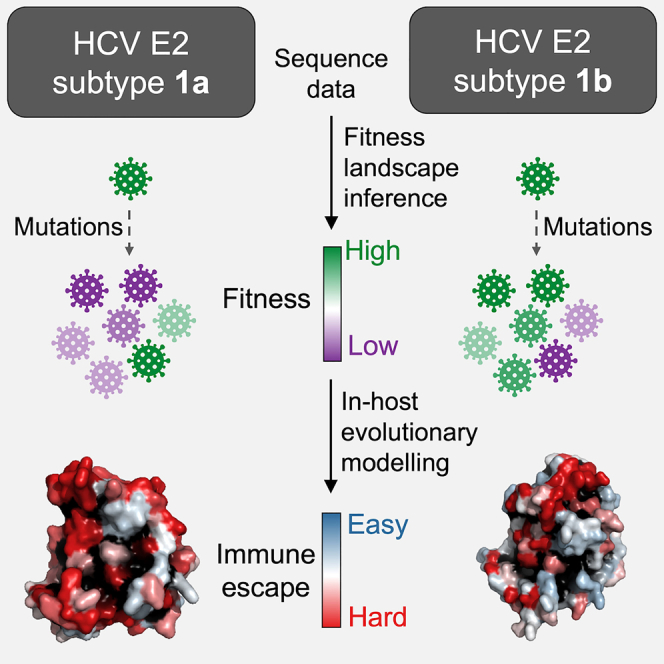
We aim to address these questions by using computational modeling, leveraging sequence data as well as available clinical and experimental data. Our approach is to first infer a model for the fitness landscape of HCV E2 (the primary target of neutralizing antibodies) for subtype 1b, which we validate against in-vitro fitness measurements, and compare it with an analogous model for subtype 1a that we had inferred previously (Quadeer et al., 2019a). Our comparative analysis suggests E2 1b to be more tolerant to mutations compared with E2 1a, indicating that it may be easier for subtype 1b viruses to evade immune responses. This is further corroborated by an analysis of the average time it takes for each subtype to escape from antibody responses, which we quantify by using population-genetics-based models of in-host viral evolution (Quadeer et al., 2019a). Our analysis, in general, points to significant differences in viral evolutionary constraints experienced by HCV subtypes 1a and 1b, which may contribute to observed subtype-specific differences in disease outcomes. We additionally employ the evolutionary models to identify potentially escape-resistant human monoclonal antibodies (HmAbs) against both subtypes 1a and 1b, which may assist in the rational design of a vaccine that is effective against the most prevalent HCV subtypes worldwide.
Results
Inference and validation of the HCV E2 1b fitness landscape
We inferred a computational model for the fitness landscape of E2 1b using the sequence data available at the HCV-GLUE database (Singer et al., 2018, 2019) (see STAR Methods for details). This involved obtaining a maximum entropy (least-biased) probabilistic model—a “prevalence landscape” that captures the probability of observing a virus with a particular E2 protein sequence in circulation. In this model, the probability of any sequence can be expressed as
| (Equation 1) |
where is the set of all fields that represent the effect of mutations at a single residue, is the set of all couplings that represent the effect of interactions between mutations at two different residues, and is a normalization factor. The quantity represents the energy of sequence , which is inversely related to its prevalence. Inferring a maximum entropy model involves choosing the model parameters (fields and couplings) such that the single and double mutant probabilities obtained from the model match with those of the multiple sequence alignment (MSA). Maximum entropy models have been used for inferring the fitness landscape of HCV polymerase protein (Hart and Ferguson, 2015) and of several proteins of HIV (Barton et al., 2016; Ferguson et al., 2013; Flynn et al., 2017; Louie et al., 2018; Mann et al., 2014), and for designing a T cell-based HIV vaccine candidate, shown to be immunogenic in rhesus macaques (Murakowski et al., 2021). The maximum entropy model inference for E2 protein is challenging as it involves estimating a large number of model parameters, a consequence of the high mutational diversity of E2 compared to other HCV proteins (Figure S1). To tackle this problem, we used an efficient computational approach introduced in (Louie et al., 2018) to infer fitness landscapes of the HIV envelope protein. The method was also applied to infer a fitness landscape for the E2 protein of HCV subtype 1a (Quadeer et al., 2019a).
The single and double mutant probabilities obtained from our inferred model for HCV E2 subtype 1b aligned well with those of the MSA (Figures S2A and S2B). Other statistics, such as triple mutant probabilities and the distribution of the number of mutations computed from our inferred model, although not explicitly included while training, also matched well with those of the MSA (Figures S2C and S2D). These results indicate that our inferred prevalence landscape model accurately captures the statistical variations in the observed E2 1b sequence data.
We curated in-vitro infectivity measurements of subtype 1b strains from four experimental studies (Esteban-Riesco et al., 2013; Naik et al., 2017; Pantua et al., 2013; Urbanowicz et al., 2015) (see Data S1 for details). Comparing fitness predictions from the inferred model with these in-vitro infectivity measurements demonstrated that the inferred landscape for E2 1b is a reasonably good representative of the underlying protein fitness landscape (Figure 1A). Specifically, a strong negative Spearman correlation (; see STAR Methods for details) was observed between the model-predicted energies (inversely related to prevalence; see Equation (1)) and measured fitness values. This accuracy is commensurate with fitness predictions reported by studies of other proteins (Ferguson et al., 2013; Flynn et al., 2017; Hart and Ferguson, 2015; Louie et al., 2018; Mann et al., 2014; Quadeer et al., 2019a).
Figure 1.

Validation of the inferred E2 1b fitness landscape
(A) Normalized experimental fitness measurements correlate strongly with the energy computed from the inferred landscape. In contrast, a much lower correlation is observed for the conservation-only model (Quadeer et al., 2019a) that does not take couplings into account (inset). References for fitness (infectivity) measurements are shown in the legend. Normalization of fitness measurements and predicted model energies were performed by subtracting the mean from each dataset and dividing by its standard deviation. In (Esteban-Riesco et al., 2013) and (Naik et al., 2017), E2 fitness measurements were reported in two different E1 backgrounds. In (Esteban-Riesco et al., 2013), one background involved methionine (M) at residue 299, while the other involved valine (V). Similarly, in (Naik et al., 2017), one background involved threonine (T) at residue 292, while the other involved alanine (A).
(B) Precision of contact predictions vs. the top x pairs according to the inferred model couplings. Precision is the proportion of top x pairs that are truly in contact. Two residues were assumed to be in contact if their carbon-alpha atoms were less than 8Å apart according to the available E2 1b crystal structure (PDB ID: 6MEI). See also Figure S3.
In addition to mutations at individual residues, interactions between mutations at different residues have been shown to be important contributors to HCV fitness (Parera and Martinez, 2014; Quadeer et al., 2019a). Thus, we compared the predictions of our model, which takes into account residue interactions via couplings, with those of a model based only on amino acid conservation (or single mutant probabilities (Quadeer et al., 2019a)). Compared to our model, the conservation-only model provided a much lower correlation () between the model-predicted energy and in-vitro infectivity measurements (Figure 1A, inset), corroborating the importance of incorporating residue interactions in determining fitness. This suggests that the fitness effect of individual mutations in HCV also depends on the sequence background in which they are introduced, a phenomenon commonly called “epistasis” in genetics. Moreover, the strong couplings of maximum entropy models are known to be informative of contacts in the protein tertiary structure (Weigt et al., 2008; Dunn et al., 2007; Morcos et al., 2011). The inferred E2 1b model couplings were also found to be good predictors of contacts in the E2 1b protein structure [PDB ID: 6MEI] (Figure 1B; see STAR Methods for details). The contact prediction from the model couplings was comparable with direct coupling analysis (DCA) (Morcos et al., 2011), a standard contact prediction method (Figure S3).
Fitness models indicate subtype 1b to be less evolutionarily constrained than subtype 1a
We compared the fitness landscape model of HCV E2 1b with an analogous model developed previously for E2 1a (Quadeer et al., 2019a) to investigate whether the observed differences in disease outcome of the two subtypes' infections may be related to the associated fitness constraints. We first compared the fitness landscapes based on two standard metrics used for quantifying landscape ruggedness: autocorrelation and neutrality (Kouyos et al., 2012; Quadeer et al., 2020; Vassilev et al., 2003). Autocorrelation quantifies the average change in fitness as one moves randomly along the landscape (see STAR Methods for details). We observed that the decay of autocorrelation of the E2 1b landscape was slower than that of 1a (Figure 2A, left panel), suggesting that the average change in fitness while moving along the landscape of 1b is less than that of 1a. The neutrality associated with each landscape quantifies the maximum number of mutation steps one can take on average without much change in fitness while moving randomly along the landscape (see STAR Methods for details). Our results showed that the E2 1b landscape was more neutral than 1a (Figure 2A, right panel), further suggesting that the average fitness cost upon mutation in 1b is lower than that of 1a. These results were qualitatively robust to the choice of the involved parameters (Figure S4).
Figure 2.

Comparison of the fitness landscapes of E2 1a and 1b
(A) (Left panel) Comparison of the autocorrelation of the sequence energies of each subtype's landscape. The x axis denotes the number of random walk steps (K) for which the autocorrelation was computed. The starting sequences were chosen within a Hamming distance (D) of 5 from the MSA (see STAR Method for details). (Right panel) Comparison of the neutrality of the landscape of each subtype. Neutrality was computed for random walk steps. A random step in the walk was accepted only when the change in fitness from the sequence at the previous step was within a small value, ε (shown on the xaxis) (see STAR Methodfor details).
(B) (Left panel) Statistically significant enrichment of almost all peak sequences in known escape mutations (listed in Table S1), for each subtype. Enrichment is the fraction of known escape mutations within all mutations in a peak sequence, and the p value measures the probability of observing by random chance at least the observed number of escape mutations among all observed mutations in a peak sequence (see STAR Methods for details). The dashed horizontal line is the cut-off for statistically significant results (pvalue ), with all peak sequences above this line being classed as statistically significant. (Middle panel) Number of peaks observed in each landscape. (Right panel) The cumulative fraction of sequences associated with the peaks versus the top x peaks in each landscape, plotted on a log-log scale. See also Figures S4–S7.
We also compared the two landscapes using a low-dimensional representation that characterizes each landscape by its local “peaks”, representing fitness maxima. Such local peaks have been shown to be informative of immune-escape pathways employed by HIV (Barton et al., 2015), and of the evolution of poliovirus under vaccine-induced and natural selective pressures (Quadeer et al., 2020). Peaks were obtained by following a steepest ascent walk from each sequence in the MSA. In the previous HIV study (Barton et al., 2015), sequences representing each peak in the landscape, referred to as “peak sequences”, were found to be strongly enriched in HLA-associated mutations driven by host immune responses. Investigating the peak sequences of each subtype for HCV E2 revealed that almost all of the peak sequences of both subtypes were statistically significantly enriched in known escape mutations from E2-specific HmAbs (Keck et al., 2008, 2009, 2016; Kato et al., 1993; Morin et al., 2012; Bailey et al., 2015; Velázquez-Moctezuma et al., 2019) (Figure 2B left panel; see STAR Methods for details). This suggests that peaks in the fitness landscapes of E2 may be representative of immune-escape pathways employed by HCV for both subtypes. This result was also qualitatively robust to the change of parameter values involved in the landscape inference (Figure S5). We observed that there were far more peaks in the landscape of 1b than 1a (Figure 2B, middle panel), suggesting that more escape pathways may be available for subtype 1b than 1a. Rank-ordering the peaks according to the fraction of MSA sequences represented by each peak revealed that the same number of top peaks in 1b comprised a smaller fraction of sequences than 1a (Figure 2B, right panel). This indicates that the sequences are more spread out across peaks in 1b, which is suggestive of relatively diverse escape pathways utilized by subtype 1b in comparison with 1a. These qualitative results were also robust to the change of parameter values involved in the landscape inference (Figure S6). We additionally confirmed, using a procedure similar to (Barton et al., 2015; Quadeer et al., 2020), that the peaks observed in each landscape are not an artifact of finite sampling, but arise owing to the interplay between mutations at different residues in each subtype (Quadeer et al., 2019a). Generally, the comparative analysis of the 1a and 1b fitness landscapes (Figure 2) was robust to finite sampling, returning qualitatively similar results even when fewer sequences were used for inferring the landscapes (Figure S7).
Overall, this comparison of fitness landscapes demonstrates that intrinsic fitness constraints governing the evolution of HCV subtypes may differ significantly. Our results are suggestive of subtype 1b to be under less fitness constraints compared with 1a, potentially making it easier for 1b to escape from antibody-mediated immune pressure. Such a difference between the two subtypes is not apparent by comparing simple sequence level statistics such as residue-wise entropies (Figure S8).
Evolutionary modeling and escape time prediction for subtype 1b
To further investigate whether the ease of escaping immune responses is subtype-dependent, we quantified the time it takes for E2 1b to escape immune responses using an in-host viral evolutionary model similar to the one we had employed previously for E2 1a (Quadeer et al., 2019a). This evolutionary model takes into account stochastic dynamics during in-host viral evolution, such as host-viral interactions, competition within the viral population, and multiple pathways that may be employed by the virus to escape from immune pressure. This was accomplished by incorporating the inferred E2 1b fitness landscape into a Wright–Fisher-like population genetics model (Ewens, 2004). The parameters involved in this model, such as mutation rate and effective size of the viral population, were set according to known values for HCV (Bull et al., 2011; Cuevas et al., 2009; Sanjuan et al., 2010), while the sequence survival probability in the population from one generation to the next was determined by the inferred E2 1b fitness landscape (see STAR Methods for details).
For each E2 1b residue, we used the evolutionary model to predict the time for an escape mutation to reach a majority in the population. Specifically, for calculating the escape time associated with a particular residue, we started the evolutionary simulation by first initializing the population with duplicates of a sequence randomly picked from the MSA having the consensus amino acid at the selected residue. Immune pressure was modeled as a fixed reduction in fitness of sequences having the consensus amino acid at the residue, thereby allowing a selective advantage to sequences that incur a mutation at that residue. We continued the simulation until the sequences having a mutation at the selected residue reached a majority in the population. We considered this as a representative marker of viral escape from the corresponding immune pressure. The number of generations for escape was recorded, and this procedure was repeated multiple times with the same initial sequence, as well as with multiple distinct initial sequences. The mean number of generations over all these simulation runs, termed “escape time”, was computed and recorded for every E2 1b residue (see STAR Methods for details).
We validated the predicted escape times for E2 1b residues using experimental and clinical data. First, we assessed the ability of our model to predict known escape mutations from multiple E2-specific HmAbs (Keck et al., 2008, 2009, 2016; Kato et al., 1993; Morin et al., 2012; Bailey et al., 2015) (listed in Table S1). Such mutations would be expected to be associated with lower escape times compared to mutations at other residues, enabling the virus to escape the associated antibody pressure. Our results demonstrated that this was indeed the case (, Mann-Whitney test, Figure 3A). We also validated the escape time metric by predicting the escape times associated with mutating the buried and exposed (surface) residues in E2 1b, respectively. Buried residues that form the protein core are more likely to be crucial for stability (Chen and Zhou, 2005), implying that mutations at these residues would be expected to be associated with higher escape times than mutations at the exposed ones. Residues of the available crystal structure of E2 1b (PDB ID: 6MEI) (Flyak et al., 2018) were classified as exposed or buried according to the standard relative solvent accessibility (RSA) metric (see STAR Methods for details). Comparing the escape times associated with these two sets of residues revealed that the mutations at buried residues were predicted to have higher escape times (, Mann-Whitney test; Figure 3B), as expected. Overall, these tests provide confidence in the capability of the evolutionary model to distinguish E2 1b residues associated with low and high escape times.
Figure 3.

Validation of E2 1b escape times predicted using the in-host evolutionary model
(A) Comparison of escape times associated with the residues at which mutation is known to assist in escape from HmAbs (listed in Table S1) and those of the remaining E2 residues.
(B) Comparison of escape times associated with the mutations at exposed and buried residues. Residues of the available crystal structure of E2 1b (PDB ID: 6MEI) (Flyak et al., 2018) were classified as exposed or buried according to the standard RSA metric (see STAR methods for details). For each box plot, the middle line indicates the median, the edges of the box represent the first and third quartiles, and whiskers extend to span a 1.5 interquartile range from the edges. All reported P values were calculated using the one-sided Mann-Whitney test.
Escape time comparison indicates subtype 1b may evade HmAbs more easily than subtype 1a
We compared the predicted escape times of the exposed residues of E2 1b (which are more likely to be targeted by HmAbs) with those previously predicted for E2 1a (Quadeer et al., 2019a). All relevant model parameters were matched to perform a fair comparison between the two subtypes (see STAR Methods for details). We found that the escape times of exposed residues of E2 1b were generally lower than those of 1a (, Mann-Whitney test; Figure 4A), suggesting that it may be easier for subtype 1b to escape from antibody responses than 1a. This was also true if all residues of both subtypes were considered (, Mann-Whitney test; Figure S9). To visualize the escape times associated with exposed residues, we superimposed them as a heatmap on the resolved crystal structure of E2 for each subtype (Figure 4B). Here, as in Figure 4A, the low concentration of residues with high escape time (colored in red) in subtype 1b compared with 1a is also evident. Moreover, we observed that few common exposed residues were associated with very high escape times for both subtypes (Figure S10). Interestingly, an epitope 412–425 comprising such residues (423 and 425) was shown in a recent study to be capable of inducing potent neutralizing antibodies against multiple HCV genotypes including 1a and 1b (Czarnota et al., 2020). Hence, targeting these residues may assist in eliciting an effective immune response against both subtypes.
Figure 4.

Escape time comparison of E2 subtype 1a and 1b
(A) Comparison of escape times associated with exposed residues of E2 of each subtype.
(B) Escape times of exposed residues superimposed on the crystal structure of E2 1a (left panel, PDB ID: 4MWF (Kong et al., 2013)) and E2 1b (right panel, PDB ID: 6MEI (Flyak et al., 2018)). Buried residues are shown in black.
(C and D) Comparison of the minimum escape time associated with mutating the binding residues of known E2-specific HmAbs for each subtype (C) taken together and (D) individually. Binding residues of an antibody were determined by global alanine scanning mutagenesis (Pierce et al., 2016; Gopal et al., 2017; Keck et al., 2019; Bailey et al., 2017), where each residue of the wild-type sequence was substituted by alanine (or glycine/serine if the residue in the wild-type was alanine). For the majority of antibodies (Pierce et al., 2016; Keck et al., 2019; Gopal et al., 2017), the fraction of the mutant sequence binding with respect to the wild-type sequence, called relative binding (RB), was reported for each residue, and we defined binding residues of any one of these antibodies as residues with RB less than or equal to 20%. For some specific antibodies (HEPC98, HEPC50, HEPC46, HEPC74, HEPC43, and HEPC3), critical binding residues as reported in (Bailey et al., 2017) were used. The dashed line for each subtype denotes the optimal cut-off value ζ (see STAR Methods for details). HmAbs predicted to be escape-resistant for any subtype are colored in red, while the remaining ones are colored in blue. The HmAbs predicted to be escape-resistant for both subtypes are shown in bold. In each box plot in (A and C), the middle line indicates the median, the edges of the box represent the first and third quartiles, and whiskers extend to span a 1.5 interquartile range from the edges. All reported P values were calculated using the one-sided Mann-Whitney test.
See also Figures S9–S11.
As the majority of the neutralizing antibodies block HCV entry (Harman et al., 2015), we also investigated the escape times of residues of each subtype that are involved in binding to CD81, the main HCV receptor (Ströh et al., 2018). We found that the escape times of these residues for subtype 1b were lower than those of 1a, suggesting that the entry of subtype 1b is more flexible and thereby making it potentially easier for 1b to escape CD81-targeting antibodies without significantly compromising viral entry (Figure S11).
We further employed our model to compare the two subtypes based on the escape resistance of known HmAbs. Specifically, we focused on HmAbs with binding residues determined using global alanine scanning experiments (Pierce et al., 2016; Keck et al., 2019; Gopal et al., 2017; Bailey et al., 2017). We compared the minimum escape time (, see STAR Method) predicted by each model for the binding residues of 35 antibodies. Similar to Figure 4A, this analysis also showed that the minimum escape times predicted for each antibody for subtype 1b were statistically significantly lower than those predicted for 1a (, Mann-Whitney test; Figure 4C).
In addition, we considered a linear classifier (Quadeer et al., 2019a) to compute an optimal escape time cut-off value ζ for each subtype to determine whether a residue is relatively escape-resistant or not. This binary classifier was designed by taking the residues with known escape mutations (listed in Table S1) as true positives and all remaining residues as true negatives (see STAR Methods for details). We then assessed each antibody by comparing the minimum escape time predicted by each model with the corresponding optimal cut-off value ζ. Based on this analysis, we observed that fewer HmAbs were predicted to be difficult to escape for 1b than 1a (colored in red in Figure 4D). All the HmAbs predicted to be difficult to escape for 1b were also predicted to be the same for 1a, except for HmAb 212.10. However, the opposite was not true, further suggesting the potentially greater ability for subtype 1b to escape from HmAbs. Nonetheless, we identified eight HmAbs, HC33-1, CBH-7, CBH-23, HC-1, HEPC50, 212.25, 212.15, and HCV1, predicted to be difficult to escape for both subtypes (shown in bold). This result is consistent with information available for these HmAbs in the literature. Specifically, multiple studies have reported HC33-1 as a potentially escape-resistant antibody for multiple genotypes (Keck et al., 2014; Pierce et al., 2016) and HCV1 as a potent cross-reactive antibody (Kong et al., 2012; Broering et al., 2009). Other studies have isolated HmAbs HEPC50, 212.25, and 212.15 from patients who have spontaneously cleared HCV (subtypes 1a, 1b, or 3a), and these antibodies have also been shown to be cross-neutralizing (Keck et al., 2019; Bailey et al., 2017). Additionally, among the HmAbs that we predicted to be relatively easy to escape for both subtypes, HmAbs CBH-4D, CBH-4G, CBH-4B, CBH-20, CBH-21, CBH-22, AR1A, AR1B, and AR2A have been reported to be non-neutralizing or isolate-specific antibodies in previous studies (Keck et al., 2005; Kong et al., 2016; Law et al., 2008). As for HmAbs AR3A, AR3B, AR3C, and AR3D, there are contradicting results in the literature. Certain studies have reported them as potent broadly neutralizing antibodies (Law et al., 2008; Merat et al., 2019), whereas other studies have found escape mutations against these antibodies (Bailey et al., 2015; Velázquez-Moctezuma et al., 2019). Our results are consistent with the latter set of studies, as both 1a and 1b models predicted these antibodies to be relatively easy to escape.
Discussion
Subtypes 1a and 1b are the two prevalent subtypes of HCV, together representing roughly half of all HCV infections worldwide. Clinical outcomes of subtype 1b infections have been shown to be considerably more severe than those of 1a, showing higher chronicity rate and higher risk of developing cirrhosis and HCC. The underlying reasons for these differences are not well understood. We reasoned that this may be due to subtype 1b being under relatively less fitness constraints than 1a, potentially enabling it to evade immune pressure more easily than 1a. We developed computational models to validate this hypothesis. First, by inferring a fitness landscape of HCV E2 1b and comparing it with an analogous model for 1a obtained previously, our analysis suggested that HCV 1b is subject to less fitness constraints. This result, taken more generally, showed that different subtypes of HCV may possess markedly distinct evolutionary properties. We incorporated the inferred fitness landscape of E2 1b into a population-genetics evolutionary model to predict the time it takes HCV to escape immune responses targeting any specific residue of E2 1b, and compared with the escape times predicted for E2 1a in our previous study (Quadeer et al., 2019a). The escape times of E2 1b were generally lower than those of 1a, suggesting that it may be easier for 1b to escape antibody responses.
The significance of incorporating interactions between mutations at different residues has been demonstrated in multiple previous studies for inferring protein fitness landscapes (Sohail et al., 2021; Hart and Ferguson, 2015; Ferguson et al., 2013; Barton et al., 2016; Flynn et al., 2017; Louie et al., 2018; Quadeer et al., 2019a, 2020) and for determining networks of residues important for mediating protein structure and function (Dahirel et al., 2011; Quadeer et al., 2014, 2018; Ahmed et al., 2019; Gaiha et al., 2019). However, residue–residue interactions, represented in the maximum entropy fitness landscape model by the coupling parameters (Equation 3 in STAR Methods), seem to be especially important for HCV E2 1b. This is evident from the significantly higher correspondence between our model predictions and in-vitro fitness (infectivity) measurements (), compared with that obtained with a model without couplings () (Figure 1A). This difference is more apparent than observed previously for models of HIV proteins (Ferguson et al., 2013; Mann et al., 2014; Flynn et al., 2017; Louie et al., 2018) or other HCV proteins (Hart and Ferguson, 2015) (as well as E2 subtype 1a (Quadeer et al., 2019a)), suggesting that collective mutational effects, including compensatory mutations, may play a particularly dominant role in shaping the evolution of E2 1b and in providing coordinated evolutionary pathways to facilitate immune escape. Importantly, the incorporation of residue–residue interactions exposed evolutionary differences between the HCV subtypes 1a and 1b (Figure 2) that were not evident with site-independent models (the single-site entropy of both subtypes appeared noticeably similar; Figure S8). Hence, the incorporation of collective mutational effects appears essential to elucidate the distinct evolutionary properties of the different HCV subtypes, which may in turn help to explain the different associated chronicity rates.
The differences in the fitness constraints of the two HCV subtypes that we report here do not seem to stem from differences in the quality of the respective model fit. This is evident from the similar statistical accuracy of the inferred model for each subtype (1a: Figure 1 in (Quadeer et al., 2019a) and 1b: Figure S2) and from nearly the same correspondence between model predictions and experimental fitness measurements (1a: −0.72 (Quadeer et al., 2019a) and 1b: −0.71 (Figure 1)). Additionally, the qualitative results related to the comparison of the two landscapes appeared robust to changes in the number of sequences as well as the specific parameter values used in inferring the landscapes (Figures S5–57).
Our results suggest that distinct fitness constraints of HCV subtype 1a and 1b may contribute to the observed differences in disease outcomes of these subtypes. However, other factors may also contribute to disparate disease outcomes, including host-specific factors such as patient's ethnicity, alcohol consumption, and co-infection with HIV (Chen and Morgan, 2006; Missiha et al., 2008; Yan and Wang, 2017). Immunogenicity of the viral subtypes may also play a role because if HCV subtype 1b was found to be less immunogenic than 1a, it could contribute to higher rates of chronic disease. However, there is little evidence to support this, with experimental studies in fact suggesting that the two subtypes have similar immunogenicity (Rodríguez-López et al., 1999). This was demonstrated by stimulating the sera from HCV-infected patients with peptides selected from regions of the HCV proteome having high inter-subtype variability. Similar immunogenicity of both subtypes is also consistent with experimentally identified HCV E2-specific B cell epitope data available from the Immune Epitope Database (IEDB; https://www.iedb.org) (Vita et al., 2018), which shows that the coverage of epitopes along the E2 primary structure is similar for both 1a and 1b (Figure S12). Based on current information, it appears unlikely that differences in immunogenicity is a key factor responsible for the disparate disease outcomes of HCV subtypes 1a and 1b.
While to our knowledge possible association between viral fitness constraints and disease outcome of HCV subtypes has not been elucidated previously, related results have been reported for HIV. Like HCV, HIV subtypes have also been shown to associate with different disease outcomes, defined based on the rate of CD4+ T cell decline and speed of progression to AIDS (Easterbrook et al., 2010; Amornkul et al., 2013; Ssemwanga et al., 2013; Kiwanuka et al., 2013; Venner et al., 2016). Clinical studies on HIV have pointed to replicative fitness of the infecting subtype as an important factor associated with disease outcome (Venner et al., 2016; Claiborne et al., 2015). For instance, in (Venner et al., 2016), by following the decay of CD4+ T cell counts in untreated patients infected with HIV subtype A, C, or D, patients infected with subtype D were reported to be associated with faster disease progression. Further, by performing dual HIV competition assays of strains isolated from patients infected with each subtype against the reference HIV-1 subtype B isolates, it was shown that the relative replicative fitness of subtype D strains was higher than that of the other two subtypes. Overall, these studies are supportive of our findings for HCV by providing evidence that associations between the viral subtype and disease progression may be explained, at least in part, by differences in subtype fitness constraints. Our study motivates targetedHCV experimental studies for further investigating the effects of subtype fitness constraints on disease outcome. These may include longitudinal studies of patients infected with HCV subtypes 1a and 1b, which compare the relative fitness (or infectivity) of the infecting strains as well as their association with chronicity or clearance.
Our escape time analysis of residues known to be important for CD81 binding showed that viral entry of subtype 1b may be more flexible than that of subtype 1a (Figure S11). That is, subtype 1b may be more capable of mutating CD81 binding residues while remaining fit. This would make it easier for subtype 1b to escape CD81-targeting antibodies, which form the majority of neutralizing antibodies (Harman et al., 2015). Thus, the potential ability of 1b to escape neutralizing antibodies while maintaining binding to CD81 may lead to more chronic infections compared with 1a. This predicted subtype-specific disparity in viral entry can be confirmed via targeted experiments such as performing site-directed mutagenesis of CD81 binding residues for subtypes 1a and 1b and measuring the difference in viral entry using CD81 binding assays.
The escape times predicted by our in-host evolutionary models identified five E2-specific HmAbs (HEPC3, HEPC74, HEPC46, HC84-20, and 212.10) that appear to be relatively easy to escape for exclusively one subtype. Investigating the escape times associated with the binding residues of these HmAbs for both subtypes revealed that this distinction was due to a single residue in the footprint of each HmAb, for which mutational escape for one subtype was predicted to be relatively easy, but not for the other subtype (Figure S13). Specifically, residue 437 for HEPC3 and HEPC74, residue 546 for HEPC46, residue 608 for HC84-20, and residue 442 for 212.10 appear to be relatively easy to escape for one subtype only. Of these, residues 437, 546, and 608 are orthologous in E2 subtypes 1a and 1b. This suggests that the distinctive association of mutations at these residues with immune escape in each subtype may be due to the different amino acids preferred at these positions along with their corresponding mutational interactions with other residues in the protein. Thus, our study provides motivation for further experiments to systematically explore whether the difference in the ability to escape neutralizing antibodies is HCV genotype/subtype-specific. For instance, the sequences representing the top peaks in the fitness landscape of each subtype—which are locally most fit and enriched in antibody escape mutations (Figure 2B)—can serve as a good representative panel of HCV E2 sequences to test the neutralization patterns of antibodies.
We also identified eight HmAbs (HC33-1, CBH-7, CBH-23, HC-1, HEPC50, 212.25, 212.15, and HCV1) that appear relatively difficult to escape for both subtypes, and also identified specific exposed residues in E2 that are associated with high escape times for both subtypes (Figure S10). Incorporating the epitopes targeted by the identified escape-resistant HmAbs into rationally designed vaccines (Bailey et al., 2018; Sohail et al., 2020) may aid in eliciting an effective immune response against both HCV subtypes 1a and 1b, and eventually help to curb their global spread. Targeting the exposed yet difficult-to-escape E2 residues, e.g., by synthetic antibodies (Muyldermans, 2013; Sormanni et al., 2015; Zimmermann et al., 2018), may also present an effective therapeutic strategy. Our study motivates further investigation of the identified escape-resistant antibodies and difficult-to-escape exposed E2 residues in the context of HCV vaccines and therapies via targeted experiments.
Limitations of the study
There are multiple limitations of our study. We used the available HCV subtype 1b sequence data and in vitro fitness measurements for the inference and validation of our fitness landscape model, respectively (Figure 1). This available data for subtype 1b is limited compared with that of 1a. Thus, further sequencing efforts and fitness studies specific to HCV subtype 1b would assist in improving its computational modeling. Moreover, while our study suggests that subtype 1b is evolving under lower fitness constraints compared with 1a, it is difficult to resolve specific underlying mechanisms that lead to the observed disparity between the outcomes of the two subtypes. This is because fitness is a broad phenomenon which can be attributed to multiple factors, including the ability of the virus to infect host cells, bind to host receptor, transmit to a new host, etc. Our analysis suggests that compared with 1a, subtype 1b is more flexible in binding to the host receptor (Figure S11). However, the two subtypes may also differ in other mechanisms. Further, targeted experimental studies are needed to investigate this aspect.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Software and algorithms | ||
| MPF-BML standalone package | Louie et al. (2018); Quadeer et al. (2020) | https://github.com/ahmedaq/MPF-BML-GUI |
| DCA | Morcos et al. (2011) | http://dca.rice.edu/portal/dca/home |
| PyMOL | Schrödinger, Inc. | https://www.pymol.org |
| Other | ||
| Fitness landscape and the mean escape time predicted by the in-host evolutionary model for each residue of E2 subtype 1a | Quadeer et al. (2019a) | https://github.com/ahmedaq/HCV-E2 |
| The E2 subtype 1b infectivity measurements, used for validating the fitness landscape model | This manuscript | Data S1 |
| Accession numbers of E2 subtypes 1b sequences used for inferring the model | This manuscript | Data S2 |
| The mean escape time predicted by the in-host evolutionary model for each residue of E2 subtype 1b | This manuscript | Data S3 |
| Data and scripts for reproducing the results reported in the manuscript | This manuscript | https://github.com/hangzhangust/HCVE21a1b |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Matthew R. McKay (matthew.mckay@unimelb.edu.au).
Materials availability
This study did not generate new unique reagents.
Method details
Data preprocessing
We downloaded 1,559 aligned HCV E2 subtype 1b amino acid sequences (genome coverage 99%) from the HCV-GLUE database (http://hcv.glue.cvr.ac.uk) (Singer et al., 2018, 2019). To exclude any outlying sequences, we constructed a pairwise similarity matrix (1559 1559) of the sequences (Strimmer and Haeseler, 2009), with each th entry representing the fraction of residues at which the sequence i and sequence j are identical. By investigating the first two principal components (PCs) of this matrix, we excluded 260 outlying sequences which appeared at more than 3 scaled median absolute deviations (Leys et al., 2013) away from the median of either the first or second PC. We also excluded 16 sequences from chimpanzees and 162 sequences having no patient information. This filtering procedure resulted in a total of sequences from patients. In addition, to control residue quality, we excluded 43 residues that were fully conserved. Thus, the final processed MSA comprised sequences and residues.
Inference of HCV E2 1b fitness landscape
We inferred a maximum entropy model, i.e., the “prevalence landscape”, for E2 1b to serve as a representative of its underlying fitness landscape. The maximum entropy model is a least-biased model that can reproduce the single and double mutant probabilities of the MSA, defined as
| (Equation 2) |
Here, is the ith residue of the kth sequence from the MSA which takes on a value from either the consensus amino acid () or the mth most frequently observed mutant () for , where denotes the number of mutants at residue i. δ is the Kronecker delta function, and is one divided by the number of sequences contributed by the patient from which sequence k was obtained. For a given sequence , the maximum entropy model assigns the probability
| (Equation 3) |
where is the set of all fields that represent the effect of mutations at a single residue, and is the set of all couplings that represent the effect of interactions between mutations at two different residues. is a normalization factor, and represents the energy of sequence . The fields and couplings are chosen such that the single and double mutant probabilities obtained from the model match the single and double mutant probabilities of the MSA, i.e.,
| (Equation 4) |
The problem of inferring the model parameters can be cast as the following convex optimization problem
| (Equation 5) |
where denotes the Kullback-Leibler divergence between probability distributions, and is the patient-weighted probability of observing strain in the MSA.
As E2 1b is a long protein and has a high mean residue entropy, the total number of parameters to estimate is very large (Figure S1). To solve this problem, we considered the inference framework, MPF-BML, introduced in (Louie et al., 2018). Specifically, we inferred the model parameters using the GUI-based software implementation of MPF-BML (Quadeer et al., 2019b). The parameters that we used for model inference are as follows: (i) The sample weights were set according to in Equation (2); (ii) both and regularization parameters were set to 30 for couplings and for fields, respectively; (iii) the termination condition was set to and ; and (iv) all other parameters were set to their default values. The statistics of the E2 1b model inferred using these parameters aligned well with those obtained from the MSA (Figure S2).
Fitness verification
We used in-vitro experimental fitness measurements compiled from the literature (Esteban-Riesco et al., 2013; Urbanowicz et al., 2015; Naik et al., 2017; Pantua et al., 2013) to validate that our inferred prevalence landscape is a good proxy of the E2 1b fitness landscape. The detailed selection procedure of the specific fitness measurements (listed in Data S1) from each study is presented below.
In (Esteban-Riesco et al., 2013), the authors compared in Figure 3A infectivity (in RLU) of subtype 1b HCVpps, subtype 1a reference strain, positive and negative controls. We selected all four measurements for subtype 1b and divided them into two groups, with the same E1 background for each group. In (Urbanowicz et al., 2015), the authors compared in Figure 2 infectivity (in RLU) of subtype 1b HCVpps. We selected all eight measurements and divided them into two groups, with the same E1 background for each group. In (Naik et al., 2017), the authors compared in Figure 1B infectivity (in RLU) of HCVpps from multiple genotypes. We selected four subtype 1b measurements, the only ones that have the same E1 background. In (Pantua et al., 2013), the authors compared in Figure 1F infectivity (in RLU) of HCVpps from multiple genotypes. We selected all five subtype 1b measurements.
For each study, normalization of fitness measurements and predicted model energies was performed by subtracting the mean from the dataset and dividing by its standard deviation. To further normalize for potential experimental biases, we considered the weighted average of Spearman correlation coefficients from different experiments. This is given by
where is the number of measurements and the Spearman correlation coefficient for experiment i, and is the total number of experiments.
Residue-residue contact prediction
To investigate if the inferred maximum entropy model couplings can predict residue-residue tertiary contacts in the E2 1b protein, we considered the Frobenius norm of the corresponding coupling matrix between residue i and j, a common measure of the strength of residue-residue connections (Weigt et al., 2008; Ekeberg et al., 2013). We then applied an average product correction (APC) to suppress phylogenetic bias and finite sampling effects (Dunn et al., 2007; Ekeberg et al., 2013). These are given by:
| (Equation 6) |
where
and
As residues in contact are likely to be strongly coupled, we expected the quantity calculated in Equation (6) between these residue pairs to be large. The true contacts were determined from the available E2 1b crystal structure (PDB ID: 6MEI) (Flyak et al., 2018), where two residues were assumed to be in contact if their carbon-alpha atoms were less than 8Å apart.
Metrics for comparing the ruggedness of fitness landscapes
We adopted two metrics to compare the fitness landscape of E2 1b with E2 1a (Kouyos et al., 2012).
Autocorrelation
Autocorrelation is used to quantify the average change in fitness as one moves randomly along the landscape. Starting with random sequences chosen within a Hamming distance from the MSA, we performed a 50-step random walk along the landscape starting from each sequence. The autocorrelation of the sequence energies at the kth step was calculated as
where is the sequence at the kth step, and is the average over sequences.
Neutrality
Neutrality quantifies the maximum number of steps (measured in terms of mutations) that one can take in a landscape without much change in fitness, i.e., the resulting difference in fitness remains within a small value ϵ. We started with random sequences chosen within a Hamming distance from the MSA. For each starting sequence , we performed an random walk along the landscape. At each step k, we accepted the new sequence generated by the random walk if the difference in fitness between the new sequence and the current sequence was less than ϵ. Otherwise, we kept the current sequence, i.e., . We calculated the Hamming distance between the sequence and the starting sequence . The neutrality was then calculated as the average over the maximum Hamming distances obtained from all random sequences for a particular ϵ, i.e.,
Comparing the fitness landscapes based on the local peaks
As the local fitness maxima of the landscape, known as local “peaks”, have been shown to be representative of immune escape pathways of HIV (Barton et al., 2015), we studied the peak structure of the inferred landscapes of subtype 1a and 1b. For each sequence present in the MSA, we obtained the corresponding local peak using the following procedure: For a given sequence of the MSA, we compared the energies of all its neighboring sequences (defined as one Hamming distance away from the sequence), and then chose the most fit sequence (i.e., the sequence with the lowest energy). We repeated the above procedure until the “peak sequence”, i.e., the sequence which has higher fitness than all of its neighbors, was reached. The obtained unique number of local maxima represented the number of local peaks in the specific landscape.
We calculated the statistical significance of enrichment of known escape mutations (listed in Table S1) in the peak sequences using a P value. The enrichment of a peak sequence is defined as the fraction of escape mutations among all mutations defining that peak sequence. The P value corresponds to the probability of observing at least i mutations out of j escape mutations in a peak sequence, where there are n total mutations defining the peak sequence out of 363 total residues of the E2 protein. Mathematically, this can be written as
We tested the null hypothesis that these i escape mutations were observed in a peak sequence by a random chance, and it was rejected if .
In-host evolutionary model
We considered a population genetics viral evolutionary model similar to (Quadeer et al., 2019a) (which drew upon an earlier work (Barton et al., 2016)) for quantifying the ease of escape from antibody responses for each residue in E2 1b. This was accomplished using the “escape time” metric, which represents the number of generations required for mutations at a residue under immune pressure to reach a frequency in a fixed-sized virus population.
Specifically, we employed a well-known population genetics Wright-Fisher model (Ewens, 2004). In this model, sequences in the population undergo mutation, selection, and random sampling steps in each generation. The virus population size was fixed at , in line with the known HCV effective population size in in-host evolution (Bull et al., 2011). For a given E2 1b residue i, we started the simulation with a homogeneous population comprising copies of a sequence randomly selected from the MSA sequences having the consensus amino acid at residue i. In the mutation step, each nucleotide in the sequences was randomly mutated to another nucleotide with a fixed probability , in line with the HCV mutation rate reported in (Cuevas et al., 2009; Sanjuan et al., 2010). In the selection step, the survival probability of each sequence in the population was calculated based on its fitness predicted from the inferred landscape (see Quadeer et al., 2019a for details). To model the immune pressure at residue i, the fitness of all sequences having the consensus amino acid at residue i was decreased by a fixed value b, thereby providing a selective advantage to the sequences having a mutation at this residue. Similar to (Quadeer et al., 2019a), b was set according to the largest value of the field parameter in the inferred landscape. In the sampling step, the new generation of the population was generated through a standard multinomial sampling process parameterized by the survival probabilities calculated in the previous step and . These three steps (mutation, selection and random sampling) were repeated until the number of sequences having a mutation at residue i reached a majority in the population and the number of generations was recorded. This number was considered a representative of the time (generations) taken by the virus to escape the immune pressure at residue i. We repeated this procedure multiple times using the same initial sequence, as well as multiple distinct initial sequences. The escape time associated with residue i was calculated by averaging all number of generations over all these simulation runs.
For a fair comparison of the predicted escape times of E2 1b with those of E2 1a, we used the same parameter values in running the evolutionary simulations for both subtypes. Specifically, we used the same value for the parameter involved in mapping the predicted fitness of a sequence to its survival probability in the population for both 1a and 1b subtypes (see Quadeer et al., 2019a). Moreover, simulations were run for both subtypes for the same number of generations (500), the same number of distinct transmitted/founder (T/F) sequences (25) for each residue, and the same number of simulation runs (100) for each T/F sequence.
Relative solvent accessibility
To determine whether a residue in the crystal structure of each subtype is exposed or buried, we used the get_area( ) function in the PyMOL software (www.pymol.org) with a 1.4 solvent radius parameter to assign each residue in the crystal structure of a subtype (subtype 1a: PDB ID: 4MWF (Kong et al., 2013); subtype 1b: PDB ID: 6MEI (Flyak et al., 2018)) with a solvent accessible surface area (SASA). We then obtained the RSA values of each residue by normalizing the respective SASA values per residue in a Gly-X-Gly tripeptide construct (Miller et al., 1987). As suggested in (Jardine et al., 2016), residues with RSA 0.2 were considered as exposed, while the remaining residues were considered buried.
Estimating escape resistance of HmAbs
To compare the escape resistance of known E2-specific HmAbs against each subtype (Figures 4C and 4D), we computed the minimum escape time associated with their binding residues (determined using global alanine scanning experiments (Pierce et al., 2016; Keck et al., 2019; Gopal et al., 2017; Bailey et al., 2017)). The minimum escape time associated with an antibody was defined as
where i is selected from the set of binding residues of that antibody. To distinguish whether an E2 residue is relatively escape-resistant or not based on its predicted escape time, we designed a binary classifier using the information of known E2-specific escape mutations (listed in Table S1). Specifically, we considered a classifier that takes the residues with known escape mutations as true positives and all remaining residues as true negatives. The classifier for subtype 1b achieved an area under the receiver operating characteristic curve (AUC) of 0.88 (Figure S14A). We chose the optimal cut-off value (Figure S14B) based on the maximum F1 score and the maximum Matthews correlation coefficient (MCC)—two commonly used metrics to evaluate the performance of a binary classifier with different thresholds. For subtype 1a, ζ was determined to be 100 using a similar statistical analysis in (Quadeer et al., 2019a). The HmAbs were classified as relatively escape-resistant for a subtype if their corresponding was greater than ζ for that subtype, and vice versa.
Quantification and statistical analysis
Statistical analyses were performed using MATLAB (R2019b). Fisher's exact test was used to compute the statistical significance associated with enrichment of escape mutations among peaks in fitness landscapes of subtypes 1a and 1b. All other P values in each figure were calculated by performing one-sided Mann-Whitney tests. A P value less than 0.05 was considered as statistically significant.
Acknowledgments
The authors were supported by the General Research Fund of the Hong Kong Research Grants Council (RGC) [Grant No. 16204519].
Author contributions
A.A.Q. and M.R.M. conceptualized the idea and designed the research. H.Z. performed the research and generated the figures and tables. All authors analyzed the data and wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: January 21, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103569.
Contributor Information
Ahmed A. Quadeer, Email: eeaaquadeer@ust.hk.
Matthew R. McKay, Email: matthew.mckay@unimelb.edu.au.
Supplemental information
Data and code availability
-
•
All data used in this work has been provided in the supplemental information and is publicly available as of the date of publication. The E2 1b infectivity measurements, used for validating the fitness landscape model, are included in Data S1. Accession numbers of E2 1b sequences used for inferring the model are listed in Data S2. The mean escape time predicted by the in-host evolutionary model for each residue of E2 1b is provided in Data S3.
-
•
Data and scripts for reproducing the results are available at GitHub: https://github.com/hangzhangust/HCVE21a1b_Hang. The GUI-based software implementation of the MPF-BML method (Louie et al., 2018), used for inferring the fitness landscape parameters, is available at GitHub: https://github.com/ahmedaq/MPF-BML-GUI (Quadeer et al., 2019b). Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Ahmed S.F., Quadeer A.A., Morales-Jimenez D., McKay M.R. Sub-dominant principal components inform new vaccine targets for HIV Gag. Bioinformatics. 2019;35:3884–3889. doi: 10.1093/bioinformatics/btz524. [DOI] [PubMed] [Google Scholar]
- Amornkul P.N., Karita E., Kamali A., Rida W.N., Sanders E.J., Lakhi S., Price M.A., Kilembe W., Cormier E., Anzala O., et al. Disease progression by infecting HIV-1 subtype in a seroconverter cohort in sub-saharan africa. AIDS. 2013;27:2775–2786. doi: 10.1097/QAD.0000000000000012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amoroso P., Rapicetta M., Tosti M.E., Mele A., Spada E., Buonocore S., Lettieri G., Pierri P., Chionne P., Ciccaglione A.R., et al. Correlation between virus genotype and chronicity rate in acute hepatitis C. Hepatology. 1998;28:939–944. doi: 10.1016/s0168-8278(98)80340-1. [DOI] [PubMed] [Google Scholar]
- Bailey J.R., Barnes E., Cox A.L. Approaches, progress, and challenges to hepatitis C vaccine development. Gastroenterology. 2018;156:418–430. doi: 10.1053/j.gastro.2018.08.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey J.R., Flyak A.I., Cohen V.J., Li H., Wasilewski L.N., Snider A.E., Wang S., Learn G.H., Kose N., Loerinc L., et al. Broadly neutralizing antibodies with few somatic mutations and hepatitis C virus clearance. JCI Insight. 2017;2:e92872. doi: 10.1172/jci.insight.92872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey J.R., Wasilewski L.N., Snider A.E., El-Diwany R., Osburn W.O., Keck Z., Foung S.K., Ray S.C. Naturally selected hepatitis C virus polymorphisms confer broad neutralizing antibody resistance. J. Clin. Invest. 2015;125:437–447. doi: 10.1172/JCI78794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton J.P., Goonetilleke N., Butler T.C., Walker B.D., McMichael A.J., Chakraborty A.K. Relative rate and location of intra-host HIV evolution to evade cellular immunity are predictable. Nat. Commun. 2016;7:11660. doi: 10.1038/ncomms11660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton J.P., Kardar M., Chakraborty A.K. Scaling laws describe memories of host-pathogen riposte in the HIV population. Proc. Natl. Acad. Sci. U S A. 2015;112:1965–1970. doi: 10.1073/pnas.1415386112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broering T.J., Garrity K.A., Boatright N.K., Sloan S.E., Sandor F., Thomas W.D., Szabo G., Finberg R.W., Ambrosino D.M., Babcock G.J., et al. Identification and characterization of broadly neutralizing human monoclonal antibodies directed against the E2 envelope glycoprotein of hepatitis C virus. Virology. 2009;83:12473–12482. doi: 10.1128/JVI.01138-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno S., Crosignani A., Maisonneuve P., Rossi S., Silini E., Mondelli M.U. Hepatitis C virus genotype 1b as a major risk factor associated with hepatocellular carcinoma in patients with cirrhosis: a seventeen-year prospective cohort study. Hepatology. 2007;46:1350–1356. doi: 10.1002/hep.21826. [DOI] [PubMed] [Google Scholar]
- Bull R.A., Luciani F., McElroy K., Gaudieri S., Pham S.T., Chopra A., Cameron B., Maher L., Dore G.J., White P.A., et al. Sequential bottlenecks drive viral evolution in early acute hepatitis C virus infection. PLoS Pathog. 2011;7:1–14. doi: 10.1371/journal.ppat.1002243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention Hepatitis C questions and answers for the public. 2018. https://www.cdc.gov/hepatitis/HCV/cfaq.htm
- Chen H., Zhou H.-X. Prediction of solvent accessibility and sites of deleterious mutations from protein sequence. Nucleic Acids Res. 2005;33:3193–3199. doi: 10.1093/nar/gki633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.L., Morgan T.R. The natural history of hepatitis C virus (HCV) infection. Int. J. Med. Sci. 2006;3:47–52. doi: 10.7150/ijms.3.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y.-K., Kim Y.N., Song B.-C. Predictors of spontaneous viral clearance and outcomes of acute hepatitis C infection. Clin. Mol. Hepatol. 2014;20:368–375. doi: 10.3350/cmh.2014.20.4.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claiborne D.T., Prince J.L., Scully E., Macharia G., Micci L., Lawson B., Kopycinski J., Deymier M.J., Vanderford T.H., Nganou-Makamdop K., et al. Replicative fitness of transmitted HIV-1 drives acute immune activation, proviral load in memory CD4+ T cells, and disease progression. Proc. Natl. Acad. Sci. U S A. 2015;112:E1480–E1489. doi: 10.1073/pnas.1421607112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevas J.M., Gonzalez-Candelas F., Moya A., Sanjuan R. Effect of ribavirin on the mutation rate and spectrum of hepatitis C virus in vivo. Virology. 2009;83:5760–5764. doi: 10.1128/JVI.00201-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czarnota A., Offersgaard A., Pihl A.F., Prentoe J., Bukh J., Gottwein J.M., Bieńkowska-Szewczyk K., Grzyb K. Specific antibodies induced by immunization with hepatitis B virus-like particles carrying hepatitis C virus envelope glycoprotein 2 epitopes show differential neutralization efficiency. Vaccines. 2020;8:1–19. doi: 10.3390/vaccines8020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahirel V., Shekhar K., Pereyra F., Miura T., Artyomov M., Talsania S., Allen T.M., Altfeld M., Carrington M., Irvine D.J., et al. Coordinate linkage of HIV evolution reveals regions of immunological vulnerability. Proc. Natl. Acad. Sci. U S A. 2011;108:11530–11535. doi: 10.1073/pnas.1105315108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn S., Wahl L., Gloor G. Mutual information without the influence of phylogeny or entropy dramatically improves residue contact prediction. Bioinformatics. 2007;24:333–340. doi: 10.1093/bioinformatics/btm604. [DOI] [PubMed] [Google Scholar]
- Easterbrook P.J., Smith M., Mullen J., O’shea S., Chrystie I., Ruiter A., Tatt I.D., Geretti A.M., Zuckerman M. Impact of HIV-1 viral subtype on disease progression and response to antiretroviral therapy. J. Int. AIDS Soc. 2010;13:4. doi: 10.1186/1758-2652-13-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekeberg M., Lövkvist C., Lan Y., Weigt M., Aurell E. Improved contact prediction in proteins: using pseudolikelihoods to infer potts models. Phys. Rev. E. 2013;87:012707. doi: 10.1103/PhysRevE.87.012707. [DOI] [PubMed] [Google Scholar]
- Esteban-Riesco L., Depaulis F., Moreau A., Bacq Y., Dubois F., Goudeau A., Gaudy-Graffin C. Rapid and sustained autologous neutralizing response leading to early spontaneous recovery after HCV infection. Virology. 2013;444:90–99. doi: 10.1016/j.virol.2013.05.037. [DOI] [PubMed] [Google Scholar]
- Ewens W.J. Springer; 2004. Mathematical Population Genetics. [Google Scholar]
- Ferguson A.L., Mann J.K., Omarjee S., Ndung’U T., Walker B.D., Chakraborty A.K. Translating HIV sequences into quantitative fitness landscapes predicts viral vulnerabilities for rational immunogen design. Immunity. 2013;38:606–617. doi: 10.1016/j.immuni.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flyak A.I., Ruiz S., Colbert M.D., Luong T., Crowe J.E., Bailey J.R., Bjorkman P.J. HCV broadly neutralizing antibodies use a CDRH3 disulfide motif to recognize an E2 glycoprotein site that can be targeted for vaccine design. Cell Host Microbe. 2018;24:703–716.e3. doi: 10.1016/j.chom.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn W.F., Haldane A., Torbett B.E., Levy R.M. Inference of epistatic effects leading to entrenchment and drug resistance in HIV-1 protease. Mol. Biol. Evol. 2017;34:1291–1306. doi: 10.1093/molbev/msx095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaiha G.D., Rossin E.J., Urbach J., Landeros C., Collins D.R., Nwonu C., Muzhingi I., Anahtar M.N., Waring O.M., Piechocka-Trocha A., et al. Structural topology defines protective CD8+ T cell epitopes in the HIV proteome. Science. 2019;364:480–484. doi: 10.1126/science.aav5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal R., Jackson K., Tzarum N., Kong L., Ettenger A., Guest J., Pfaff J.M., Barnes T., Honda A., Giang E., et al. Probing the antigenicity of hepatitis C virus envelope glycoprotein complex by high-throughput mutagenesis. PLoS Pathog. 2017;13:e1006735. doi: 10.1371/journal.ppat.1006735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gower E., Estes C., Blach S., Razavi-Shearer K., Razavi H. Global epidemiology and genotype distribution of the hepatitis C virus infection. J. Hepatol. 2014;61:S45–S57. doi: 10.1016/j.jhep.2014.07.027. [DOI] [PubMed] [Google Scholar]
- Harman C., Zhong L., Ma L., Liu P., Deng L., Zhao Z., Yan H., Struble E., Virata-Theimer M.L., Zhang P., et al. A view of the E2-CD81 interface at the binding site of a neutralizing antibody against hepatitis C virus. J. Virol. 2015;89:492–501. doi: 10.1128/JVI.01661-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart G.R., Ferguson A.L. Empirical fitness models for hepatitis C virus immunogen design. Phys. Biol. 2015;12:066006. doi: 10.1088/1478-3975/12/6/066006. [DOI] [PubMed] [Google Scholar]
- Hwang S.-J., Lee S.-D., Lu R.-H., Chu C.-W., Wu J.-C., Lai S.-T., Chang F.-Y. Hepatitis C viral genotype influences the clinical outcome of patients with acute posttransfusion hepatitis C. J. Med. Virol. 2001;65:505–509. [PubMed] [Google Scholar]
- ICTV Table 1 - Confirmed HCV Genotypes/subtypes (May 2019) 2019. https://talk.ictvonline.org/ictv_wikis/flaviviridae/w/sg_flavi/634/table-1---confirmed-hcv-genotypes-subtypes-may-2019
- Jardine J.G., Sok D., Julien J.-P., Briney B., Sarkar A., Liang C.-H., Scherer E.A., Henry Dunand C.J., Adachi Y., Diwanji D., et al. Minimally mutated HIV-1 broadly neutralizing antibodies to guide reductionist vaccine design. PLoS Pathog. 2016;12:1–33. doi: 10.1371/journal.ppat.1005815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato N., Sekiya H., Ootsuyama Y., Nakazawa T., Hijikata M., Ohkoshi S., Shimotohno K. Humoral immune response to hypervariable region 1 of the putative envelope glycoprotein (gp70) of hepatitis C virus. Virology. 1993;67:3923–3930. doi: 10.1128/jvi.67.7.3923-3930.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck Z.-Y., Angus A.G.N., Wang W., Lau P., Wang Y., Gatherer D., Patel A.H., Foung S.K.H. Non-random escape pathways from a broadly neutralizing human monoclonal antibody map to a highly conserved region on the hepatitis C virus E2 glycoprotein encompassing amino acids 412-423. PLoS Pathog. 2014;10:1–13. doi: 10.1371/journal.ppat.1004297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck Z.-Y., Girard-Blanc C., Wang W., Lau P., Zuiani A., Rey F.A., Krey T., Diamond M.S., Foung S.K.H. Antibody response to hypervariable region 1 interferes with broadly neutralizing antibodies to hepatitis C virus. Virology. 2016;90:3112–3122. doi: 10.1128/JVI.02458-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck Z.-Y., Li S.H., Xia J., von Hahn T., Balfe P., McKeating J.A., Witteveldt J., Patel A.H., Alter H., Rice C.M., et al. Mutations in hepatitis C virus E2 located outside the CD81 binding sites lead to escape from broadly neutralizing antibodies but compromise virus infectivity. Virology. 2009;83:6149–6160. doi: 10.1128/JVI.00248-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck Z.-Y., Li T.-K., Xia J., Bartosch B., Cosset F.-L., Dubuisson J., Foung S.K.H. Analysis of a highly flexible conformational immunogenic domain a in hepatitis C virus E2. Virology. 2005;79:13199–13208. doi: 10.1128/JVI.79.21.13199-13208.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck Z.-Y., Olson O., Gal-Tanamy M., Xia J., Patel A.H., Dreux M., Cosset F.-L., Lemon S.M., Foung S.K.H. A point mutation leading to hepatitis C virus escape from neutralization by a monoclonal antibody to a conserved conformational epitope. Virology. 2008;82:6067–6072. doi: 10.1128/JVI.00252-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck Z.-Y., Pierce B.G., Lau P., Lu J., Wang Y., Underwood A., Bull R.A., Prentoe J., Velázquez-Moctezuma R., Walker M.R., et al. Broadly neutralizing antibodies from an individual that naturally cleared multiple hepatitis C virus infections uncover molecular determinants for E2 targeting and vaccine design. PLoS Pathog. 2019;15:e1007772. doi: 10.1371/journal.ppat.1007772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiwanuka N., Ssetaala A., Mpendo J., Wambuzi M., Nanvubya A., Sigirenda S., Nalutaaya A., Kato P., Nielsen L., Kaleebu P., et al. High HIV-1 prevalence, risk behaviours, and willingness to participate in HIV vaccine trials in fishing communities on Lake Victoria, Uganda. J. Int. AIDS Soc. 2013;16:18621. doi: 10.7448/IAS.16.1.18621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L., Giang E., Nieusma T., Kadam R.U., Cogburn K.E., Hua Y., Dai X., Stanfield R.L., Burton D.R., Ward A.B., et al. Hepatitis C virus E2 envelope glycoprotein core structure. Science. 2013;342:1090–1094. doi: 10.1126/science.1243876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L., Giang E., Robbins J.B., Stanfield R.L., Burton D.R., Wilson I.A., Law M. Structural basis of hepatitis C virus neutralization by broadly neutralizing antibody HCV1. Proc. Natl. Acad. Sci. U S A. 2012;109:9499–9504. doi: 10.1073/pnas.1202924109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L., Lee D.E., Kadam R.U., Liu T., Giang E., Nieusma T., Garces F., Tzarum N., Woods V.L., Ward A.B., et al. Structural flexibility at a major conserved antibody target on hepatitis C virus E2 antigen. Proc. Natl. Acad. Sci. U S A. 2016;113:12768–12773. doi: 10.1073/pnas.1609780113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouyos R.D., Leventhal G.E., Hinkley T., Haddad M., Whitcomb J.M., Petropoulos C.J., Bonhoeffer S. Exploring the complexity of the HIV-1 fitness landscape. PLoS Genet. 2012;8:e1002551. doi: 10.1371/journal.pgen.1002551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law M., Maruyama T., Lewis J., Giang E., Tarr A.W., Stamataki Z., Gastaminza P., Chisari F.V., Jones I.M., Fox R.I., et al. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 2008;14:25–27. doi: 10.1038/nm1698. [DOI] [PubMed] [Google Scholar]
- Lee M.-H., Yang H.-I., Lu S.-N., Jen C.-L., You S.-L., Wang L.-Y., L’Italien G., Chen C.-J., Yuan Y., the REVEAL-HCV Study Group Hepatitis C virus genotype 1b increases cumulative lifetime risk of hepatocellular carcinoma. Int. J. Cancer. 2014;135:1119–1126. doi: 10.1002/ijc.28753. [DOI] [PubMed] [Google Scholar]
- Leys C., Ley C., Klein O., Bernard P., Licata L. Detecting outliers: do not use standard deviation around the mean, use absolute deviation around the median. J. Exp. Social Psychol. 2013;49:764–766. [Google Scholar]
- Louie R.H.Y., Kaczorowski K.J., Barton J.P., Chakraborty A.K., Mckay M.R. Fitness landscape of the human immunodeficiency virus envelope protein that is targeted by antibodies. Proc. Natl. Acad. Sci. U S A. 2018;115:E564–E573. doi: 10.1073/pnas.1717765115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magiorkinis G., Magiorkinis E., Paraskevis D., Ho S.Y.W., Shapiro B., Pybus O.G., Allain J.-P., Hatzakis A. The global spread of hepatitis C virus 1a and 1b: a phylodynamic and phylogeographic analysis. PLoS Med. 2009;6:1–12. doi: 10.1371/journal.pmed.1000198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann J.K., Barton J.P., Ferguson A.L., Omarjee S., Walker B.D., Chakraborty A., Ndungu T. The fitness landscape of HIV-1 Gag: Advanced modeling approaches and validation of model predictions by in vitro testing. PLoS Comput. Biol. 2014;10:e1003776. doi: 10.1371/journal.pcbi.1003776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merat S.J., Bru C., Berg D.V.D., Molenkamp R., Tarr A.W., Koekkoek S., Kootstra N.A., Prins M., Ball J.K., Bakker A.Q., et al. Cross-genotype AR3-specific neutralizing antibodies confer long-term protection in injecting drug users after HCV clearance. Hepatology. 2019;71:14–24. doi: 10.1016/j.jhep.2019.02.013. [DOI] [PubMed] [Google Scholar]
- Messina J.P., Humphreys I., Flaxman A., Brown A., Cooke G.S., Pybus O.G., Barnes E. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology. 2014;61:77–87. doi: 10.1002/hep.27259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S., Janin J., Lesk A.M., Chothia C. Interior and surface of monomeric proteins. J. Mol. Biol. 1987;196:641–656. doi: 10.1016/0022-2836(87)90038-6. [DOI] [PubMed] [Google Scholar]
- Missiha S.B., Ostrowski M., Heathcote E.J. Disease progression in chronic hepatitis C: modifiable and nonmodifiable factors. Gastroenterology. 2008;134:1699–1714. doi: 10.1053/j.gastro.2008.02.069. [DOI] [PubMed] [Google Scholar]
- Morcos F., Pagnani A., Lunt B., Bertolino A., Marks D.S., Sander C., Zecchina R., Onuchic J.N., Hwa T., Weigt M. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc. Natl. Acad. Sci. U S A. 2011;108:E1293–E1301. doi: 10.1073/pnas.1111471108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin T.J., Broering T.J., Leav B.A., Blair B.M., Rowley K.J., Boucher E.N., Wang Y., Cheslock P.S., Knauber M., Olsen D.B., et al. Human monoclonal antibody HCV1 effectively prevents and treats HCV infection in chimpanzees. PLoS Pathog. 2012;8:e1002895. doi: 10.1371/journal.ppat.1002895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakowski D.K., Barton J.P., Peter L., Chandrashekar A., Bondzie E., Gao A., Barouch D.H., Chakraborty A.K. Adenovirus-vectored vaccine containing multidimensionally conserved parts of the HIV proteome is immunogenic in rhesus macaques. Proc. Natl. Acad. Sci. U S A. 2021;118 doi: 10.1073/pnas.2022496118. e2022496118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muyldermans S. Nanobodies: natural single-domain antibodies. Annu. Rev. Biochem. 2013;82:775–797. doi: 10.1146/annurev-biochem-063011-092449. [DOI] [PubMed] [Google Scholar]
- Naik A.S., Owsianka A., Palmer B.A., O’Halloran C.J., Walsh N., Crosbie O., Kenny-Walsh E., Patel A.H., Fanning L.J. Reverse epitope mapping of the E2 glycoprotein in antibody associated hepatitis C virus. PLoS One. 2017;12:1–20. doi: 10.1371/journal.pone.0175349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osella A.R., Misciagna G., Guerra V., Elba S., Buongiorno G., Cavallini A., Di Leo A., Sonzogni L., Mondelli M.U., Silini E.M. Hepatitis C virus genotypes and risk of cirrhosis in southern Italy. Clin. Infect. Dis. 2001;33:70–75. doi: 10.1086/320887. [DOI] [PubMed] [Google Scholar]
- Pantua H., Diao J., Ultsch M., Hazen M., Mathieu M., McCutcheon K., Takeda K., Date S., Cheung T.K., Phung Q., et al. Glycan shifting on hepatitis C virus (HCV) E2 glycoprotein is a mechanism for escape from broadly neutralizing antibodies. J. Mol. Biol. 2013;425:1899–1914. doi: 10.1016/j.jmb.2013.02.025. [DOI] [PubMed] [Google Scholar]
- Parera M., Martinez M.A. Strong epistatic interactions within a single protein. Mol. Biol. Evol. 2014;31:1546–1553. doi: 10.1093/molbev/msu113. [DOI] [PubMed] [Google Scholar]
- Petruzziello A., Marigliano S., Loquercio G., Cozzolino A., Cacciapuoti C. Global epidemiology of hepatitis C virus infection: an up-date of the distribution and circulation of hepatitis C virus genotypes. World J. Gastroenterol. 2016;22:7824. doi: 10.3748/wjg.v22.i34.7824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce B.G., Keck Z.-Y., Lau P., Fauvelle C., Gowthaman R., Baumert T.F., Fuerst T.R., Mariuzza R.A., Foung S.K.H. Global mapping of antibody recognition of the hepatitis C virus E2 glycoprotein: implications for vaccine design. Proc. Natl. Acad. Sci. U S A. 2016;113:E6946–E6954. doi: 10.1073/pnas.1614942113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadeer A.A., Barton J.P., Chakraborty A.K., Mckay M.R. Deconvolving mutational patterns of poliovirus outbreaks reveals its intrinsic fitness landscape. Nat. Commun. 2020;11:377. doi: 10.1038/s41467-019-14174-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadeer A.A., Louie R.H.Y., Mckay M.R. Identifying immunologically-vulnerable regions of the HCV E2 glycoprotein and broadly neutralizing antibodies that target them. Nat. Commun. 2019;10:2073. doi: 10.1038/s41467-019-09819-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadeer A.A., Louie R.H.Y., Shekhar K., Chakraborty A.K., Hsing I.-M., McKay M.R. Statistical linkage analysis of substitutions in patient-derived sequences of genotype 1a hepatitis C virus nonstructural protein 3 exposes targets for immunogen design. J. Virol. 2014;88:7628–7644. doi: 10.1128/JVI.03812-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadeer A.A., McKay M.R., Barton J.P., Louie R.H.Y. MPF-BML: a standalone GUI-based package for maximum entropy model inference. Bioinformatics. 2019;36:2278–2279. doi: 10.1093/bioinformatics/btz925. [DOI] [PubMed] [Google Scholar]
- Quadeer A.A., Morales-Jimenez D., McKay M.R. Co-evolution networks of HIV/HCV are modular with direct association to structure and function. PLoS Comput. Biol. 2018;14:e1006409. doi: 10.1371/journal.pcbi.1006409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimondi S., Bruno S., Mondelli M.U., Maisonneuve P. Hepatitis C virus genotype 1b as a risk factor for hepatocellular carcinoma development: a meta-analysis. Hepatology. 2009;50:1142–1154. doi: 10.1016/j.jhep.2009.01.019. [DOI] [PubMed] [Google Scholar]
- Rodríguez-López M., Riezu-Boj J.I., Ruiz M., Berasain C., Civeira M.P., Prieto J., Borrás-Cuesta F. Immunogenicity of variable regions of hepatitis C virus proteins: selection and modification of peptide epitopes to assess hepatitis C virus genotypes by ELISA. J. Gen. Virol. 1999;80:727–738. doi: 10.1099/0022-1317-80-3-727. [DOI] [PubMed] [Google Scholar]
- Rosen H.R. Clinical practice. Chronic hepatitis C infection. N. Engl. J. Med. 2011;364:2429–2438. doi: 10.1056/NEJMcp1006613. [DOI] [PubMed] [Google Scholar]
- Rosenthal E.S., Graham C.S. Price and affordability of direct-acting antiviral regimens for hepatitis C virus in the United States. Infect. Agents Cancer. 2016;11:24. doi: 10.1186/s13027-016-0071-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi C., Butt Z.A., Wong S., Buxton J.A., Islam N., Yu A., Darvishian M., Gilbert M., Wong J., Chapinal N., et al. Hepatitis C virus reinfection after successful treatment with direct-acting antiviral therapy in a large population-based cohort. Hepatology. 2018;69:1007–1014. doi: 10.1016/j.jhep.2018.07.025. [DOI] [PubMed] [Google Scholar]
- Sanjuan R., Nebot M.R., Chirico N., Mansky L.M., Belshaw R. Viral mutation rates. Virology. 2010;84:9733–9748. doi: 10.1128/JVI.00694-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silini E., Bottelli R., Asti M., Bruno S., Candusso M., Brambilla S., Bono F., Iamoni G., Tinelli C., Mondelli M., et al. Hepatitis C virus genotypes and risk of hepatocellular carcinoma in cirrhosis: a case-control study. Gastroenterology. 1996;111:199–205. doi: 10.1053/gast.1996.v111.pm8698200. [DOI] [PubMed] [Google Scholar]
- Singer J., Thomson E., Hughes J., Aranday-Cortes E., Mclauchlan J., Filipe A.D.S., Tong L., Manso C., Gifford R., Robertson D., et al. Interpreting viral deep sequencing data with GLUE. Viruses. 2019;11:323. doi: 10.3390/v11040323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer J.B., Thomson E.C., McLauchlan J., Hughes J., Gifford R.J. GLUE: a flexible software system for virus sequence data. BMC Bioinformatics. 2018;19:532. doi: 10.1186/s12859-018-2459-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohail M.S., Louie R.H.Y., McKay M.R., Barton J.P. MPL resolves genetic linkage in fitness inference from complex evolutionary histories. Nat. Biotechnol. 2021;39:472–479. doi: 10.1038/s41587-020-0737-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohail M.S., Quadeer A.A., McKay M.R. How genetic sequence data can guide vaccine design. IEEE Potentials. 2020;39:31–37. [Google Scholar]
- Sormanni P., Aprile F.A., Vendruscolo M. Rational design of antibodies targeting specific epitopes within intrinsically disordered proteins. Proc. Natl. Acad. Sci. U S A. 2015;112:9902–9907. doi: 10.1073/pnas.1422401112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ssemwanga D., Nsubuga R.N., Mayanja B.N., Lyagoba F., Magambo B., Yirrell D., Van der Paal L., Grosskurth H., Kaleebu P. Effect of HIV-1 subtypes on disease progression in rural Uganda: a prospective clinical cohort study. PLoS One. 2013;8:1–7. doi: 10.1371/journal.pone.0071768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strimmer K., Haeseler A.V. In: The Phylogenetic Handbook: A Practical Approach to DNA and Protein Phylogeny. Lemey P., Salemi M., Vandamme A.-M., editors. Cambridge University Press; 2009. Genetic distances and nucleotide substitution models; pp. 112–113. [Google Scholar]
- Ströh L.J., Nagarathinam K., Krey T. Conformational flexibility in the CD81-binding site of the hepatitis C virus glycoprotein E2. Front. Immunol. 2018;9:1396. doi: 10.3389/fimmu.2018.01396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanowicz R.A., Mcclure C.P., Brown R.J.P., Tsoleridis T., Persson M.A.A., Krey T., Irving W.L., Ball J.K., Tarr A.W. A diverse panel of hepatitis C virus glycoproteins for use in vaccine research reveals extremes of monoclonal antibody neutralization resistance. Virology. 2015;90:3288–3301. doi: 10.1128/JVI.02700-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev V.K., Fogarty T.C., Miller J.F. Smoothness, ruggedness and neutrality of fitness landscapes: from theory to application. Nat. Comput. Ser. Adv. Evol. Comput. 2003:3–44. [Google Scholar]
- Velázquez-Moctezuma R., Galli A., Law M., Bukh J., Prentoe J. Hepatitis C virus escape studies of human antibody AR3A reveal a high barrier to resistance and novel insights on viral antibody evasion mechanisms. Virology. 2019;93:e01909–e01918. doi: 10.1128/JVI.01909-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venner C.M., Nankya I., Kyeyune F., Demers K., Kwok C., Chen P.-L., Rwambuya S., Munjoma M., Chipato T., Byamugisha J., et al. Infecting HIV-1 subtype predicts disease progression in women of sub-saharan africa. EBioMedicine. 2016;13:305–314. doi: 10.1016/j.ebiom.2016.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vita R., Mahajan S., Overton J.A., Dhanda S.K., Martini S., Cantrell J.R., Wheeler D.K., Sette A., Peters B. The immune epitope database (IEDB): 2018 update. Nucleic Acids Res. 2018;47:D339–D343. doi: 10.1093/nar/gky1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigt M., White R.A., Szurmant H., Hoch J.A., Hwa T. Identification of direct residue contacts in protein-protein interaction by message passing. Proc. Natl. Acad. Sci. U S A. 2008;106:67–72. doi: 10.1073/pnas.0805923106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization Hepatitis C, fact sheet. 2021. https://www.who.int/news-room/fact-sheets/detail/hepatitis-c
- Wyles D.L., Luetkemeyer A.F. Understanding hepatitis C virus drug resistance: clinical implications for current and future regimens. Top. Antivir. Med. 2017;25:103–109. [PMC free article] [PubMed] [Google Scholar]
- Yan Z., Wang Y. Viral and host factors associated with outcomes of hepatitis C virus infection. Mol. Med. Rep. 2017;15:2909–2924. doi: 10.3892/mmr.2017.6351. [DOI] [PubMed] [Google Scholar]
- Zimmermann I., Egloff P., Hutter C.A., Arnold F.M., Stohler P., Bocquet N., Hug M.N., Huber S., Siegrist M., Hetemann L., et al. Synthetic single domain antibodies for the conformational trapping of membrane proteins. eLife. 2018;7:e34317. doi: 10.7554/eLife.34317. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data used in this work has been provided in the supplemental information and is publicly available as of the date of publication. The E2 1b infectivity measurements, used for validating the fitness landscape model, are included in Data S1. Accession numbers of E2 1b sequences used for inferring the model are listed in Data S2. The mean escape time predicted by the in-host evolutionary model for each residue of E2 1b is provided in Data S3.
-
•
Data and scripts for reproducing the results are available at GitHub: https://github.com/hangzhangust/HCVE21a1b_Hang. The GUI-based software implementation of the MPF-BML method (Louie et al., 2018), used for inferring the fitness landscape parameters, is available at GitHub: https://github.com/ahmedaq/MPF-BML-GUI (Quadeer et al., 2019b). Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.