

Summary
Human respiratory viruses are of vastly different virulence, giving rise to symptoms ranging from common cold to severe pneumonia or even death. Although this most likely impacts molecular evolution of the corresponding viruses, the specific differences in their evolutionary patterns remain largely unknown. By comparing structural and nonstructural genes within respiratory viruses, greater similarities in codon usage bias (CUB) between nonstructural genes and humans were observed in weakly virulent viruses, whereas in strongly virulent viruses, it was structural genes whose CUBs were more similar to that of humans. Further comparisons between genomes of weakly and strongly virulent coronaviruses revealed greater similarities in CUBs between strongly virulent viruses and humans. Finally, using phylogenetic independent contrasts, dissimilation of viral CUB from that of humans was observed in SARS-CoV-2. Our work revealed distinct CUB evolutionary patterns between weakly and strongly virulent viruses, a previously unrecognized interaction between CUB and virulence in respiratory viruses
Subject areas: Phylogenetics, Virology, Microbial genomics, Evolutionary biology
Graphical abstract
Highlights
-
•
Nonstructural genes and human CUBs are more similar in weakly virulent viruses
-
•
Structural genes and human CUBs are more similar in strongly virulent viruses
-
•
Strongly virulence coronaviruses tend to have greater CUB similarities to humans
-
•
CUB of SARS-CoV-2 appear gradually dissimilating from that of humans
Phylogenetics; Virology; Microbial genomics; Evolutionary biology
Introduction
Human mastadenoviruses (HAdVs), human rhinoviruses (HRVs), human respiratory syncytial viruses (HRSVs), common human coronaviruses (CH-CoVs), and new human coronaviruses (NH-CoVs) are the most frequent causative agents of disease in humans and have significant impacts on morbidity and mortality worldwide (Boncristiani et al., 2009; Greenberg, 2016; Pillaiyar et al., 2020; Zhang et al., 2020). HAdVs and HRVs are usually associated with lower mortality common colds, involving rhinitis, pharyngitis, sneezing, hoarseness, and/or cough (Greenberg, 2011; Jacobs et al., 2013; Zhang et al., 2020). HRSVs and CH-CoVs may frequently cause serious lower respiratory tract illness or even death in children and elderly people (Alansari and Potgieter, 1994; Tognarelli et al., 2019). NH-CoVs (SARS-CoV, MERS-CoV, and SARS-CoV-2) are highly pathogenic emerging and reemerging viruses, and SARS-CoV-2 has posed a great threat to global public health since the end of 2019 (Choi et al., 2003; Esposito et al., 2020; Jiang et al., 2020; Pillaiyar et al., 2020; Wang et al., 2020). To predict the next occurrence of new large-scale public health emergencies caused by another NH-CoV, similar to SARS-CoV-2, it is important to systematically analyze patterns of coevolution between respiratory viruses and humans to better understand the specific evolutionary pattern of this particular virus.
As the majority of mammalian viruses do not encode any tRNA, the translational efficiency of viral proteins is mainly determined by the similarity between the synonymous codon usage of viral genes and the tRNA supply of the host (Albers and Czech, 2016; Bahir et al., 2009; Chen et al., 2020; Tian et al., 2018). Many previous works have demonstrated that the dependence of viral gene expression on tRNA resources, in competition with host genes, will retain the virus whose CUB is more similar to that of host (Bahir et al., 2009; Lucks et al., 2008). In contrast, our preceding work suggested that overly increased viral expression would prevent excessive assimilation between viral and host CUBs, so as not to increase the translation load on hosts (Chen et al., 2020). The competition for host tRNA resources because of translational selection had been revealed in some respiratory tract viruses (Alonso and Diambra, 2020; Hernandez-Alias et al., 2021; Rice et al., 2021). However, the specific differences in the evolutionary pattern among different human respiratory tract viruses remain largely unknown.
In this work, we systematically analyzed CUB evolutionary patterns for four types of respiratory tract viruses and their relationship with human CUB. In weakly virulent human respiratory viruses (HAdVs and HRVs), it was usually nonstructural genes whose CUBs were more similar to that of humans, whereas in strongly virulent human respiratory viruses (HRSVs, CH-CoVs, and NH-CoVs), structural genes tended to have more human-like CUBs. By comparing the CUB of weakly virulent CH-CoVs and that of strongly virulent NH-CoVs to the tRNA supply of humans, we found greater similarities in CUBs between NH-CoVs and humans. More importantly, dissimilation of CUB (gradual change of the viral CUB to become less similar to the host CUB) has been observed in SARS-CoV-2 since its emergence in humans.
Results
The CUB of early genes is more similar to that of humans in most serotypes of HAdVs
In this work, our goal is to compare the CUB evolutionary patterns of respiratory viruses with weak or strong virulence. To achieve this goal, we first defined the virulence of each respiratory virus according to the symptoms of humans during viral infection (See STAR methods). Then, we focused on one of the weakly virulent respiratory viruses (Human mastadenoviruses, HAdVs), whose genes are expressed in three different stages of viral infection (gene categories are listed on Table S1).
During the first stage of HAdVs infection, early genes optimize the cellular milieu for viral replication to benefit the expression of intermediate and late genes in the second and final stage of viral infection, respectively (Nevins, 1987). Thus, the mRNA abundance of early genes at the initial stage of infection will be much lower than that of intermediate and late genes in the other two stages of infections. On that basis, we expected that the translational efficiencies of early genes need to be increased, for example, by increasing the similarities of their CUBs to the tRNA supply of the host, so as to increase the viability of the virus. In contrast, intermediate and late genes might be under weaker translational selection pressure, because the optimized cellular environment would enable sufficiently high translational efficiency. To test this hypothesis, we calculated the deviation from proportionality (Chen et al., 2020) (DP, see STAR methods) of synonymous codon usage of viral genes to the tRNA supply of the host for each gene of HAdV species downloaded from NCBI Virus (Hatcher et al., 2017). In HAdV-A, HAdV-C, HAdC-D, HAdV-E, and HAdV-F, the DP values of early genes were smaller than those of late genes (HAdV-A: Wilcoxon rank-sum test P = 0.0039; HAdV-C: Wilcoxon rank-sum test P = 1.1 × 10−107; HAdV-D: Wilcoxon rank-sum test P = 1.5 × 10−188; HAdV-E: Wilcoxon rank-sum test P = 1.8 × 10−109; HAdV-F: Wilcoxon rank-sum test P = 7.3 × 10−8; Figure 1); the opposite was true in HAdV-B and HAdV-G (HAdV-B: Wilcoxon rank-sum test P = 0; HAdV-G: Wilcoxon rank-sum test P = 2.6 × 10−5; Figure 1). The same patterns were observed between early and intermediate genes (HAdV-A: Wilcoxon rank-sum test P = 3.5 × 10−10; HAdV-B: Wilcoxon rank-sum test P = 8.1 × 10−11; HAdV-C: Wilcoxon rank-sum test P = 2.0 × 10−116; HAdV-D: Wilcoxon rank-sum test P = 6.9 × 10−133; HAdV-E: Wilcoxon rank-sum test P = 1.4 × 10−34; HAdV-F: Wilcoxon rank-sum test P = 0.26; HAdV-G: Wilcoxon rank-sum test P = 0.17; Figure 1). Five out of seven serotypes of HAdVs showed smaller DP values of early genes (binomial P = 0.063), indicating that the translational selection in early genes were marginally stronger than that in intermediate and late genes.
Figure 1.

CUB evolutionary patterns among early, intermediate, and late genes of HAdVs
Early genes and human CUBs were more similar than CUBs of intermediate or late genes to that of humans. The difference between viral gene CUBs and human tRNA supply was measured by DP values. The p values represent comparisons among the DP values of early, intermediate, and late genes by the Wilcoxon rank-sum test. E (red), I (green), and L (blue) represent early, intermediate, and late genes, respectively. The dots represent the mean of DP values in each group. The numbers of genes in each group were listed in Table S1.
Intermediate genes upregulate the transcriptional activity of major late promoters and drive the expression of late genes and the synthesis of the corresponding structural proteins (Lutz and Kedinger, 1996; Lutz et al., 1997). Accordingly, the mRNA abundance of intermediate genes in the second stage of infection may be lower than that of late genes in the final stage. To further increase the expression of late genes, the translational efficiency of intermediate genes seems to be increased through increasing the similarities of CUBs to the tRNA supply of humans. However, it is not clear whether it is necessary for the similarities in CUBs between intermediate genes and humans to be further increased when the cellular surroundings for viral replication have been improved by early genes. To examine this problem, we compared the DP values of intermediate and late genes. We observed a skewed CUB evolution pattern between intermediate genes and late genes in different serotypes of HAdVs (Figure 1). In HAdV-D, HAdV-E, and HAdV-F, intermediate genes displayed more human-like CUBs comparing to late genes (HAdV-D: Wilcoxon rank-sum test P = 0.77; HAdV-E: Wilcoxon rank-sum test P = 0.31; HAdV-F: Wilcoxon rank-sum test P = 0.74; Figure 1); the opposite was true in HAdV-A, HAdV-B, HAdV-C, and HAdV-G (HAdV-A: Wilcoxon rank-sum test P = 7.7 × 10−5; HAdV-B: Wilcoxon rank-sum test P = 1.1 × 10−49; HAdV-C: Wilcoxon rank-sum test P = 2.6 × 10−21; HAdV-G: Wilcoxon rank-sum test P = 0.068; Figure 1). Compared to late genes, smaller DP values of intermediate genes were only observed in three out of seven serotypes of HAdVs (binomial P = 0.50), indicating no difference in the translational selection between intermediate and late genes.
The CUBs of nonstructural genes are more similar to that of humans in weakly virulent respiratory viruses
Among early, intermediate and late genes, the CUBs of early genes were more similar to that of humans in most serotypes of HAdVs. Early genes coded for nonstructural genes, whereas intermediate and late genes usually coded for structural genes (Fabry et al., 2005; Vellinga et al., 2005). Thus, we expected that nonstructural genes would have more human-like CUBs. To test it, we compared the DP values between nonstructural and structural genes of HAdVs (gene categories were listed in Table S1). As expected, smaller DP values were observed in nonstructural genes of HAdV-A, HAdV-C, HAdV-D, HAdV-E, and HAdV-F (HAdV-A: Wilcoxon rank-sum test P = 9.7 × 10−5; HAdV-C: Wilcoxon rank-sum test P = 1.9 × 10−125; HAdV-D: Wilcoxon rank-sum test P = 5.1 × 10−192; HAdV-E: Wilcoxon rank-sum test P = 1.4 × 10−97; HAdV-F: Wilcoxon rank-sum test P = 9.2 × 10−7; Figure 2A); the opposite was true in HAdV-B and HAdV-G (HAdV-B: Wilcoxon rank-sum test P = 0; HAdV-G: Wilcoxon rank-sum test P = 0.0018; Figure 2A).
Figure 2.

CUB evolutionary patterns between nonstructural and structural genes in weakly virulent respiratory virus
(A and B) Greater similarities in CUB between nonstructural genes and humans were observed in weakly virulent HAdVs (A) and HRVs (B). The difference between viral gene CUBs and human tRNA supply was measured by DP. The p values represent comparisons between the DP values of structural and nonstructural genes by the Wilcoxon rank-sum test. Nonstructural and structural genes are colored with red and cyan, respectively. The dots represent the mean of DP values in each group. The numbers of genes in each group were listed in Table S1.
As CUB evolutionary patterns may be vastly different among viruses, we wondered whether the same or opposite evolutionary patterns could be observed in another weakly virulent respiratory virus (human rhinoviruses, HRVs). This problem was checked by comparing the DP values between nonstructural and structural genes of HRVs (gene categories were listed in Table S1), and smaller DP values in nonstructural genes of all HRVs were observed (HRV-A: Wilcoxon rank-sum test P = 1.9 × 10−10; HRV-B: Wilcoxon rank-sum test P = 1.5 × 10−7; HRV-C: Wilcoxon rank-sum test P = 1.7 × 10−48; Figure 2B). Among ten serotypes of HAdVs and HRVs, eight showed smaller DP values in nonstructural genes (binomial P = 0.011), indicating greater similarities in CUBs between nonstructural genes and humans in weakly virulent respiratory viruses.
The CUBs of structural genes are more similar to that of humans in strongly virulent respiratory viruses
The results above showed greater similarities in CUBs between nonstructural genes and humans in HAdVs and HRVs. As these two types of viruses were defined as weakly virulent respiratory viruses (See STAR methods), we wondered whether the same or opposite patterns would be observed in human respiratory syncytial viruses (HRSVs: HRSV-A and HRSV-B) and human coronaviruses (H-CoVs: 229E, NL63, OC43, HKU1, SARS-CoV, MERS-CoV, and SARS-CoV-2), which are defined as strongly virulent respiratory viruses (See STAR methods). We examined this problem through comparing the DP values between nonstructural and structural genes of HRVs and H-COVs (gene categories were listed in Table S1), and observed smaller DP values in structural genes of all these two kinds of viruses (HRSV-A: Wilcoxon rank-sum test P = 7.3 × 10−86; HRSV-B: Wilcoxon rank-sum test P = 1.6 × 10−19; HKU1: Wilcoxon rank-sum test P = 3.1 × 10−31; NL63: Wilcoxon rank-sum test P = 1.9 × 10−126; 229E: Wilcoxon rank-sum test P = 4.5 × 10−11; OC43: Wilcoxon rank-sum test P = 0.12; SARS2: Wilcoxon rank-sum test P = 0; SARS: Wilcoxon rank-sum test P = 3.9 × 10−6; MERS: Wilcoxon rank-sum test P = 1.7 × 10−58; Figure 3). These results revealed that structural genes and humans CUBs seemed to be more similar in strongly virulent respiratory viruses (binomial P = 0), which is opposite to the evolutionary patterns in respiratory viruses with weak virulence.
Figure 3.

CUB evolutionary patterns between nonstructural and structural genes in strongly virulent respiratory viruses
(A and B) Greater similarities in CUBs between structural genes and humans were observed in strongly virulent HRSVs (A) and H-CoVs (B). The difference between viral gene CUBs and human tRNA supply was measured by DP. The p values represent comparisons between the DP values of structural and nonstructural genes by the Wilcoxon rank-sum test. Nonstructural and structural genes are colored with red and cyan, respectively. The dots represent the mean of DP values in each group. The numbers of genes in each group were listed in Table S1.
Dissimilated CUB evolutionary patterns in the coevolution of SARS-CoV-2 and humans
The opposite CUB evolutionary patterns between weakly and strongly virulent respiratory viruses indicated an association between viral virulence and CUBs. To further investigate this relationship, we focused on human coronaviruses, which were derived from numerous animal coronavirus species and entered humans through accidental human–animal contact. Although they may have similar origins, the symptoms of the host after their infections may be quite different. According to the symptoms and mortality of viral infection, the common human coronavirus (CH-CoVs: 229E, NL63, OC43, and HKU1) were defined as weakly virulent coronaviruses, whereas new human coronavirus (NH-CoVs: SARS-CoV, MERS-CoV, and SARS-CoV-2) were defined as strongly virulent coronaviruses (see STAR methods). It was previously shown that for some viruses, greater similarities between viral CUBs and the tRNA supply of host may lead to higher viral expression and translational load on host, therefore more severe symptoms of host (Chen et al., 2020). Based on that, we expected that the similarities in CUBs between NH-CoVs and humans may be greater than that in CUBs between CH-CoVs and humans. To test this hypothesis, the DP values of each human coronavirus downloaded from NCBI Virus were calculated. As expected, smaller DP values were observed in NH-CoVs comparing to CH-CoVs (Figure 4A), indicating that the symptoms of human coronavirus infection may be associated with the similarities in CUBs between the virus and the host because of different competitive abilities on host tRNA resources (Alonso and Diambra, 2020; Chen et al., 2020; Hernandez-Alias et al., 2021). Among twelve pairs of comparisons between NH-CoVs and CH-CoVs, eleven pairs showed smaller DP values in NH-CoVs (Figure 4B; binomial P = 0.00024), further supporting our hypothesis. The exception in the comparisons between SARS2 and 229E may indicate that the CUBs of coronaviruses are also associated with other selective pressures.
Figure 4.

Dissimilation of codon usage bias in the co-evolution of SARS-CoV-2 and humans
(A) Greater similarities in CUB between new human coronaviruses and humans were observed. The p values represent comparisons between the DP values of common and new human coronaviruses by Wilcoxon rank-sum test.
(B) The distance of seven kinds of human coronaviruses CUBs to human tRNA supply. The p values represent comparisons between the DP values of each pair of human coronaviruses by Wilcoxon rank-sum test. ∗∗∗: P < 0.001.
(C) The CUBs of SARS-CoV-2 and humans have significantly dissimilated since December 23, 2019 inferred by original values of DP and collection date. Pearson's rank correlation coefficients and p values between the original values of DP and collection dates of each viral genome are shown. Each dot (N = 715,835) represents a genome of SARS-CoV-2.
(D) The CUBs of SARS-CoV-2 and humans have significantly dissimilated since the COVID-19 pandemic inferred by phylogenetic independent contrasts (PIC). Pearson's rank correlation coefficients and p values between the PIC of DP and collection dates are shown.
To enable long-term coexistence of the virus and its host, the CUB of a virus must be similar to host tRNA supply, but not too similar such that the host becomes sick (Chen et al., 2020). On that basis, we expected that NH-CoVs and human CUBs should be gradually dissimilated as they mostly cause severe symptoms. We first tested this hypothesis by calculating the Pearson's correlation coefficients using the original values of DP and collection date. As expected, the difference between SARS-CoV-2 and human CUBs has significantly increased since its emergence in humans (Pearson's R value = 0.0024, P = 0.031, slope = 2.2 × 10−8; Figure 4C). As the DP values of all genomes are not statistically independent observations because of shared ancestry, we recalculated the correlation of DP values and collection dates using phylogenetic independent contrasts (PIC). The result still supported the dissimilation of SARS-CoV-2 and human CUBs (Pearson's R value = 0.018, P = 1.6 × 10−50, slope = 7.2 × 10−7; Figure 4D). No significantly increasing or decreasing tendencies were observed in other human coronaviruses because of too few genomes or lack of collection date (Table S2). These results revealed a gradual trend of dissimilation of CUBs between SARS-CoV-2 and humans. Nevertheless, the dissimilation of SARS-CoV-2 and human CUBs observed here could be a result of recent evolution but not necessarily representative in the long term.
Discussion
In the current study, we carried out a systematic analysis of CUB evolutionary patterns in different virulent human respiratory viruses. We found greater similarities in CUBs between nonstructural genes and humans in weakly virulent HAdVs and HRVs, but between structural genes and humans in strongly virulent HRSVs, CH-CoVs, and NH-CoVs. Further analysis revealed greater similarity in CUBs between NH-CoVs and humans compared to that in CUBs between CH-CoVs and humans. More importantly, SARS-CoV-2 and human CUBs have tended to dissimilate since the COVID-19 pandemic.
There are two caveats worth discussion in regard to the relationship between virulence and CUB. First, although these viruses are quite different in genome structure, gene number, gene length, and so on (Table S1 and Figure S1), the symptoms of the host after infections are all related to the respiratory system. After the definition of viral virulence according to the symptoms of host, these viruses were comparable to some level. Second, the mechanism behind this relationship may be that greater similarities in CUBs between virus and host will increase the expression level of viruses, thus exacerbating the symptoms of the host. In this work, our results strongly support the relationship between virulence and CUB without rejecting the link between other selective pressures and CUB, such as the ZAP antiviral protein of host that may selectively reduce CpG dinucleotides in the SARS-CoV-2 genome (Kmiec et al., 2020; MacLean et al., 2021).
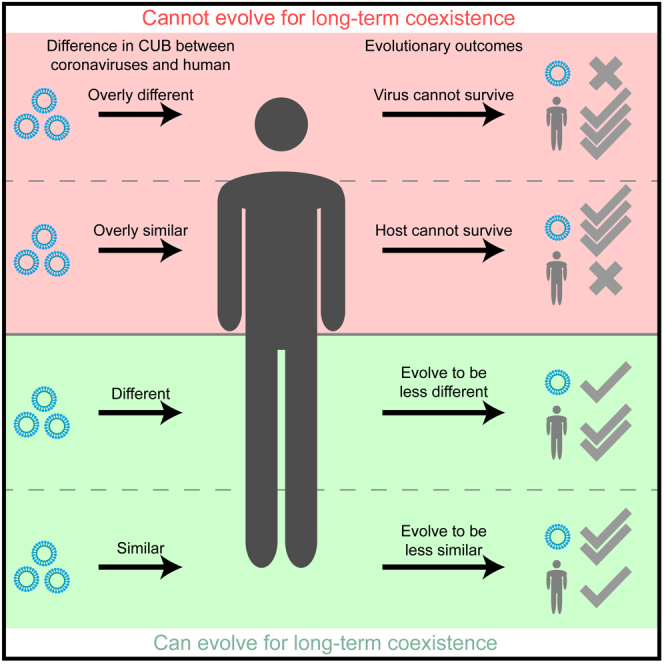
Based on the results of this work, we modified our previous virus-host coevolution model (Chen et al., 2020) to better characterize the possible evolution patterns of strongly virulent NH-CoVs compared to weakly virulent CH-CoVs. Animal coronaviruses may have evolved to be optimal for long-term coexistence with animal species. By chance, infection with animal coronaviruses whose CUB is overly similar to (red area, left; Figure 5) or overly different from (red area, right; Figure 5) human tRNA supply may cause death of the host or the virus, respectively. Infection with an animal coronavirus whose CUB is not overly similar (green area, left; Figure 5) to the human tRNA supply may cause severe symptoms and form new member of NH-CoVs, such as SARS-CoV, MERS-CoV, or SARS-CoV-2. Infection with an animal coronavirus whose CUB is not overly different from (green area, right; Figure 5) the human tRNA supply may cause mild symptoms and form a member of CH-CoVs, such as 229E, NL63, OC43, or HKU1. To evolve for long-term coexistence, the CUBs of weakly virulent human coronaviruses will be converged with that of humans to produce more copies of the virus (white arrow from right; Figure 5), whereas the CUBs of strongly virulent human coronaviruses (especially in SARS-CoV-2 and human coevolution; Figures 4C and 4D) will be diverged from that of humans, causing a reduced translation load and toxicity to humans (white arrow from left; Figure 5).
Figure 5.

Schematic diagram of the different CUB evolutionary patterns in common and new human coronaviruses
Infections of an animal coronavirus whose CUB is not overly similar (green, left part) to the human tRNA supply may cause severe symptoms and form a new human coronavirus, such as SARS-CoV, MERS-CoV, or SARS-CoV-2, whereas infection of an animal coronavirus whose CUB is not overly different (green, right part) from the human tRNA supply may cause mild to moderate symptoms and form a common human coronavirus, such as 229E, NL63, OC43, or HKU1. For long-term coexistence, common human coronaviruses with CUBs similar to that of the host were retained for highly viral expression levels (white, right arrow). In contrast, to evolve for low host toxicity, the expression levels of new human coronaviruses were decreased through dissimilating their CUBs from the tRNA supply of the host (white, left arrow).
On that basis, our work has potential practical implications. Considering the obvious association between CUBs and virulence, our approach can be used to screen all animal coronavirus mutants to predict the virulence through calculating viral and host CUBs. This may provide new methods for predicting the most dangerous animal coronaviruses or wild animal hosts and even for predicting the next occurrence of new large-scale public health emergencies. Besides, the dissimilation of SARS-CoV-2 CUBs to human tRNA supply may indicate lower translational efficiencies per mRNA, and therefore delayed onset or weakened symptoms of host than before. Thus, before the CUB difference of SARS-CoV-2 and humans become optimal for long-term coexistence, the lengthening incubation period of SARS-CoV-2 infection may further challenge our fight against this large-scale public health emergency.
Limitations of the study
There are limitations in the modeling of CUB evolutionary patterns in viruses with different virulence. On the one hand, there is no experiment data that can clearly quantify the viral virulence of each species. Indeed, we can only estimate the viral virulence using symptoms and mortality reported by the World Health Organization and Centers for Disease Control. Although this classification of viral virulence was rough, most patterns in each section were consistent by binomial test. On the other hand, the evolution of a virus was affected by both virulence and transmission capacity. The frequency of each species can be largely affected by the transmission capacity that may be less affected by CUBs. Although the transmission capacities of different species were not equal, dissimilation of CUB evolutionary patterns can still be observed in SARS-CoV-2 since its emergency in humans, indicating an important role of the interaction between CUB and virulence in the evolution of SARS-CoV-2.
STAR★Methods
Key resource table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Human mastadenoviruses A | NCBI Virus | HAdV-A |
| Human mastadenoviruses B | NCBI Virus | HAdV-B |
| Human mastadenoviruses C | NCBI Virus | HAdV-C |
| Human mastadenoviruses D | NCBI Virus | HAdV-D |
| Human mastadenoviruses E | NCBI Virus | HAdV-E |
| Human mastadenoviruses F | NCBI Virus | HAdV-F |
| Human mastadenoviruses G | NCBI Virus | HAdV-G |
| Human rhinoviruses A | NCBI Virus | HRV-A |
| Human rhinoviruses B | NCBI Virus | HRV-B |
| Human rhinoviruses C | NCBI Virus | HRV-C |
| Human respiratory syncytial viruses A | NCBI Virus | HRSV-A |
| Human respiratory syncytial viruses B | NCBI Virus | HRSV-B |
| Human coronavirus 229E | NCBI Virus | 229E |
| Human coronavirus HKU1 | NCBI Virus | HKU1 |
| Human coronavirus NL63 | NCBI Virus | NL63 |
| Human coronavirus OC43 | NCBI Virus | OC43 |
| Severe acute respiratory syndrome coronavirus 2 | NCBI Virus | SARS2 |
| Severe acute respiratory syndrome coronavirus | NCBI Virus | SARS |
| Middle East respiratory syndrome-related coronavirus | NCBI Virus | MERS |
| Deposited data | ||
| Key codes and datasets | This paper | https://github.com/chenfengokha/CUBofSARS2 |
| Features of human respiratory virus | This paper | Table S1 |
| CUB evolutionary patterns between coronaviruses and humans inferred by PIC | This paper | Table S2 |
| Software and algorithms | ||
| R Studio 1.2.5 | RStudio, Inc. | https://www.rstudio.com |
| R 3.6.1 | R Foundation for Statistical Computing | https://www.R-project.org |
Resource availability
Lead contact
Further requests for resources should be directed to and will be fulfilled by the lead contact, Feng Chen (chenfeng5@mail.sysu.edu.cn).
Materials availability
This study did not generate new materials.
Method details
The definition of viral virulence among human respiratory viruses
To define viral virulence, we compared the symptoms and mortality caused by the infections of human respiratory viruses. In details, most of HAdVs and HRVs are usually associated with upper respiratory tract disease, such as lower mortality common colds, involving rhinitis, pharyngitis, sneezing, hoarseness, and/or cough (Fendrick et al., 2003; Greenberg, 2011; Jacobs et al., 2013; Zhang et al., 2020). HRSVs may frequently cause serious lower respiratory tract illness and even death in children below the age of five (Azar and Landry, 2018; Tognarelli et al., 2019; Zhang et al., 2020). CH-CoVs (229E, NL63, OC43, and HKU1) are associated with both upper and lower respiratory tract disease, and even death in children and elderly people (Kahn and McIntosh, 2005). NH-CoVs (SARS-CoV, MERS-CoV, and SARS-CoV-2) infections usually result severe lower respiratory tract disease, and all these three viruses have posed great threats to global public health (Choi et al., 2003; Esposito et al., 2020; Jiang et al., 2020; Pillaiyar et al., 2020; Wang et al., 2020).
Upper respiratory tract infections are usually associated with lower mortality and mild symptoms, including common cold, tonsillitis, sinusitis, laryngitis. On the other hand, lower respiratory tract infections usually result higher mortality and server symptoms, including pneumonia, bronchitis, and bronchiolitis. In this work, we defined viral virulence in the category of human respiratory viruses or human coronaviruses. In the first category, HAdVs and HRVs, which usually result upper respiratory tract, were defined as weakly virulent respiratory viruses. On contrast, HRSVs, CH-CoVs, and NH-CoVs were defined as strongly virulent respiratory viruses. In the category of human coronaviruses, NH-CoVs infections usually lead to more severe symptoms comparing to CH-CoVs. Based on that, NH-CoVs were defined as strongly virulent coronaviruses, while CH-CoVs were defined as weakly virulent coronaviruses.
Data sets used in this study
All reference and mutant sequences were downloaded from NCBI Virus (Hatcher et al., 2017). These sequences are derived from HAdVs (HAdV-A, HAdV-B, HAdV-C, HAdV-D, HAdV-E, HAdV-F, and HAdV-G), HRVs (HRV-A, HRV-B, and HRV-C), HRSVs (HRSV-A, HRSV-B), and human coronaviruses (229E, NL63, OC43, HKU1, SARS-CoV, MERS-CoV, and SARS-CoV-2).
The genomes of HAdV-A (GenBank: NC_001460.1), HAdV-B (GenBank: NC_011203.1), HAdV-C (GenBank: NC_001405.1), HAdV-D (GenBank: NC_010956.1), HAdV-E (GenBank: NC_003266.2), HAdV-F (GenBank: NC_001454.1), HAdV-G (GenBank: NC_006879.1), HRV-A (GenBank: NC_038311.1), HRV-B (GenBank: NC_038312.1), and HRV-C (GenBank: EF186077.2), HRSV-A (GenBank: NC_038235.1), HRSV-B (GenBank: NC_001781.1), 229E (GenBank: NC_002645.1), NL63 (GenBank: NC_005831.2), OC43 (GenBank: NC_006213.1), HKU1 (GenBank: NC_006577.2), SARS-CoV (GenBank: NC_004718.3), MERS-CoV (GenBank: NC_019843.3), and SARS-CoV-2 (GenBank: NC_045512.2) were used as references for identifying the coding region of each species, respectively.
Calculation of viral codon frequency
All sequences were aligned to the reference sequence using BLASTN with default parameters (Camacho et al., 2009). Six types of open reading frames were chosen for each gene to maintain the highest amino acid identity with the reference sequence and to avoid including a stop codon until the end. Otherwise, these sequences were manually confirmed and removed. Then, the codon frequencies of each gene were scaled so that all groups of synonymous codons added up to 1 (Chen et al., 2020).
Viral and host CUB calculation
In this work, we used the metric DP, which means the deviation from proportionality of synonymous CUBs between viral genes and the tRNA supply of the host (Chen et al., 2020), to estimate the difference between viral and human CUBs. Specifically, we first calculated the Euclidean distance in synonymous codon usage between the viral genes and the tRNA supply of the host for each of the 18 amino acids with at least two synonymous codons.
Here, ni is the number of synonymous codons for amino acid i, Yij is the fraction of codon j among the synonymous codons for amino acid i in the viral gene, and Xij is the tRNA supply represented by the fraction of codon j among the synonymous codons in the host transcriptome. The DP value of each viral gene is defined as the weighted geometric mean of all 18 Di values.
Phylogenetic independent contrasts
The collection date values of each virus were transformed to numeric using “timeDate” Package in R. The phylogenetic trees of all viruses were directly generated and downloaded from NCBI virus (Hatcher et al., 2017). Phylogenetic independent contrasts (PIC) were calculated in R using normalized DP and collection date inferred by the phylogenetic tree of all genomes.
Quantification and statistical analysis
p values of less than 0.001 that calculated by Wilcoxon rank-sum tests were indicated by ∗∗∗.
Acknowledgments
We thank Yao Liu, Zizhang Li, Wenjing Yang, and Peng Wu for their comments on the manuscript. This work was supported by the National Natural Science Foundation of China (grant number 32000401 to F.C.), National Natural Science Foundation of China (grant numbers 32122022, 31871320 and 81830103 to J.-R. Y.).
Author contributions
J.-R.Y. and F.C. conceived the idea, designed and supervised the study. F.C. acquired and analyzed data. F.C. and J.-R.Y. wrote the paper.
Declaration of interests
The authors declare no conflict of interest.
Published: January 21, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103682.
Contributor Information
Feng Chen, Email: chenfeng5@mail.sysu.edu.cn.
Jian-Rong Yang, Email: yangjianrong@mail.sysu.edu.cn.
Supplemental information
P values of less than 0.001 were indicated by ∗∗∗.
Data and code availability
The present manuscript did not generate any sequencing data and custom software. All the datasets were downloaded from NCBI Virus using the virus names listed in key resource table. Custom R codes were used to analyze the viral genome. All R codes and the final datasets required to reanalyze the data reported in this paper are available from the lead contact upon request. The key codes and datasets are available on GitHub (https://github.com/chenfengokha/CUBofSARS2).
References
- Alansari H., Potgieter L.N.D. Molecular cloning and sequence analysis of the phosphoprotein, nucleocapsid protein, matrix protein and 22K (M2) protein of the ovine respiratory syncytial virus. J.Gen. Virol. 1994;75:3597–3601. doi: 10.1099/0022-1317-75-12-3597. [DOI] [PubMed] [Google Scholar]
- Albers S., Czech A. Exploiting tRNAs to boost virulence. Life. 2016;6:4. doi: 10.3390/life6010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso A.M., Diambra L. SARS-CoV-2 codon usage bias downregulates host expressed genes with similar codon usage. Front. Cell. Dev. Biol. 2020;8:831. doi: 10.3389/fcell.2020.00831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azar M.M., Landry M.L. Detection of influenza A and B viruses and respiratory syncytial virus by use of clinical laboratory improvement amendments of 1988 (clia)-waived point-of-care assays: a paradigm shift to molecular tests. J. Clin. Microbiol. 2018;56:00367–00368. doi: 10.1128/JCM.00367-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahir I., Fromer M., Prat Y., Linial M. Viral adaptation to host: a proteome-based analysis of codon usage and amino acid preferences. Mol. Syst. Biol. 2009;5:13. doi: 10.1038/msb.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boncristiani H.F., Criado M.F., Arruda E. Respiratory viruses. Encycl. Microbiol. Third Edition. 2009:500–518. doi: 10.1016/B978-012373944-5.00314-X. [DOI] [Google Scholar]
- Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden T.L. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F., Wu P., Deng S., Zhang H., Hou Y., Hu Z., Zhang J., Chen X., Yang J.-R. Dissimilation of synonymous codon usage bias in virus–host coevolution due to translational selection. Nat. Ecol. Evol. 2020;5:589–600. doi: 10.1038/s41559-020-1124-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K.W., Chau T.N., Tsang O., Tso E., Chiu M.C., Tong W.L., Lee P.O., Ng T.K., Ng W.F., Lee K.C., et al. Outcomes and prognostic factors in 267 patients with severe acute respiratory syndrome in Hong Kong. Ann. Intern. Med. 2003;139:715–723. doi: 10.7326/0003-4819-139-9-200311040-00005. [DOI] [PubMed] [Google Scholar]
- Esposito S., Noviello S., Pagliano P. Update on treatment of COVID-19: ongoing studies between promising and disappointing results. Infez. Med. 2020;28:198–211. [PubMed] [Google Scholar]
- Fabry C.M., Rosa-Calatrava M., Conway J.F., Zubieta C., Cusack S., Ruigrok R.W., Schoehn G. A quasi-atomic model of human adenovirus type 5 capsid. Embo J. 2005;24:1645–1654. doi: 10.1038/sj.emboj.7600653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendrick A.M., Monto A.S., Nightengale B., Sarnes M. The economic burden of non-influenza-related viral respiratory tract infection in the United States. Arch. Intern. Med. 2003;163:487–494. doi: 10.1001/archinte.163.4.487. [DOI] [PubMed] [Google Scholar]
- Greenberg S.B. Update on rhinovirus and coronavirus infections. Semin. Respir. Crit. Care. 2011;32:433–446. doi: 10.1055/s-0031-1283283. [DOI] [PubMed] [Google Scholar]
- Greenberg S.B. Update on human rhinovirus and coronavirus infections. Semin. Respir. Crit. Care. 2016;37:555–571. doi: 10.1055/s-0036-1584797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatcher E.L., Zhdanov S.A., Bao Y., Blinkova O., Nawrocki E.P., Ostapchuck Y., Schäffer A.A., Brister J.R. Virus variation resource - improved response to emergent viral outbreaks. Nucleic Acids Res. 2017;45:D482–D490. doi: 10.1093/nar/gkw1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Alias X., Benisty H., Schaefer M.H., Serrano L. Translational adaptation of human viruses to the tissues they infect. Cell Rep. 2021;34:108872. doi: 10.1016/j.celrep.2021.108872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs S.E., Lamson D.M., St George K., Walsh T.J. Human rhinoviruses. Clin. Microbiol. Rev. 2013;26:135–162. doi: 10.1128/CMR.00077-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S., Hillyer C., Du L. Neutralizing antibodies against SARS-CoV-2 and other human coronaviruses. Trends Immunol. 2020;41:355–359. doi: 10.1016/j.it.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn J.S., McIntosh K. History and recent advances in coronavirus discovery. Pediatr. Infect. Dis. J. 2005;24:60. doi: 10.1097/01.inf.0000188166.17324.60. [DOI] [PubMed] [Google Scholar]
- Kmiec D., Nchioua R., Sherrill-Mix S., Stürzel C.M., Heusinger E., Braun E., Gondim M.V.P., Hotter D., Sparrer K.M.J., Hahn B.H., et al. CpG frequency in 5' third of the env gene determines sensitivity of primary HIV-1 strains to the zinc-finger antiviral protein. mBio. 2020;11:e02903. doi: 10.1128/mBio.02903-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucks J.B., Nelson D.R., Kudla G.R., Plotkin J.B. Genome landscapes and bacteriophage codon usage. PLoS Comput. Biol. 2008;4:1000001. doi: 10.1371/journal.pcbi.1000001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz P., Kedinger C. Properties of the adenovirus IVa2 gene product, an effector of late-phase-dependent activation of the major late promoter. J. Virol. 1996;70:1396–1405. doi: 10.1128/jvi.70.3.1396-1405.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz P., Rosa-Calatrava M., Kedinger C. The product of the adenovirus intermediate gene IX is a transcriptional activator. J. Virol. 1997;71:5102–5109. doi: 10.1128/jvi.71.7.5102-5109.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean O.A., Lytras S., Weaver S., Singer J.B., Boni M.F., Lemey P., Kosakovsky Pond S.L., Robertson D.L. Natural selection in the evolution of SARS-CoV-2 in bats created a generalist virus and highly capable human pathogen. PLoS Biol. 2021;19:e3001115. doi: 10.1371/journal.pbio.3001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevins J.R. Regulation of early adenovirus gene expression. Microbiol. Rev. 1987;51:419–430. doi: 10.1128/mr.51.4.419-430.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillaiyar T., Meenakshisundaram S., Manickam M. Recent discovery and development of inhibitors targeting coronaviruses. Drug Discov. Today. 2020;25:668–688. doi: 10.1016/j.drudis.2020.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice A.M., Castillo Morales A., Ho A.T., Mordstein C., Mühlhausen S., Watson S., Cano L., Young B., Kudla G., Hurst L.D. Evidence for strong mutation bias toward, and selection against, U content in SARS-CoV-2: implications for vaccine design. Mol. Biol. Evol. 2021;38:67–83. doi: 10.1093/molbev/msaa188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L., Shen X., Murphy R.W., Shen Y. The adaptation of codon usage of +ssRNA viruses to their hosts. Infect. Genet. Evol. 2018;63:175–179. doi: 10.1016/j.meegid.2018.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tognarelli E.I., Bueno S.M., González P.A. Immune-modulation by the human respiratory syncytial virus: focus on dendritic cells. Front. Immunol. 2019;10:810. doi: 10.3389/fimmu.2019.00810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellinga J., Van der Heijdt S., Hoeben R.C. The adenovirus capsid: major progress in minor proteins. J. Gen. Virol. 2005;86:1581–1588. doi: 10.1099/vir.0.80877-0. [DOI] [PubMed] [Google Scholar]
- Wang L., Wang Y., Ye D., Liu Q. Review of the 2019 novel coronavirus (SARS-CoV-2) based on current evidence. Int. J. Antimicrob. Agents. 2020;55:105948. doi: 10.1016/j.ijantimicag.2020.105948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N., Wang L., Deng X., Liang R., Su M., He C., Hu L., Su Y., Ren J., Yu F., et al. Recent advances in the detection of respiratory virus infection in humans. J. Med. Virol. 2020;92:408–417. doi: 10.1002/jmv.25674. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
P values of less than 0.001 were indicated by ∗∗∗.
Data Availability Statement
The present manuscript did not generate any sequencing data and custom software. All the datasets were downloaded from NCBI Virus using the virus names listed in key resource table. Custom R codes were used to analyze the viral genome. All R codes and the final datasets required to reanalyze the data reported in this paper are available from the lead contact upon request. The key codes and datasets are available on GitHub (https://github.com/chenfengokha/CUBofSARS2).